
Towards a Unitary Hypothesis of Alzheimer’s Disease Pathogenesis
Abstract
The “amyloid cascade” hypothesis of Alzheimer’s disease (AD) pathogenesis invokes the accumulation in the brain of plaques (containing the amyloid-β protein precursor [AβPP] cleavage product amyloid-β [Aβ]) and tangles (containing hyperphosphorylated tau) as drivers of pathogenesis. However, the poor track record of clinical trials based on this hypothesis suggests that the accumulation of these peptides is not the only cause of AD. Here, an alternative hypothesis is proposed in which the AβPP cleavage product C99, not Aβ, is the main culprit, via its role as a regulator of cholesterol metabolism. C99, which is a cholesterol sensor, promotes the formation of mitochondria-associated endoplasmic reticulum (ER) membranes (MAM), a cholesterol-rich lipid raft-like subdomain of the ER that communicates, both physically and biochemically, with mitochondria. We propose that in early-onset AD (EOAD), MAM-localized C99 is elevated above normal levels, resulting in increased transport of cholesterol from the plasma membrane to membranes of intracellular organelles, such as ER/endosomes, thereby upregulating MAM function and driving pathology. By the same token, late-onset AD (LOAD) is triggered by any genetic variant that increases the accumulation of intracellular cholesterol that, in turn, boosts the levels of C99 and again upregulates MAM function. Thus, the functional cause of AD is upregulated MAM function that, in turn, causes the hallmark disease phenotypes, including the plaques and tangles. Accordingly, the MAM hypothesis invokes two key interrelated elements, C99 and cholesterol, that converge at the MAM to drive AD pathogenesis. From this perspective, AD is, at bottom, a lipid disorder.
THE PROBLEM
Alzheimer’s disease (AD) is the most common, and arguably the most devastating, neurodegenerative dementia of the elderly [1]. The two main histopathological hallmarks of AD are the accumulation of extracellular neuritic plaques containing amyloid-β (Aβ) and of intracellular neurofibrillary tangles consisting mainly of hyperphosphorylated forms of the microtubule-associated protein tau. The vast majority of AD is sporadic (SAD), but a genetic variant of apolipoprotein E (APOE), APOE4, predisposes people to the disease [2]. Mutations in three genes have been identified in the autosomal-dominant familial form (FAD): amyloid-β precursor protein (APP), presenilin-1 (PS1; gene PSEN1), and presenilin-2 (PS2; gene PSEN2). Presenilins are components of the γ-secretase complex that, together with α- and β-secretase (BACE1), processes AβPP [3].
In the “non-amyloidogenic” pathway (Fig. 1A, left), full-length AβPP (∼700 aa) is first cleaved by α-secretase at the plasma membrane (PM) to produce a long soluble N-terminal fragment (NTF; sAβPPα) and a short membrane-bound 83-aa C-terminal fragment (CTF), called C83 (α-CTF). C83 is then cleaved by the γ-secretase complex to produce two peptides, P3 and the AβPP intracellular domain (AICD), a nucleus-targeted transcription factor. In the alternative “amyloidogenic” pathway (Fig. 1A, right), full-length AβPP is first cleaved by BACE1 within endosomes to produce a slightly shorter soluble N-terminal fragment (sAβPPβ), and a correspondingly slightly longer 99-aa membrane-bound C-terminal fragment, called C99 (β-CTF) [4]. C99 is then cleaved by the γ-secretase complex to produce two peptides, Aβ (predominantly 40-aa long [Aβ40]) and AICD [5].
Fig. 1
AβPP processing. A) Processing intermediates. Yellow dot, cholesterol-binding domain (CBD); orange dot, sphingolipid-binding domain (SBD). Adapted from [3], with permission (doi.org/10.1016/j.nbd.2020.105062). B) Human C99 sequence; important residues for SBD [149] and CBD (based on [117] and [118]) binding, and the YENPTY motif, in bold. Cleavage sites for α-, β-, and γ-secretase are indicated (arrows). See text for details.
![AβPP processing. A) Processing intermediates. Yellow dot, cholesterol-binding domain (CBD); orange dot, sphingolipid-binding domain (SBD). Adapted from [3], with permission (doi.org/10.1016/j.nbd.2020.105062). B) Human C99 sequence; important residues for SBD [149] and CBD (based on [117] and [118]) binding, and the YENPTY motif, in bold. Cleavage sites for α-, β-, and γ-secretase are indicated (arrows). See text for details.](https://content.iospress.com:443/media/jad/2024/98-4/jad-98-4-jad231318/jad-98-jad231318-g001.jpg)
In the most widely-accepted pathogenetic model of AD, both SAD and FAD arise when AβPP is misprocessed to produce a slightly longer, aggregation-prone, form of Aβ (Aβ42) that accumulates in the plaques [1]. The Aβ is toxic to cells, and the resulting stress stimulates tau hyperphosphorylation, leading to the tangles. The senile plaques are proposed to be the cause of the disease. The overall process has been called the “amyloid cascade” [1, 6, 7]. However, there are other data that challenge this perspective. For example, plaques are prevalent in the brains of aged subjects without dementia [8]. Moreover, an AD-protective isoform of APOE3, known as the “Christchurch” variant, was recently shown to delay AD symptoms in an FADPS1 patient in spite of unusually high brain plaque levels [9].
Numerous in vitro studies have shown that high levels of soluble or oligomerized Aβ can induce cell toxicity through multiple mechanisms, such as membrane damage, mitochondrial dysfunction, and tau phosphorylation [10]. However, these results could not be replicated in animal models of AD [11], casting doubt on what looked initially to be a straightforward hypothesis. Nevertheless, the field continued to produce data further cementing amyloid’s putative role as the main cause of neuronal death in AD, which, in turn, stimulated a search for compounds to inhibit Aβ production [12]. Disappointingly, most anti-Aβ strategies and the use of BACE1 inhibitors [13] have had limited success in ameliorating clinical outcome [14], and γ-secretase inhibitors, rather than improving disease outcomes, made them worse [15].
IMPLICATIONS OF THE AMYLOID CASCADE
When amyloid was observed to be abundant in plaques in AD brains, the logical conclusion was that its production was augmented, and that a pathological hallmark as conspicuous as plaques had to be relevant to the disease mechanism. Later, the discovery that γ-secretase is the only enzyme responsible for Aβ production [16] led some to the conclusion that its activity in AD was increased [17], resulting in toxic higher concentrations of Aβ that were deposited in the plaques, and became the source of the idea that AD pathogenesis was caused by altered γ-secretase enzymatic function [18].
However, it is important to note that the efficiency of an enzyme’s function is defined by the rate at which it converts substrate (e.g., C99) to product(s) (e.g., Aβ + AICD). Thus, measuring only the levels of the amount of product as the readout of enzyme activity can lead to misinterpretations. For example, if a given enzyme under normal conditions consumes 1 unit of substrate to produce 1 unit of product per minute (i.e., a conversion efficiency of 1.0), producing 1.5 units of product in a minute could be interpreted as a 50% increase in enzymatic activity. Yet, if those 1.5 units of product were derived from 2 units of substrate (i.e., an efficiency of 0.75), we would conclude the opposite, namely, a 25% decrease in enzyme activity.
It is true that in AD altered AβPP cleavage causes an increase in total Aβ and a tendency towards the production of longer neurotoxic species of amyloid (e.g., Aβ42 rather than Aβ40) that are more prone to aggregate into senile plaques [19]. But it is also true that while elevations in Aβ are highly variable among patients, AD tissues typically present with significant increases in the levels of C99 (the substrate for Aβ production) compared to the total amount of amyloid produced [20–22], a result consistent with the idea that γ-secretase activity in most AD patients is decreased, not increased [23]. For example, using an in vitro reconstitution assay admittedly prone to artefacts [24, 25], Sun et al. showed that ∼90% of 138 FAD mutations in PS1 presented with increases in Aβ42:Aβ40 ratio but had reduced total amyloid production [26] (but to be fair, ∼10% showed the opposite result [26, 27], implying that different γ-secretase activities are associated with different PS1 mutations [28]). However, as noted above, enzymatic activity should be reported at the ratio of product:substrate; merely reporting the product (e.g., ratio of Aβ42:Aβ40 [26]), does not say anything about the activity of γ-secretase. Similar results, although not the goal of those studies, were found in other reports [29].
As a result, we can define AD not only by an elevated ratio of longer:shorter Aβ species [30], but also by an increased ratio of C99:total Aβ [31]. This implies that in AD, defects in γ-secretase result in reductions both in the quantity of cleaved product (i.e., Aβ) relative to the amount of available C99 and in the “quality” of its processing towards longer, more aggregation-prone, Aβ species. From this point of view, we can redefine AD as a partial loss of function of γ-secretase [32–35] due to inherited genetic mutations in the case of FAD [36–38], or to undetermined causes in the case of SAD. Based on this, the buildup of uncut C99 could be considered an early pathological hallmark that could elicit many of the molecular features seen in AD cells [28]. This is hardly a new idea, as C99 has been suggested to be a pathogenic trigger of disease by many groups [39–42]. For example, some authors have shown that in animal models of AD with pathogenic mutations in PSEN1 or APP, C99 accumulation occurs in AD-relevant brain areas (cortex and hippocampus) at early stages of development, long before the appearance of any detectable increases in Aβ [39, 40, 42]. These data also highlight the contribution of C99 accumulation to some of the symptoms of AD [32, 41], including endosomal dysfunction, cognitive impairment, and hippocampal degeneration [21, 43–45]. Interestingly, other groups found C99 to be especially toxic to neurons [46, 47], and that the accumulation of C99, rather than alterations in Aβ or AICD, caused symptoms of dementia [41, 48].
