
Refinement of a neuronal differentiation protocol predominantly yields human iPS cell-derived dopaminergic neurons for the investigation of neurodegenerative pathomechanisms in vitro
Abstract
BACKGROUND:
Major pathomechanisms underlying neurodegenerative diseases, such as Parkinson’s Disease, are still not well understood. Induced human pluripotent and rodent embryonic stem cells provide powerful disease models to address neurodegeneration-inducing pathomechanisms on a molecular and cellular level.
OBJECTIVE:
Our aim is to establish a refined protocol to generate healthy and patient donor stem cell-derived dopaminergic neurons to investigate neurodegenerative events in vitro.
METHODS:
Human healthy donor- and patient-derived induced pluripotent stem cells were differentiated into stable dopaminergic progenitor cell lines and further differentiated into dopaminergic neurons. Induced pluripotent stem cells, neuronal progenitors and terminally differentiated neurons were characterized by confocal laser microscopy-based immunofluorescence analysis, live cell imaging demonstrating dopamine transporter-specific uptake of a fluorescent substrate and transcriptome analysis.
RESULTS:
Based on our immunofluorescence analysis, dopaminergic differentiation approaches predominantly yield dopaminergic neurons and GFAP-expressing glial cells. We detected a small partition of GABAergic neurons, yet neither serotonergic nor glutamatergic neurons. Dopaminergic neurons were successfully stained for pre- and postsynaptic and mitochondrial markers. Live cell imaging experiments verified dopamine transporter-dependent uptake of the fluorescent monoamine transporter substrate ASP+.
CONCLUSION:
Human stem cell-derived dopaminergic neurons are a suitable cellular system for fluorescence-based experimental approaches to address neurodegenerative events in vitro.
1Introduction
Dopamine (DA) releasing neurons, together with noradrenergic and serotonergic neurons, constitute the monoaminergic neurotransmitter system of the central nervous system (CNS). DA, as the other monoaminergic neurotransmitters, is involved in a variety of physiological processes, ranging from learning and memory, movement to emotional processes, including arousal, reinforcement and reward [1–3]. As for serotonergic neurons, located to the raphé nuclei, the number of DA neurons is small compared to other types of neurons, and amount for up to 400,000 cells located to distinct brain regions of the human CNS. From these regions, DA neurons build axonal networks to other brain areas, where they exert their neuromodulatory effect via G-protein coupled receptors on their target neurons [4–6]. The main DA signaling pathways are the mesocorticolimbic projections and the nigrostriatal pathway. The mesocorticolimbic projections are built by the mesolimbic and mesocortical pathway. DA neurons of the mesolimbic pathway are located to the ventral tegmental area (VTA) of the midbrain and project their axons to the nucleus accumbens inside the ventral striatum. The mesocortical pathway is formed by DA neurons of the VTA that project their axons to the prefrontal cortex. DA signaling of these pathways has been shown to contribute to reward-related cognition [7] and a dysfunctional mesocorticolimbic DA signaling is associated with addiction, attention deficit hyperactivity disorder and schizophrenia [6, 8, 9]. The nigrostriatal pathway is maintained by DA neurons located to the substantia nigra pars compacta in the midbrain, which grow their axons to the caudate nucleus and putamen inside the dorsal striatum. DA signaling of these neurons contributes to reward cognition, associative learning and movement. Regarding this signaling pathway, disruption of DA neurotransmission is associated with psychiatric diseases, including addiction, and most importantly with neurodegenerative disorders, predominantly Parkinson Disease [3, 10, 11].
Parkinson’s Disease (PD) is a slowly progressing neurodegenerative disease with rising incidences during the past years. PD affected about 0.3% of the entire population of industrialized countries, and 1–3.5% of people aged 60 years and older [12–14]. Currently, PD is the second most common neurodegenerative disorder, only surpassed by Alzheimer’s Disease [15, 16]. PD is characterized by a progressive cell death of DA neurons in the substantia nigra pars compacta, thereby depleting the striatum of sufficient DA signalling required to maintain physiological processes. This continuous DA neurodegeneration strongly affects the dorsolateral putamen in the striatum, causing the typical clinical PD phenotype symptoms such as tremor, rigidity, bradykinesia and postural instability [17]. Despite displaying an almost homogenous clinical phenotype, the onset of PD has been shown to be multifactorial. On the genetic level, several genes were identified, which, if mutated, are associated with PD. The gene SNCA, for example, encodes for alpha-synuclein, a cytoplasmic protein that is predominantly found in presynaptic terminals of CNS neurons. In the disease state, point mutations in the SNCA gene cause alpha-synuclein to also form stable β-sheets instead of alpha-helical structures. As a consequence, misfolded proteins can produce protein aggregates, protofibrils and ultimately large fibrils that accumulate as large deposits called Lewy bodies. On the long run, these accumulations of large aggregates lead to cell death and extensive neuronal loss. [18–20]. In addition to genetic causes, neurobiochemical abnormalities are implied to play key roles in PD pathogenesis by contributing to a progressive degradation of DA neurons. Here, mitochondrial dysfunction, neuroinflammation and free radical-induced damage are considered as major pathomechanisms underlying PD pathogenesis [21–23]. With regard to mitochondrial dysfunction and oxidative stress, rodent in vivo and in vitro models were established to gain insight into yet not well understood molecular and cellular mechanisms of each pathomechanism on its own and especially regarding their interaction. One of these models relies on systemic application of MPTP (1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine), which is metabolized to MPP+ (methyl-4-phenylpyridinium) and subsequently taken up by DA neurons. Once taken up into DA neurons, MPP+ exerts a neurotoxic effect by inhibition of complex I of the mitochondrial respiratory chain, which ultimately leads to a PD-related phenotype based on selective DA cell death [24–27].
With the advent of human induced pluripotent stem (hiPS) cells, human stem cell-derived models for neurodegenerative diseases are available, which have the potential to significantly add to the insights gained by non-human in vivo and in vitro research models [28–32]. With regard to PD, hiPS cell-derived DA neurons featuring the genetics of healthy donors and patients provide the opportunity to invesitgate disease-related pathomechanisms in a yet unexploited way. The main obstacle in generating human stem cell-derived DA neurons is to establish reliable and efficient differentiation protocols [29, 33–35]. Generally, dopaminergic differentiation protocols, relying on growth factor-based selection of DA precursors, yield an average of 8 to 40% of stem cell-derived DA neurons (reviewed in [29]). In addition to a comparatively small yield of DA neurons, only a few studies were able to demonstrate physiological functionality, including the recording of spontaneous action potentials or calcium ion oscillations [36–38]. In 2011, Kriks and colleagues published a differentiation protocol, which significantly increased the yield of human embryonic stem cell-derived DA neurons to nearly 80% after 50 days of terminal differentiation. Furthermore, these stem cell-derived neurons were efficiently engrafted in animal models of PD [39]. In this study, CHIR99201, a potent GSK3b inhibitor strongly activating WNT signalling [40], was successfully applied to enhance the expression of dopaminergic phenotype-specific transcription factors. A slightly altered differentiation protocol was established, which reduced the duration of terminal differentiation to 28 days, yet decreased the yield of DA neurons to 35% of all cells and 70% of all neurons [41]. Recently, Li and colleagues, published a protocol for the rapid induction and long-term self-renewal of neuronal progenitors derived from human embryonic stem cells [42]. These progenitors can be successfully differentiated into neurons, however, yielded only up to 10% DA neurons. For our study, we have adapted the generation of neuronal progenitors to establish healthy donor- and PD patient donor-derived neuronal DA precursor cell lines. These cell lines were then terminally differentiated into mature DA neurons using a set of selected growth factors and small molecules that were applied to generate mature, functional human DA neurons in vitro [39, 41]. We have characterized the neuronal population generated by this protocol regarding their yield of DA and non-DA neurons, neuronal maturity and synaptic marker proteins. In addition, we show that hiPS cell-derived neurons can be stained for mitochondrial markers, that allow tracking mitochondria morphology, and furthermore applied for live cell imaging experiments. In summary, although the differentiation protocol does not yield a homogenous DA neuronal population, the amount of DA-producing neurons generated demonstrate the suitability of this human microscopy-based cellular model to investigate neurodegenerative events in vitro.
2Material and methods
2.1Human induced pluripotent stem cell lines tissue culture
HiPS cell line B7_051 derived from a healthy donor was generated using a Sendai Virus reprogramming kit (Cytotune 2.0, Life Technologies). All steps for iPS cell production were performed as described in the protocols provided by the manufacturer. Briefly, human skin fibroblasts obtained via skin biopsy were transfected with a Sendai virus reprogramming vector (Cytotune 2.0, Life Technologies) containing sequences for the four Yamanaka factors Oct4, Sox2, Kfl4 and c-Myc [28, 43] (Ethics Committee II of Medical Faculty Mannheim of Heidelberg University approval no. 2014-626N-M). HiPS cell lines were cultured in feeder-free condition with mTeSR™-1 medium (Stemcell Technologies) on human embryo stem cell-qualified Matrigel (Corning) and split weekly with a ratio of 1 : 6. Culture medium was changed daily. The Parkinson patient-derived induced pluripotent stem (hiPSPD) cell line with an alpha-synuclein gene triplication was obtained from EBiSC (European Bank for induced pluripotent Stem Cells; Edinburgh; UK) and has previously been genomically characterised [44].
The loss of the Sendai Virus reprogramming vector in the established hiPS cell line was verified by PCR analysis.
2.2Transcriptome analysis of hiPS cells, neuronal progenitors and terminally differentiated neurons
Total RNA was extracted using an RNeasy kit (Qiagen). RNA quality was controlled by Nanodrop ND-1000, Agilent 2100 Bioanalyzer. For gene expression analysis, total RNA was processed by the German Cancer Research Center (DKFZ Heidelberg) Genomics and Proteomics core facility and hybridized on Illumina Human ref-12 bead arrays according to the specifications of the manufacturer. Normalized Data was exported and microarray data analysis was performed using a commercially available software package (Graphpad Prism 7.0).
