
Study 329 continuation phase: Safety and efficacy of paroxetine and imipramine in extended treatment of adolescent major depression
Abstract
OBJECTIVE: This is an analysis of the unpublished continuation phase of Study 329, the primary objective of which was to compare the efficacy and safety of paroxetine and imipramine with placebo in the treatment of adolescents with unipolar major depression. The objectives of the continuation phase were to assess safety and relapse rates in the longer term. The objective of this publication, under the Restoring Invisible and Abandoned Trials (RIAT) initiative, was to see whether access to and analysis of the previously unpublished dataset from the continuation phase of this randomized controlled trial would have clinically relevant implications for evidence-based medicine.
METHODS: The study was an eight-week double-blind randomized placebo-controlled trial with a six month continuation phase. The setting was 12 North American academic psychiatry centres, from 20 April 1994 to 15 February 1998. 275 adolescents with major depression were originally enrolled in Study 329, with 190 completing the eight-week acute phase. Of these, 119 patients (43%) entered the six-month continuation phase (paroxetine n = 49; imipramine n = 39; placebo n = 31), in which participants were continued on their current treatment, blinded. As per the protocol, we have looked at rates of relapse (based on Hamilton Depression Scale scores) across both acute and continuation phases, and generated a safety profile for paroxetine and imipramine compared with placebo for up to six months.
ANOVA testing (generalized linear model) using a model including effects of site, treatment and site x treatment interaction was applied. Otherwise we used only descriptive statistics.
RESULTS: Of patients entering the continuation phase, 15 of 49 for paroxetine (31%), 12 of 39 for imipramine (31%) and 12 of 31 for placebo (39%) completed as responders. Across the study, 25 patients on paroxetine relapsed (41% of those showing an initial response), 15 on imipramine (26%), and 10 on placebo (21%). In the continuation and taper phases combined there were 211 adverse events in the paroxetine group, 147 on imipramine and 100 on placebo. The taper phase had a higher proportion of severe adverse events per week of exposure than the acute phase, with the continuation phase having the fewest events.
CONCLUSIONS: The continuation phase did not offer support for longer-term efficacy of either paroxetine or imipramine. Relapse and adverse events on both active drugs open up the risks of a prescribing cascade. The previously largely unrecognised hazards of the taper phase have implications for prescribing practice and need further exploration.
1Background
In 2013, in response to concerns about selective reporting of outcomes of randomized controlled trials, an international group of researchers called on funders and investigators of abandoned (unpublished) or misreported trials to publish undisclosed outcomes or correct misleading publications [1]. This initiative was dubbed ‘restoring invisible and abandoned trials’ (RIAT). The researchers identified many trials requiring restoration, and emailed the appropriate parties, asking them to signal their intention to publish the unpublished trials or publish corrected versions of misreported trials.
The RIAT researchers approached GlaxoSmithKline (GSK) (formerly SmithKline Beecham: SKB) and asked whether it intended to restore Study 329, a double-blinded randomized controlled trial comparing paroxetine and imipramine with placebo in the treatment of adolescent major depression. GSK did not signal any such intent.
The acute phase of Study 329 was originally reported in the Journal of the American Academy of Child and Adolescent Psychiatry in 2001 [2]. It was reanalysed and published under the RIAT initiative in 2015 in BMJ (Le Noury et al.) [3].
The acute phase of Study 329 was followed by a six-month continuation phase. This article represents a RIAT publication of the continuation phase.
The 1993/1994/1996 trial protocol [4] stated that the objectives for the continuation phase were:
• To provide information on the safety profile of paroxetine and imipramine when these agents are given to adolescents for an extended period of time;
• To estimate the rate of relapse among paroxetine, imipramine, and placebo responders who were maintained on treatment (p. 000547).
The clinical study report (CSR) [5] stated that the continuation phase ‘was not designed to determine whether paroxetine or imipramine are superior to placebo in preventing relapse’, but instead ‘to provide information on the relapse rates of responders over an extended period’ (p. 000023).
Study enrolment took place between April 1994 and March 1997. The final date on which the last patient took study medication during the continuation phase was 3 September 1997. In a small number of patients, 30-day follow-up data were collected into February 1998.
2Methods
We have reanalysed Study 329 according to the RIAT recommendations. To this end, we have used the CSR [5], including Appendices A-G (publically available on the GSK website [http://www.gsk.com/en-gb/media/resource-centre/paroxetine/paroxetine-paediatric-and-adolescent-patients/]), other publically available documents, [6] and the individual participant level data accessed through SAS Solutions OnDemand website, [7] on which GSK subsequently also posted some Study 329 documents (available only to users approved by GSK). Following negotiation, [8] GSK posted approximately 77,000 pages of de-identified individual case report forms (CRFs, Appendix H) on that website. A table of sources of data consulted in preparing each part of this paper is available as RIAT Appendix 1–RIAT Audit Record (RIATAR).
Except where indicated, in accordance with RIAT recommendations, our methods are those set out in the protocol, [4] as outlined in RIAT Appendix 1.
2.1Participants
The acute phase participants comprised 275 adolescents between the ages of 12 and 18 years, meeting DSM-IV criteria [9] for a current episode of major depression of at least eight weeks’ duration(see Le Noury et al. [3] for details of eligibility criteria, standardization across sites, randomization, blinding, recruitment, screening, consent, demographic and baseline characteristics). In the continuation phase, patients who had responded to treatment were eligible to continue on the same medication at the same dosage for an additional six months.
Some participants were not able to progress to the continuation phase because of a shortage of study medication supplies, resulting from a slower-than-expected rate of enrollment, which led to some of the medication expiring before use (CSR, p. 000027), Amendment 2 of the Study 329 protocol (approved 28 October 1996) provided two options for these patients: treatment by a third party, who was provided with the identity of the study medication, or open-label paroxetine treatment for up to six months (after a one-week down-titration and washout period) by the study physician (p. 000538).
2.2Interventions
Study medication was provided to patients in weekly blister packs. Patients were instructed to take the medication twice daily. There were six dosing levels. Over the first four weeks of the acute phase, all patients were titrated to level 4, corresponding to paroxetine 20 mg or imipramine 200 mg, regardless of response. Non-responders (those failing to reach responder criteria) could be titrated over the following four weeks up to level 5 or 6, corresponding to a maximum dose of 60 mg paroxetine 60 mg and 300 mg imipramine.
Medication compliance was evaluated based on the number of capsules dispensed, taken, and empty blister packs returned. Non-compliance was defined as taking less than 80% or more than 120% of the number of capsules expected to be returned at two consecutive visits, and resulted in withdrawal from the study. Any patient missing two consecutive visits was also withdrawn.
Patients were provided with 45-minute weekly sessions of supportive psychotherapy, [10] primarily for the purpose of assessing the treatment effects.
2.3Taper phase
A discontinuation taper phase was recommended for all patients, whether terminating the study early (during both the acute and continuation phase), or completing the acute phase but not continuing, or completing the six-month continuation phase. If the patient accepted a taper phase, the protocol recommended tapering medication/placebo in a linear fashion over a seven to 17 day period, with patient, family, and clinical and research personnel all remaining blind to medication assignment.
Not all patients agreed to a taper phase. For those who did taper their medication, it was difficult in some cases to be certain of the exact duration of exposure, because the date of last dose was left incomplete. We have used the exact number of days where available, and for other patients we have assumed an average taper phase of 2 weeks, unless there were indications to the contrary. The taper phase includes patients tapering during the acute phase as well as the continuation phase, so there are more patients in the taper phase than in the continuation phase.
2.4Outcomes
Patients were evaluated every four weeks from week 12 to week 32 during the six months of the continuation phase.
2.4.1Efficacy Endpoints
a. Percentage of Patients Who Relapsed
The protocol defined patients as relapsed if they no longer met the criteria for response. The protocol definition of response was having a Hamilton Depression Scale (HAM-D) score ≤8, or a 50% or greater reduction in HAM-D score relative to the baseline score (such patients were defined as ‘responders’ in the CSR). The CSR added a second indicator of response, ‘remission’, defined as having a HAM-D score ≤8, ‘in order to provide a rigorous anchor point in analyzing relapses in the continuation phase’ (p. 000050). We have accepted this departure from the protocol and applied this more conservative remission criterion to our analyses of relapses. This reduces the number of relapses.
