
Long Non-Coding RNA Profile in Genetic Symptomatic and Presymptomatic Frontotemporal Dementia: A GENFI Study
Abstract
Background:
Long non-coding RNAs (lncRNAs) play crucial roles in gene regulation and are implicated in neurodegenerative diseases, including frontotemporal dementia (FTD). However, their expression patterns and potential as biomarkers in genetic FTD involving Chromosome 9 Open Reading Frame (C9ORF72), Microtubule Associated Protein Tau (MAPT), and Progranulin (GRN) genes are not well understood.
Objective:
This study aimed to profile the expression levels of lncRNAs in peripheral blood mononuclear cells collected within the GENetic Frontotemporal dementia Initiative (GENFI).
Methods:
Fifty-three lncRNAs were analyzed with the OpenArray Custom panel, in 131 patients with mutations in C9ORF72, MAPT, and GRN, including 68 symptomatic mutation carriers (SMC) and 63 presymptomatic mutation carriers (PMC), compared with 40 non-carrier controls (NC).
Results:
Thirty-eight lncRNAs were detectable; the relative expression of NEAT1 and NORAD was significantly higher in C9ORF72 SMC as compared with NC. GAS5 expression was instead significantly lower in the GRN group versus NC. MAPT carriers showed no significant deregulations. No significant differences were observed in PMC. Disease duration did not correlate with lncRNA expression.
Conclusions:
NEAT1 and NORAD are upregulated in C9ORF72 SMC and GAS5 levels are downregulated in GRN SMC, underlining lncRNAs’ relevance in FTD and their potential for biomarker development. Further validation and mechanistic studies are crucial for clinical implications.
INTRODUCTION
Long non-coding RNAs (lncRNAs) are RNA molecules encompassing more than 200 nucleotides. They do not possess canonical open reading frames1 and are not the product of alternative splicing of a coding gene.2 LncRNA regulates gene expression in neurodegenerative diseases. The majority of studies have been carried out in Alzheimer’s disease (AD). For example, it has been shown that the lncRNA amyloid-β cleaving enzyme-1 antisense, named BACE1-AS, is transcribed from the opposite strand of BACE1 gene, and can form an RNA duplex. It binds to the BACE1 mRNA to improve its stability and translation, positively regulating the expression of BACE1 protein and promoting an increase in the cleavage of the amyloid-β protein precursor.3 Moreover, the lncRNA Brain Cytoplasmic 200 (BC200) was found to be upregulated in brain tissues from patients with AD and it has been shown that it enhances the production of amyloid protein through the regulation of BACE1 expression.4 Few data on the role of lncRNAs in other neurodegenerative disorders, including frontotemporal dementia (FTD) are available, although it is known that they are involved in a number of common mechanisms, such as inflammation, oxidative damage and synaptic dysfunction (see5 for review).
FTD encompasses several clinical syndromes. The most common is the behavioral variant (bv)FTD, characterized by the development of behavioral disturbances, aggressiveness, lack of empathy, and decline in social conduct, followed by non-fluent variant primary progressive aphasia (nfvPPA) and semantic variant (sv)PPA. About 50% of FTD cases display a family history for dementia, often with dominant traits.6 At histopathology, all syndromes described are collectively classified as frontotemporal lobar degeneration (FTLD). According to the type of protein depositing, FTLD is classified into FTLD-Tau, FTLD-TAR DNA Binding protein (TDP)43, and FTLD fused in Sarcoma (FUS).7 Three major causal genes responsible for autosomal dominant inherited FTD have been discovered so far, including microtubule associated protein tau (MAPT), characterized by the deposition of tau protein in the brain, progranulin (GRN) and chromosome 9 open reading frame 72 (C9ORF72), both characterized by deposition of TDP-43. MAPT carriers quite often develop bvFTD with parkinsonism, whereas GRN mutations are associated with phenotypic heterogeneity, including the classical syndromes but also atypical presentations such as corticobasal syndrome (CBS) and progressive supranuclear palsy (PSP).8 The C9ORF72 expansion instead may present not only with FTD but also with amyotrophic lateral sclerosis (ALS), or both, and is often associated with late onset psychosis.9
In FTD and ALS the lncRNAs nuclear paraspeckle assembly transcript 2 (NEAT2) and Metastasis Associated Lung Adenocarcinoma Transcript 1 (MALAT1) co-localize at nuclear paraspeckles with TDP-43 and FUS proteins.10 Moreover, the binding to TDP-43 is markedly higher in brains from demented patients. 10 The expression of 84 lncRNAs was analyzed in serum samples from genetic and sporadic FTD. Despite the statistical threshold was not reached due to limited sample size, the results showed a generalized deregulation of lncRNA expression levels in both genetic and sporadic FTD as compared with non-demented controls. In detail, a trend toward downregulation was observed in GRN and C9ORF72 patients, whereas a trend toward upregulation was observed in MAPT mutation carriers. Notably, a few lncRNAs, including hepatocellular carcinoma upregulated EZH2-associated (HEIH), Eosinophil Granule Ontogeny Transcript (EGOT), and NEAT1, were downregulated in all groups.11 The origin of circulating lncRNAs is not known and no studies on lncRNA in circulating cells are available so far.
