
Activation of Pink1/Parkin-mediated mitochondrial autophagy alleviates exertional heat stroke-induced acute lung injury in rats
Abstract
OBJECTIVE:
To investigate the role of Pink1/Parkin-mediated mitochondrial autophagy in exertional heat stroke-induced acute lung injury in rats.
METHODS:
Sixty SD rats were divided into four groups: normal group (CON group), normal Parkin overexpression group (CON + Parkin group), exertional heat stroke group (EHS group), and exertional heat stroke Parkin overexpression group (EHS + Parkin group). Adeno-associated virus carrying the Parkin gene was intravenously injected into the rats to overexpress Parkin in the lung tissue. An exertional heat stroke rat model was established, and survival curves were plotted. Lung micro-CT was performed, and lung coefficient and pulmonary microvascular permeability were measured.
RESULTS:
Compared with the EHS group, the survival rate of rats in the EHS + Parkin overexpression group was significantly increased, lung coefficient and pulmonary microvascular permeability were reduced, and pathological changes such as exudation and consolidation were significantly reduced. The levels of inflammatory factors IL-6, IL-1β, TNF- α, and ROS were significantly decreased; the degree of mitochondrial swelling in type II alveolar epithelial cells was reduced, and no vacuolization was observed. Lung tissue apoptosis was reduced, and the colocalization fluorescence of Pink1 and Parkin, as well as LC3 and Tom20, were increased. The expression of Parkin and LC3-II/LC3-I ratio in lung tissue were both increased, while the expression of P62, Pink1, MFN2, and PTEN-L was decreased.
CONCLUSION:
Impairment of Pink1/Parkin-mediated mitochondrial autophagy function is one of the mechanisms of exertional heat stroke-induced acute lung injury in rats. Activation of the Pink1/Parkin pathway can alleviate acute lung injury caused by exertional heat stroke.
1Introduction
Climate change has led to an increase in global warming year by year, the frequency and duration of extreme hot weather, and an increase in the incidence of heat stroke year by year [1]. Heat stroke (HS) is a heat illness associated with high temperature and humidity and has a high rate of disability and death if not treated effectively. It is divided into “classic heat stroke” and “exertional heat stroke” depending on the cause of heat [2]. Exertional heat stroke (EHS) occurs in people who work and train outdoors in summer and has a high mortality rate [3]. It is characterized by a core body temperature > 40°C and is associated with central nervous system dysfunction, which progresses to multi-organ impairment in severe cases [2]. The lungs, as an important organ for regulating heat dissipation in the body, are susceptible to damage from heat shock, which leads to altered ventilation and the development of acute lung injury (ALI)/or acute respiratory distress syndrome (ARDS) [4, 5]. Thermogenic organ damage is associated with intracellular mitochondrial damage [5]. Mitochondria are important organelles in eukaryotic cells and are responsible for energy synthesis and metabolism, cell differentiation, and apoptosis, and can be referred to as the “motor of the cell” [6]. Studies have confirmed that heat shock can directly cause damage to mitochondria and activate apoptosis [6, 7]. In addition, damage to mitochondria generates large amounts of reactive oxygen species (ROS), which enhance intracellular oxidative stress and induce an inflammatory response [8]. In turn, oxidative stress and inflammatory responses further erode telomeres and damage mitochondria [9], thus creating a vicious cycle that amplifies the degree of oxidative stress in a cascade and eventually generates systemic inflammatory response syndrome (SIRS), leading to organ failure [10]. Therefore, effective and selective clearance of damaged mitochondria is essential to adapt to the current environment of the organism and to maintain the health of the body [11].
Mitochondrial autophagy is the process by which damaged, aged, and dysfunctional mitochondria can be recognized by specific autophagic vesicles and then selectively transported to lysosomes to complete degradation. Mitochondrial autophagy plays a crucial role in controlling mitochondrial quality and maintaining mitochondrial dynamic homeostasis [12]. Enhancement of mitochondrial autophagy facilitates the clearance of dysfunctional mitochondria and prevents excessive cellular damage. The Pink1/Parkin pathway is a classical pathway that regulates mitochondrial autophagy. This pathway is involved in the development of several diseases. It was found that Parkin translocated from the cytoplasm to mitochondria during sepsis and induced the onset of the mitochondrial autophagy process. Animal experiments confirmed that in a mouse model of sepsis, Pink1 and Parkin knockout mice had a greater degree of intracellular mitochondrial damage and a higher number of organ failures and mortality [13–15]. However, the role of the Pink1/Parkin pathway and mitochondrial autophagy in pulmonary injury in heat stroke is still lacking in research. Since the pathophysiological mechanism of exertional heat stroke has similarities with sepsis, both of which have mitochondrial damage and uncontrolled SIRS, this study investigates the role of Pink1/Parkin pathway in EHS lung injury by establishing an EHS rat model and observing the effect of Parkin overexpression on HS rat lung tissue and provides a corresponding theoretical basis for the rescue and treatment of EHS lung injury.
2Materials and methods
2.1Materials
2.1.1Animals
Sixty healthy male SD rats, weighing 300–350 g, 8-week-old, were purchased from Spelford (Beijing) Biotechnology Co. Ltd. (Beijing, China). They were maintained in the experimental animal center of the PLA General Hospital with a room temperature of 25°C, 50% –60% relative humidity, and a 12 h light-dark cycle. The experimental animal protocol was approved by the Animal Ethics Committee of the Eighth Medical Center of the PLA General Hospital with approval number 20208141030.
2.1.2Main instruments and reagents
The six-channel small animal runner (XR-PT-10A, Shanghai Xinsoft Information Technology Co., Ltd.) was placed in a transparent simulated high temperature and high humidity environment chamber [13], which can precisely control the temperature and humidity inside the chamber and maintain them constantly. Adeno-associated virus (AAV-Lung-Parkin) with Parkin gene was purchased from OBiO Technology (Shanghai, China); Evans blue (EB) was purchased from MCE, USA; rabbit anti-glyceraldehyde phosphate dehydrogenase (GPHD) was purchased from MCE, USA. The rabbit anti-rat glyceraldehyde phosphate dehydrogenase (GAPDH) monoclonal antibody, rabbit anti-rat P62 monoclonal antibody, mouse anti-rat Tom20 monoclonal antibody, rabbit anti-rat Pink1 polyclonal antibody, rabbit anti-rat mitofusin-2 (MFN2) polyclonal antibody were purchased from Abcam, UK. The rabbit anti-rat microtubule-associated protein 1 light chain 3 (LC3) polyclonal antibody, mouse anti-rat Parkin monoclonal antibody, mouse anti-rat PTEN polyclonal antibody was purchased from CST, US; the mouse anti-rat PTEN-α monoclonal antibody was purchased from CST, US; TdT-mediated dUTP nick-end labeling (TUNEL) kit was purchased from Roche, Switzerland; ELISA reagents for IL-1β and IL-6 were purchased from CST, US. TNF-α ELISA kit was purchased from Abcam (UK); the ROS kit was purchased from Shanghai Yaji Biotechnology Co. Ltd., Shanghai, China. All other reagents were domestic analytical purity.
2.2Methods
2.2.1Animal grouping and model construction
Sixty healthy male SD rats were divided into 4 groups according to the random number table method, including the control group (CON group), normal Parkin overexpression group (CON + Parkin group), heat stroke group (HS group), and heat stroke Parkin overexpression group (HS + Parkin group), (n = 15). Before HS modeling, experimental rats were placed in a thermal environmental chamber and acclimatized at room temperature (24–26°C) using the step training method for a period of 7 d. In rats overexpressing Parkin in the lung tissue of the experimental group, we used tail vein injection of AAV-Lung-Parkin (5×1011 μg) 300 NL [16]. After 4 weeks of injection, we performed a heat stress experiment and closely observed the consciousness and mental changes in the rats [17]. In this experiment, we did not perform mRNA level assays to verify the expression of Parkin in lung tissue.
For the construction details of Parkin-related viral vectors, we used the following method of construction. First, the Parkin gene fragment was cloned into a viral vector, and then the vector was amplified in cells to finally prepare a viral vehicle containing the Parkin gene. During the injection process, we introduced the viral vector into the rats by tail vein injection to achieve overexpression of Parkin in lung tissue. As for the frequency of Parkin vector injection, it depended on the specific experimental situation. Generally, the expression of the target genes can be observed after one injection. However, to ensure the accuracy and reproducibility of the experiment, it was recommended to perform multiple observations and experiments within an appropriate period after injection.
After 7 d of acclimatization, rats were injected with AAV-Lung-Parkin (5×1011 μg) 300 NL via tail vein to overexpress Parkin in rat lung tissue [16], and heat stress experiments were performed 4 weeks later. Rats were fasted for 12 h before the heat stress experiments, and drinking water was not restricted. The rats were weighed 30 min before the start of the modeling and water intake was prohibited (the rats were stimulated to defecate before the operation by gently pressing their lower abdomen with our finger). When the temperature of the experimental chamber reached 39.5±0.3°C and the relative humidity was 55% ±5%, the rats were put into the running platform and started to run at an initial speed of 5 m/min (the slope was 0), and the speed was increased by 1 m/min every 2 min, and then the speed was increased to 15 m/min for 20 min and kept at a constant speed until fatigue occurred (the rats could not continue to run even after appropriate repulsion). The consciousness and mental changes of the rats were closely observed throughout the experiment [17]. After the rats reached the diagnostic criteria for EHS, they were removed from the thermal chamber, heat exposure was stopped, weighed, and cooled naturally at room temperature (24–26°C), and their mental status was continued to be monitored until 5 h after modeling. The diagnostic criteria for EHS were that the rats showed signs of central nervous system dysfunction, i.e., no voluntary activity for more than 5 s (mild painless stimulation could not drive the animals to crawl or change position).
2.2.2Observation of 5-h survival rate of rats
Sixty rats were randomly selected from each group, and their survival was observed and recorded at 0, 1, 2, 3, 4, and 5 h after modeling and survival curves were plotted to calculate the survival rate.
2.2.3Micro-CT of rat lungs
At 5 h after modeling, the rats were anesthetized by isoflurane inhalation (2 ml/100 g, 200 mg/kg, induced concentration 2% –2.5%, maintained concentration 1% –1.5%), and the imaging of rat lungs was recorded using a Micro-CT instrument (Aloka Latheta LCT-200, Hitachi, Japan) with the following measurement parameters: 120 s of film time, 90 kV of voltage, 160μA of current, and 40μm of photo pixels.