In addition we have regarded as relapses any patients who, having previously responded, were discontinued from the study because of a suicide-related event. A number of these patients were hospitalized or discontinued from the study immediately at that point without a further HAM-D being completed. We have regarded such cases as relapses even though the most recently undertaken HAM-D score (prior to the event) might have been < 8. This seems to us a necessary modification of the protocol as such an outcome was in all likelihood not anticipated when the protocol was developed or at any point prior to the analysis of the data. The data are available for other researchers to analyse using other approaches.
b. Percentage of patients withdrawing because of lack of efficacy
The protocol called for the percentage of patients withdrawing because of lack of efficacy to be evaluated at the end of the continuation phase. We have included in this category those patients whose final HAM-D scores were consistent with a lack of efficacy, even if the stated reason for withdrawal was non-compliance or protocol violation or adverse events other than suicide-related events.
2.4.2Safety endpoints
An adverse experience/event was defined in the protocol (p. 000564) as:
‘any noxious, pathologic or unintended change in anatomical, physiologic or metabolic functions as indicated by physical signs, symptoms and/or laboratory changes occurring in any phase of the clinical trial whether associated with drug or placebo and whether or not considered drug related.
This includes an exacerbation of pre-existing conditions or events, intercurrent illnesses, drug interaction or the significant worsening of the disease under investigation that is not recorded elsewhere in the case report form under specific efficacy assessments.’
Adverse events were to be elicited by the investigator asking a non-leading question such as: ‘Do you feel different in any way since starting the new treatment/the last assessment?’ (p. 000565). Details of treatment-emergent adverse events, including their severity, any change in study drug administration, investigator attribution to study drug, any corrective therapy given, and outcome status were documented. Attribution or relationship to study drug was judged by the investigator to be ‘unrelated’, ‘probably unrelated’, ‘possibly related’ or ‘related’ (p. 000565).
2.5Sources of safety data
Adverse event data come from the CSR of the continuation phase lodged on GSK’s website, primarily Appendix D: Patient Data Listings of Adverse Experiences. Appendix B provides details of concomitant medications. Additional information was available from the summary narratives in the body of the CSR for patients who had adverse events that were designated as serious or led to withdrawal. However, a number of other patients discontinued because of adverse events that were not regarded as serious, or discontinued because of lack of efficacy or protocol violations, did not generate patient narratives.
The tables in Appendix D of the CSR report the verbatim terms used by the blinded investigators along with preferred terms as coded by SKB using the Adverse Drug Events Coding System (ADECS) dictionary. Appendix D also includes ratings of severity and ratings of relatedness. We used the MedicalDictionary for Regulatory Activities (MedDRA®) to code the verbatim terms provided in CSR Appendix D. MedDRA terminology is the international medical terminology developed under the auspices of the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) www.meddra.org). It has been endorsed by the United States Food and Drug Administration (FDA) and is now used by GSK [11].
Our analysis of the acute phase [3] had established that there are significant adverse event data missing from CSR Appendix D, so all CRFs for all patients entering the continuation phase were scrutinised for all adverse events occurring during the continuation phase. These adverse events were compared with those reported in CSR Appendix D. This review process identified additional adverse events that had not been recorded as verbatim terms in CSR Appendix D. It also led to recoding of a number of the reasons for discontinuation. The new adverse events and the reasons for changing discontinuation category are recorded in RIAT Appendix 2 accompanying this paper.
2.6Coding of adverse events
The protocol (p. 000574) stated that adverse events were to be coded by body system and preferred terms, and compared using descriptive statistics, but did not specify a coding dictionary. The CSR (written after the study concluded) specified that adverse events were coded using the Adverse Drug Experience Coding System (ADECS), which SKB used at the time (p. 000044). ADECS was derived from a coding system developed by the FDA, Coding Symbols for a Thesaurus of Adverse Reaction Terms (COSTART), but ADECS is not itself a recognized system and is no longer available.
We coded adverse events using MedDRA, which has replaced COSTART for the FDA, because it is by far the most commonly used coding system today. For coding purposes, we have taken the original terms used by the clinical investigators as transcribed into the CSR Appendix D, and applied MedDRA codes to these descriptions. Information from Appendix D was transcribed into spreadsheets (available at Restoring Study329.org). The verbatim terms and the ADECS coding terms were transcribed first into these sheets, allowing all coding to be done before the drug names were added in. The transcription was carried out by a research assistant who was a MedDRA trained coder, but took no part in the actual coding. All coding was carried out by JLN, and checked by DH, or vice versa.
All of our coding from the verbatim terms in the CSR Appendix D was done blind, as was coding from the CRFs.
In general, MedDRA coding stays closer to the original clinician description of the event than ADECS. Most coding was straightforward. The vast majority of the verbatim terms simply mapped onto coding terms in MedDRA. The main coding challenges arose in relation to suicide-related events in the acute phase; these are covered in Le Noury et al. [3].
2.7Analysis of safety data
As in our acute study re-analysis, in analysing the safety data, we present all adverse events rather than only those happening at a particular rate. Secondly, we have grouped events into broader system-organ-class (SOC) groups–psychiatric, cardiovascular, gastrointestinal, respiratory and other. Thirdly, we break down events by severity, selecting adverse events coded as severe, and utilising the listing in the CSR (Appendix G) of patients who discontinued for any reason.
In our acute study re-analysis [3], we laid out our categorization of suicidal events in detail. These were events whose coding in our opinion should have included a suicide-related code. There were also a considerable number of events coded under headings such as nightmares or abnormal thoughts. At the time this study was conceived and executed, few investigators or patients were familiar with the phenomenon of treatment-induced behavioral change, up to and including suicidality, and no rating instruments were included to ensure that such events were recorded systematically. Investigators and patients were apparently not briefed on this possibility. It is possible that a number of treatment induced behavioral disturbances would have been communicated obliquely. We have therefore taken events that might conceal a suicide-related event and presented these together in Table 12a, along with more clearly suicide-related events. The details of all patients included in this way are laid out in Table 12b.
As the acute and continuation phases are of very different duration, and a significant number of patients dropped out in the course of the continuation phase, a simple listing of the adverse events from each phase risks misleading. We have therefore presented the total number of events but also estimated the rate at which events occurred by duration of exposure.
2.8Data access
We have made available on Study329.org all the data we have used, with the exception of the actual CRFs, which we do not have permission to share.
2.9Statistical methods
No formal hypothesis testing was planned for the continuation phase. We applied ANOVA testing (generalized linear model) using a model including effects of site, treatment, and site×treatment interaction as per the Study 329 protocol. Otherwise we used only descriptive statistics.
3Results
Attrition due to non-response, dropout and relapse is shown in Table 1. Demographic data are laid out in Table 2. Table 3 has response and relapse data. Tables 4–13 have safety data.
Table 1 shows that only 43 of 275 patients completed the continuation phase.
Table 2 shows the demographic characteristics of those who entered the continuation phase.
3.1Efficacy
The overall profiles of HAM-D scores for observed cases in both acute and continuation phases for all three arms of the study are presented in Fig. 1.
Although efficacy could be assessed for the acute phase, the dropout rates between the acute and continuation phases and within the continuation phase were too high to allow a standard efficacy analysis. In the continuation phase, the dropout rates were 30/49 [61%] for paroxetine; 27/39 [69%] for imipramine; and 18/31 [58%] for placebo.
Table 3 shows response (remission) and relapse rates, which ranged from 21% for placebo to 41% for paroxetine. We have included in Table 3 a category of potential completers, to take into account 13 patients who dropped out of the study although their HAM-D scores were well within the responder range (HAM-D of 2 or 3). Some of them were discontinued because further blinded treatment was unavailable; for others, no clear explanation for discontinuation was given.
3.2Discontinuations
During the continuation phase, 75 patients discontinued (30 from paroxetine, 27 from imipramine and 18 from placebo). The reasons for discontinuation are given in Table 4. Table 4 in RIAT Appendix 2 gives a breakdown of when these dropouts occurred.
Following a review of the codes given for reasons for withdrawal from the study that were found in the CSR (Appendix G), along with a review of patient narratives and CRFs where applicable, we proposed changes to these reasons for withdrawal in a proportion of those discontinued. These proposed changes can be found in Table 5 in RIAT Appendix 2.