Here, we show results of profiling of lncRNAs in peripheral blood mononuclear cells (PBMC) from the very well characterized genetic GENFI cohort. To our knowledge, a large study of these molecules has not been carried out yet in genetic FTD, which represent the best model of the disease as the pathology can be predicted in life.
MATERIALS AND METHODS
Population
All demographic and clinical data, as well as samples included in the study, were collected within the Genetic frontotemporal dementia initiative (GENFI), a natural history study of genetic FTD involving several research centers across Europe and Canada (http://www.genfi.org.uk).12 The GENFI study was performed in accordance with the Declaration of Helsinki, reviewed and approved by all countries’ respective Ethics Committees and all participants signed an informed consent to take part in the research. This research study was performed in Italy, Ethics Committee Milano Area 2, parere 882_2022 del 13-9-22.
Variables included were: age at sampling, age at onset, Gender, mutation group (symptomatic mutation carriers, SMC; presymptomatic mutation carriers, PMC; non-carrier family members considered as controls, NC), mutated gene (MAPT, GRN, C9ORF72). One hundred and seventy-one PAX gene samples were collected, including 68 SMC (22 MAPT, 22 GRN, and 24 C9ORF72), 63 PMC (20 MAPT, 22 GRN, and 21 C9ORF72) and 40 NC. Carriers of other rare FTD causing mutations were not included. Demographics of the population are shown in Table 1.
Table 1
Demographic and clinical characteristics of the study population
| Non carriers (NC) | Presymptomatic mutation carriers (PMC) | Symptomatic mutation carriers (SMC) | |
| Subjects (n) | 40 | 63 | 68 |
| Age, mean years (SD) | 53 (15) | 42 (11) | 65 (8) |
| Age at onset, mean years (SD) | – | – | 61 (8) |
| Gender distribution, females:males | 27 : 13 | 36 : 27 | 28 : 40 |
| Mutated gene n (%) | |||
| GRN | 22 (35) | 22 (32) | |
| C9ORF72 | 21 (33) | 24 (36) | |
| MAPT | 20 (32) | 22 (32) | |
| Age at onset, mean years (SD) | |||
| GRN | 64 (7) | ||
| C9ORF72 | 59 (9) | ||
| MAPT | 54 (6) | ||
| NfLpg/ml (SEM) | 8.53 (0.18) | 16.21 (2.83) | 56.05 (3.81)* |
| GFAP pg/ml (SEM) | 103.15 (10.21) | 107.42 (6.12) | 175.44 (9.32)** |
*p = 0.002; **p < 0.001.
Sample processing
PAX gene tubes were stored and frozen according to the manufacturer. RNA was extracted using the PAX Gene blood miRNA kit that enable the extraction of total RNA including small ncRNA, according to the protocol of the manufacturer (Qiagen).
The quality and quantity of the extracted RNA were assessed using a Bioanalyzer 2100. The RNA Integrity Number (RIN) was determined, with values ranging from 7.9 to 8.4 across the samples, indicating that the RNA is of sufficient quality for further analysis (Supplementary Figure 1). Extracted RNA was stored at –80°C until use.
Retrotranscription and real-time PCR
Total RNA was retrotranscribed with the SuperScript VILO cDNA Synthesis Kit. The OpenArray Custom panel, comprising a total of 56 lncRNA genes (53 target genes and three housekeeping genes: B2M, ACTB and GAPDH, Supplementary Table 1) was used following the manufacturer’s instructions (Thermofisher, 2012). The reproducibility between real-time PCR experiments was assessed by including the same cDNA sample in each run.
Plasma neurofilament light chain and glial fibrillary acidic protein
Plasma neurofilament light chain (NfL) and glial fibrillary acidic protein (GFAP) levels were measured on the SIMOA HD-1 Analyzer as previously described.13
Statistical analysis
Data were analyzed using the statistical spreadsheets Jamovi v 2.5.6.0 (https://www.jamovi.org). LncRNA expression levels (expressed as replicate 2–ΔCt values for each gene) and gender were considered continuous variables and expressed as mean±standard deviation (SD). Conversely, gender, biological group (no mutations, MAPT, C9ORF72, and GRN, respectively) and genetic status (NC, PMC, and SMC, respectively) were all considered categorical variables. Significant statistical threshold was set at 0.05. Shapiro–Wilk’s test of normality was performed for continuous variables, and intergroup comparisons were carried out using ANCOVA test (gender and age as covariates). Post hoc analyses for multiple comparisons were performed with Tukey’s correction. The Pearson test was applied for correlations between deregulated lncRNAs and NFL and GFAP protein levels.
RESULTS
Thirty-eight lncRNAs out of 53 were detectable. Among those, the relative expression of two lncRNAs, NEAT 1 and NORAD, were upregulated in C9ORF72 biological group.