2.3Generation of ventral midbrain DA neuronal progenitors and terminal dopaminergic differentiation
2.3.1Neural induction of hiPS cells into ventral midbrain DA progenitors
For the generation of a stable DA neural precursor cell (NPC) line, the protocol of Li et al. [42] was modified. Briefly, 7-10 hiPS cell clones were selected and plated on Matrigel-coated tissue dishes and cultured in mTeSR™-1 medium with daily medium change. After 3 days medium was changed to neural induction medium: DMEM/F12 GlutaMax™:Neurobasal® (1 : 1), 1xB27 and 1xN2 supplement, 1% MEM non-essential amino acids, 1% GlutaMAXTM supplement and Penicillin-Streptomycin (100 U/mL and 100μg/mL, respectively; medium and supplements from Thermo-Fisher Scientific), 3μM of the GSK3-inhibitor CHIR99021 (Tocris Bioscience), 2μM SB431542 (Sigma-Aldrich) and 10 ng/mL human leukemia inhibitory factor (hLIF, Sigma-Aldrich). Medium was changed every other day. After 10 days, cells were split 1 : 3 once per week for 6 passages using Accutase (Thermo-Fisher Scientific). After this period, cells where characterized regarding their NPC phenotype and subsequently maintained in neuronal induction medium (weekly splitted in a ratio of 1 : 10).
2.3.2Differentiation of hiPS cell-derived NPC into DA neurons
The first stage of terminal neuronal differentiation comprised a dopaminergic selection phase for 2 weeks: NPC were seeded on Matrigel-coated glass coverslips with a density of approx. 4500 cells/cm2. Next day NPC culture medium was changed to SHH medium consisting of DMEM/F12 GlutaMax™:Neurobasal® (1 : 1), 1xB27 and 1xN2 supplement, 1% MEM non-essential amino acids, 1% GlutaMAXTM, 1% Penicillin-Streptomycin (100 U/mL and 100μg/mL, respectively), 20 ng/mL epidermal growth factor (EGF), 20 ng/mL fibroblast growth factor 8b (FGF-8b), 25 ng/mL human sonic hedgehog protein (SHH; all Miltenyi Biotec) and 100μM L-ascorbic acid (Roth). Medium was changed every other day. After 2 weeks, the next stage of terminal differentiation was initiated by withdrawing SHH medium and starting culture in terminal differentiation medium, consisting of DMEM/F12 GlutaMax™:Neurobasal® (1 : 1), 1xB27 and 1xN2 supplement, 1% MEM non-essential amino acids, 1% GlutaMAXTM, 1% Penicillin-Streptomycin (100 U/mL and 100μg/mL, respectively), 5 ng/mL brain-derived neurotrophic factor (BDNF), 5 ng/mL glial-derived neurotrophic factor (GDNF; both Miltenyi Biotec), 100μM L-ascorbic acid (Roth) and 0.5 mg/mL N6,2′-O-Dibutyryladenosine 3′,5′-cyclic monophosphate (dbcAMP; Sigma-Aldrich). Medium was changed every other day. Mature neurons were found after 50 days of culture in terminal differentiation medium.
2.4Immunofluorescence analysis of hiPS cells, NPC, and stem cell-derived neurons
Immunofluorescence analysis was performed at several cellular stages of various differentiation stages (see figure legends). Prior to permeabilization of cells and antibody incubation, cells grown on glass cover slips or in culture dishes (Ibidi) were fixed with 4% para-formaldehyde (PFA, Sigma-Aldrich) in 1x phosphate buffered saline (PBS) for 10–15 min at room temperature (RT) and subsequently washed three times with 1xPBS. Depending on the antibodies applied, two different permeabilization methods were applied: after fixation cells were either permeabilized with 0.1% Triton X-100 in 0.1% sodium citrate (Thermo-Fisher Scientific) for 5 min at 4 °C or with 0.1% saponin in 1x PBS (Sigma Aldrich) at RT for 15 min. After permeabilization with Triton X-100, cells were blocked in blocking solution containing 10% horse serum (Thermo-Fisher Scientific) and 0.2% fish skin gelatine (Sigma-Aldrich) in 1xPBS for 1 hour at RT. Primary antibodies were then applied simultaneously diluted in blocking solution for 45 min at RT, followed by three washing steps with blocking solution. Fluorochrome-conjugated secondary antibodies were diluted and incubated with the cells for 45 min in the dark. Finally, cells were washed three times with 1xPBS and once with ddH2O before mounting them with fluorescent mounting medium (Dako Cytomation) onto microscopy slides. After permeabilization with saponin, cells were incubated with primary antibodies. Primary antibodies were simultaneously diluted in antibody solution (0.2% fish skin gelatine and 0.01% saponin in 1xPBS) and 100μL of antibody solution was applied per cover slip for 45 min at RT. Coverslips were then washed three times with blocking solution, followed by secondary antibody incubation for 45 min at RT in the dark. Before mounting coverslips with fluorescent mounting medium (Dako Cytomation) on microscopy slides, cells were washed three times with 1xPBS and once with ddH2O. All primary antibodies are listed in Table 1 with corresponding dilutions, permeabilization solution and manufacturer. All secondary antibodies were Alexa Fluor-conjugated secondary antibodies (Thermo Fisher Scientific) and diluted in a ratio of 1 : 1000.
Table 1
Primary antibodies including host species used for immunofluorescence experiments with corresponding dilution and permeabilization protocol
| Primary Antibody (host) | Dilution | Manufacturer (Cat. No.) | Permeabilized with |
| Oct3/4 (rat) | 1 : 100 | R&D Systems (MAB1759) | Triton-X |
| SOX2 (goat) | 1 : 200 | Santa Cruz Biotechnology (sc-17320) | Triton-X |
| PAX6 (mouse) | 1 : 200 | Synaptic Systems (153 011) | Triton-X |
| Nestin (rabbit) | 1 : 250 | abcam (ab105389) | Triton-X/Saponin |
| Map2a (mouse) | 1 : 200 | Synaptic Systems (188 011) | Triton-X/Saponin |
| Tau (guinea pig) | 1 : 200 | Synaptic Systems (314 004) | Triton-X/Saponin |
| NeuN (mouse) | 1 : 200 | Merck Millipore (MAB377) | Triton-X |
| TuJ (rabbit) | 1 : 200 | Synaptic Systems (302 304) | Triton-X/Saponin |
| GFAP (mouse) | 1 : 500 | Synaptic Systems (173 111) | Triton-X/Saponin |
| TH (rabbit) | 1 : 1000 | Merck Millipore (AB152) | Saponin |
| DAT extracellular (rat) | 1 : 200 | ATS Bio (AB-N17) | Triton-X |
| vMAT2 (rabbit) | 1 : 100 | Synaptic Systems (138 302) | Triton-X |
| GAD (rabbit) | 1 : 200 | Synaptic Systems (198 003) | Saponin |
| vGLUT (guinea pig) | 1 : 250 | Synaptic Systems (135 307) | Triton-X |
| TPH2 (rabbit) | 1 : 200 | Dianova (ABR-38586) | Saponin |
| SERT (mouse) | 1 : 200 | ATS Bio (AB-N40) | Saponin |
| Synapsin (mouse) | 1 : 200 | Synaptic Systems (106 011) | Triton-X/Saponin |
| Homer (guinea pig) | 1 : 200 | Synaptic Systems (160 004) | Triton-X/Saponin |
| Bassoon (rabbit) | 1 : 200 | Synaptic Systems (141 002) | Saponin |
| AIF (mouse) | 1 : 1000 | Acris (BM4114) | Triton-X |
| Mfn2 (rabbit) | 1 : 200 | Sigma-Aldrich (M6444) | Triton-X |
2.5Image acquisition of immunostained hiPS cell-derived neurons
All images were acquired with a confocal Leica TCS SP5 imaging system attached to a DM IRE2 microscope equipped with an acusto-optical beam splitter and a HCX PL APO 63x oil planchromat lens. Excitation lasers included an argon ion laser (458 – 514 nm), a DPSS laser (561 nm) and a HeNe laser (633 nm). Confocal z-stacks were acquired with sections taken every 0.1 to 0.5μm and exported as tiff-files. Image processing was performed with NIH ImageJ software version 1.51n (NIH Bethesda). If not indicated otherwise in figure legends, images are maximum projections of confocal z-stacks.
2.6ASP+ live cell imaging of hiPS cell-derived dopaminergic neurons
Functional dopamine transporter (DAT) molecules located to the cell surface of DA neurons allow to selectively label DA neurons with the fluorescent monoamine transporter substrate 4-(4-(dimethylamino)styryl)-N-methylpyridinium iodide (ASP+, Life Technologies). After DAT-dependent uptake, ASP+ accumulates in mitochondria [45, 46]. HiPS cell-derived DA neurons were washed with ND medium and exposed to 10μM ASP+ for 60 sec. Then, cells were washed again with ND medium and imaged in dye-free ND medium on a Leica TCS SP5 imaging system attached to a DM IRE2 [47]. Excitation laser was an argon ion laser running at 488 nm. Confocal z-stacks were acquired with sections taken every 0.5μm. Images were imported to and processed using NIH ImageJ version 1.51 (NIH Bethesda).
3Results and discussion
3.1Comparison of pluripotency and neuronal phenotype markers in healthy donor- and patient donor-derived hiPS cells and NPCs
For generation of hiPS cells, human skin fibroblast from a healthy donor were obtained via skin biopsy and transfected with a Sendai virus construct coding for four pluripotency genes Oct4, Sox2, Klf4 and c-Myc (Cytotune 2.0, Life Technologies). After emergence of cell clusters, hiPS cells were selected and expanded according to their morphology. Figure 1A shows exemplary images of donor fibroblasts and hiPS cell clusters. Healthy donor-derived hiPS cells displayed a normal karyotype after being kept undifferentiated for 20 passages (data upon request), and were positively immunostained for the pluripotency markers Oct4 and Sox2 (Fig. 1B). For transcriptome analyses of both hiPS cell lines and healthy donor-derived fibroblasts, quantitative RT-PCR was performed for several genes associated with pluripotency (Fig. 1D). Expression levels of mRNA transcripts were related to the transcriptional level of the housekeeping gene beta-actin (ACTB), which did not significantly differ between cell lines. Fibroblasts showed very low expression levels for all analyzed pluripotency genes, except for c-Myc. Compared to fibroblasts’ expression profile, both hiPS cell lines showed an increased expression level of pluripotency-associated transcription factors Oct4, Sox2, Lin28, Nanog and ZFP42. Only in case of c-Myc and Klf4, hiPS cell lines expression levels were not increased compared to fibroblasts. In case of c-Myc, this may be explained by the fact that this transcription factor is not a core pluripotency factor and not necessarily required for reprogramming of fibroblasts [48–51] and similar expression levels may be related to c-Myc’s role in cell growth [52–54]. In contrast to c-Myc, Klf4, Oct4 and Sox2 are considered to be essential for the induction of pluripotency (reviewed in [54]). The low expression profile of Kfl4 observed in our analysis may be compensated by the high expression levels of Oct4, Sox2, Lin28, Nanog and the other pluripotency transcription factors tested, which constitute a core pluripotency network and are partially able to bind to promoter and enhancer regions of other pluripotency-inducing transcription factors [48, 49, 54].
