
NADPH-cytochrome P450 reductase expression and enzymatic activity in primary-like human hepatocytes and HepG2 cells for in vitro biotransformation studies
Abstract
BACKGROUND:
Human hepatocyte in vitro cell culture systems are important models for drug development and toxicology studies in the context of liver xenobiotic metabolism. Often, such culture systems are used to elucidate the biotransformation of xenobiotics or drugs and further investigate drug and drug metabolite effects on biological systems in terms of potential therapeutic benefit or toxicity. Human hepatocytes currently used for such in vitro studies are mostly primary cells or cell lines derived from liver cancers. Both approaches have limitations such as low proliferation capacity and progressive dedifferentiation found in primary cells or lack of liver functions in cell lines, which makes it difficult to reliably predict biotransformation of xenobiotics in patients. In order to overcome these limitations, HepaFH3 cells and Upcyte® hepatocytes representing primary-like hepatocytes of the first and second generation are increasingly used. Based on primary human hepatocyte cells transduced for stable expression of Upcyte® proliferation genes, they are mitotically active and exhibit liver functions over an extended period, making them comparable to primary human hepatocytes. These hepatocyte models show active liver metabolism such as urea and glycogen formation as well as biotransformation of xenobiotics. The latter is based on the expression, activity and inducibility of cytochrome P450 enzymes (CYP) as essential phase I reaction components. However, for further characterisation in terms of performance and existing limitations, additional studies are needed to elucidate the mechanisms involved in phase I reactions. One prerequisite is sufficient activity of microsomal NADPH-cytochrome P450 reductase (POR) functionally connected as electron donor to those CYP enzymes.
OBJECTIVE:
For Upcyte® hepatocytes and HepaFH3 cells, it is so far unknown to what extent POR is expressed, active, and may exert CYP-modulating effects. Here we studied POR expression and corresponding enzyme activity in human hepatoblastoma cell line HepG2 and compared this with HepaFH3 and Upcyte® hepatocytes representing proliferating primary-like hepatocytes.
METHODS:
POR expression of those hepatocyte models was determined at mRNA and protein level using qRT-PCR, Western Blot and immunofluorescence staining. Kinetic studies on POR activity in isolated microsomes were performed by a colorimetric method.
RESULTS:
The investigated hepatocyte models showed remarkable differences at the level of POR expression. Compared to primary-like hepatocytes, POR expression of HepG2 cells was 4-fold higher at mRNA and 2-fold higher at protein level. However, this higher expression did not correlate with corresponding enzyme activity levels in isolated microsomes, which were comparable between all cell systems tested. A tendency of higher POR activity in HepG2 cells compared to HepaFH3
(p = 0.0829) might be present. Compared to primary human hepatocyte microsomes, POR activity was considerably lower in all hepatocyte models.
CONCLUSION:
In summary, our study revealed that POR expression and activity were clearly detectable in all in vitro hepatocyte models with the highest POR expression in cancer cell line HepG2. However, POR activity was lower in tested hepatocyte models when compared to human primary hepatocyte microsomes. Whether this was caused by e.g. polymorphisms or metabolic differences of investigated hepatocyte models will be target for future studies.
1Introduction
The liver as one of human’s largest organs is a main location for metabolisation and clearance of pharmaceuticals or xenobiotics due to its high expression of phase I and II enzymes [1, 2]. For this reason, hepatocytes are intensively studied for drug development, diagnostics and therapeutic applications to save costs and reduce drug side effects. There is still a great need for the development of reliable in vitro hepatocyte metabolism models for preclinical screening of drug conversion, clearance and potential hepatotoxicity.
A clear understanding of the enzymatic interplay to enable complete liver phase I and phase II reactions is crucial for the prediction of drug pharmacokinetics. This can be affected by dynamic variability within and between individuals, age-related modifications as well as by genetic polymorphisms of relevant enzymes [2–4]. In phase I metabolism, cytochrome P450 monooxygenases (CYPs) represent the most prominent enzyme family for oxidative biotransformation of drugs and other lipophilic xenobiotics [5, 6]. From the 57 known human CYPs only about a handful enzymes, mostly belonging to CYP-families 1, 2 and 3, are responsible for the metabolisation of more than three quarters of FDA-approved drugs [7, 8].
Preclinical evaluation of novel drug candidates and scientific investigation of already used drugs rely on physiologically relevant in vitro models of human hepatocytes for metabolism, biotransformation and toxicology studies. Currently, primary human hepatocytes (pHHs) are the “gold standard” for in vitro studies on hepatic metabolism, clearance, hepatotoxicity and drug-drug interaction [9]. However, this research is still restricted by pHH scarcity, donor variability and their rapid dedifferentiation in vitro [10–14]. An inflammatory response by endotoxin contamination [15, 16] originating from bacterial collagenase preparations, loss of normal cell polarity when dissolving them from liver tissue or down-regulation of liver-specific transcription factors influencing phase I/II protein expression were discussed as possible causes [17–19].
To overcome these limitations, several liver cancer-derived cell lines such as HepaRG and HepG2 were developed to serve as surrogate for pHHs. Advantages are their unlimited availability, convenient handling and proliferative capacity [20–24]. A clear disadvantage is their genetic instability due to their cancer origin, which makes them almost unusable for clinical applications such as disease-related liver repopulation. Widely used HepG2 cells, a human hepatoblastoma cell line, exhibit only low expressions and activities of nearly all phase I CYP enzymes and therefore have limited value for prediction of hepatic biotransformation. However, several approaches to increase HepG2 liver function were described in the literature [25, 26]. These include genetic engineering of the cell line to enhance CYP expression. Recently published data demonstrated the successful generation of a genetically modified CYP3A4-overexpressing HepG2 clone, showing considerable enzymatic activity for this specific CYP [27].
A promising approach to obtain a more relevant surrogate of pHHs for in vitro biotransformation and toxicology studies is the use of primary-like hepatocytes such as HepaFH3 cells and Upcyte® hepatocytes, which are now available in the second generation [28, 29]. These cell strains are proliferation-competent by lentivirus-mediated transduction of defined proliferation genes (Upcyte® factors). They show some improved liver functions compared to cell lines such as HepG2 or HepaRG, but perform still less than freshly isolated pHHs. Data on the expression and inducibility of CYP enzymes show that essential components of the phase I and phase II reactions are present and active [29–33].
