
Nomenclature of humanized mAbs: Early concepts, current challenges and future perspectives
Abstract
Nomenclature of monoclonal antibodies traditionally followed a strict scheme indicating target and species information. Because of the rapid advances in this field, emphasized by approval of four humanized and six human antibodies in 2017, the International Nonproprietary Name of new antibodies was updated profoundly by removing the species substem completely. In this review we give an overview about what developments led to the preference of the scientific community towards human-like antibodies. We summarize the major updates in naming schemes that tried to classify antibodies according to their humanization technique or to the final primary sequence and how this led to the erroneous perception to indicate expected immunogenicity. Following the new 2017 nomenclature update, there will not be any information available about the species origin in the names of new antibodies, which emphasizes the need for providing additional supplemental information to the scientific community and develop tools to accurately estimate and control the safety of new monoclonal antibody molecules.
1.Introduction
Monoclonal antibodies (mAbs) have been available for nearly four decades now and are the logical and further development of the serum therapy applied already in 1896 for diphtheria for which the first Nobel Prize in Physiology or Medicine was awarded to Emil von Behring [18]. In 1975, the breakthrough-technology for production of monoclonal antibodies was published by Köhler and Milstein [36] enabling the immortalization of B-cells in order to use them as cell factory for mAb production, for which another Nobel Prize was awarded. Such hybridoma cells are generated most often from the isolated spleen of a vaccinated mouse followed by fusion with immortal myeloma cells of the Sp2/0 or the NS0 lineage. This led to the first therapeutic mAb muromonab-CD3 approved by the FDA in 1986 [60]. The hybridoma technology for the development of new and highly specific mouse antibodies with nano- to picomolar affinities still represents a robust, readily available and fast method by means of well-established protocols [67]. Such mouse-derived mAbs are often applied in basic research with an appreciable number finding its way into clinical application. The downside of mouse-derived sequences in human therapy is elicitation of anti-mouse antibodies in patients and hence various humanization strategies were developed to predict and avoid immunogenic peptides. Humanized mAbs are engineered to replace mouse related epitopes or immunogenic structures in the variable region by human sequences, but simultaneously preserve binding capacity of mAbs as such to retain therapeutic effects of the original mouse mAb. To specify a humanized mAb, different naming nomenclatures and guidelines were defined by the World Health Organization (WHO) or the American Medical Association (AMA) to distinguish human from humanized, chimeric and mouse mAbs in order to characterize them already by their name. However, especially newly emerging techniques for humanization caused significant confusion about the usage of the term humanization and therefore a new naming nomenclature was introduced in 2017 [30, 44]. In this review we discuss the historical background of the name definition and elaborate reasons for critical voices in the scientific community enforcing the development of the new 2017 naming nomenclature.
2.Immunogenic risk of ‘non-human’ antibodies
The human immune system recognizes non-human structures present on foreign antigens, thus reacts to mouse derived peptides on therapeutic mAbs by humoral and also very prominently by cellular immunity envisaged by inflammatory symptoms. Therefore, special attention on adverse immune reactions needs to be paid during therapeutic application of mAbs containing xeno- or neo-epitopes. The presence of various immunogenic risk factors is indicated by high titers of anti-drug antibodies (ADA) comprising human anti-mouse antibodies (HAMA) [55], human anti-chimeric antibodies (HACA) [2] or even human anti-human antibodies (HAHA) [41]. The development of human anti-drug antibodies directed against a fully human antibody can be explained by ‘intrinsic factors’ e.g. unique CDR sequence patterns and individual CDRH3 diversity [21] or various post-translational modifications, as well as, to some extend to certain ‘extrinsic factors’ such as protein aggregation or the fact that human germline genes for VH and VL show allelic polymorphism [5, 63, 64]. Therefore a therapeutic and fully human antibody derived from a V-gene allelic variant that is not present in particular patient groups, might also represent an increased immunogenic risk. Consequently, antibodies targeting any biotherapeutic molecule of interest are the prevailing analyte molecules measured from patient blood samples typically by ELISA-based assays to detect and estimate drug immunogenicity [35, 37]. The development of high titers of such ADAs in the human plasma usually reduces efficacy when blocking the antigen binding cleft and/or alter pharmacokinetic properties leading to faster clearance of the therapeutic protein [24]. The initiation and extent of immune reactions against the therapeutic protein might also be the result of process-related factors such as chemical modifications induced by purification, formulation and storage conditions [26]. Additionally, impurities by host-cell-proteins (HCP), as well as the route and dose of delivery and the patient’s health status play a significant role.
New data are constantly collected to accumulate knowledge about factors determining protein immunogenicity within a human subject, but the development of highly demanded in-silico, in-vitro and ex-vivo tools [19, 53] with predictive power of the actual in-vivo immunogenicity still needs to mature to be considered for routine use in non-clinical studies to prove low immunogenicity [52]. Difficulties in establishing such concepts arise from the multifactorial cause of immunogenicity and a combination of process related secondary immunogenic risk factors and the primary antibody sequence itself that might be identified as non-self.
3.Other factors determining clinical immunogenicity
One major driver of immunogenicity of biotherapeutics is the route of drug administration. There is evidence that intravenous administration bears lower risk of immune reactions than intramuscular or subcutaneous administration [22, 23, 66]. Furthermore, a constant and regular drug dosing schedule (‘maintenance treatment’) was shown to be more beneficial than fluctuating blood levels in intermittent therapies (‘episodic treatment’) for low serum ADAs [57, 61]. In contrast, many therapeutic anti-cancer antibodies are applied only for a defined time period to prepare the patient for subsequent bone marrow transplantation (see ongoing clinical trials for Iomab-B) or to kill tumor cells. It was also reported that higher drug doses are less likely to provoke an immune reaction than lower mAb concentrations, an effect known as ‘high-zone tolerance’ [22].
Additionally, the patient’s immune competence plays a significant role for mAb immunogenicity, which can be controlled by co-administration of immunosuppressive drugs such as methotrexate (MTX) or azathioprine (AZA) [61]. One key study indicated that immunocompromised multiple myeloma patients on intensive chemotherapy show up reduced induction of ADAs against recombinant human granulocyte macrophage colony-stimulating factor (rhGM-CSF) compared to immune-competent patients [50].
The intended clinical indication of a mAb therapeutic is another factor for determining the immunogenic risk profile. Many mAbs directed against immune cells (e.g. anti-CD19/CD3 bispecific for Blinatumomab, anti-CD20 for Rituximab) aim to destroy malignant immune cells as their primary mechanism of action. However, also healthy cells are targeted leading to the suppression of the patient’s capability to elicit an adaptive immune response including formation of ADAs.
Over the years, physicians have learned to control such secondary factors triggering adverse immune reactions. This is the reason why mouse and chimeric antibodies can still be found in late-stage clinical trials (see Table 4 in ref. [31]) for oncological indications. Furthermore, it is possible to safely administer novel and modified mAb constructs [4, 10, 58] based on synthetic IgG-scaffolds, comprising various Fv- or Fc-derived mAb fragments, bispecific constructs [6] and therapeutic fusion proteins including immunotoxins and immunocytokines. All of these synthetic constructs have the potential to carry novel T-cell epitopes (‘neo-epitopes’) determined by a non-self primary amino acid sequence. In the case of fusion proteins, those immune epitopes may be concentrated in the linker sequence, the drug conjugate (toxin, interleukin or enzyme) and in the junctions between several connected biomolecules.
Another important aspect is the presence or modification of antibody Fc-moieties which has not been captured in any systematic name of antibodies, but has significant effect on pharmacological behavior of the mAb molecule and its immunogenicity. Today we know how to engineer the Fc to influence the half-life, the assembly of heterologous heavy chains, the ligand (protein A) or receptor binding and effector functions including antibody-dependent cell-mediated cytotoxicity (ADCC), antibody-dependent cellular phagocytosis (ADCP) and complement-dependent cytotoxicity (CDC). This knowledge is now used to create tailor-made Fc constructs by different glycoengineering approaches to generate afucosylated mAbs for increased ADCC, traditional site-directed mutagenesis to enhance half-life by enhanced binding to neonatal Fc receptor (FcRn) or domain exchange for construction of bispecific molecules, to name just a few examples [54, 62].