Fig.1
Generation of hiPS cells from adult human skin fibroblasts by sendaiviral reprogramming: (A) Exemplary images showing morphological differences between healthy donor human skin fibroblasts, healthy donor-derived hiPS cells (hiPSCcontrol), and patient donor-derived hiPS cells (hiPSCPD), the latter established by Divine and colleagues [44]. Scale bars: 200μm. (B) Immunofluorescence analysis of hiPSCcontrol for the pluripotency transcription factors Oct4 and Sox2. Scale bar: 200μm. (C) Exemplary images of healthy donor (DANPC) and patient donor (DANPC - PD) hiPS cell-derived DA progenitors. Scale bars: 200μm. (D) Expression analysis of mRNA of pluripotency markers in healthy donor-derived fibroblast, hiPSCcontrol and hiPSCPD. The analysis shows genotype-independent up-regulation of pluripotency related genes in hiPS cells compared to adult human skin fibroblasts. Graph shows mean±SD.
![Generation of hiPS cells from adult human skin fibroblasts by sendaiviral reprogramming: (A) Exemplary images showing morphological differences between healthy donor human skin fibroblasts, healthy donor-derived hiPS cells (hiPSCcontrol), and patient donor-derived hiPS cells (hiPSCPD), the latter established by Divine and colleagues [44]. Scale bars: 200μm. (B) Immunofluorescence analysis of hiPSCcontrol for the pluripotency transcription factors Oct4 and Sox2. Scale bar: 200μm. (C) Exemplary images of healthy donor (DANPC) and patient donor (DANPC - PD) hiPS cell-derived DA progenitors. Scale bars: 200μm. (D) Expression analysis of mRNA of pluripotency markers in healthy donor-derived fibroblast, hiPSCcontrol and hiPSCPD. The analysis shows genotype-independent up-regulation of pluripotency related genes in hiPS cells compared to adult human skin fibroblasts. Graph shows mean±SD.](https://content.iospress.com:443/media/jcb/2017/3-1/jcb-3-1-jcb179007/jcb-3-jcb179007-g001.jpg)
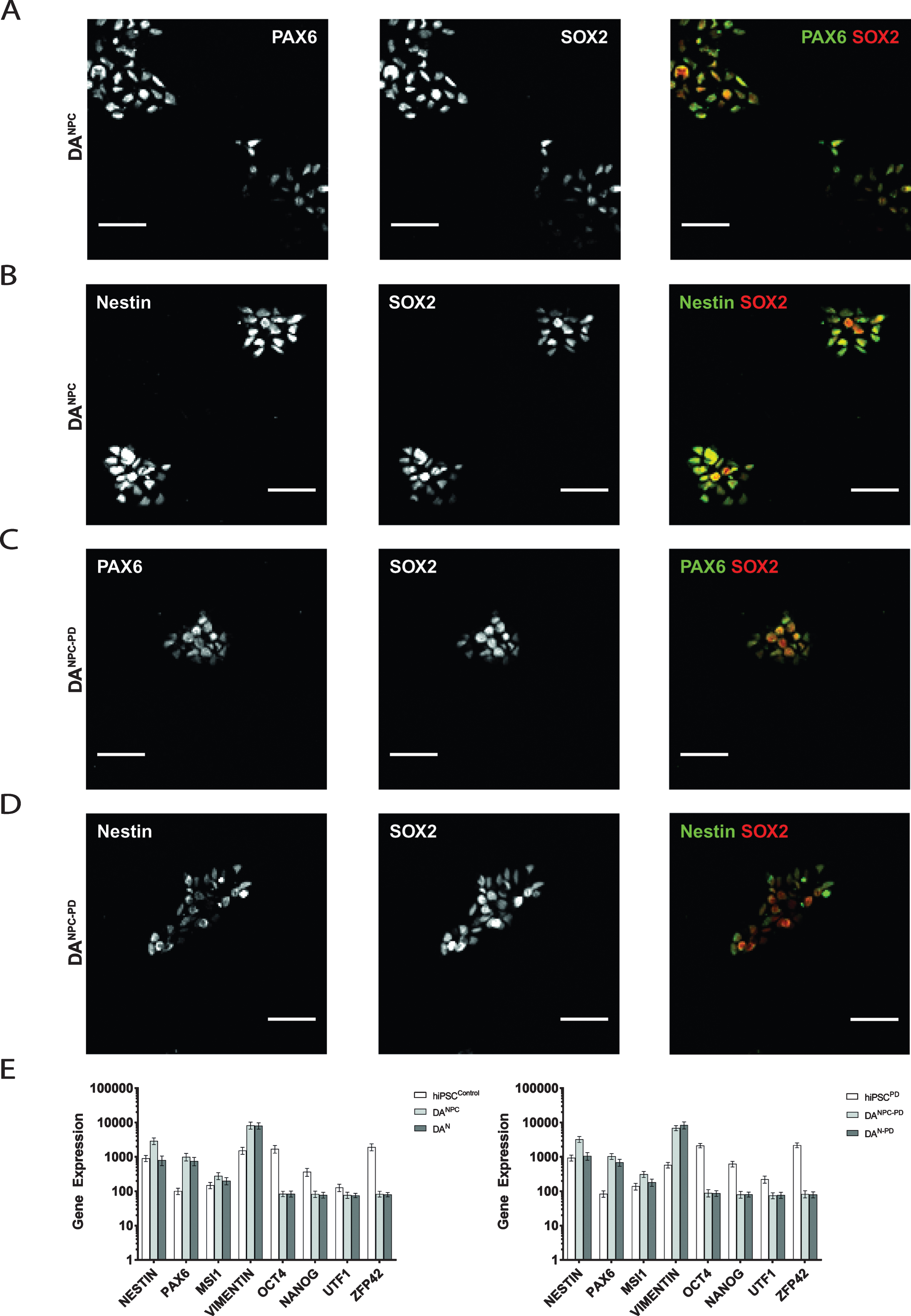
In order to generate stable DA NPC lines, hiPS cells of both genotypes were induced along neural fate pathway (see 2.3). NPC lines allow long-term cell culture and provide a convenient starting point for terminal differentiation into functional DA neurons [39, 42]. Exemplary images of our stable dopaminergic NPC lines are shown in Fig. 1C. Our immunofluorescence analysis showed that both genotypes of hiPS cell-derived DA progenitors express the neuronal progenitor markers (Fig. 2). Heathy donor-derived NPC (DANPC) displayed predominantly compact nucleus-shaped, overlapping staining patterns for Pax6 and Sox2 (Fig. 2A and 2B). In DANPC we observed a cytosolic localization for Nestin (Fig. 2B and 3A). Compared to DANPC, patient donor-derived DA progenitors (DANPC - PD) exhibited similar immunostainings for Nestin, Pax6 and Sox2 (Fig. 2C and 2D). In addition to immunofluorescence analysis, NPC lines of both genotypes were analyzed for transcription of a subset of differentiation-state associated genes in order to trace the phenotypic transition from pluripotency to neuronal stages. Upon neuronal induction, both hiPS cell-derived neuronal precursor cell lines show down-regulation of the pluripotency genes Oct4, Nanog, UTF1 and ZFP42 (Fig. 2E). In contrast to this, genes associated with neuronal phenotype, Nestin, Pax6, MSI1 and Vimentin, are up-regulated after successful generation of hiPS cell-derived NPC lines. With the exception of Nestin, a common marker for neuronal progenitors, all neuronal phenotype-related genes were still expressed at similar rates in hiPS cell-derived neurons. This may reflect the circumstance that terminal differentiation is not a simultaneously occurring process and late stage cultures likely still contain a significant number of proliferating progenitors. In summary, the gene set selected for our transcriptome analysis did not reveal genotype-specific differences at pluripotency and later neuronal stages. Although a differential gene analysis would provide a more complete insight into transcriptional activities, our findings showed that no aberrant gene expression interfered with successful neuronal differentiation using our protocol.
Fig.2
Generation of genotype-specific DA progenitor cell lines. Immunofluorescence analysis of healthy donor hiPS cell-derived DANPC showed co-distributed immunostainings for Pax6 and Sox2 (A) as well as for Nestin and Sox2 (B). Similar distributions of antibody signals for Pax6 and Sox2 (C), and Nestin and Sox2 (D) were found in DANPC - PD. Scale bars: 50μm. (E) Expression analysis of pluripotency and neuronal phenotype genes in hiPS cells, NPC and neurons of both genotypes. Pluripotency markers (Oct4, Nanog, UTF1, ZFP42) are downregulated in dopaminergic progenitors (DANPC and DANPC - PD) and terminally differentiated neuronal cultures (DAN and DAN - PD). Genes associated with acquisition of a neuronal phenotype (Nestin, Vimentin, Pax6, MSI1) are equally upregulated. Graphs show mean±SD.