In particular, the ANCOVA analysis of NEAT1 showed that the overall model yielded a significant result (p = 0.003), indicating that these variables collectively explain a significant portion of the variance in 2–ΔCt. Biological group showed a significant effect (F(3,96) = 5.35, p = 0.003, ἠ2p = 0.143), suggesting differences in 2–ΔCt levels among the groups. Specifically, the C9ORF72 group differed significantly from others in post hoc comparisons (0.098±0.01 versus 0.04±0.05-fold regulation, t(96) = –2.66, p = 0.043) compared to NC. As expected, age also had a significant influence (p = 0.046), indicating that older age was associated with altered 2-Δ Ct levels, but the interaction between biological group and age was not statistically significant (p = 0.256). Stratifying results according to genetic status, the comparison between C9ORF72 SMC and NC showed a statistically increased of NEAT1 2–ΔCt values (0.12±0.01 versus 0.04±0.01-fold regulation versus NC, t(43) = –2.49, p = 0.043, Fig. 1).
Fig. 1
Box plots of NEAT1 profiling in PBMC from SMC and PMC carrying mutations in C9ORF72, GRN, MAPT as compared with NC. Data are expressed as 2-ΔCt fold regulation.
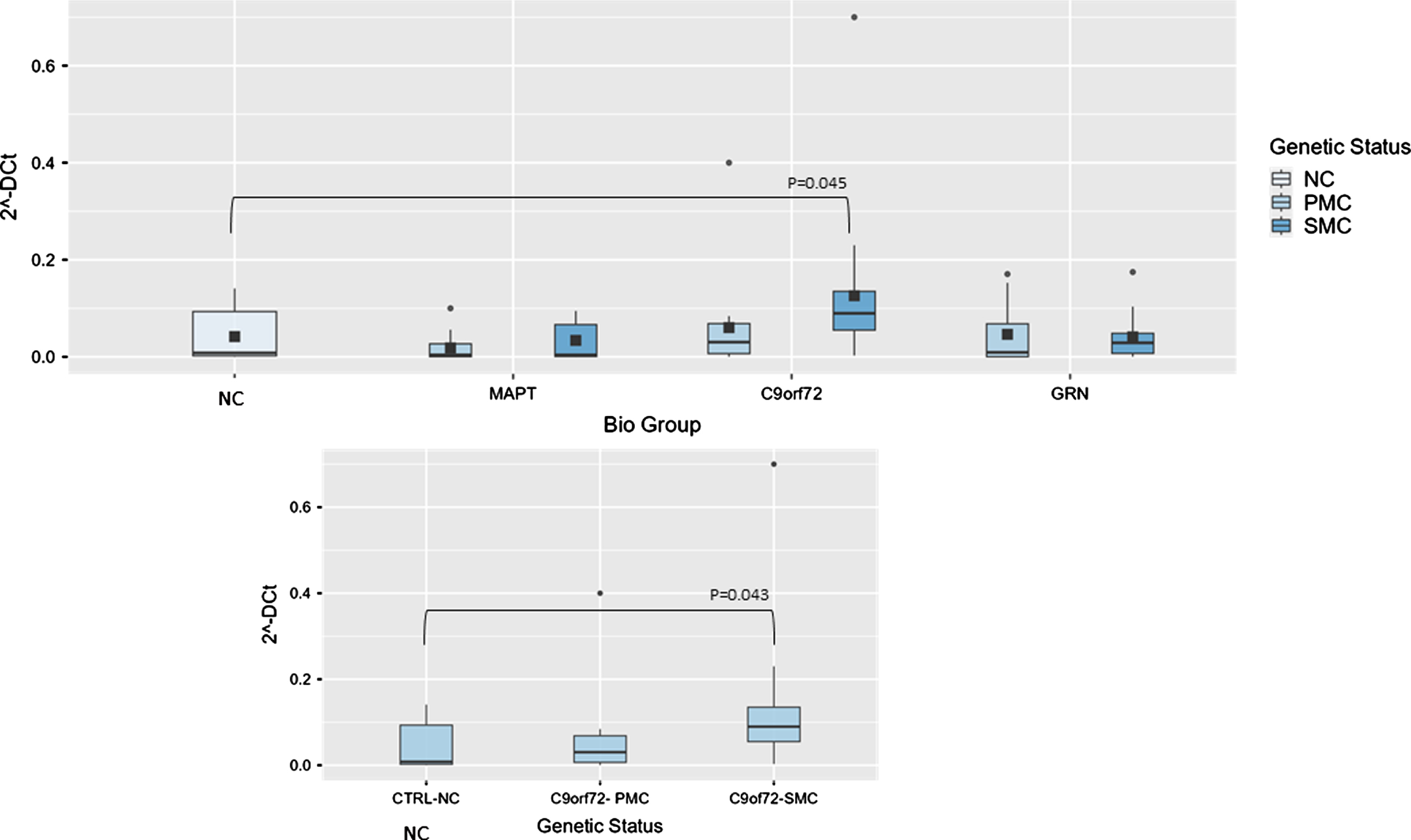
Fig. 2
Box plots of NORAD profiling in PBMC from SMC and PMC carrying mutations in C9ORF72, GRN, MAPT as compared with NC. Data are expressed as 2-ΔCt fold regulation.
