
Assessment of specific human antibodies against SARS-CoV-2 receptor binding domain by rapid in-house ELISA
Abstract
BACKGROUND:
The recently emerged SARS-CoV-2 caused a global pandemic since the last two years. The urgent need to control the spread of the virus and rapid application of the suitable health measures raised the importance of available, rapid, and accurate diagnostic approaches.
OBJECTIVE:
The purpose of this study is to describe a rapid in-house optimized ELISA based on the expression of the receptor binding domain (RBD) of the SARS-CoV-2 spike protein in a prokaryotic system.
METHODS:
We show the expression of the 30 kDa recombinant SARS-CoV-2 RBD-6
RESULTS:
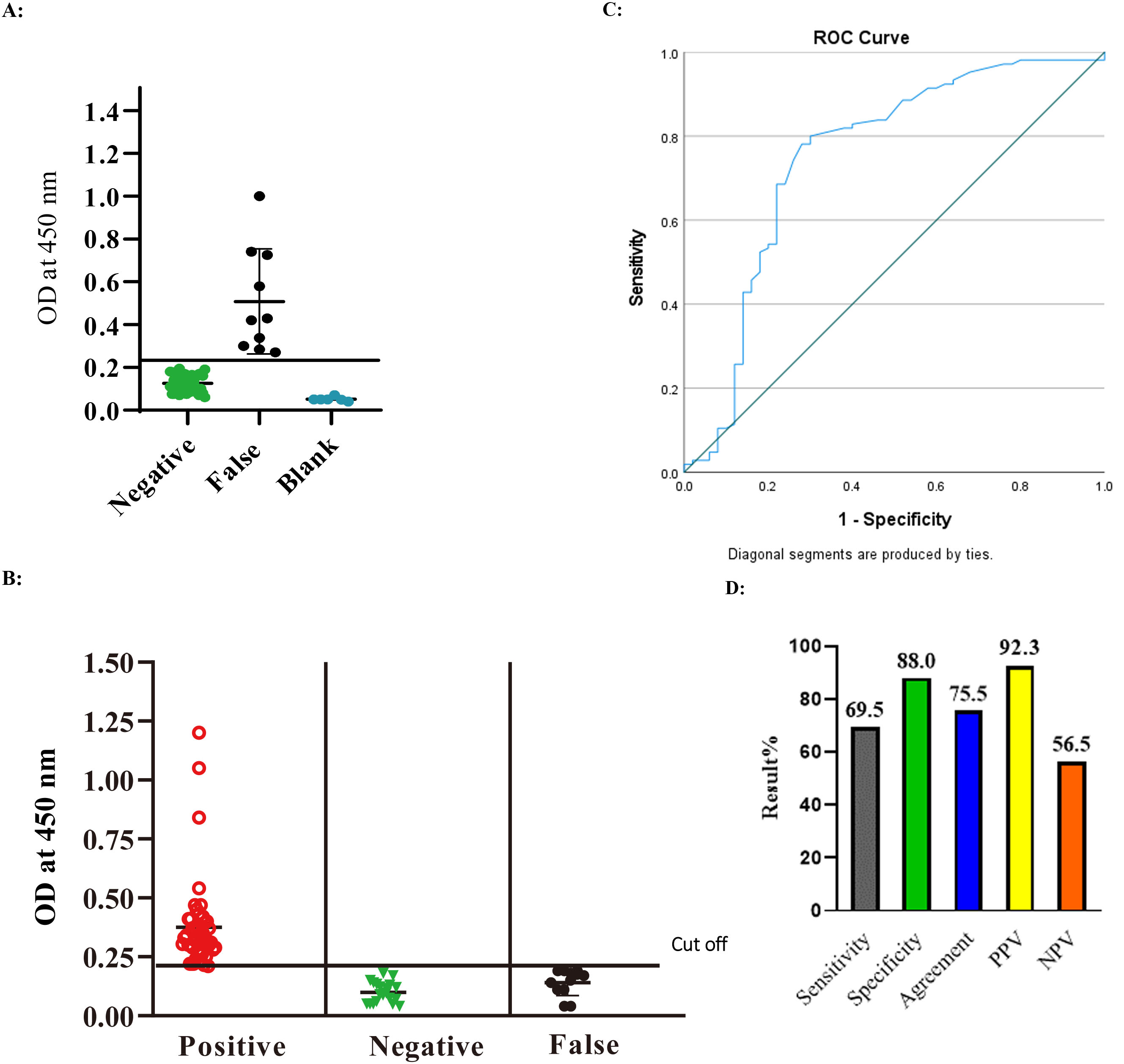
The assessment was carried out using a total of 155 human sero-positive and negative SARS-CoV-2 antibodies. The ELISA showed 69.5% sensitivity, 88% specificity, 78.5% agreement, a positive predictive value (PPV) of 92.3%, and a negative predictive value of 56.5%. Moreover, the optical density (OD) values of positive samples significantly correlated with the commercial kit titers.
CONCLUSIONS:
Specific human antibodies against SARS-CoV-2 spike protein were detected by rapid in-house ELISA in sera of human COVID-19-infected patients. The availability of this in-house ELISA protocol would be valuable for various diagnostic and epidemiological applications, particularly in developing countries. Future studies are planned for the use of the generated SARS-CoV-2 rRBD-6
1.Introduction
Since December 2019, the world witnessed a rapidly escalating outbreak of SARS-CoV-2 virus, the causative agent of COVID-19 disease. SARS-CoV-2 virus is a positive strand RNA enveloped virus. It causes sudden acute respiratory syndrome and has spread over the world with high speed and contagious potential. The SARS-CoV-2 virus epidemic outperformed the previous outbreaks caused by either Severe Acute Respiratory Syndrome (SARS) and Middle East Respiratory Syndrome (MERS) during the last twenty years, both also members of human coronaviruses (HCoVs) family. The rapid assault of SARS-CoV-2 in December 2019 has left healthcare institutions throughout the world baffled. To limit the rapid impact on health, several health measures and methods have been implemented, starting with the quarantine of clinically suspected persons, cancellation of social and professional meetings, application of WHO recommendations, and finally, country lock down. Within a few weeks, the virus had wrecked devastation on social, economic, and even political levels on a daily basis [1].