3.3Safety
Table 5 gives the number of adverse events reported by SKB in their continuation phase CSR. The report only provides data on events happening at a 5% or greater rate.
This can be contrasted with the data in Table 6, which presents all adverse events found in both CSR Appendix D and the CRFs, summarised by System Organ Class (SOC). In MedDRA, some adverse events always fall within a particular SOC; others require that the coder choose between SOCs. A full summary and full breakdown of adverse events can be found in Tables 1 and 2 in RIAT Appendix 2.
3.4Severity ratings
Designating adverse events as serious hinged on the judgement of the clinical investigator. We are therefore not able to make comparable judgements of seriousness, but there are two other methods to approach the issue of severity of adverse events. One is to look at those rated as severe rather than moderate or mild at the time of the event (see Table 7).
A full breakdown of the severe adverse events within each SOC can be found in Table 3 of RIAT Appendix 2. A second method is to look at those that led to dropouts (Table 4).
3.5Adverse events by exposure
As there were a large number of discontinuations in the continuation phase, a simple listing of adverse events may tell a different story from an analysis of these events in proportion to the duration of exposure.
Table 8 shows the weeks of exposure for each group in each phase. Tables 9 and 10 show the numbers of adverse events and severe adverse events in each phase, with the rates per 100 weeks exposure displayed in Figs. 2 and 3.
We have also looked at all behavioral adverse events in each phase (Table 11 and Fig. 4). These events include: agitation, aggravated depression, akathisia, abnormal dreams, depersonalisation,disinhibition, feelings of despair, hallucinations, impulsive behavior, negative thoughts, neurosis, paranoia, psychosis, suicide attempt, suicidal ideation, suicidal gesture, self-harm, and self-injury.
Table 12 gives the numbers of suicidal adverse events in each phase, with the rates per 100 weeks exposure displayed in Fig. 5. A full listing of all patients who experienced potentially suicidal events is presented in Table 13.
3.6Adverse event profile of patients entering the continuation phase
Patients who completed acute and continuation phases may belong to different cohorts. Accordingly we analysed the acute-phase adverse event profiles of patients who entered the continuation phase, compared with those who did not, shown in Table 13. A full breakdown of all these adverse events can be found in Table 12 in RIAT Appendix 2.
4Discussion
The original Study 329 investigators are to be commended for undertaking a study that included a continuation phase for the purposes of providing longitudinal data on the treatment of adolescents with major depression. As one of the few bodies of data offering information on longer term treatment of adolescents with mood disorders, the study data are of value.
We analysed and reported the continuation phase according to the original Study 329 protocol (with approved amendments). RIAT Appendix 1 shows the sources of information used in preparing this paper, which should aid other researchers who wish to access the data, either to check our analysis or to interrogate it in other ways. We draw minimal conclusions regarding efficacy and harms, inviting others to offer their own analysis.
The number of patients relapsing was designated a secondary outcome in Study 329. The results, however, remain unpublished. In our analysis, although we used more stringent criteria (i.e. remission) for response, we found higher rates of response in all three treatment groups than were reported in Keller et al. or in the text of the CSRs prepared by SKB, because we followed the classification in the CSR Appendix D, where response is based on HAM-D scores only, regardless of whether the patient or investigator violated the protocol.
Our analysis revealed higher relapse numbers in the active treatment groups than in the placebo group. This is in part determined by our decision to include in the relapse category patients who had a significant adverse event in the behavioral domain, but the higher numbers hold whether or not these patients are included.
Relapse was not a primary endpoint of the trial, and cannot be analysed in a way that would allow a definitive statement about rates of relapse compared to placebo. Furthermore it can be difficult to distinguish between apparent relapse and an adverse drug reaction, requiring caution in the case of patients who fail to respond to active treatment. Some of the patients in this study appear to have become paranoid or manic, or to have had a depressive relapse, all of which might lead to further diagnoses and/or prescriptions (a prescribing cascade) when in fact the wisest course of action might be to withdraw treatment.
The data on adverse events controlled for duration of exposure points to the taper phase as the riskiest period of treatment. It was difficult to be confident of the exact duration of exposure in the taper phase in some patients, but our estimates of duration are not likely to have inflated adverse event figures.
The CSR argued that simply looking at relapses is not a good way to establish long-term comparative efficacy (p. 000023). It proposed a randomized discontinuation design as the best way forward(p. 000023). However, the data from this study point to a discontinuation syndrome associated with paroxetine use. If this is the case, a randomized discontinuation design would not work, and we would be left with a more naturalistic option like the present study.
With regard to adverse events, the continuation phase of the study stands out as a phase where fewer adverse events either happened or were recorded. This to some extent is not surprising. It might be expected that the acute phase would weed out those patients not suited to the treatment they were on. But simple explanations like this may not fully account for the data, in that the patients entering the continuation phase appeared to have as many adverse events during the acute phase as those patients who did not opt to continue with treatment.
There are no other studies in this age group that we are aware of with which this study can be compared.
In our reporting of the acute phase of Study 329, we suggested that researchers and clinicians should recognise the potential biases in published research, including the potential barriers to accurate reporting of harms to which this study pointed. We also urged regulatory authorities to mandate access to trial data. This analysis of the continuation phase of Study 329 adds further weight to this recommendation.
It also adds weight to our invitation to others to access the data we have used. We are very clear that the analyses offered here are not the only ones possible. Our understanding of this dataset can only be enhanced by input from others who may make differing calls regarding coding and/or apply different analytic tools to the data.
Trial Registration
Registration number and name of trial register: SmithKline Beecham study 29060/329.
Trial Protocol
SmithKline Beecham study 29060/329, Final Clinical Report (Acute Phase) [5], Appendix A, Protocol (from p. 000531) [4].
Trial Funding
SmithKline Beecham study.
Ethical approval
“The protocol and statement of informed consent were approved by an Institutional Review Board (IRB) prior to each center’s initiation, in compliance with 21 United States Code of Federal Regulations (CFR) Part 56. Written informed consent was obtained from each patient prior to entry into the study, in compliance with 21 CFR Part 50. Case report forms were provided for each patient’s data to be recorded.” (Final Clinical Report page 000030). The sample informed consent is provided in Appendix C of the protocol (pp. 000590-000594). No further information is available regarding the particular IRB that approved the study.
Funding for RIAT reanalysis
No funding received.
Data analysis protocol for RIAT reanalysis
Submitted to GSK on 28 October 2013. Approved by GSK on 4 December 2013.
Authorship
All authors meet ICMJE authorship criteria. Conception/design of the work: Healy, Jureidini, Nardo. Acquisition of data: Jureidini (negotiation with GSK); Tufanaru and Abi-Jaoude (RIATAR); Nardo (efficacy data using GSK online remote system); Le Noury (harms data using GSK online remote system). Data analysis: Nardo (efficacy); Le Noury and Healy (harms). Data interpretation: all authors. Drafting the work and revising it critically for important intellectual content, final approval of the version to be published: all authors. Agreement to be accountable for all aspects of the work: all authors (guarantor Jureidini).
12Appendices
RIAT Appendix 1 – RIAT audit record (RIATAR)
RIAT Appendix 2 – Adverse event tables
Appendices
Detailed data tables and other relevant documents are available at http://study329.org.