HOW DOES ELEVATED C99 RESULT IN LONGER Aβ SPECIES?
As mentioned above, C99 is cleaved in an intracellular compartment by γ-secretase to produce two peptides, Aβ and AICD (Fig. 1). It is important to note that γ-secretase is an unusual “rhomboid-like” protease that cleaves substrates located within membrane domains [49], especially within lipid rafts [50]. Lipid rafts are transient membrane domains formed by local increases in cholesterol, sphingomyelin, and saturated phospholipids, resulting in a more rigid and “liquid-ordered” membrane “island” (i.e., the raft) located within the surrounding “liquid-disordered” membrane [51, 52]. These local elevations in cholesterol (by ∼45% in mouse embryonic fibroblasts [MEFs] [53]) and sphingomyelin (by ∼40% in mouse liver [54]) create highly ordered membrane microdomains that passively segregate and enrich for lipid-binding proteins, thereby facilitating protein-protein interactions for the regulation of numerous signaling pathways [52]. Moreover, the membrane of a lipid raft is thicker than the surrounding liquid-disordered region [55, 56], due mainly to local increases in straight and long saturated phospholipid acyl chains [55–57]. This modification in the structure of the membrane induces changes in the conformation of those transmembrane (TM) proteins embedded within them and modulates their enzymatic activities. Relevant to AD, it has long been known that γ-secretase activity resides in lipid rafts [58, 59], and in fact it has now been shown that these rafts are located intracellularly, in subdomains of the ER [60–62]. Moreover, increases in intracellular cholesterol are known to induce the association between C99 and γ-secretase in membranes, and are required to activate the production of Aβ [63]. Importantly, γ-secretase cleavage is activated by the formation of these lipid rafts and by the regulation of their lipid composition [64].
Equally important, cleavage by γ-secretase is not sequence-specific: it “cuts what it sees” [65] (Fig. 2). Under normal circumstances, following formation of the lipid raft, γ-secretase processes C99 in a “product line” consisting of an initial cleavage event followed by sequential ∼3-aa “bites” to produce a distribution of successively shorter fragments, ranging in length from ∼36 - ∼48 amino acids (i.e., Aβ36-Aβ48) [30, 66–70]. Under these conditions, there is “hydrophobic matching” between the length of C99’s α-helical transmembrane domain [71] and the thickness of the [lipid-raft] membrane in which it resides [72]. In normal cells, the initial cleavage of C99 by γ-secretase [73] favors the production of Aβ40 (Fig. 2A). In AD cells, however, the lipid composition of the raft is altered by increases in the levels of ceramide derived from elevated sphingomyelinase activity [35], which generates a significantly longer [74] and slightly thinner [75–77] lipid raft (but still thicker than the surrounding “free” ER (i.e., our term for ER not apposed to other intracellular organelles) [78–80]), forcing the C99 TM domain to maintain hydrophobic matching by tilting [73, 81, 82]. This tilt changes the alignment of C99 with γ-secretase and alters the positioning/conformation of the initial γ-secretase cleavage site [24, 83, 84] on C99 such that more Aβ42 and less Aβ40 is produced (Fig. 2B) [85–87]. This model is consistent with an altered lipid milieu playing a role in determining the physical apposition of PS1 to C99 [81], and implies that the A β42:A β40 ratio in AD is fundamentally a surrogate marker of the thickness of the membrane in which C99 cleavage occurs [88, 89]. It turns out that Aβ42 happens to have the unfortunate property of being more aggregation-prone, more prevalent in plaques, and more toxic to neurons than is Aβ40 [90], influencing the field to focus on Aβ42 more as a pathogenic culprit and therapeutic target while focusing less on its biophysical implications.
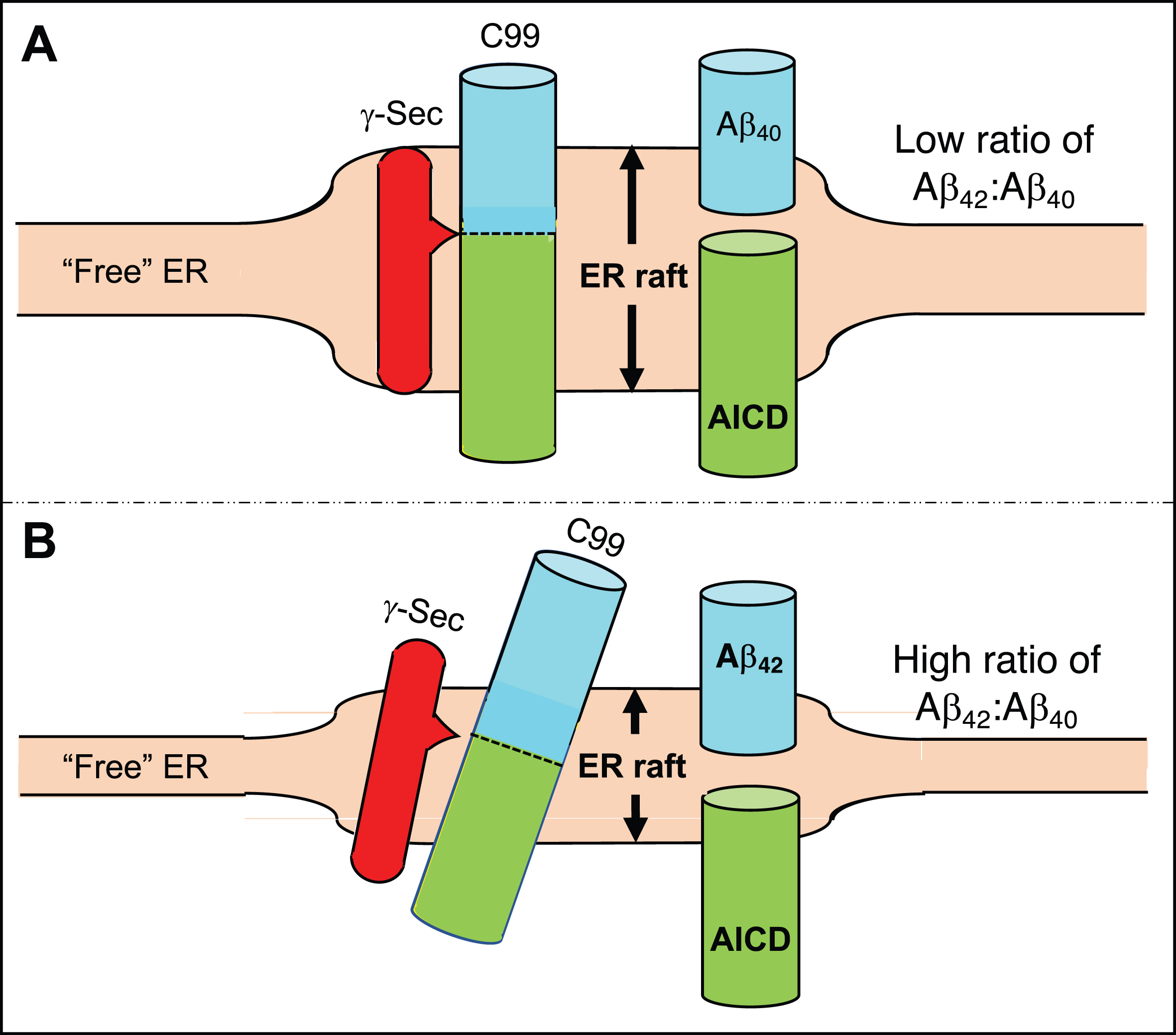
Fig. 2
Generation of Aβ40 versus Aβ42. A) In the normal situation the γ-secretase complex aligns with C99 in the ER raft such that its initial cleavage favors a product line that generates Aβ40, and the Aβ42:Aβ40 ratio is relatively low. B) In AD, the ER raft is thinner than normal (shorter double-headed arrow) but is still thicker than the “free” ER. Hydrophobic matching of C99’s α-helical region within the thinner membrane causes it (and perhaps the γ-secretase complex as well) to tilt, resulting in the initial cleavage of C99 displaced by ∼2 amino acids, generating a higher Aβ42:Aβ40 ratio. See text for details.
C99 REGULATES Aβ PRODUCTION
Pathogenic mutations in AβPP [33, 91] and PS1 [92] promote an increase in the Aβ42:Aβ40 ratio associated with increased levels of C99, suggesting that, ironically, the accumulation of C99 is to blame for the decrease in the ability of γ-secretase to cleave its own substrate, namely C99 itself. In support of this idea, in vitro studies have shown that upon exceeding a concentration threshold, C99 peptides associate with each other in membranes [69], inducing a conformational change in them that precludes, or at least delays, their cleavage by γ-secretase [25, 93]. This decrease in enzymatic activity by substrate saturation could explain why inhibition of γ-secretase favors the accumulation of C99 fragments [94]. Taken together, we can conclude that the degree of C99 accumulation is a major determinant of amyloid production via the modulation of its own processing. The mechanism by which C99 affects its own cleavage, ultimately resulting in an increased Aβ42:Aβ40 ratio, is addressed below.