However, for primary-like hepatocytes of both generations little is known about the expression, activity and perhaps associated CYP-modulating effects of the NADPH-cytochrome P450 reductase (POR or CPR). This 77 kDa microsomal flavoprotein, which is present in almost all tissues to various extent, plays a vital role both in biotransformation and clearance of xenobiotics as well as for biosynthesis and metabolism of miscellaneous endogenous substances such as cholesterol, steroid hormones and bile acids [34]. POR is an indispensable part of oxidative processes catalysed by several oxygenase enzymes [2]. For microsomal cytochrome P450 monooxygenases (CYPs), which are strongly involved in the hepatic drug metabolism, POR acts as obligatory electron donor and is therefore crucial for biotransformation at all. Via its FMN/FAD flavogroups, POR mediates the transfer of two single electrons originating from NADPH to the CYP’s prosthetic haem iron. Stoichiometrically, liver POR expression is up to 10 times lower compared to microsomal CYPs, suggesting that POR could be a limiting factor for the activity of CYP monooxygenases [35, 36]. Indeed it was shown, that the amount of POR in human liver as well as the enzyme activity of a particular polymorphic variant impact expression and activity levels of several relevant CYP enzymes [13, 37, 38]. Interindividual variability in POR expression and activity can be caused by influences on gene regulation at the transcriptional level by transcription factors like PXR or CAR, by hormonal influences or polymorphism in the POR gene itself [3, 4, 39]. Genetically-caused POR deficiency in vivo can lead to dysfunctions in steroid genesis, ambiguous genitalia and Antley-Bixler syndrome [40, 41]. More than 50 polymorphic mutations in the human POR gene have been described so far, modulating the activity of various CYP enzymes in varying degrees of severity [4, 42, 43]. However, the mutual influence strongly depends on the interplay between POR gene polymorphism that affect the enzyme activity, CYP isoforms and the substrate to be metabolised. Hence, the interaction of a specific POR variant with a particular CYP enzyme does not predict its interaction with other CYPs.
For the assessment of a hepatocyte model system to be relevant for biotransformation of pharmaceuticals and to uncover potential limitations, a comprehensive characterisation of all relevant liver phase I/II components is indispensable. In this study, we therefore compared the expression and corresponding POR activity between HepG2, HepaFH3 and Upcyte® hepatocytes.
2Methods
2.1Cell culture
Commercially available human hepatocellular carcinoma (HepG2) cells (HB-8065, ATCC, Manassas, VA, USA) were cultured under standard conditions (37°C, 5% CO2) in polystyrene-based tissue culture flasks (SARSTEDT AG & Co. KG, Nümbrecht, Germany) in Dulbecco’s minimal essential medium (D-MEM) supplemented with 10% fetal bovine serum (FBS) superior, 6 mM L-alanyl-L-glutamine and 49.2 g/L NaHCO3, all purchased from Biochrom GmbH (Berlin, Germany). HepaFH3 cells [29] and second generation Upcytes® (donor 653-03, Upcyte® technologies GmbH, Hamburg, Germany) [33] were cultivated under standard conditions on collagen I (Corning Inc., Corning, NY, USA) coated culture vessels in Hepatocyte Culture Medium (Upcyte® technologies GmbH, Hamburg, Germany).
For POR expression experiments, cells from three consecutive passages were used. More precisely, HepG2 cells from passages 19 to 21, HepaFH3 from passages 15 to 17 and second generation Upcyte® hepatocytes from passages 8 to 10 were used, whereby cell culture medium was replaced every second day. For either cell passaging or experimental seeding, hepatocytes were harvested by trypsin/EDTA treatment (0.05% v/v Trypsin and 0.02% v/v EDTA in water, Biochrom GmbH, Berlin, Germany). Prior to any experiment, morphological evaluation of the hepatocytes was performed using an Olympus CKX41 inverted microscope (Olympus Corporation, Tokyo, Japan).
2.2Quantitative real-time PCR
Total RNA was extracted from 2 × 106 cells with innuPREP RNA Mini Kit (Analytik Jena AG, Jena, Germany) according to manufacturer’s instructions. RNA amount and purity were examined by spectrophotometric analysis with a NanoDrop™ 1000 (Thermo Fisher Scientific Inc., Waltham, MA, USA). Subsequently, DNase digestion was realised with the DNA-free kit (Ambion, Life Technologies AG, Carlsbad, CA, USA) to eliminate genomic DNA, followed by verification of RNA integrity (28S and 18S rRNA bands) by agarose gel electrophoresis and ethidium bromide staining. For reverse transcription reaction with Revert Aid H Minus Reverse Transcriptase (RT; 10 U/μL, Thermo Fisher Scientific Inc., Waltham, MA, USA), 2 μg of isolated RNA were used. The reaction was supplemented with Oligo(dT)18 primers (25 ng/μL, Thermo Fisher Scientific Inc., Waltham, MA, USA) and dNTPs (1 mM, Carl Roth GmbH + Co. KG, Karlsruhe, Germany). Additionally, for assessment of contamination with genomic DNA, a reverse transcriptase minus control (–RT) was prepared. Finally, cDNA (+ and –RT) was diluted 1:10 in DEPC water (Carl Roth GmbH + Co. KG, Karlsruhe, Germany) and stored at –80°C until use. Samples for qRT-PCR were prepared with Maxima Probe qPCR Master Mix (Thermo Fisher Scientific Inc., Waltham, MA, USA) and intercalating dye Evagreen (Biotium Inc., Fremont, CA, USA). Primers obtained from BioTeZ Berlin Buch GmbH (Berlin, Germany) were used in a final concentration of 0.25 μM with the sequences shown in Table 1.
Table 1
Primer sequences for qRT-PCR
| Primer name | Sequence (5’⟶ 3’) | Amplicon size [bp] |
| POR Forward | AAGGCGGTGCCCACATCTAC | 151 |
| POR Reverse | TAGCGGCCCTTGGTCATCAG | |
| GAPDH Forward | TGCACCACCAACTGCTTAGC | 87 |
| GAPDH Reverse | GGCATGGACTGTGGTCATGAG | |
| SDHA Forward | CGAACGTCTTCAGGTGCTTT | 147 |
| SDHA Reverse | AAGAACATCGGAACTGCGAC |
For gene expression analysis of POR by quantitative real-time (qRT)-PCR, the CFX96 Real-Time System was used (Bio-Rad Laboratories, Inc., Hercules, CA, USA). The expression of GAPDH and SDHA was determined in addition to POR for normalisation of POR expression by 2(–ΔΔCt) method. All samples were analysed in duplicates except of – RT and NTC (no template control). For verification of gene specific amplicons, melting curve and agarose gel analyses were performed after qRT-PCR.