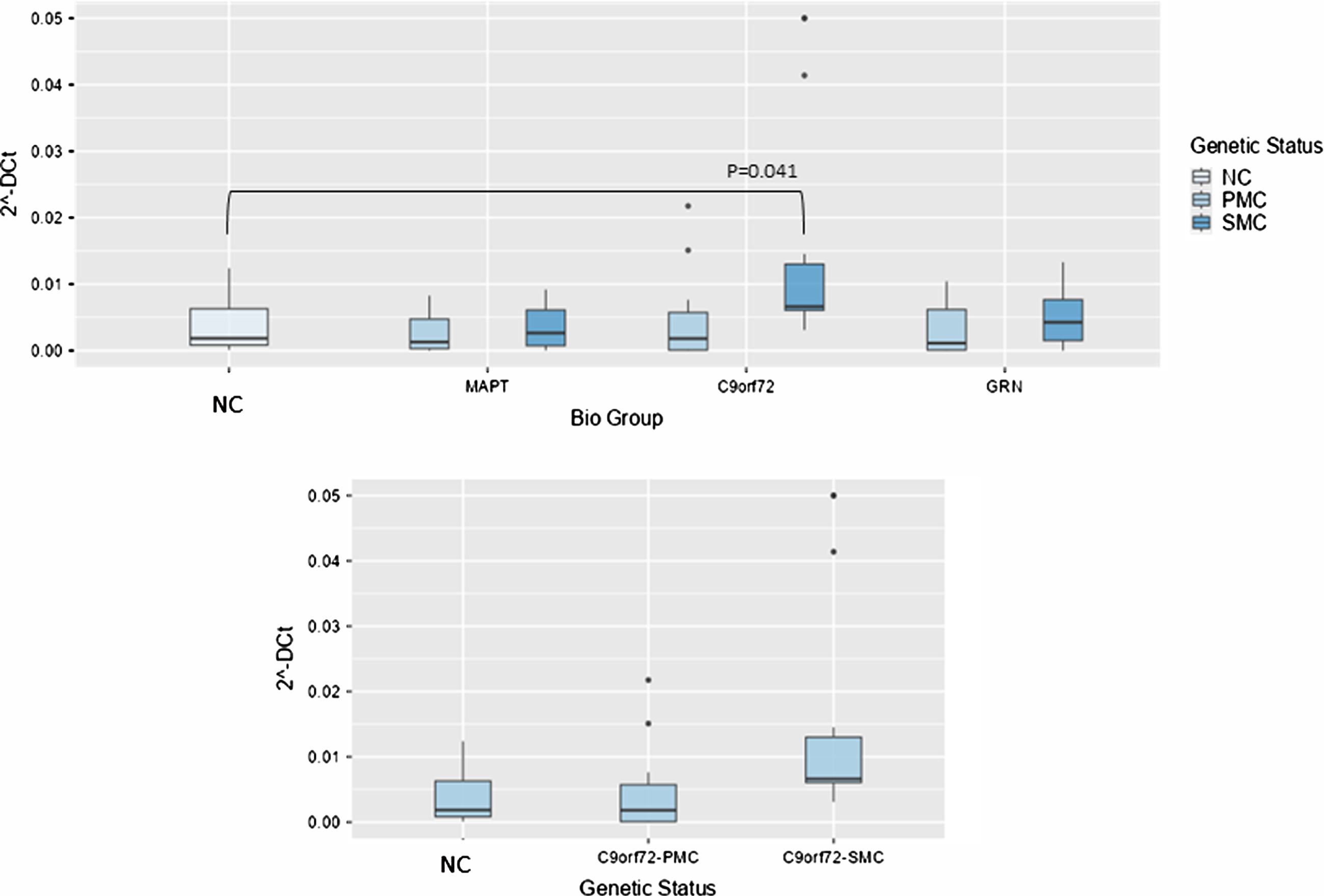
Concerning NORAD analysis, the overall model was significant (P < 0.001), indicating substantial variability explained by these variables. Biological group significantly impacted 2–ΔCt levels (F(3,99) = 5.61, p = 0.001, ἠ2p=0.145), highlighting variability across NC, MAPT, C9ORF72, and GRN groups. Post hoc tests revealed that NORAD was significantly more abundant in C9ORF72 biological group as compared with NC (0.014±0.01 versus 0.0038±0.00391-fold regulation, t(99) = –2.49, p = 0.041), but stratifying according to genetic status, the comparison between NC and C9ORF72 SMC showed a trend towards significance (p = 0.065), suggesting potentially meaningful differences (Fig. 3).
Fig. 3
Box plots of GAS5 profiling in PBMC from SMC and PMC carrying mutations in C9ORF72, GRN, MAPT as compared with NC. Data are expressed as 2-ΔCt fold regulation.