2.2.4Histopathological observation of rat lung
First, pentobarbital sodium was used at a concentration of 3 mg/ml with a concentration of 30–50 mg/kg, and then 1 ml of liquid medicine was injected into rats by intravenous injection so that they were decapitated under anesthesia. After the rats were killed, a small piece of tissue from the lower lobe of the right lung was taken for observation, and then it was cut and soaked in 4% paraformaldehyde solution for fixation, dehydrated in gradient ethanol, sectioned after conventional paraffin embedding (5μm), stained with hematoxylin-eosin (HE) after dewaxing, and dehydrated to observe histopathological changes under light microscopy. Ten fields of view were randomly selected under a DM4000B light microscope (×200). The lung histopathological damage was scored concerning the observation and analysis of Hong et al. criteria [18]. The scoring items included alveolar cavity congestion, neutrophil infiltration, fibrin exudation, and alveolar septal widening, and the score of each item was divided into 0, 1, 2, and 3 points according to the degree of damage, which were no damage, mild damage, moderate and severe damage, respectively, and the mean value was calculated. In addition, the mean of the scores of the HE-stained sections for the degree of pulmonary edema was calculated according to the degree of edema in the interstitium and alveoli, with scores of 0, 1, 2, and 3 for none, mild, moderate and severe, respectively [19].
2.2.5LW/BW
LW/BW is an index to assess the severity of pulmonary edema, where LW denotes wet lung weight and BW denotes body weight. Higher values of LW/BW indicate more severe pulmonary edema. The rats were given a 12-h fasting and allowed to drink unlimited water. Before the start of the experiment, the rats were weighed and defecated to stimulate them, and their water intake was prohibited. When the temperature of the experimental chamber reached 39.5±0.3°C and the relative humidity was 55% ±5%, the rats were put into the running table. The rats started to run at an initial speed of 5 m/min, and the speed was increased by 1 m/min every 2 min, and then the speed was increased to 15 m/min in 20 min and kept at a constant speed until they were fatigued (unable to continue running after appropriate repulsion) and stopped running. The consciousness and mental changes of the rats were closely observed throughout the experiment. At the end of the experiment, the rats were executed, and their lung tissues were removed and measured for LW/BW. The lung tissues were gently dried with absorbent paper and then weighed and recorded as wet weight. Afterward, the lung tissues were dried at 70°C to a stable weight, weighed again, and the dry weight was recorded.
The formula for calculating LW/BW is: LW/BW = (wet weight –dry weight) / body weight.
2.2.6Measurement of pulmonary vascular permeability in rats
The lung tissue permeability of rats was measured using the EB dye exudation technique [20]. EB was dissolved in saline in a 2% solution and injected from the tail vein of rats at 2 mL/kg. The EB dye was observed to turn blue in both ears and eyes of the rats, and it was determined that the EB dye had been uniformly distributed in the rats. The EB dye was circulated in vivo for 2 h, the right ear was cut open after thoracotomy, and the pulmonary circulation was flushed with 10 mL of phosphate-buffered saline (PBS) via the left ventricle. The whole lung tissue was taken and soaked in 2 mL formamide solution (10 mL/kg), then incubated with formamide at 60°C for 16 h. After all the pigments were extracted from the lung tissue, the tissue was removed, and the supernatant was centrifuged at 7,000×g for 10 min, the absorbance (A) was measured, and the EB content in the tissue was calculated.
2.2.7ELISA method for the determination of IL-6, IL-1β, TNF-α and ROS in lung tissues
The contents of cytokines, IL-6, IL-1β, TNF-α, and ROS in lung tissues were detected by double antibody sandwich ELISA method using rat IL-6, IL-1β, TNF-α, and ROS ELISA kits, and the instructions of the ELISA kits were strictly followed.
2.2.8Mitochondrial morphology in type II epithelial cells of lung tissue by transmission electron microscopy
The tissue was fixed with 2.5% glutaraldehyde solution and rinsed three times with PBS for 15 min each time. The tissue was fixed again with 1% osmium acid for 2 h and rinsed through PBS three times. The tissue was dehydrated in an ethanol gradient, embedded and polymerized to make resin blocks, and then sectioned by an ultra-thin sectioning machine and double stained with uranyl acetate –lead citrate (3%) to make copper mesh sections, which were observed and analyzed under transmission electron microscope (JEM-1230) (×30000).
2.2.9Detection of apoptosis by TUNEL method
Paraffin sections of lung tissues were taken, dewaxed with gradient ethanol, rinsed with tap water, dripped with protease working solution, incubated at 37°C for 20 min, washed with PBS; dripped with TUNEL solution, incubated at 37°C for 1 h, washed with PBS; dripped with TUNEL solution again, incubated at room temperature for 20 min, rinsed with PBS and then stained with diaminobenzidine (DAB), re-stained with hematoxylin, dehydrated with gradient ethanol. The cells were transparent, sealed, and observed under a light microscope. Positive cells, i.e., apoptotic cells, were brownish-yellow, and normal cells were blue. Image J image analysis software was used to analyze the images of each group of TUNEL-stained sections, and five fields of view (200×) were randomly selected under the microscope of each section to calculate the apoptosis index. Apoptosis index = number of positive cells/total number of cells×100%.
2.2.10Western blot
The protein concentration was determined by the diquinolinic acid (BCA) kit after protein extraction from the supernatant, and the protein samples were electrophoresed in an electrophoresis apparatus, followed by electrophoresis, closed by skim milk powder solution, and the antibody working solution was prepared according to the instructions (Pink1, Parkin, P62 and LC3 antibodies were all expressed at 1:2). The primary antibody was incubated overnight at 4°C in a shaker, and the corresponding secondary antibody (1:5000) was incubated at room temperature for 1 h. The signal intensity was detected by the ECL chemiluminescence imaging system. The band optical density values were analyzed by ImagePro Plus 6. 0 software and the relative expression was calculated by the ratio of absorbance with GAPDH internal reference.
2.2.11Immunofluorescence detection
Paraffin sections of rat lung tissue were dewaxed with gradient ethanol, dehydrated, blown dry, washed three times with PBS, and 1% Triton-X-100 mixed with 2% sheep serum was closed at room temperature for 1 h. After washing with PBS, the following I antibodies [Pink1 (1:50), Parkin (1:50), LC3 (1:50 and Tom20 (1:100)] were added and incubated overnight at 4°C. After washing I antibody, goat anti-rabbit fluorescent II antibody (DyLight 594, 1:500) or goat anti-mouse fluorescent II antibody (DyLight 488, 1:500) was added and incubated at room temperature for 30 min. After washing with PBS, the nuclei were stained with DAPI. The co-staining of Pink1 with Parkin and LC3 with om20 in lung tissue was observed by confocal microscopy (Olympus FV3000).
2.3Statistical processing
GraphPad Prism 9 software was used for statistical analysis. Data were expressed as mean±standard deviation (SD). One-way ANOVA was used for comparison between multiple groups, and the SNK-q test was used for comparison between groups. Differences were considered statistically significant at P < 0.05.
3Results
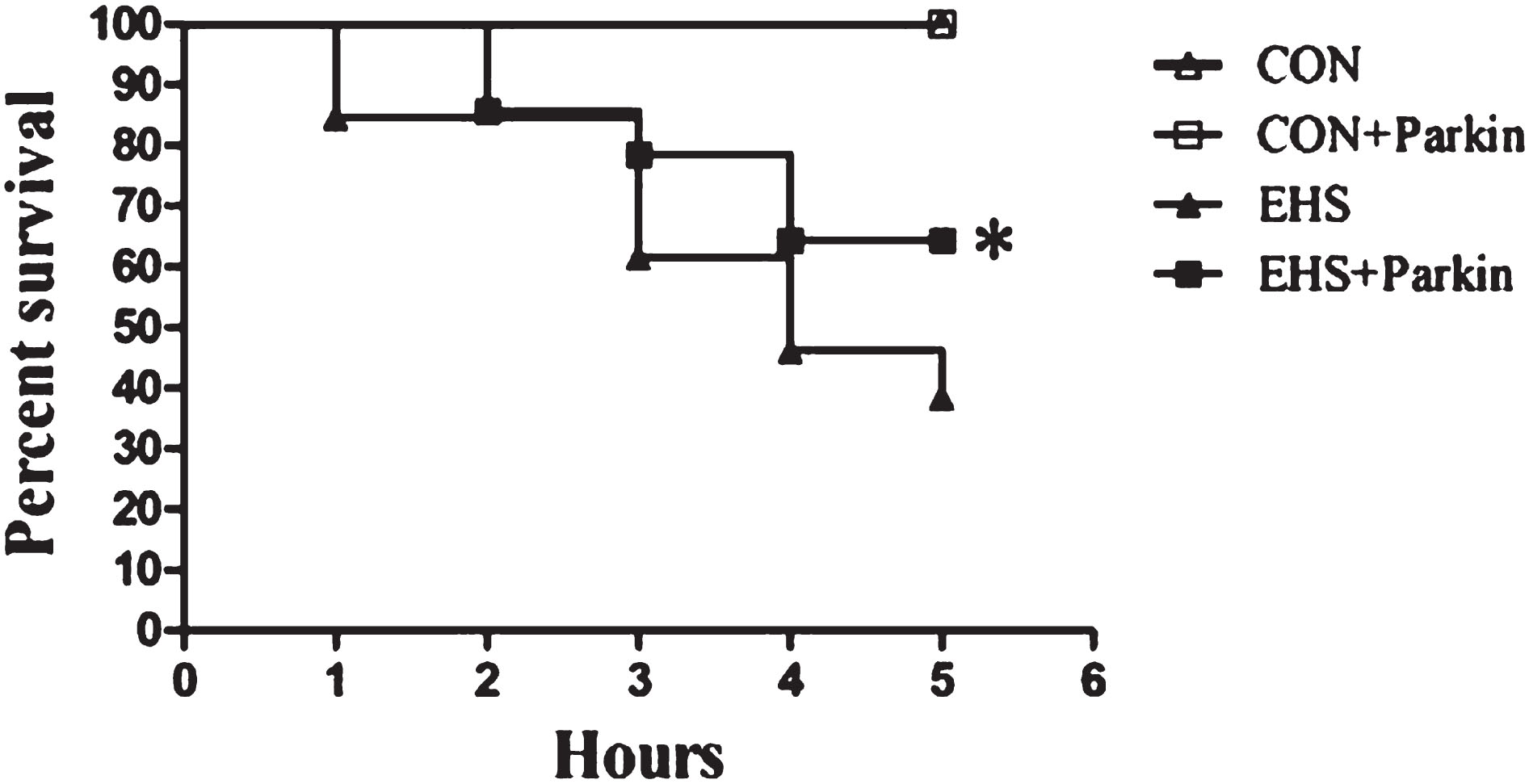
3.1Effect of Parkin overexpression on the survival rate of EHS rats
As shown in Fig. 1, the survival rate of rats in the EHS group was about 87% (13/15) at 1 h, 53% (8/15) at 3 h, and 33% (5/15) at 5 h after heat shock. Compared with them, the survival rates of rats in the EHS + Parkin group were significantly higher (P < 0.05), except for the 2-h survival rate, which was essentially the same in both groups. there was no difference in the survival rates of rats in the CON group and CON + Parkin group.