2.3Western blot
Protein extraction for Western blot analysis was performed from frozen cell pellets (2×106 cells) by 15 min incubation in RIPA buffer on ice, supplemented with 1 mM PMSF (Carl Roth GmbH + Co. KG, Karlsruhe, Germany). Samples were subsequently centrifuged for 10 min at 10,000 rpm and 4°C. Supernatant’s protein content was quantified by Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific Inc., Waltham, MA, USA) according to manufacturer’s instructions. Protein extracts were diluted 1:6 in 6× Laemmli buffer, denatured for 5 min at 95°C and stored at – 20°C until use. SDS-PAGE was performed according to Laemmli et al. (1970) [44]. Briefly, 20 μg protein extract were loaded onto a 10 % resolving gel. For molecular weight assignment the MagicMark™ XP Western Protein Standard was used (Thermo Fisher Scientific Inc., Waltham, MA, USA). After blotting the proteins on a PVDF membrane (Carl Roth GmbH + Co. KG, Karlsruhe, Germany), unspecific binding sites were blocked by incubation for 1 hour at ambient temperature in 2% BSA (Carl Roth GmbH + Co. KG, Karlsruhe, Germany) in PBS. Immunodetection of POR was achieved by using a polyclonal rabbit-anti-POR IgG as primary antibody (1 mg/mL, Abcam, Cambridge, UK) diluted 1:1000 in 1% BSA in PBS. Primary antibodies of the loading controls GAPDH and α-tubulin were monoclonal mouse-anti-GAPDH IgG (0.5 mg/mL, antibodies-online GmbH, Aachen, Germany) and monoclonal mouse-anti-α-tubulin IgG1/κ (0.2 mg/mL, Thermo Fisher Scientific Inc., Waltham, MA, USA) in 1:5000 and 1:,2000 dilution, respectively. Incubation with primary antibodies occurred at 4°C overnight. Incubation of the blots with peroxidase conjugated secondary antibodies goat-anti-mouse IgG and goat-anti-rabbit IgG (both 1:2000 in 1% BSA solved in PBS, Sigma Aldrich, St. Louis, MO, USA) for 1 hour at ambient temperature facilitated protein detection by enhanced chemiluminescence (Hewitt, #20) reaction using the Ashersham Prime Western Blotting Detection Reagent (GE Healthcare, IL, USA) in combination with a biostep Celvin® S 420 chemiluminescence imaging system (biostep GmbH, Burkhardtsdorf, Germany).
2.4Immunofluorescence
Cells growing on glass slides were fixed for 30 min in 5% formaldehyde solution, followed by 10 min permeabilisation in 0.2% Triton-X100 solution. Blocking of unspecific binding sites occurred for 30 min in 3% BSA solved in PBS. All steps were performed at ambient temperature. Visual detection of POR protein expression was realised using polyclonal rabbit-anti-POR IgG as primary antibody (1 mg/mL, Abcam) at 1:100 dilution and a Cy3 conjugated goat-anti-rabbit IgG (1:200, Dianova GmbH, Hamburg, Germany) as secondary antibody. Incubation of the samples with the primary antibody occurred over night at 4°C and with the secondary antibody for 1 hour at ambient temperature. For assessment of background binding, samples were stained with a primary antibody of the same isotype (polyclonal rabbit IgG, 1 mg/mL, Abcam) but without target binding properties serving as isotype control. Staining of genomic DNA was realised by 5 min incubation of the samples with 4’,6-Diamidino-2-phenylindole dihydrochloride (DAPI, 0.2 μg/mL in PBS, Carl Roth GmbH + Co. KG, Karlsruhe, Germany). High resolution pictures were taken by using a LSM800 confocal Laser Scanning Microscope system and ZEN software for picture post processing (Carl Zeiss Microscopy GmbH, Jena, Germany).
2.5POR enzyme activity
Human hepatocyte microsomal fractions were extracted from 1×107 freshly harvested cells. Cell lysis occurred by sonication with a Sonopuls ultrasonic homogenizer equipped with an UW3100 ultrasonic probe (BANDELIN electronic GmbH & Co. KG, Berlin, Germany) with 3×10 s pulses at 40% amplitude intermitted by 20 s breaks to prevent heating of the samples. Microsome extraction was then performed with the Microsome Isolation Kit (BioVision Inc., Milpitas, CA, USA) according to manufacturer’s protocol, and the microsomal fractions were eluted in 200 μL cold storage buffer. Subsequently, an aliquot was diluted 1:5 in storage buffer and quantified by Pierce™ BCA Protein Assay Kit against a BSA standard (Carl Roth GmbH + Co. KG, Karlsruhe, Germany). Remaining microsomal extract was aliquoted and stored at – 80°C until use.
For determination of POR activity in microsomal fractions, the Cytochrome P450 Reductase Activity Kit (Abnova Corporation, Taipei City, Taiwan) was used. In addition to the positive control included in the kit (recombinant human POR; rhPOR), commercially available human hepatocyte microsomes (HHM, Thermo Fisher Scientific Inc., Waltham, MA, USA) pooled from biopsies of 50 healthy donors served as reference. As a control for POR activity in non-hepatic cells, microsomal fractions of Chinese hamster ovary cells (CHO cells) were used. Prior to the POR kinetic studies with microsomal fractions, a standard curve with the substrate glucose-6-phosphate (G6P) was separately prepared for calculation of POR activity. POR activity in microsomal fractions was measured in duplicates, whereby 5 μg protein were used per sample. For distinguishing between POR and background activity of remaining substrate reducing enzymes, microsomal fractions were treated with 0.1 μM Diphenyleneiodonium Chloride (DPI) as POR inhibitor. Kinetics of POR-mediated substrate reduction were measured for 30 min by OD460nm detection with a FLUO Star Omega microplate reader (Software version: 3.00 R2, BMG LABTECH GmbH, Ortenberg, German), followed by data analysis by MARS Data Analysis Software (Version: 2.41).
2.6Statistical analysis
One-way ANOVA with Holm-Sidak’s multiple comparison test was used to test for significant differences between groups using Prism 6 (GraphPad Software, San Diego, CA, USA). Statistical significance was assumed for p < 0.05 for all statistical analysis.
3Results
Here we were interested to investigate and compare POR expression and activity in cell cultures of HepG2, HepaFH3 and Upcyte® cells.