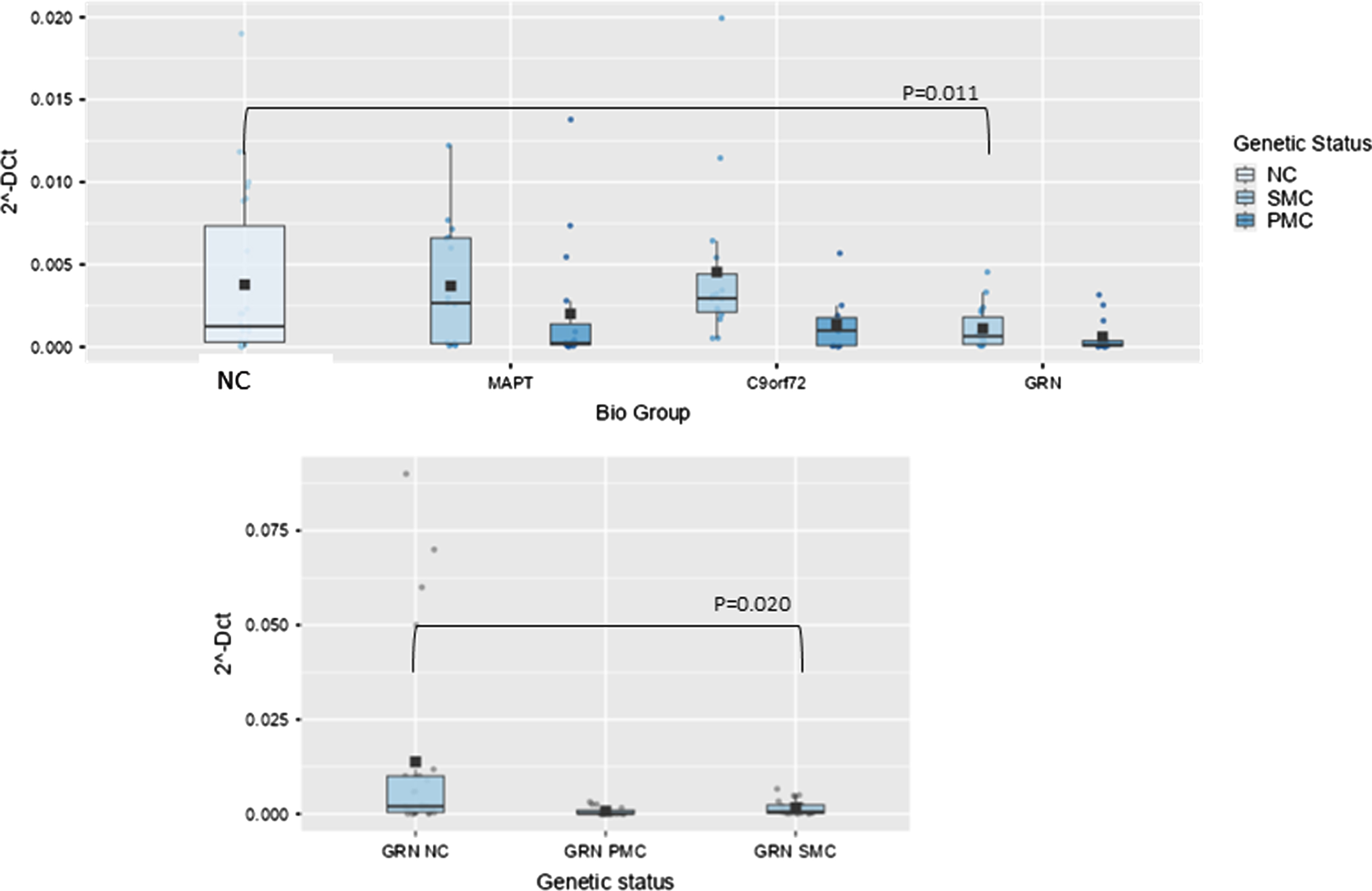
In GRN biological group, one lncRNA, namely Growth Arrest Specific (GAS)5 was significantly lower than NC. The overall model was significant, indicating that the predictors collectively explain a significant portion of the variance in the dependent variable (p < 0.001). Age had a significant effect (p < 0.001), as did the biological group (F(3,105) = 3.65, p = 0.015, ἠ2p=0.095). However, nor did the interaction between biological group and age (p = 0.142). Post hoc comparisons using the Tukey test revealed that the GRN group significantly differed from NC (9.20×10–4 ± 0.0011 versus 0.037×10–2 ± 0.1×10–3, fold regulation, t(105) = 3.14, p = 0.025). In GRN biological group GAS5 was significantly lower, also stratifying results according to genetic status and after post hoc analysis (SMC versus NC; 0.00164±0.0019 versus 0.013±0.024, fold regulation, t(55) = 2.78, p = 0.020, Fig. 3).
Conversely, no significant lncRNA deregulations were observed in MAPT carriers, both SMC and PMC, versus NC.
No correlations between disease duration in symptomatic individuals and lncRNA relative expression was found (p > 0.05).