Fig. 1
The over-expression of Parkin increases the survival rate of EHS rats (n = 15).
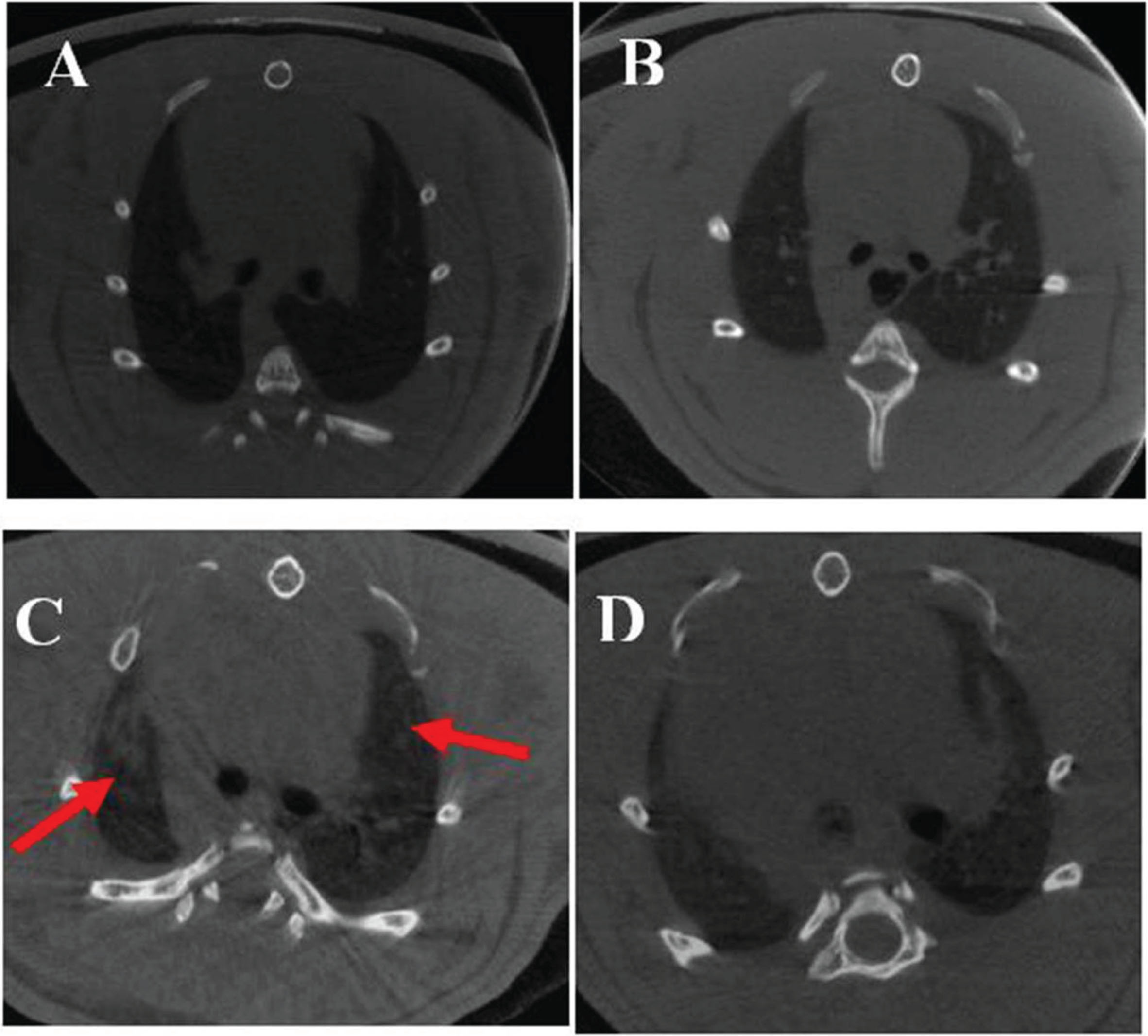
3.2Effect of Parkin overexpression on lung imaging in EHS rats
Mi.cro-CT of rat lungs showed patchy exudate and increased lung texture in the EHS group (shown by red arrows in Fig. 2). There was no significant difference in the lung imaging changes between the CON and CON + Parkin overexpression groups.
Fig. 2
Micro-CT images of rats in each group. A: CON group; B: CON + Parkin group; C: EHS group; D: HS + Parkin group red arrow: patchy exudation.
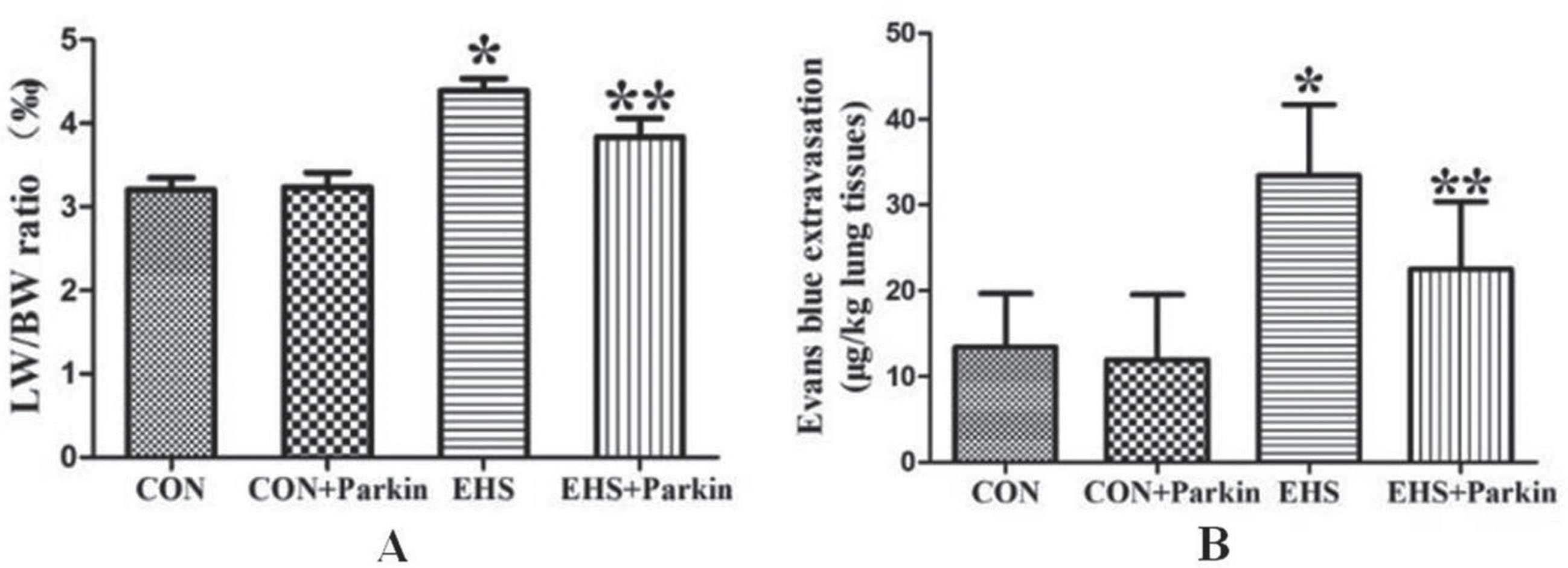
3.3Effect of Parkin overexpression on lung coefficient and pulmonary vascular permeability in EHS rats
Compared with CON, the lung coefficient and pulmonary vascular permeability were significantly higher in the EHS group, and the difference was statistically significant (P < 0.05, Fig. 3). In contrast, the lung coefficient and pulmonary vascular permeability of the rats in the heat stroke Parkin overexpression group decreased significantly compared with those in the EHS group, and the difference was statistically significant (P < 0.05). there was no significant difference in the lung coefficient and pulmonary vascular permeability in the CON group compared with the CON + Parkin group (P > 0.05).
Fig. 3
The over-expression of Parkin decreased the pulmonary index and vascular permeability of HS rats (N = 5). A: CON group; B: CON + Parkin group; C: EHS group; D: EHS + Parkin group.
