
Renal ischaemia-reperfusion injury is promoted by transcription factor NF-kB p65, which inhibits TRPC6 expression by activating miR-150
Abstract
AIM:
To investigate the mechanism by which NF-κB p65 activates miR-150 to suppress TRPC6 expression and promote renal ischemia-reperfusion injury.
METHODS:
To assess the transcription of miR-150, NF-B p65, and TRPC6 in HK-2 cells treated with hypoxia reperfusion and rat kidney tissue damaged by ischemia-reperfusion (I/R), qPCR was implemented. The protein production of NF-κB p65 and TRPC6 was assessed by Western blot (WB) analysis. The histological score of rat kidney tissue was assessed using H&E (hematoxylin and eosin) staining. To assess the rate of apoptosis of renal tissue cells following I/R injury, we used the TACS TdT In Situ Apoptosis Detection Kit. To find out the impairment of renal function, blood levels of creatinine (Cr) and blood urea nitrogen (BUN) were tested in rats. Concentrations of inflammatory cytokines, including IL-1β, IL-10, and TNF-α, were detected in HK-2 cells and rat renal tissue cells utilizing ELISA kits. FITC and CCK-8 were employed to analyze the death rate and cellular proliferation of HK-2 cells. To analyse the mechanism of engagement between NF-κB p65 and the miR-150 promoter, coupled with the detrimental impact of miR-150 on TRPC6, we adopted the dual-luciferase reporter assay. To confirm the activating effect of NF-κB p65 on miR-150,we implemented the ChIP assay.
RESULTS:
NF-κB p65 expression was significantly upregulated in rat renal tissue following IRI. Applying the dual-luciferase reporter assay, we demonstrated that the specific attachment of NF-B p65 with the miR-150 promoter location is viable, resulting in the promotion of the activity of the promoter. When miR-150 was overexpressed, we observed a notable reduction in cell proliferation. And it notably increased the rate of cellular apoptosis rate and amounts of the proinflammatory cytokines IL-1β, IL-10, and TNF-α. Employing the dual-luciferase reporter assay, we demonstrated that miR-150 transfection diminished the function of luciferase in the TRPC6-WT group, whereas luciferase activity in the TRPC6-MUT group remained unchanged, indicating that miR-150 is a targeted inhibitor of TRPC6. In the rat renal I/R model, when miR-150 was inhibited or TRPC6 was overexpressed in the rat kidney I/R model, the histological score of rat kidney tissue significantly decreased, so did the quantities of proinflammatory cytokines IL-1β, IL-10, TNF-α, creatinine (Cr) and blood urea nitrogen (BUN) contents and the rate of cell apoptosis in kidney tissue.
CONCLUSION:
Activation of miR-150 by NF-κB p65 results in downregulation of TRPC6 expression and promotion of IRI in the kidney.
Highlights
1. NF-κB p65 and miR-150 are highly expressed in renal ischemia-reperfusion tissues.
2. The binding of NF-κB p65 and miR-150 in the promoter region enhances the activity of the promoter.
3. Overexpression of miR-150 promotes H/R injury in HK-2 cells.
4. Inhibition of TRPC6 expression by miR-150 promotes H/R injury in HK-2 cells.
5. Activation of miR-150 by NF-κB p65 inhibits the expression of TRPC6 and promotes renal ischemia-reperfusion injury in rats.
1Introduction
The term “Renal Ischemia-Reperfusion Injury” (IRI) is a common clinical syndrome that can lead to renal dysfunction and increase the risk of chronic kidney disease. A variety of signaling pathways are activated due to oxygen deprivation and metabolic derangements, causing cellular damage and inflammatory reactions [1]. IRI is a very diverse and widespread clinical condition that frequently causes decreased renal function, subsequently progressing to chronic kidney disease with a high morbidity and mortality [2]. Despite the various treatment approaches that have been attempted, effective treatments for IRI are still being investigated. Therefore, advancement of new methods to improve patient prognosis is urgently required [3].
NF-kB is a fundamental controller of autoimmune diseases and modulates inflammation in pathogens and cancer cells [4]. Small noncoding RNAs known as miRNAs have been found to be essential for controlling cellular homeostasis. Cellular and organ damage could occur from their disease. Many studies have suggested that miRNAs are involved in the pathophysiologic processes of diseases including acute kidney injury, chronic kidney disease, and graft injury [5, 6]. Moreover, NF-kB p65 can bind directly to the miR-150 promoter in order to promote its expression [7]. TRCP6 is a cation channel that can be expressed in multiple tissues of the kidney [8]. It is important to note that numerous studies have demonstrated that TRPC6 plays a helpful function in the therapy of IRI [9]. Research in recent years has found that stimulation of the TLR4/NF-kB p65 signaling pathway can enhance IRI development in the kidney [10].
Recently, increasing evidence has shown that circular RNAs (circRNAs) are extensively involved in the pathogenesis of ischemic stroke and other diseases. Circular RNAs participate in the pathogenesis of ischemic diseases by regulating miRNAs and downstream target genes [11]. Atherosclerosis (AS) is a disease that severely threatens human health, and circRNAs are also implicated in its pathogenesis [12–14]. Circ_0033596 mediates ox-LDL-induced endothelial cell injury through the miR-637/GRB2 pathway [15]. These findings provide insights into investigating the mechanism of the NF-kB/miR-150/TRPC6 axis in renal IRI. The exact mechanism by which this pathway promotes kidney IRI is still not fully understood. According to current studies available, we think that NF-kB p65 can promote IRI of the kidney by suppressing the expression of TRPC6 via the activation of miR-150. In this study, we will extensively explore this topic and provide new ideas for the creation of IRI therapies in coming years.
2Materials and methods
2.1A rat model of renal I/R injury
This study has been approved by the Ethics Committee of The Third Affiliated Hospital of Qiqihar Medical University (QMU-AECC-2022-69). All animal experiments were performed following the guidelines issued by National Institutes of Health. The experimental mice used in the present investigation weighed between 180 to 220 grams and they were male Sprague-Dawley rats with an age of 21 days old. The rats were acquired from Shanghai, China-based Slac Laboratory Animal Center. They were reared in a specific pathogen-free animal facility with a temperature range of 22–25°C and a relative humidity range of 60–65% under control of a 12-hour light and dark cycle. The mice had open access to water and food. After a week of acclimatization, the rats were randomized into 3 groups: (1) Sham group (n = 10) as control group; (2) I/R group (n = 10) as model group; and (3) lentivirus injection group (n = 40, each group consisted of 10 rats) as experimental group. Before the experiment, the rats were in abrosia 12 hours to establish a rat model of renal IRI. They were then intraperitoneally injected with 1% pentobarbital sodium (50 mg/kg; Sigma-Aldrich, St.Louis, MO, USA) to be anesthetized. Their left renal pedicle was clamped lasting 45 minutes, subsequently the clamp was released to enable blood flow to return. The experiment additionally encompassed a control group that had the identical surgical treatment without any vascular blood vessel obstruction. The experimental treatment of injecting lentiviral vectors was consistent with IR group, and lentiviral vectors carrying NF-kB p65-siRNA or miR-150-mimics were injected into the rat tail vein respectively. After 45 minutes of ischemia, the clamp was loosened and they were allowed to recover for 24 hours before being sacrificed. Their eyeballs were also collected for analysis and blood samples were taken at the same time [16].