ELEVATED LEVELS OF C99 ARE A LIKELY CAUSE OF AD
Based on the above analysis, we propose that increased C99 is a more accurate indicator of disease progression than is increased amyloid or plaque burden. The correlations between Aβ production, Aβ42:Aβ40 levels, and plaque/tangle burden and the progression of dementia are not significant enough to blame any one of these as the sole cause or indicator of disease [95]. In fact, brain samples from cognitively normal older adults often show significant amyloid deposition [96]. On the other hand, many groups have shown a significant association between C99 levels and the appearance of cognitive impairment, both in animal models and in AD patients [97]. Moreover, an “AD-protective” mutation in AβPP (“Icelandic”) causes a substantial decrease not only in amyloid but also in C99. Conversely, some pathogenic mutations in PSEN1 associated with aggressive forms of AD [98] do not cause higher amyloid levels, senile plaques, or neurofibrillary tangle formation [99, 100].
This raises a conundrum: if C99 is so important in AD pathogenesis, why hasn’t the field come to the conclusion that C99, and not Aβ, is the culprit, or at least concluded that γ-secretase activity is reduced, not increased, in AD? One explanation is that experimental limitations, as well as the definition of AD itself, have contributed to difficulties in data interpretation.
With respect to experimental design, we note that C99 encompasses Aβ (at its N-terminal “half”) and AICD (at its C-terminal “half”), and therefore contains the same [partial and/or overlapping] amino acid sequences as are present on both fragments (see Fig. 1). Thus, in the absence of detailed controls, it is easy to envision difficulties in interpreting experimental data that might unequivocally implicate any particular AβPP fragment. In fact, many antibodies used to detect Aβ in cells or tissues by imaging approaches or by enzyme-linked immunosorbent assay (ELISA) cannot discriminate between C99 and other AβPP fragments [101], a pitfall that raises the interesting possibility that, guided by the amyloid cascade hypothesis, some histological studies had interpreted elevations in C99 as deposition of Aβ [102, 103].
Second, AD is defined operationally: to receive a diagnosis of AD requires the presence of a threshold number of plaques and tangles in postmortem brain samples from patients with cognitive impairment [104]. Therefore, those cases diagnosed with AD-like dementia, but presenting with low numbers of extracellular deposits of Aβ, will not be diagnosed as AD after histological analysis, even if they carry mutations in PSEN1 [36, 105, 106].
That C99, and not Aβ, is a trigger of disease can also explain why treatments aimed at inhibiting γ-secretase activity or minimizing Aβ production/size have yielded disappointing results [107]. However, at this point, a logical question arises: if C99 is so bad, why have anti-BACE1 therapies (that prevent C99 formation in the first place) not been successful either? There are really only two responses to this question: (1) The C99 idea is wrong, or (2) C99 and/or the regulation of AβPP cleavage to produce the “right” amount of C99 is necessary to fulfill a currently-unknown cellular function. Which brings us to the question of:
WHAT IS THE FUNCTION OF C99?
Full-length AβPP has been proposed to play a role in neurite outgrowth [108] and cell signaling [109], while the C99 fragment has been shown to modulate voltage-gated potassium channels [110] and to play a role in mitophagy [111]. However, the most frequent and supported hypothesis for the normal function of AβPP (and by inference, of C99) is that it is associated with the regulation of cellular cholesterol [112]. The expression, processing, and activity of AβPP are modulated by changes in the levels and distribution of cholesterol [113, 114]. Conversely, alterations in the expression, distribution, and cleavage of AβPP results in changes in cholesterol homeostasis [115]. Extensive data suggest that one function of AβPP could be to serve as a cholesterol sensor [116] and/or a regulator of cholesterol distribution in the cell [35, 53]. To make the connection between AβPP and cholesterol even more direct, AβPP contains a cholesterol-binding domain (CBD; yellow dot in Fig. 1A and highlighted sequence in Fig. 1B) consisting of tandem GxxxG motifs located within C99 immediately upstream of the γ-secretase cleavage site [116–119], and whose abrogation prevents AβPP’s and C99’s ability to regulate this lipid [53]. (The GxxxG motifs have also been proposed to play an alternative role, as a mediator of C99 homodimerization, but this is controversial [120, 121].) Thus, a likely role of C99 is to maintain cellular cholesterol homeostasis. But how is this accomplished?
Cholesterol is produced within animal cells, in the endoplasmic reticulum (ER) [122], but is also taken up from outside the cell, primarily from circulating lipoproteins containing free cholesterol and cholesteryl esters (CEs), with most of the cholesterol released to the plasma membrane [122]. Compared to the PM, the ER contains very low, but biologically important, levels of cholesterol [123]. For example, the amount of cholesterol in ER membranes regulates the activation of the sterol-regulatory element binding proteins (SREBP1 and 2), transcription factors that are localized to the ER membrane in an inactive state. When the level of intracellular cholesterol is low, SREBP is released and eventually travels to the nucleus, where it switches on genes, such as those encoding the low-density lipoprotein receptor (LDLR) and 3-hydroxy-3-methylglutary-CoA reductase (HMGCR), that cause the cell to generate more cholesterol. Conversely, when the level of ER cholesterol is high, SREBP remains within the ER and cannot translocate to the nucleus, thereby reducing the production of cholesterol by reducing HMGCR levels [124].
These pathways imply that normal cholesterol homeostasis in the cell is maintained by the crosstalk between the PM, where most of the cholesterol resides, and the ER, where most of the cholesterol-regulatory enzymes reside [124]. This PM-ER communication is a regulated process, with cholesterol trafficking mediated by both vesicular (e.g., via the LDL receptor and endosomes) [125] and non-vesicular (e.g., via Aster/GRAMD [GRAM domain-containing protein] proteins [126]) [127, 128] processes; we will focus here mainly on the vesicular pathway. Excess cholesterol in a PM-localized raft induces the invagination of the plasma membrane and the formation of a cholesterol-rich endosome that pinches off from the PM, enters the cell proper, and eventually fuses with a lysosome [129]. The lower pH in the resulting endolysosome induces the activation of cholesterol-regulatory enzymes, such as Niemann-Pick type C protein 1 (NPC1) [130], which contributes to the fusion of the endolysosome with the ER (likely via inter-organelle contact sites [131]) and the release of cholesterol to ER membranes [132].
We note in this context that problems in endosome function [133] and in lysosome acidification [134], driven by elevated levels of C99 [135], play a key role in AD pathogenesis [136]. In particular, C99 accumulates in lysosomes that are defective in acidification [134]. This result seems counter-intuitive, as elevated endolysosomal pH should decrease, not increase, BACE1 enzymatic activity and hence C99 production. Remarkably, however, the vacuolar ATP-synthase (v-ATPase) required for endosomal acidification is inhibited by phosphorylation of C99 (at Tyr-682 in the highly-conserved YENPTY motif [135] [see Fig. 1]). Thus, it is possible that the degree of BACE1-induced production of C99 [43, 137] is regulated by v-ATPase-mediated lysosome acidification via an equilibrium between phosphorylated and dephosphorylated C99 [138].
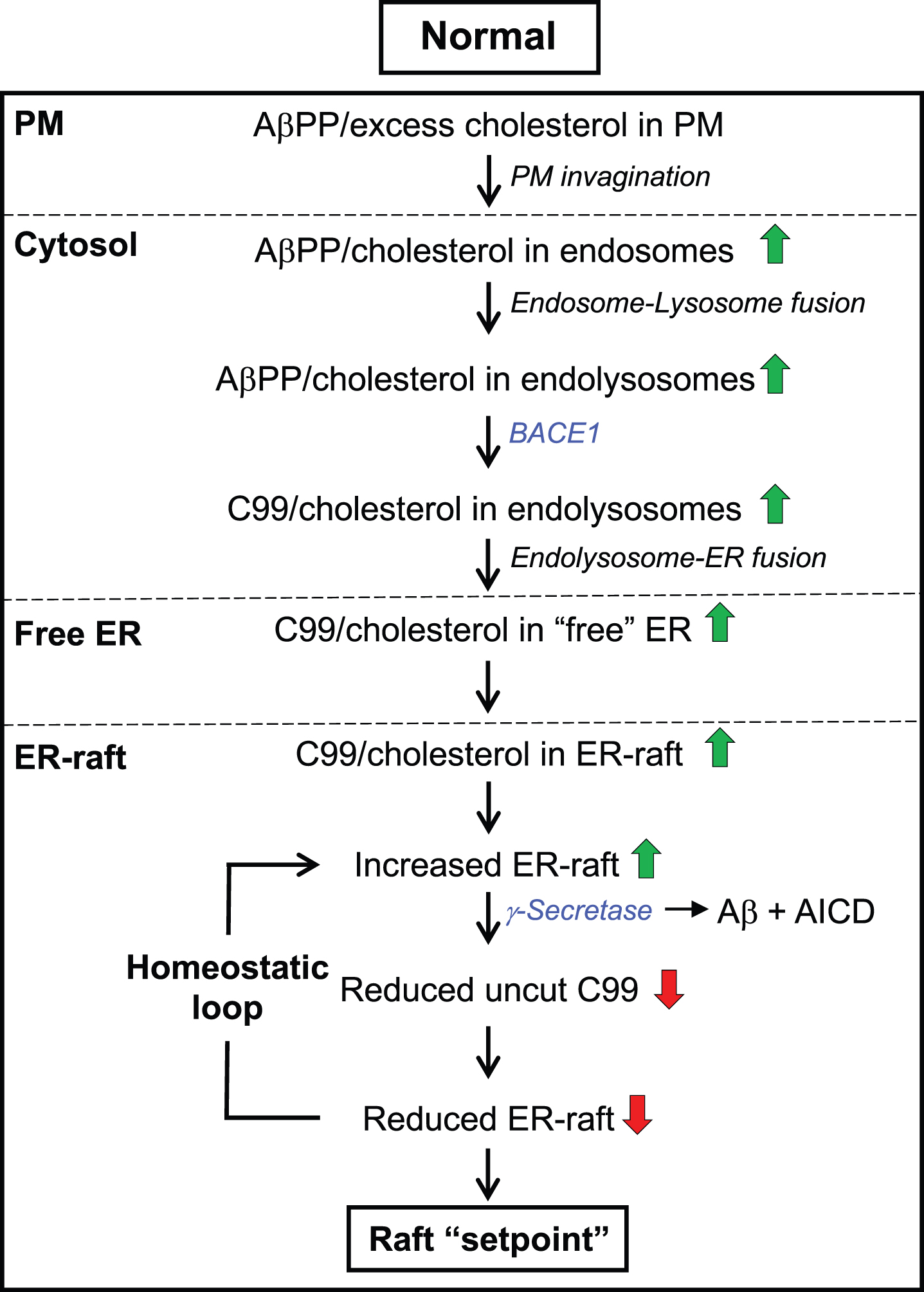
Importantly, this PM-derived cholesterol changes the structure of the ER membrane by forming an intracellular lipid raft (Fig. 3) [139]. Formation of the raft is maintained by proteins with affinity for cholesterol, in particular C99, that are capable of clustering cholesterol into local islands. This membrane reorganization results in changes in the distribution and conformation of ER proteins embedded within these islands (i.e., rafts) and in the modulation of the activities they regulate. For example, C99-mediated raft formation induces the activity of acyl-CoA:cholesterol acyltransferase (ACAT1; gene SOAT1), the enzyme that converts cholesterol into cholesteryl esters that are then deposited as lipid droplets (LDs), in order to detoxify the cell of excess cholesterol and return cholesterol to normal levels in a homeostatic loop [53, 140]. Alterations in the regulation of any step in this pathway leads to changes in the distribution of cholesterol and in the composition of lipid membranes, leading to altered raft “setpoints” (discussed below).