Since the declaration of SARS-CoV-2 outbreak a pandemic on March 11
Reverse transcriptase polymerase chain reaction (RT-PCR) with primer/probe combinations is the gold standard approach for diagnosis of SARS-CoV-2 infection, allowing detection of the viral nucleocapsid (N), RNA-dependent RNA polymerase (RdRP), and envelope (E) genes [2]. To avoid false negative results, consider factors such as viral load, sample collection and transportation, RNA extraction protocol, type of enzyme inhibitors, and RT-PCR method when performing RT-PCR [3, 4]. Traditional viral culture is useful, but it takes time and necessitates biosafety level-3 facilities, which are not readily available [5]. Novel technologies such as clustered regularly interspersed short palindromic repeats (CRISPR) and loop-mediated isothermal amplification (LAMP) were also used to build rapid diagnostic tests [6], however their use is still limited [7].
Serology testing is a reliable, straightforward, and cost-effective method for detecting infections both directly and indirectly. It has been successfully used in epidemiological surveillance studies in the past. Several serology-based rapid point-of-care assays were also developed to shorten assay turnaround times [8]. Within the context of the SARS-CoV-2 pandemic, assessing antibody response with a fast, reliable and easy to handle assay is a corner stone in determining previous infections among unvaccinated groups. In addition, SARS-CoV-2 serological assay can quantitatively and qualitatively monitor immune response and identify convalescent individuals who showed strong anti SARS-CoV-2 response thus potentially serving as therapeutic plasma donors.
SARS-CoV-2 spike protein receptor binding domain (RBD) is a promising candidate for the development of SARS-CoV-2 serological tests. Formed of 224 amino acids residues, it is the key entry point of SARS-CoV-2 to the host cells through binding with angiotensin converting enzyme (ACE-2). It harbors the unique mutations differentiating SARS-CoV-2 from other members of HCoVs [9]. Most studies attempting the use of SARS-CoV-2 RBD in serological assays relied on the expression of RBD from mammalian expression systems. Albeit its usefulness and numerous advantages, mammalian expression is time consuming, expensive, and requires sophisticated laboratory settings. Only a few studies reported RBD expression in prokaryotic expression systems for investigating its structural and binding characteristics [10, 11]. Less than a handful of studies reported the reactivity of RBD produced in E. coli with human sera of infected SARS-CoV-2 [12].
Here, we report the expression and purification of SARS-CoV-2 spike RBD from E. coli suitable for reactivity with human anti-SARS-CoV-2 IgG in human sera. Our study provides an improved immunoassay format for assessment of human anti-SARS-CoV-2 spike protein RBD through rapid in-house ELISA protocol.
2.Materials and methods
2.1Plasmids, bacterial strains and chemicals
The expression vector pET-30b(+) and bacterial strains used as host cells; E. coli DH5
2.2Human immune sera
A total of 155 preserved human serum samples (105 seropositive and 50 seronegative for SARS-CoV-2 antibody determined by commercial SARS-COV-2 IgG kit) (Abbott, Ireland), were tested. These sera were previously collected from patients in the first five months of the pandemic and before emerging of SARS-CoV-2 vaccines. The use of the serum samples was approved by the NRC ethical committee (Reference # NRC-20192).
2.2.1Construction of SARS-CoV-2 RBD expression vector
The nucleotide sequence encoding RBD domain retrieved from NCBI database (NC_045512.2) of severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, was used to construct an expression system. The region sandwiched between nucleotides 955 and 1623 in spike gene ORF, was codon optimized for expression in E. coli BL21(DE3) using IDT (Integrated DNA Technologies) codon optimization tools. The codon-optimized RBD sequence was cloned in frame with an N-terminal histidine tag in pET30b(+) vector (Novagen) between the restriction sites KpnI and BamH1 at the 5’ and 3’ ends, respectively. The resulting peptide corresponds to the region of R319-F541 designated by [13, 14] as the RBD of SARS-CoV-2 spike protein. The recombinant plasmid was first transformed into chemically competent E. coli DH5
Then, the recombinant expression vector was transformed into four different chemically competent E. coli expression strains: E. coli BL21(DE3) pLysS, E. coli BL21(DE3) Arctic RIL, E. coli BL21(DE3) C43 and E. coli BL21(DE3) Rosetta Gami.