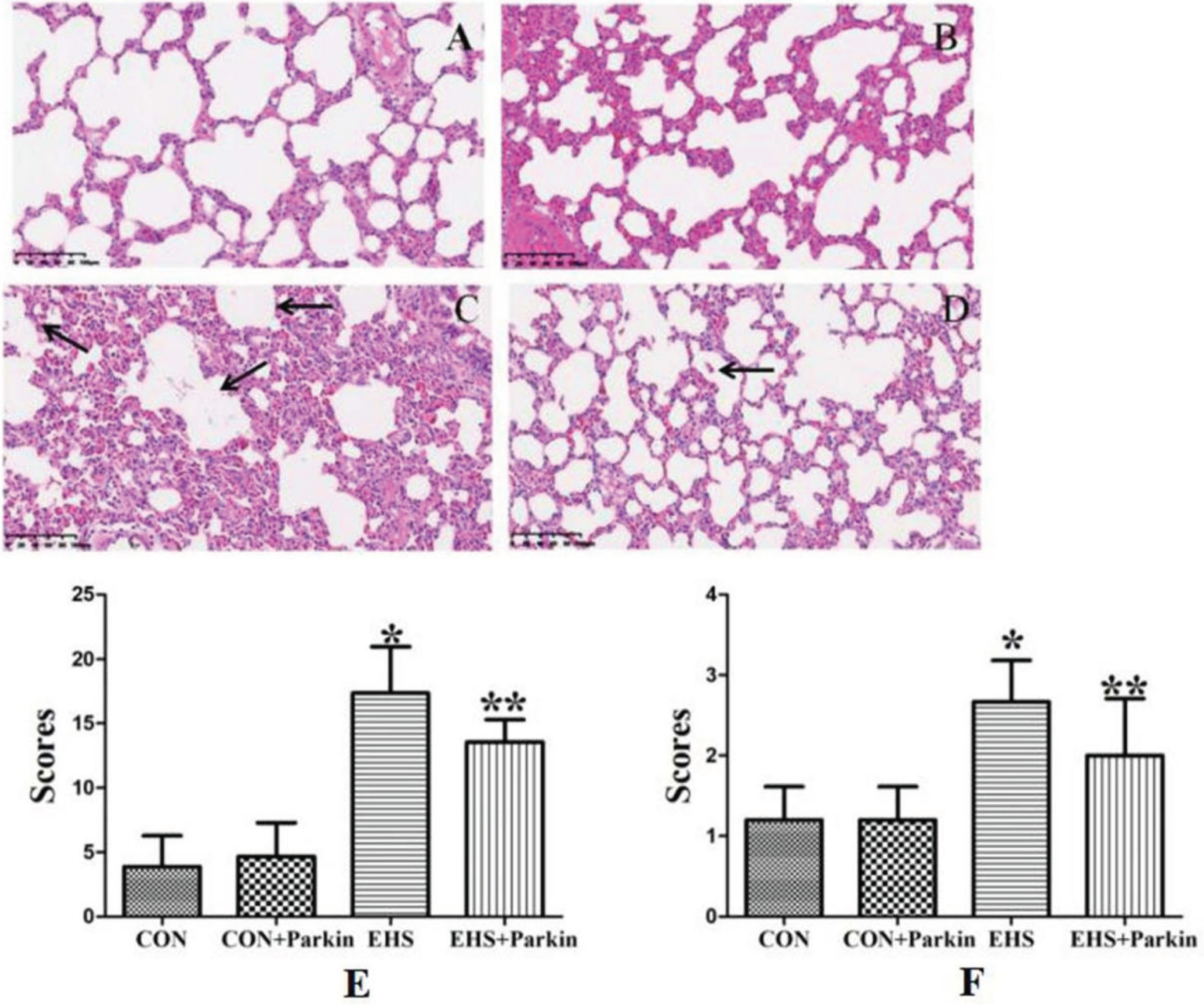
3.4Histological pathological changes of the lung in EHS rats by Parkin overexpression
As shown in Fig. 4, rats in the CON and CON + Parkin overexpression groups had clear lung tissue structure, smooth alveolar walls, and no fluid exudation from the alveolar cavity; rats in the HS group showed alveolar collapse, a large number of erythrocytes, inflammatory cells, and plasma-like material exuded from the alveolar cavity, and a significant increase in lung pathology score and pulmonary edema score (P < 0.05); compared with the HS group, the alveolar collapse was reduced in the heat stroke Parkin overexpression group, the degree of inflammatory infiltration was low, no significant exudate material was seen in the alveoli, and the pulmonary pathology score and pulmonary edema score were significantly lower compared with the HS group (P < 0.05).
Fig. 4
The Parkin over-expression rats resist the HS-induced pathological changes of the lung (N = 5). A: CON group; B: CON + Parkin group; C: EHS group; D: EHS + Parkin group. Arrows indicate edema. E: The histologic scoring of lung injury; F: The histologic scoring of lung edema.
3.5Effect of Parkin overexpression on the levels of IL-6, IL-1β, TNF-α, and ROS in lung tissues of rats with pyrexia
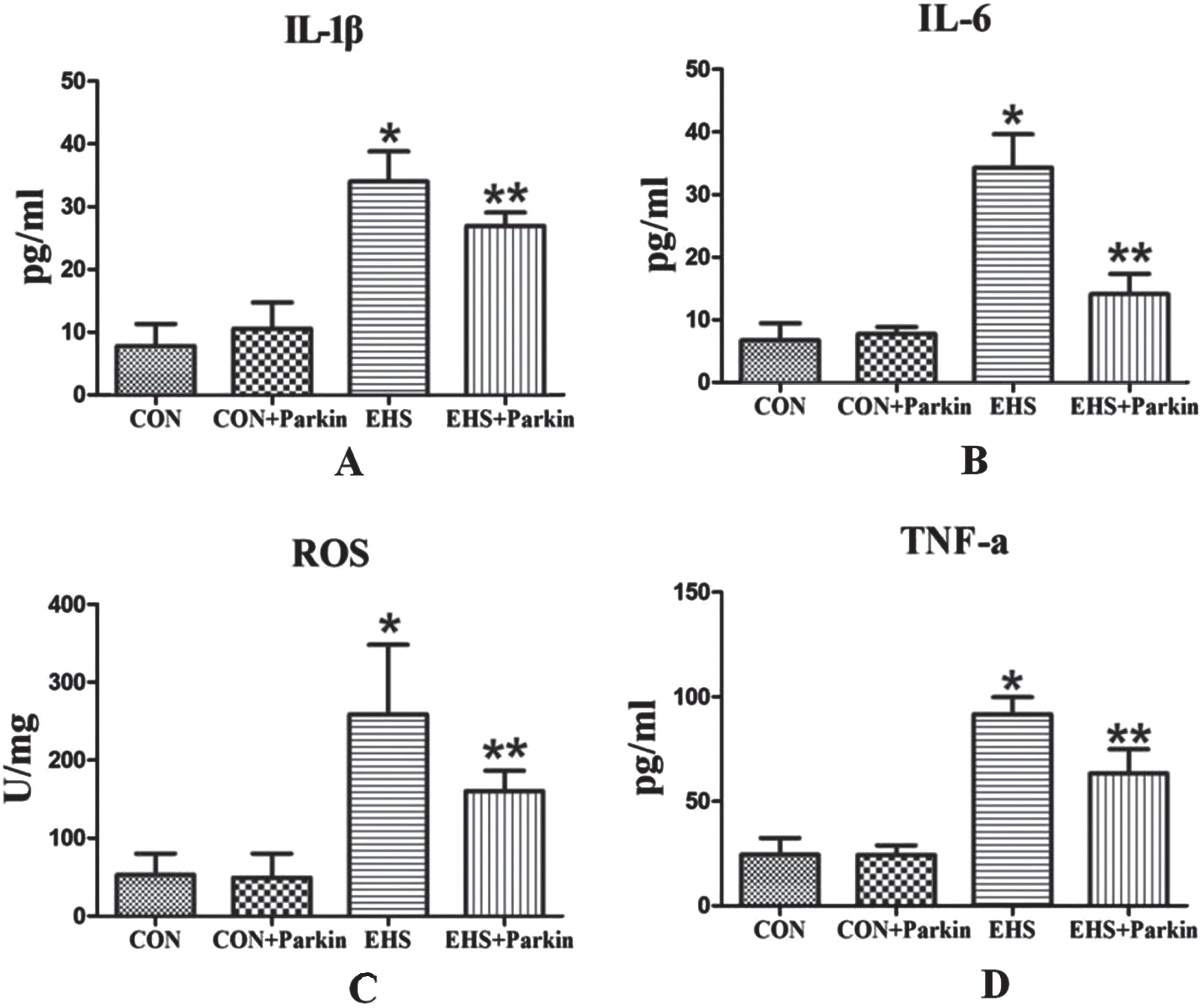
The levels of IL-6, IL-1β, TNF-α, and ROS were significantly increased in the lung tissues of rats in the EHS group compared with the CON group (P < 0.05, Fig. 5), while the levels of all the above cytokines were significantly decreased in the lung tissues of rats in the EHS + Parkin group compared with the EHS group (P < 0.05).
Fig. 5
The Parkin over-expression rats attenuated the HS-induced increasing level of IL-1β, IL-6, TNF-α, and ROS in the lung (N = 5). A: IL-1β; B: IL-6; C: ROS; D: TNF-α. *P < 0.05 vs Control group; **P < 0.05 vs EHS group.
3.6Effect of Parkin overexpression on mitochondrial morphology in lung epithelial cells
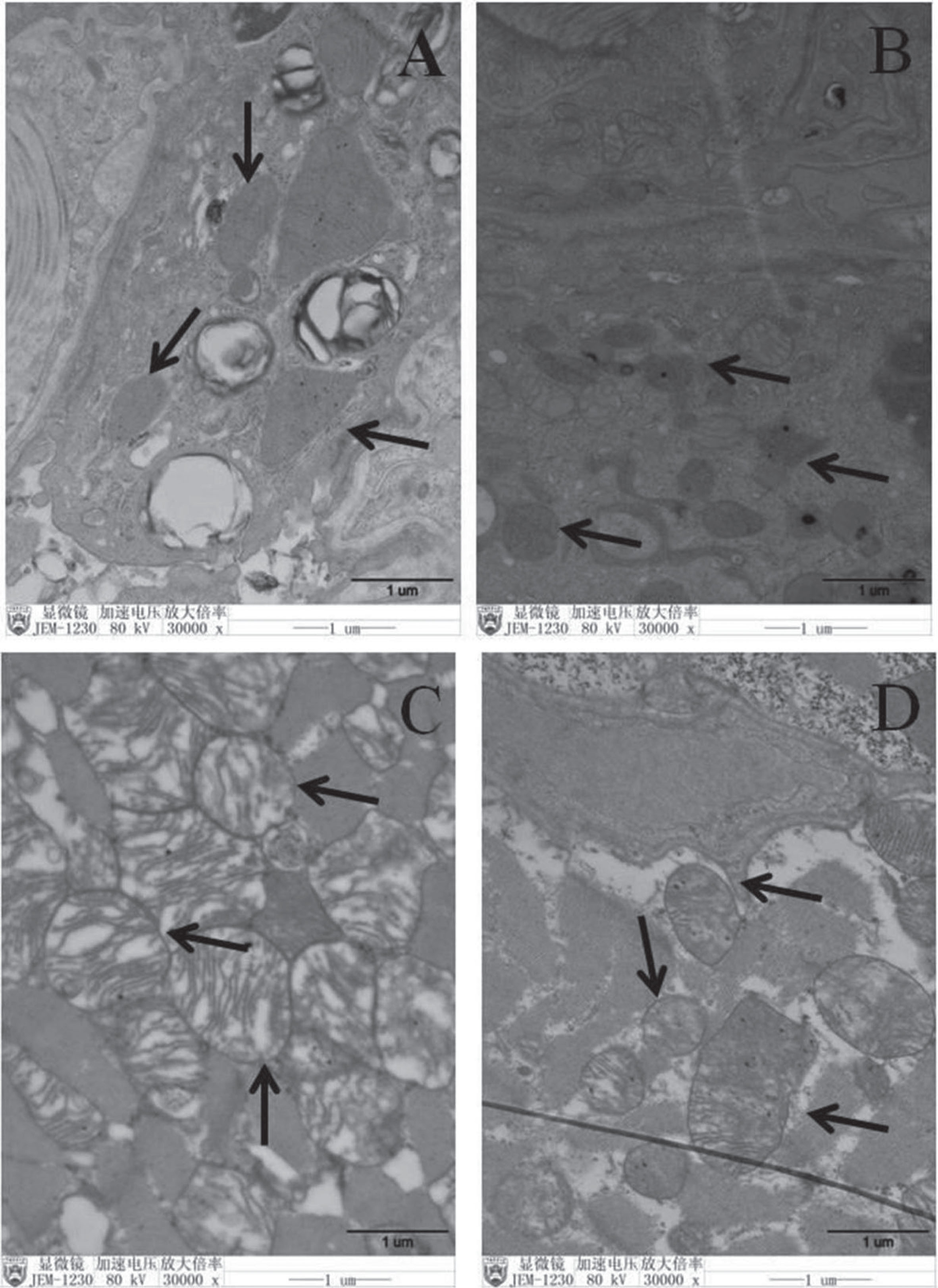
The mitochondrial morphology in the lung type II epithelial cells of the CON and CON + Parkin groups was regular, and the mitochondrial cristae were tightly arranged and visible, while the mitochondria in the lung type II epithelial cells of the EHS group were swollen, the mitochondrial cristae were broken, and most of the mitochondria were vacuolated. In contrast, the mitochondria in the lung type II epithelial cells of EHS + Parkin rats were slightly enlarged, the swelling of mitochondria was less than that in the EHS group, and the mitochondrial cristae were intact and did not show vacuolization (Fig. 6).