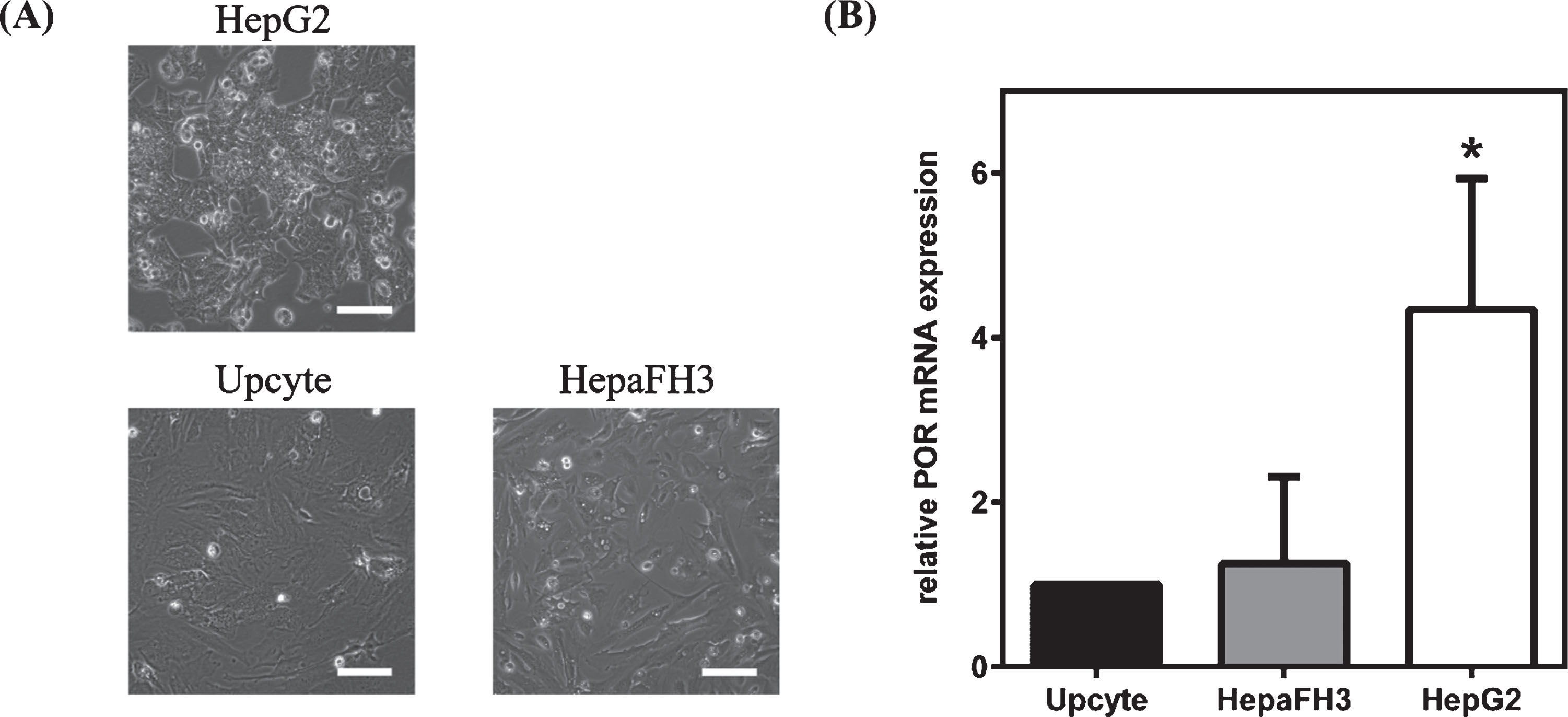
Two days after seeding, morphological evaluation revealed differences between HepG2 hepatoblastoma cell line and primary-like hepatocytes (Fig. 1A). HepG2 cells were considerably smaller and showed distinct formation of cell aggregates. HepaFH3 and second generation Upcyte® hepatocytes were of similar morphology, larger than HepG2, more spread, and they showed pronounced migration. Analysis of POR expression was carried out at both the intra- (passage-dependent) and the inter-cellular (cell line-dependent) level. The data from three consecutive passages revealed no significant variation in POR expression at the mRNA level within all hepatocyte culture systems. In contrast to the intra-cellular comparison, a normalisation to reference gene GAPDH expression was not possible for inter-cellular studies because there were substantial differences in GAPDH mRNA (data not shown) and protein (Fig. 2A) expression levels between the cell types. Therefore, SDHA, which showed no significant differences in mRNA expression between cell types, was used as a reference for determination of inter-cellular variability in POR gene expression. HepG2 cells showed a significantly 4-fold higher POR mRNA expression (p < 0.0001) compared to both primary-like hepatocyte systems (Fig. 1B). No significant difference in POR expression was detectable between HepaFH3 and Upcyte® cells at mRNA levels.
Fig. 1
Morphology and POR mRNA expression in HepG2, HepaFH3 and Upcyte® cells. (A) Exponentially growing cells were analysed by phase contrast microscopy. Pictures taken with Olympus CKX41 at 10× primary magnification. Scale bar: 100 μm. (B) POR mRNA expression analysis by qRT-PCR of exponentially growing cell cultures. Data shown as arithmetic mean±standard deviation; *p < 0.05; *significant changes compared to Upcyte cells (n = 9).