2.3Expression of SARS-CoV-2 rRBD-6×
E. coli BL21(DE3) pLysS, E. coli BL21(DE3) Arctic RIL, E. coli BL21(DE3) C43 and E. coli BL21(DE3) Rosetta Gami transformed with recombinant plasmid pET30b(+) rRBD were grown in LB liquid medium containing 25
2.3.1Testing solubility of SARS-CoV-2 rRBD-6×
Induced bacterial pellets of different E. coli expression hosts described above were resuspended in phosphate-buffered saline and subjected to lysis by 5 repeated cycles of freezing (in frozen alcohol bath) and thawing (at 42
2.3.2Purification and renaturation of SARS-CoV-2 rRBD-6×
For purification of SARS-CoV-2 rRBD-6
Fractions containing highest concentration of purified rRBD-6
2.4Development and optimization of SARS-CoV-2 RBD human IgG ELISA
The in-house generated SARS-CoV-2 rRBD-6
2.5Statistical analysis
Graphs were generated using Graphpad prism software (version 8). Statistical analysis was carried out using the SPSS program and the calculations were as follows:
The cut-off value of the developed assay was determined as follow:
The sensitivity, specificity, agreement, PPV and NPV were calculated as follow:
3.Results
3.1Establishment of SARS-CoV-2 recombinant RBD construct in pET-30b
The receptor binding domain of SARS-CoV-2 spike protein is known to play a pivotal role in binding to ACE2 in the host cell [18]. Anti-SARS-CoV-2 spike protein RBD IgG is known to be present following SARS-CoV-2 infection. Accordingly, we aimed to express SARS-CoV-2 spike protein RBD in prokaryotic expression system to ensure its low cost, rapid and efficient production for detection of anti-SARS-CoV-2 IgG in human sera. We retrieved the nucleotide sequence of the full-length SARS-CoV-2 spike ORF from the NCBI database (NC_045512.2 Severe acute respiratory syndrome coronavirus 2 isolate Wuhan-Hu-1, complete genome) [19]. The region sandwiched between nucleotides 955 and 1623 was codon optimized for expression in E. coli BL21(DE3) (corresponding to R319 to F541 of SARS-CoV-2 spike protein). The nucleotide sequence was cloned in frame with an N-terminal histidine tag in pET-30b(+) vector (Novagen) between the restriction sites KpnI and BamH1 at the 5’ and 3’ ends, respectively. Fused to the N-terminal histidine tag, the 6
Figure 1.
Confirmation of the presence of SARS-CoV-2 rRBD insert in pET-30b(+) by colony PCR from E. coli DH5
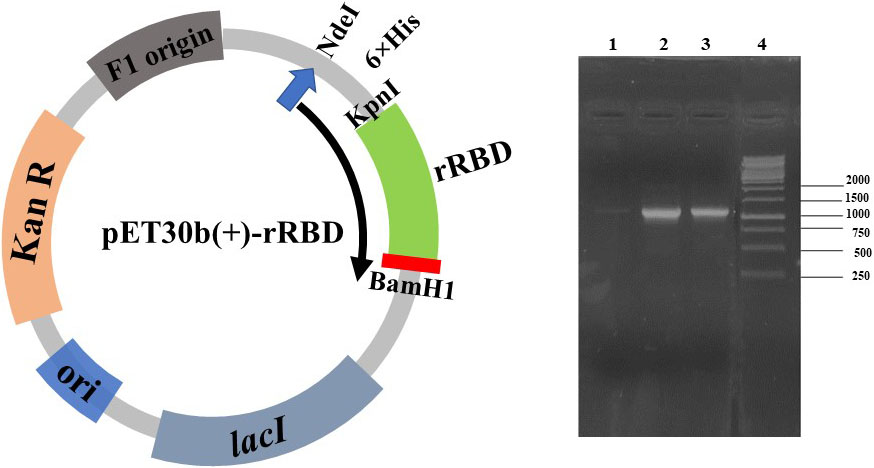
3.2Expression and purification of recombinant SARS-CoV-2 RBD-6× ×
SARS-CoV-2 rRBD-6
Figure 2.
Expression of SARS-CoV-2 rRBD-6

Next, we investigated the solubility of SARS-CoV-2 rRBD-6
Figure 3.
Purification and renaturation of SARS-CoV-2 rRBD-6
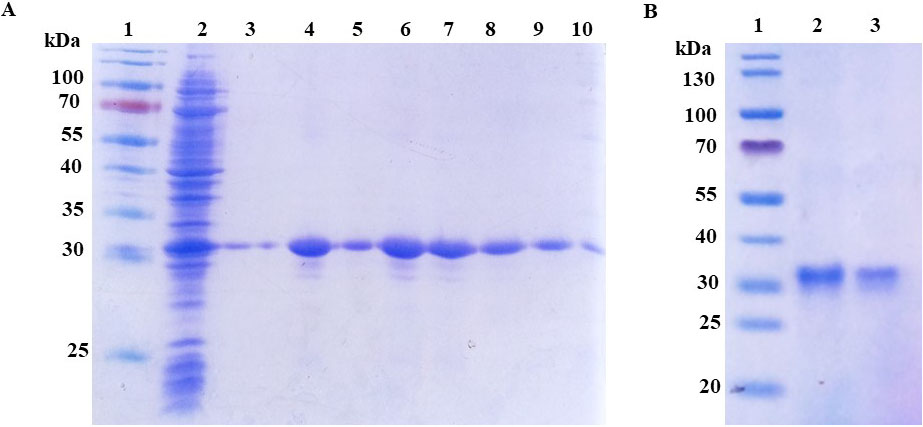
Figure 4.
Optimization of indirect ELISA utilizing different concentrations of purified SARS-CoV-2 rRBD-6
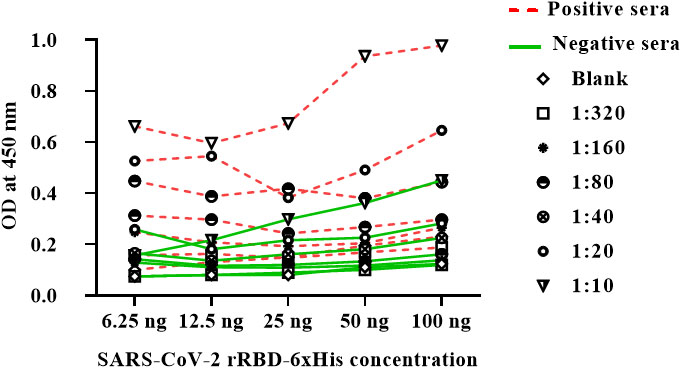
3.3Assessment of the antigenicity of SARS-CoV-2 rRBD-6×
In order to optimize the ELISA assay conditions, the protocol should offer enough antigen to bind antibodies, but with low non-specific backgrounds. To determine the optimal antigen-antibody concentration, we began by assaying different SARS-CoV-2 rRBD-6
Figure 5.