2.2H/R damage was inflicted on HK-2 cells after they were grown
The Cell Bank of the Chinese Academy of Sciences is where the human kidney tubular epithelial cells HK-2 were bought. They were raised in DMEM/F12 medium coupled with a fetal bovine serum content of 10% and kept at 37 degrees Celsius with a CO2 concentration of 5% [17]. Two groups of cells were selected at random. Control cultures were raised in normoxic circumstances, whereas H/R cultures were treated to 6 hours of low oxygen levels before receiving 6 hours of oxygen supply with 95% air and 5% carbon dioxide. Following that, the two groups underwent treatment for 6 hours to 1% oxygen, 5% carbon dioxide, and 94% nitrogen, subsequently undergoing reoxygenation for varied durations of time (0, 2, 6, and 12 hours) with the addition of 21% oxygen, 5% carbon dioxide, and 74% nitrogen.
2.3Cell transfection
RiBoBio (Guangzhou, China) created the miR-150 inhibitor, mimic, and negative control (NC). The pcDNA-NF-kB p65, pcDNA-Vector, si-NF-kB p65 (sequence: GATCAATGGCTACACAGGA), and si-NC (sequence: TTCTCCGAACGTGTCACGT) were provided by Santa Cruz Biotechnology. These cell transfections were conducted as per directions provided by the maker for Lipofectamine 2000. The cells were extracted 48 hours after transfection for use in in vitro experiments [18].
2.4TACS TdT in situ apoptosis detection
The TACS TdT In Situ Apoptosis Detection Kit was used to analyze the rate of cell apoptosis in rat kidney slices based on the directions provided by the manufacturer, and staining was used in the analysis [19]. Deparaffinized and hydrated kidney tissue sections were then processed with proteinase K at 24°C for 15 minutes. Endogenous peroxidase activity was declined after washing the slides with a 3% H2O2 methanol solution. The TdT-labeled reaction mixture was then combined with the slides and raised for 1 hour at 37°C, before being treated with horseradish peroxidase-conjugated streptavidin for 1 hour at 37°C. Color was produced on the slides employing TACS blue label substrate solution. Following that, the slides were cleaned, counterstained and fixed by means of Permount solution. The number of positively labeled cells was counted using photomicrographs.
2.5ELISA
PBS buffer (pH 7.4) was employed to homogenize mouse kidney tissue, which was then centrifuged at 14,000 g for 15 minutes at 4°C. ELISA assay kits from Pierce Biotechnology were employed to quantify the amounts of the cytokines TNF-α, IL-1β, and IL-10, as per the producer’s procedure [20].
2.6Renal function analysis
Rats were euthanized after a 24-hour reperfusion period, and blood was extracted from the eye’s angular vein. To separate the serum, the blood was centrifuged (4,500 rpm/min). The creatinine (Cr) and blood urea nitrogen (BUN) levels were utilized as indicators to assess the severity of kidney injury [21].
2.7Histological evaluation
After being fixed in paraffin, rat kidney tissue was sliced into sections that were 4μm thick. Following attachment to slides made of glass, the individual parts were stripped of paraffin with xylene, hydrated with gradient ethanol, and finally stained with H&E (hematoxylin and eosin). The optical microscope was used to examine stained sections at a magnification of 200 times, and parameters such as hemorrhage, cytoplasmic vacuolation, tubular dilatation, and tubular necrosis were used to evaluate kidney injury scores. The scores represented the degree of damage: 0 = normal kidney; 1 = minimal necrosis involving less than 5%; 2 = mild necrosis involving 5–25%; 3 = moderate necrosis involving 25–75%; 4 = severe necrosis involving more than 75% [22].
2.8FITC
The cells were planted at a density of 2×10∧4 cells/ml, then placed in plates with 96 wells with a volume of 100μL each well and fostered in a cell culture incubator for 24 hours. To identify cell apoptosis, the FITC Annexin V Cell Apoptosis Detection Kit was employed [23].
2.9Cell proliferation analysis
The Cell Counting Kit-8 Cell was adopted to analyze proliferation. Each of the groups included four replicates, with cells planted at a volume of 5×10∧3 cells per well in plates with 24 wells. The cultures were cultured for 4 hours at 37°C after 10μl of CCK-8 solution was incorporated to the culture medium. To assess cell growth, the absorbance at 450 nm was assessed [24].
2.10Dual-luciferase assay
To create the pcDNA-NF-kB p65 plasmid, the full-length human NF-kB p65 sequence of coding was subcloned into a pcDNA vector. The pri-miR-150 promoter sequence, miR-150 and TRPC6 binding locations, and their associated MUT 3’UTRs were created utilizing GeneCopoeia. They then were inserted into the pGL3-promoter vector to create the constructions pGL3-miR-150, WT pGL3-TRPC6-3’UTR, and MUT pGL3-TRPC6-3’UTR. To encourage adhesion and proliferation, HK-2 cells were put to a plate with 96 wells and grown for a overnight period. HK-2 cells were transfected with pcDNA-vector or pcDNA-NF-kB p65 and co-transfected with 200 ng of GL3-miR-150 luciferase reporter vector for NF-kB p65 and miR-150 investigations. HK-2 cells were initially transfected with NC-mimic or miR-150 mimic, followed by 200 ng of either WT pGL3-TRPC6-3’UTR or MUT pGL3-TRPC6-3’UTR luciferase reporter vectors for miR-150 and TRPC6 investigations. The Dual-Luciferase Reporter Assay System was implemented to measured the performance of luciferase after a 48-hour transfection period [25].
2.11ChIP
The ChIP analysis was carried out following the guidance enclosed with the ChIP kit. The cells were collected in SDS buffer, centrifuged, and then suspended in IP buffer. Immunoprecipitation of cross-linked chromatin samples was conducted over a period of overnight incubation using an NF-kB p65 antibody. A technical control was established using non-specific IgG. qPCR analysis was performed on the immunoprecipitated DNA using specific primers designed for the miR-150 promoter region: forward 5’-TCTCCCAACCCTTGTACCAGTG-3’ and reverse 5’-CAGTGCGTGTCGTGGAGT-3’. ImageJ analysis software was used for image quantification [26].
2.12Western blot
The cells and tissue samples were then homogenized in RIPA buffer with a protease inhibitor cocktail (1 : 100) at 4°C after being rinsed once using ice-cold phosphate-buffered saline. After that, the homogenates were centrifuged at 12,000 rpm. The protein content was identified utilizing the BioRad Bradford protein assay kit. The proteins were purified by SDS-PAGE electrophoresis, put onto PVDF membranes, and kept alive for a overnight period at 4°C in TBST buffer containing 5% nonfat dry milk with primary antibodies targeting NF-kB p65, TRPC6, and GAPDH. After washing the membranes three times with TBST, they were treated for an hour with secondary antibodies conjugated to horseradish peroxidase. Three rinses were then conducted on the membranes with TBST before immunoreactive bands were identified utilizing the ECL detection method [27].
2.13qPCR
The miRNeasy Mini Kit was applied to extract total RNA from the samples, and the TaqMan MicroRNA Reverse Transcription Kit was employed to create cDNA. The TaqMan MicroRNA Assay Kit was utilized for real-time qPCR, and the SYBR GreenERTM Two-Step Kit was employed for detection. All procedures were conducted in line with the supplier’s directions. The 7500 Real-Time PCR System was employed to conduct real-time qPCR, with the following procedures: 2 minutes at 50°C, 10 minutes at 95°C, 40 15-second cycles at 95°C, and 1 minute at 60°C. Three replicates of each sample were analyzed [28].