Table 1
Naming scheme of antibodies according to the international nonproprietary naming (INN) program by the WHO before 2014 and current (2018) revisions excluding the species substem B
| Pre-2014 INN: | |||||
| 1. Prefix | 2. Target substem A | 3. Species substem B | 4. Universal stem | ||
| random | -b(a)- | bacterial | -a- | rat | -mab |
| -am(i)- | serum amyloid protein | -axo- | rat-mouse (pre-substem) | ||
| (SAP)/amyloidosis (pre-substem) | -e- | hamster | |||
| -c(i)- | cardiovascular | -i- | primate | ||
| -f(u)- | fungal | -o- | mouse | ||
| -gr(o)- | skeletal muscle mass related growth | -u- | human | ||
| factors and receptors (pre-substem) | -vet- | veterinary use (pre-substem) | |||
| -k(i)- | interleukin | -xi- | chimeric | ||
| -l(i)- | immunomodulating | -xizu- | chimeric-humanized | ||
| -N(e)- | neural | -zu- | humanized | ||
| -s(o)- | bone | ||||
| -tox(a)- | toxin | ||||
| -t(u)- | tumour | ||||
| -v(i)- | viral | ||||
| Post-2017 INN | |||||
| 1. Prefix | 2. Target substem (currently under revision) | 3. Universal stem | |||
| random | -ba- | bacterial | -mab | ||
| -ami- | serum amyloid protein | ||||
| (SAP)/amyloidosis (pre-substem) | |||||
| -ci- | cardiovascular | ||||
| -fung- | fungal | ||||
| -gros- | skeletal muscle mass related growth | ||||
| factors and receptors (pre-substem) | |||||
| -ki- | interleukin | ||||
| -li- | immunomodulating | ||||
| -ne- | neural | ||||
| -os- | bone | ||||
| -toxa- | toxin | ||||
| -ta- | tumour | ||||
| -vet- | veterinary use (pre-stem) | ||||
| -vi- | viral | ||||
4.mAb nomenclature in the light of the number of INN designations versus EMA/FDA approvals
Over the years, different international and national organizations like the World Health Organization (WHO) or the American Medical Association (AMA) have established guidelines for implementing a consistent nomenclature system for mAbs resulting in the International Nonproprietary Name (INN) or the United States Adopted Name (USAN) scheme, respectively [30, 44]. The goal of the INN program was to provide a ‘unique name that is globally recognized and is public property’. Such INNs are assigned to mAbs upon request to the WHO and positively passing an INN selection procedure by an INN expert group [70]. Until 2017, the INN of mAbs was assembled from a prefix that could be freely chosen followed by a substem A indicating the target (e.g. -l(i)- for immunomodulating or -t(u)- for tumor) (Table 1). Another substem B indicated the different stages of humanization from the mouse derived mAb indicated by the suffix ‘-omab’, to chimeric mAbs with the suffix ‘-ximab’ and the humanized mAbs ending with ‘-zumab’. Human derived mAbs were identified by the suffix ‘-umab’. Information about target and species were then followed by the universal stem ‘-mab’ (Table 1). This nomenclature was used until 2017 but in future all monoclonal antibodies will again start with an individual prefix preceding a single target substem only, that was slightly modified, which is then followed by the ‘-mab’ stem. The substem B containing species information was dropped completely by the WHO/INN and AMA/USAN program. One first example of this kind is the anti-PD1 antibody receiving the proposed INN Cemiplimab, which was under regulatory review in late 2017 for the treatment of cutaneous squamous cell carcinoma.
Fully human mAbs can be developed from human derived B cells by cloning techniques in combination with a cell enrichment or immortalization step. The majority of fully human antibodies in clinical use, however, were isolated either from immunized mice transgenic for the human IgG locus or from sophisticated combinatorial libraries by phage display [7, 9, 17, 42]. Application of all such techniques are often expensive or impractical since ethical restrictions or even IP rights limit the access to human tissue or human antibody libraries. This might explain why it took 16 years from the first therapeutic use of a murine mAb in 1986 (muromonab-CD3 targeting the T-cell co-receptor CD3-
First chimerization approaches [40] paved the way for intense upcoming efforts to make antibodies more human-like for best immunogenic tolerance resulting in optimal patient safety standards. The first therapeutic antibody of this kind was ReoPro
As a next step, instead of using the entire variable region of a mouse antibody, promising results were achieved by just identifying the complementary determining regions (CDR) and grafting them onto human framework (FR) acceptor sequences also designated as ‘FR scaffolds’ [28]. In such a humanization process by ‘CDR-grafting’, the fraction of murine sequences was further decreased, leading to the first humanized antibody Daclizumab, targeting CD25 (Interleukin-2 receptor alpha chain) and approved for therapeutic use in 1997. Daclizumab was developed as a human-like derivative of the mouse antibody ‘anti-Tac’ [49] by using the human mature antibody EU as FR scaffold (Supplement 1). In a best-fit approach the FRs of EU were selected based on high sequence identity to the FR residues of the murine mAb. In-depth analysis of the sequence together with the first use of a structure model allowed to define important amino acids based on the spatial proximity to CDR loops or the possibility of contacting the antigen directly (Supplement 1, red arrows). In case of massive deviation from the original amino acid in terms of size and charge the human amino acid was replaced by the original mouse derived amino acid in a process called (murine) ‘backmutation’. Additionally, certain unusual EU residues were replaced by the murine counterpart (Supplement 1, orange arrows). Introduction of murine backmutations at critical positions within the human FR perfectly restored the wild-type canonical structure of CDRH1 (class 1) and CDRH2 (class 2) (Supplement 1, green arrows). It was stated that these backmutations make the humanized antibody slightly less human, but it was presumed that the original CDR loop conformations would be better represented. Finally, the humanized variant (Ka
Figure 1.
Chronological summary of cumulative WHO-INN assignments (A) and EMA/FDA approvals (B). Antibody names were extracted from recommended WHO-INN lists or IMGT database (accessible on November 20th, 2017) and sorted according to their source infix. For each year, the number of new antibody names or approvals is explicitly stated. For 2017 only recommended INNs from list number 77 were included and does not represent all new INNs assigned in 2017. Note: the IMGT database states 1992 as the first approval year for the first approved (murine) therapeutic mAb Muromonab-CD3 although it was already approved by the FDA in 1986.