References
[1] | Doshi P , Dickersin K , Healy D , Vedula SS , Jefferson T . Restoring invisible and abandoned trials: A call for people to publish the findings. BMJ. (2013) ;346: :f2865–f2865. |
[2] | Keller MB , Ryan ND , Strober M , et al. Efficacy of paroxetine in the treatment of adolescent major depression: A randomized, controlled trial. J Am Acad Child Adolesc Psychiatry. (2001) ;40: (7):762–72. |
[3] | Le Noury JC , Nardo JM , Healy D , et al. Restoring Study 329: Efficacy and harms of paroxetine and imipramine in treatment of major depression in adolescence. BMJ. (2015) ;351: :h4320. |
[4] | SmithKline Beecham, A multi-center, double-blind, placebo controlled study of paroxetine and imipramine in adolescents with unipolar major depression. Protocol number 29060/329. 1993, amended 1994, 1996. [Internet]. [Accessed 29 February 2016]. Available from: http://www.gsk.com/media/360485/329-AppA.PDF. |
[5] | SmithKline Beecham. A multi-center, double-blind, placebo controlled study of paroxetine and imipramine in adolescents with unipolar major depression-acute phase, Final clinical report. [Internet]. [Accessed 29 February 2016]. Available from: http://www.gsk.com/media/389566/depression_329_full.pdf, via http://www.gsk.com/en-gb/media/resource-centre/paroxetine/paroxetine-paediatric-and-adolescent-patients/ |
[6] | Healthy Skepticism International News, Paxil Study 329: Paroxetine vs Imipramine vs Placebo in Adolescents, [Internet]. 2010 [Accessed 29 February 2016]. Available from: http://www.healthyskepticism.org/global/news/int/hsin2010-01. |
[7] | SAS Solutions OnDemand. [Internet]. [Accessed 29 February 2016]. Available from: https://www.ondemand.sas.com/sam/?sid=1393012805&rid=DqqHX0rCqAWZ7TJlCIPijRTscQ. |
[8] | Correspondence between Jureidini and GSK, Rapid Responses to Putting GlaxoSmithKline to the test over paroxetine. BMJ. 2013;347:f6754[Internet]. [Accessed 29 February 2016]. Available from: http://www.bmj.com/content/347/bmj.f6754/rapid-responses |
[9] | American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 3rd Edition-Revised (DSMIII-R). Washington, DC: American Psychiatric Association; (1987) . |
[10] | Fawcett J , Epstein P , Fiester SJ , et al. Clinical management–imipramine/placebo administration manual. NIMH Treatment of Depression Collaborative Research Program. Psychopharmacol Bull. (1987) ;23: (2):309–24. |
[11] | Winter C. MedDRA in clinical trials-industry perspective SFDA-ICH MedDRAWorkshop, Beijing, 13-14 May 2011. https://www.meddra.org/sites/default/files/page/documents_insert/christina_winter_2_meddra_in_clinical_trials_industry_perspective.pdf. |
Figures and Tables
Fig.1
Least Squares Mean ± SEM for the HAM-D scores of the observed cases. The upper eight-week values are from the remaining acute phase cohort while the lower values are from those selected to enter the continuation phase. ANOVA testing (generalised linear model) using a model including effects of site, treatment, and site×treatment interaction, as per the protocol, revealed no statistical significance at any week in either phase.
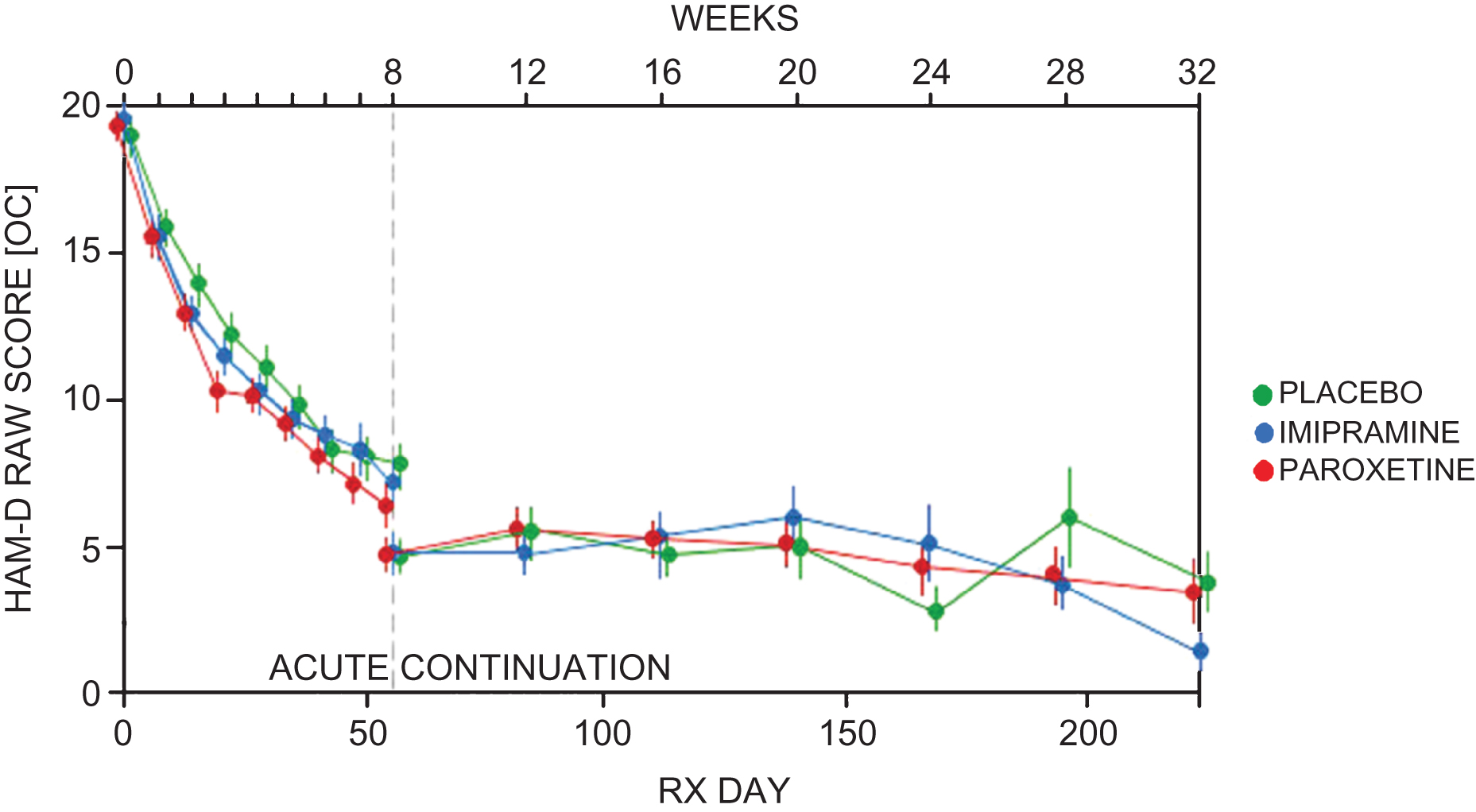
Fig.2
Total adverse events per 100 weeks exposure.
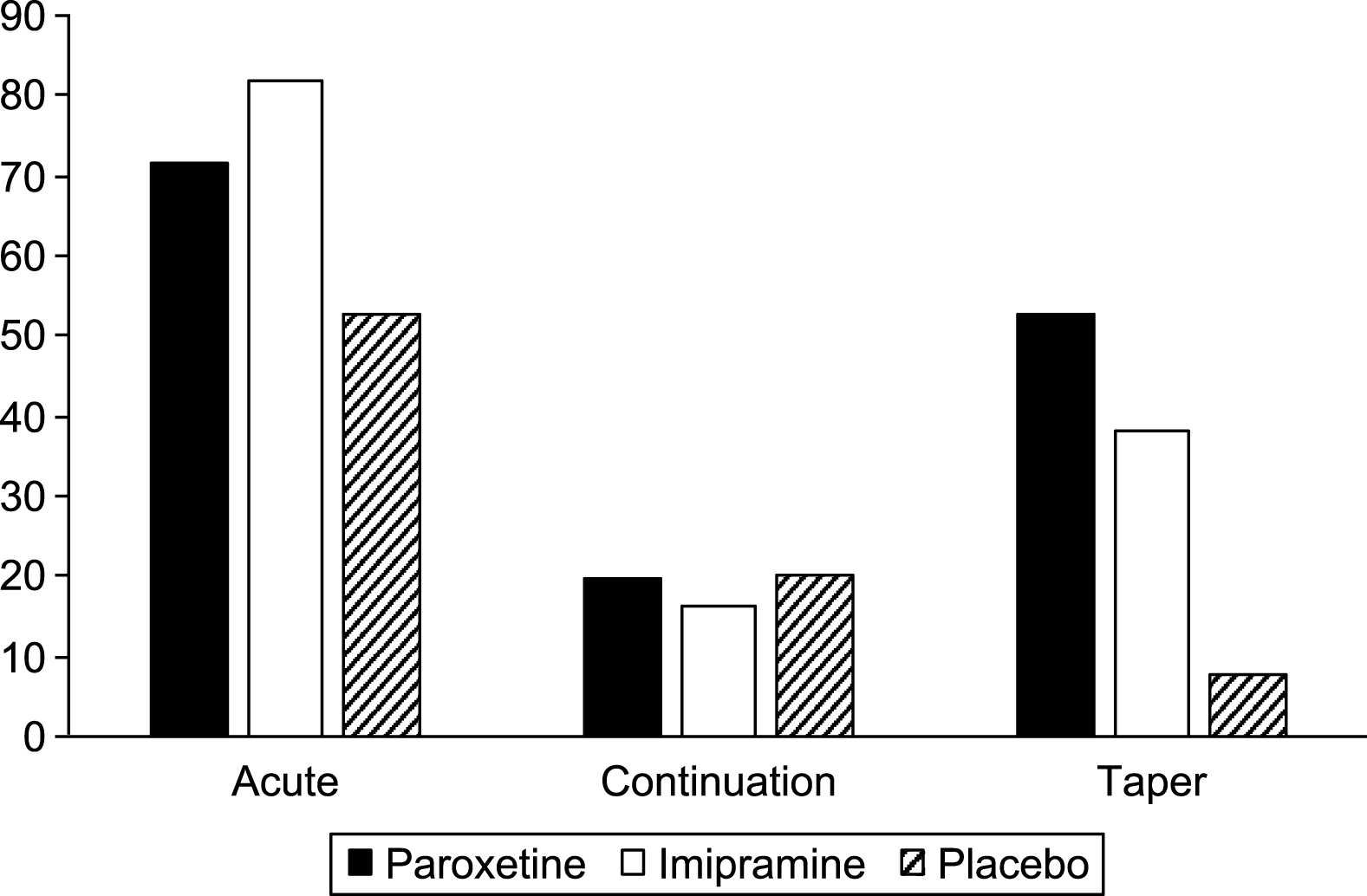
Fig.3
Severe adverse events per 100 weeks exposure.