Optimization of indirect ELISA utilizing different dilutions of positive and negative human sera.
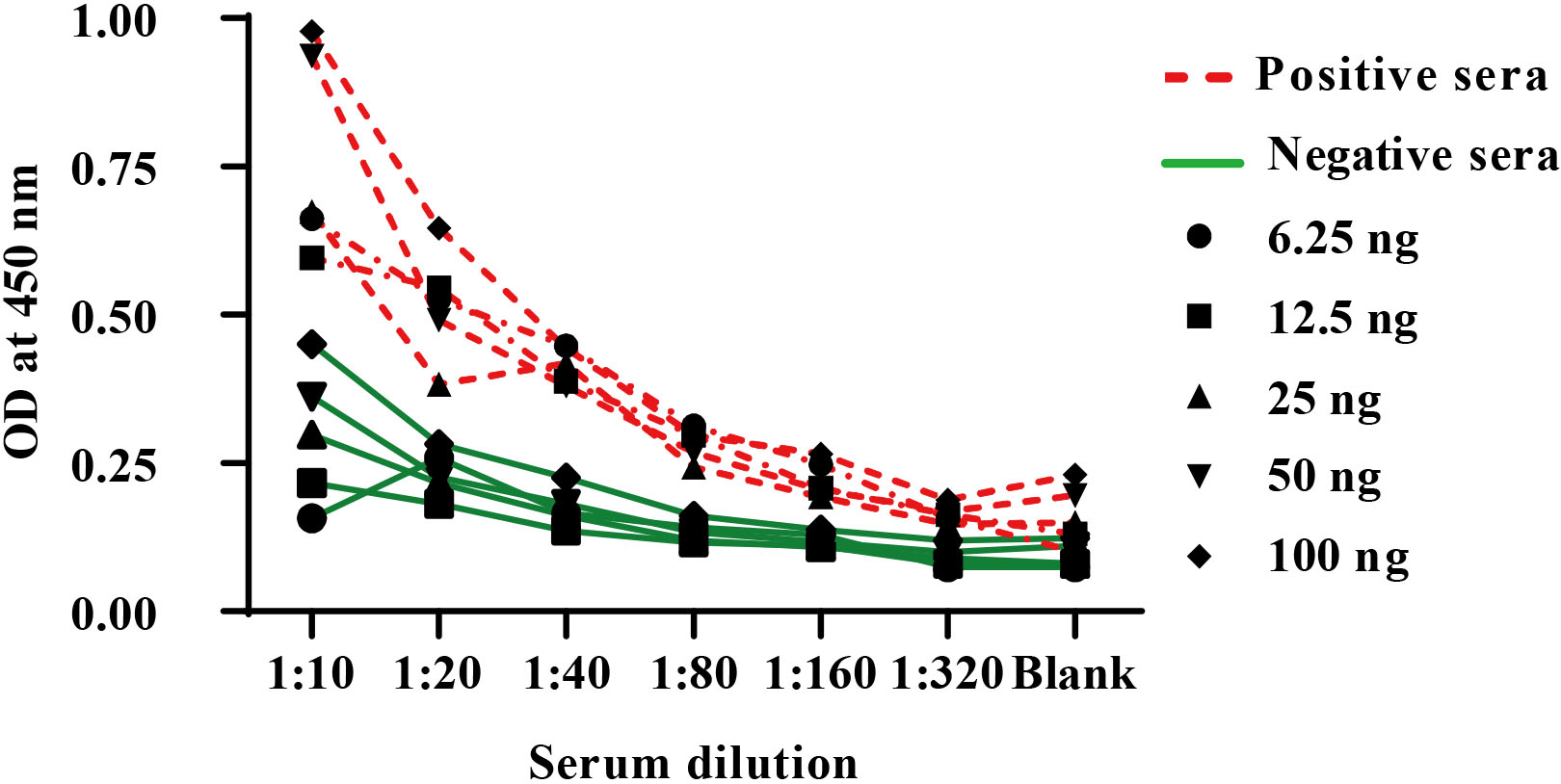
Figure 6.
Optimization of an indirect ELISA utilizing different dilutions of protein A peroxidase conjugate.