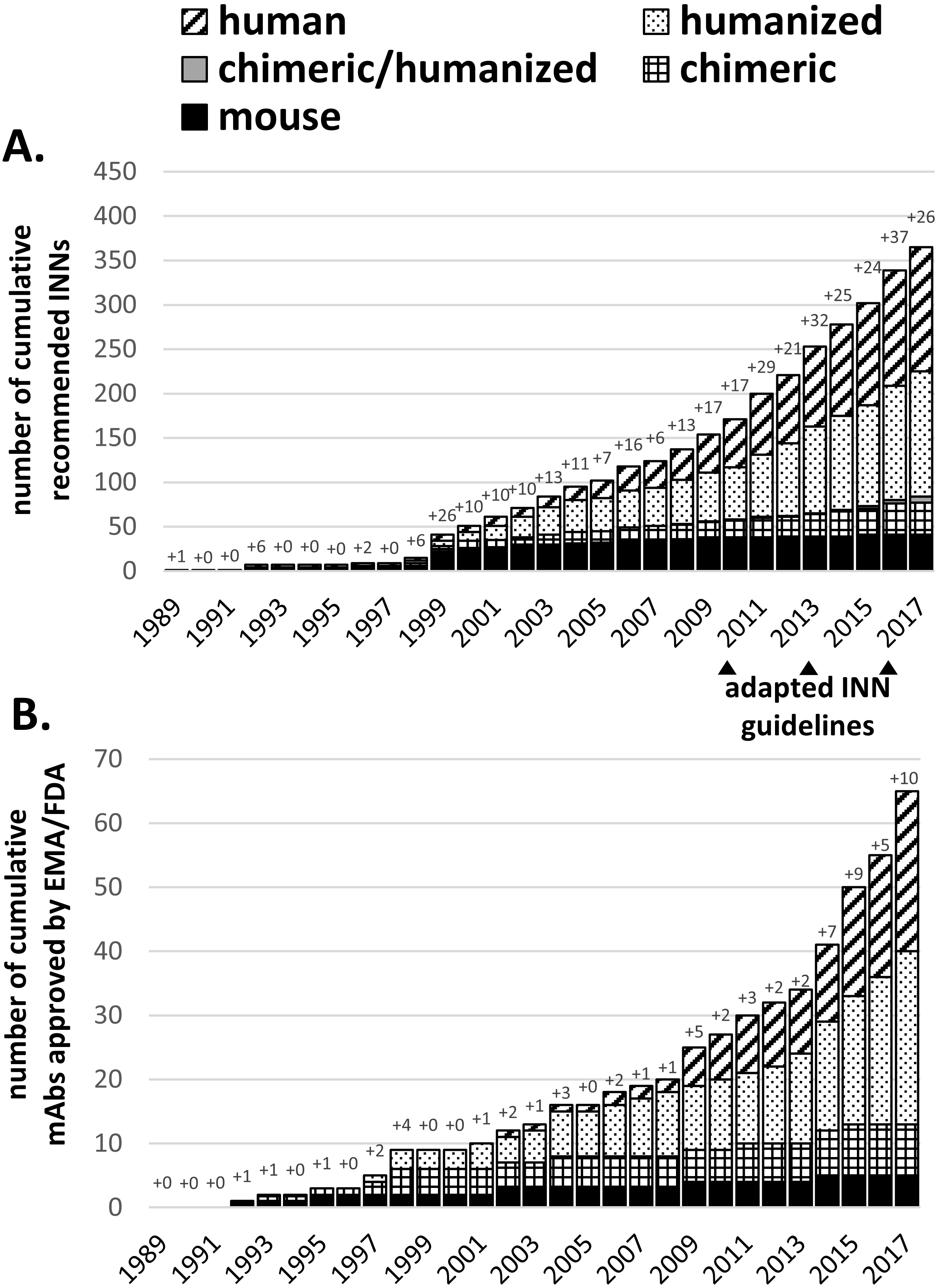
INNs are granted by the WHO often during the time course of clinical studies and are therefore over-representing the number of EMA/FDA approved mAbs. Figure 1A shows the increasing number of granted INNs starting in 1989 which can be matched with concomitant increase in EMA/FDA approvals (Fig. 1B). In Fig. 1 mouse-derived mAbs are indicated in black, stagnating in new assignments and approvals since 2003, while in the same period a significant upturn was seen for human and humanized mAbs (hatched and dotted bars). Grey bars in Fig. 1A indicate mAb variants with one chain being humanized and one chain being chimeric. Three bispecific antibodies Catumaxomab (EMA 2009), Blinatumomab (FDA 2014) and Emicizumab (FDA 2017) were approved for human therapy (indicating the new generation of artificial therapeutics reaching the market). In the future, the increasing number of mAbs in clinical trials will further translate into higher numbers of approvals (6–8 per year or more, [31]). Until 1997, only mouse or chimeric mAbs were released for clinical application, but after the first humanized antibody Daclizumab was approved in 1997, there was virtually a ‘humanization boom’ initiated (Fig. 1, dotted bars), leading to various INN designations and EMA/FDA approvals. The success of mAbs can be evaluated by mentioning that approximately every sixth mAb receiving an INN was also approved for human therapy in the past. Today humanized and human mAbs have become the most accepted mAb format for therapeutic use. Until now, 2017 represented the year with the highest numbers of mAb approvals in history, counting already ten mAbs in total in November 2017 with additional nine mAbs under regulatory review. Interestingly, INNs indicate that all approved mAbs are human or humanized antibodies (Table 2).
5.The naming schemes defined antibody categories
Unlike the first approved mAbs that received regulatory approval before an INN was defined (e.g. Muromonab-CD3 or Daclizumab), nowadays companies usually apply for an INN before EMA/FDA approval is granted. Adalimumab exemplifies the evolution of a typical mAb nomenclature. First developed by Cambridge Antibody Technology and BASF, this antibody was named D2E7 in the lab based on an internal subclone number [32]. Then it was decided to apply for an INN for which the name ‘Adalimumab’ was granted in 2001 according to the WHO guidelines at that time. Only one year later, the antibody was approved under
Table 2
Summary of all ten mAbs approved in 2017 by the FDA or EMA
| International non- proprietary name (INN) | INN recommended list no. | Brand name | Company | Development technology | Format | Target | Clinical indication | Closest human VH germline gene | Closest human VL germline gene |
|---|---|---|---|---|---|---|---|---|---|
| Avelumab | 75 (2016) | Bavencio | EMD Serono | Human antibody phage display library | human IgG1/lambda | PD-L1 | Oncology (Merkel cell carcinoma, MCC) | IGHV3-23*01 (90.8%) | IGLV2-14*01 (99.0%) |
| Benralizumab | 64 (2010) | Fasenra | AstraZeneca | POTELLIGENT technology (afucosylation) | humanized IgG1/kappa | IL-5R | Respiratory diseases (asthma) | IGHV1-46*01 (78.6%) | IGKV1-10*01 (87.6%) |
| Brodalumab | 67 (2012) | Siliq | Valeant Pharmaceuticals | Abgenix’s XenoMouse | human IgG2/kappa | IL-17R | Immunology (plaque psoriasis) | IGHV1-18*01 (96.9%) | IGKV3-15*01 (93.7%) |
| Dupilumab | 70 (2013) | Dupixent | Regeneron Pharmaceuticals Inc. | VelocImmune | human IgG4/kappa | IL-4R | Immunology (Atopic dermatitis) | IGHV3-23*04 (92.9%) | IGKV2-28*01 (96.0%) |
| Durvalumab | 74 (2015) | Imfinzi | AstraZeneca | Abgenix’s XenoMouse | human IgG1/kappa | PD-L1 | Oncology | ||
| (urothelial carcinoma) | IGHV3-7*01 (99.0%) | IGKV3-20*01 (96.9%) | |||||||
| Emicizumab | 75 (2016) | Hemlibra | Genentech | not stated | bispecific humanized IgG4/kappa | coagulation factor IX/X | Hematology (hemophilia A) | IGHV3-23*04 (87.8%) IGHV1-2*02 (75.5%) | IGKV1-39*01 (80.0%) |
| Guselkumab | 71 (2014) | Tremfya | Janssen Biotech Inc. | MorphoSys’s HuCAL phage library | human IgG1/lambda | IL-23 | Immunology (plaque psoriasis) | IGHV5-51*01 (93.9%) | IGLV1-40*01 (91.8%) |
| Inotuzumab ozogamicin | 54 (2005) | Besponsa | Wyeth Pharmaceuticals Inc. | not stated | humanized IgG4/kappa (ADC | CD22 | Oncology (acute lymphoblastic leukemia, ALL) | IGHV1-46*01 (82.7%) | IGKV1-6*01 (73.7%) |
| Ocrelizumab | 56 (2006) | Ocrevus | Genentech | not stated | Humanized IgG1 | CD20 | Immunology (multiple sclerosis) | IGHV3-23*04 (81.6%) | IGKV1-39*01 (83.2%) |
| Sarilumab | 68 (2012) | Kevzara | Regeneron Pharmaceuticals Inc. | VelocImmune | human IgG1/kappa | IL-6R | Immunology (Rheumatoid arthritis) | IGHV3-9*01 (94.9%) | IGKV1-12*01 (96.8%) |
the same INN name and ‘Humira’ as the chosen trade (company) name. Examples of such trade names are Humira
In 2011, the WHO defined an antibody as humanized (i.e. ‘-zumab’) when the CDRs were originating from a species other than human or when CDRs were generated synthetically, whereas the remaining chain is of human origin. They acknowledged that ‘more recent protocols’ for humanization are allowed. Obscurity arose when guidelines for the required definition of CDRs were not explicitly stated, since several concepts are available based on sequence variability- (Kabat), structural- (Chothia) or combined (IMGT) analysis (compare Supplementary Fig. 1). Furthermore, the introduction of murine backmutations and additional residues for affinity maturation was not regulated, regarding number and location of amino acids. Providing a consistent naming scheme was more and more impossible for novel humanization techniques generating antibody sequences that did not fit unambiguously into one of the -o-, -xi-, -zu- and -u- categories.
