
Practical considerations for creating a strategic and proactive clinical safety and pharmacovigilance organization for the future
Abstract
BACKGROUND:
In 2012, Patient Safety (PS) in AstraZeneca was facing a situation with multiple challenges, scientifically and structurally.
OBJECTIVE:
To meet these and support AstraZeneca’s ambition to return to growth after years of patent expiry, we undertook a project to fundamentally revisit ways of working to create an organisation set up to provide strategic safety in support of drug project decision-making.
METHOD:
In this paper, we describe the challenges we faced, the project to deliver changes to respond to them, and the methodology used. The project had two main components: creating a new operating model and simplifying the procedural framework.
RESULTS:
It was delivered in a focused effort by internal PS resources with cross-functional input. The framework simplification resulted in a 71% reduction in procedural documents and a survey of PS staff revealed an increase in satisfaction of 10%–20% across all scores.
CONCLUSIONS:
With >3 years of observation time, this project has provided AstraZeneca with a PS organisation able to provide strategic safety, supporting successful portfolio delivery, while ensuring patient safety and maintaining compliance with global pharmacovigilance regulations. It has driven efficiency and set the foundation for continued organisational evolution to meet future business needs in an everchanging environment.
1.Introduction
The realisation that medicines may carry risks was known both in ancient Western and Eastern cultures [1]. In more recent times, it became evident that even pure, carefully prepared products may carry unpredictable risks. The famous case of 15-year-old Hannah Greener who died in 1848 on receipt of the newly discovered chloroform anaesthesia serves as a good example. The event triggered one of the first structured and focused international surveillance programmes for a pharmaceutical product [2]. This reactive approach to safety surveillance changed fundamentally with the thalidomide disaster in the early 1960s [3]. In its aftermath, new legislation triggered two principles that shape the pharmaceutical industry to the present day: the structured and regulated preclinical testing of new pharmaceuticals before clinical trials; and the proactive safety surveillance of such products during clinical development and on the market.
While preclinical safety departments in the pharmaceutical industry could build on well-established principles of toxicology, newly formed clinical drug safety, patient safety (PS), or pharmacovigilance (PV) departments had to start from scratch [4]. Now, almost half a century later, the basic operational principles of PV can still trace their roots to the 1970s. Much of their focus remains on meeting the demands of regulatory authorities; in particular, the processing of individual cases to report adverse events (ICSRs), data quality to ensure effective signal detection, and on various forms of aggregate safety and benefit–risk reporting at predefined intervals (e.g. periodic safety reports).
With these operational aspects of PV now well established, an increasing focus in recent years has been on further strategic contributions. Beginning in the 1980s, the Council for International Organization of Medical Sciences (CIOMS) initiated a series of working groups to help codify such strategic PV principles [5].
The objective of this article is to describe how AstraZeneca has reorganised and focused its PS function towards becoming a strategic and scientifically competent driver of drug product safety and, thus, safety for patients.
1.1.Context of PS in the pharmaceutical industry
As comparatively newer functional groups, PS departments initially settled at the periphery of influence within pharmaceutical companies with some variability in the remit regarding pre-clinical safety, clinical development safety, and/or post-marketing safety, especially in those companies where the PS department’s remit was clinical development and post-marketing safety. We consider this was largely due to the early focus on operational matters such as a compliance-driven processing of ICSRs and associated aggregate reports. Within the industry, such activities are referred to as license-to-operate activities, as they adhere to regulatory requirements. PS departments are generally proficient at performing such routine PV tasks.
However, strictly meeting license-to-operate requirements did not meet the increased expectations of a modern PS department, which include prioritisation of safety concerns in order to de-risk pharmaceutical product development along with characterisation of the combined benefit–risk profile and associated risk minimisation activities.
At the same time, PV and drug development are becoming increasingly heavily regulated by health authorities, with new countries, for example China and nations in the Middle East, setting up comprehensive PV regulatory frameworks. These are mainly based on well-established models like the EU Good Vigilance Practice (GVP), for instance in Brazil and Mexico, or the United States Food and Drug Administration (FDA) Regulation, but the requirements could be adapted or amended which call for separate, often complex, solutions. Some National Authorities, for example Health Canada, have significantly increased their activity level in the PV area, which means increased demands on pharmaceutical companies to supply advanced compilations and analyses.
Even if some countries are adopting frameworks building on the EU GVP, there is also the notice of a growing divergence between existing regulatory authority legislative frameworks, illustrated by the contrast between the FDA and the European Medicines Agency (EMA) on the principles for causality assessment of potential Suspected Unexpected Serious Adverse Reaction (SUSARs) in a clinical trial setting. This nonconformity obviously causes a need for parallel processes.