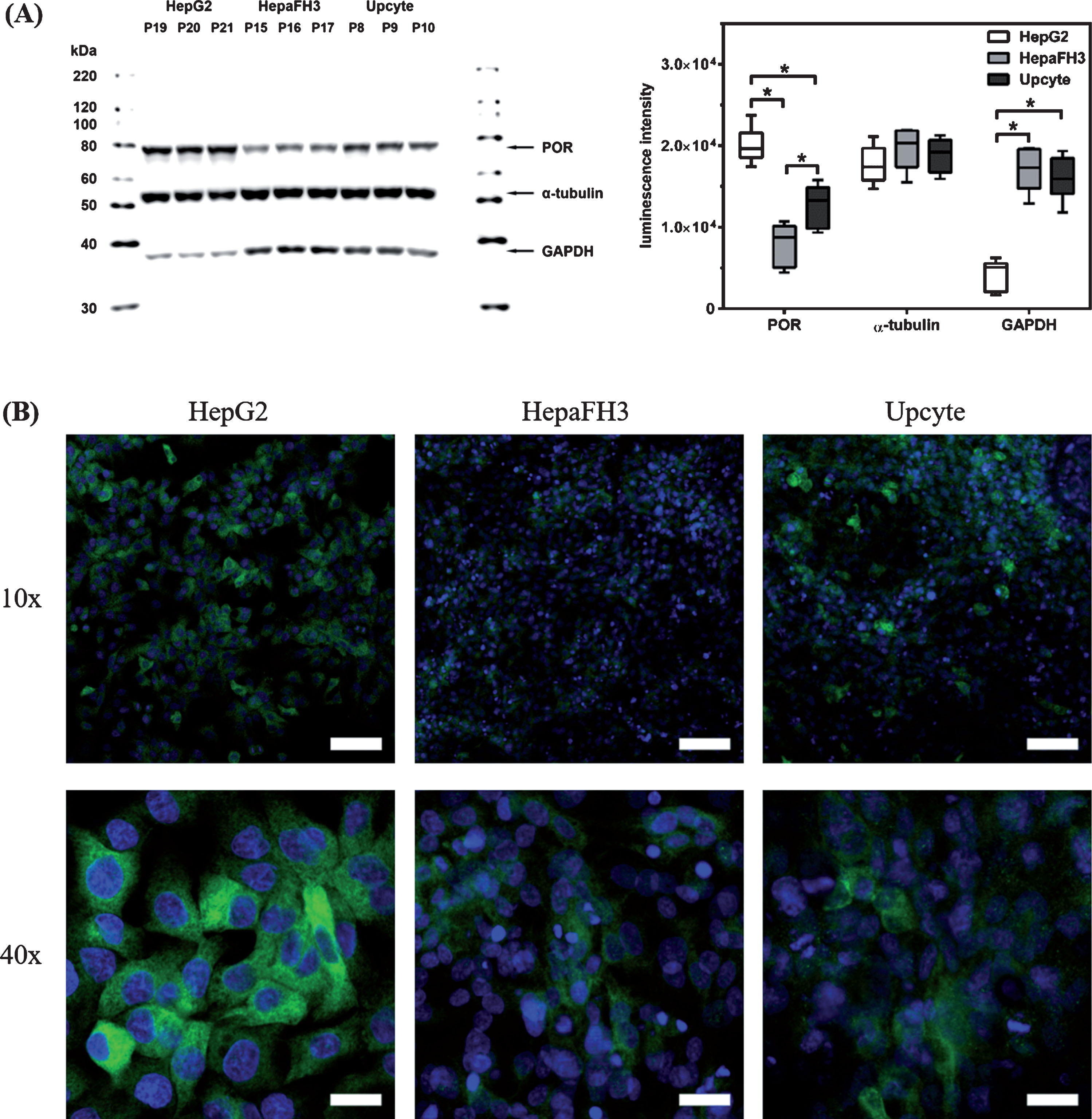
As found for POR mRNA levels, there was no passage number-dependent fluctuation of protein expression levels within each cell culture system (Fig. 2A). However, there were clear inter-cell type differences in POR protein expressions. Semi-quantitative analyses of Western blot signals revealed significantly increased POR luminescence in HepG2 compared to primary-like hepatocytes (for both p < 0.0001), with HepaFH3 having the lowest POR signal (HepaFH3 vs. Upcytes® with p = 0.0002). As found for mRNA expression, there were also different expression levels of GAPDH protein in HepG2 and primary-like hepatocytes. Therefore, α-tubulin was selected as loading control to check for equal protein amounts, because it showed a similar protein expression over several passages within the same culture and between the different hepatocyte culture systems. The higher POR expression in HepG2 compared to primary-like hepatocytes was confirmed by immunofluorescence staining (Fig. 2B). Further, protein expression appeared to be slightly higher in second generation Upcyte® hepatocytes compared to HepaFH3 cells.
Fig. 2
Intra- and inter-cellular analysis of POR protein expression for HepG2, HepaFH3 and Upcyte® hepatocytes. (A) Exponentially growing cells were processed for SDS-PAGE and Western blot (left). Western blot signals were analysed semiquantitatively based on luminescence intensities (right). Blots were probed with anti-POR, anti-α-tubulin and anti-GAPDH antibodies as indicated. Quantitative Western blot data are shown as arithmetic mean±standard deviation; * p < 0.05; n = 9 for POR and GAPDH; n = 6 for α-tubulin. (B) Immunofluorescence staining of POR protein expression in exponentially growing hepatocyte cultures, as indicated. Nuclei were stained with DAPI (blue). Pictures taken with a confocal laser scanning microscope (Zeiss) at 10× and 40× primary magnification. Scale bars: 10× ⟶ 100 μm, 40× ⟶ 25 μm).
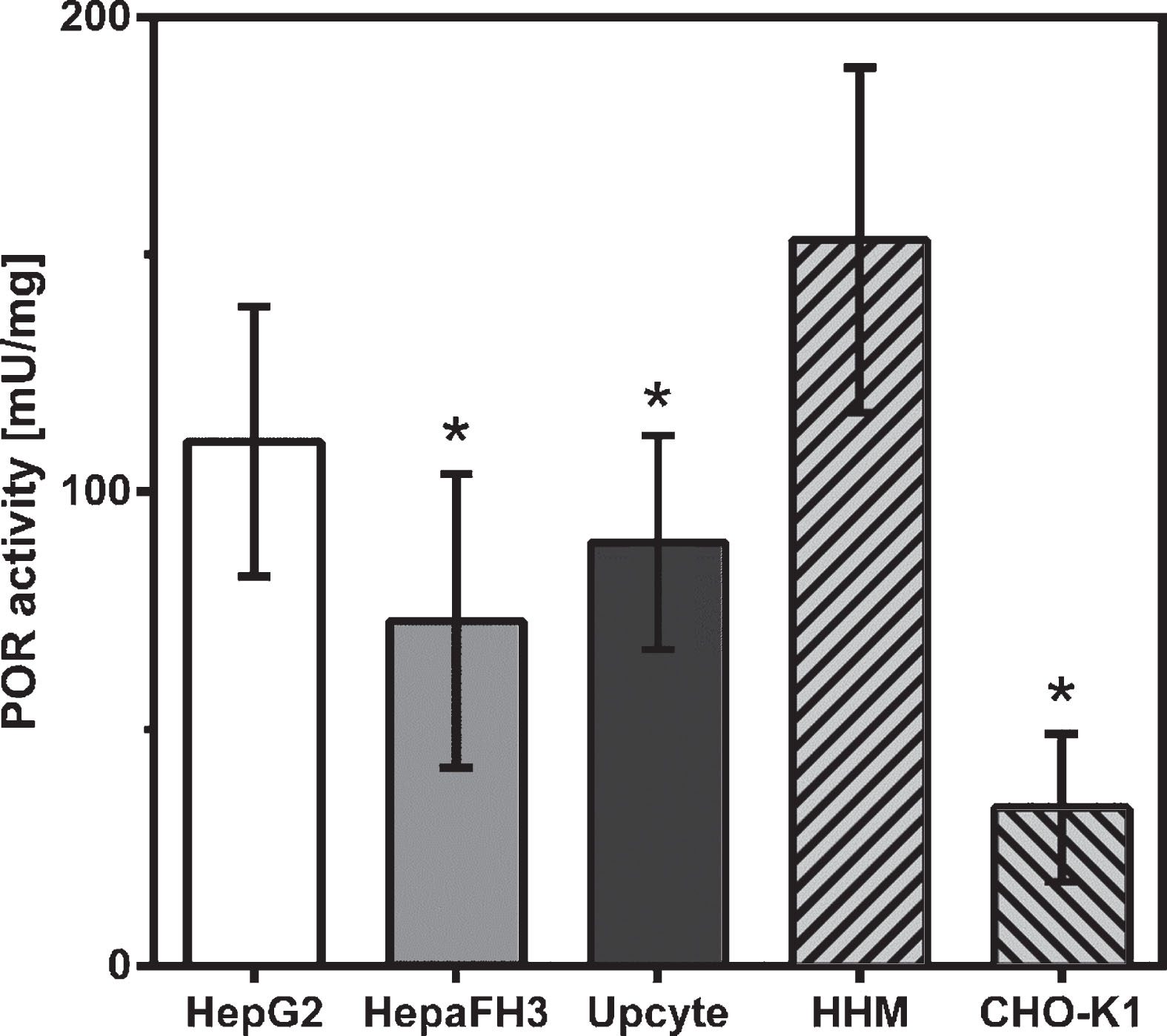
Now we were interested to see whether observed inter-cell type specific differences in POR expression were also detectable at enzyme activity levels. Kinetics of the NADPH-dependent substrate reduction into a coloured product with an absorbance maximum at 460 nm were determined in the presence or absence of 0.1 μM Diphenyleneiodonium Chloride (DPI). This is a specific inhibitor for NADPH-dependent flavoproteins and used to exclude extraneous reductases capable of reducing the same substrate. As demonstrated in Fig. 3, we found that POR activity was not significantly different between the three cell lines, but there was a tendency of slightly lower activity in HepaFH3 (72.8±31.0 mU/mg, p = 0.0829) and second generation Upcyte® hepatocytes (89.3±22.6 mU/mg; p = 0.3537), when compared to HepG2 (110.6±28.4 mU/mg). Surprisingly, these findings do not indicate a direct correlation between POR expression at mRNA and protein levels with corresponding enzyme activity levels. Compared to human microsomes isolated from liver biopsies (HHM), all tested hepatocyte models showed lower POR activity, which was significant for HepaFH3 (p = 0.0003) and Upcyte® (p = 0.0037) but less pronounced for HepG2 (p = 0.0669). As a control for POR activity in non-hepatic cells, microsomal fractions of Chinese hamster ovary cells (CHO cells) were used. These cells showed the lowest POR enzyme activity (Fig. 3).
Fig. 3
POR activity levels in hepatocyte cultures and non-hepatocyte CHO cells. Exponentially growing cells were processed for microsome isolations and quantitative determinations of POR enzyme activity. Human hepatocyte microsomes (HHM) isolated from liver biopsies served as positive control, CHO-K1 cells as negative control. Data shown as arithmetic mean±standard deviation; *p < 0.05; *significant changes compared to HHM; n = 6.