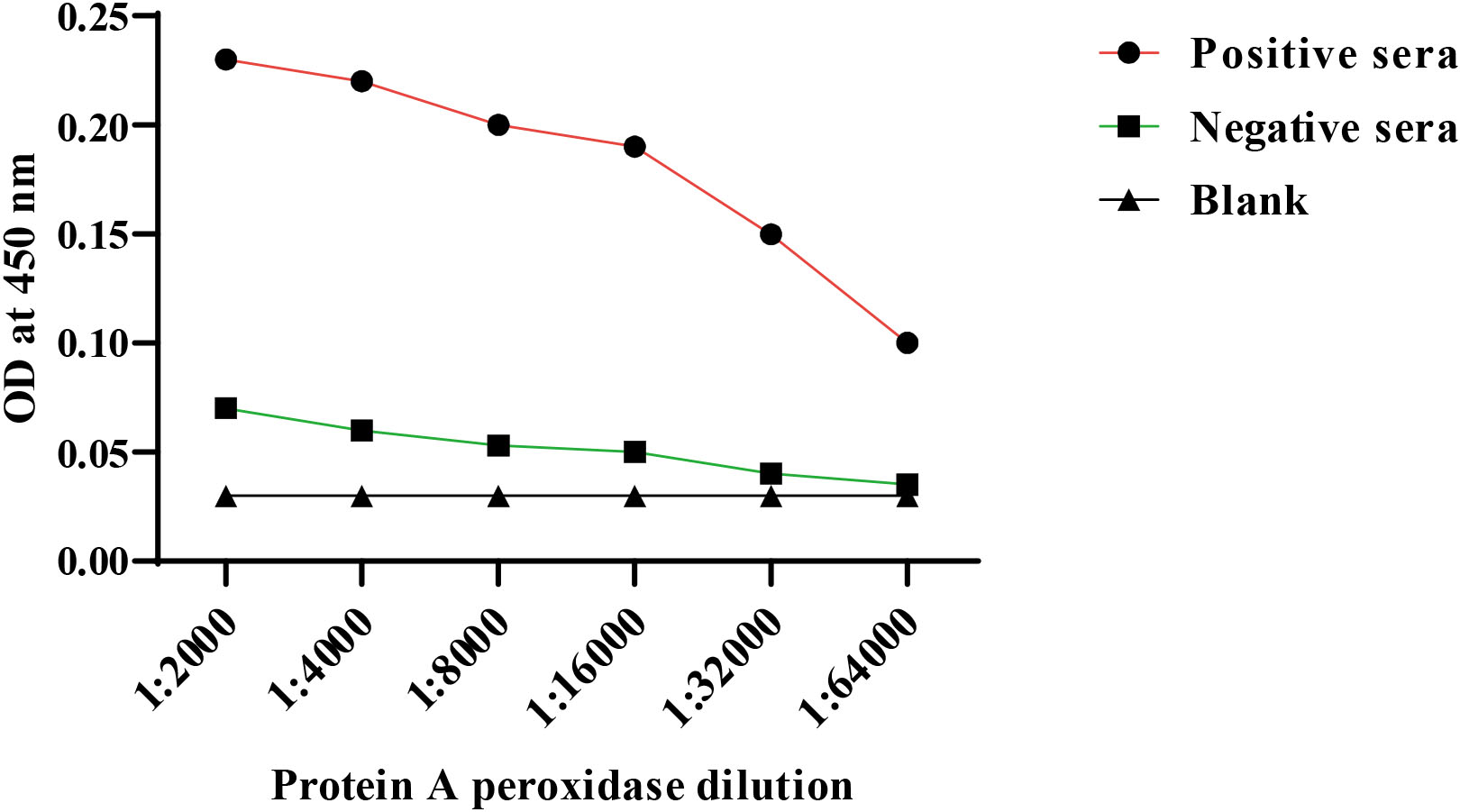
3.4Proof of concepts and in-house validation of anti- SARS-Cov-2 IgG ELISA assay
Negative samples (
Figure 7.
A: The Cut off value of the ELISA assay. B: Testing of human sera for the presence of IgG antibody directed to SARS-CoV-2 RBD protein using the optimized ELISA conditions. C: Receiver operating characteristics (ROC) analysis of the optimized developed indirect ELISA. D: Sensitivity, Specificity, Agreement, PPV and NPV of the developed ELISA utilizing in-house expressed purified SARS-CoV-2 rRBD-6
4.Discussion
Coronavirus disease 2019 (COVID-19) is an emerging disease caused by the coronavirus 2 (SARS-CoV-2) that causes severe acute respiratory syndrome. Antibody testing is currently focused on the spike (S) and nucleocapsid (N) structural components of SARS-CoV-2. The S protein is formed of two subunits (S1 and S2), and the RBD in S1 is responsible for recognizing angiotensin converting enzyme 2, a human cell surface receptor [20]. Antibodies that target the RBD in S1 can neutralize SARS-CoV-2 by preventing it from infecting host cells, hence anti-RBD antibodies serve as both immunological and neutralizing antibodies (nAbs) [21]. Several successful vaccines based on the S protein are currently available and immunization campaigns have begun around the world [22, 23, 24]. Anti-RBD antibodies and nAbs serological tests are becoming increasingly important for assessing humoral immunity against SARS-CoV-2 not only after vaccination but also in the assessment of previous infections.
Antibodies to SARS-CoV-2 can now be tested using a range of methods, including point-of-care lateral flow-based devices, high-throughput immunoassay analyzers, and manual approaches like ELISA. The nucleocapsid protein (N), full-length spike protein (S), S1 subunit, and receptor binding domain (RBD) of the S protein are among the antigens studied and used to generate kits for serological testing [25]. These kits were approved by FDA, applied after using specimens of 60 to 120, and showed between 80 to 98% sensitivity. Because SARS-CoV-2 antibody assays use a variety of techniques and antigenic targets, they must be thoroughly assessed before being used in clinical settings [26].
Similar to numerous developing countries, Egypt relies on the import of serological kits, which is an expensive and time consuming strategy. In addition, a sustainable supply of imported kits cannot be guaranteed in case of flight restrictions, border closures or urgent need of the kits by the manufacturer’s countries. Accordingly, developing a local SARS-CoV-2 serological assay is of particular importance for the current pandemic and may represent a prototype for the rapid diagnosis of future emerging pathogens.