Considering NfL and GFAP levels, as expected, significant increases were observed between SMC and NC: NfL mean levels±SEM were 56.05±3,81 versus 8.53±0.18 pg/ml, p = 0.002 and GFAP were 175.44±9.32 versus 103.15±10.21 pg/ml, p < 0.0001, respectively (Table 1). These differences in mean values remained significant even when stratifying symptomatic patients by the mutated gene (p < 0.001).
Considering instead the levels of NfL or GFAP and the lncRNAs, no significant correlations emerged (p > 0.05).
DISCUSSION
Herein, we showed that the relative expression of NORAD and NEAT1 lncRNAs was higher in C9ORF72 SMC, whereas relative levels of GAS5 were less abundant in GRN SMC. No significant differences were instead observed in PMC, although this group was very heterogenous as regards the age at sampling, and many subjects were still far from the mean age at disease onset in their families. Therefore, we cannot rule out whether the change observed in SMC occur in proximity of symptom manifestation.
LncRNA NORAD modulates the genome stability. It is expressed in cells after DNA damage and, when deleted, it expresses chromosomal instability and aneuploidy.14 Notably, C9ORF72 expansions are known to cause R-loops, in turn increasing genomic instability and DNA damage, and generate dipeptide repeat proteins, that lead to DNA damage and impairment of the DNA damage response.15 Therefore, DNA damage may play a role in the pathogenesis of the disease in carriers of the hexanucleotide expansion. Here, we considered only patients with FTD, therefore we cannot rule out whether the same modifications are present also in carriers developing ALS. It is interesting to note that dendritic cells isolated from C9ORF72–/– mice showed a marked early activation of the type I interferon response, and C9ORF72–/– myeloid cells were selectively hyperresponsive to activators of the stimulator of interferon genes (STING) protein, key regulator of the innate immune response.16
As regards lncRNA NEAT1, in samples from patients with AD, it was shown that NEAT1 expression is upregulated in different brain regions related to the disease.17 Nevertheless, there are still controversial data on the role of this lncRNA (damaging versus protective) in AD and other neurodegenerative diseases. (see18 for review)
LncRNA GAS5 has been shown to be downregulated in brain samples from patients with AD compared to age-matched healthy controls.19 In line with this observation, a previous human transcriptomic analysis and microarray data of six brain regions from AD patients also showed that GAS5 is downregulated in AD.20
In a previous study, circulating levels of lncRNA were evaluated but, despite trends toward increased or decreased levels were observed, no significant data were obtained.11 It has to be considered however that the source of circulating lncRNA cannot be determined and that there are no study comparing PBMC and circulating lncRNA levels in the same individuals.
A strength of the study is represented by NC from the same families, reducing the genetic background variability. A weakness of the study is instead that we did not compare the same subjects from the asymptomatic to the symptomatic phase. As regards the longitudinal analysis of PMC, the follow up of participants in GENFI is ongoing and these findings could be confirmed longitudinally in the next future.
Lastly, it has to be acknowledged that, despite the extracted RNA being of good quality for subsequent expression analysis, several long non-coding RNAs were not detected. The lack of detection of long non-coding RNAs (lncRNAs) in blood can be attributed to several factors.
Firstly, the presence of ribonuclease enzymes in blood can lead to the rapid degradation of RNA molecules, including lncRNAs, making them difficult to detect.21
Additionally, many lncRNAs are expressed at intrinsically low levels, which may render them below the detection limit of standard real time PCR.22 Another significant factor is the tissue-specific expression of many lncRNAs: if a lncRNA is predominantly expressed in specific tissues such as the brain or liver, it is unlikely to be detected in the blood.23 RNA stability is another crucial element to consider, as some lncRNAs may be inherently unstable in blood, despite protective mechanisms such as encapsulation in exosomes or association with proteins, leading to their rapid degradation.24 These combined factors can explain the difficulty in detecting lncRNAs in blood. Therefore, we cannot rule out whether these lncRNAs play a role in neurodegeneration, and the present results need to be validated in an independent cohort of patients, possibly also including sporadic cases.
Conclusions
Relative expression of NORAD and NEAT1 lncRNAs is more abundant in PBMC from C9ORF72 SMC, whereas relative transcription of GAS5 is less abundant in PBMC from GRN SMC, whereas no significant changes have been observed in PMC. This study emphasizes the importance of lncRNAs in FTD and their potential for guiding genetic stratification and developing biomarkers. Further validation and mechanistic investigations are essential to facilitate clinical application.