2.14Statistical analysis
For statistical analysis, WSPSS software (version 20.0) was utilized. The mean plus or minus the standard deviation is provided as the result. Inter-group comparisons were made utilizing paired t-tests. Where appropriate, one-way or two-way ANOVA with Bonferroni’s post-hoc test was employed. Statistical significance was defined as an AP-value less than 0.05.
3Results
3.1Following I/R injury, NF-kB p65 was shown to be strongly expressed in the renal tissues of rats
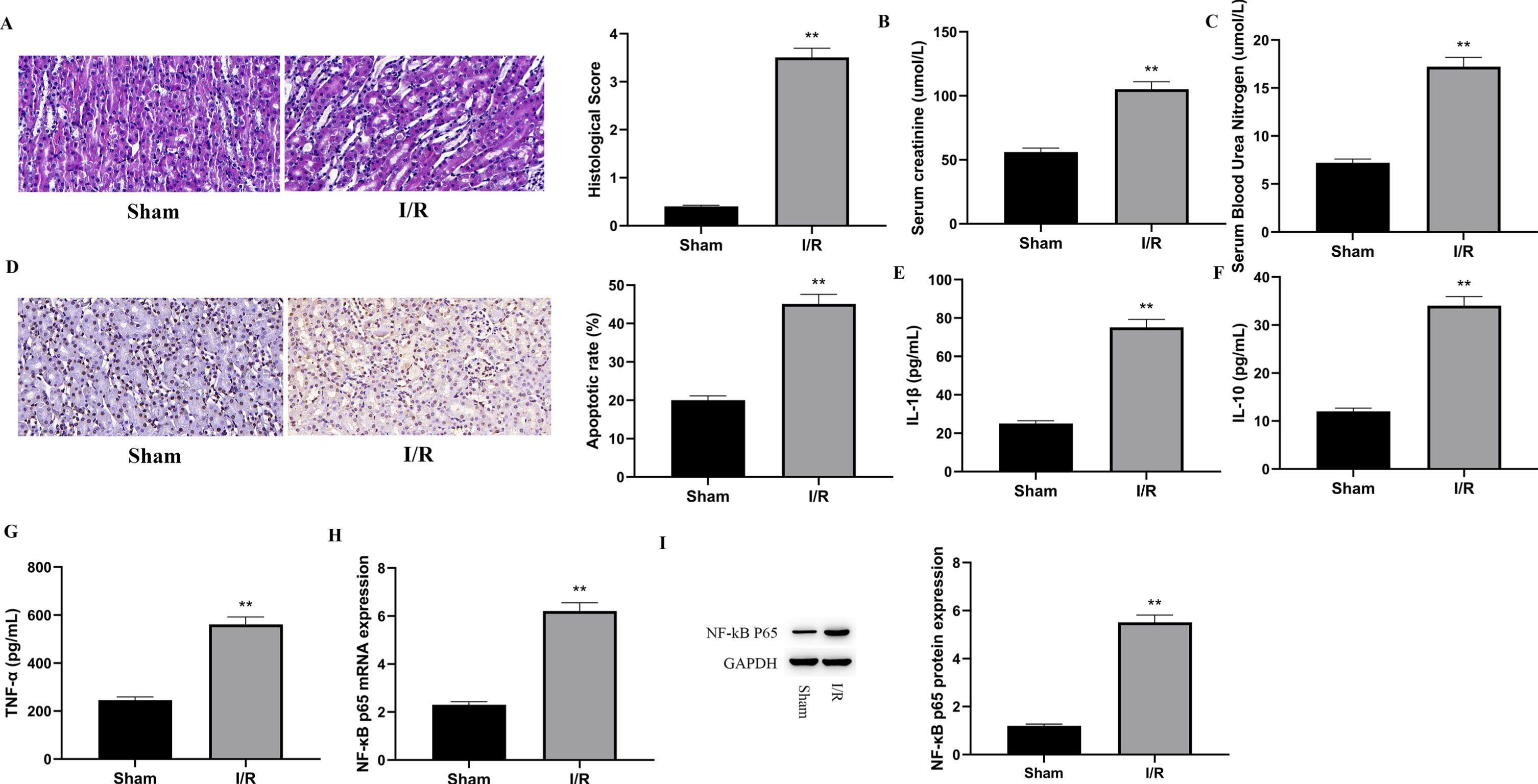
This study revealed that the stimulation of the TLR4/NF-kB p65 signaling pathway enhances renal I/R damage in rats. The goal of this study was to see how much NF-kB p65 was expressed in rat kidney tissue after I/R injury. The first step was to create a renal I/R damage rat model. The renal tissue H&E (hematoxylin and eosin) staining and histological evaluation of the Sham and I/R groups are shown in Fig. 1A. The I/R group showed cytoplasmic vacuolization, tubular dilation, and increased cytoplasmic space but not the Sham group. As a result, the I/R group’s histology score was considerably greater than the Sham group’s (P < 0.01). The rats’ renal function was monitored by measuring the levels of creatinine (Cr) and blood urea nitrogen (BUN). As illustrated in Fig. 1B and 1C, greater creatinine (Cr) and blood urea nitrogen (BUN) levels in the I/R group than in the Sham group (P < 0.01), demonstrating that I/R caused renal damage. The apoptosis rate in rat kidney tissues following I/R was measured, as illustrated in Fig. 1D. Comparing the I/R group to the Sham group, the apoptosis rate in the I/R group was noticeably higher (P < 0.01). Tissue I/R can activate the innate immune system, then induce an inflammatory response, ultimately leading to damaged cells. To evaluate the amounts of inflammatory factors in rat renal tissue following I/R, ELISA was employed to measure IL-1, IL-10, and TNF-, as illustrated in Fig. 1E, 1F, and 1G. The percentages of IL-1, IL-10, and TNF- in the I/R group were much greater than in the Sham group (P < 0.01). Furthermore, the degree of NF-kB p65 expression in rat kidney tissues was examined utilizing qPCR and WB. As demonstrated in Fig. 1 H and 1I, both mRNA and protein content of NF-kB p65 were much greater in the I/R group than in the Sham group (P < 0.01), indicating that NF-kB p65 was notably expressed in rat renal tissues subjected to IR injury.
Fig. 1
Following I/R injury, NF-kB p65 was shown to be strongly expressed in the renal tissues of rats.