Since the RBD of SARS-CoV-2 spike protein is one of the important domains of the virus used in vaccine trials, drug development, and diagnostic purposes, several mammalian expression systems were applied to produce it [27, 28]. Compared to prokaryotic expression systems, mammalian systems are costly, time consuming, sometimes have low production yield. However, mammalian systems produce efficiently folded and post-translationally modified proteins. To match our goal of developing a local SARS-CoV-2 serological assay, we based our assay on RBD cloned with an N-terminal 6
So far, we report for the first time the successful expression of SARS-CoV-2 rRBD in four different E. coli expression strains: E. coli BL21(DE3) pLysS, E. coli BL21(DE3) Arctic RIL, E. coli BL21(DE3) C43 and E. coli BL21(DE3) Rosetta Gami. Our results corroborate with Gao and colleagues work of successful expression of RBD with a C-terminal histidine tag in E. coli BL21 (DE3) Rosetta Gami from pET-28a(+) using 1 mM IPTG as an inducer [11]. The same study reported that the renatured RBD could efficiently bind to ACE2 as was also shown by He and colleagues for RBD expressed from E. coli (BL21) [10]. Our study adds an extra evidence of the feasibility of RBD expression in E. coli at different IPTG concentrations, induction durations, and temperatures. We successfully expressed the 30 kDa RBD protein using a low concentration of IPTG (0.25 mM) at 28
To the best of our knowledge, this study is among the unique studies establishing a diagnostic immunoassay using SARS-CoV-2 rRBD protein produced in E. coli. Our optimized ELISA test was used to detect IgG in human sera of infected COVID-19 patients, and showed sensitivity and specificity of 69.6% and 88%, respectively. Recently, Villafañe et al. [30], showed 100% serological concordance, using an in-house ELISA test based on recombinant RBD expressed in mammalian system, with commercial test based on the full-length spike protein (COVIDAR). However, the sensitivity of COVIDAR test, reported for IgG detection was 72% to 74% between 2 and 3 weeks from the onset of symptoms and seroconversion increased up to 90.4% after 3 weeks [31]. Moreover, Márquez-Ipiña and colleagues showed the reactivity of a shorter RBD fragment (N318-V510) expressed in E. coli BL21 strain C41 with 55 sera samples obtained from COVID-19 convalescent individuals [12]. However, the direct ELISA protocol reported was longer in time (including an 8 hours incubation for coating) and the assay time was only reduced by using a sandwich ELISA protocol [12].
Interestingly, we have tested the same sera samples used in this study, using the chemiluminescence kit from Abbott which uses the nucleocapsid protein (NP) as a coating antigen. These samples showed high IgG titer compared to their reactivity to our rRBD. Our results may correlate with the conclusion of Fafi-Kremer et al. [32] that a subset of patients may have an insufficient humoral immune response against a specific domain of the SARS-CoV-2 compared to their high immune reaction to another protein of the same virus.
To date, numerous mutations were identified in different antigens of the SARS-CoV-2 virus. Some of these mutations affected its interactions with the host cells and susceptibility to the disease. Barton et al. [33] concluded that mutations in the RBD enable the escape of immune responses such as K417N/T and the mutation E484K augments both ACE2 viral binding and also immune escape. This hypothesis may explain the discrepancy in the results within the samples we tested.
Although, both sensitivity and specificity are essential parameters for an assay, assessing the negative predictive values (NPV) and positive predictive values (PPV) of each assay are more valuable in determining how the assay can best be applied and interpreted. For any assay results, both PPV and NPV, respectively point to the probability that an assay can successfully determine whether individuals do or do not have a specific condition. Upon screening a percent of the population, both metrics are more significant than are sensitivity and specificity [34]. In our ELISA assay using the prokaryotic rRBD, PPV and NPV were 92.3% and 56.5%, respectively. The PPV value indicates reliable testing of the optimized in-house ELISA test.
We believe that our study had two limitations. First, sera were collected from 155 patients just before starting the vaccination protocols. Therefore, it was not possible to increase the number of samples especially that most vaccines available to date use the mRNA of S protein including the RBD. Second, more investigations are required to test the reactivity of our SARS-CoV-2 rRBD-6xHis with anti-SARS-CoV-2 IgM for diagnosis of early infection.
In conclusion, we successfully developed a rapid and sensitive in-house ELISA assay based on SARS-CoV-2 rRBD-6xHis expressed and purified from E. coli. Our in-house optimized assay detects anti SARS-CoV-2 IgG in human sera is an economical system that can be used in serological tests, functional and vaccine studies.
Author contribution
N.H. and E.A. carried out protein expression and purification. A.S., I.A., M.E., and M.S. conceived data assembly. A.E., A.T., and As.E. performed the immunological assays. A.T., A.E., N.H., Y.S. carried out manuscript writing. Y.S. was responsible of funding acquisition. Y.S., N.H., A.E., and A.T. designed the experiments. Y,S. demonstrated cohort management and supervision.
Funding
We would like to acknowledge the STDF- Egypt/Science and Technology Development Fund (Grant No. 43703) to Yasser SHAHEIN for financially supporting the current research. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ethical Approval
All the steps were approved by the Ethical Review Board of the National Research Center, Egypt (NRC#20192) according to Helsinki Declaration (1975).
Supplementary data
The supplementary files are available to download from http://dx.doi.org/10.3233/HAB-220003.