To stay competitive, a modern PS department needs to be ready to embrace advancements in scientific, technical, and digital capabilities. For investigational products, examples include increasing expertise in translational aspects of preclinical testing (such as the drug-induced liver injury modeling tool, DILIsym®, to predict liver injury), the use of exploratory biomarkers (such as glutamate dehydrogenase and sorbitol dehydrogenase to detect liver injury), proactive PK/PD (pharmacokinetic/pharmacodynamic) modelling for certain safety topics, as well as internally generated novel working tools to better quantify the concepts of signal strength and medical importance. For marketed products, examples include predictive drug safety exploiting modelling of available datasets, the power of big data for signal evaluation, and robotic process automation for case processing.
Lastly, it is widely recognised that patients, physicians, national healthcare providers, and insurance companies demand the use of safer pharmaceutical products. In fact, the safety of these products is a competitive, differentiating factor in their utilisation [6–8].
1.2.Internal setting
AstraZeneca, a large-cap pharmaceutical company with a long history of delivering small drug molecules to market, has subsequently acquired several small and mid-sized biotechnology companies, e.g. MedImmune, which have largely operated as quasi-independent subsidiaries. For instance, the PS groups at AstraZeneca and its biologics subsidiary MedImmune operated cooperatively in parallel (e.g. sharing critical systems), with each focusing on their respective organisation’s product remit. The organisational structures in AstraZeneca and MedImmune PS respectively were, however, quite different.
By 2012, these parallel PS groups were facing multiple challenges, from a scientific and structural perspective:
An internal analysis revealed the early development failure rate within AstraZeneca to be alarmingly high, with safety a significant contributing factor for the attrition [9]. A follow up, internal analysis of safety failures identified a potential for earlier assessment of safety concerns and their incorporation into drug project decision-making as one key area to improve portfolio success rates.
The AstraZeneca pipeline was undergoing strategic transformation. The focus was shifting from the previous broad mix of primary care prescription drugs across multiple therapy areas to products for oncology, cardiovascular, metabolic, and respiratory diseases, many focused on specialty care. In addition, new approaches, including dual or triple drug regimens, are becoming increasingly more common. The pipeline was undergoing rapid growth and maturation following significant R&D investment with several pipeline assets initiating phase III development in multiple therapeutics areas.
The drive to fill the pipeline and an increasing emphasis on sharing business risk led to the development of a greater number of partnerships with other pharmaceutical companies. Such collaborations frequently carried a shared responsibility for safety, which is associated with its own set of complexities.
An increase in the number of ongoing trials, particularly late-phase trials contributed to the receipt of a greater number of ICSRs from clinical studies. Combined with the increasing focus on specialty care products, this trend led to more complex cases to process and an increase in demand for resources.
Cross-functional safety management teams were introduced after recommendation by CIOMS [10]. Successful adaptation had been a gradual process that was further complicated by different ways of working in the AstraZeneca and MedImmune legacy organisations.
In addition, our PS departments had received criticism from company stakeholders for a lack of transparent processes and clear lines of accountability.
In 2013, the groundwork for a strategic reorganisation and uplift of the PS function was formulated under the name of the “One Patient Safety” (OnePS) initiative. With the overall context of merging the AstraZeneca and MedImmune Patient Safety organisations, the initiative consisted of two pillars:
Redefining ways of working in a new operating model.
Fundamentally simplifying the procedural framework.
There were three purposes of the OnePS initiative: (i) to facilitate the combined operational safety administration of the legacy AstraZeneca and MedImmune PS organisations; (ii) to move from a reactive safety management-focused organisation to a proactive, strategy-focused organisation; and (iii) to adapt the organisation to the changing context of the AstraZeneca R&D business described above.
The objective of this article is to describe how AstraZeneca has reorganised and focused its PS function towards becoming a strategic and scientifically competent driver of drug product safety for patients.
2.Process
2.1.OnePS operating model
To achieve the objectives described above, the initial step was to develop upon existing ways of working and procedural guidances that would add true value to projects. The OnePS project was delivered with internal resources (except for limited vendor support with interviews and communications). This included internal change and project management expertise and leveraged the considerable subject matter expertise within the PS departments. Five focus groups were established concentrating on specific procedural areas as described below. Group members were selected to provide a diversity of representation with regards to geographic region, as well as level of experience and organisational background. In all groups, cross-functional participation was sought to ensure input from key interacting functions (e.g. clinical, biostatistics, regulatory, and toxicology) and other internal stakeholders (e.g. local affiliates/marketing companies and project leadership).
The five focus groups explored the following topics:
Clinical safety risk assessment including pharmacoepidemiology.
Safety in clinical programme and trial design, and interpretation.
Integration of preclinical–clinical safety support.
Local affiliate/marketing company interfaces and end-to-end PV system including post-marketing PV.
Competencies build of the personnel in PS.
For some of these focus areas, pre-existing analyses and initiatives were already identified as part of previous improvements. Examples of pre-existing analyses include a root cause analysis of safety failures in early clinical studies carried out as part of the analysis of early development performance in AstraZeneca as already described [9]. The focus group that was concentrating on competency build in PS incorporated previously established competency and training grids into their work.