Fig. 3
The C99-cholesterol axis in the normal situation. Role of cholesterol: Excess plasma membrane (PM) cholesterol derived from extracellular lipoproteins induces the invagination of the PM and the formation of cholesterol-rich endosomes, which pinch off from the PM, enter the cell, and eventually fuse with lysosomes. The resulting endolysosomes fuse with the ER and release cholesterol to ER membranes, forming an intracellular lipid raft. Cholesterol-regulatory proteins detoxify the cell of excess cholesterol and return cholesterol to normal levels in a homeostatic loop. Role of C99: At the same time, the excess PM cholesterol binds to AβPP (via the CBD) that is also internalized in endolysosomes, where it is cleaved by BACE1 at low pH, forming C99. C99 is then delivered to the ER raft where it is cleaved by γ-secretase in an amount sufficient to reduce the initial raft C99 content to a “setpoint” level. Once C99 is cleaved (releasing Aβ containing the CBD), clustering of cholesterol in lipid rafts is reduced dramatically. Note that the ability of C99 to bind cholesterol (via the CBD) allows for a homeostatic loop to be set up in which the equilibrium between the amount of cut versus uncut C99, and the corresponding amount of cholesterol bound to uncut C99, determines the raft setpoint. Green/red arrows, increased/decreased levels. See text for details.
This trafficking of cholesterol is paralleled by a similar trafficking of AβPP and C99 (Fig. 3). As noted above, in the amyloidogenic pathway AβPP is internalized in cholesterol-rich endosomes [141], where it is then cleaved by BACE1 [142, 143], an enzyme activated at low pH [144]. This results in the production of C99 that is then delivered from endosomes to the ER (by a currently unknown mechanism [131], but possibly via endosome-ER contacts [145–147] and/or the retromer complex that recycles proteins from endosomes to the plasma membrane via the Golgi [148]), where it “attracts” cholesterol [116, 117] and sphingomyelin [149] (see below), resulting in the formation of the lipid raft domain described above. In the raft, C99 associates with γ-secretase (by physical proximity [63]) and is cleaved into Aβ (which is then exported from the raft via exosomes [150, 151]) and AICD. Once C99 is cleaved, releasing Aβ containing the CBD (see below and Fig. 1), clustering of cholesterol in lipid rafts is reduced dramatically. Thus, like cholesterol, C99, and raft formation, is regulated in a homeostatic loop that is mediated by the level of C99 and the degree of its cleavage by γ-secretase (i.e., C99 controls raft formation and hence γ-secretase levels; γ-secretase controls C99 levels and hence raft formation [53]). In indirect support of this scenario, we note that of the 114 documented pathogenic point mutations in AβPP (https://www.alzforum.org) 25 are located in the C99 region, but none are at the critical glycines in the CBD GxxxG motifs (see Fig. 1B), implying that the presumed loss of C99’s ability to bind cholesterol disrupts the relationship between C99 and cholesterol to the point where C99 is no longer pathogenic (a conclusion also supported by mutagenesis of the CBD [53]).
In broad view, both cholesterol trafficking and AβPP trafficking follow essentially the same topographical pathway (PM → endosome → endolysosome → ER → lipid raft) (Fig. 3). We believe that this is not a coincidence: the fact that the cholesterol-internalization and AβPP-cleavage pathways operate in parallel could help explain the effects of cholesterol on AβPP metabolism [152] and the fact that cholesterol turnover [153, 154]—mainly the oxidation of cholesterol to 24(S)-hydroxycholesterol by CYP46A1 (cytochrome P450 46A1; cholesterol 24-hydroxylase) [155] and its negative regulator ATAD3A (ATPase family AAA domain-containing protein 3A) [156], both of which are localized to ER rafts [156, 157]—is necessary for Aβ production [115, 158–160]. Moreover, the “superposition” of both pathways reveals a picture in which AβPP is a reporter of the lipid environment of the cell, by regulating the transport of cholesterol between PM and ER, and that AβPP’s internalization and cleavage by BACE1 and γ-secretase are essential steps in the regulation of cellular cholesterol and overall lipid homeostasis.
Accordingly, we propose that C99 is required to maintain intracellular cholesterol and raft homeostasis. Specifically, in the normal situation (Fig. 3), after a threshold is exceeded, the excess of cholesterol in the plasma membrane [161] binds to full-length AβPP [162, 163] and induces its internalization by the formation of cholesterol-rich endosomes. Following cleavage of AβPP by BACE1 within endolysosomes to produce C99, C99 traffics to the ER where it “attracts” cholesterol (at 1:1 stoichiometry [117]) [116, 117] and sphingomyelin [149], resulting in the formation of a lipid raft domain. Formation of the raft promotes the association of γ-secretase with C99 and its cleavage into Aβ and AICD. Once C99 is cleaved and Aβ is released, clustering of cholesterol in lipid rafts is reduced dramatically.
Thus, the interplay between C99 and cholesterol—a “C99-cholesterol axis”—sets up a self-correcting homeostatic feedback loop such that, under normal circumstances, the amount of uncleaved C99 is maintained at extremely low levels, in order to help maintain intracellular cholesterol and raft levels at a desired “setpoint”, both quantitatively (e.g., raft amount) and qualitatively (e.g., raft function) (Fig. 3). Moreover, this setpoint likely varies both spatially (e.g., in different cells and tissues) and temporally (e.g., during development or in response to changing environmental conditions). In AD, however, the steady-state levels of C99 and cholesterol are increased significantly, disrupting this feedback loop and setting off a chain of events that results in the disease, as described below.
FAMILIAL AND SPORADIC AD: A SHARED PHENOTYPE WITH DISTINCT TRIGGERS
As in FAD, tissues from SAD patients also have increased C99 levels [97], by various proposed mechanisms, including lysosomal dysfunction [134] and altered activation of γ-secretase [164]. Relevantly, increased exposure of cells to high levels of extracellular cholesterol can induce an increased rate of cholesterol internalization and AβPP cleavage, and an increase in C99 and in the Aβ42:Aβ40 ratio [165, 166]. Therefore, any condition or gene variant that provokes an elevation in cholesterol uptake and traffic to the ER will be capable of inducing high levels of C99 in sporadic forms of the disease [167].
Notably, the ɛ4 allele of APOE (ApoE4) is one of these SAD genetic risk factors, as it is recycled from endolysosomes much less efficiently than is ApoE3 [168–170]. This results in an accumulation of intracellular cholesterol, thereby stimulating an increase in C99, similar to what was described above for FAD. In fact, of the 34 AD risk-factor genes with coding-region non-synonymous mutations identified by us (Table 1), nine are associated with altered cholesterol trafficking or metabolism (marked in red in Table 1; see also [171]).