4Discussion
The aim of this study was to determine POR expression and activity in three different hepatocyte cell cultures, namely HepG2, HepaFH3 and second generation Upcyte® hepatocytes. First, we studied POR expression over several passages in order to validate expression and functionality in this cultivation period and to identify possible time-dependent discontinuities. Second, POR expression and activity was analysed for comparison of the different cell cultures. According to current knowledge, POR as the exclusive electron donor represents a basic prerequisite for CYP-dependent phase I reactions in the context of liver biotransformation and clearance of drugs and other xenobiotics [2, 34]. Analyses of POR expression and activity in hepatoblastoma cell line HepG2 and primary-like hepatocytes generated by the Upcyte® technology thus represent a further validation step for the use of these cell cultures as surrogates of primary human hepatocytes.
Passage number-dependent analysis of variations in intra-cellular POR expression at mRNA and protein levels and corresponding kinetic activity studies revealed no significant differences within the hepatocyte in vitro models over the studied time period. As a cell line originating from a human hepatoblastoma this was not quite clear for HepG2, since cells could adapt their phenotype due to genetic instability typical for cancer cells. For primary-like hepatocytes, however, this observation supports the finding from earlier studies that these cell models remain functionally active over a longer cultivation period than is the case for cultivated primary human hepatocytes [12, 13, 17]. Interestingly, cancer cell line HepG2 cells showed significantly increased POR expression at both gene and protein levels compared to primary-like hepatocytes. At the functional level, however, this did not manifest significantly in an increased POR activity. Second generation Upcyte® hepatocytes showed only about 25% of POR mRNA expression detected in HepG2, but the enzyme activity was nearly the same. HepaFH3 showed a lower POR expression at the protein level than Upcyte® hepatocytes of the second generation, but enzyme activities were not significantly different between these cells. All investigated cell systems showed lower POR activity than found in microsomes from human liver tissue.
Variability in POR expression level and activity between the analysed hepatocyte models may be due interindividual genetic differences of donors [21, 24, 28, 29]. Huang et al. were able to detect 140 SNPs by sequencing the POR gene of 842 persons from four ethnic groups, of which 43 were present with a frequency of >1% [45]. In recent studies it was further shown that different amounts of POR protein correlated with the expression and metabolic activity of several CYPs, suggesting a direct dependency and possible limitation by POR [38, 46].
Aside from non-genetic POR activity influencing factors, the POR gene is highly polymorphic and the individual functional activity of the enzyme can vary greatly, with consequent influences on electron transfer capacity to reaction partners [38]. Since HepG2, HepaFH3 and second generation Upcyte® hepatocytes clearly originated from different donors, possible influences by POR polymorphisms need to be investigated in future studies. These could explain significantly higher POR expression of HepG2 compared to primary-like hepatocytes, which, however, was not that much reflected by higher enzyme activities in the cancer cell line (Fig. 3).