Consequently, in 2014 the WHO updated their INN scheme to account for these issues and to be consistent with emerging novel antibody development techniques [44]. The goal was that classification of antibodies should solely be based on sequence analysis of the newly constructed final V region ‘as a whole’ and excluding the D- and J-regions, no matter how this antibody sequence was generated and modified. An engineered mAb was assigned a ‘-ximab’ stem (for chimeric mAb) when sequence comparison to mammalian germline genes gave a hit closest to a non-human species. According to previous USAN definition, a humanized mAb would have to meet a distinct identity to the closest related V-germline gene. The anti-CD19 antibody Coltuximab was humanized by resurfacing [51] but received a typical chimeric (‘-xi-’) species substem [68] because it shows just 75% human VH identity. Other examples are the resurfaced Mirvetuximab [1] and Vadastuximab which also received a chimeric (‘-xi-’) substem. When this concept is also applied to mature V regions of somatically mutated human antibodies the procedure may lead to the curious case that an originally human mAb was defined as chimeric (Supplement 2).
The INN nomenclature indicating the species origin was initially intended to provide clinicians with information on immunogenicity. However, experimental data demonstrated that many different recombinant strategies to make antibodies more human like can result in quite similar clinical benefits. This quickly rendered the discrimination between chimeric, humanized and human obsolete. However, the INN names influenced the commercial value as they suggest different clinical effects. For example, an engineered human(ized) antibody traditionally implicates low immunogenicity, but by receiving a chimeric (i.e. ‘-ximab’) name the commercial position compared to a competitor ‘-zumab’ molecule would be significantly impaired. Although it was never intended by the INN program, a ‘-zumab’ name is often used by companies for marketing purposes to indicate low but never proved immunogenicity. Despite the fact that one goal of mAb engineering is to reduce clinical immunogenicity, the 2014 nomenclature may have triggered companies to engineer mAbs for reaching a humanized INN without clear immunogenic benefit. An detailed overview about previous updates on the INN definition is reviewed in ref. [30].
Table 3
Concepts and techniques for generating novel antibody sequences
| Method | Short description | Examples |
|---|---|---|
| Chimerization [40] | Replacement of murine with human antibody constant regions | Infliximab |
| CDR-grafting [28] by selection of – fixed – best-fit – consensus human FR sequence | Transfer of mouse CDRs onto human framework acceptor sequences | Daclizumab |
| Specificity determining region (SDR)-grafting [45] | Grafting of only the residues within CDRs that are actually interacting with the antigen and residues supporting the correct loop conformation | |
| Resurfacing [51]/veneering [43] | Removal of conformational B-cell epitopes or removal of murine residues from the accessible surface of the antibody variable region based on molecular antibody models | Coltuximab |
| Superhumanization [33, 59] | Transfer of murine CDRs onto human germline framework regions defining the same canonical structure classes of the hypervariable loops and based on highest CDR sequence homology | |
| Germlinization (germline humanization) [46] | A follow-up method to superhumanization in which frameworks of antibodies of non-human primate origin were changed to the closest human germline in order to increase the germinality index (GI) | |
| Deimmunization [8, 29] | Removal of potential or actual T-cell epitopes | |
| Removal of apparently ‘random’ somatic mutations [65] | Replacement of somatic mutations not required for binding by germline residues | |
| Humaneering [15] | Transfer of sub-CDR structures into human partial V region library | Ifabotuzumab |
| Framework shuffling [11] | Murine CDRs are combined with a pool of human germline framework genes | |
| Guided selection phage display [27] | Sequential or parallel replacement of non-human VL and VH with human gene segments from a shuffled library by antigen panning and selection | Adalimumab |
| Human framework selection (HFS) adaptation (HFA) [16] | Frameworks from the same human germline are combined in a library | |
| n-CoDeR library [56] | Use of site-specific combination to incorporate different in-vivo formed CDRs (from multiple healthy individuals) into a single antibody framework scaffold. Individual parental CDRs are isolated from immune system and transferred into another scaffold | Lupartumab amadotin |
| Transgenic mice (e.g. XenoMouse [38]) | Mice modified with human humoral immune repertoire | Panitumumab |
| Ribosome or mRNA display library [20, 39] | Combination of diversification and selection by linking mRNA to encoded protein either by stalled ribosomes or covalent bondings. | |
| Bispecific mAbs [6] | Combining specificities to two different antigens into one molecule | Duvortuxizumab, navicixizumab, pasotuxizumab |
| Antibody fragments [6] | Multitude of antibody fragments and comboniations such as e.g., Fab fragments, scFv constructs | Abciximab (Fab), blinatumomab (BiTE |
| Antibody-based fusion proteins [58] | e.g. Fc-fusion constructs | Abatacept (Fc-fusion) |
| Fc engineering | Modification by glycosylation (Potelligent | Mogamulizumab (afucosylation by Potelligent |
6.The WHO closes down the INN species substem in 2017
The first criterion for mAb functionality is the efficacy, which is primarily determined by its affinity, specificity and effector functions. State-of-the-art therapeutic proteins must further show highest safety standards regarding the quality attributes arising from manufacturing and formulation processes to prevent product contamination, aggregation and impurities. Furthermore, certain immunogenic risk factors [3, 47] such as distinct glycosylation patterns, glycation, deamidation and oxidation of amino acid side chains or presence of T-cell epitopes are inherent to the biomolecule’s primary sequence and have to be eliminated during antibody engineering by removing certain amino acid sequences responsible for the potential immunogenicity.
Assuming that during evolution antibody sequences were optimized for minimal, although not completely suppressed immunogenicity, virtually all humanization procedures minimize the number of non-human sequence patterns by using traditional humanization procedures and supplementing them by specialized and sophisticated techniques. Examples for such procedures are ‘humaneering’ [15] or specificity-determining region (SDR) grafting [45], where sub-CDR sequences are defined and combined with a human V germline library to generate human germline-like antibodies. Beside the novel antibody development techniques, new structural formats are continuously evolving. Bispecific antibody variants might use combinations of both chimeric and humanized antibody chains generating hybrid molecules indicated by an atypical ‘-xizu-’ species substem (e.g. Duvortuxizumab, Navicixizumab or Pasotuxizumab). To illustrate the complexity of engineering approaches we have summarized various new aspects that are jointly responsible for closing down the old INN species substem in Table 3.
Enforced by the development of various new humanization techniques and inconsistencies provoked by attempts towards a standardized naming scheme using traditional species substems, the WHO and AMA decided to drop the species substem completely in 2017. This process was initiated to a large extent by ‘The Antibody Society’ (Janice Reichert) who put the majority of new naming concepts and discussion in the seminal paper by Jones et al. [30]. Both the INN (WHO) and USAN (AMA) programs harmonized their guidelines by removing the species substem, but previously assigned names will still remain effective. According to the 2017 guidelines, all mAbs will receive a chosen prefix and a target substem followed by the universal ‘-mab’ stem only (Table 1). In order to prevent confusion, also some target substems were updated. For example cancer targeting antibodies receive the stem ‘-tamab’ since under the old target designation it would receive ‘-tumab’ which can easily be confused with human antibodies defined by the pre-2017 antibody classification.