Of the main outputs from these five focus areas, three were applied to the new operating model, specifically: a value-stream map defining eight discrete steps with a description of key activities per step; a renewed PS product/project team structure to support this; and a definition of roles and responsibilities for teams and key members (resulting from a Responsible, Accountable, Consulted, and Informed [RACI] analysis [11]). These are further described below.
2.2.Eight value-adding steps
To establish an output structure, Lean Six Sigma methodology [12] was applied. An overall value-stream mapping analysis was performed with the aim to embrace all aspects of clinical safety input into drug projects and products. From this analysis, eight discrete value-adding steps were defined (see Fig. 1) covering all phases, from preclinical to post-marketing activities for mature brands.
Fig. 1.
PS high-level value stream map covering drug development, life-cycle management, and maintenance: Eight discrete value-adding steps. Duplicate activities indicated by ‘a’ in the clinical development space and ‘b’ for post-marketing, in which data sources differ. To describe key activities per step, the steps were split. However, the majority of marketed products have ongoing clinical development/life-cycle management activities, so ‘a’ and ‘b’ activities will be ongoing in parallel. BRAT: benefit-risk action team; CDS: core data sheet; CTD: common technical document; DSUR: development safety update report; Dvt: development; EHR: electronic health record; HCP: healthcare professional; IB: investigator’s brochure; ICSR: individual case safety report; ISS: industrial safety services; PBRER: periodic benefit-risk evaluation reports; PS: patient safety; PSUR: periodic safety update reports; Reg: regulatory; RMP: risk management plan.
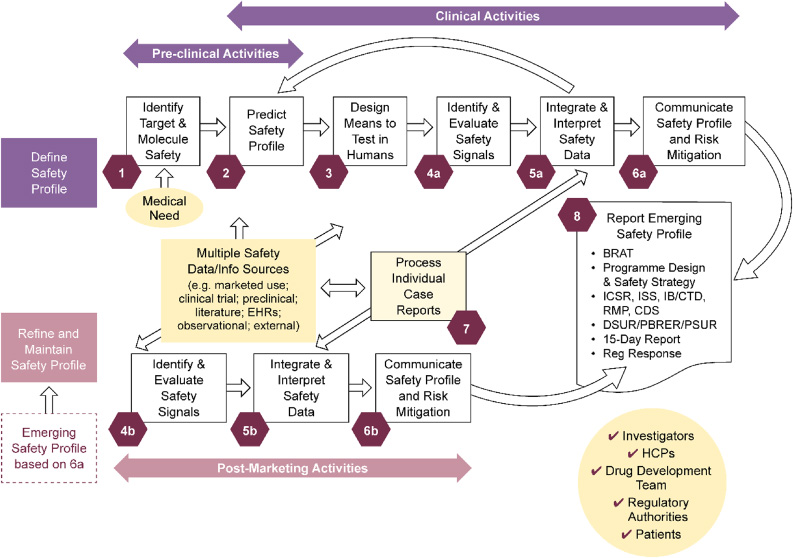
For each of these value-adding steps, for all representing areas where contributions from PS are impactful for drug projects and products, the teams worked up a succinct description of the key activities per step to drive value and visibility of Patient Safety’s contributions. An example of such an output is provided in Textbox 1; a full graphical representation of these activities is available as Supplementary Fig. S1 in the supplementary material.

2.3.Safety product/project team structure
To deliver the identified value-adding activities, the PS drug project team structure was reassessed. A revised team structure was proposed taking into consideration lessons learned from prior implementation. As the PS team would need to work across all phases of drug development, from preclinical to post-marketing for mature brands, a lean and flexible team structure was needed. To fulfil this, an operating model was established by which the clinical safety/PV work would be delivered via two teams. This means that tasks commonly described as safety science and risk management for a project/product are the focus for the two teams. It is worth noting that routine safety work, such as case handling, basic literature monitoring, and signal detection, as well as periodic safety report management, is performed in partnership with a third-party vendor. The activities performed by the vendor were not impacted by the change in the revised team structure.
The two team structures, the Safety Strategy and Management Team (SSaMT) and Safety Surveillance Team (SST), and their respective membership are presented in Fig. 2.
Fig. 2.
Clinical drug safety/PV project/product work delivered via two teams: Lean team structure with agenda-driven memberships, same for all phases/science units. Experts indicate agenda-driven membership from across functions as needed, e.g. epidemiology/real-world evidence, biostatistics, clinical pharmacology, clinical operations, and quality assurance. GCL: global clinical lead; GRL: global regulatory lead (key member for late-phasing project); GSP: global safety physician; PS: Patient Safety; PVS: pharmacovigilance scientist; Study Phys: clinical study physician/medical monitor; SSaMT: safety strategy and management team; TPL: toxicology project lead (key member for early phasing projects).