Table 1
AD risk genes associated with coding-region non-synonymous mutations
| Gene | Name | MAM hypothesis-related function? |
| APOE | Apolipoprotein E [367] | Cholesterol efflux [169]; ApoE4 upregulates MAM function [166] |
| ABI3 | ABI gene family member 3 [368] | Reg plasma cholesterol in Abi3-KO mice [369] |
| CR1 | Complement receptor type 1 (CD35) [370] | Binds ApoE4 [371] |
| DOC2A | Double C2-like domain-containing protein α [372] | Cholesterol required for DOC2A-RAB27A interaction [373] |
| ABCA1 | Phospholipid-transporting ATPase [181] | Reg sterol sensing [374]; exports cholesterol [180] |
| ABCA7 | Phospholipid-transporting ATPase [370] | Cholesterol (?)/lipid transport [375], transports ceramide [376] |
| ATP8B4 | Phospholipid transporting ATPase [377] | Reg sterol transport in C. elegans homolog tat-2 [378] |
| MAPT | Microtubule-associated protein tau[379] | Reg by cholesteryl esters [261] |
| AKAP9 | A-kinase anchor protein 9 [380] | Reg phospho-tau [381] via cholesterol [293, 382] |
| ADAM10 | Disintegrin &metalloprotease domain-cont prot 10 [383] | α-Secretase; increases BACE1 and C99 [384] |
| BIN1 | Myc box-dependent-interacting protein 1 [370] | With CD2AP, reg endosomal recycling of BACE1 [385] |
| CD2AP | CD2-associated protein [370] | With BIN1, reg endosomal recycling of BACE1 [385] |
| GGA3 | ADP-ribosylation factor-binding protein [386] | Negative regulator of BACE1 [386] |
| NOTCH3 | Neurogenic locus Notch homolog 3 [387] | Cleavage regulated by γ-secretase [388] |
| TM2D3 | TM2 domain-containing protein 3 [389] | Reg Notch/presenilin signaling in fly homolog almondex |
| SHARPIN | SHANK-associated RH domain interacting protein [391] | Reg ferroptosis, mitophagy [392, 393] via MAM [394, 395] |
| MME | Neprilysin [396] | In lipid rafts [397]; degrades Aβ [398]; reg by AICD [399] |
| UNC5C | Netrin receptor [400] | In lipid rafts [401] |
| TREM2 | Triggering receptor expressed on myeloid cells 2 [368] | Reg by PS1 [402]; senses lipids [403], cholesterol [404] |
| CD33 | Myeloid cell surface antigen CD33 [405] | Reg TREM2 [406] |
| MS4A4A | Membrane-spanning 4-domains subfam A memb 4A [407] | Modulates TREM2 [407] |
| MS4A6A | Membrane-spanning 4-domains subfam A memb 6A [370] | Modulates TREM2 [407] |
| ADAM17 | Tumor necrosis factor-α converting enzyme [408] | α-Secretase; reg APP levels [408]; cleaves TREM2 [409] |
| CLU | Clusterin (ApoJ) [370] | In MAM [410]; binds ApoER2/VLDLR [411]; exports chol [412] |
| PLCG2 | Phospholipase C-gamma-2 (PLCγ2) [368] | Synthesis of DAG and IP3, P522R mutant protective in AD [413] |
| PLD3 | Phospholipase D; 5’⟶3’ exonuclease [414] | Reg C99, cholesterol levels via degraded mtDNA [415] |
| GRN | Progranulin [416] | Reg lysosomes and lipids (polyunsaturated TAGs) [417] |
| SORL1 | Sortilin-related receptor (SorLA) [418] | APP sorting [419]; reg BACE1 [420]; cut by γ-secretase [421] |
| PICALM | PtdIns-binding clathrin assembly protein [370] | C99 autophagy [422, 423]; reg BACE1 [424], chol, LDLR [425] |
| EPHA1 | Ephrin type-A receptor 1 [370] | No obvious connection |
| GCAT | Glycine acetyltransferase [426] | No obvious connection |
| NME8 | Thioredoxin domain-containing protein 8 [427] | No obvious connection |
| PILRA | Paired Ig-like type 2 receptor α [428] | No obvious connection |
| ZCWPW1 | Zinc finger CW-type PWWP domain protein 1 [429] | No obvious connection |
Gene functions connected to cholesterol (entries in red), MAM (blue), both cholesterol and MAM (green), or neither (black).
Thus, an increase in the steady-state level of C99 in AD can come from many sources (Table 1), including: 1) reduced cleavage of C99 by γ-secretase [32]; 2) increased BACE1 cleavage of AβPP [172–174], as is found in the AβPP “Swedish” mutation [175]; 3) increased steady-state levels of AβPP, as is seen in Down syndrome [176] (APP is on chromosome 21) or in patients with mutations in the sortilin-related receptor (SORL1; an AD risk factor [167]) that reduces AβPP recycling via the retromer pathway [177, 178]; 4) increased intracellular cholesterol levels [165, 179] due to, for example, mutations in ATP-binding cassette sub-family A member 1 (ABCA1), a cholesterol exporter [180] and AD risk factor [181]; and 5) increased intracellular cholesterol due to inefficient recycling of ApoE4-derived lipoproteins [166]. Clearly, elevated C99 is toxic to cells, no matter the source. Thus, another question immediately arises: what is the nature of that toxicity? To answer this question, we must take a slight detour into the structure and function of the lipid raft subdomain of the ER.
MAM: A LIPID RAFT IN THE ER
All the evidence marshalled above points to elevated C99 as the trigger of the cellular defects seen in AD, mediated by C99’s association with cholesterol (and, a fortiori, with sphingomyelin; see below). In fact, C99’s toxicity is proportional to its capacity to form lipid rafts in the ER [53]. These ER-localized rafts have a name reflective of their special nature and special status in the cell: mitochondria-associated ER membranes, or MAM [182].
MAM is a highly dynamic and transient subdomain of the ER that communicates, both physically and biochemically, with mitochondria [51, 182]. MAM lies in close apposition to mitochondria (ranging from ∼10–80 nm [183–185]) (Fig. 4A), facilitating bidirectional crosstalk between the two organelles, with specific molecules travelling relatively short distances from the ER to mitochondria for some molecules and in the opposite direction for others. This apposition is mediated physically by pairs of tethers, one on the ER cytoplasmic face and one on the mitochondrial outer membrane (MOM), that help bring the two organelles together. For example, mitofusin-2 (MFN2), located both at the ER and the MOM, dimerizes to align the two organelles [186], as do the pairs VAPB (vesicle-associated membrane protein-associated protein B/C) - PTPIP51 (protein tyrosine phosphatase interacting protein 51) [187], and SIGMAR1 (sigma-1 receptor) - VDAC (voltage-dependent anion channel) [51], with tethering regulated, in an unknown fashion, by MAM-localized PACS2 (phosphofurin acidic cluster sorting protein 2) [188, 189]. By contrast, presenilins are not physical tethers but are negative regulators of apposition such that pathogenic mutations modify presenilin structure to promote tethering via alteration in the lipid composition of the MAM, as described here. Importantly, both C99 [35] and γ-secretase activity [60–62] are localized to the MAM, as is γ-secretase activating protein (GSAP) [190]. Thus, the final step in the amyloidogenic pathway to produce A β can take place only in the MAM [191].
MAM functions include phospholipid synthesis [182, 192], cholesterol and steroid metabolism [193, 194], sphingolipid (e.g., sphingomyelin and ceramide) metabolism [35], glucose homeostasis [195], calcium trafficking [51, 196, 197], and mitochondrial oxidative phosphorylation (OxPhos) [35] and dynamics [186, 198]. MAM is also central to the modulation of various other key cellular processes, including the regulation of hypoxia [199], iron homeostasis [199, 200], ER stress [201], autophagy [202], and inflammasome signaling [203]. We note that all of these processes are disturbed in AD [28], implying an intimate relationship between MAM function and AD pathology [89, 204]. For example, calcium trafficking between the ER and mitochondria, which is perturbed in AD [205], involves more than 30 MAM-localized proteins [206]. Of these, perhaps the most prominent is the sigma-1 receptor (gene SIGMAR1) [207], which is a key “gatekeeper” for the transfer of Ca2 + from the ER to mitochondria [208], with an interorganellar gap distance of only ∼7–15 nm required for maximum transfer [196]. Similarly, 9 of the 10 enzymes of glycolysis, which is impaired in AD [209], are localized to MAM [210] and the tenth, hexokinase, is localized to the mitochondrial outer membrane [211]. Autophagy is another cellular function intimately reliant on MAM [202, 212]: not only are at least 5 autophagy-related proteins localized to MAM, but MAM is required for the synthesis of the phosphatidylethanolamine (see below) that is required to build the phagophore [202]. Finally, mitochondrial bioenergetics [35] and organelle morphology and dynamics—most notably the balance between organellar fusion and fission—which are also altered in AD [213, 214], are MAM-mediated functions. Also relevant to mitochondrial function, hypoxia, which is a feature of AD [215], upregulates BACE1 at the transcriptional level [216]. Given this multiplicity of functions, it is highly likely that MAM composition, not only its constituent proteins and lipids, but even microRNAs [217], differs in different cell types, at different times, and in response to different environments [218].
With respect to AD, C99 induces the formation of MAM via its ability to bind and “cluster” cholesterol, forming the raft, as described above. In turn, several proteins are recruited to the MAM so as to co-regulate multiple homeostatic pathways, some of which have been described above (e.g., cholesteryl ester synthesis via ACAT1 [219]). Alterations in the regulation of MAM formation, as occurs in AD, perturbs these pathways. As examples, we will focus on three key MAM functions relevant to AD: regulation of phospholipids, sphingolipids, and cholesterol.
Altered phospholipid metabolism is a feature of AD [220, 221]. Most of the cell’s phosphatidylethanolamine (PE) is synthesized in a salvage pathway located at the ER-mitochondrial interface (i.e., MAM): phosphatidylserine (PS) is synthesized in the MAM by phosphatidylserine synthases [222]; PS then translocates to the mitochondria [223, 224] where it is decarboxylated by phosphatidylserine decarboxylase (PISD) [225] to form PE; PE then travels back to the MAM, where it traffics to the rest of the cell or undergoes further modifications [226] (see Fig. 4B). The trafficking of PS from MAM to mitochondria is a well-recognized measure of MAM function [227]. In cells from both SAD and FAD patients this pathway, and cellular PS and PE production, are upregulated significantly [74].