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
[1] | WHO, Preventing and mitigating COVID-19 at work. Available from: file:///C:/Users/tiger/AppData/Local/Temp/WHO-2019-nCoV-Workplace-actions-Policy-brief-2021.1-eng.pdf. |
[2] | G. Pascarella, A. Strumia, C. Piliego, F. Bruno, R. Del Buono, F. Costa, S. Scarlata and F.E. Agrò, COVID-19 diagnosis and management: A comprehensive review, J Intern Med 288: ((2020) ), 192–206. doi: 10.1111/joim.13091. |
[3] | L.M. Kucirka, S.A. Lauer, O. Laeyendecker, D. Boon and J. Lessler, Variation in False-Negative Rate of Reverse Transcriptase Polymerase Chain Reaction-Based SARS-CoV-2 Tests by Time Since Exposure, Ann Intern Med 173: ((2020) ), 262–267. doi: 10.7326/M20-1495. |
[4] | S. Woloshin, N. Patel and A.S. Kesselheim, False Negative Tests for SARS-CoV-2 Infection-Challenges and Implications, N Engl J Med 383: ((2020) ): e38. doi: 10.1056/NEJMp2015897. |
[5] | W. Bain, J.S. Lee, A.M. Watson and M.S. Stitt-Fischer, Practical Guidelines for Collection, Manipulationand Inactivation of SARS-CoV-2 and COVID-19 Clinical Specimens, Curr Protoc Cytom 93: ((2020) ): e77. doi: 10.1002/cpcy.77. |
[6] | J.P. Broughton, X. Deng, G. Yu, C.L. Fasching, V. Servellita, J. Singh, X. Miao, J.A. Streithorst, A. Granados, A. Sotomayor-Gonzalez et al., CRISPR-Cas12-based detection of SARS-CoV-2, Nat Biotechnol 38: ((2020) ), 870–874. doi: 10.1038/s41587-020-0513-4. |
[7] | R. Augustine, A. Hasan, S. Das, R. Ahmed, Y. Mori, T. Notomi, B.D. Kevadiya and A.S. Thakor, Loop-Mediated Isothermal Amplification (LAMP): A Rapid, Sensitive, Specific, and Cost-Effective Point-of-Care Test for Coronaviruses in the Context of COVID-19 Pandemic, Biology 9: ((2020) ), 182. doi: 10.3390/biology9080182. |
[8] | R.W. Peeling, C.J. Wedderburn, P.J. Garcia, D. Boeras, N. Fongwen, J. Nkengasong, A. Sall, A. Tanuri and D.L. Heymann, Serology testing in the COVID-19 pandemic response, Lancet Infect Dis 20: ((2020) ), e245–e249. doi: 10.1016/S1473-3099(20)30517-X. |
[9] | N. Kaur, R. Singh, Z. Dar, R.K. Bijarnia, N. Dhingra and T. Kaur, Genetic comparison among various coronavirus strains for the identification of potential vaccine targets of SARS-CoV2, Infect Genet Evol 89: ((2021) ), 104490. doi: 10.1016/j.meegid.2020.104490. |
[10] | Y. He, J. Qi, L. Xiao, L. Shen, W. Yu and T. Hu, Purification and characterization of the receptor-binding domain of SARS-CoV-2 spike protein from Escherichia coli, Eng Life Sci 21: (6) ((2021) ), 453–460. doi: 10.1002/elsc.202000106. |
[11] | X. Gao, S. Peng, S. Mei, K. Liang, M. Saleem, I. Khan, E. Vong and J. Zhan, Expression and functional identification of recombinant SARS-CoV-2 receptor binding domain (RBD) from E. coli system, Prep Biochem Biotechnol ((2021) ), 1–7. doi: 10.1080/10826068.2021.1941106. |
[12] | A.R. Márquez-Ipiña, E. González-González, I.P. Rodríguez-Sánchez, I.M. Lara-Mayorga, L.A. Mejía-Manzano, M.G. Sánchez-Salazar, J.G. González-Valdez, R. Ortiz-López, A. Rojas-Martínez, G. Trujillo-de Santiago and M.M. Alvarez, Serological Test to Determine Exposure to SARS-CoV-2: ELISA Based on the Receptor-Binding Domain of the Spike Protein (S-RBDN318-V510) Expressed in Escherichia coli, Diagnostics (Basel) 11(2): ((2021) ), 271. doi: 10.3390/diagnostics11020271. |
[13] | J. Lan, J. Ge, J. Yu, S. Shan, H. Zhou, S. Fan, Q. Zhang, X. Shi, Q. Wang, L. Zhang and X. Wang, Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor, Nature 581: (7807) ((2020) ), 215–220. doi: 10.1038/s41586-020-2180-5. |
[14] | F. Amanat, D. Stadlbauer, S. Strohmeier, T.H.O. Nguyen, V. Chromikova, M. McMahon, K. Jiang, G.A. Arunkumar, D. Jurczyszak, J. Polanco et al., A serological assay to detect SARS-CoV-2 seroconversion in humans, Nat Med 26: (7) ((2020) ), 1033–1036. doi: 10.1038/s41591-020-0913-5. |
[15] | M.R. Green and J. Sambrook, The Hanahan Method for Preparation and Transformation of Competent Escherichia coli: High-Efficiency Transformation, Cold Spring Harb Protoc 3: ((2018) ). doi: 10.1101/pdb.prot101188. |
[16] | M.M. Bradford, A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding, Anal Biochem 72: ((1976) ), 248–254. doi: 10.1006/abio.1976.9999. |
[17] | T.A. Alandijany, S.A. El-Kafrawy, A.M. Tolah, S.S. Sohrab, A.A. Faizo, A.M. Hassan, T.L. Alsubhi, N.A. Othman and E.I. Azhar, Development and optimization of in-house ELISA for detection of Human IgG antibody to SARS-CoV-2 full length spike protein, Pathogens 9: (10) ((2020) ), 803. doi: 10.3390/pathogens9100803. |