3.2In HK-2 cells, NF-kB p65 can enhance miRNA-150 expression
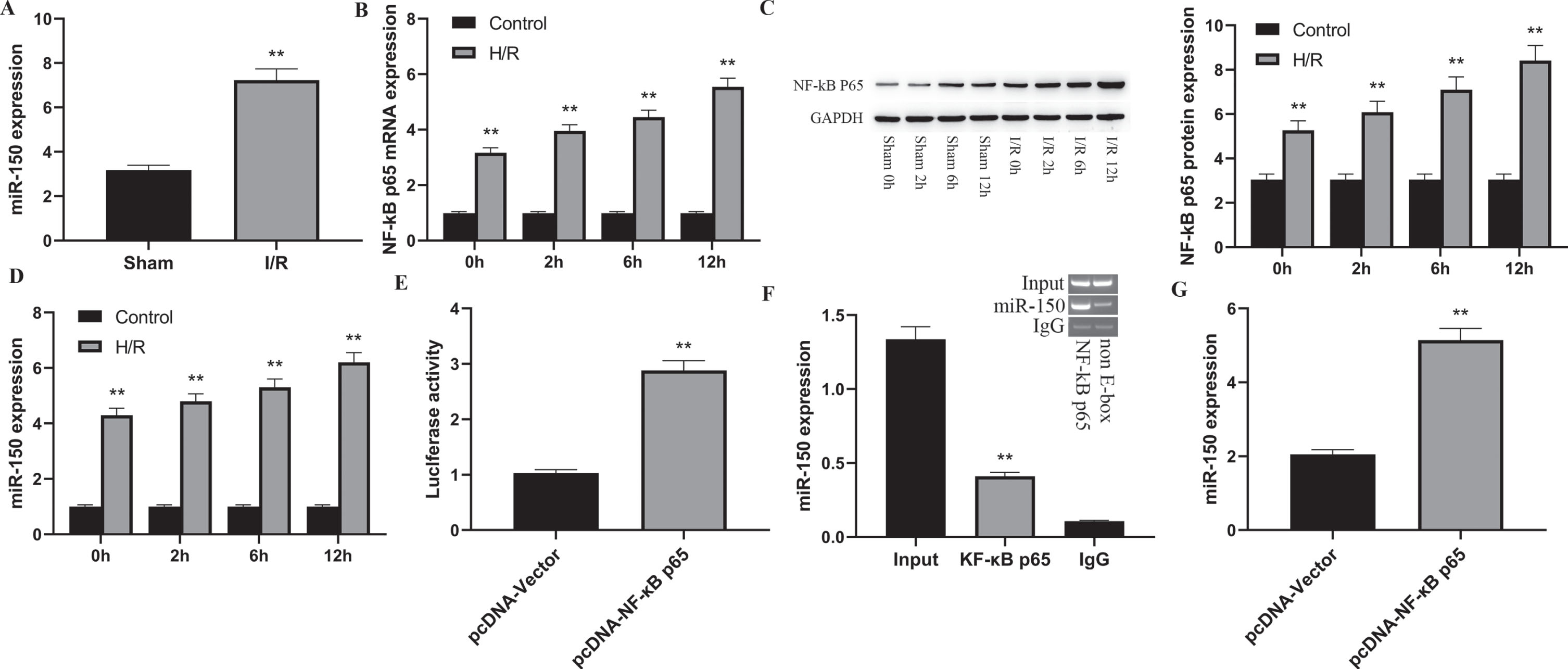
The research found that NF-kB p65 can bind directly to the promoter of miR-150 and enhance its expression. In this investigation, the amount of production of miR-150 in rat renal tissues treated to I/R injury was assessed to see if NF-kB p65 enhances renal I/R injury by triggering miR-150. The transcription of miR-150 was considerably greater in the I/R group than in the Sham group (P < 0.01), as demonstrated in Fig. 2A. In addition, a in vitro I/R HK-2 cell model was established by subjecting cells to hypoxia followed by reoxygenation for 0 h, 2 h, 6 h, and 12 h to further detect the production of NF-kB p65 and miR-150. The analyses demonstrated that the levels of NF-kB p65 (Fig. 2B and C, P < 0.01) and miR-150 (Fig. 2D) were much greater in the H/R group than in the control group, and the concentrations of NF-kB p65 and miR-150 rose as the reoxygenation time increased, displaying a manner varied in time. To confirm that NF-kB p65 can promote miR-150 production, a luciferase reporter gene plasmid containing a upstream sequence of the miR-150 promoter region was established and co-transfected into HK-2 cells with pcDNA-NF-kB p65. The luciferase reporter gene assay showed that, when compared to the control group, transfection of pcDNA-NF-kB p65 led to an impressive rise in luciferase expression in miR-150-WT group cells (p < 0.01), while activity in miR-150-MUT group cells was unaffected (Fig. 2E). In addition, as illuatrated in Fig. 2F and 2 G, ChIP analysis showed that the amount of miR-150 pushed down by NF-kB p65 was much greater than IgG (p < 0.01). Moreover, when NF-kB p65 was overexpressed, there was a substantial rise in miR-150 transcript in the pcDNA-NF-kB p65 group in comparison to the pcDNA-Vector group (p < 0.01). The results presented here demonstrate that NF-kB p65 may interact with the promoter region of miR-150 in HK-2 cells, increasing its activity.
Fig. 2
NF-kB p65 can enhance miRNA-150 expression.
3.3The impact of miR-150 on HK-2 cells injured by H/R
An in vitro model of HK-2 cells with varying durations of hypoxia followed by reoxygenation was created to examine the influence of miR-150 on renal I/R damage. Overexpression and suppression of miR-150 were achieved employing transfection of miR-150-mimic and miR-150-inhibitor. As illustrated in Fig. 3A, there was an enormous rise in miR-150 production in the miR-150-mimic group compared to the NC-mimic group (p < 0.01). As demonstrated in Fig. 3G, the miR-150-inhibitor group had a substantial drop in miR-150 expression (p < 0.01). However, regardless of overexpression or inhibition of miR-150, miR-150 transcription in HK-2 cells rose in a manner that correlated with extended reoxygenation time. The death rate in HK-2 cells was analyzed, and it was discovered that, with regard to the NC-mimic group, the apoptosis rate in the miR-150-mimic group was considerably greater (Fig. 3B, p < 0.01), whereas inhibiting miR-150 expression contributed to a significantly lower cell apoptosis rate (Fig. 3H, p < 0.01). Observation of HK-2 cell proliferation was also carried out. The outcomes demonstrated that excessive expression of miR-150 reduced HK-2 cell proliferation markedly (Fig. 3C, p < 0.01), whereas suppression of miR-150 expression raised the proliferation of cells notably (Fig. 3I, p < 0.01). However, when the time of reoxygenation increased in both groups, HK-2 cell proliferation decreased.
Fig. 3
The impact of miR-150 on HK-2 cells injured by H/R.