Fig. 4
Schematic representation of the MAM subdomain of the ER. A) EM of rat hepatocytes showing MAM compared to “free” ER (adapted from [185], with permission). B) Key MAM functions. ACAT1, acyl-CoA cholesterol acyltransferase-1; DGAT2, diacyglycerol-O-acyltransferase-2; IP3R, inositol triphosphate receptor; MFN2, mitofusin-2; MCU, mitochondrial calcium uniporter; PISD, phosphatidylserine decarboxylase; SCD1, stearoyl-CoA desaturase-1; VDAC, voltage-dependent anion channel. See text for details.
![Schematic representation of the MAM subdomain of the ER. A) EM of rat hepatocytes showing MAM compared to “free” ER (adapted from [185], with permission). B) Key MAM functions. ACAT1, acyl-CoA cholesterol acyltransferase-1; DGAT2, diacyglycerol-O-acyltransferase-2; IP3R, inositol triphosphate receptor; MFN2, mitofusin-2; MCU, mitochondrial calcium uniporter; PISD, phosphatidylserine decarboxylase; SCD1, stearoyl-CoA desaturase-1; VDAC, voltage-dependent anion channel. See text for details.](https://content.iospress.com:443/media/jad/2024/98-4/jad-98-4-jad231318/jad-98-jad231318-g004.jpg)
Sphingolipid pathways are also altered in AD [228, 229]. In particular, the ceramide content in brains and cells from AD patients and in AD cell models is increased [55, 230–233] as a result of the upregulation of de novo ceramide synthesis [234] and of the activity of sphingomyelinases (SMases) that convert sphingomyelin (SM) to ceramide [231, 235]. MAM is involved in the regulation of sphingolipid metabolism [236] and at least one neutral SMase in mice likely resides at the MAM [237] (although the homologous gene in humans is apparently a pseudogene [238]). The ceramide that is enriched at MAM [35, 239] (by ∼25% in mouse liver [54]), can displace raft cholesterol [240, 241] while still maintaining the liquid-ordered nature of these rafts [240]. In AD, MAM-localized SMase activity is upregulated [35], with a concomitant local increase in ceramide [35] that increases MAM [242] and Aβ42 [243] formation and reduces mitochondrial bioenergetics [35, 244–248].
Finally, elevated serum cholesterol [249] and altered cholesterol metabolism [230, 249–251] have been well-documented in AD, as have been the effects of increased cholesterol on AβPP metabolism [152], including increased Aβ production [165, 252]. Importantly, elements of the cellular cholesterol homeostatic machinery, including SREBP1 [253], which interacts with C99 [254], and ACAT1 [255] are localized in MAM. In cells from both SAD and FAD patients, ACAT1 activity is upregulated significantly [74], as is the accumulation of lipid droplets [74, 256, 257], including LDs within neurons [258]. Remarkably, ACAT1 activity is required for Aβ production [115, 158–160, 259, 260]. Moreover, ACAT1-derived CEs regulate not only Aβ production [159, 261] but also tau production, by inhibiting the degradation of phosphorylated tau [261]. Thus, MAM-localized cholesterol metabolism plays a fundamental role in the development of the two key neuropathological hallmarks of AD: plaques and tangles.
The parallel alteration of cholesterol and sphingolipids is not coincidental, as the levels of these two classes of lipids are positively co-regulated in the cell [256, 262–265] and help to establish the formation of lipid rafts [56]. Specifically, sphingomyelin forms an “umbrella” around cholesterol (at a 1:1-1:2 ratio [263]), protecting it from interactions with water at the membrane/cytoplasm interface [266, 267], thereby initiating lipid raft formation (e.g., MAM). This relationship between sphingolipids and sterols is relevant to AD, as AβPP processing has been shown to occur preferentially in lipid rafts [268] that also contain C99 [269].
As mentioned above, C99 contains a cholesterol-binding domain. Perhaps of equal significance, C99 also contains a sphingolipid-binding domain (SBD; orange dot in Fig. 1A and sequence in Fig. 1B) that binds to sphingomyelin [149]. Thus, these two lipid-binding domains can potentially cooperate to promote raft formation by attracting equal amounts of cholesterol and sphingomyelin to C99. Moreover, and perhaps not coincidentally, in keeping with the dynamic nature of raft assembly and disassembly, both the CBD and the SBD are located within the N-terminal region of C99 such that cleavage of C99 by γ-secretase releases both domains as part of Aβ (see Fig. 1). From this point of view, C99 helps assemble the raft, whereas γ-secretase, via C99 cleavage and Aβ release and export [151], helps disassemble it. Perhaps Aβ has its own role to play in the cell after all [116, 270, 271].
GENETIC RISK FACTORS IN SAD ARE ALSO CONSISTENT WITH ALTERED MAM FUNCTION
It is clear that C99 levels can be elevated in the familial form of AD, due to genetic mutations in PSEN1, PSEN2, and APP that conspire to increase C99, and hence cholesterol levels. But what about sporadic AD, where these genes are normal? What is the underlying pathogenetic process in SAD, and does it differ from that in FAD [28]?
Besides the mutations in the nine genes associated with cholesterol metabolism noted above (marked in red in Table 1), there are mutations in nine additional SAD risk genes that are associated with MAM function (marked in blue in Table 1) and in eleven other genes associated with both cholesterol metabolism and MAM function (marked in green in Table 1). Thus, of the 34 SAD risk factor genes on the list, 29 are consistent with the mechanism of AD pathogenesis that we propose here.
THE C99-CHOLESTEROL AXIS IN AD
These findings have led us to propose a model that invokes perturbations in C99 and cholesterol (i.e., disruption of the “C99-cholesterol axis”) as potential players in a linked pathogenetic mechanism leading to AD (Fig. 5). As in the normal situation (Fig. 3), after a threshold is exceeded [272], excess PM cholesterol binds to full-length AβPP and induces its internalization by endosomes [162] and cleavage by BACE1 within endolysosomes to produce C99 which, owing to increased BACE1 activity [273–275], is now elevated above normal levels [97]. At this point, the pathogenic pathways resulting in AD diverge, but not along classical FAD-SAD lines, as is commonly described. Rather, the divergence is between the early-onset (EOAD) and late-onset (LOAD) forms of the disease.
Fig. 5
The C99-cholesterol axis in AD. In AD, cholesterol and AβPP enter the cell as in the normal situation (see Fig. 3). However, (A) in C99-driven early-onset AD (EOAD) - both FAD and those forms of SAD associated with underlying C99-related risk factors (see Table 1) - impaired γ-secretase activity is insufficient to cleave all of the MAM-localized C99, leaving “excess” uncut C99 to accumulate in the MAM raft above the “setpoint” level. The elevated uncut C99 attracts more cholesterol (and sphingomyelin), enlarging the raft that, in turn, attracts even more C99 that, too, is cleaved sub-optimally. In this way, MAM homeostasis is disrupted in a vicious cycle in which the raft grows inexorably and relatively rapidly (a “runaway” raft). B) In cholesterol-driven late-onset AD (LOAD) associated with underlying cholesterol-related risk factors (see Table 1), the raft also grows, but for a different reason: the accumulation of excess intracellular cholesterol binds to C99 and causes the raft to grow. Although γ-secretase is functioning relatively normally and efficiently, it is present in an amount insufficient to cleave all the C99 present in the MAM. The excess uncut C99 attracts more cholesterol, enlarging the raft while also generating yet more uncut C99, resulting in the same type of vicious cycle seen in EOAD, albeit in a more slowly-growing raft. Green/red arrows, increased/decreased levels. See text for details.