It is likely, that POR gene polymorphisms can have a strong influence on phase I drug metabolism via the CYP system. However, this strongly depends on the interaction of POR genotype, CYP variant and substrate. A detailed characterisation of POR polymorphisms in HepG2 and primary-like hepatocytes would be useful to further elucidate the overall regulation between POR and the CYP system in the investigated cell models. Likewise, other POR expression and activity modulating parameters such as the NADPH level and kinetics must be also taken into account as factors within the investigated hepatocyte models.
Altogether, we will further focus on POR as an important element to understand CYP-dependent biotransformation of drugs and xenobiotics.
Acknowledgments
This work was funded by grants of the Ministerium für Wirtschaft, Forschung und Kultur (MWFK, state of Brandenburg, Germany) for the Fraunhofer Project Group “Pilzbasierte zellfreie Synthese-Plattformen – PZ-Syn” (project number 22-F241-03-FhG/005/001). This work was further supported by the European Fonds for Regional Development (EFRE, Brandenburg, Germany; project “Entwicklung eines physiologisch relevanten Testsystems zur In-vitro-Erfassung von Hepatotoxizität im Hochdurchsatz” (project number: 85009748). We acknowledge Susanne Steinbrecht for her constructive support in completing this article.
References
[1] | Bell CC , Hendriks DF , Moro SM , Ellis E , Walsh J , Renblom A , et al. Characterization of primary human hepatocyte spheroids as a model system for drug-induced liver injury, liver function and disease. Scientific Reports. (2016) ;6: :25187. |
[2] | Zanger UM , Schwab M . Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacology & Therapeutics. (2013) ;138: (1):103–41. |
[3] | Tee MK , Huang N , Damm I , Miller WL . Transcriptional regulation of the human P450 oxidoreductase gene: Hormonal regulation and influence of promoter polymorphisms. Molecular Endocrinology. (2011) ;25: (5):715–31. |
[4] | Miller WL , Agrawal V , Sandee D , Tee MK , Huang N , Choi JH , et al. Consequences of POR mutations and polymorphisms. Molecular and Cellular Endocrinology. (2011) ;336: (1-2):174–9. |
[5] | Guengerich FP . Cytochrome p450 and chemical toxicology. Chemical Research in Toxicology. (2007) ;21: (1):70–83. |
[6] | Zanger UM , Turpeinen M , Klein K , Schwab M . Functional pharmacogenetics/genomics of human cytochromes P450 involved in drug biotransformation. Analytical and Bioanalytical Chemistry. (2008) ;392: (6):1093–108. |
[7] | Samer CF , Lorenzini KI , Rollason V , Daali Y , Desmeules JA . Applications of CYP450 testing in the clinical setting. Molecular Diagnosis & Therapy. (2013) ;17: (3):165–84. |
[8] | Lynch TJ , Price AL . The effect of cytochrome P450 metabolism on drug response, interactions, and adverse effects. Am Fam Physician. (2007) ;76: :391–6. |
[9] | Hewitt NJ , Gómez Lechón MJ , Houston JB , Hallifax D , Brown HS , Maurel P , et al. Primary hepatocytes: Current understanding of the regulation of metabolic enzymes and transporter proteins, and pharmaceutical practice for the use of hepatocytes in metabolism, enzyme induction, transporter, clearance, and hepatotoxicity studies. Drug Metabolism Reviews. (2007) ;39: (1):159–234. |
[10] | Gomez-Lechon M , Donato M , Castell J , Jover R . Human hepatocytes in primary culture: The choice to investigate drug metabolism in man. Current Drug Metabolism. (2004) ;5: (5):443–62. |
[11] | Rijntjes P , Moshage H , Van Gemert P , De Waal R , Yap S . Cryopreservation of adult human hepatocytes: The influence of deep freezing storage on the viability, cell seeding, survival, fine structures and albumin synthesis in primary cultures. Journal of Hepatology. (1986) ;3: (1):7–18. |
[12] | Bhatia SN , Underhill GH , Zaret KS , Fox IJ . Cell and tissue engineering for liver disease. Science Translational Medicine. (2014) ;6: (245):245sr2-sr2. |
[13] | Fang Y , Gao N , Tian X , Zhou J , Zhang H-F , Gao J , et al. Effect of P450 oxidoreductase polymorphisms on the metabolic activities of ten cytochrome P450s varied by polymorphic CYP genotypes in human liver microsomes. Cellular Physiology and Biochemistry. (2018) ;47: (4):1604–16. |
[14] | den Braver-Sewradj SP , den Braver MW , van Dijk M , Zhang Y , Dekker SJ , Wijaya L , et al. Inter-individual variability in activity of the major drug metabolizing enzymes in liver homogenates of 20 individuals. Current Drug Metabolism. (2018) ;19: (4):370–81. |
[15] | Krüger-Genge A , Fuhrmann R , Franke R-P , Jung F . Effect of lipopolysaccharide on the adherence of human umbilical vein endothelial cells (HUVEC) on a natural substrate. Clin Hemorheol Microcirc. (2019) ;71: (2):175–81. |
[16] | Zhou J , Yang H , Lehmann C . Inhibition of GPR 55 improves dysregulated immune response in experimental sepsis. Clin Hemorheol Microcirc. (2018) ;70: (4):553–561. |
[17] | Paine AJ , Andreakos E . Activation of signalling pathways during hepatocyte isolation: Relevance to toxicology in vitro. Toxicology in Vitro. (2004) ;18: (2):187–93. |
[18] | Elaut G , Henkens T , Papeleu P , Snykers S , Vinken M , Vanhaecke T , et al. Molecular mechanisms underlying the dedifferentiation process of isolated hepatocytes and their cultures. Current Drug Metabolism. (2006) ;7: (6):629–60. |
[19] | Borlak J , Singh PK , Rittelmeyer I . Regulation of liver enriched transcription factors in rat hepatocytes cultures on collagen and EHS sarcoma matrices. PloS One. (2015) ;10: (4):e0124867. |
[20] | Hewitt N , Lecluyse E , Ferguson S . Induction of hepatic cytochrome P450 enzymes: Methods, mechanisms, recommendations, and in vitro–in vivo correlations. Xenobiotica. (2007) ;37: (10-11):1196–224. |
[21] | Xuan J , Chen S , Ning B , Tolleson WH , Guo L . Development of HepG2-derived cells expressing cytochrome P450s for assessing metabolism-associated drug-induced liver toxicity. Chemico-Biological Interactions. (2016) ;255: :63–73. |