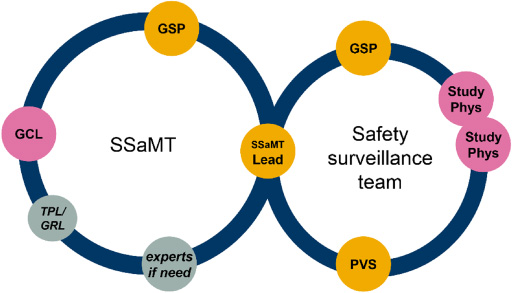
The SSaMT:
Sets the safety strategy at project/product level.
Addresses key safety decisions and emerging safety issues.
Is PS led, flexible, and cross functional.
Has a small core membership (only three core members, the SSaMT Lead, the Global Safety Physician, and the Global Clinical Lead, other members invited on an agenda-driven basis).
In summary, the SSaMT is the main body for individual product safety decision making, for EU marketed products, together with the European Union Qualified Person for Pharmacovigilance (EU QPPV, see below).
The SST is a PS-led team consisting of the Global Safety Physician, the SSaMT Leader and, depending on the size of the programme, additional safety/clinical physicians and scientists, with the core remit of reviewing safety data from all sources at project level. Depending on phase of development and complexity, one SSaMT and Surveillance Team can handle one or many compounds, the latter common in early development (Phase I–IIa) and for mature, established brands.
2.4.PS Centre of Excellence
In addition to these two drug-project safety teams, the PS Centre of Excellence (CoE) was an integral part of the reorganisation. The CoE is a cutting-edge scientific consultative hub proactively working to advance the science of clinical drug safety and PV. In particular, the CoE hub is supporting:
Safety science, for example, focusing on the management of organ toxicities, identifying safety biomarkers, and collecting health informatics, including real-world evidence.
Implementation of state-of-the-art PV and risk management policies and practices.
In the new merged organisation, the office of the EU QPPV, responsible for the PV system including all EU marketed products, is also located within the PS CoE.
2.5.Safety operations, process, and compliance
Support for safety operations, process, and compliance was provided by two organisational sections. These two sections collectively supported:
PV and clinical drug safety process and compliance.
Local affiliate/marketing company PV interface.
Licensing partnerships.
Vendor management, including the vendor for case handling.
Safety systems and analytics, including a safety database.
2.6.Roles and responsibilities for teams and key roles
As the final step to creating the operating model and to provide a basis for authoring the new procedural documents for the integrated PV system, a RACI analysis was performed. The RACI covered all the activities identified under the eight value-adding steps in the value-stream mapping. In the RACI analysis, Responsibility (R), Accountability (A), Consulted (C), and Informed (I) were assigned both at team level (e.g. SSaMT, surveillance, global project, and clinical project teams), and also for key roles inside and outside the PS departments (e.g. Global Safety Physician, SSaMT Leader, EU QPPV, Global Clinical Lead, and Global Regulatory Lead). The full analysis resulted in a 258-row (activities) by 36-column (teams and roles) table. Fig. 3 demonstrates how the operating model and RACI relate to the build of the OnePS procedural framework and integrated PV system.
Fig. 3.
Building blocks for an integrated PV system. Following the definition of the project approach, a pre-work phase was performed to (i) create an inventory of all current AstraZeneca and MedImmune PS department-owned procedural documents; (ii) collate all key global PV regulations, directives, and guidance (clinical trial and post-marketing); (iii) create the OnePS workstream book; and (iv) create the OnePS procedural document templates. PS: patient safety; PV: pharmacovigilance; QA: quality assurance; RACI: Responsible, Accountable, Consulted, and Informed.

2.7.OnePS procedural framework
Following the creation of the OnePS operating model, an updated set of procedural documents was needed. A procedural framework is critical in pharmacovigilance as it sets the foundation for transparent and traceable ways of working and processes, of importance not only to demonstrate compliance with legal and/or regulatory requirements, but also to ensure quality and consistency of activities and communicate best practice across an organisation. Certain elements were considered fundamental to its delivery, for example, it was recognised that the existing documents may not represent the most efficient and simplified documentation of the PV system and new ways of working. Standard operating procedures had been amended over time as a result of business or regulatory changes/inspections and had become lengthy to review and maintain. In view of this, OnePS represented a unique opportunity to start afresh and develop a new suite of PV procedures.
Existing global PV regulations and guidelines, including European GVP modules, FDA regulations, and CIOMS guidances, together with analyses of AstraZeneca/MedImmune regulatory authority inspection findings, were the foundational building blocks for the new procedures. Other building blocks were the new PS operating model, along with the associated and detailed RACI document. These building blocks informed the key principles and guided the framework for the ultimate delivery of the new standards, procedures, and guidelines, as illustrated in Fig. 3.
2.8.Planning tool – workstream book
A list of all activities and deliverables required from the PS department to meet the global regulatory requirements was created, detailing each specific requirement with the following information mapped out:
Regulatory reference(s).
Activity(ies).
Categorisation for future functional ownership, identifying those activities in scope for PS procedures versus other functional groups.
Whether the activity was currently in scope of legacy PS department-owned procedural documents.
Key interfacing functions for the activity.
Current standard operating procedures or other documentation covering the activity.