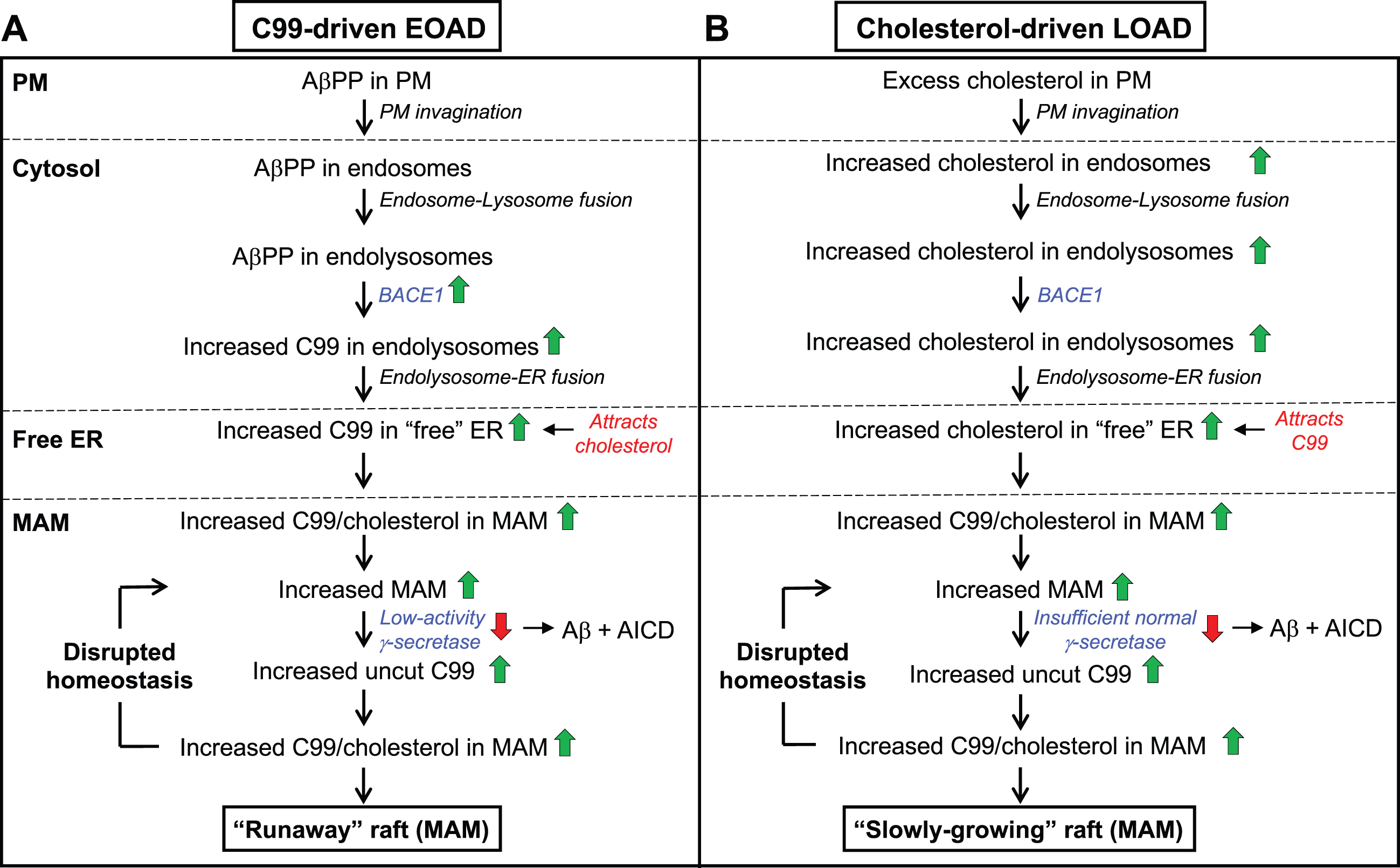
In EOAD (Fig. 5A), which includes all forms of FAD (including Down syndrome) and those forms of SAD associated with risk factors affecting C99 (e.g., the “blue” genes in Table 1), elevated C99 traffics to the ER-raft (i.e., MAM) where, owing to FAD mutations or to C99-related SAD risk factors, its cleavage is incomplete (i.e., the mismatch between C99 level and γ-secretase activity is such that uncut C99 accumulates above the desired setpoint level). The excess of uncut C99 now attracts more cholesterol (by ∼40% in PS1/PS2-double knockout [DKO] MEFs compared to wild-type MEFs [53]), resulting in the formation of a more extensive raft domain containing more “incoming” C99 that is also cleaved inefficiently, Moreover, the chronic formation of MAM and the increased absolute amount of Aβ triggers the upregulation of SMase activities [270], resulting in reduced sphingomyelin and elevated ceramide levels [35], and a concomitant reduction in cholesterol at the MAM, leading to the formation of longer [74] and thinner [75, 276] MAM structures. Thus, in “C99-driven” EOAD, the normal homeostatic loop is replaced by a vicious cycle in which excess C99 drives raft formation while the growing “runaway” raft recruits more γ-secretase that is unable (or in the case of Down syndrome, insufficient) to cleave all of the C99 presented to it.
In LOAD (Fig. 5B), the C99-cholesterol axis is also perturbed, but in a slightly different way. In the majority of these patients, the disease is “cholesterol-driven,” that is, risk factors (e.g., ApoE4) conspire to increase the level of intracellular cholesterol above normal levels [53, 277] and a subsequent increase in MAM-localized cholesterol (by ∼100% in PS1/PS2-DKO MEFs [53]) that in turn provokes an increase in raft-localized C99. Note that in LOAD, γ-secretase is structurally normal but, coupled with a potential insufficiency in its amount, activity, and/or orientation within the lipid raft, is nevertheless unable to cleave the large amount of cholesterol-recruited C99 present, resulting in a slow but steady accumulation of uncut C99. Once again there is a mismatch between C99 level and γ-secretase activity, only this time it results in a “slowly-growing” raft that exceeds the desired setpoint level.
While we believe that C99 plays a key role in delivering cholesterol to the MAM via the vesicular pathway, we note that a number of proteins involved in the non-vesicular pathway for cholesterol trafficking from the plasma membrane to the ER [272, 278, 279] are also localized to the MAM. These include ADP-ribosylation factor 1 (ARF1) [280], oxysterol-binding protein-related proteins-5 (ORP5) and –8 (ORP8) [223, 281], and Aster-B/GRAMD1B [282] (which is abundant in brain [278]). Moreover, the Aster proteins rely on phosphatidylserine for their function [223, 283, 284], implying that no matter how cholesterol enters the cell, MAM (and therefore C99) likely plays a central role in intracellular cholesterol trafficking [285].
Note that this analysis supports the idea that FAD and SAD (or more properly, EOAD and LOAD) are essentially two subtypes of the same disease [286], and that both disorders share a common pathomechanism that differs only in age of onset, severity of symptoms, and rate of progression. In sum, from a mechanistic point of view, F AD and SAD are the same disease.
In both cases, the cell makes heroic efforts to reduce the inexorable increase in raft size (e.g., by shutting down de novo cholesterol synthesis [53]; by upregulating SMase activity [35] to both increase ceramide and reduce sphingomyelin levels (thereby reducing cholesterol levels); by converting cholesterol to cholesteryl esters that are stored in lipid droplets [74]; by oxidizing cholesterol [287] [at least in the early stages [288], if not the late stages [289], of the disease]; by exporting cholesterol [290]), and succeeds admirably in staving off the inevitable for decades.
WHAT ABOUT TAU?
Hyperphosphorylated tau, a common but inconsistent feature of AD (similar to Aβ), can aggregate into tangles and disrupt cytoskeletal stability and axonal trafficking. Cholesterol appears to play a key role in inducing tau hyperphosphorylation, but the exact mechanism is unclear, as it was reported that tau hyperphosphorylation was stimulated by levels of intracellular cholesterol that were both increased (e.g., via the MAM-localized [291] transient receptor potential cation channel TRPV4 [292]) and decreased (e.g., via the cholesterol biosynthesis enzyme 24-dehydrocholesterol reductase [DHCR24; also called Seladin-1] [293]). Conversely, promotion of cholesterol efflux reduced tau phosphorylation [294], as did reduced MAM-mediated [255] cholesterol esterification [261, 295]. More recently, it was found that tau hyperphosphorylation disrupts ER-mitochondria contacts at MAM [296]. Furthermore, inhibition of BACE1, but not γ-secretase, reduced phosphorylated tau levels in FAD neurons [297, 298], indicating that C99 was at the heart of this effect. While the exact details still need to be worked out, these results clearly speak to important roles for cholesterol metabolism and MAM functionality in promoting and/or responding to tau pathology.
AN OVERALL MODEL OF AD PATHOGENESIS: THE “MAM HYPOTHESIS”
Based on the data and arguments presented above, we propose a “MAM hypothesis” of AD pathogenesis [89, 204, 299–301]. It posits that in both familial and sporadic AD, the functional cause of the disease is massively increased ER-mitochondrial communication and up-regulated MAM function that, in turn, results in the phenotypes seen in AD, including the plaques and tangles. The biochemical cause of AD is MAM-mediated lipid dyshomeostasis driven by increased intracellular cholesterol levels that result from increased steady-state levels of the MAM-localized cholesterol sensor C99 (in the case of EOAD) or from the unregulated accumulation of intracellular cholesterol that interacts with C99 (in the case of LOAD).
An overall model of the MAM hypothesis of AD pathogenesis is shown in Fig. 6. Specifically, in FAD, AβPP processing is affected directly, with a profound interrelationship between AβPP misprocessing (i.e., increased C99 levels) and deranged cholesterol metabolism, as described above. In SAD, the same relationship between C99 and cholesterol also holds true, but in a different way, as the genetic predisposition to develop SAD runs on two parallel tracks, one promoting the increase in cellular C99 levels (causing EOAD, as in FAD) and the other promoting elevated intracellular cholesterol (causing LOAD). As in FAD, both perturbations operate together and interact with each other to generate the downstream MAM-mediated phenotypes, including the plaques and tangles [261].
Fig. 6
A model of AD pathogenesis. A) Inherited mutations in FAD (affecting C99 [in blue]), or genetic risk factors in SAD (affecting either C99 [in blue] or intracellular cholesterol levels [in red], or both [in green]; colors as in Table 1), converge to increase ER-mitochondrial communication at the MAM. B) These perturbations eventually give rise to the features of AD, as shown. See text for further details.
![A model of AD pathogenesis. A) Inherited mutations in FAD (affecting C99 [in blue]), or genetic risk factors in SAD (affecting either C99 [in blue] or intracellular cholesterol levels [in red], or both [in green]; colors as in Table 1), converge to increase ER-mitochondrial communication at the MAM. B) These perturbations eventually give rise to the features of AD, as shown. See text for further details.](https://content.iospress.com:443/media/jad/2024/98-4/jad-98-4-jad231318/jad-98-jad231318-g006.jpg)
Note that both elevated C99 and elevated intracellular cholesterol conspire to increase ER-mitochondrial communication at the MAM (Fig. 6A) that, in turn, gives rise to the biochemical, morphological, and clinical features of the disease (Fig. 6B). Importantly, all of those features, including the plaques and tangles, are downstream consequences of the primary functional problem, which is upregulated MAM function. This model helps explain why clinical trials based on removing plaques and/or Aβ have fared so poorly.