3.2Terminal neuronal differentiation along the dopaminergic pathway
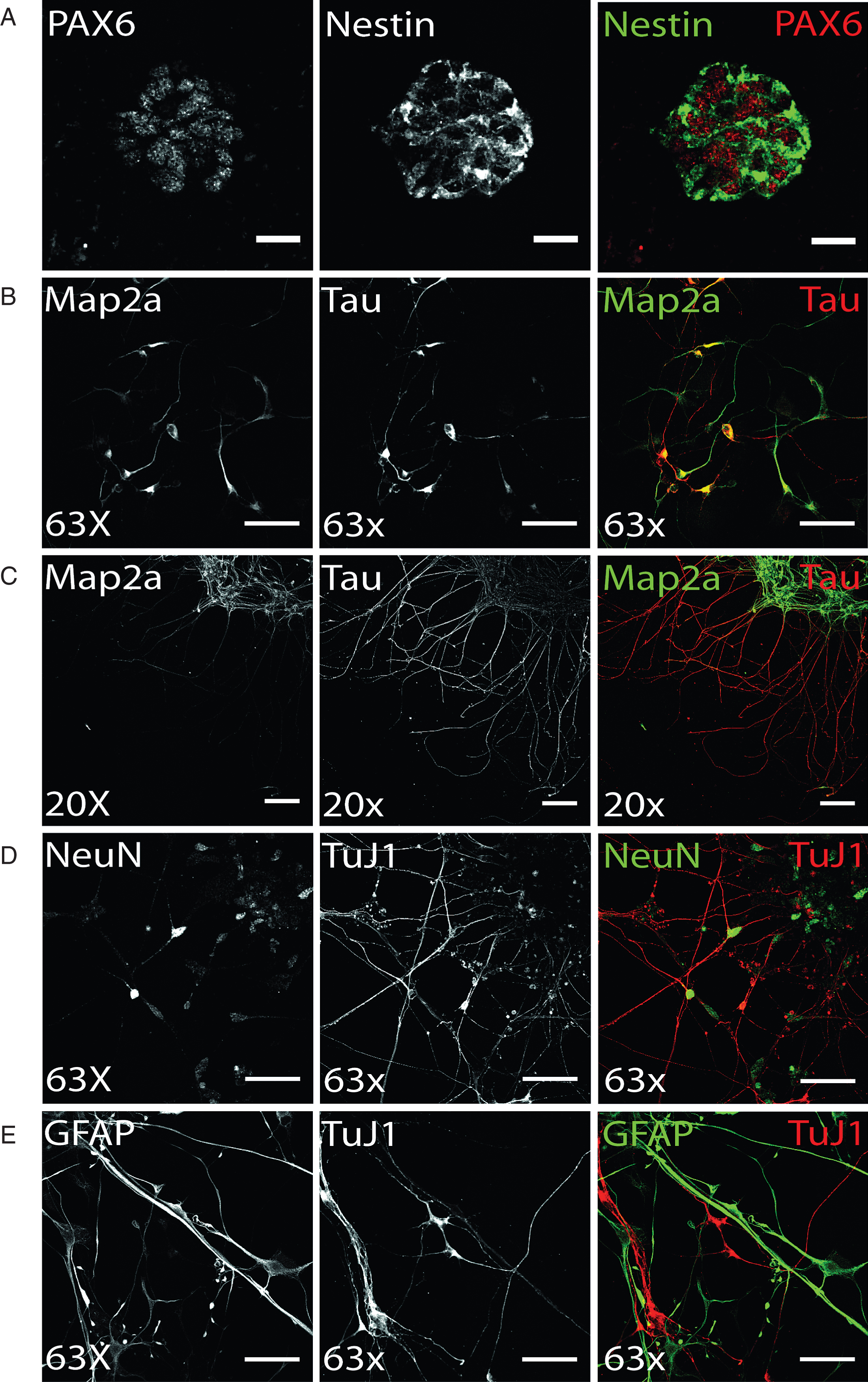
Terminal dopaminergic differentiation was started by a two week selection of ventral midbrain dopaminergic progenitors in order to increase the yield of DA neurons (see 2.3.). Following this selection stage, terminal differentiation of preselected NPC was induced and already after 14 days of terminal differentiation, hiPS cell-derived neurons could be identified based on immunostainings (Fig. 3). The axonal marker protein Tau and the somatodendritic protein Map2a were already detectable on day 14 of terminal differentiation (Fig. 3B), yet still found to colocalize in immature human stem cell-derived neurons. Upon aging of neurons, the level of neuronal polarity increases and immunosignals of both proteins were clearly restricted to distinct cellular locations. On d50 of terminal neuronal differentiation, neuronal polarity is fully established in in vitro-generated neurons and was verified by immunofluorescence analysis (Fig. 3C). Accordingly, on d50 of terminal neuronal differentiation, expression of the neuronal maturation marker NeuN can be found in approximately 80% of all cells, which are also positively immunostained for the neuronal microtubuli protein TuJ1 (Fig. 3D). However, induction of terminal differentiation does not result in a pure neuronal culture: in addition to TuJ1-positive hiPS cell-derived neurons, immunostaining for GFAP revealed a similar partition of glial cells (Fig. 3E).
Fig.3
Differentiation of ventral midbrain DA NPCs into neural cells: (A) Exemplary Pax6 and Nestin immunostaining of DANPC, the starting point for terminal differentiation. PAX6 (red) shows nuclear expression of protein, whereas Nestin (green) signal is restricted to cytoplasm. Scale bar: 25μm. (B-D) Neuronal phenotype of hiPS cell-derived neurons was verified by immunofluorescence analysis for Map2a, Tau, NeuN, and TuJ1. (B) On d14 of terminal differentiation, neuronal polarity was only partially established, indicated by co-localized signal of axonal TAU and somatodendritic MAP2a. Scale bar: 50μm. (C) On d15 of terminal differentiation, neuronal polarity was established as shown by exclusively TAU-positive axonal and Map2a-positive dendritic networks. Scale bar: 100μm. (D) Exemplary immunostaining of neuronal maturation marker NeuN and neuronal structural marker TuJ1. Scale bar: 50μm. (E) Terminal differentiation of NPC gave also rise to glial cells, as shown by cells showing GFAP-positive, TuJ1-negative immunostaining (d50). Scale bar: 50μm.
3.3Verification of the dopaminergic phenotype
In order to confirm the dopaminergic identity of developing hiPS cell-derived neurons, immunofluorescence analyses were performed for various dopaminergic neuron-specific marker proteins, including (1) tyrosine hydroxylase (TH), the rate-limiting enzyme for dopamine synthesis, (2) dopamine transporter (DAT), which was detected in subcellular compartments as well as located to the cell surface, and (3) vesicular monoamine transporter (vMAT2), which is required to load dopamine into synaptic vesicles.
On d14 of terminal differentiation, quantification of immunostainings revealed up to 40% GFAP-positive cells and 30% Tau-positive cells in both cell lines (data not shown). Of the latter, cells were predominantly positively stained by DAT and TH antibodies, and the amount of DA neurons was within the expected range of previously published data by Reinhardt and colleagues [41]. TH immunofluorescence showed granular intracellular staining, which can be found in perinuclear somatic area as well as on neurites (Fig. 4A). Cells that were found to be negative for neuronal markers did not display TH immunostaining on a detectable level. Similarly, expression of DAT was found to be present in the majority of hiPS cell-derived neurons (Fig. 4B), with localization in the somatic region and TAU-positive axons. In addition, on d50 of terminal differentiation, DAT molecules were also detected on the cell surface of hiPS cell-derived DA neurons (Fig. 4C), indicating the ability to take up DA or other monoamine transporter substrates [55]. Immunostaining pattern for cell surface-located DAT molecules displayed a more granular staining compared to the vesicular tightly packed intracellularly located DAT. Furthermore, neurons were analyzed for the presence of vMAT2. Here, the majority of neurons was also positively stained and revealed a globular staining pattern along neurites and perinuclear regions (Fig. 4D). Genotype-specific differences in immunostainings regarding dopaminergic markers were not observed. The immunofluorescence analysis for dopaminergic marker proteins also showed that not all cells identified as neurons by immunostaining for Map2a, TuJ1 and/or Tau displayed a dopaminergic phenotype.
Fig.4
Verification of dopaminergic identity. Exemplary images of terminally differentiated hiPS cell-derived DA neurons on d50 are shown. (A) Expression of rate-limiting enzyme for DA synthesis TH. Immunostaining shows cells positive for Map2a (green) were also found to be positive for TH, with signal being mainly located in somatic areas (arrow heads), but also present in neurites. (B, C) As for TH, DAT expression could be verified by immunocytochemistry. For intracellular DAT molecules (B), dense immunosignals were found in perinuclear regions and on neurites. Cell surface-located DAT was especially present on neurites (C) and displayed more granular immunosignals compared to whole cell staining patterns. (D) Exemplary immunofluorescence images for vMAT2. Scale bar: 50μm. (E) Gene expression analysis of genes encoding for alpha-synuclein (SNCA) and DA signaling (TH, DAT, VMAT2, catechol-O-methyltransferase (COMT), monoamino oxidase A (MAOA), dopamine receptors D1-D4 (DRD1-DRD4). (F) Gene expression analysis of genes encoding for alpha-synuclein (SNCA) and presynaptic scaffolding proteins Piccolo (PCLO), Bassoon (BSN), postsynaptic scaffolding proteins Homer1 and Shank3, and the synaptic vesicle-associated proteins Synapsin 1 (SYN1), Synaptobrevin-1 (VAMP1), Syntaxin 1A (STX1A), and SNAP25.