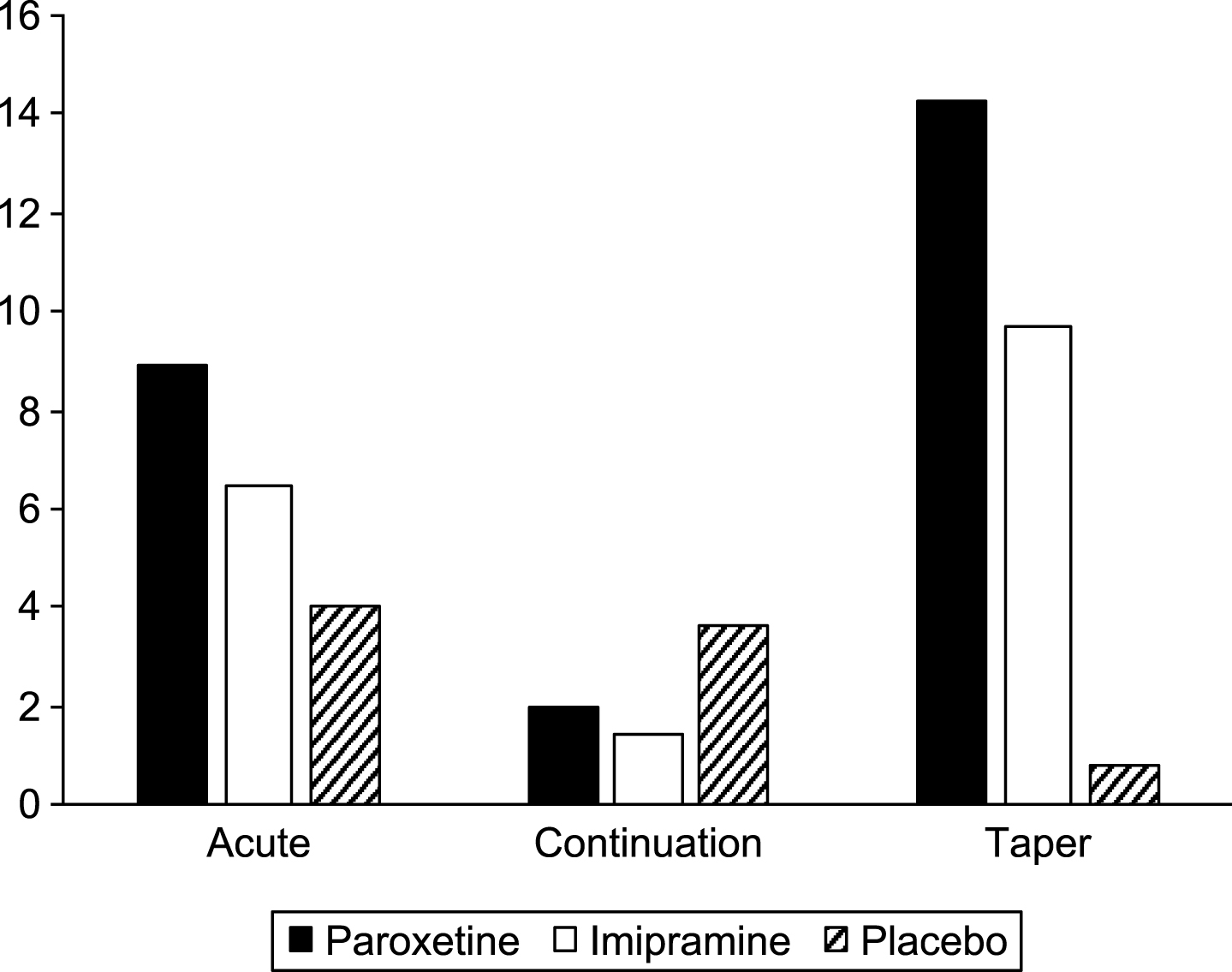
Fig.4
Behavioral events per 100 weeks exposure.
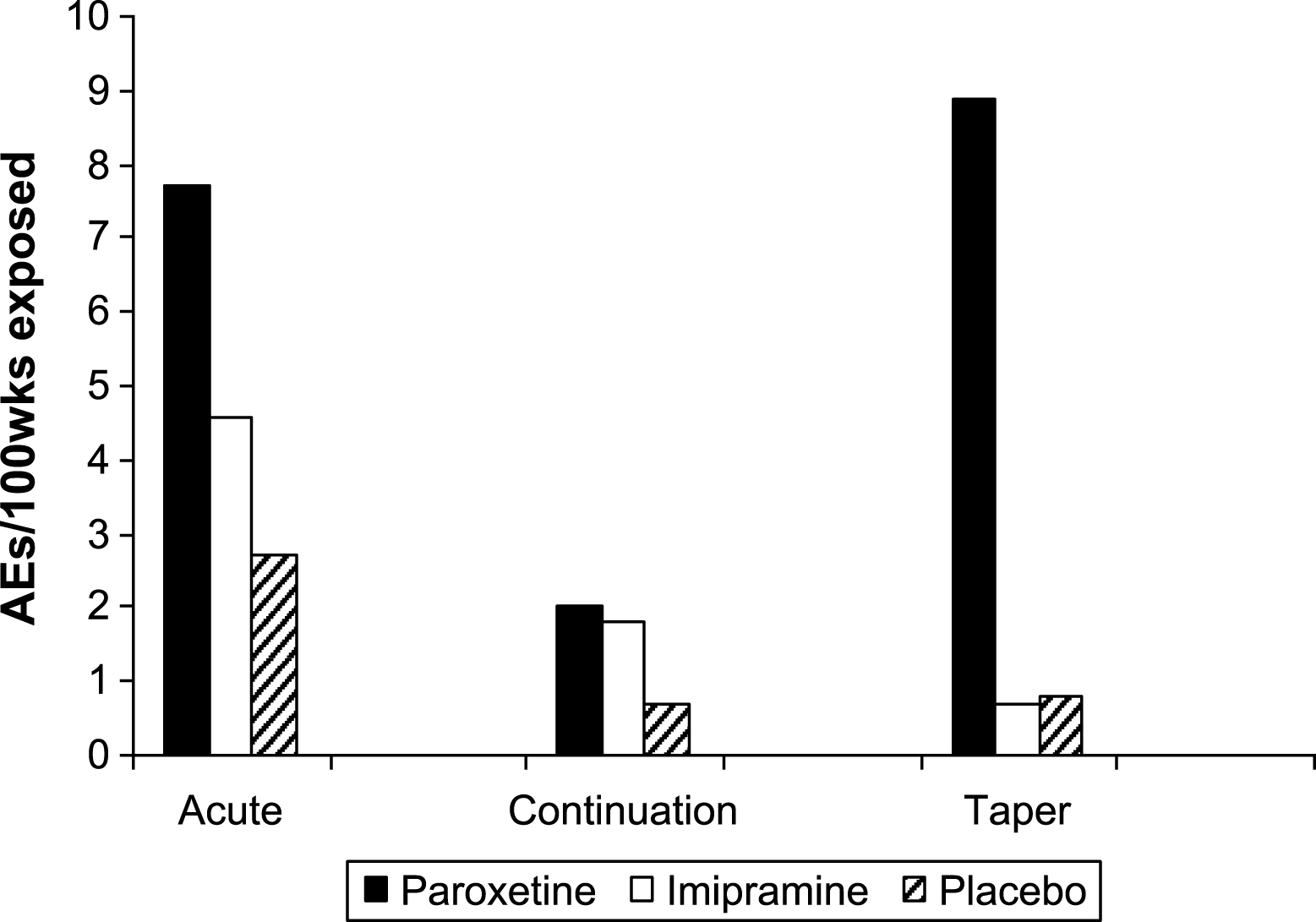
Fig.5
Suicidal adverse events per 100 weeks exposure.