Table 4
Suggested entries in a supplemental antibody information sheet
| Suggested entries | Available tools |
|---|---|
| 1. Naming system | |
| INN name and date | MedNet-INN |
| Trade name | |
| National nomenclature (e.g. USAN) | |
| 2. Biological function | |
| 3. Development | |
| Company | |
| Source | |
| Patent information | Espacenet patent search |
| 4. Sequence information | |
| Isotype/subtype | |
| Germline alignment | IMGT/DomainGapAlign |
| Canonical structure class | Abysis |
| FR/CDR definition and lengths | Abysis |
| – Kabat | |
| – Chothia | |
| – IMGT | IMGT/DomainGapAlign |
| 5. Protein/formulation properties | |
| Binding properties | |
| – Antigen specificity | |
| – Affinity (KD) | |
| – Binding rates | |
| Biophysical properties | |
| – Thermal/chem. stability | |
| – pI | Expasy/ProtParam |
| – Aggregation propensity (hydrophobic patches) | |
| PTMs | |
| – Glycosylation sites | NetNGlyc |
| – Terminal lysine modifications | |
| Crystal structure | |
| Expression system | |
| 6. Immunogenic risk assessment | |
| In-silico analysis | |
| – T cell epitopes | IEDB |
| – Humanness | Abysis |
| – Human string content (HSC) | |
| in-vitro tests | |
| – T-cell activation assays | |
| Clinical | |
| – Anti-drug antibody (ADA) titer | |
| 7. Other clinical data | |
| Therapeutic area/ indication | IMGT/mAb-DB |
| EMA/FDA approval year | |
| Prescription information | FDA drug database (drugs@FDA) |
| Biological half-life |
To advance the information flow from benchtop to bedside accompanying the mAb nomenclature and approval process, it becomes more critical that supplemental sources of information now provide details about the used humanization method, sequence information, number and location of murine backmutations or known T cell epitope content. Thus, information packages summarized in Table 4 should help basic research scientists, marketers, physicians and last but not least the patient itself to understand how the therapeutic ‘-mab’ molecule was generated, modified and how it compares to other biotherapeutic proteins already on the market. Furthermore, such supplemental information provide a comprehensive resource of documentation and an archive to extract information, built connections and generate knowledge after new evidence about immunogenicity and safety factors were identified in future clinical observations.
7.What defines an antibody as humanized or human?
While the nomenclature suggested that immunogenicity of non-human antibodies could be reduced in a sequential order from murine to chimeric to humanized mAbs by increasing the fraction of ‘human sequences’, experimental data supporting this was missing, and any proof of a clear trend in reduction of immunogenicity between these humanization techniques was not always unambiguously translated into the clinics. Novel immunogenic epitopes might be generated at newly emerging synthetic human FR/murine CDR interfaces of humanized variants. Also somatic mutations in fully human antibodies have the potential to trigger immunogenic reactions. So the question arises what defines a sequence as human or human-like? Attempts were made to define a threshold to distinguish murine and humanized antibody variable regions. Ideally, this threshold truly correlates with in-vivo immunogenicity. Consequently, achieving such a threshold by a new mAb would establish an acceptance criterion for immunogenic tolerance in a human subject, even though the immunological status of a single patient might differ significantly.
Figure 2.
Sequence identity of all murine germline CDRH1 and CDRH2 to the closest human germline CDRs (A) and correlation of the number of mutations in FR (black) and CDR (grey) regions of recommended humanized mAbs (list 77; 2017) with germinality of the closest related human germline V genes (B).
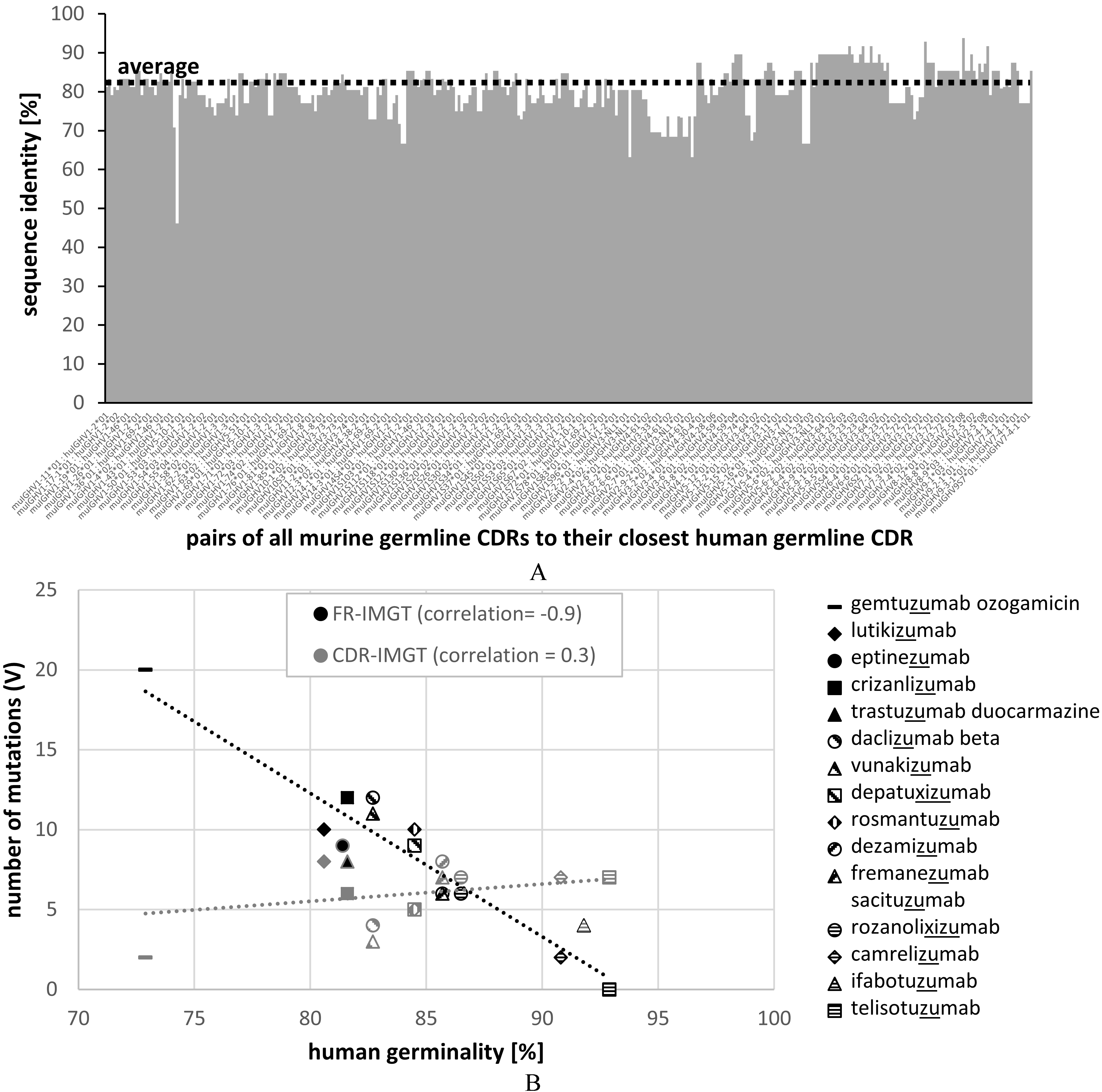
Evaluation of mAb’s immunogenicity could be done by analyzing large datasets of murine and human antibodies and infer certain statistics from these analyses. Analyzed parameters might include average numbers and position-weighted somatic mutations of human and non-human antibodies or the presence of T-cell epitopes. Such a threshold of mutations was defined by the USAN scheme in the past. To designate a mAb the -zumab or the -umab stem, the human germinality identity of VH and VL was required to be at least 85%. Problematic enough, the rationale behind this threshold has never been clearly explained to the scientific community [30, 44]. Additionally this threshold was only applied on the V gene segment and did not include any evaluation of the J region or the heavy chain D region. Many approved humanized antibodies did not reach the 85% threshold, but already contained a ‘zumab’ designation from previous applications [30]. It was also realized that extensively somatically mutated antibodies directly isolated from human subjects (e.g. HIV patients) do not reach the clause, although being clearly a human antibody per se (Supplement 2). Especially in case of anti-HIV mAbs it was found that only highly somatically mutated mature mAbs have neutralizing capacity [25, 34]. This highlights the problem in setting a threshold as an absolute criterion. Therefore, the INN scheme refused the use of thresholds for decision finding in the INN application process.