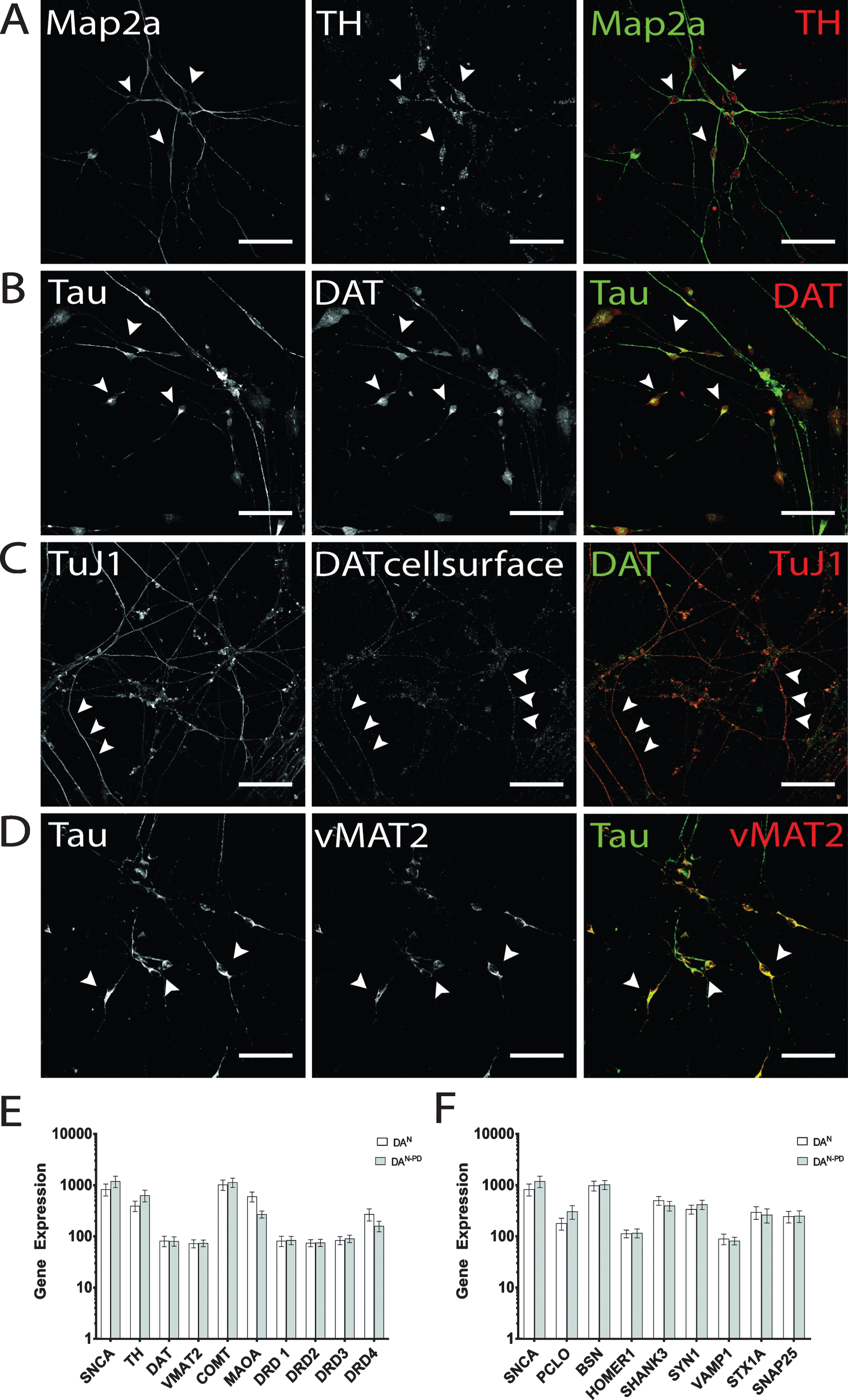
Regarding their mRNA expression profiles, we have compared the transcription levels of genes associated with dopaminergic neurotransmission (Fig. 4E) and synapse formation (Fig. 4F) in hiPS cell-derived neuronal cultures of both genotypes. As expected, SNCA transcripts were enhanced in neurons derived from hiPSPD cells (1200 vs 837) [44]. With the exception for TH (397 in hiPSC neurons vs 636 in hiPSCPD neurons), the dopamine receptor D4 mRNA transcripts (DRD4; 272 in hiPSC neurons vs 160 in hiPSCPD neurons), and monoamino oxidase A (MAOA, 604 in hiPSC neurons vs 271 in hiPSCPD neurons), no differences in transcription levels were found for genes involved in DA signaling (Fig. 4E). Of note, absolute values of mRNA transcripts received may be regarded low, since the overall number of DA neurons among the cellular population is comparatively small. Therefore, to strictly restrict such analysis to DA neurons, separation of DA neurons is required. The comparison of randomly selected synaptic protein mRNA transcripts revealed that the transcription levels of the presynaptic scaffolding protein Piccolo was enhanced in hiPSPD neurons (PCLO, 180 in hiPSC neurons vs 370 in hiPSCPD neurons), while the presynaptic scaffolding protein Bassoon did not differ in transcription levels (Fig. 4F). A similar observation was made for Synapsin1 (SYN1; 341 in hiPSC neurons vs 423 in hiPSCPD neurons), which was also enhanced in the PD genotype neurons, while other synaptic vesicle protein-encoding genes did not differ in transcription levels. The comparison of the postsynaptic density genes Homer1 and Shank3 showed that in hiPSCPD neurons Shank3 transcripts were downregulated compared to healthy control hiPSC neurons (504 in hiPSC neurons vs 399 in hiPSCPD neurons). Again, these data were acquired by performing an mRNA extraction of complete cell culture dishes without separating DA neurons from other cells arising during terminal differentiation (see below). Therefore absolute transcription levels of synaptic genes may be compromised by the lack of purity.
In order to identify the neuronal phenotype of the non-dopaminergic neuronal population derived from hiPS cell lines using our differentiation protocol, immunostaining for other neuronal phenotype markers were performed. On d50 of terminal differentiation, co-immunofluorescence analysis of neuronal phenotype markers Map2a and TuJ1 in combination with markers for either GABA-ergic, glutamatergic or serotonergic neurons was performed.
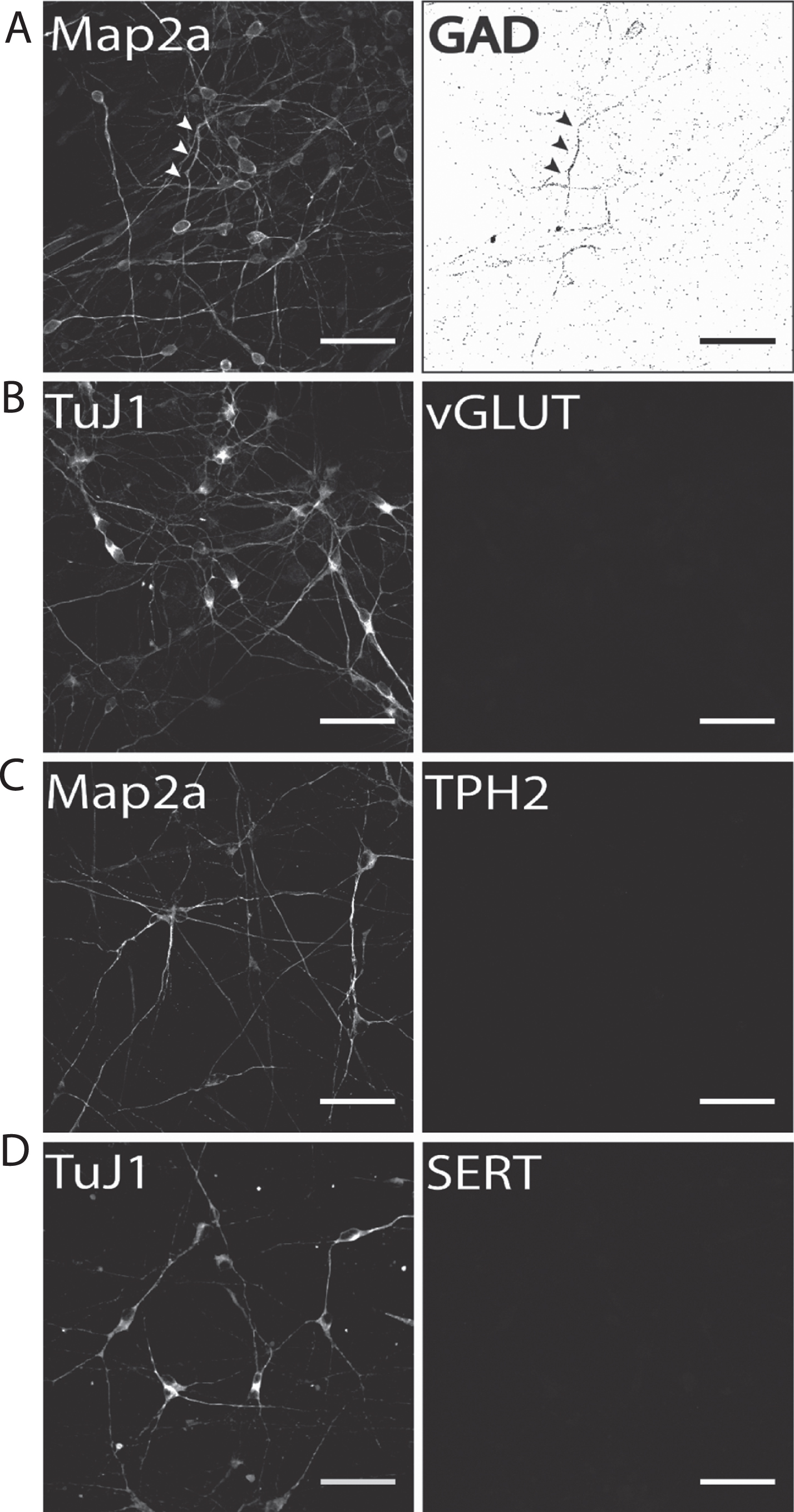
Our results show that on d50 of terminal neuronal differentiation a small proportion of neurons were positively immunostained for the GABA-ergic marker protein glutamate decarboxylase-1 (GAD1; Fig. 5A). Therefore, it is particularly important to either distinctly distinguish or separate DA from GABAergic neurons to prevent compromising experimental results when addressing DA neurons. In contrast to GAD1 immunostaining, no neuronal subpopulation of vGLUT-positive glutamatergic neurons was found (Fig. 5B). The same holds true for immunofluorescence analysis for two serotonergic markers tryptophan hydroxylase 2 (TPH2; Fig. 5C), which is the rate-limiting enzyme for serotonin synthesis in serotonergic CNS neurons, and the serotonin transporter (SERT, Fig. 5D). In both cases, no hiPS cell-derived neurons were found to be positively immunostained. This is particularly important for two reasons: (1) fluorescent dyes specifically taken up by monoamine transporters, such as ASP+ [47, 56], can be applied to selectively label hiPS cell-derived DA neurons when no other monoaminergic neurons are present and (2) hiPS cell-derived neuronal populations generated according to our protocol may be applied to selectively investigate the effect of Parkinsonian drugs, for example MPP+, on dopaminergic neurotransmission [55] in human stem cell-derived dopaminergic neurons.
Fig.5
Identification of potential subpopulations in hiPS cell-derived neuronal cultures. Exemplary images of terminally differentiated hiPS cell-derived neurons are shown (d50). (A) GAD1 as GABA-ergic marker, (B) vGLUT1 as a glutamatergic marker, and (C) TPH2 and (D) SERT as marker proteins for serotonergic subpopulations. Except for GAD1, no cells were positively stained for any of the other tested markers, indicating a predominantly DA neurons phenotype for neurons generated with our protocol. Scale bar: 50μm.