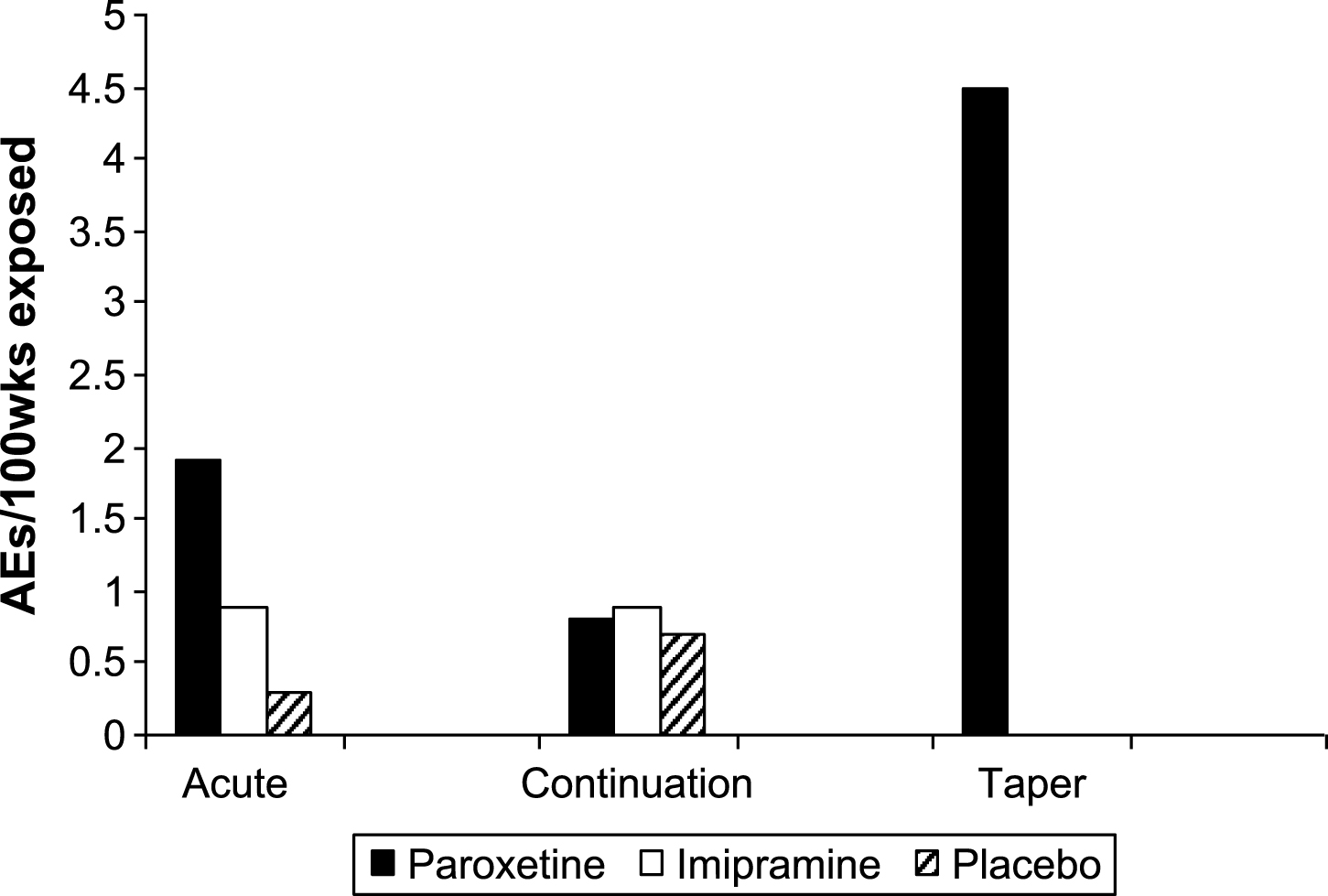
Table 1
Allocations and discontinuations in acute and continuation phasesi
| Paroxetine N = 93 | Imipramine N = 95 | Placebo N = 87 | ||||
| SKB | RIAT | SKB | RIAT | SKB | RIAT | |
| Acute phase dropout | 26 | 26 | 38 | 38 | 21 | 21 |
| Acute phase completion | 67 | 67 | 57 | 57 | 66 | 66 |
| Pre-continuation phase dropouts | 15 | 18* | 17 | 18** | 33 | 35# |
| Continuation phase entry | 52 | 49 | 40 | 39 | 33 | 31 |
| Continuation phase dropouts | 34 | 31∧ | 27 | 27 | 20 | 18 |
| Continuation phase completion | 18 | 18 | 13 | 12 | 13 | 13 |
*Differs from Fig. 1 of Le Noury et al. because during the CRF checks, two additional patients (329.003.00292 and 329.011.00283) were found to have completed the acute phase but never returned for the week 12. They were reclassified by us as ‘post acute discontinuations’. ∧Patient 329.0002.00058 discontinued during the taper phase of continuation for AE suicidal overdose. **Patient 329.008.00161 was originally thought to have entered the continuation phase. However, the CRF check showed the patient did not return for the week 12 visit, and the notes state that the week 8 medications were never dispensed. #Again during the CRF check, two placebo patients (329.007.00144 and 329.011.00210) were found to have completed the acute phase but never returned after their week 8 visit. In addition, for patient 329.005.00334 there was a note that no study meds were available, so the patient could not continue into the continuation phase. ∼ Patient 329.003.00076 was originally classed as ‘completed’. However, review of the CRF revealed that the patient stopped study meds approximately one week prior to the week 32 visit but still attended to complete study paperwork. No down-titration or follow up. This patient was moved to Discontinuations for protocol violation.
Table 2
Continuation phase demographics
| Paroxetine N = 49 | Imipramine N = 39 | Placebo N = 31 | |
| Mean age (+/–sd) | 14.8 (+/–1.7) | 14.8 (+/–1.8) | 15.0 (+/–1.6) |
| % female | 65% | 46% | 68% |
| Caucasian | 41 | 36 | 25 |
| Black | 3 | 0 | 1 |
| Hispanic | 2 | 2 | 2 |
| Other | 3 | 1 | 3 |
Table 3
Rates of response, relapse and non-response (*)
| Paroxetine N = 93 | Imipramine N = 95 | Placebo N = 87 | |
| Response at some point | 61 | 57 | 47 |
| Completed responders (+ potential completed responders) | 15 (+3) | 12 (+1) | 12 (+9) |
| Lack of efficacy: acute phase | 29 | 36 | 38 |
| Lack of efficacy: continuation phase | 3 | 2 | 2 |
| Acute phase dropouts | 9 | 16 | 14 |
| Continuation phase dropouts | 12 | 14 | 11 |
| Acute phase relapse | 6 | 5 | 3 |
| Continuation phase relapse | 19 | 10 | 7 |
| Total relapses | 25 (41%) | 15 (26%) | 10 (21%) |
*The term response here uses the more conservative remission criterion defined in the protocol (HAM-D < 8).