Current tools utilised for the activity.
Input was sought from additional cross-functional representatives to aid in completion of the full details described above.
2.9.Document production
Each workstream was responsible for planning the delivery of new procedural documents required to cover the scope that was allocated via the workstream planner. Drafts of new procedural documents were prepared after identifying the workstream deliverables. Document deliverables were identified and tracked across all workstreams. Draft documents authored using the OnePS procedural document templates were reviewed internally by the workstream before being released for official document review.
An approach was agreed for the standardised review of project deliverables by PS representatives outside of the production workstream and by appropriate functions/groups outside of PS. For each new procedural document, the affected functional groups (PS and non-PS) who needed to review the documents were documented and were centrally recorded on the overall delivery plan.
2.10.Consistency check
Once the reviews were completed and final versions of the documents had been created, these were submitted for a final consistency check. A consistency check process was established and performed by the AstraZeneca EU QPPV, deputy EU QPPV and a member of the OnePS implementation team to review all new procedural documents produced by the workstreams to ensure:
End-to-end completeness of the documents across and between workstreams, i.e. no gaps in regulatory requirements.
No duplication of activities or information between different documents.
No duplication of text within documents (simplification).
A consistent approach as to the use of templates, presentation style, and level of detail.
Once complete, the documents were submitted for approval and publishing via the AstraZeneca quality management system. Upon approval of the new procedural documents, legacy documents were retired from internal systems. Standard “read and sign” training was assigned to staff via the company’s learning management system.
2.11.Training
Following the delivery of the new procedural documents, a curriculum of live training sessions was created to supplement the standard “read and sign” training approach that had been used internally. This live training focused on key changes from the legacy procedural documents and provided an overview of the process(es), implementation plans, and toolkit items. Each training session covered one or more procedural documents from a given workstream with one or more sessions per workstream.
As part of the creation of the new procedural framework, a decision was made to procedurally formalise the administration of clinical safety across all science units by creating an internal Standard. Within the AstraZeneca clinical quality framework, the internal Standard represents a high level, widely applicable and mandatory document. A key element was to newly define the SSaMT (i.e., safety strategy and management team), and to create a new AstraZeneca executive safety board (chaired by the Chief Medical Officer, together with the Head of Global Regulatory Affairs, PS, and Quality Assurance [GRAPSQA] as the co-chair).
The standard also brought new clarity to the definitions of safety strategy and the overarching purpose of the PS function within our organisation: “… safety strategy implies the anticipation and prioritisation of potential safety concerns, understanding their possible consequences, and the proactive development of appropriate management plans to address those concerns and consequences”. Similarly, the standard clarified the goals of PS: “PS primary functional goals (are): robust and fit-for-purpose, license-to-operate activities, preferred-partner, gold-standard level of strategic PS expertise, and the careful identification, characterisation, communication, and management of safety risks”.
Given the growing recognition of the need to focus PV oversight into developmental programmes, a new foundational document, the SSaMT Plan, managed by the safety function, was introduced. It was recognised that the older developmental risk management plans, which were generally based on the EU risk management plan template that focused on approved products, did not always adequately address the unique challenges associated with the development of investigational products. While the SSaMT plan contains a risk log, it has additional modules to ensure consistency of safety messaging within projects, memorialisation of safety-related decisions and signal tracking, and provides a comprehensive checklist of strategic safety considerations for various development milestones along with instructions on extrapolating these considerations into potential consequences, such as labelling or health authority pushback.
Another new, key element in this procedural transformation was an increased focus for PS input and oversight newly focused at the time of certain developmental milestones: first-in-human clinical trials, as well as investment decisions for phases II and III, and commercialisation. Review by a newly created internal PS peer review board is required at these milestones to review key safety risks within the SSaMT Plan to better inform key milestone decisions.
3.Quantitative measures
Based on the challenges and drivers for the integration, the aim of the OnePS initiative was not merely to move to one set of aligned procedural documents, but to simplify and build in more strategic, efficient, and cost-effective ways of working. This has been successful. The key quantitative measures of success selected to document this include (i) procedural document simplification, and (ii) internal customer feedback.
3.1.Procedural document simplification
Procedural documents remain a fundamental part of the PV quality system. PS departments dedicate significant effort to ensure all employees receive appropriate training on procedural documents and in maintaining these documents based on changes in regulation and in alignment with other internal documents. Quantitative measures of the simplification effort were felt to be important in measuring not only the impact to staff but all ancillary activities stemming from procedural documents. Parameters to assess workload on staff and effort to maintain documents included: overall number of documents, number of pages, and total word counts. Data were collected for the legacy documents (AstraZeneca and MedImmune) and for the new OnePS procedural documents.
Table 1 presents the number of procedural documents covering various topics from the old PS departments, the associated pages and word counts from those documents before the OnePS initiative, and the positive results of simplification gained from combining the two legacy document systems into one updated system (‘New OnePS Docs’). In short, there was a 71% decrease in the total number of procedural documents using the OnePS approach, as well as a 64% decrease in the number of pages and a 56% decrease in total word count, all exceeding the 50% reduction expected from halving the numbers of two joined frameworks. These measures of simplification were estimated to also result in a saving of five FTEs annually for document maintenance activities.