3.4Detection of synapses in healthy donor hiPS cell-derived DA neurons
One goal in establishing robust dopaminergic differentiation protocols is to provide reproducible cellular human disease models, for example to address synaptic connectivity and DA signaling in vitro. To demonstrate the potential to analyze human dopaminergic synapses in vitro, immunostainings were performed for presynaptic Synapsin1, one of the most specific markers for CNS synapses and the postsynaptic marker Homer1, which is found to be concentrated at postsynaptic densities. In order to identify dopaminergic neurons, cells were counterstained for TH expression (data not shown). Immunofluorescence of hiPS cell-derived DA neurons revealed that both synaptic proteins were expressed and appeared in punctual staining patterns along the neurites (Fig. 6A). Comparison of immunostaining signals showed that Homer1- and Synapsin1-derived signals localized to similar structures, indicating spatial proximity that provides the cornerstone of synapse formation (indicated by white arrows in Fig. 6A). Of note, DA neurons were shown to rely on volume transmission in addition to synaptic transmission [57, 58] explaining why presynaptic proteins localize to axonal varicosities along the axons of DA neurons, partially without a corresponding postsynaptic counterpart. To ensure that functional synapses are addressed, such synapses may be verified by activity-dependent synaptic uptake and release of fluorescent dyes [59, 60].
Fig.6
Visualization of presynaptic proteins and mitochondrial marker AIF hiPS cell-derived DA neurons (d50). Exemplary images of terminally differentiated healthy donor stem cell-derived neurons are shown. (A) Immunostaining for postsynaptic marker Homer1 (green) and presynaptic marker protein Synapsin1 (red). Merge images show partial co-localization, indicating spatial proximity of both synaptic proteins required for synapse formation in dopaminergic hiPS cell-derived neurons (arrow heads). Yellow asterisk indicates area of zoom images shown in the second panel. Scale bar: 25μm, Zoom images: 10μm. (B) Additional expression of presynaptic scaffolding protein bassoon (green) was assessed in dopaminergic TuJ1 positive neurons (red) and found to be present in somatic and neuritic regions (arrow heads). Scale bar: 50μm. (C) Co-staining of bassoon (red) and mitochondrial marker AIF (green) shows extensive colocalization in bassoon positive presynapses of hiPS cell-derived neurons. Scale bar: 50μm.
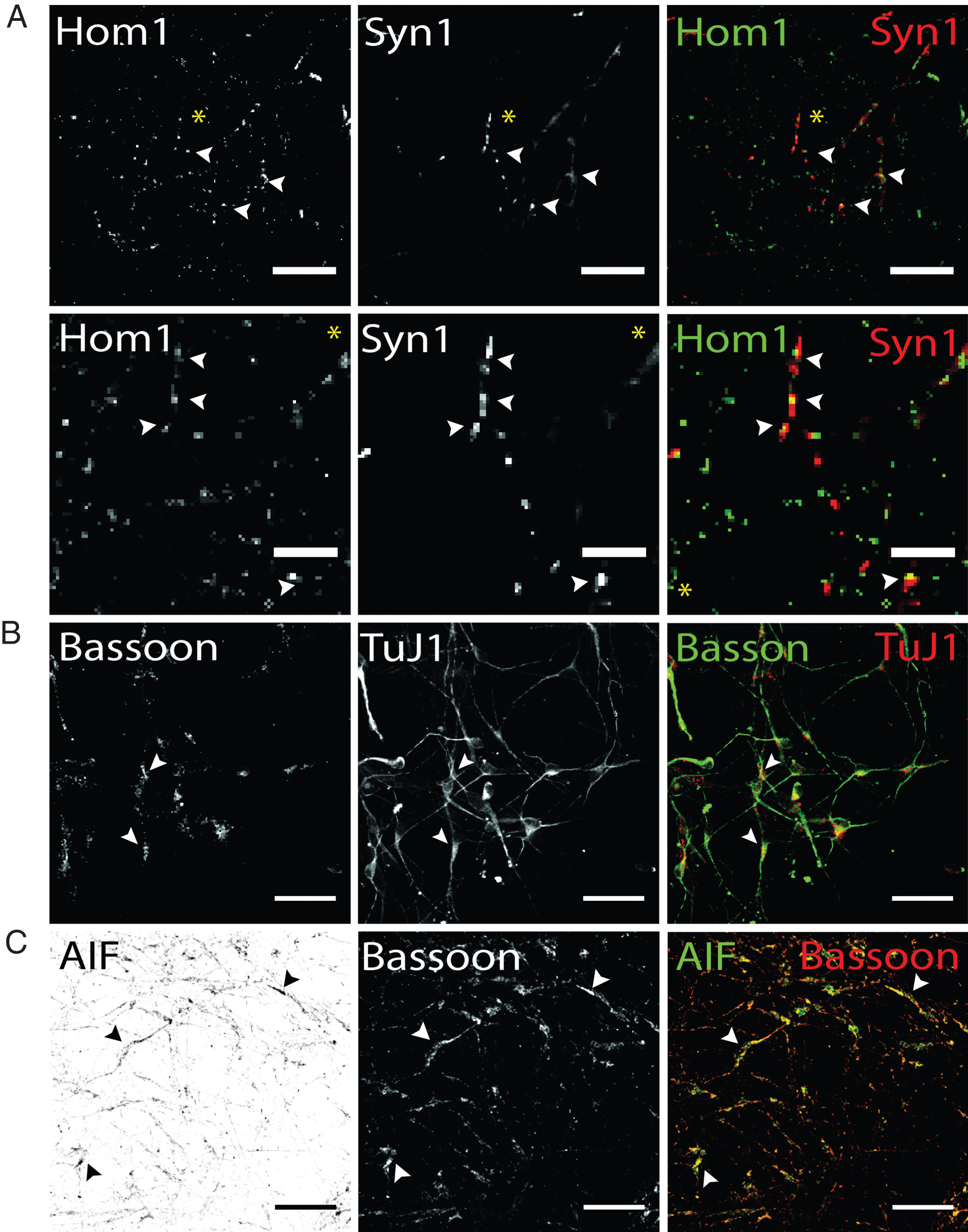
In addition, hiPS cell-derived neurons were analyzed for the localization of the presynaptic scaffolding protein Bassoon, essential for maintenance and assembly of the presynaptic active zone of vertebrate synapses (45). Bassoon has been shown to be involved in recruitment of voltage-gated Ca2 + channels, priming of synaptic vesicles and structural integrity of synapses (46). Immunostaining hiPS cell-derived neuronal networks showed localization of Bassoon signals cell soma-close as well as TuJ1-positive neurites (Fig. 6B). TuJ1-deficient, Bassoon-positive cellular structures could not be observed. Regarding Parkinson’s disease, an aberrant synaptic mitochondrial activity was reported (47). To detect presynaptically located mitochondria, antibody co-staining of Bassoon and the mitochondrial marker apoptosis-initiating-factor (AIF), which is located in the intermembrane space of mitochondria, was performed. AIF has not only been shown to be involved in pro-apoptotic processes, but also in maintaining a functional complex I in the respiratory chain of mitochondria (48). Our results of immunostaining for presynaptic bassoon and AIF showed a granular staining for both proteins and extensive colocalization in Bassoon-positive subcellular structures. To summarize, immunofluorescence analysis using antibodies targeted against synaptic and mitochondria marker proteins enables to address certain aspects of synaptic connectivity, including synaptic density in hiPS cell-derived dopaminergic networks or synaptic mitochondrial density.
3.5Visualization of mitochondria in healthy donor hiPS cell-derived DA neurons
A disturbed mitochondria metabolism has been shown to be involved in the neurodegenerative pathology of Parkinson’s disease. Mitochondria constantly undergo fusion and fission processes and move along cytoskeletal tracks to maintain physiological homeostasis. Especially the former two occur in a delicate balance and disturbance can result in neurodegenerative pathology, as observed in PD, and are mediated by mitofusins (Mfn) 1 and 2 [61]. For the latter one, in vivo studies performed in mice have shown a connection between the loss of Mfn2 and retrograde degeneration of DA neurons, suggesting a role for Mfn2 in PD pathology [62]. Given this example, analysis of mitochondrial localization and function by microscopic approaches, such as immunofluorescence analyses or live cell imaging experiments, provide a fast high-potential approach to investigate mitochondria metabolism in human DA neurons in vitro. Such experimental approaches were recently applied to investigate differential effects of the Parkinsonian toxic MPP+ in mouse embryonic stem cell-derived monoaminergic neurons [55].
In addition to the visualization of mitochondrial density at synapses (Fig. 6C), the localization, and thereby the density of mitochondria, can be tracked combining mitochondrial AIF and axonal TAU staining to visualize mitochondrial trafficking along dopaminergic axonal networks (Fig. 7A). Similarly mitochondrial tracking in hiPS cell-derived DA neurons can be performed by application of Mfn2 antibodies (Fig. 7B). Compared to AIF (Fig. 6C and 7A), Mfn2 staining appeared evenly distributed throughout cells, with no clear subcellular distribution patterns. Staining for intracellular mitochondria markers and neuronal phenotype markers, including Tau and TuJ1, may also be combined with staining for cell surface-located DAT molecules (Fig. 7B). This enables to combine the analysis of mitochondrial aspects of interest with the availability of DAT molecules that are able to take up DA or other substrates. Generally, since AIF and Mfn2 immunostaining is not restricted to hiPS cell-derived dopaminergic neurons, co-staining with a dopaminergic phenotype marker, either TH or DAT, is required to restrict analysis to dopaminergic neurons.
Fig.7
Immunocytochemical analysis of mitochondria distribution in hiPS cell-derived DA neurons and DAT specific uptake of fluorescent dye ASP+. (A) Immunostaining of mitochondrial intermembrane protein AIF in dopaminergic neurons after 50 days of terminal differentiation. AIF (green) is found to be present in cell soma and neurites (arrow heads). Dopaminergic neurons can be identified by co-localized signals of Tau (red) and TH (magenta). Scale bar: 50μm. (B) Mitochondrial fusion protein Mfn2 (green) can be visualized by immuno-cytochemistry and is evenly distributed throughout cells. Since not only neuronal cells are positively stained for Mfn2, co-staining with neuronal marker (here: TuJ1, red) and dopaminergic marker DAT was applied to identify Mfn-signals exhibited by DA neurons. Scale bar: 50μm. (C) Live cell imaging experiments in hiPS cell-derived DA neurons show DA-specific uptake of fluorescent ASP+ dye into neurons and accumulation in mitochondria. Zoom images show distribution of mitochondria along neurites and neurite endings, with mitochondrial morphology being round and punctual (arrow heads), whereas mitochondria located in cell somas are more elongated (arrows). Scale bar: 25μm, Zoom images: 5μm.