Table 4
Reasons for discontinuation during continuation phase
| Reason for withdrawal | Paroxetine N = 31* | Imipramine N = 27 | Placebo N = 18 | |||
| SKB | RIAT | SKB | RIAT | SKB | RIAT | |
| Adverse events | 2 | 5* | 8 | 9 | 4 | 4 |
| Lack of efficacy | 7 | 1 | 6 | 1 | 6 | 2 |
| Relapse | 0 | 4 | 0 | 3 | 0 | 4 |
| Withdrawn consent | 7 | 9 | 5 | 5 | 2 | 2 |
| Protocol violation–non-compliance | 11 | 9 | 6 | 6 | 4 | 3 |
| Protocol violation–by investigator | 0 | 1 | 0 | 2 | 0 | 0 |
| Lost to follow-up | 2 | 2 | 1 | 1 | 2 | 3 |
| Other (“Feeling well”) | 1 | 0 | 0 | 0 | 0 | 0 |
*Includes case 58 who discontinued in taper phase of continuation phase.
Table 5
Adverse events reported in SKB’s clinical study report (CSR)
| Body system | Paroxetine N = 52 | Imipramine N = 40 | Placebo N = 33 |
| Body as a whole | 29 | 14 | 33 |
| Cardiovascular | 1 | 2 | 0 |
| Digestive | 14 | 14 | 25 |
| Psychiatric | 0 | 0 | 0 |
| Nervous | 20 | 7 | 6 |
| Respiratory | 16 | 4 | 9 |
| All other body systems | 6 | 4 | 6 |
| Total | 86 | 45 | 79 |
Table 6
RIAT Summary of all adverse events during continuation phase
| Paroxetine N = 49 | Imipramine N = 39 | Placebo N = 31 | ||||
| System Organ Class (MedDRA) | Reported in | Further AEs | Reported in | Further AEs | Reported in | Further AEs |
| CSR (App. D) | found in | CSR (App. D) | found in | CSR (App. D) | found in | |
| only | CRF | only | CRF | only | CRF | |
| Cardiovascular disorders | 18 | – | 6 | 1 | 6 | – |
| Gastrointestinal/digestive disorders | 19 | 3 | 19 (+2)* | 3 | 9 | 2 |
| Psychiatric disorders | 15 | 9 | 9 (+1)* | 11 | 6 | 2 |
| Nervous system disorders | 35 | 3 | 11 | 3 | 23 | 1 |
| Respiratory disorders | 20 | – | 6 | – | 11 | 1 |
| All other SOCs | 29 | 1 | 18 (+1)* | 1 | 26 (+1)* | 2 |
| Total AEs | 136 | 16 | 69 (+4)* | 19 | 81 (+1)* | 8 |
| Grand total AEs | 152 | 92 | 90 | |||
*Additional adverse events (AEs) found in patient narratives but missing from CSR Appendix D.
Table 7
Summary of all adverse events rated as severe during continuation phase
| Paroxetine N = 49 | Imipramine N = 39 | Placebo N = 31 | |
| System Organ Class (MedDRA) | CSR only (Appendix D) | CSR only (Appendix D) | CSR only (Appendix D) |
| Gastrointestinal disorders | 1 | 1 | 1 |
| Psychiatric disorders | 4 | 3 | 2 |
| Nervous system disorders | 6 | 2 | 7 |
| Respiratory disorders | – | – | 3 |
| General disorders | – | 1 | – |
| All other SOCs | 4 | 1 | 3 |
| Total severe AEs | 15 (9.9%) | 8 (8.7%) | 16 (17.8%) |
Table 8
Total weeks exposure in all phases
| Paroxetine | Imipramine | Placebo | |
| Acute phase (8 weeks) | 640 (n = 93) | 634 (n = 95) | 626 (n = 87) |
| Continuation phase (six months) | 771 (n = 49) | 566 (n = 39) | 445 (n = 31) |
| Taper phase (2 weeks) | 112 (n = 56) | 144 (n = 72) | 128 (n = 64) |
Table 9
RIAT figures for adverse events in each phase
| Paroxetine | Imipramine | Placebo | |
| Acute phase | 457 | 520 | 330 |
| Continuation phase | 152 | 92 | 90 |
| Taper phase | 59 | 55 | 10 |
| Total AEs | 668 | 667 | 430 |
Table 10
RIAT figures for severe adverse events in each phase
| Paroxetine | Imipramine | Placebo | |
| Acute phase | 57 | 41 | 25 |
| Continuation phase | 15 | 8 | 16 |
| Taper phase | 16 | 14 | 1 |
| Total AEs | 88 | 63 | 42 |
Table 11
RIAT behavioral events
| Paroxetine | Imipramine | Placebo | |
| TOTAL AEs | 74 | 40 | 21 |
| Acute phase | 49 | 29 | 17 |
| Continuation phase | 15 | 10 | 3* |
| Taper phase | 10 | 1 | 1 |
| TOTAL episodes | 53 | 31 | 18 |
| Acute phase | 34 | 22 | 14 |
| Continuation phase | 13 | 8 | 3 |
| Taper phase | 6 | 1 | 1 |
| Total no. patients | 39 | 27 | 16 |
*One case either suicidal gesture or trauma.
Table 12
RIAT suicidality and suicide-related events
| Paroxetine | Imipramine | Placebo | |
| TOTAL AEs | 23 | 11 | 5 |
| Acute phase | 12 | 6 | 2 |
| Continuation phase | 6 | 5 | 3* |
| Taper phase | 5 | 0 | 0 |
| TOTAL episodes | 20 | 9 | 5 |
| Acute phase | 9 | 4 | 2 |
| Continuation phase | 6 | 5 | 3 |
| Taper phase | 5 | 0 | 0 |
| Total no. patients | 15 | 9 | 5 |
*Includes one case which could potentially be classed as ‘trauma’.