Fig. 6
Pulmonary mitochondrial injury was attenuated to a greater extent morphologically in HS + Parkin group rats. A: CON group; B: CON + Parkin group; C: EHS group; D: EHS + Parkin group. Arrows indicate mitochondrion.
3.7Effect of Parkin overexpression on apoptosis in rat lung tissues
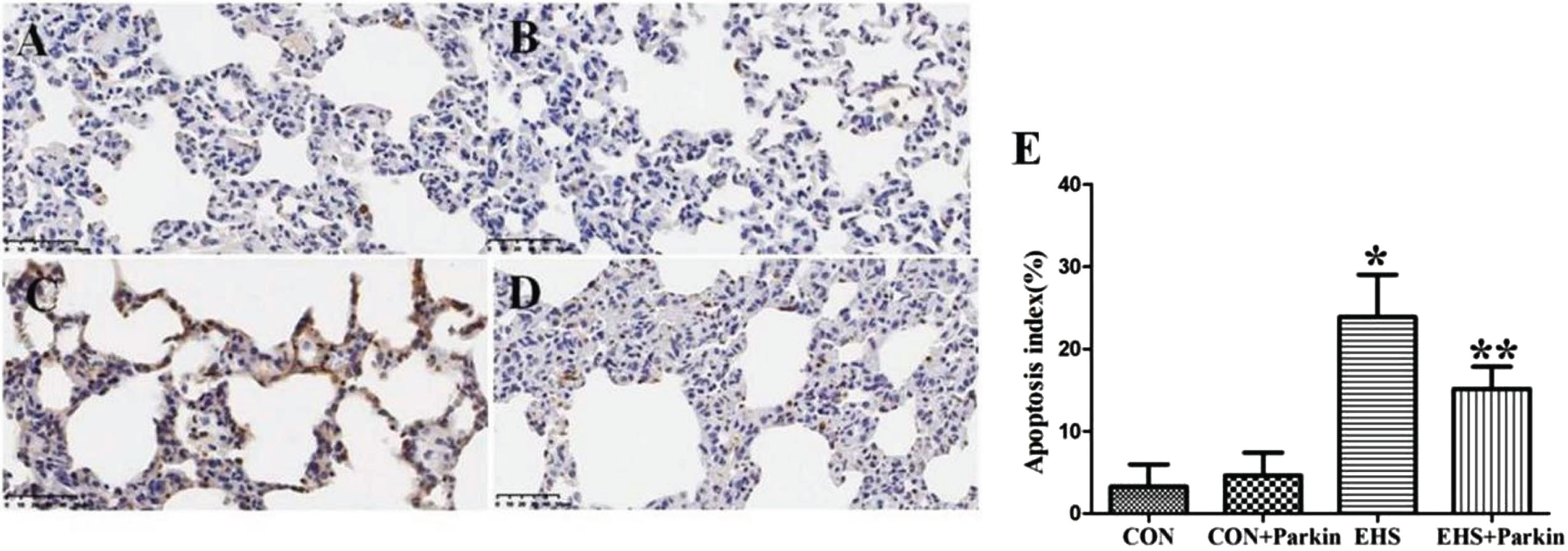
Compared with the CON group, the number of apoptotic cells in the lung tissues of rats in the EHS group increased and the apoptotic index was significantly higher (P < 0.05); compared with the EHS group, the apoptotic cells in the lung tissues of rats in the EHS + Parkin group were significantly reduced and the apoptotic index was significantly lower (P < 0.05). there was no significant difference in apoptosis in the lung tissues of the CON and CON + Parkin groups (Fig. 7).
Fig. 7
Pulmonary apoptosis was attenuated to a greater extent in the EHS + Parkin group (N = 5). A: CON group; B: CON + Parkin group; C: EHS group; D: EHS + Parkin group. E: Apoptosis index. *P < 0.05 vs Control group; **P < 0.05 vs EHS group.
3.8Effect of Parkin overexpression on the level of mitochondrial autophagy in lung tissues of rats with heat stroke
As shown in Fig. 8, the expression of the Parkin protein in the lung tissues of both CON + Parkin and EHS + Parkin groups was significantly increased after tail vein injection of the adeno-associated virus carrying the Parkin gene. Compared with the CON group, the expression of Parkin was increased in the lung tissues of the HS group (P < 0.05), the autophagy level marker LC3-II/LC3-I ratio was significantly lower (P < 0.05), and the expression of P62 was increased (P < 0.05). Compared with the EHS group, the LC3-II/LC3-I ratio was increased and P62 expression was decreased in the lung tissues of EHS + Parkin rats (P < 0.05).
Fig. 8
Parkin over-expression rats exhibited an increasing level of mitochondrial autophagy in their lung (N = 5). A: The protein levels of Parkin, P62, and LC3 in lung tissues were performed by immunoblotting. B-D: The statistical analysis of Parkin, P62 expression and LC3-I/LC3-IIratio. *P < 0.05 vs Control group; **P < 0.05 vs EHS group. E: Immunofluorescence staining against LC3 and Tom 20 was performed and observed by fluorescent microscopy. LC3 (green), Tom20 (red), The co-staining (orange) of LC3 and Tom 20 was showed by a white arrow, bar = 50μm.
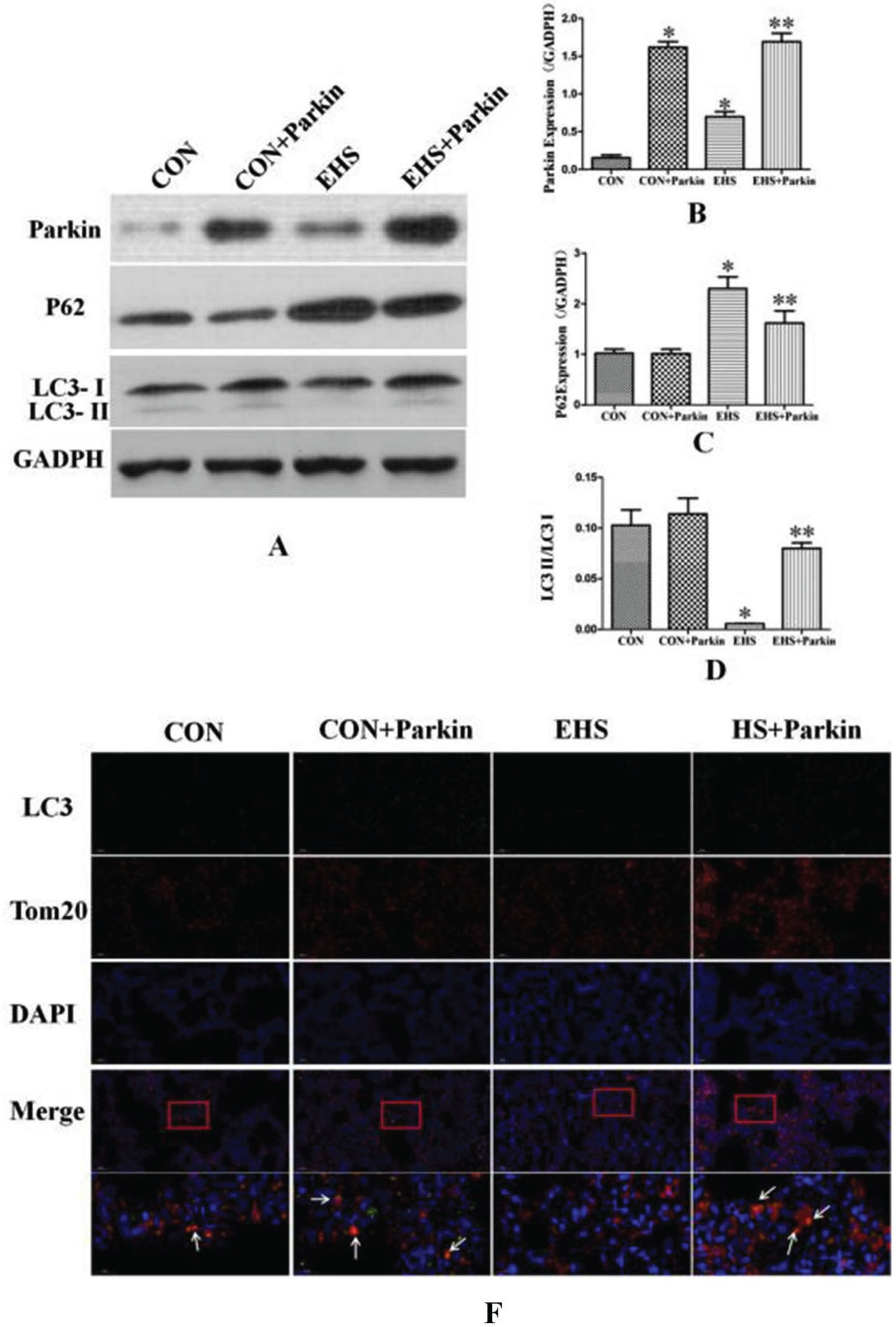
Immunofluorescence results showed that the LC3 fluorescence intensity (green fluorescence) and the co-localization of LC3 with Tom20 (red fluorescence) in lung tissues of rats in the EHS group was reduced (orange fluorescence) compared with that in the CON group; the LC3 fluorescence intensity (green fluorescence) was significantly enhanced in lung tissues of rats in the EHS + Parkin group compared with that in the EHS group, and the co-localization of LC3 with Tom20 was also significantly enhanced. The LC3-II/LC3-I ratio was slightly increased in the lung tissues of the rats in the
expression of P62 was not significantly different, and the LC3 fluorescence intensity (green fluorescence) and LC3 co-localization with Tom20 (orange fluorescence) were also enhanced in the lung tissues of the rats in the EHS + Parkin group.