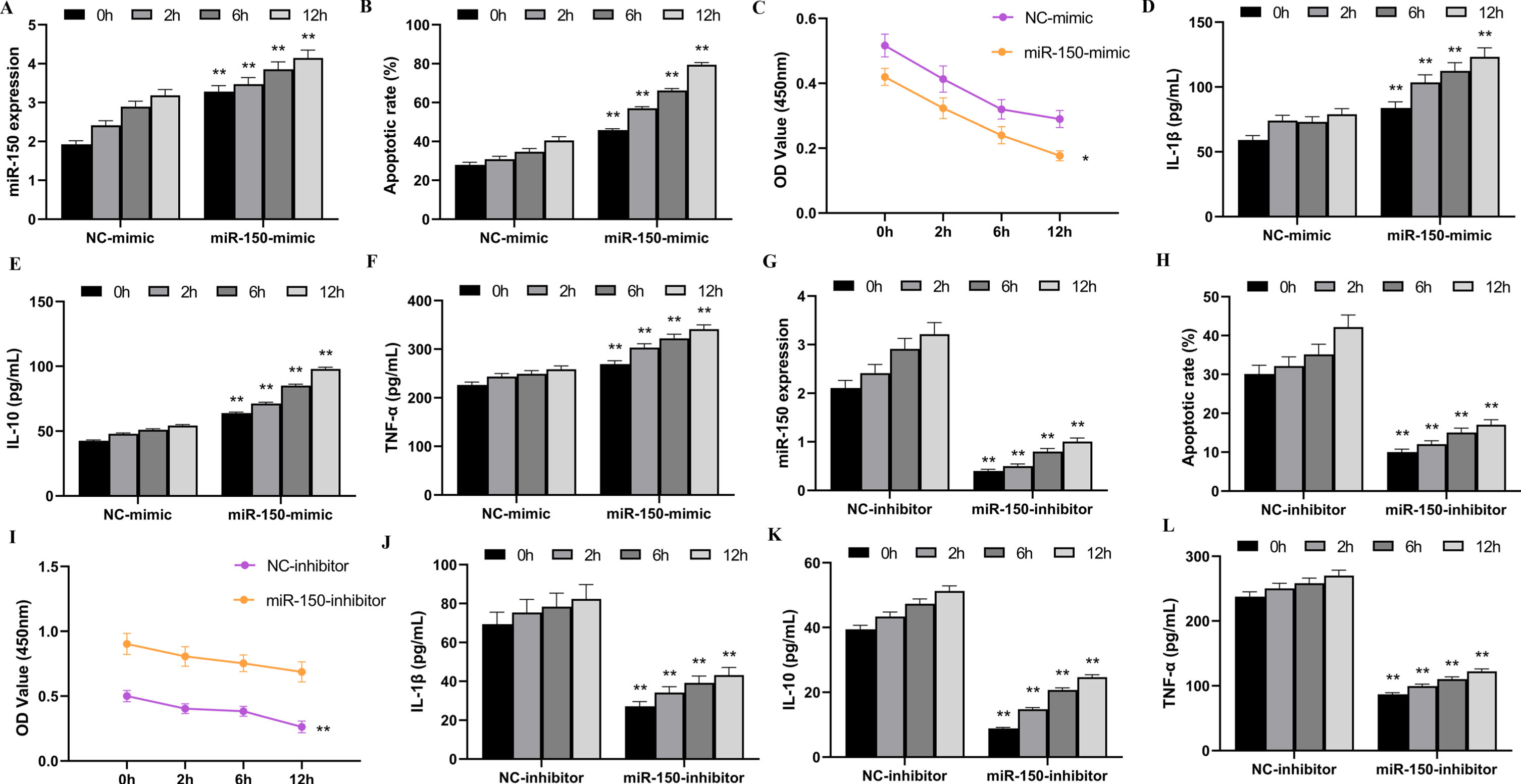
The amounts of inflammatory factors in HK-2 cells following H/R treatment, including IL-1β, IL-10, and TNF-α was also assessed using ELISA. The findings indicated that the significant increase in IL-1β, IL-10, and TNF-α amounts was observed due to miR-150 overexpression (Fig. 3D, E, and F, p < 0.01), while miR-150 inhibition reduced the quantity of these inflammatory factors (Fig. 3J, K, and L, p < 0.01). The results presented demonstrate that miR-150 can cause damage in HK-2 cells when placed under hypoxia-reoxygenation.
3.4MiR-150 can promote H/R injury in HK-2 cells by targeting and inhibiting TRPC6
According to the TargetScan database, TRPC6 was discovered as a putative desired gene of miR-150 (Fig. 4A). There is evidence from studies suggesting that TRPC6 appears to have a beneficial impact on renal I/R damage. To see if miR-150 affects TRPC6, HK-2 cells overexpressing miR-150 were co-transfected with pcDNA-TRPC6 3’-UTR or pcDNA-TRPC6 mut-3’-UTR mutant. The dual-luciferase reporter gene test corroborated the findings. As illustrated by Fig. 4B, the excessive expression of miR-150 drastically lowered the luciferase activity of TRPC6-WT cells in comparison to control cells (p < 0.01), while the luciferase activity of TRPC6-MUT cells remained unchanged. The above results demonstrate that miR-150 is able to target the TRPC6’s 3’-UTR region, suppressing transcription and regulating expression, implying that miR-150 targets TRPC6 mRNA through its 3’-UTR.
Fig. 4
miR-150 promotes the release of inflammatory factors and H/R injury in HK-2 cells by acting as a targeted inhibitor of TRPC6.
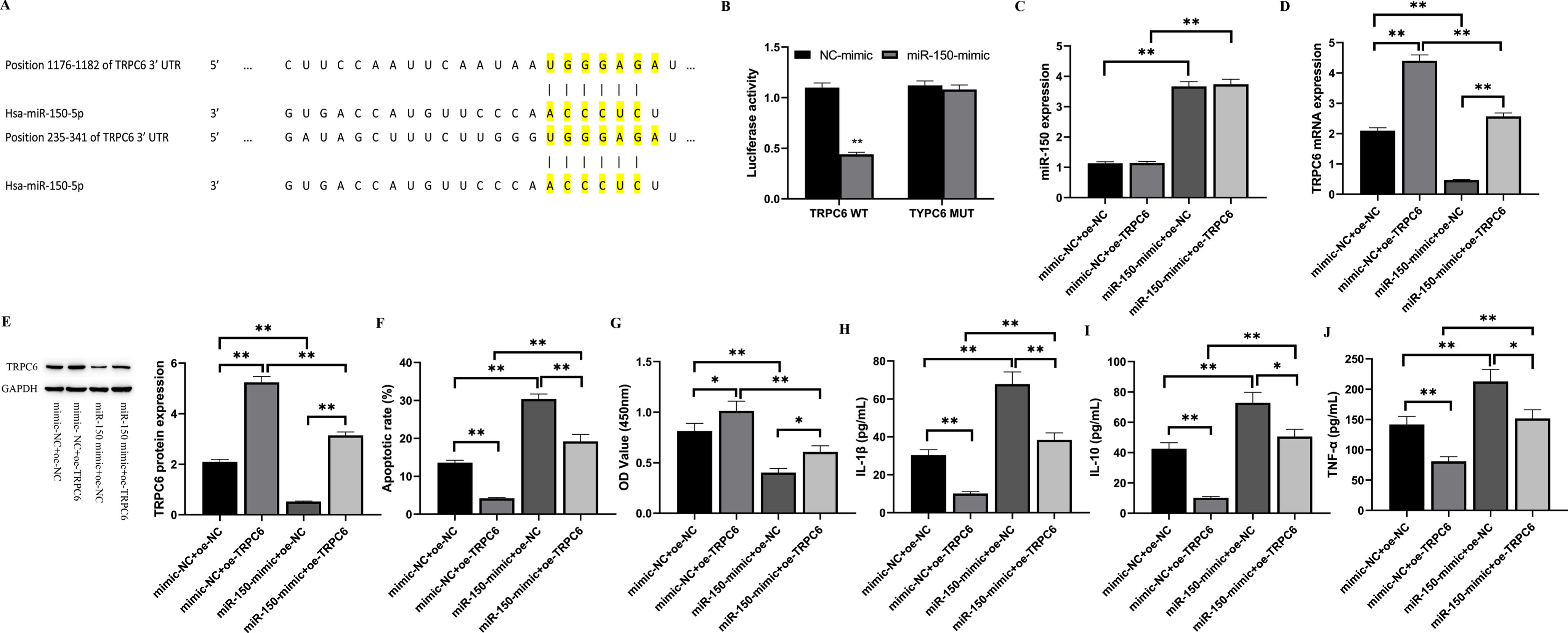
Furthermore, in the current investigation, we examined the implications of changing TRPC6 transcription on HK-2 cells undergoing I/R damage. The data in Fig. 4C indicates that the transcript levels of miR-150 was considerably greater in the miR-150-mimic+oe-NC group than in the mimic-NC+oe-NC group (p < 0.01). TRPC6 mRNA and protein expression levels were considerably reduced in the miR-150-mimic+oe-NC group in comparison to the mimic-NC+oe-NC group (p < 0.01), indicating that miR-150 targets and inhibits TRPC6 expression. Additionally, the effect of miR-150 and TRPC6 on HK-2 cell death and proliferation was studied. Based on the data presented in Fig. 4F, it can be inferred that the excessive expression of miR-150 contributed to a important rise in the apoptosis rate of HK-2 cells in the miR-150-mimic+oe-NC group compared to the mimic-NC+oe-NC group (p < 0.01), while the TRPC6 overexpression notably lowered the apoptosis rate in the mimic-NC+oe-TRPC6 group (p < 0.01). The apoptosis rate of cells in the miR-150-mimic+oe-TRPC6 group was considerably lower than that of the miR-150-mimic+oe-NC group (p < 0.05). In addition, cell proliferation was tested, and Fig. 4G exhibits the outcomes, which indicates that the overexpression of miR-150 significantly decreased HK-2 cell proliferation (p < 0.01), while TRPC6 overexpression significantly promoted cell proliferation. The impact of miR-150/TRPC6 on inflammatory factors in cells was also investigated in this study. Figures 4H, I, and J show the levels of IL-1β, IL-10, and TNF-α, respectively. The levels of IL-1β, IL-10, and TNF-α (p < 0.01) had a significant increase in the miR-150-mimic+oe-NC group, compared to the mimic-NC+oe-NC group. TRPC6 overexpression exhibited a substantial decline inflammatory factor levels in the mimic -NC+oe-TRPC6 group (p < 0.01). Furthermore, inflammatory factor levels in cells were considerably lower in the miR-150-mimic+oe-TRPC6 group than in the miR-150-mimic+oe-NC group (p < 0.05). These results indicate that TRPC6 can protect I/R injury in HK-2 cells and reduce the level of inflammatory markers following I/R treatment. In conclusion, miR-150 can target and inhibit TRPC6 in HK-2 cells, promoting I/R damage.