Table 13
All potential suicidal events – outline of events per patient
| Patient ID No | Sex &age | Study phase | Day of | Total AEs within |
| event onset | event | |||
| Paroxetine | ||||
| 329.001.00063 | F 14 | Acute | 34 | Disinhibition |
| 329.001.00065 | M 14 | Acute | 14 | Suicidal ideation |
| Aggravated depression | ||||
| Self injury | ||||
| Feeling of despair | ||||
| 329.001.00205 | M 12 | Acute | 4 | Disinhibition |
| 329.002.00055 | M 13 | Continuation | 84 | Self harm |
| Continuation | 140 | Suicidal ideation | ||
| 329.002.00058 | F 16 | Taper during continuation | 122 | Suicide attempt |
| 329.002.00099 | M 15 | Continuation | 121 | Akathisia |
| 329.002.00106 | F 15 | 30 day discontinuation | 51 | Aggravated depression |
| Suicidal gesture | ||||
| 329.002.00245 | F 14 | Acute | 14 | Suicide attempt |
| 329.003.00075 | F 17 | Acute | 1 | Akathisia (teeth chattering) |
| Akathisia (tremulous) | ||||
| Acute | 51 | Akathisia (nervous jittery) | ||
| Continuation | 170 | Akathisia (hyperkinesia) | ||
| 329.003.00089 | F 14 | Acute | 29 | Disinhibition |
| Acute | 56 | Suicidal ideation | ||
| Agitation | ||||
| Impulsive behaviour | ||||
| 329.003.00250 | F 15 | Acute | 37 | Suicide attempt |
| Taper during continuation | 75 | Suicide attempt | ||
| 329.003.00313 | M 18 | Acute | 12 | Suicide attempt |
| Suicidal ideation | ||||
| Hallucinations | ||||
| Psychosis | ||||
| Psychosis | ||||
| 329.004.00015 | F 16 | Acute | 31 | Suicide attempt |
| Suicidal ideation | ||||
| Continuation | 73 | Suicidal ideation | ||
| 329.004.00017 | F 16 | Acute | 7 | Akathisia |
| Continuation | 163 | Disinhibition | ||
| 329.004.00214 | F 15 | Acute | 5 | Akathisia |
| Continuation | 84 | Suicidal ideation | ||
| 329.005.00008 | M 12 | Acute | 37 | Disinhibition |
| Continuation | 77 | Disinhibition | ||
| 329.005.00011 | F 16 | Continuation | 156 | Suicide attempt |
| 329.005.00257 | F 12 | Continuation | 225 | Akathisia |
| 329.005.00333 | F 16 | Acute | 19 | Abnormal dreams |
| Aggravated depression | ||||
| 30 day discontinuation | 37 | Suicidal ideation | ||
| 329.006.00038 | F 15 | Acute | 57 | Suicide attempt |
| 329.006.00039 | F 15 | Acute | 18 | Suicide attempt |
| 42 | Suicidal ideation | |||
| Aggravated depression (more depressed) | ||||
| Aggravated depression (nervousness/irritable mood) | ||||
| Suicidal ideation | ||||
| Continuation | 84 | Aggravated depression | ||
| 329.007.00145 | M 16 | Continuation | 113 | Akathisia |
| 329.007.00268 | F 13 | Continuation | 169 | Akathisia |
| 329.007.00309 | F 18 | Acute | 1 | Akathisia |
| 329.008.00160 | F 14 | Acute | 20 | Akathisia |
| 329.008.00188 | F 17 | Acute | 4 | Akathisia |
| 329.008.00271 | F 13 | Acute | 24 | Akathisia (feelings of shakiness/ tremor) |
| Acute | 63 | Akathisia (increased irritability/nervousness) | ||
| Continuation | 65 | Akathisia (shaky/tremor) | ||
| 329.009.00130 | M 15 | Acute | 32 | Akathisia |
| 329.009.00170 | F 14 | Continuation | 237 | Negative thoughts |
| Suicidal ideation | ||||
| Agitation | ||||
| Akathisia | ||||
| 329.009.00173 | F 12 | Acute | 40 | Akathisia |
| 329.009.00193 | F 12 | Acute | 4 | Akathisia |
| 329.009.00201 | M 14 | Acute | 58 | Akathisia |
| Paranoia | ||||
| 329.009.00240 | M 14 | Acute | 48 | Aggravated depression |
| 329.009.00324 | F 13 | Acute | 15 | Abnormal dreams (nightmares) |
| 29 | Abnormal dreams (increased dreaming) | |||
| 329.009.00329 | M 14 | Acute | 35 | Aggravated depression |
| 329.010.00278 | F 17 | Acute | 24 | Akathisia |
| 329.011.00283 | M 17 | Acute | 27 | Akathisia |
| 329.011.00288 | M 15 | Acute | 48 | Agitation (also AE of mutism at same time. Patient sent to psychiatric unit and removed from study) |
| 329.012.00025 | F 14 | Acute | 1 | Akathisia |
| Imipramine | ||||
| 329.001.00070 | M 12 | Acute | 7 | Akathisia |
| 329.002.00057 | F 15 | Acute | 20 | Akathisia |
| 329.002.00100 | M 15 | Continuation | 84 | Self harm |
| 329.002.00103 | F 14 | Continuation | 224 | Suicidal ideation |
| Aggravated depression | ||||
| 329.002.00243 | F 15 | Acute | 9 | Akathisia |
| 329.002.00244 | F 17 | Acute | 3 | Depersonalisation |
| 329.003.00247 | M 16 | Acute | 45 | Akathisia |
| 329.004.00215 | F 14 | Acute | 37 | Akathisia |
| Abnormal dreams | ||||
| Hallucinations | ||||
| 329.005.00003 | F 13 | Acute | 8 | Akathisia |
| 329.005.00006 | M 17 | Continuation | 84 | Suicidal ideation |
| Continuation | 116 | neurosis | ||
| 329.005.00007 | F 15 | Continuation | 145 | Abnormal dreams |
| 329.005.00113 | F 15 | Acute | 32 | Suicidal gesture |
| Suicidal ideation | ||||
| (pt taken to ER) | ||||
| 329.005.00117 | F 16 | Acute | 29 | Akathisia |
| 329.005.00255 | F 15 | Acute | 2 | Akathisia (shakiness) |
| Acute | 33 | Akathisia (hand tremors) | ||
| 329.005.00295 | F 13 | Acute | 23 | Suicide attempt |
| 329.006.00040 | F 18 | Acute | 7 | Aggravated depression |
| 329.006.00041 | M 18 | Acute | 12 | Aggravated depression |
| 329.007.00146 | F 15 | Continuation | 172 | Akathisia |
| 329.008.00161 | M 17 | Acute | 1 | Akathisia |
| 329.009.00172 | F 13 | Acute | 36 | Abnormal dreams |
| Agitation | ||||
| 329.009.00194 | M 12 | Acute | 3 | Abnormal dreams (increased dreaming) |
| Abnormal dreams (nightmares) | ||||
| 329.009.00264 | F 14 | Acute | 11 | Akathisia |
| 329.009.00325 | F 15 | Acute | 35 | Abnormal dreams |
| Continuation | 112 | Aggravated depression | ||
| Self harm | ||||
| 329.009.00326 | M 13 | 30 day discontinuation | 26 | Akathisia |
| 329.010.00279 | F 13 | Acute phase | 33 | Suicidal ideation |
| 329.012.00221 | M 17 | Acute | 30 | Disinhibition |
| Continuation | 132 | Suicide attempt | ||
| 329.012.00223 | F 13 | Acute | 26 | Suicidal ideation |
| Suicide attempt | ||||
| Aggravated depression | ||||
| Placebo | ||||
| 329.001.00123 | F 16 | Acute | 46 | Suicidal ideation |
| Aggravated depression | ||||
| 329.002.00241 | M 15 | Continuation | 108 | Suicidal ideation |
| 329.003.00252 | F 14 | Acute | 37 | Akathisia |
| 329.005.00120 | M 13 | Acute | 21 | Akathisia (nervousness) dy21 |
| Akathisia (nervousness) dy23 | ||||
| Disinhibition (laughing a lot/euphoria) dy21 | ||||
| 329.005.00331 | F 15 | Acute | 3 | Akathisia |
| Acute | 18 | Depersonalisation | ||
| 329.007.00266 | F 12 | Acute | 5 | Akathisia |
| 329.008.00158 | F 16 | Continuation | 112 | Suicidal ideation |
| 329.008.00191 | F 17 | Acute | 28 | Akathisia |
| 329.009.00129 | M 16 | Acute | 35 | Suicidal ideation |
| 329.009.00135 | F 17 | Acute | 42 | Aggravated depression |
| 329.009.00169 | M 13 | Acute | 64 | Disinhibition |
| 329.009.00197 | M 12 | Continuation | 172 | Suicide attempt (or trauma) |
| 329.009.00198 | F 17 | Acute | 2 | Abnormal dreams |
| Acute | 15 | Akathisia | ||
| 329.009.00238 | F 16 | Acute | 15 | Abnormal dreams |
| 329.012.00027 | M 15 | Acute | 16 | Akathisia |
| 329.012.00217 | F 14 | 30 day discontinuation | 30 | Aggravated depression |
Table 14
Total (and severe) adverse events occurring during the acute phase for acute phase only patients versus continuing patients
| Paroxetine | Imipramine | Placebo | ||||
| System Organ Class (MedDRA) | Acute phase | Continuing | Acute phase | Continuing | Acute phase | Continuing |
| only patients | patients | only patients | patients | only patients | patients | |
| n = 44 | n = 49 | n = 56 | n = 39 | n = 56 | n = 31 | |
| Cardiac and vascular disorders | 14 (1) | 26 | 91 (3) | 36 | 22 | 10 |
| Gastrointestinal disorders | 45 (5) | 62 (16) | 74 (9) | 59 (7) | 45 (2) | 32 (2) |
| Psychiatric disorders | 58 (18) | 42 (6) | 39 (4) | 24 | 20 (4) | 7 (1) |
| Nervous system disorders | 31 (1) | 63 (5) | 73 (9) | 34 (3) | 55 (3) | 22 (4) |
| Respiratory and thoracic disorders | 18 (1) | 21 (1) | 15 (1) | 7 | 27 (2) | 13 (2) |
| All other SOCs | 24 | 53 (3) | 44 (4) | 24 (1) | 51 (1) | 26 (4) |
| Total AEs | 190 (26) | 267 (31) | 336 (30) | 184 (11) | 220 (12) | 110 (13) |