3.9Parkin overexpression activated the Pink1/Parkin pathway in lung tissues of EHS rats
Western results showed that the expression of Pink1, Parkin, MFN2, and PTEN-L in the lung tissues of rats in the HS group was significantly increased compared with the CON group. Compared with the HS group, the protein levels of MFN2 and PTEN-L in the HS + Parkin group decreased (P < 0.05), and the level of Pink1 also tended to decrease. In contrast, there was no difference in the expression of Pink1, MFN2, and PTEN-L in the CON + Parkin group compared with the CON group (P > 0.05). There was also no significant difference in the expression of PTEN in the lung tissue of rats in each group (P > 0.05, Fig. 9).
Fig. 9
Parkin over-expression rats activate the Pink1/Parkin pathway in their lung (N = 5). A: The protein levels of Pink1, MFN2, PTEN-L, and PTEN in lung tissues were performed by immunoblotting. B-D: The statistical analysis of Pink1, MFN2, PTEN-L, and PTEN expression. *P < 0.05 vs Control group; **P < 0.05 vs EHS group. E: Immunofluorescence staining against Pink1 and Parkin was performed and observed by fluorescent microscopy. Pink (green), Parkin (red), The co-staining (orange) of Pink1 and Parkin was showed by the white arrow, bar = 50μm.
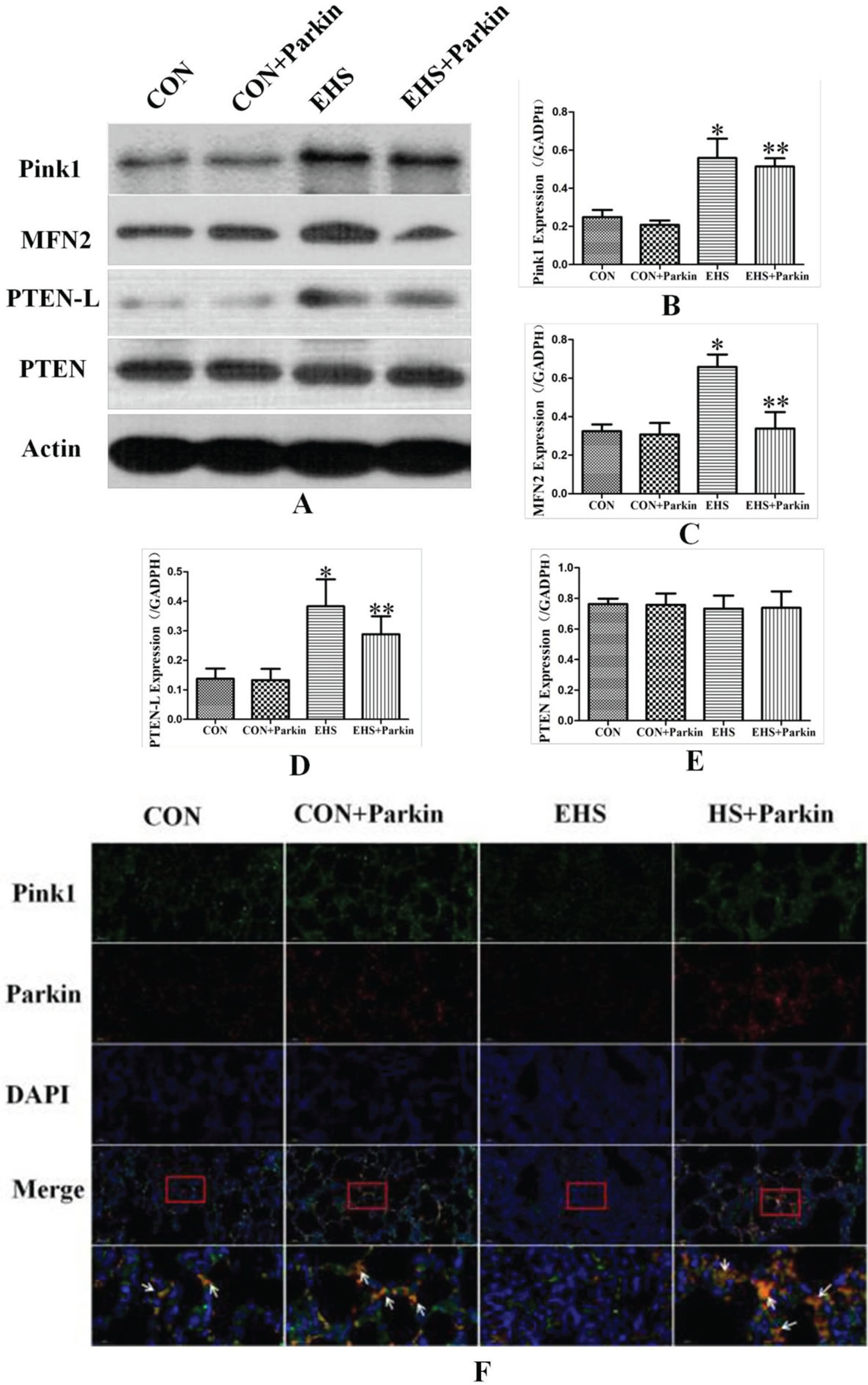
Immunofluorescence results showed that the intensity of Parkin fluorescence was significantly enhanced (red fluorescence) in the lung tissues of rats in both CON and CON + Parkin groups after tail vein injection of the adeno-associated virus carrying Parkin gene. The intensity of Pink1 (green fluorescence) and Parkin (red fluorescence) co-localized in lung tissues of rats in the EHS group was reduced (orange fluorescence) compared with that in the CON group, whereas the intensity of Pink1 (green fluorescence) and Parkin (red fluorescence) co-localized in lung tissues of rats in the HS + Parkin group was significantly enhanced compared with that in the EHS group. The intensity of Pink1 fluorescence (green fluorescence) in lung tissue was unchanged and the intensity of Pink1 (green fluorescence) and Parkin (red fluorescence) co-localization fluorescence (orange fluorescence) in lung tissue was slightly enhanced compared with the CON group.
4Discussion
Heat stroke is a fatal disease caused by thermal injury factors acting on the organism, and its pathophysiology is characterized by multi-organ failure. Among them, hyperthermia-induced acute lung injury or ARDS is one of its common complications [21]. We found, by establishing an exertional heat stroke rat model, that the core temperature in rats performing high-intensity exercise in a high-temperature and high-humidity external environment increased dramatically, and obvious pathological changes occurred in the lungs, manifested by progressively increasing interstitial blood vessels with significant dilatation and congestion, massive alveolar cavity hemorrhage, and blurred alveolar structures, which led to elevated lung coefficients and increased pulmonary vascular permeability, and a large number of apoptotic cells were visible in the lungs. CT of the lungs showed a ground glass-like shadow with parenchymal congestion with edema in mottled lung tissue. This is consistent with the results of previous studies [4, 9, 22].
The mechanism of lung injury due to heat radiation is not fully understood. It is currently believed that direct heat shock and secondary systemic inflammatory response are the pathophysiological basis [23]. It was found that in ARDS due to heat stroke, significantly elevated concentrations of endotoxins as well as inflammatory cytokines were detected in lung tissue, along with inflammatory cell infiltration and increased alveolar cavity macrophages, indicating a significant inflammatory response in the lungs of patients with heat stroke [24]. Our study likewise found that the levels of IL-6, IL-1β, and TNF-α were significantly elevated in the lung tissues of heat-shot rats, confirming that heat shock caused an inflammatory response in the lungs of rats. In addition, heat stroke not only caused local inflammatory responses in tissues but also led to the development of SIRS. An animal study of heat stroke found that the levels of inflammatory factors such as IL-1β, IL-6, and TNF-α were significantly elevated in the blood of animal models of heat stroke, with IL-6 showing a significant elevation in the early stages of SIRS. Therefore, the immune response is dysregulated in the heat-shot disease state, and immune cells and inflammatory factors can induce an inflammatory storm through a cascade of amplified responses, leading to more severe organ and tissue damage.
Oxidative stress likewise plays an important role in the pathological changes caused by heat stroke. Liu et al. found a large number of oxidative stress genes highly expressed in the lungs of rats with heat stroke (e.g., Aqp3, Cygb, SOD2, Hmox1, etc.) by high-throughput sequencing technology, and inhibition of oxidative stress response could attenuate the heat stroke-induced pulmonary inflammatory response and improve lung function [24]. Our results suggest that the levels of reactive oxygen species (ROS) clusters in the lungs of heat-shot rats are significantly elevated, further confirming that heat shock can lead to excessive activation of oxidative stress responses in lung tissue.
Mitochondria are one of the main sources of ROS, and mitochondrial dysfunction occurs to induce enhanced intracellular oxidative stress and increased ROS expression [25]. We found by transmission electron microscopy that swelling of mitochondria, breakage of mitochondrial cristae, and vacuolization of most mitochondria occurred in lung type II epithelial cells of heat-shot rats, confirming that heat shock caused severe damage to mitochondria in lung tissue cells. The damage to mitochondria enhanced oxidative stress, and the production of ROS in turn further aggravated the damage to mitochondria, forming a vicious circle, resulting in a cascade amplification of ROS content, which leads to a “waterfall” inflammatory response, causing apoptosis and necrosis of cells, and eventually progressing to organ failure [26]. Therefore, timely removal of damaged mitochondria plays a positive role in protecting organ function.