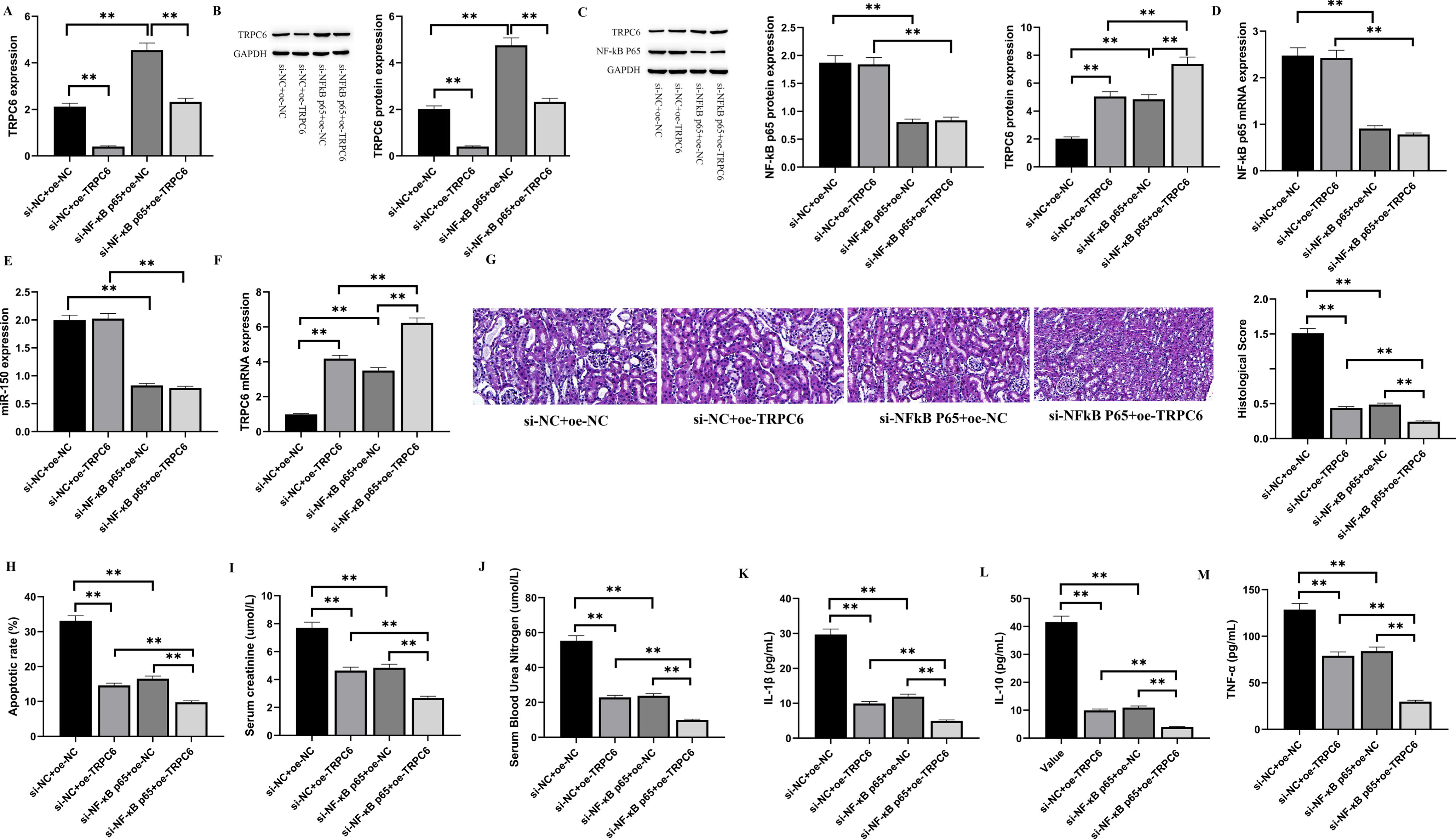
3.5NF-kB p65 activates miR-150 to target and inhibit TRPC6 and promotes renal I/R injury in rats
To see if NF-kB p65 exerts its biological function through miR-150/TRPC6, we carried out a rescue experiment and discovered that the knockdown of NF-kB p65 caused a large rise in TRPC6 expression in the tissue of rat renal I/R injury. The observed outcome could be reversed by introducing a miR-150 mimic, as illustrated in Fig. 5A and 5B. Therefore, we concluded that NF-kB p65 can regulate TRPC6 transcription by miR-150. We designed lentiviral vectors carrying si-NC, si-NF-kB p65, oe-NC, and oe-TRPC6 to investigate the biological process that explains how NF-kB p65 encourages rat renal I/R damage by triggering miR-150 to decrease TRPC6, then injected them into the rat models of renal I/R through tail vein. After detecting the levels of NF-kB p65, miR-150, and TRPC6 in all groups (Fig. 5C to F), it was found that wiping out NF-kB p65 greatly lowered the levels of NF-kB p65 and miR-150. However, wiping out NF-kB p65 or overexpressing TRPC6 resulted in a important upregulation of TRPC6 expression. This research discloses that NF-kB p65 regulates TRPC6 expression via miR-150. Figure 5G shows the morphological and histological evaluation of the kidney tissues of rats after renal I/R surgery. The si-NC+oe-NC group showed cytoplasmic vacuoles of renal tissue cells, growth of renal tubules, and evident spatial changes in the cytoplasm, but the si-NC+oe-TRPC6, si-NF-kB p65+oe-NC, and si-NF-kB p65+oe-TRPC6 groups did not. Therefore, the histological score of the si-NC+oe-NC group was higher (P < 0.01). In addition, the apoptotic rate of rat renal tissues after I/R was determined. As shown in Fig. 5H, both silencing NF-kB p65 and overexpressing TRPC6 might diminish cell apoptosis produced by renal I/R in rats. Creatinine (Cr) and blood urea nitrogen (BUN) were detected to monitor renal function in rats. As illustrated in Figs. 5I and 5J, relative to the si-NC+oe-NC group, the levels of blood creatinine and BUN in the si-NF-kB p65+oe-NC group and the si-NC+oe-TRPC6 group had a significant reduction (p < 0.01). To evaluate the concentration of inflammatory factors in rat kidneys after I/R, ELISA was employed to measure the amounts of IL-1β, IL-10, and TNF-α in rat renal tissues. As shown in Figs. 5K, 5L, and 5M, both silencing NF-kB p65 and overexpressing TRPC6 could reduce the levels of inflammatory factors produced by renal I/R in rats. Compared with the si-NC+oe-TRPC6 group and the si-NF-kB p65+oe-NC group, the concentration of inflammatory factors in the si-NF-kB p65+oe-TRPC6 group was considerably lower (p < 0.05). These findings indicate that inhibiting NF-kB p65 and overexpressing TRPC6 both have an encouraging impact on rat renal I/R damage.