[22] | Kanebratt KP , Andersson TB . HepaRG cells as an in vitro model for evaluation of cytochrome P450 induction in humans. Drug Metabolism and Disposition. (2008) ;36: (1):137–45. |
[23] | Tascher G , Burban A , Camus S , Plumel M , Chanon S , Le Guevel R , et al. In-Depth Proteome Analysis Highlights HepaRG Cells as a Versatile Cell System Surrogate for Primary Human Hepatocytes. Cells. (2019) ;8: (2):192. |
[24] | Wilkening S , Stahl F , Bader A . Comparison of primary human hepatocytes and hepatoma cell line Hepg2 with regard to their biotransformation properties. Drug Metabolism and Disposition. (2003) ;31: (8):1035–42. |
[25] | Steinbrecht S , Kammerer S , Küpper J-H . HepG2 cells with recombinant cytochrome P450 enzyme overexpression: Their use and limitation as in vitro liver model. Journal of Cellular Biotechnology. (2019) ;5: (1):55–64. |
[26] | Kammerer S , Küpper J-H . Human hepatocyte systems for in vitro toxicology analysis. Journal of Cellular Biotechnology. (2018) ;3: (2):85–93. |
[27] | Herzog N , Katzenberger N , Martin F , Schmidtke K-U . Generation of cytochrome P450 3A4-overexpressing HepG2 cell clones for standardization of hepatocellular testosterone 6β-hydroxylation activity. Journal of Cellular Biotechnology. (2015) ;1: (1):15–26. |
[28] | Burkard A , Dähn C , Heinz S , Zutavern A , Sonntag-Buck V , Maltman D , et al. Generation of proliferating human hepatocytes using upcyte® technology: Characterisation and applications in induction and cytotoxicity assays. Xenobiotica. (2012) ;42: (10):939–56. |
[29] | Herzog N , Hansen M , Miethbauer S , Schmidtke KU , Anderer U , Lupp A , et al. Primary-like human hepatocytes genetically engineered to obtain proliferation competence display hepatic differentiation characteristics in monolayer and organotypical spheroid cultures. Cell Biology International. (2016) ;40: (3):341–53. |
[30] | Nörenberg A , Heinz S , Scheller K , Hewitt NJ , Braspenning J , Ott M . Optimization of upcyte® human hepatocytes for the in vitro micronucleus assay. Mutation Research/Genetic Toxicology and Environmental Mutagenesis. (2013) ;758: (1-2):69–79. |
[31] | Schaefer M , Schänzle G , Bischoff D , Süssmuth RD . Upcyte human hepatocytes: A potent in vitro tool for the prediction of hepatic clearance of metabolically stable compounds. Drug Metabolism and Disposition. (2016) ;44: (3):435–44. |
[32] | Tolosa L , Gómez-Lechón MJ , López S , Guzmán C , Castell JV , Donato MT , et al. Human upcyte hepatocytes: Characterization of the hepatic phenotype and evaluation for acute and long-term hepatotoxicity routine testing. Toxicological Sciences. (2016) ;152: (1):214–29. |
[33] | Ramachandran SD , Vivarès A , Klieber S , Hewitt NJ , Muenst B , Heinz S , et al. Applicability of second-generation upcyte® human hepatocytes for use in CYP inhibition and induction studies. Pharmacology Research & Perspectives. (2015) ;3: (5):e00161. |
[34] | Riddick DS , Ding X , Wolf CR , Porter TD , Pandey AV , Zhang Q-Y , et al. NADPH–cytochrome P450 oxidoreductase: Roles in physiology, pharmacology, and toxicology. Drug Metabolism and Disposition. (2013) ;41: (1):12–23. |
[35] | Hart SN , Zhong X-B . P450 oxidoreductase: Genetic polymorphisms and implications for drug metabolism and toxicity. Expert Opinion on Drug Metabolism & Toxicology. (2008) ;4: (4):439–52. |
[36] | Gomes AM , Winter S , Klein K , Turpeinen M , Schaeffeler E , Schwab M , et al. Pharmacogenomics of human liver cytochrome P450 oxidoreductase: Multifactorial analysis and impact on microsomal drug oxidation. (2009) . |
[37] | Flück CE , Mullis PE , Pandey AV . Reduction in hepatic drug metabolizing CYP3A4 activities caused by P450 oxidoreductase mutations identified in patients with disordered steroid metabolism. Biochemical and Biophysical Research Communications. (2010) ;401: (1):149–53. |
[38] | Zhang H-F , Li Z-H , Liu J-Y , Liu T-T , Wang P , Fang Y , et al. Correlation of cytochrome P450 oxidoreductase expression with the expression of 10 isoforms of cytochrome P450 in human liver. Drug Metabolism and Disposition. (2016) ;44: (8):1193–200. |
[39] | Maglich JM , Stoltz CM , Goodwin B , Hawkins-Brown D , Moore JT , Kliewer SA . Nuclear pregnane x receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Molecular Pharmacology. (2002) ;62: (3):638–46. |
[40] | Flück CE , Tajima T , Pandey AV , Arlt W , Okuhara K , Verge CF , et al. Mutant P450 oxidoreductase causes disordered steroidogenesis with and without Antley-Bixler syndrome. Nature Genetics. (2004) ;36: (3):228. |
[41] | McCammon KM , Panda SP , Xia C , Kim J-JP , Moutinho D , Kranendonk M , et al. Instability of the human cytochrome P450 reductase A287P variant is the major contributor to its Antley-Bixler syndrome-like phenotype. Journal of Biological Chemistry. (2016) ;291: (39):20487–502. |
[42] | Chen X , Pan LQ , Naranmandura H , Zeng S , Chen SQ . Influence of various polymorphic variants of cytochrome P450 oxidoreductase (POR) on drug metabolic activity of CYP3A4 and CYP2B6. PloS One. (2012) ;7: (6):e38495. |
[43] | Udhane SS , Parween S , Kagawa N , Pandey AV . Altered CYP19A1 and CYP3A4 activities due to mutations A115V, T142A, Q153R and P284L in the human P450 oxidoreductase. Frontiers in Pharmacology. (2017) ;8: :580. |
[44] | Laemmli UK . Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. (1970) ;227: (5259):680. |
[45] | Huang N , Agrawal V , Giacomini KM , Miller WL . Genetics of P450 oxidoreductase: Sequence variation in 842 individuals of four ethnicities and activities of 15 missense mutations. Proceedings of the National Academy of Sciences. (2008) ;105: (5):1733–8. |
[46] | Backes WL , Kelley RW . Organization of multiple cytochrome P450s with NADPH-cytochrome P450 reductase in membranes. Pharmacology & Therapeutics. (2003) ;98: (2):221–33. |