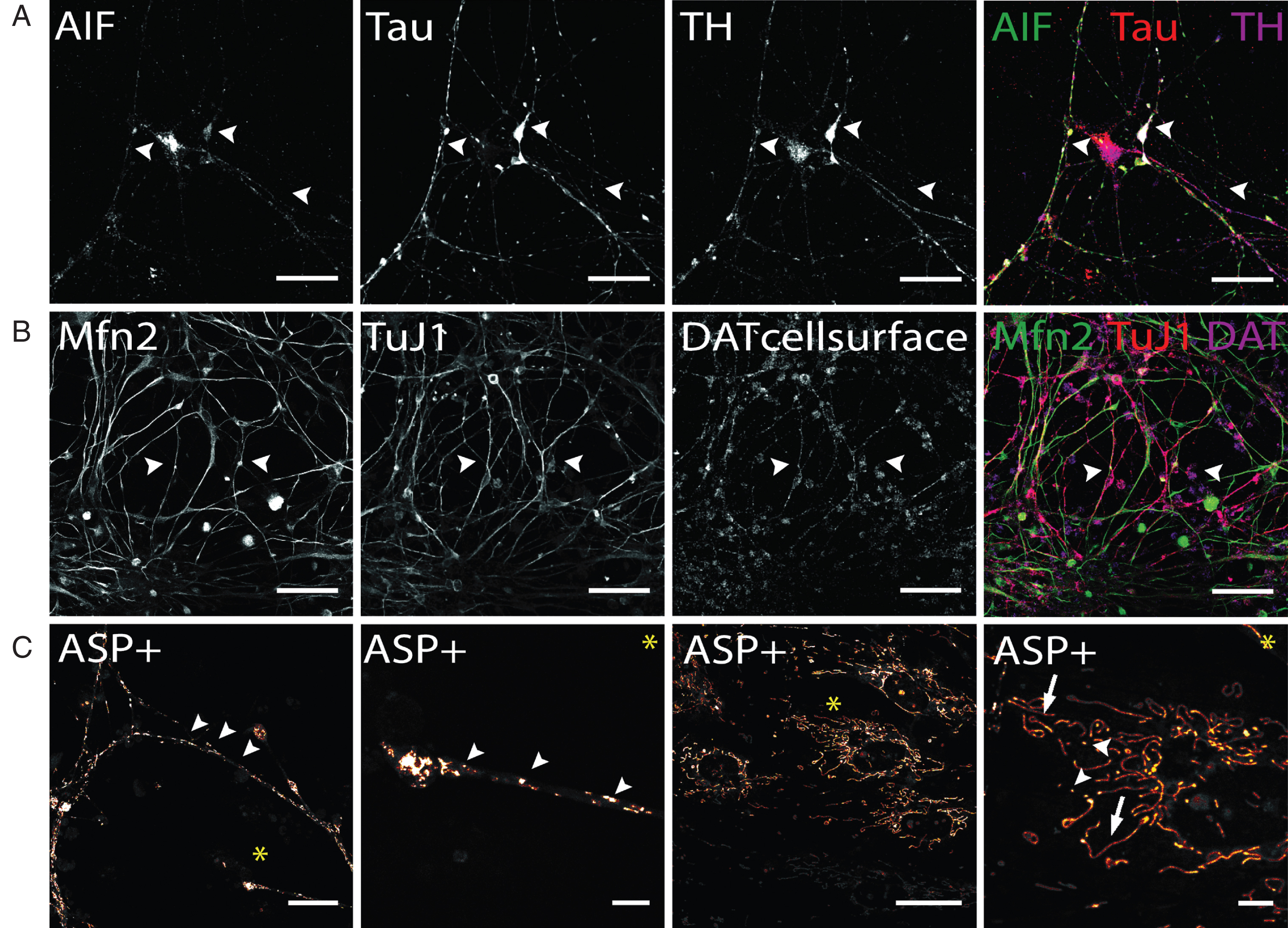
Regarding live cell imaging approaches, the application of MPTP-derived fluorescent dyes, which are selectively taken up by monoamine transporters expressed in stem cell-derived neurons [47, 55, 56] and label mitochondria [45, 46], provide a suitable tool to analyze morphology, fusion and fission of mitochondria in hiPS cell-derived DA neurons even if the neuronal population contains non-dopaminergic neurons [55]. Figure 7C demonstrates that ASP+ taken up DAT-dependently into hiPS cell-derived DA neurons enables to selectively label mitochondria on neurites and cell bodies of these neurons without staining mitochondrial structures inside non-dopaminergic cells. In neurites and neurite endings, mitochondria appear to be predominantly of small and round shape and can be differentiated from each other. In cell soma, mitochondria distribution is less dense compared to neurites. A large proportion of mitochondria appear elongated implicating extensive fission and fusion processes occurring especially in somatic areas. In addition to investigating mitochondria based on MPTP-derived fluorescent dyes, synaptic activity of DA neurons can also be selectively investigated in a mixed hiPS cell-derived neuronal population. Here, the application of fluorescent false neurotransmitters (FFN) enables to selectively track DA containing synaptic vesicles of hiPS cell-derived DA neurons, since their transport into synaptic vesicle depends on vMAT2, expressed in DA neurons [63]. In summary, both, morphological studies based on immunofluorescence analysis and live cell imaging using DAT- or vMAT2-dependent uptake of fluorescent dyes, can be easily applied to investigate mitochondria density, trafficking and fusion-fission as well as synaptic activity of human dopaminergic neurons in vitro.
4Conclusion
Mature, human stem cell-derived DA neurons can be generated from preselected DA progenitors within 50 days. The obtained neuronal cultures are devoid of other monoaminergic neurons, enabling to perform live cell experiments by selectively label DA neurons using fluorescent transporter substrates such as ASP+ or fluorescent false neurotransmitters. Immunostaining of synapses and mitochondria still requires co-staining with DA phenotype markers, to restrict respective analysis on DA neurons, and avoid compromising results by the high number of glia cells and small number of GABAergic neurons growing out of neuronal progenitors. Taken together, hiPS cell-derived DA neurons are a most suitable experimental system for a confocal laser scanning microscope-based experimental approaches to investigate DA signaling in vitro. Thereby human in vitro-generated DA neurons constitute an easily accessible cellular model to study core pathomechanisms and will significantly contribute to our understanding of DA signaling in the context of neurodegenerative diseases.
Declaration of interest
Authors declare that there is no conflict of interest.
Acknowledgments
YM and PS were funded by Marie-Sklodowska-Curie action project No. 608381, funding scheme FP7-MC-ITN. The work of YM and TL is funded by the H.W. & J. Hector Stiftung. EN is funded by the State Ministry of Baden-Wuerttemberg for Sciences, Research and Arts (Cooperative Research Training Group “Tissue analytics for stem cell based diagnostics and therapy”). The work of EN and MH is funded by the German Federal Ministry of Research (BMBF, Innovation Partnership M2Aind, project No. M2OGA (03FH8I02IA). SH and AML were funded by the Deutsche Forschungsgemeinschaft (SFB636 to AML) and the FOUNDATION ROGER DE SPOELBERCH (to AML). The funders were not involved in design of the study or writing of the manuscript. The authors thank Gina Tillmann and Isabell Moskal for their excellent technical assistance.