Fig. 5
NF-kB p65 promotes renal I/R injury in rats by inhibiting TRPC6 expression via miR-150.
4Discussion
When interpreting the results of this study, we can draw scientific conclusions and clinical applications for the future: NF-kB p65 can suppress TRPC6 expression by activating miR-150, thus promoting kidney IRI. The study revealed a new molecular mechanism and therapeutic strategy, providing a theoretical and practical basis for further understanding IRI.
Research conducted before has demonstrated that quercetin has a protective effect on myocardial IRI, as well as its molecular mechanisms. This chemical has been proven to prevent cardiac damage induced by IRI by suppressing the NF-kB pathway [29]. Despite this, the link between NF-kB and kidney injury resulting from IRI remains unproven. Our study revealed that NF-kB p65/miR-150 was overexpressed in rat kidney I/R injury tissue, which is the same as previous studies, but the mechanism of NF-kB in kidney I/R injury still needs further investigation. Researchers have also found that some miR promoter regions contain elements that NF-kB can bind [30] and the expression of miR-150 can be induced by direct binding of NF-kB p65 to its promoter [27], as demonstrated by our study. Furthermore, it was discovered over a decade ago that the existence of TRPC6 gene mutations was linked to the development of familial renal illness [31]. Then TRPC6, as a potential therapeutic target, is being widely studied. In recent years, TRPC6 has been discovered to improve kidney I/R injury [32]. Combined with our findings, we conclude that miR-150 can target and inhibit TRPC6, promoting the I/R injury in HK-2 cells. It not only sheds light on the molecular process underlying renal I/R injury, but also provides crucial clues for novel options for therapy.
Although the results of this study have potential clinical value, we have to be aware of limitations. Using HK-2 cells for experiments can not show the whole process of kidney IRI injury. Further research is required to validate the results of this study and to provide additional data support for the development of novel treatment approaches for kidney IRI.
In summary, this study reveals the molecular mechanisms of kidney IRI by exploring the NF-kB p65/miR-150/TRPC6 signaling pathway. In the future, we will further explore the interaction between NF-kB and other genes, as well as its potential role in various physiological and pathological processes of other organs, providing a more comprehensive treatment approach for different types of tissue I/R injury.
Ethical statement
This study has been approved by the Ethics Committee of The Third Affiliated Hospital of Qiqihar Medical University on June 6, 2022 (QMU-AECC-2022-69). All animal experiments were performed following the guidelines issued by National Institutes of Health.
Conflict of interest
The authors have stated that they have no conflict of interest.
Data availability statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Funding
Health Commission of Heilongjiang Province (20220202050966).
Authorship
(1) Conceptualization and design: Lei Pang, Shuangyu Li
(2) Administrative support: Zengwei Ma
(3) Provision of study materials or patients: Shuangyu Li, Shanshan Sun
(4) Data collection and curation: Chunlei Yu, Baohua Gu
(5) Data analysis and interpretation: Ying Shi, Qiubo Ma, Xiaoyan Yu
(6) Manuscript writing: All authors
(7) Final approval of manuscript: All authors
References
[1] | Pefanis A , Ierino FL , Murphy JM , Cowan PJ . Regulated necrosis in kidney ischemia-reperfusion injury. Kidney Int. (2019) ;96: (2):291–301. doi: 10.1016/j.kint.2019.02.009. |
[2] | Sharfuddin AA , Molitoris BA . Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. (2011) ;7: (4):189–200. doi: 10.1038/nrneph.2011.16. |
[3] | Malek M , Nematbakhsh M . Renal ischemia/reperfusion injury; from pathophysiology to treatment. J Renal Inj Prev. (2015) ;4: (2):20–7. doi: 10.12861/jri2015.06. |
[4] | Dorrington MG , Fraser IDC . NF-κB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front Immunol. (2019) ;10: :705. doi: 10.3389/fimmu.2019.00705. |
[5] | Mahtal N , Lenoir O , Tinel C , Anglicheau D , Tharaux PL . MicroRNAs in kidney injury and disease. Nat Rev Nephrol. (2022) ;18: (10):643–62. doi: 10.1038/s41581-022-00608-6. |
[6] | Zhao XQ , Ao Y , Chen HY , Wang H . The role of miRNA in kidney development. Yi Chuan. (2020) ;42: (11):1062–72. Chinese. doi: 10.16288/j.yczz.20-112. |
[7] | Ghose J , Bhattacharyya NP . Transcriptional regulation of microRNA-100, -146a, and -150 genes by p53 and NFκB p65/RelA in mouse striatal STHdh(Q7)/ Hdh(Q7) cells and human cervical carcinoma HeLa cells. RNA Biol. (2015) ;12: (4):457–77. doi: 10.1080/15476286.2015.1014288. |
[8] | Dryer SE , Kim EY . The Effects of TRPC6 Knockout in Animal Models of Kidney Disease. Biomolecules. (2022) ;12: (11):1710. doi: 10.3390/biom12111710. |
[9] | Shen B , He Y , Zhou S , Zhao H , Mei M , Wu X . TRPC6 May Protect Renal Ischemia-Reperfusion Injury Through Inhibiting Necroptosis of Renal Tubular Epithelial Cells. Med Sci Monit. (2016) ;22: :633–41. doi: 10.12659/msm.897353. |
[10] | Li YW , Zhang Y , Zhang L , Li X , Yu JB , Zhang HT , Tan BB , Jiang LH , Wang YX , Liang Y , Zhang XS , Wang WS , Liu HG . Protective effect of tea polyphenols on renal ischemia/reperfusion injury via suppressing the activation of TLR4/NF-κB p65 signal pathway. Gene. (2014) ;542: (1):46–51. doi: 10.1016/j.gene.2014.03.021. |