To maintain mitochondrial homeostasis, a strict quality control system exists within the cell to identify and repair damaged mitochondria. If mitochondrial damage is too severe to be repaired, or if there are too many mitochondria to be an operational burden, the cell removes the damaged or excess mitochondria through the mitochondrial autophagy pathway. Mitochondrial autophagy is an important regulatory mechanism to maintain the balance of mitochondrial quantity and quality and to maintain the dynamic balance of the intracellular mitochondrial network [11]. Mitochondrial gene mutations, high intracellular ROS levels, and the chemical factor antimycin may lead to mitochondrial damage and induce a mitochondrial autophagic cascade [27]. Incomplete or inhibited mitochondrial autophagy can lead to impaired clearance of damaged mitochondria, increased ROS production, enhanced inflammatory response, increased apoptosis, and impaired organ function. Conversely, enhanced mitochondrial autophagy can clear damaged mitochondria, reduce ROS production, decrease oxidative stress, and reduce lung injury [26, 27]. Fu et al. found that a decrease in the number of mitochondria and consequently low ATP production during mitochondrial injury can induce mitochondrial autophagy, reduce apoptosis, and play a protective role in the injury [28]. In our study, we found that mitochondria appeared swollen, increased in size, and loss of mitochondrial cristae as well as mitochondrial vacuolization in lung tissue of heat-shot rats. Immunoblotting showed that the autophagy level marker LC3-II/LC3-I ratio would be low and the expression of P62 was increased in lung tissues of heat shot rats. Immunofluorescence results showed decreased binding of LC3 to mitochondria suggesting a decrease in autophagic vesicles encapsulating mitochondria. The above results suggest that heat shock-induced a decrease in the level of mitochondrial autophagy in rat lung tissue. Therefore, heat shock inhibition of mitochondrial autophagy levels may be one of the mechanisms causing inflammatory responses and cellular damage in lung tissue.
Pink1/Parkin-mediated mitochondrial autophagy is one of the classical pathways of mitochondrial autophagy. When mitochondrial damage under the influence of the external environment leads to depolarization of the inner membrane, Pink1 accumulates on the inner mitochondrial membrane and recruits and binds to Parkin molecules through autophosphorylation activation, and the autophagosome wraps around the damaged mitochondria under the guidance of LC3 junction protein, and mitochondrial autophagy occurs [29]. The interaction between Pink1 and Parkin is the key link of Pink1/Parkin pathway activation is a key link. Mitochondrial outer membrane fusion protein 2 (mitofusion2, MFN2) is a downstream molecule of the Pink1/Parkin pathway. mFN2 inhibits mitochondrial fusion and provides autophagic signals [30]. Pinkl phosphorylates MFN2 to deliver impaired mitochondrial signals to bind to Parkin and activated Parkin further ubiquitinates and modifies MFN2, thereby recruiting more Parkin and ubiquitination cycles, and this positive cycle amplifies the molecular signal for mitochondrial autophagy, generating a cascade of amplified mitochondrial autophagic processes [31]. We found that although heat shock caused an increase in Pink1 and Parkin protein expression in rat lung tissue (Figs. 8 and 9), the degree of binding between Pink1 and Parkin was decreased (Fig. 9), suggesting that heat shock inhibits the level of mitochondrial autophagy associated with blocked intermolecular interactions between Pink1 and Parkin. Exertional heat stroke inhibits Pink1/Parkin pathway activation, resulting in a decrease in both mitochondrial autophagy vesicle production and degradation, leading to a decrease in mitochondrial autophagy levels in lung tissue. The abnormal mitochondrial autophagy process further causes impaired degradation of Pink1, Parkin, and MFN2 [32], which accumulate in lung tissues and eventually manifest as increased expression of Pink1, Parkin, and MFN2 in lung tissues of rats with heat stroke (Figs. 8 and 9).
PTEN-L, an isoform of PTEN, is localized to the outer mitochondrial membrane. PTEN-L is a protein phosphatase that inhibits the phosphorylation of ubiquitin Ser65-Ub by Pink1.Ser65-Ub phosphorylation is a key component of Pink1-mediated translocation of Parkin to damaged mitochondria and activation of Parkin’s E3 ubiquitin ligase. PTEN-L expression enhancement prevents Parkin translocation to mitochondria, inhibits Parkin’s E3 ubiquitin ligase activity, and prevents its induction of mitochondrial autophagy [33]. Our study found that PTEN-L expression was increased in the lung tissue of rats in the heat-shock group (Fig. 9), while the level of PTEN was unchanged. upregulation of PTEN-L, a negative regulator of the Pink1/Parkin pathway, attenuates Pink1 binding to Parkin and inhibits the mitochondrial autophagy pathway, which ultimately leads to the onset of lung injury. This may be the reason why heat shock induces increased expression of Pink1 and Parkin molecules in lung tissue but decreases mitochondrial autophagy levels.
Pink1/Parkin-mediated mitochondrial autophagy protects cells from damage induced by different stimuli. Studies have demonstrated that activation of the Pink1/Parkin pathway has a protective effect against sepsis-induced lung injury, kidney injury, and liver injury [34–36]. Rat lung tissue overexpressed Parkin protein by tail vein injection of adeno-associated virus carrying Parkin gene. After administration of heat shock, we found that the survival rate of rats overexpressing Parkin molecules was significantly increased (Fig. 1). Pulmonary vascular permeability was reduced (Fig. 3), lung imaging (Fig. 2) and lung histopathological changes were attenuated (Fig. 4), lung tissue exudation was reduced, inflammatory cell infiltration was decreased, and apoptosis was significantly reduced (Fig. 7). The levels of inflammatory factors IL-6, IL-1β, and TNF-α in lung tissue were decreased, and the levels of oxidative stress products ROS were all significantly reduced (Fig. 5). Transmission electron microscopy revealed reduced mitochondrial swelling in lung epithelial cells, and no vacuolization was detected (Fig. 6). Immunohistochemistry and immunofluorescence results showed that Parkin overexpression caused a significant increase in Parkin expression in rat lung tissue (Fig. 8), enhanced Pink1-Parkin interaction (Fig. 9), increased LC3 binding to mitochondria (Fig. 8), increased autophagy protein LC3-II/LC3-I ratio, and decreased P62 expression (Fig. 8). These results suggest that Parkin overexpression can partially counteract the inhibition of the Pink1/Parkin pathway by PTEN-L when lung injury occurs in HS rats, activate the pathway, promote mitochondrial autophagy to clear damaged mitochondria, maintain the effectiveness of mitochondrial function, and maintain cellular homeostasis, thus alleviating the inflammatory response and excessive activation of oxidative stress, mitigating cellular damage, and ultimately reducing HS acute lung injury in rats and improve the prognosis of heat shot rats. In addition, Parkin overexpression enhanced the level of mitochondrial autophagy and then attenuated the excessive accumulation of Pink1 and MFN2 in lung tissue (Figs. 8 and 9). Therefore, activation of the Pink1/Parkin pathway and enhancement of mitochondrial autophagy have a protective effect on heat-shot lung injury.
In conclusion, this study demonstrated that HS spreads to the respiratory system and causes lung injury, that the inhibition of mitochondrial autophagy level received in lung tissue is one of the mechanisms by which HS causes the occurrence of lung injury, and that lung injury in EHS rats can be attenuated by activating Pink1/Parkin-mediated mitochondrial autophagy. The shortcoming of this assay may be that the Pink1/Parkin pathway is only one of the pathways that affect mitochondrial autophagy. Mitochondrial membrane proteins such as Nip3-like protein X (Nix) can also be involved in Parkin-dependent mitochondrial autophagy [37]. FUN14 domain-containing protein 1 (FUNDC1) receptor [38] and Bcl-2-like protein 13 (Bcl-2-like protein 13, BCL2L13) [39] induce mitochondrial autophagy in a Parkin non-dependent manner, whether these regulatory mitochondrial pathways are involved in the development of lung injury in heat stroke still needs to be further explored.
Ethical approval
The authors assert that all procedures contributing to this work comply with the ethical standards of the relevant national guides on the care and use of laboratory animals and have been approved by the Ethics Committee of the Eighth Medical Center of Chinese PLA General Hospital (approval number: 20208141030). This study was reported following ARRIVE guidelines.
Data availability
The datasets used or analyzed during the current study are available from the corresponding author upon reasonable request.
Conflict of interest
None.
Funding
This study was supported by the Special Tasks for military health and epidemic prevention in 2020 ([2021]208) and the Subject in the Hospital (2016ZD-008).
Author contributions
Guarantor of integrity of the entire study: Jiaxin Wang, Yuxiang Zhang
Study concepts: Yuxiang Zhang, Yan Gu
Study design: Jiaxin Wang, Yuxiang Zhang, Zhengzhong Sun, Yan Gu
Definition of intellectual content: Jiaxin Wang, Yuxiang Zhang, Zhengzhong Sun, Liya Jiang
Experimental studies: Jiaxin Wang, Yuxiang Zhang, Zhengzhong Sun, Liya Jiang, Lyv Xuan
Data acquisition: Jiao Wang
Statistical analysis: Jiaxin Wang, Zhengzhong Sun, Liya Jiang
Manuscript preparation: Jiaxin Wang, Zhengzhong Sun, Liya Jiang
Manuscript editing: Jiaxin Wang, Zhengzhong Sun, Liya Jiang
Manuscript review: Yuxiang Zhang, Yan Gu
All authors have read and approved this article.