Table 1
Quantitative results from the OnePS procedural framework
| Systems | ICSRs | Signals | Risk | Periodics | Q&C | Documents | Pages | Words | |
| Total old docs, n | 8 | 58 | 28 | 24 | 11 | 26 | 155 | 1,878 | 442,766 |
| Old MedI docs, n | 5 | 27 | 12 | 3 | 6 | 16 | 69 | 701 | 122,082 |
| Old AZ docs, n | 3 | 31 | 16 | 21 | 5 | 10 | 86 | 1,177 | 320,684 |
| New OnePS docs, n | 5 | 13 | 9 | 6 | 4 | 8 | 45 | 685 | 195,718 |
| Reduction, n (%) | 3 (38) | 45 (78) | 19 (68) | 18 (75) | 7 (64) | 18 (69) | 110 (71) | 1,193 (64) | 247,048 (56) |
[i] ICSR: individual case safety reports; Old AZ: legacy AstraZeneca; Old MedI: legacy MedImmune; OnePS: One Patient Safety; Q&C: quality and compliance.
3.2.Internal customer feedback
As the project set out to implement new ways of working through the establishment of a new operating model, the PS department staff were integral in providing feedback on the project. Research was facilitated by a third-party vendor for this purpose, measuring employee engagement and message retention of global PS staff. The same vendor was used for assessment of all activities and the feedback was used to shape further communication to all staff following the roll-out of the OnePS process.
Face-to-face focus groups and phone interviews of several dozen therapeutic area (TA)-aligned safety staff were arranged across the major geographic footprint sites (US, UK, and Sweden) supporting global R&D activities before the full OnePS roll-out to establish a baseline and, subsequently, after the roll-out to assess interval progress. The question sets predominantly used open-ended questions focusing on OnePS communications and understanding; as such, results were primarily qualitative.
Baseline evaluation found that global PS staff characterised the department as having dedicated staff with good technical and collaboration skills; this remained true with interval follow-up. The most relevant interval changes noted after roll-out of the PS operating model were an increase in the feeling of empowerment and efficiency of safety staff. However, feelings of being overwhelmed and confused by the many changes were also expressed. The feedback suggested that continued effort on change management would be needed to enable adaptation of staff to new ways of working. Word clouds highlight the most frequently recorded individual qualitative concepts summarising this internal change is shown in Fig. 4 (A representing positive associations and B negative associations).
Fig. 4.
OnePS qualitative feedback word maps post-implementation. (a) Positive associations. (b) Negative associations. OnePS: One Patient Safety.

In addition to this survey of global safety staff, a more quantitative survey focusing on the usefulness of the updated OnePS procedural documents was sent to a larger audience (n = 148) of both global PS staff (TA and delivery/enablement) and local PS staff located in various local affiliate/marketing company settings. As with the pulse check, a third-party vendor was used. Questions were rated on a scale of 1–10 with aggregate average responses tracked. A consistent 10–20% increase in average scores was noted with an average of 0.96 indicating a real improvement in the usefulness of the updated procedures. This was viewed as a meaningful change indicator for the procedural documents when considering the initial feedback on the OnePS operating model and timing of this assessment which was performed 6 weeks after the release of the new procedural documents. Table 2 shows the questions used, as well as the results.
Table 2
OnePS quantitative survey of safety staff
| Question | Old1 score | New2 score | Change | Increase, % |
| Ease of working out which procedural document to refer to for a given activity/process? | 6.0 | 7.1 | 1.09 | 18 |
| Ease of locating the current version of a specific document in the applicable official repository (e.g. AZDoc3 or AEGIS4)? | 5.3 | 6.1 | 0.77 | 14 |
| How appropriate do you think the level of detail is in the documents? | 6.3 | 7.1 | 0.85 | 14 |
| How easy do you find the overall layout of the documents in terms of readability (how easy is it to read the overall documents)? | 6.1 | 7.3 | 1.20 | 20 |
| How easy do you find the overall layout of the documents in terms of understanding (how easy it is to understand the overall documents)? | 6.4 | 7.2 | 0.79 | 12 |
| How easy do you find the document in terms of understanding your specific role in a process? | 6.3 | 7.0 | 0.66 | 10 |
| How easy do you find it to locate specific information in a document (e.g. on a part of the process or a certain situation)? | 6.2 | 7.0 | 0.84 | 14 |
| How consistent do you find the overall procedural documents (e.g. consistency between 1 procedure and another)? | 6.3 | 7.4 | 1.07 | 17 |
| How helpful is the information in the procedural documents in terms of understanding the key principles and context for the processes? | 6.4 | 7.4 | 0.99 | 15 |
| Overall, how would you rate the quality of the procedural documents? | 6.5 | 7.4 | 0.92 | 14 |
| Overall | 6.2 | 7.1 | 0.90 | 15 |
[i] Score is on a scale of 1–10, with 10 being best. Distribution of rating scores for each question is shown in Supplementary Fig. S2 in the supplementary material. 1Old, old procedural documents. 2New, new procedural documents. 3AstraZeneca’s quality system for procedural documents. 4AstraZeneca’s document management software to perform key tasks in the controlled document life cycle process. OnePS: One Patient Safety.