[11] | Zhou Z , Hu Q , Guo H , Wang X . CircSEC11A knockdown alleviates oxidative stress and apoptosis and promotes cell proliferation and angiogenesis by regulating miR-29a-3p/SEMA3A axis in OGD-induced human brain microvascular endothelial cells (HBMECs). Clin Hemorheol Microcirc. (2023) ;84: (3):247–62. |
[12] | Zhu L , Zhao P , Meng X , Jin H , Tuo B . Circ_0091822 aggravates ox-LDL-induced endothelial cell injury through targeting the miR-661/RAB22A axis. Clin Hemorheol Microcirc. (2023) ;83: (1):47–59. |
[13] | Zhang P , Wang W , Li M . Role and mechanism of circular RNA circ_0050486 in regulating oxidized low-density lipoprotein-induced injury in endothelial cells. Clin Hemorheol Microcirc. (2022) ;82: (2):107–24. |
[14] | He D , Li Z , Chen Y , Huang M . Circular RNA circ_0029589 promotes ox-LDL-induced endothelial cell injury through regulating RAB22A by serving as a sponge of miR-1197. Clin Hemorheol Microcirc. (2023) ;83: (4):359–76. |
[15] | Teng Y , Ren F , Wang Y , Xu H , Song H . Circ_0033596 depletion ameliorates oxidized low-density lipoprotein-induced human umbilical vein endothelial cell damage. Clin Hemorheol Microcirc. (2023) ;84: (1):53–70. |
[16] | Shen B , Mei M , Pu Y , Zhang H , Liu H , Tang M , Pan Q , He Y , Wu X , Zhao H . Necrostatin-1 Attenuates Renal Ischemia and Reperfusion Injury via Meditation of HIF-1a/mir-26a/TRPC6/PARP1 Signaling. Mol Ther Nucleic Acids. (2019) ;17: :701–13. doi: 10.1016/j.omtn.2019.06.025. |
[17] | Shen B , Mei M , Pu Y , Zhang H , Liu H , Tang M , Pan Q , He Y , Wu X , Zhao H . Necrostatin-1 Attenuates Renal Ischemia and Reperfusion Injury via Meditation of HIF-1a/mir-26a/TRPC6/PARP1 Signaling. Mol Ther Nucleic Acids. (2019) ;17: :701–13. doi: 10.1016/j.omtn.2019.06.025. |
[18] | Slater SC , Jover E , Martello A , Mitic T , Rodriguez-Arabaolaza I , Vono R , Alvino VV , Satchell SC , Spinetti G , Caporali A , Madeddu P . MicroRNA-532-5p Regulates Pericyte Function by Targeting the Transcription Regulator BACH1 and Angiopoietin-1. Mol Ther. (2018) ;26: (12):2823–37. doi: 10.1016/j.ymthe.2018.08.020. |
[19] | Ranganathan P , Jayakumar C , Tang Y , Park KM , Teoh JP , Su H , Li J , Kim IM , Ramesh G . MicroRNA-150 deletion in mice protects kidney from myocardial infarction-induced acute kidney injury. Am J Physiol Renal Physiol. (2015) ;309: (6):F551–8. doi: 10.1152/ajprenal.00076.2015. |
[20] | Sun X , Wei Z , Li Y , Wang J , Hu J , Yin Y , Xie J , Xu B . Renal denervation restrains the inflammatory response in myocardial ischemia-reperfusion injury. Basic Res Cardiol. (2020) ;115: (2):15. doi: 10.1007/s00395-020-0776-4. |
[21] | He Z , Tang H , You X , Huang K , Dhinakar A , Kang Y , Yu Q , Wu J . BAPTA-AM Nanoparticle for the Curing of Acute Kidney Injury Induced by Ischemia/Reperfusion. J Biomed Nanotechnol. (2018) ;14: (5):868–83. doi: 10.1166/jbn.2018.2532. |
[22] | Liu F , Ni W , Zhang J , Wang G , Li F , Ren W . Administration of Curcumin Protects Kidney Tubules Against Renal Ischemia-Reperfusion Injury (RIRI) by Modulating Nitric Oxide (NO) Signaling Pathway. Cell Physiol Biochem. (2017) ;44: (1):401–11. doi: 10.1159/000484920. Epub 2017 Nov 13. |
[23] | Lan F , Yue X , Ren G , Li H , Ping L , Wang Y , Xia T . miR-15a/16 enhances radiation sensitivity of non-small cell lung cancer cells by targeting the TLR1/NF-κB signaling pathway. Int J Radiat Oncol Biol Phys. (2015) ;91: (1):73–81. doi: 10.1016/j.ijrob2014.09.021. |
[24] | Mao A , Zhao Q , Zhou X , Sun C , Si J , Zhou R , Gan L , Zhang H . MicroRNA-449a enhances radiosensitivity by downregulation of c-Myc in prostate cancer cells. Sci Re. (2016) ;6: :27346. doi: 10.1038/srep27346. |
[25] | Wu H , Huang T , Ying L , Han C , Li D , Xu Y , Zhang M , Mou S , Dong Z . MiR-155 is Involved in Renal Ischemia-Reperfusion Injury via Direct Targeting of FoxO3a and Regulating Renal Tubular Cell Pyroptosis. Cell Physiol Biochem. (2016) ;40: (6):1692–705. doi: 10.1159/000453218. Epub 2016 Dec 23. |
[26] | Slater SC , Jover E , Martello A , Mitic T , Rodriguez-Arabaolaza I , Vono R , Alvino VV , Satchell SC , Spinetti G , Caporali A , Madeddu P . MicroRNA-532-5p Regulates Pericyte Function by Targeting the Transcription Regulator BACH1 and Angiopoietin-1. Mol Ther. (2018) ;26: (12):2823–37. doi: 10.1016/j.ymthe.2018.08.020. |
[27] | Qi M , Zheng L , Qi Y , Han X , Xu Y , Xu L , Yin L , Wang C , Zhao Y , Sun H , Liu K , Peng J . Dioscin attenuates renal ischemia/reperfusion injury by inhibiting the TLR4/MyD88 signaling pathway via up-regulation of HSP70. Pharmacol Res. (2015) ;100: :341–52. doi: 10.1016/j.phrs.2015.08.025. |
[28] | Ranganathan P , Jayakumar C , Tang Y , Park KM , Teoh JP , Su H , Li J , Kim IM , Ramesh G . MicroRNA-150 deletion in mice protects kidney from myocardial infarction-induced acute kidney injury. Am J Physiol Renal Physiol. (2015) ;309: (6):F551–8. doi: 10.1152/ajprenal.00076.2015. |
[29] | Liu X , Yu Z , Huang X , Gao Y , Wang X , Gu J , Xue S . Peroxisome proliferator-activated receptor γ (PPAR γ) mediates the protective effect of quercetin against myocardial ischemia-reperfusion injury via suppressing the NF-κB pathway. Am J Transl Res. (2016) ;8: (12):5169–86. |
[30] | Yang Z , Fang S , Di Y , Ying W , Tan Y , Gu W . Modulation of NF-κB/miR-21/PTEN pathway sensitizes non-small cell lung cancer to cisplatin. PLoS One. (2015) ;10: (3):e0121547. doi: 10.1371/journal.pone.0121547. |
[31] | Hall G , Wang L , Spurney RF . TRPC Channels in Proteinuric Kidney Diseases. Cells. (2019) ;9: (1):44. doi: 10.3390/cells9010044. |
[32] | Pu Y , Zhao H , Shen B , Zhou Q , Xie P , Wu X . TRPC6 ameliorates renal ischemic reperfusion injury by inducing Zn2+ influx and activating autophagy to resist necrosis. Ann Transl Med. (2022) ;10: (5):249. doi: 10.21037/atm-21-5837. |