References
[1] | The Lancet Health in a world of extreme heat. Lancet. (2021) ;398: (10301):641. doi: 10.1016/S0140-6736(21)01860-2. |
[2] | Bouchama A , Abuyassin B , Lehe C , Laitano O , Jay O , O’Connor FG , et al. Classic and exertional heatstroke. Nat Rev Dis Primers. (2022) ;8: (1):8. doi: 10.1038/s41572-021-00334-6. |
[3] | Belval LN , Casa DJ , Adams WM , Chiampas GT , Holschen JC , Hosokawa Y , et al. Consensus Statement- Prehospital Care of Exertional HeatStroke. Prehosp Emerg Care. (2018) ;22: (3) 392–397. doi: 10.1080/10903127.2017.1392666. |
[4] | Liu Z , Chen J , Hu L , Li M , Liang M , Chen J , et al. Expressionprofiles of genes associated with inflammatory responses andoxidative stress in lung after heat stroke. Biosci ReBSR. (2020) ;40: (6):BSR20192048. doi: 10.1042/BSR20192048. |
[5] | Varghese GM , John G , Thomas K , Abraham OC , Mathai D Predictors ofmulti-organ dysfunction in heatstroke, Emerg Med J. (2005) ;22: (3) 185–7. doi: 10.1136/emj.2003.009365. |
[6] | Zhang Y , Wang S , Wang X , Zan Q , Yu X , Fan L , et al. Monitoring ofthe decreased mitochondrial viscosity during heat stroke with amitochondrial AIE probe. Anal Bioanal Chem. (2021) ;413: (14):3823–3831. doi: 10.1007/s00216-021-03335-2. |
[7] | Wen Y , Zhang W , Liu T , Huo F , Yin C Pinpoint Diagnostic Kit forHeat Stroke by Monitoring Lysosomal pH. Anal Chem. (2017) ;89: (21):11869–11874. doi: 10.1021/acs.analchem.7b03612. |
[8] | Chen Y , Tong H , Pan Z , Jiang D , Zhang X , Qiu J , et al. Xuebijinginjection attenuates pulmonary injury by reducing oxidative stressand proinflammatory damage in rats with heat stroke. Exp Ther Med. (2017) ;13: (6):3408–3416. doi: 10.3892/etm.2017.4444. |
[9] | Wang L , Lu Z , Zhao J , Schank M , Cao D , Dang X , et al. Selectiveoxidative stress induces dual damage to telomeres and mitochondriain human T cells. Aging Cell. (2021) ;20: (12):e13513. doi: 10.1111/acel.13513. |
[10] | Zhou Z , Hu Q , Guo H , Wang X CircSEC11A knockdown alleviatesoxidative stress and apoptosis and promotes cell proliferation andangiogenesis by regulating miR-29a-3p/SEMA3A axis in OGD-inducedhuman brain microvascular endothelial cells (HBMECs). Clin HemorheolMicrocirc. (2023) ;84: (3) 247–262. doi: 10.3233/CH-221689. |
[11] | Wei H , Liu L , Chen Q Selective removal of mitochondria viamitophagy: distinct pathways for different mitochondrial stresses. Biochim Biophys Acta. (2015) ;1853: (10 Pt B):2784–90. doi: 10.1016/j.bbamcr.2015.03.013. |
[12] | Roca-Portoles A , Tait SWG Mitochondrial quality control: frommolecule to organelle. Cell Mol Life Sci. (2021) ;78: (8) 3853–3866. doi: 10.1007/s00018-021-03775-0. |
[13] | Chen H , Lin H , Dong B , Wang Y , Yu Y , Xie K Hydrogen alleviates celldamage and acute lung injury in sepsis via PINK1/Parkin-mediatedmitophagy. Inflamm Res. (2021) ;70: (8):915–930. doi: 10.1007/s00011-021-01481-y. |
[14] | Zhang Y , Chen L , Luo Y , Wang K , Liu X , Xiao Z , et al. Pink1/Parkin-Mediated Mitophagy Regulated the Apoptosis of DendriticCells in Sepsis. Inflammation. (2022) ;45: (3):1374–1387. doi: 10.1007/s10753-022-01628-x. |
[15] | Kim M , Nikouee A , Sun Y , Zhang QJ , Liu ZP , Zang QS Evaluation ofParkin in the Regulation of Myocardial Mitochondria-AssociatedMembranes and Cardiomyopathy During Endotoxemia. Front Cell DevBiol. (2022) ;10: 796061. doi: 10.3389/fcell.2022.796061. |
[16] | Wei J , Ni N , Meng W , Gao Y Early urine proteome changes in theWalker-256 tail-vein injection rat model. Sci Rep. (2019) ;9: (1):13804. doi: 10.1038/s41598-019-50301-1. |
[17] | Iwaniec J , Robinson GP , Garcia CK , Murray KO , de Carvalho L , Clanton TL , et al. Acute phase response to exertional heat stroke in mice. Exp Physiol.222-232. (2021) ;106: (1):222–232. doi: 10.1113/EP088501. |
[18] | Hong SB , Koh Y , Lee IC , Kim MJ , Kim WS , Kim DS , et al. Inducedhypothermia as a new approach to lung rest for the acutely injuredlung. Crit Care Med. (2005) ;33: (9):2049–2055. doi: 10.1097/01.ccm.0000178186.37167.53. |
[19] | Ma C , Dong L , Li M , Cai W Qidonghuoxue Decoction AmelioratesPulmonary Edema in Acute Lung Injury Mice through the Upregulationof Epithelial Sodium Channel and Aquaporin-1. Evid Based ComplementAlternat Med. (2020) ;2020: 2492304. doi: 10.1155/2020/2492304. |
[20] | Sendo T , Kataoka Y , Takeda Y , Furuta W , Oishi R Nitric oxideprotects against contrast media-increased pulmonary vascularpermeability in rats. Invest Radiol. (2000) ;35: (8):472–8. doi: 10.1097/00004424-200008000-00003. |
[21] | Liu SY , Song JC , Mao HD , Zhao JB , Song Q;; ,Expert Group of Heat Stroke Prevention and Treatment of the People’s Liberation Army, and People’s Liberation Army Professional Committee of Critical Care Medicine. Expert consensus on the diagnosis and treatment of heatstroke in China. Mil Med Res. (2020) ;7: (1) 1. |
[22] | Lin CH , Tsai CC , Chen TH , Chang CP , Yang HH Oxytocin maintains lunghistological and functional integrity to confer protection in heatstroke. Sci Rep. (2019) ;9: (1):18390. doi: 10.1038/s41598-019-54739-1. |
[23] | Lim CL Heat Sepsis Precedes Heat Toxicity in the Pathophysiology ofHeat Stroke-A New Paradigm on an Ancient Disease. Antioxidants(Basel). (2018) ;7: (11) 149. doi: 10.3390/antiox7110149. |
[24] | Liu Z , Chen J , Hu L , Li M , Liang M , Chen J , et al. Expressionprofiles of genes associated with inflammatory responses andoxidative stress in lung after heat stroke. Biosci ReBSR. (2020) ;40: (6):BSR20192048. doi: 10.1042/BSR20192048. |
[25] | Szechyńska-Hebda M , Ghalami RZ , Kamran M , Van Breusegem F , Karpiński S To Be or Not to Be? Are Reactive Oxygen Species,Antioxidants, and Stress Signalling Universal Determinants of Lifeor Death? Cells. (2022) ;11: (24):4105. doi: 10.3390/cells11244105. |
[26] | Zhao Y , Huang S , Liu J , Wu X , Zhou S , Dai K , Kou Y MitophagyContributes to the Pathogenesis of Inflammatory Diseases. Inflammation. (2018) ;41: (5):1590–1600. doi: 10.1007/s10753-018-0835-2. |
[27] | Yao RQ , Ren C , Xia ZF , Yao YM Organelle-specific autophagy ininflammatory diseases: a potential therapeutic target underlying thequality control of multiple organelles. Autophagy. (2021) ;17: (2) 385–401. doi: 10.1080/15548627.2020.1725377. |
[28] | Fu HR , Li XS , Zhang YH , Feng BB , Pan LH Visnagin amelioratesmyocardial ischemia/reperfusion injury through the promotion ofautophagy and the inhibition of apoptosis. Eur J Histochem. (2020) ;64: (s2):3131. doi: 10.4081/ejh.2020.3131. |
[29] | Silvian LF PINK1/Parkin Pathway Activation for MitochondrialQuality Control -Which Is the Best Molecular Target for Therapy? Front Aging Neurosci. (2022) ;14: 890823. doi: 10.3389/fnagi.2022.890823. |
[30] | McLelland GL , Goiran T , Yi W , Dorval G , Chen CX , Lauinger ND , et al. Mfn2 ubiquitination by PINK1/parkin gates the p97-dependent releaseof ER from mitochondria to drive mitophagy. Elife. (2018) ;7: e32866. doi: 10.7554/eLife.32866. |
[31] | Wojtyniak P , Boratynska-Jasinska A , Serwach K , Gruszczynska-BiegalaJ , Zablocka B , Jaworski J , et al. Mitofusin 2 IntegratesMitochondrial Network Remodelling, Mitophagy and Renewal ofRespiratory Chain Proteins in Neurons after Oxygen and GlucoseDeprivation. Mol Neurobiol. (2022) ;59: (10) 6502–6518. doi: 10.1007/s12035-022-02981-6. |
[32] | Kawajiri S , Saiki S , Sato S , Sato F , Hatano T , Eguchi H , et al. PINK1 is recruited to mitochondria with parkin and associates withLC3 in mitophagy. FEBS Lett. (2010) ;584: (6):1073–9. doi: 10.1016/j.febslet.2010.02.016. |
[33] | Wang L , Cho YL , Tang Y , Wang J , Park JE , Wu Y , et al. PTEN-L is anovel protein phosphatase for ubiquitin dephosphorylation to inhibitPINK1-Parkin-mediated mitophagy. Cell Res. (2018) ;28: (8) 787–802. doi:10.1038/s41422-018-0056-0. |
[34] | Chen H , Lin H , Dong B , Wang Y , Yu Y , Xie K Hydrogen alleviates celldamage and acute lung injury in sepsis via PINK1/Parkin-mediatedmitophagy. Inflamm Res. (2021) ;70: (8) 915–930. doi: 10.1007/s00011-021-01481-y. |
[35] | Lin Q , Li S , Jiang N , Shao X , Zhang M , Jin H , et al. PINK1-parkinpathway of mitophagy protects against contrast-induced acute kidneyinjury via decreasing mitochondrial ROS and NLRP3 inflammasomeactivation. Redox Biol. (2019) ;26: 101254. doi: 10.1016/j.redox.2019.101254. |
[36] | Wang S , Tao J , Chen H , Kandadi MR , Sun M , Xu H , et al. Ablation ofAkt2 and AMPKα2 rescues high fat diet-induced obesity andhepatic steatosis through Parkin-mediated mitophagy. Acta Pharm Sin B. (2011) ;11: (11):3508–3526. doi: 10.1016/j.apsb.2021.07.006. |
[37] | Xu D , Chen P , Wang B , Wang Y , Miao N , Yin F , et al. NIX-mediatedmitophagy protects against proteinuria-induced tubular cellapoptosis and renal injury. Am J Physiol Renal Physiol. (2019) ;316: (2) F382–F395. doi: 10.1152/ajprenal.00360.2018. |
[38] | Zhang Y , Lian Y , Lian X , Zhang H , Chen Y , Sheng H , et al. FUNDC1Mediated Mitophagy in Epileptic Hippocampal Neuronal Injury Inducedby Magnesium-Free Fluid. Neurochem Res. (2023) ;48: (1) 284–294. doi: 10.1007/s11064-022-03749-z. |
[39] | Fang Q , Zheng S , Chen Q , Chen L , Yang Y , Wang Y , et al. Theprotective effect of inhibiting mitochondrial fission on thejuvenile rat brain following PTZ kindling through inhibiting theBCL2L13/LC3 mitophagy pathway. Metab Brain Dis. (2023) ;38: (2):453–466. doi: 10.1007/s11011-022-01077-3. |