4.Discussion
In a world of few absolutes and many diverging opinions, it can be agreed by all individuals and institutions that access to safe medicines is a top priority. In the pharmaceutical paradigm existing in the early 21st century, the primary accountability for this commitment to safe medicines is shared by pharmaceutical companies in close collaboration with a variety of regulatory authorities. The aim of this paper was to describe how AstraZeneca has updated the clinical safety operating model and procedural framework to meet the growing expectation for the pharmaceutical industry to develop a culture of more proactive and more strategic PV.
For this project, we chose to deliver the OnePS operating model process completely with internal resources (except for some limited vendor support with interviews and communications), in order to leverage the considerable subject matter expertise within the PS department. As with any improvement project, the resources to accomplish this process needed to be carefully managed to ensure that other ongoing business needs were met; for example, delivering the portfolio and maintaining compliance with global regulations.
From an inspection readiness and license to operate perspective, the simplification and consolidation of process documents is deemed to be an essential prerequisite for effectively managing, incorporating, and implementing the increasingly widespread, rapid, and complex changes in global PV regulations and requirements, thus ensuring continuous high quality and compliance within the total PV system.
When we devised the new model, we took care to ensure that it was flexible enough to enable organisational changes to occur without big overhauls of the operating model and procedural framework. This has been effective; the model and framework have proven to be future proof, as evidenced by not needing any significant changes (other than role names in procedural documents) to allow two re-organisations during the following years.
As with every fundamental change, to leverage efficiency, changes to behaviours are essential. To address this, as part of the implementation, a set of face-to-face sessions hosted by senior PS leaders across all of AstraZeneca’s R&D sites were conducted. These were arranged in addition to workshops around the key process areas and the required formal training. Leadership closely monitored behaviours through line management, as well as through the above-described focus groups and surveys. Despite this, based on feedback from the focus groups interviews and the above workshops, as well as spontaneous feedback from staff, it was recognised that further work with behaviours, roles, and responsibilities is necessary. A series of interactive sessions and workshops focusing on key areas have been arranged and, together with the above described efforts, these have been critical to successfully achieve the necessary changes to behaviours.
As pharmaceutical organisations engage in the complex and high-risk development of drugs, a clinical drug safety and pharmacovigilance organisation, with an organisational positioning to ensure independence, and robust, transparent processes and compliance tracking is considered essential to ensure the safety of patients, as well as to ensure a robust assessment of benefit/risk when developing new medicines to meet unmet medical needs. This is an expectation from regulatory authorities, as repeatedly evidenced at inspections, and nowadays the vast majority of pharmaceutical companies adheres to these principles. Still, finding the correct visibility and voice for PS groups within the matrixed, cross-functional environment of a large organisation can be challenging. On the one hand, as noted above, there is an expectation that safety decision-making is firewalled from more commercial considerations; on the other hand, there are frequently opportunities – and needs – for the PS function to help drive cross-functional decision-making to enable the right internal decisions for the business, and to deliver new medicines to patients with unmet medical need while protecting patients and ensuring the basic component of regulatory compliance. At AstraZeneca, the OnePS initiative described in this paper has helped the PS group become a contributor to strategic project decision-making, while simultaneously gaining efficiencies during the merger of two different internal safety departments. This was accomplished by providing clear definitions of the safety strategy in practice, raising expectations regarding the scientific level of expertise and understanding of drug development, and by revising our internal procedural framework to better define roles and place more accountability on project-level safety teams. This change, as noted above, has given an improvement of scientific quality as well as an increased efficiency. Together this has served to continue to ensure the safety of patients and study participants and support AstraZeneca’s delivery of new medicines to patients with unmet medical needs. As such, this change to a strategic, value-contributing function has been a necessary part of AstraZeneca’s change journey. Success rates have improved from candidate drug nomination to phase III completion from 4% in 2005–2010 to 19% in 2012–2016, bringing more new medicines to patients and doctors [13]. The company has returned to growth, with double-digit growth for key products over the last 3 years, and AstraZeneca has moved from number 15 in 2017 to number 1 in the Pharmaceutical Innovation Index ranking for 2018 – an index measuring a company’s ability to launch successfully – to add value to pipeline molecules [14]. After a dip in 2019, falling out of the top 10, AstraZeneca is in the 2020 ranking back as number 6. Interestingly, launched in 2019 by the same group, the Pharmaceutical Invention Index examines pipeline novelty by the breadth and depth of novel agents, providing a more forward-looking view of who is developing medicines that matter, embracing science and innovations, and AstraZeneca has been number 1 in this ranking in both 2019 and 2020 [15].