8.Germline identity is differently influenced by CDR and FR
To evaluate the similarities of murine to human antibodies in more detail, we related all murine germline CDRH1 and CDRH2 sequences to the closest human germline CDR sequences (both described in the IMGT databank) (Fig. 2A). In average, murine germline CDRs showed 80% identity to the closest human germline CDR sequences ranging from 46% to 94% identity. In a typical humanization process, murine CDRs are transferred onto selected human framework regions. Assuming 22 amino acids as the typical length of CDRH1 (5 residues) plus CDRH2 (17 residues), this would mean that in average four to five murine CDR residues are transferred onto the human framework acceptor sequence. Considering a typical VH gene length of 98 amino acids, this translates into an average loss of germinality of around 5% that we have to accept by simply transferring murine CDRs onto human FRs. The human germinality of the resulting humanized mAb can be further decreased by the presence of certain somatic FR mutations if mature FRs are selected as scaffold. Backmutations in the FRs or additional somatic mutations present in the murine CDR regions further decrease the germinality of the antibody but are required for optimized binding affinity. For reaching an 85% threshold, this means that in average only nine such additional mutations are allowed as murine FR backmutations to restore binding. Humanization of murine mAbs with initially low CDR identity to human germline genes will challenge the engineering process to reach the 85% clause defined by previous USAN antibody naming rules. Contrary, murine CDRs with high human germline identities would allow the introduction of more murine backmutations in the FRs for improved binding affinity without respect to immunogenicity. This illustrates that the natural antibody repertoire available for humanization under the 85% clause is limited by the sequence of murine CDRs significantly.
To overlook the contribution of FR and CDR mutations to germline identity, we extracted the amino acid sequences of the latest INN list of recommended humanized mAbs published in 2017 (15 individual mAbs). For each of the 15 individual mAbs we identified the closest IMGT human germline heavy chain variable region and calculated the percentage of identity. Then we plotted the number of mutations of FRs and CDRs separately against the percentage of identity. A correlation of
9.Conclusion
MAbs are applied in human beings for different purposes ranging from diagnostics to long-term therapy. The long-term therapy is the most challenging regarding immunogenic side reactions because of recurrent exposure with foreign protein sequences. Thereby, the immune system of the patient will gain attention to non-human peptides and activate different fates to defend against the normally beneficial mAb. Consequently, in some instances the therapeutic protein has to be applied at higher dose or more frequent intervals to counteract the suppressed efficacy provoked by neutralizing immune reactions. In other cases, even more life-threatening mechanisms are triggered within a patient’s body after receiving the mAb infusion.
First attempts to control drug-related immunogenicity risk factors were successfully applied by traditional humanization approaches, which accumulated in a total of 365 INN assignments and 65 FDA/EMA approvals. Humanization nowadays represents a standard technique of the antibody engineer’s repertoire (also see ‘low-hanging fruit’ in ref. [10]) that should be considered as the gold-standard to reduce expected immunogenicity of new mAb constructs by design. However, increasing evidence suggests that actual clinical immunogenicity does not always gradually decrease from murine to chimeric to humanized to fully human antibody sequences [21]. This highlights the fact that factors other than the primary amino acid sequence guide the actual immunogenic outcome of a therapeutic antibody. Furthermore, we want to stress out that currently no universally applicable assay is available that captures the immunogenic risk of mAb therapeutics in a predictive manner, although rough guidelines are continually being established and updated by the regulatories [13, 14, 69] to evaluate the immunogenicity of biotherapeutics in a retrospective manner, after the drug was applied to human patients. One reason for the difficulties in establishing predictive assays is the multifactorial cause of observed clinical immunogenicity, which is not fully understood yet [12]. Despite this fact different preventive strategies could be developed.
In parallel to the development of humanization techniques a meaningful naming system for mAbs was developed which should reflect the human likeness of mAbs. But over the years it was more and more challenging and finally nearly impossible to assign mAbs as humanized since the definition of the WHO from 2011 was too imprecise. Therefore the authorities decided to define a humanized antibody not by the protein engineering technique, which developed into a broad spectrum of approaches (Table 3) but rather calculated the percentage of identity to the nearest related germline, which must be of human origin. This approach was defined in 2014 but the scientific community voted rightly against this projection [30, 44, 48] since also originally human antibodies might not assign the name human (i.e. ‘umab’) or humanized (i.e. ‘zumab’) since somatic mutations are too frequent (Supplement 2). Additionally, this system does not tell anything about the immunological status of the mAb. Therefore, the WHO closed down the INN species substem in 2017 and in the future all mAbs will be entitled ‘mab’, no matter how they were engineered or from where they originate. This implies that information about species source and expected immunogenicity must be documented in suitable forms other than the mAb’s name. This is already under discussion by WHO experts [44]. We believe that in the near future a better system for prediction of immunogenicity needs to be defined enabling the classification of mAbs.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/HAB-347.
References
[1] | O. Ab, K.R. Whiteman, L.M. Bartle, X. Sun, R. Singh, D. Tavares, A. LaBelle, G. Payne, R.J. Lutz, J. Pinkas, V.S. Goldmacher, T. Chittenden and J.M. Lambert, IMGN853, a Folate Receptor-α (FRα) – Targeting Antibody – Drug Conjugate, Exhibits Potent Targeted Antitumor Activity against FRα-Expressing Tumors, Mol Cancer Ther 14: ((2015) ), 1605–1613. |
[2] | W. Afif, E.V. Loftus, W.A. Faubion, S.V. Kane, D.H. Bruining, K.A. Hanson and W.J. Sandborn, Clinical Utility of Measuring Infliximab and Human Anti-Chimeric Antibody Concentrations in Patients With Inflammatory Bowel Disease, Am J Gastroenterol 105: ((2010) ), 1133–1139. |
[3] | M.D.F.S. Barbosa, S. Kumar, H. Loughrey and S.K. Singh, Biosimilars and biobetters as tools for understanding and mitigating the immunogenicity of biotherapeutics, Drug Discov Today 17: ((2012) ), 1282–1288. |