AUTHOR CONTRIBUTIONS
Serpente Maria (Data curation; Methodology; Validation; Writing – review & editing); Chiara Fenoglio (Conceptualization; Data curation; Formal analysis; Writing – review & editing); Marina Arcaro (Investigation; Methodology); Tiziana Carandini (Resources); Luca Sacchi (Resources); Manuela Pintus (Resources); Emanuela Rotondo (Project administration; Resources); Vittoria Borracci (Project administration; Resources); Laura Ghezzi (Resources); Arabella Bouzigues (Project administration; Resources); Lucy Russel (Project administration; Resources); Phoebe Foster (Resources); Eve Ferry-Bolder (Project administration; Resources); John van Swieten (Resources); Lize Jiskoot (Resources); Harro Seelaar (Resources); Raquel Sanchez-Valle (Resources); Robert laforce (Resources); Caroline Graff (Resources); Rik Vandenberghe (Resources); Alexandre de Mendonca (Resources); Pietro Tiraboschi (Resources); Isabel Santana (Resources); Alexander Gerhard (Resources); Johannes Levin (Resources); Sandro Sorbi (Resources); Markus Otto (Resources); Florence Pasquier (Resources); Simon Ducharme (Resources); Chris Butler (Resources); Isabelle Le Ber (Resources); Elizabeth Finger (Resources); Maria Carmela Tartaglia (Resources); Mario Masellis (Resources); James Rowe (Resources); Matthis Synofzik (Resources); Fermin Moreno (Resources); Barbara Borroni (Resources); Jonathan Rohrer (Project administration; Supervision); Andrea Arighi (Supervision; Writing – review & editing); Daniela Galimberti (Conceptualization; Project administration; Supervision; Writing – original draft; Writing – review & editing).
ACKNOWLEDGMENTS
We would like to thank the participants and their families for contributing to the study.
FUNDING
DG was supported by grants from the Italian Ministry of Health (Ricerca Corrente) and Fondazione Gigi & Pupa Ferrari Onlus. MS is supported by the Italian Ministry of Health, grant GR-2019-12369100. JCVS, LCJ and HS was supported by the Dioraphte Foundation grant 09-02-03-00, Association for Frontotemporal Dementias Research Grant 2009, Netherlands Organization for Scientific Research grant HCMI 056-13-018, ZonMw Memorabel (Deltaplan Dementie, project number 733051042), ZonMw Onderzoeksprogramma Dementie (YOD-INCLUDED, project number10510032120002), EU Joint Programme-Neurodegenerative Disease Research-GENFI-PROX. Alzheimer Nederland and the Bluefield Project. RS-V. is supported by Alzheimer’s Research UK Clinical Research Training Fellowship (ARUK-CRF2017B-2) and has received funding from Fundació Marató de TV3, Spain (grant no. 20143810). C.G. received funding from EU Joint Programme-Neurodegenerative Disease Research-Prefrontals Vetenskapsrådet Dnr 529-2014-7504, EU Joint Programme-Neurodegenerative Disease Research-GENFI-PROX, Vetenskapsrådet 2019-0224, Vetenskapsrådet 2015-02926, Vetenskapsrådet 2018-02754, the Swedish FTD Inititative-Schörling Foundation, Alzheimer Foundation, Brain Foundation, Dementia Foundation and Region Stockholm ALF-project. R.V. has received funding from the Mady Browaeys Fund for Research into Frontotemporal Dementia. JL received funding for this work by the Deutsche Forschungsgemeinschaft German Research Foundation under Germany’s Excellence Strategy within the framework of the Munich Cluster for Systems Neurology (EXC 2145 SyNergy—ID 390857198). MO has received funding from Germany’s Federal Ministry of Education and Research (BMBF). EF has received funding from a Canadian Institute of Health Research grant #327387. MM has received funding from a Canadian Institute of Health Research operating grant and the Weston Brain Institute and Ontario Brain Institute. JBR has received funding from the Wellcome Trust (103838) and is supported by the Cambridge University Centre for Frontotemporal Dementia, the Medical Research Council (SUAG/051 G101400) and the National Institute for Health Research Cambridge Biomedical Research Centre (BRC-1215-20014). FM is supported by the Tau Consortium and has received funding from the Carlos III Health Institute (PI19/01637). JDR is supported by the Bluefield Project and the National Institute for Health and Care Research University College London Hospitals Biomedical Research Centre and has received funding from an MRC Clinician Scientist Fellowship (MR/M008525/1) and a Miriam Marks Brain Research UK Senior Fellowship. Several authors of this publication (JCVS, MS, RV, AdM, MO, RV and JDR) are members of the European Reference Network for Rare Neurological Diseases (ERN-RND)—Project ID No 739510. This work was also supported by the EU Joint Programme—Neurodegenerative Disease Research GENFI-PROX grant (2019-02248; to JDR, MO, BB, CG, JCVS and MS. RL is supported by the Canadian Institutes of Health Research and theChaire de Recherche sur les Aphasies Primaires Progressives Fondation Famille Lemaire.
CONFLICT OF INTEREST
Chiara Fenoglio, Barbara Borroni, and Daniela Galimberti are Editorial Board Members of this journal but were not involved in the peer-review process of this article nor had access to any information regarding its peer-review.
All other authors have no conflict of interest to report.
DATA AVAILABILITY
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JAD-240557.