References
[1] | Denenberg VH , Kim DS , Palmiter RD . The role of dopamine in learning, memory, and performance of a water escape task. Behav Brain Res (2004) ;148: (1-2):73–8. |
[2] | Parker NF , Cameron CM , Taliaferro JP , Lee J , Choi JY , Davidson TJ , et al. Reward and choice encoding in terminals of midbrain dopamine neurons depends on striatal target. Nat Neurosci (2016) ;19: (6):845–54. |
[3] | Wise RA . Dopamine, learning and motivation. Nat Rev Neurosci (2004) ;5: (6):483–94. |
[4] | Bjorklund A , Dunnett SB . Dopamine neuron systems in the brain: An update. Trends Neurosci (2007) ;30: (5):194–202. |
[5] | Dahlstroem A , Fuxe K . Evidence for the existence of monoamine-containing neurons in the central nervous system. I. Demonstration of monoamines in the cell bodies of brain stem neurons. Acta Physiol Scand Suppl SUPPL (1964) ;232: :1–55. |
[6] | DiMaio S , Grizenko N , Joober R . Dopamine genes and attention-deficit hyperactivity disorder: A review. J Psychiatry Neurosci (2003) ;28: (1):27–38. |
[7] | Schultz W . Dopamine neurons and their role in reward mechanisms. Curr Opin Neurobiol (1997) ;7: (2):191–7. |
[8] | Grace AA . Dysregulation of the dopamine system in the pathophysiology of schizophrenia and depression. Nat Rev Neurosci (2016) ;17: (8):524–32. |
[9] | Weiss F , Maldonado-Vlaar CS , Parsons LH , Kerr TM , Smith DL , Ben-Shahar O . Control of cocaine-seeking behavior by drug-associated stimuli in rats: Effects on recovery of extinguished operant-responding and extracellular dopamine levels in amygdala and nucleus accumbens. Proc Natl Acad Sci U S A (2000) ;97: (8):4321–6. |
[10] | Hodaie M , Neimat JS , Lozano AM . The dopaminergic nigrostriatal system and Parkinson’s disease: Molecular events in development, disease, and cell death, and new therapeutic strategies. Neurosurgery (2007) ;60: (1):17–28; discussion -30. |
[11] | Wise RA . Roles for nigrostriatal–not just mesocorticolimbic–dopamine in reward and addiction. Trends Neurosci (2009) ;32: (10):517–24. |
[12] | Clarke CE . Parkinson’s disease. BMJ (2007) ;335: (7617):441–5. |
[13] | de Lau LM , Breteler MM . Epidemiology of Parkinson’s disease. Lancet Neurol (2006) ;5: (6):525–35. |
[14] | Sveinbjornsdottir S . The clinical symptoms of Parkinson’s disease. Journal of Neurochemistry (2016) ;139: :318–24. |
[15] | Ascherio A , Schwarzschild MA . The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol (2016) ;15: (12):1257–72. |
[16] | Pringsheim T , Jette N , Frolkis A , Steeves TD . The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Movement disorders: Official Journal of the Movement Disorder Society (2014) ;29: (13):1583–90. |
[17] | Dauer W , Przedborski S . Parkinson’s disease: Mechanisms and models. Neuron (2003) ;39: (6):889–909. |
[18] | Kim WS , Kågedal K , Halliday GM . Alpha-synuclein biology in Lewy body diseases. Alzheimer’s Research & Therapy (2014) ;6: (5):73. |
[19] | Klein C , Westenberger A . Genetics of Parkinson’s Disease. Cold Spring Harbor Perspectives in Medicine (2012) ;2: (1):a008888. |
[20] | Stefanis L . α-Synuclein in Parkinson’s Disease. Cold Spring Harbor Perspectives in Medicine (2012) ;2: (2):a009399. |
[21] | Blesa J , Trigo-Damas I , Quiroga-Varela A , Jackson-Lewis VR . Oxidative stress and Parkinson’s disease. Front Neuroanat (2015) ;9: :91. |
[22] | Kim GH , Kim JE , Rhie SJ , Yoon S . The role of oxidative stress in neurodegenerative diseases. Exp Neurobiol (2015) ;24: (4):325–40. |
[23] | Schapira AH . Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol (2008) ;7: (1):97–109. |
[24] | JohnsonRG Jr. Accumulation of biological amines into chromaffin granules: A model for hormone and neurotransmitter transport. Physiol Rev (1988) ;68: (1):232–307. |
[25] | Langston JW , Ballard P , Tetrud JW , Irwin I . Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science (1983) ;219: (4587):979–80. |
[26] | Moratalla R , Quinn B , DeLanney LE , Irwin I , Langston JW , Graybiel AM . Differential vulnerability of primate caudate-putamen and striosome-matrix dopamine systems to the neurotoxic effects of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Proc Natl Acad Sci U S A (1992) ;89: (9):3859–63. |
[27] | Singer TP , Ramsay RR . Mechanism of the neurotoxicity of MPTP. An update. FEBS Lett (1990) ;274: (1-2):1–8. |
[28] | Takahashi K , Tanabe K , Ohnuki M , Narita M , Ichisaka T , Tomoda K , et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell (2007) ;131: (5):861–72. |
[29] | Engel M , Do-Ha D , Munoz SS , Ooi L . Common pitfalls of stem cell differentiation: A guide to improving protocols for neurodegenerative disease models and research. Cell Mol Life Sci (2016) ;73: (19):3693–709. |
[30] | Okano H , Yamanaka S . iPS cell technologies: Significance and applications to CNS regeneration and disease. Molecular Brain (2014) ;7: :22. |
[31] | Takahashi K , Yamanaka S . A decade of transcription factor-mediated reprogramming to pluripotency. Nat Rev Mol Cell Biol (2016) ;17: (3):183–93. |
[32] | Torrent R , De Angelis Rigotti F , Dell’Era P , Memo M , Raya A , Consiglio A . Using iPS Cells toward the Understanding of Parkinson’s Disease. J Clin Med (2015) ;4: (4):548–66. |
[33] | Li W , Chen S , Li JY . Human induced pluripotent stem cells in Parkinson’s disease: A novel cell source of cell therapy and disease modeling. Progress in Neurobiology (2015) ;134: :161–77. |
[34] | Xiao B , Ng HH , Takahashi R , Tan EK . Induced pluripotent stem cells in Parkinson’s disease: Scientific and clinical challenges. J Neurol Neurosurg Psychiatry (2016) ;87: (7):697–702. |
[35] | Xu X , Huang J , Li J , Liu L , Han C , Shen Y , et al. Induced pluripotent stem cells and Parkinson’s disease: Modelling and treatment. Cell Prolif (2016) ;49: (1):14–26. |
[36] | Brennand KJ , Simone A , Jou J , Gelboin-Burkhart C , Tran N , Sangar S , et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature (2011) ;473: (7346):221–5. |
[37] | Hartfield EM , Yamasaki-Mann M , Ribeiro Fernandes HJ , Vowles J , James WS , Cowley SA , et al. Physiological characterisation of human iPS-derived dopaminergic neurons. PLoS One (2014) ;9: (2):e87388. |
[38] | Perrier AL , Tabar V , Barberi T , Rubio ME , Bruses J , Topf N , et al. Derivation of midbrain dopamine neurons from human embryonic stem cells. Proc Natl Acad Sci U S A (2004) ;101: (34):12543–8. |
[39] | Kriks S , Shim JW , Piao J , Ganat YM , Wakeman DR , Xie Z , et al. Dopamine neurons derived from human ES cells efficiently engraft in animal models of Parkinson’s disease. Nature (2011) ;480: (7378):547–51. |
[40] | Lyashenko N , Winter M , Migliorini D , Biechele T , Moon RT , Hartmann C . Differential requirement for the dual functions of beta-catenin in embryonic stem cell self-renewal and germ layer formation. Nat Cell Biol (2011) ;13: (7):753–61. |
[41] | Reinhardt P , Glatza M , Hemmer K , Tsytsyura Y , Thiel CS , Hoing S , et al. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS One (2013) ;8: (3):e59252. |
[42] | Li W , Sun W , Zhang Y , Wei W , Ambasudhan R , Xia P , et al. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proceedings of the National Academy of Sciences of the United States of America (2011) ;108: (20):8299–304. |
[43] | Fusaki N , Ban H , Nishiyama A , Saeki K , Hasegawa M . Efficient induction of transgene-free human pluripotent stem cells using a vector based on Sendai virus, an RNA virus that does not integrate into the host genome. Proc Jpn Acad Ser B Phys Biol Sci (2009) ;85: (8):348–62. |
[44] | Devine MJ , Ryten M , Vodicka P , Thomson AJ , Burdon T , Houlden H , et al. Parkinson’s disease induced pluripotent stem cells with triplication of the alpha-synuclein locus. Nat Commun (2011) ;2: :440. |
[45] | Mason JN , Farmer H , Tomlinson ID , Schwartz JW , Savchenko V , DeFelice LJ , et al. Novel fluorescence-based approaches for the study of biogenic amine transporter localization, activity, and regulation. J Neurosci Methods (2005) ;143: (1):3–25. |
[46] | Schwartz JW , Blakely RD , DeFelice LJ . Binding and transport in norepinephrine transporters. Real-time, spatially resolved analysis in single cells using a fluorescent substrate. The Journal of Biological Chemistry (2003) ;278: (11):9768–77. |
[47] | Lau T , Proissl V , Ziegler J , Schloss P . Visualization of neurotransmitter uptake and release in serotonergic neurons. J Neurosci Methods (2015) ;241: :10–7. |
[48] | Chen X , Xu H , Yuan P , Fang F , Huss M , Vega VB , et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell (2008) ;133: (6):1106–17. |
[49] | Kim J , Woo AJ , Chu J , Snow JW , Fujiwara Y , Kim CG , et al. A Myc network accounts for similarities between embryonic stem and cancer cell transcription programs. Cell (2010) ;143: (2):313–24. |
[50] | Nakagawa M , Koyanagi M , Tanabe K , Takahashi K , Ichisaka T , Aoi T , et al. Generation of induced pluripotent stem cells without Myc from mouse and human fibroblasts. Nat Biotechnol (2008) ;26: (1):101–6. |
[51] | Wernig M , Meissner A , Cassady JP , Jaenisch R . c-Myc is dispensable for direct reprogramming of mouse fibroblasts. Cell Stem Cell (2008) ;2: (1):10–2. |
[52] | Lin CY , Loven J , Rahl PB , Paranal RM , Burge CB , Bradner JE , et al. Transcriptional amplification in tumor cells with elevated c-Myc. Cell (2012) ;151: (1):56–67. |
[53] | Nie Z , Hu G , Wei G , Cui K , Yamane A , Resch W , et al. c-Myc is a universal amplifier of expressed genes in lymphocytes and embryonic stem cells. Cell (2012) ;151: (1):68–79. |
[54] | Papp B , Plath K . Epigenetics of reprogramming to induced pluripotency. Cell (2013) ;152: (6):1324–43. |
[55] | Martí Y , Matthaeus F , Lau T , Schloss P . Methyl-4-phenylpyridinium (MPP+) differentially affects monoamine release and re-uptake in murine embryonic stem cell-derived dopaminergic and serotonergic neurons. Molecular and Cellular Neurosciences (2017) ;83: :37–45. |
[56] | Matthaeus F , Schloss P , Lau T . Differential Uptake Mechanisms of Fluorescent Substrates into Stem-Cell-Derived Serotonergic Neurons. ACS Chem Neurosci (2015) ;6: (12):1906–12. |
[57] | Fuxe K , Agnati LF , Marcoli M , Borroto-Escuela DO . Volume transmission in central dopamine and noradrenaline neurons and its astroglial targets. Neurochem Res (2015) ;40: (12):2600–14. |
[58] | Rice ME , Patel JC . Somatodendritic dopamine release: Recent mechanistic insights. Philos Trans R Soc Lond B Biol Sci (2015) ;370: (1672):20140185. |
[59] | Iwabuchi S , Kakazu Y , Koh JY , Harata NC . Evaluation of the effectiveness of Gaussian filtering in distinguishing punctate synaptic signals from background noise during image analysis. J Neurosci Methods (2014) ;223: :92–113. |
[60] | Vaithianathan T , Matthews G . Visualizing synaptic vesicle turnover and pool refilling driven by calcium nanodomains at presynaptic active zones of ribbon synapses. Proc Natl Acad Sci U S A (2014) ;111: (23):8655–60. |
[61] | Santel A . Get the balance right: Mitofusins roles in health and disease. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research (2006) ;1763: (5):490–9. |
[62] | Pham AH , Meng S , Chu QN , Chan DC . Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Human Molecular Genetics (2012) ;21: (22):4817–26. |
[63] | Gubernator NG , Zhang H , Staal RG , Mosharov EV , Pereira DB , Yue M , et al. Fluorescent false neurotransmitters visualize dopamine release from individual presynaptic terminals. Science (2009) ;324: (5933):1441–4. |