[4] | A. Beck, T. Wurch, C. Bailly and N. Corvaia, Strategies and challenges for the next generation of therapeutic antibodies, Nat Rev Immunol 10: ((2010) ), 345–352. |
[5] | S.D. Boyd, B.A. Gaëta, K.J. Jackson, A.Z. Fire, E.L. Marshall, J.D. Merker, J.M. Maniar, L.N. Zhang, B. Sahaf, C.D. Jones, B.B. Simen, B. Hanczaruk, K.D. Nguyen, K.C. Nadeau, M. Egholm, D.B. Miklos, J.L. Zehnder and A.M. Collins, Individual Variation in the Germline Ig Gene Repertoire Inferred from Variable Region Gene Rearrangements, J Immunol 184: ((2010) ), 6986–6992. |
[6] | U. Brinkmann and R.E. Kontermann, The making of bispecific antibodies, MAbs 9: ((2017) ), 182–212. |
[7] | M. Brüggemann, M.J. Osborn, B. Ma, J. Hayre, S. Avis, B. Lundstrom and R. Buelow, Human Antibody Production in Transgenic Animals, Arch Immunol Ther Exp (Warsz.) 63: ((2015) ), 101–108. |
[8] | F.J. Carr, G. Carter, A. Hamilton, T. Jones and S. Williams, Method for identification of t-cell epitopes and use for preparing molecules with reduced immunogenicity, EP1366455 A2, (2003) . |
[9] | P.J. Carter, Potent antibody therapeutics by design, Nat Rev Immunol 6: ((2006) ), 343–357. |
[10] | P.J. Carter and G.A. Lazar, Next generation antibody drugs: pursuit of the “high-hanging fruit”, Nat Rev Drug Discov 17: ((2018) ), 197–223. |
[11] | W.F. Dall’Acqua, M.M. Damschroder, J. Zhang, R.M. Woods, L. Widjaja, J. Yu and H. Wu, Antibody humanization by framework shuffling, Methods 36: ((2005) ), 43–60. |
[12] | M. Deehan, S. Garcês, D. Kramer, M.P. Baker, D. Rat, Y. Roettger and A. Kromminga, Managing unwanted immunogenicity of biologicals, Autoimmun Rev 14: ((2015) ), 569–574. |
[13] | EMA, Guideline on Immunogenicity assessment of therapeutic proteins, EMEA/CHMP/BMWP/14327/2006 Rev 1, 18 May 2017, ((2017) ). |
[14] | FDA, Guidance for Industry: Immunogenicity Assessment for Therapeutic Protein Products, ((2014) ). |
[15] | P. Flynn, K. Luehrsen, R. Balint, J.-H. Her, C. Bebbington and G. Yarranton, Antibody specificity transfer using minimal essential binding determinants, US20050255552 A1, (2005) . |
[16] | J. Fransson, A. Teplyakov, G. Raghunathan, E. Chi, W. Cordier, T. Dinh, Y. Feng, J. Giles-Komar, G. Gilliland, B. Lollo, T.J. Malia, W. Nishioka, G. Obmolova, S. Zhao, Y. Zhao, R.V. Swanson and J.C. Almagro, Human Framework Adaptation of a Mouse Anti-Human IL-13 Antibody, J Mol Biol 398: ((2010) ), 214–231. |
[17] | A. Frenzel, T. Schirrmann and M. Hust, Phage display-derived human antibodies in clinical development and therapy, MAbs 8: ((2016) ), 1177–1194. |
[18] | B.S. Graham and D.M. Ambrosino, History of Passive Antibody Administration for Prevention and Treatment of Infectious Diseases, Curr Opin HIV AIDS 10: ((2015) ), 129–134. |
[19] | F. Groell, O. Jordan and G. Borchard, In vitro models for immunogenicity prediction of therapeutic proteins, Eur J Pharm Biopharm 130: ((2018) ), 128–142. |
[20] | J. Hanes and A. Plückthun, In vitro selection and evolution of functional proteins by using ribosome display, Proc Natl Acad Sci USA 94: ((1997) ), 4937–4942. |
[21] | F.A. Harding, M.M. Stickler, J. Razo and R.B. DuBridge, The immunogenicity of humanized and fully human antibodies, MAbs 2: ((2010) ), 256–265. |
[22] | P. Hindryckx, G. Novak, N.V. Casteele, R. Khanna, D. Laukens, J. Vipul and B.G. Feagan, Incidence, Prevention and Management of Anti-Drug Antibodies Against Therapeutic Antibodies in Inflammatory Bowel Disease: A Practical Overview, Drugs 77: ((2017) ), 363–377. |
[23] | M.B. Hock, K.E. Thudium, M. Carrasco-Triguero and N.F. Schwabe, Immunogenicity of Antibody Drug Conjugates: Bioanalytical Methods and Monitoring Strategy for a Novel Therapeutic Modality, AAPS J 17: ((2014) ), 35–43. |
[24] | R.G.E. Holgate and M.P. Baker, Circumventing immunogenicity in the development of therapeutic antibodies, IDrugs Investig Drugs J 12: ((2009) ), 233–237. |
[25] | J.K. Hwang, C. Wang, Z. Du, R.M. Meyers, T.B. Kepler, D. Neuberg, P.D. Kwong, J.R. Mascola, M.G. Joyce, M. Bonsignori, B.F. Haynes, L.-S. Yeap and F.W. Alt, Sequence intrinsic somatic mutation mechanisms contribute to affinity maturation of VRC01-class HIV-1 broadly neutralizing antibodies, Proc Natl Acad Sci USA 114: ((2017) ), 8614–8619. |
[26] | V. Jawa, L.P. Cousens, M. Awwad, E. Wakshull, H. Kropshofer and A.S. De Groot, T-cell dependent immunogenicity of protein therapeutics: Preclinical assessment and mitigation, Clin Immunol Orlando Fla 149: ((2013) ), 534–555. |
[27] | L.S. Jespers, A. Roberts, S.M. Mahler, G. Winter and H.R. Hoogenboom, Guiding the selection of human antibodies from phage display repertoires to a single epitope of an antigen, Biotechnology 12: ((1994) ), 899–903. |
[28] | P.T. Jones, P.H. Dear, J. Foote, M.S. Neuberger and G. Winter, Replacing the complementarity-determining regions in a human antibody with those from a mouse, Nature 321: ((1986) ), 522–525. |
[29] | T. Jones, L. Crompton, F. Carr and M. Baker, Deimmunization of Monoclonal Antibodies, in: A.S. Dimitrov (Ed.), Ther. Antibodies, Humana Press, (2009) , pp. 405–423. |
[30] | T.D. Jones, P.J. Carter, A. Plückthun, M. Vásquez, R.G.E. Holgate, I. Hötzel, A.G. Popplewell, P.W.H.I. Parren, M. Enzelberger, H.J. Rademaker, M.R. Clark, D.C. Lowe, B.I. Dahiyat, V. Smith, J.M. Lambert, H. Wu, M. Reilly, J.S. Haurum, S. Dübel, J.S. Huston, T. Schirrmann, R.A.J. Janssen, M. Steegmaier, J.A. Gross, A.R.M. Bradbury, D.R. Burton, D.S. Dimitrov, K.A. Chester, M.J. Glennie, J. Davies, A. Walker, S. Martin, J. McCafferty and M.P. Baker, The INNs and outs of antibody nonproprietary names, MAbs 8: ((2015) ), 1–9. |
[31] | H. Kaplon and J.M. Reichert, Antibodies to watch in 2018, MAbs ((2018) ), 1–21. |
[32] | J. Kempeni, Preliminary results of early clinical trials with the fully human anti-TNFα monoclonal antibody D2E7, Ann Rheum Dis 58: ((1999) ), I70–I72. |
[33] | W.Y. Khee Hwang, J.C. Almagro, T.N. Buss, P. Tan and J. Foote, Use of human germline genes in a CDR homology-based approach to antibody humanization, Methods 36: ((2005) ), 35–42. |
[34] | F. Klein, R. Diskin, J.F. Scheid, C. Gaebler, H. Mouquet, I.S. Georgiev, M. Pancera, T. Zhou, R.-B. Incesu, B.Z. Fu, P.N.P. Gnanapragasam, T.Y. Oliveira, M.S. Seaman, P.D. Kwong, P.J. Bjorkman and M.C. Nussenzweig, Somatic mutations of the immunoglobulin framework are generally required for broad and potent HIV-1 neutralization, Cell 153: ((2013) ), 126–138. |
[35] | C. Kloks, C. Berger, P. Cortez, Y. Dean, J. Heinrich, L. Bjerring Jensen, V. Koppenburg, S. Kostense, D. Kramer, S. Spindeldreher and H. Kirby, A fit-for-purpose strategy for the risk-based immunogenicity testing of biotherapeutics: a European industry perspective, J Immunol. Methods 417: ((2015) ), 1–9. |
[36] | G. Kohler and C. Milstein, Continuous cultures of fused cells secreting antibody of predefined specificity, Nature 256: ((1975) ), 495–497. |