[18] | L. Cantuti-Castelvetri, R. Ojha, L.D. Pedro, M. Djannatian, J. Franz, S. Kuivanen, F. van der Meer, K. Kallio, T. Kaya, M. Anastasina, T. Smura, L. Levanov, L. Szirovicza, A. Tobi, H. Kallio-Kokko, P. Oesterlund, M. Joensuu, F.A. Meunier and M. Simons, Neuropilin-1 facilitates SARS-CoV-2 cell entry and infectivity, Science 370: (6518) ((2020) ), 856–860. doi: 10.1126/science.abd2985. |
[19] | F. Wu, S. Zhao, B. Yu, Y.M. Chen, W. Wang et al., A new coronavirus associated with human respiratory disease in China, Nature 579: ((2020) ), 265–269. doi: 10.1038/s41586-020-2202-3. |
[20] | F. Li, W. Li, M. Farzan and S.C. Harrison, Structure of SARS coronavirus spike receptor-binding domain complexed with receptor, Science 309: ((2005) ), 1864–1868. doi: 10.1126/science.1116480. |
[21] | B. Ju, Q. Zhang, J. Ge et al., Human neutralizing antibodies elicited by SARS-CoV-2 infection, Nature 584: ((2020) ), 115–119. doi: 10.1038/s41586-020-2380-z. |
[22] | F.P. Polack, S.J. Thomas, N. Kitchin et al., Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine, N Engl J Med 383: ((2020) ), 2603–2615. doi: 10.1056/NEJMoa2034577. |
[23] | L.R. Baden, H.M. ElSahly, B. Essink et al., Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine, N Engl J Med 384: ((2021) ), 403–416. doi: 10.1056/NEJMoa2035389. |
[24] | D.Y. Logunov, I.V. Dolzhikova, D.V. Shcheblyakov et al., Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: an interim analysis of a randomised controlled phase 3 trial in Russia, Lancet 397: ((2021) ), 671–681. doi: 10.1016/S0140-6736(21)00234-8. |
[25] | S. Alpdagtas, E. Ilhan, E. Uysal, M. Sengor, C.B. Ustundag and O. Gunduz, Evaluation of current diagnostic methods for COVID-19, APL Bioengineering 4: (4) ((2020) ), 041506. doi: 10.1063/5.0021554. |
[26] | A. Hussain, A. Hasan, M.M. NejadiBabadaei, S.H. Bloukh, M.E.H. Chowdhury, M. Sharifi, S. Haghighat and M. Falahati, Targeting SARS-CoV2 Spike Protein Receptor Binding Domain by Therapeutic Antibodies, Biomed Pharmacother 130: ((2020) ), 110559. doi: 10.1016/j.biopha.2020.110559. |
[27] | Argentinian AntiCovid Consortium, Structural and functional comparison of SARS-CoV-2-spike receptor binding domain produced in Pichia pastoris and mammalian cells, Sci Rep 10: ((2020) ), 21779. doi: 10.1038/s41598-020-78711-6. |
[28] | K. Rattanapisit, B. Shanmugaraj, S. Manopwisedjaroen, P.B. Purwono, K. Siriwattananon, N. Khorattanakulchai, O. Hanittinan, W. Boonyayothin, A. Thitithanyanont, D.R. Smith, et al., Rapid Production of SARS-CoV-2 Receptor Binding Domain (RBD) and Spike Specific Monoclonal Antibody CR3022 in Nicotiana benthamiana, Sci Rep 10: ((2020) ), 17698. doi: 10.1038/s41598-020-74904-1. |
[29] | L. Du, G. Zhao, C.C. Chan, S. Sun, M. Chen, Z. Liu, H. Guo, Y. He, Y. Zhou, B.J. Zheng and S. Jiang, Recombinant receptor-binding domain of SARS-CoV spike protein expressed in mammalian, insect and E. coli cells elicits potent neutralizing antibody and protective immunity, Virology 393: (1) ((2009) ), 144–50. doi: 10.1016/j.virol.2009.07.018. |
[30] | L. Villafañe, L.G. Vaulet, F.M. Viere, L.I. Klepp, M.A. Forrellad, M.M. Bigi, M.I. Romano, G. Magistrelli, M.R. Fermepin and F. Bigi, Development and evaluation of a low cost IgG ELISA test based in RBD protein for COVID-19, J Immunol Methods 500: ((2022) ), 113182. doi: 10.1016/j.jim.2021.113182. |
[31] | D.S. Ojeda, M.M.G. Lopez Ledesma, H.M. Pallarés, G.S. Costa Navarro, L. Sanchez, B. Perazzi et al., Emergency response for evaluating SARS-CoV-2 immune status, seroprevalence and convalescent plasma in Argentina, PLoS Pathog 17: (1) ((2021) ), e1009161. doi: 10.1371/journal.ppat.1009161. |
[32] | S. Fafi-Kremer, T. Bruel, Y. Madec et al., Serologic responses to SARSCoV-2 infection among hospital staff with mild disease in eastern France, EBioMedicine 59: ((2020) ), 102915. doi: 10.1016/j.ebiom.2020.102915. |
[33] | M.I. Barton, S.A. MacGowan, M.A. Kutuzov, O. Dushek, G.J. Barton and P.A. van der Merwe, Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics, Elife 10: ((2021) ), e70658. doi: 10.7554/eLife.70658. |
[34] | R. Trevethan, Sensitivity, Specificity, and Predictive Values: Foundations, Pliabilities, and Pitfalls in Research and Practice, Front Public Health 5: ((2017) ), 307. doi: 10.3389/fpubh.2017.00307. |