REFERENCES
1. | Mattick JS , Amaral PP , Carninci P , et al. Long non-coding RNAs: Definitions, functions, challenges and recommendations. Nat Rev Mol Cell Biol (2023) ; 24: : 430–447. |
2. | Iyer MK , Niknafs YS , Malik R , et al. The landscape of long noncoding RNAs in the human transcriptome. Nat Genet (2015) ; 47: : 199–208. |
3. | Faghihi MA , Modarresi F , Khalil AM , et al. Expression of a noncoding RNA is elevated in Alzheimer’s disease and drives rapid feed-forward regulation of beta-secretase. Nat Med (2008) ; 14: : 723–730. |
4. | Li H , Zheng L , Jiang A , et al. Identification of the biological affection of long noncoding RNA BC200 in Alzheimer’s disease. Neuroreport (2018) ; 29: : 1061–1067. |
5. | Teixeira LCR , Mamede I , Luizon MR , et al. Role of long non-coding RNAs in the pathophysiology of Alzheimer’s disease and other dementias. Mol Biol Rep (2024) ; 51: : 270. |
6. | Pottier C , Ravenscroft TA , Sanchez-Contreras M , et al. Genetics of FTLD: Overview and what else we can expect from genetic studies. J Neurochem (2016) ; 138: (Suppl 1), 32–53. |
7. | Swift IJ , Rademakers R , Finch N , et al. A systematic review of progranulin concentrations in biofluids in over 7,000 people-assessing the pathogenicity of GRN mutations and other influencing factors. Alzheimers Res Ther (2024) ; 16: : 66. |
8. | Rademakers R , Neumann M , Mackenzie IR . Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol (2012) ; 8: : 423–434. |
9. | Galimberti D , Fenoglio C , Serpente M , et al. Autosomal dominant frontotemporal lobar degeneration due to the C9ORF72 hexanucleotide repeat expansion: Late-onset psychotic clinical presentation. Biol Psychiatry (2013) ; 74: : 384–391. |
10. | Riva P , Ratti A , Venturin M . The long non-coding RNAs in neurodegenerative diseases: Novel mechanisms of pathogenesis. Curr Alzheimer Res (2016) ; 13: : 1219–1231. |
11. | Fenoglio C , Serpente M , Visconte C , et al. Circulating non-coding RNA levels are altered in autosomal dominant frontotemporal dementia. Int J Mol Sci (2022) ; 23: : 14723. |
12. | Rohrer JD , Nicholas JM , Cash DM , et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal dementia Initiative (GENFI) study: A cross-sectional analysis. Lancet Neurol (2015) ; 14: : 253–262. |
13. | Heller C , Foiani MS , Moore K , et al. Plasma glial fibrillary acidic protein is raised in progranulin-associated frontotemporal dementia. J Neurol Neurosurg Psychiatry (2020) ; 91: : 263–270. |
14. | Huarte M . The emerging role of lncRNAs in cancer. Nat Med (2015) ; 21: : 1253–1261. |
15. | Kok JR , Palminha NM , Dos Santos Souza C , et al. DNA damage as a mechanism of neurodegeneration in ALS and a contributor to astrocyte toxicity. Cell Mol Life Sci (2021) ; 78: : 5707–5729. |
16. | McCauley ME , O’Rourke JG , Yáñez A , et al. C9orf72 in myeloid cells suppresses STING-induced inflammation. Nature (2020) ; 585: : 96–101. |
17. | Wu J , Chen L , Zheng C , et al. Co-expression network analysis revealing the potential regulatory roles of lncRNAs in Alzheimer’s disease. Interdiscip Sci (2019) ; 11: : 645–654. |
18. | Li K , Wang Z . lncRNA NEAT Key player in neurodegenerative diseases. Ageing Res Rev (2023) ; 86: : 101878. |
19. | Patel RS , Lui A , Hudson C , et al. Small molecule targeting long noncoding RNA GAS5 administered intranasally improves neuronal insulin signaling and decreases neuroinflammation in an aged mouse model. Sci Rep (2023) ; 13: : 317. |
20. | Liang WS , Dunckley T , Beach TG , et al. Gene expression profiles in anatomically and functionally distinct regions of the normal aged human brain. Physiol Genomics (2007) ; 28: : 311–322. |
21. | Statello L , Guo C-J , Chen L-L , et al. Gene regulation by long non-coding RNAs and its biological functions. Nat Rev Mol Cell Biol (2021) ; 22: : 96–118. |
22. | Dehling E , Volkmann G , Matern JCJ , et al. Mapping of the communication-mediating interface in nonribosomal peptide synthetases using a genetically encoded photocrosslinker supports an upside-down helix-hand motif. J Mol Biol (2016) ; 428: : 4345–4360. |
23. | Baldock RA , Day M , Wilkinson OJ , et al. ATM localization and heterochromatin repair depend on direct interaction of the 53BP1-BRCT2 domain with γH2AX. Cell Rep (2015) ; 13: : 2081–2089. |
24. | Managadze D , Rogozin IB , Chernikova D , et al. Negative correlation between expression level and evolutionary rate of long intergenic noncoding RNAs. Genome Biol Evol (2011) ; 3: : 1390–1404. |