[37] | E. Koren, H.W. Smith, E. Shores, G. Shankar, D. Finco-Kent, B. Rup, Y.-C. Barrett, V. Devanarayan, B. Gorovits, S. Gupta, T. Parish, V. Quarmby, M. Moxness, S.J. Swanson, G. Taniguchi, L.A. Zuckerman, C.C. Stebbins and A. Mire-Sluis, Recommendations on risk-based strategies for detection and characterization of antibodies against biotechnology products, J Immunol Methods 333: ((2008) ), 1–9. |
[38] | R. Kucherlapati and A. Jakobovits, Method of making transgenic mice lacking endogenous heavy chains, US5939598 A, (1999) . |
[39] | D. Lipovsek and A. Plückthun, In-vitro protein evolution by ribosome display and mRNA display, J Immunol Methods 290: ((2004) ), 51–67. |
[40] | S.L. Morrison, M.J. Johnson, L.A. Herzenberg and V.T. Oi, Chimeric human antibody molecules: mouse antigen-binding domains with human constant region domains, Proc Natl Acad Sci USA 21: ((1984) ), 6851–6855. |
[41] | A. Nechansky, HAHA – nothing to laugh about. Measuring the immunogenicity (human anti-human antibody response) induced by humanized monoclonal antibodies applying ELISA and SPR technology, J Pharm Biomed Anal 51: ((2010) ), 252–254. |
[42] | A.L. Nelson, E. Dhimolea and J.M. Reichert, Development trends for human monoclonal antibody therapeutics, Nat Rev Drug Discov 9: ((2010) ), 767–774. |
[43] | E.A. Padlan, A possible procedure for reducing the immunogenicity of antibody variable domains while preserving their ligand binding properties, Mol Immunol 28: ((1991) ), 489–498. |
[44] | P.W.H.I. Parren, P.J. Carter and A. Plückthun, Changes to International Non-Proprietary Names for antibody therapeutics 2017 and beyond of mice, men and more, MAbs 9: ((2017) ), 898–906. |
[45] | R.D. Pascalis, M. Iwahashi, M. Tamura, E.A. Padlan, N.R. Gonzales, A.D. Santos, M. Giuliano, P. Schuck, J. Schlom and S.V.S. Kashmiri, Grafting of “Abbreviated” Complementarity-Determining Regions Containing Specificity-Determining Residues Essential for Ligand Contact to Engineer a Less Immunogenic Humanized Monoclonal Antibody, J Immunol 169: ((2002) ), 3076–3084. |
[46] | T. Pelat, H. Bedouelle, A.R. Rees, S.J. Crennell, M.-P. Lefranc and P. Thullier, Germline Humanization of a Non-human Primate Antibody that Neutralizes the Anthrax Toxin, by in Vitro and in Silico Engineering, J Mol Biol 384: ((2008) ), 1400–1407. |
[47] | C. Pineda, G. Castañeda Hernández, I.A. Jacobs, D.F. Alvarez and C. Carini, Assessing the Immunogenicity of Biopharmaceuticals, Bio Drugs Clin Immunother Biopharm Gene Ther 30: ((2016) ), 195–206. |
[48] | J. Pottier, R. Chastang, C. Dumet and H. Watier, Rethinking the INN system for therapeutic antibodies, MAbs 9: ((2017) ), 5–11. |
[49] | C. Queen, W.P. Schneider, H.E. Selick, P.W. Payne, N.F. Landolfi, J.F. Duncan, N.M. Avdalovic, M. Levitt, R.P. Junghans and T.A. Waldmann, A humanized antibody that binds to the interleukin 2 receptor., Proc Natl Acad Sci USA 86: ((1989) ), 10029–10033. |
[50] | P. Ragnhammar, H.J. Friesen, J.E. Frodin, A.K. Lefvert, M. Hassan, A. Osterborg and H. Mellstedt, Induction of anti-recombinant human granulocyte-macrophage colony-stimulating factor (Escherichia coli-derived) antibodies and clinical effects in nonimmunocompromised patients, Blood 84: ((1994) ), 4078–4087. |
[51] | M.A. Roguska, J.T. Pedersen, C.A. Keddy, A.H. Henry, S.J. Searle, J.M. Lambert, V.S. Goldmacher, W.A. Blättler, A.R. Rees and B.C. Guild, Humanization of murine monoclonal antibodies through variable domain resurfacing, Proc Natl Acad Sci 91 (1994) 969–973. |
[52] | A.S. Rosenberg and Z.E. Sauna, Immunogenicity assessment during the development of protein therapeutics, J Pharm Pharmacol 70: ((2017) ), 584–594. |
[53] | Z.E. Sauna, D. Lagassé, J. Pedras-Vasconcelos, B. Golding and A.S. Rosenberg, Evaluating and Mitigating the Immunogenicity of Therapeutic Proteins, Trends Biotechnol, (2018) . |
[54] | A. Saxena and D. Wu, Advances in Therapeutic Fc Engineering – Modulation of IgG-Associated Effector Functions and Serum Half-life, Front Immunol 7: ((2016) ), 580. |
[55] | R.W. Schroff, K.A. Foon, S.M. Beatty, R.K. Oldham and A.C. Morgan, Human Anti-Murine Immunoglobulin Responses in Patients Receiving Monoclonal Antibody Therapy, Cancer Res 45: ((1985) ), 879–885. |
[56] | E. Söderlind, L. Strandberg, P. Jirholt, N. Kobayashi, V. Alexeiva, A.-M. Åberg, A. Nilsson, B. Jansson, M. Ohlin, C. Wingren, L. Danielsson, R. Carlsson and C.A.K. Borrebaeck, Recombining germline-derived CDR sequences for creating diverse single-framework antibody libraries, Nat. Biotechnol 18: ((2000) ), 852–856. |
[57] | V. Strand, A. Balsa, J. Al-Saleh, L. Barile-Fabris, T. Horiuchi, T. Takeuchi, S. Lula, C. Hawes, B. Kola and L. Marshall, Immunogenicity of Biologics in Chronic Inflammatory Diseases: A Systematic Review, Bio Drugs 31: ((2017) ), 299–316. |
[58] | W.R. Strohl, Current progress in innovative engineered antibodies, Protein Cell 9: ((2018) ), 86–120. |
[59] | P. Tan, D.A. Mitchell, T.N. Buss, M.A. Holmes, C. Anasetti and J. Foote, “Superhumanized” antibodies: reduction of immunogenic potential by complementarity-determining region grafting with human germline sequences: application to an anti-CD28, J Immunol Baltim Md 1950 169: ((2002) ), 1119–1125. |
[60] | G. Tridente, Muromonab, in: Adverse Events Biomed, Springer, Milano, (2014) , pp. 263–265. |
[61] | S. Vermeire, M. Noman, G. Van Assche, F. Baert, G. D’Haens and P. Rutgeerts, Effectiveness of concomitant immunosuppressive therapy in suppressing the formation of antibodies to infliximab in Crohn’s disease, Gut 56: ((2007) ), 1226–1231. |
[62] | X. Wang, M. Mathieu and R.J. Brezski, IgG Fc engineering to modulate antibody effector functions, Protein Cell 9: ((2018) ), 63–73. |
[63] | C.T. Watson, J. Glanville and W.A. Marasco, The Individual and Population Genetics of Antibody Immunity, Trends Immunol 38: ((2017) ), 459–470. |
[64] | C.T. Watson, K.M. Steinberg, J. Huddleston, R.L. Warren, M. Malig, J. Schein, A.J. Willsey, J.B. Joy, J.K. Scott, T.A. Graves, R.K. Wilson, R.A. Holt, E.E. Eichler and F. Breden, Complete haplotype sequence of the human immunoglobulin heavy-chain variable, diversity, and joining genes and characterization of allelic and copy-number variation, Am J Hum Genet 92: ((2013) ), 530–546. |
[65] | G. Winter, F.J. Carr and W.J. Harris, Altered Antibodies, Products and Processes Relating Thereto, EP0629240 (A1) Abstract of corresponding document: WO9317105 (A1), (1994) . |
[66] | L. Yin, X. Chen, P. Vicini, B. Rup and T.P. Hickling, Therapeutic outcomes, assessments, risk factors and mitigation efforts of immunogenicity of therapeutic protein products, Cell Immunol 295: ((2015) ), 118–126. |
[67] | C. Zhang, Hybridoma technology for the generation of monoclonal antibodies, Methods Mol Biol Clifton NJ 901: ((2012) ), 117–135. |
[68] | WHO Drug Information Vol. 28, No. 1, 2014 - Recommended International Nonproprietary Names, List 71: ((2014) ). |
[69] | Assay Development and Validation for Immunogenicity Testing of Therapeutic Protein Products; Revised Draft Guidance for Industry; Availability, Fed. Regist. ((2016) ). |
[70] | WHO |