With continued focus on our operating model, value to drug projects, efficiency of safety delivery, and developing our people, we see a strong foundation for future changes in clinical drug safety and PV to support AstraZeneca’s long-term goals. We are also suggesting that the strategic changes we have implemented could be applied by the wider pharma industry in order to improve safety and PV in the pharmaceutical industry, even if in our case, the trigger for these strategic changes was the merging of the PS groups and PV systems of two companies. The changes have created a more focused and adaptive culture within the organisation that bodes well for the development of new therapeutic modalities addressing unmet medical need in our core TAs, as well as adopting emerging technologies, such as artificial intelligence, robotic process automation, and machine learning, which are currently being piloted and implemented in many companies.
5.Conclusion
Following 3 years of observation, we can conclude that although substantial effort was required to develop and implement a new operating model, and establish a completely new, simplified procedural framework, the result has been worthwhile. It has provided AstraZeneca with a PS organisation that is able to deliver strategic safety, supporting drug project decision-making and contributing to AstraZeneca’s increased portfolio success rate, while ensuring compliance by PS and AstraZeneca, and their license to operate, protecting patient safety and driving delivery of new medicines to patients with unmet medical need. It has also provided simplification, driving cost efficiency. Finally, and as important, it has allowed a setting of the foundation for future changes to the organisation to drive business needs in an everchanging environment.
Acknowledgements
The authors wish to thank Joachim Forsgren, Steffen Ernst, Nick Blundell, Ceri Deveney, and Jane Feron for their invaluable contributions to the OnePS project and/or to this article. Medical writing support was provided by Jessica Gorrill, MSc and editorial support was provided by Rachael Cazaly all of Core Medica, London, UK, supported by AstraZeneca according to Good Publication Practice guidelines.
Conflict of interest
Magnus Nord, Magnus Ysander, Tim Sullivan and Mayur Patel are employees of AstraZeneca.
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/JRS-200082.
References
[1] | Jones J, Kingery E. History of pharmacovigilance Mann’s Pharmacovigilance. In: Andrews E, Moore N (eds) Chichester, West Sussex: John Wiley & Sons, Ltd; (2014) . |
[2] | Knight PR, Bacon DR. An unexplained death: Hannah Greener and chloroform. Anesthesiology. (2002) ;96: (5):1250–3. |
[3] | Kim JH, Scialli AR. Thalidomide: The tragedy of birth defects and the effective treatment of disease. Toxicol Sci. (2011) ;122: (1):1–6. |
[4] | Andrews E, Moore N. Mann’s Pharmacovigilance. Chichester, West Sussex: John Wiley & Sons, Ltd; (2014) . |
[5] | Lindquist M. VigiBase, the WHO Global ICSR database system: Basic facts. Ther Innov Regul Sci. (2008) ;42: (5):409–19. |
[6] | Hovstadius B, Hovstadius K, Astrand B, Petersson G. Increasing polypharmacy – an individual-based study of the Swedish population 2005–2008. BMC Clin Pharmacol. (2010) ;10: :16. |
[7] | Quinn KJ, Shah NH. A dataset quantifying polypharmacy in the United States. Sci Data. (2017) ;4: :170167. |
[8] | Dwyer LL, Han B, Woodwell DA, Rechtsteiner EA. Polypharmacy in nursing home residents in the United States: Results of the 2004 National Nursing Home Survey. Am J Geriatr Pharmacother. (2010) ;8: (1):63–72. |
[9] | Cook D, Brown D, Alexander R, March R, Morgan P, Satterthwaite G. Lessons learned from the fate of AstraZeneca’s drug pipeline: a five-dimensional framework. Nat Rev Drug Discov. (2014) ;13: (6):419–31. |
[10] | CIOMS Working Group VI. Management of safety information from clinical trials Geneva 2005 [Available from: https://cioms.ch/wp-content/uploads/2017/01/Mgment_Safety_Info.pdf]. |
[11] | Smith M, James E. Role & Responsibility Charting (RACI), Project Management Forum 2005 [Available from: https://www.lxcouncil.com/wp-content/uploads/2017/12/raci_charting.pdf]. |
[12] | Swank CK. The lean service machine. Harv Bus Rev. (2003) ;81: (10):123–9, 38. |
[13] | Morgan P, Brown DG, Lennard S, Anderton MJ, Barrett JC, Eriksson U Impact of a five-dimensional framework on R&D productivity at AstraZeneca. Nat Rev Drug Discov. (2018) ;17: (3):167–81. |
[14] | Rea M. Elephants can’t jump? Endpoint News 2018 [Available from: https://endpts.com/elephants-cant-jump-ideas-mike-rea-says-some-big-pharma-players-are-crushing-old-beliefs/]. |
[15] | IDEA Pharma. Pharmaceutical Invention and Innovation Indices 2020 2020 [Available from: https://www.ideapharma.com/pii/]. |