We noted above that the SAD risk factors (Fig. 6A) fell into two categories: those that mainly affect MAM function and those that mainly affect cholesterol metabolism. Thus, one might expect that clinically, SAD patients expressing MAM-related risk factor genes would resemble FAD patients, with a relatively early age of onset and a rapid disease progression (i.e., EOAD), whereas those expressing cholesterol-related risk factors would have a later age of onset and slower progression (i.e., LOAD). Remarkably, this may actually be the case, as it was recently shown that the distribution of the age of onset in ɛ4-negative SAD patients is indeed bimodal, with one onset peaking at age ∼57 years (i.e., EOAD) and the other peaking at age ∼77 years (i.e., LOAD) [302], consistent with, but of course not proof of, this supposition.
Thus, the MAM hypothesis invokes two key interrelated elements that drive AD pathogenesis, C99 and cholesterol, that converge at the MAM. Taken together, we propose that both FAD and SAD are caused by the same underlying and linked pathogenetic processes—elevated C99 and increased intracellular cholesterol. From this perspective, AD is, at bottom, a lipid disorder, similar to what has been proposed by others [115, 119, 168, 303–305] but described here in somewhat greater detail.
CLINICAL IMPLICATIONS
How does upregulated MAM function lead to a neurological disorder? In other words, how does MAM upregulation impair neuronal functionality to the point where cognitive functions are compromised? We believe that the answer to this question lies in MAM’s role as a center of cellular lipid metabolic regulation, where multiple lipid classes are metabolized by MAM-localized enzymes. Lipids regulate multiple basic brain functions [306], including action potentials, synaptic vesicle release, endocytic transport, cellular signaling, and pathogen recognition [307]. Many of these roles arise by virtue of the fact that lipids are the principal constituent of cellular membranes, where their different charges and shapes modulate membrane properties. As such, lipids can control the recruitment, conformation, and interaction of membrane-associated proteins like receptors, channels, and enzymes [308]. Lipids also regulate membrane curvature, important for the budding and fusion of vesicles containing neurotransmitters and other cargo [309, 310]. They also regulate membrane permeability and fluidity, which are largely dependent on the fatty acid composition and degree of saturation of constituent phospholipids [311]. Furthermore, lipids, and cholesterol in particular, are the primary constituents of myelin [312], which ensheathes axons and enables efficient conductance of action potentials. Finally, lipids represent a source of energy storage and transport and can serve as reservoirs of bioactive signaling molecules. It is therefore not surprising that lipid alterations can be drivers of neurodegenerative diseases [313], which we propose to be the case in AD.
THE MAM HYPOTHESIS HAS EXPLANATORY POWER
The MAM hypothesis answers or explains many (but by no means all) of the features of AD that are not easily explained by the amyloid cascade. This includes the elevated Aβ42:Aβ40 ratio, the altered cholesterol, phospholipid, sphingolipid, calcium, and glucose levels, the increased lipid droplet formation, the reduced bioenergetics and altered mitochondrial morphology, the role of many SAD genetic risk factors and of Down syndrome, the different severities of various AβPP and presenilin mutations, the partial elucidation of the dominant nature of the FAD mutations and whether FAD and SAD are manifestations of a single underlying disease process, the secondary role of plaques and tangles, and the poor track record of amyloid-based clinical trials.
The MAM hypothesis could also provide insight regarding the preponderance of affected females over males [314]. Both estrogen and progesterone decrease profoundly in post-menopausal women [315], and also decline in elderly men, but to a lesser degree [316]. Notably, steroid synthesis, like phospholipid synthesis, is a collaboration between MAM and mitochondria: MAM-localized cholesterol translocates to mitochondria [194, 317, 318], where it is converted to pregnenolone. Pregnenolone then translocates back to the MAM for synthesis of the various steroid hormones [309], including estrogen and progesterone, and for the synthesis of pregnenolone esters by ACAT1 [319]. It turns out that estrogen inhibits the transcription of BACE1 [320, 321], so its loss de-represses BACE1 activity [322], increasing C99. Similarly, progesterone inhibits the expression of ACAT1 and reduces MAM apposition length [323, 324], so its loss de-represses ACAT1 activity, thereby perturbing cholesterol homeostasis and also increasing C99. Thus, the age-related decline in both hormones in brain [325, 326] in women (and to a lesser degree in men) can upregulate MAM, triggering AD in a “sex-biased” manner, with obvious implications for hormone replacement therapy in AD, a highly contentious topic [327, 328]. Moreover, the decline in these hormones might help explain why advancing age is the single most important determinant of AD susceptibility [329].
Finally, the MAM hypothesis helps explain why inhibiting either BACE1 [13] or γ-secretase activity [15] as approaches to treat AD would be ill-advised. In the former case, BACE1 inhibition would result in a severe reduction in C99 levels, thereby constraining normal raft production, likely with deleterious consequences. In the latter case, inhibiting γ-secretase would increase C99 production, the very outcome that we should be trying to avoid. On the other hand, if AD is caused by disruption of the C99-cholesterol axis, reducing or preventing the accumulation of intracellular cholesterol might be of therapeutic value, for example, via the use of statins, a controversial topic [330, 331]. However, we believe that statins will likely be of limited utility, given that they are designed to inhibit the activity of HMGCR, which we have shown is already down-regulated in AD cells [53]; also, statins must cross the blood-brain barrier in order to be effective.
As is with works in progress, the MAM hypothesis currently does not explain other features of the disease. These include the relative paucity of FAD mutations in PS2 compared to those in PS1; the loss of olfaction as an early indicator of AD; the brain-specificity and brain subregion-specificity (e.g., hippocampus versus cerebellum; motor neurons versus cortical neurons) of the clinical phenotype (but see below); and the identification of risk genes that currently do not fit into the MAM paradigm (e.g., entries in black in Table 1), to name but a few. Nevertheless, it is reasonable, if optimistic, to state that it is not inconceivable that they too will eventually be explained in a manner consistent with the MAM hypothesis.
For example, for currently unclear reasons, there is a discrepancy between the number of pathogenic mutations in PS1 (more than 100) compared to those in PS2 (only ∼5) (https://www.alzforum.org). Although PS1 and PS2 are highly similar in structure (64% amino acid identity), they differ in their γ-secretase activities, substrates, and functions [28, 332]. Moreover, contrary to what is seen in the vast majority of PS1 mutations [26], pathogenic mutations in PS2 increase γ-secretase activity [28] and presumably reduce C99 levels, thereby posing a challenge to the MAM hypothesis. However, PS2, but not PS1, binds to MFN2, thereby promoting tethering of ER to mitochondria, and FAD-mutant PS2 binds MFN2 even more avidly and enhances the degree of tethering above normal levels [333, 334]. Thus, mutations in PS2, coupled with the potential interactions of PS2 with full-length MFN2 and/or its splice variants [335], could indeed mimic the effects of PS1 on enhanced ER-mitochondrial communication and on the C99-cholesterol axis (brain cholesterol levels are elevated in FADPS2 patients [336]), albeit via a more circuitous route. Taken together, these findings could explain why pathogenic mutations in PS2 are not only relatively rare, but also result in a “milder” form of AD compared to mutations in PS1 [28].
TISSUE SPECIFICITY
One problem that has dogged the field is that of tissue specificity: APP, PSEN1, and PSEN2 are housekeeping genes that are expressed in essentially all tissues [257, 337, 338] (see also genecards.org), and yet, FAD-causative mutations in these genes cause overt pathology only in the brain. In one sense, the issue of tissue specificity may be a red herring, as lipid droplets accumulate in peripheral blood cells from AD patients [257]. Moreover, AD-model mice expressing ApoE4 exclusively in their livers evinced enhanced AD-like amyloid pathology compared to mice expressing ApoE3 [339], implying that there is communication between the periphery and the brain in AD, likely via a breach in the blood-brain barrier. In addition, while MAM contacts ER “loosely” (gap distance of ≥10 nm), liver mitochondria are also contacted by tightly-adhering ER (“wrappER”) that is distinct from MAM [340], with essentially no gaps between the two organelles. WrappER mediates the biogenesis of lipids and lipoproteins [341] and plays an important role in cholesterol synthesis and trafficking via three-way ER-mitochondria-peroxisome contacts [342]. Taken together, these results are not only consistent with the concept that AD is indeed a lipid disorder, but also imply that in the context of AD pathology, the C99-cholesterol axis may not be confined to the brain.
And even within the brain there is regional specificity, as, for example, cerebellum is far less affected in AD than is hippocampus [343], with tau pathology spreading outwards from the entorhinal cortex in a prion-like manner [344, 345]. Can the MAM hypothesis shed any light on this paradox? One clue may lie in the emphasis of the MAM hypothesis on lipids, and especially on the role of cholesterol and sphingomyelin in lipid raft formation. We note that two of the earliest signs of AD are deficits in olfaction [346] and episodic memory [347]. While the two phenomena are apparently unconnected, there may be a relationship between them at the cellular level, as the olfactory bulb and the hippocampus are apparently the only loci in the adult human brain that are capable of renewal by stem cells [348] (a subject of some debate [349]). Given that 1) these newly-born cells are the only ones that require de novo myelination by oligodendrocytes, 2) two of the most abundant components of myelin are cholesterol and sphingolipids [350], and 3) both cholesterol biosynthesis [351] and progesterone [352] are required for myelination, it is possible that the aberrant MAM function described here is particularly pronounced in oligodendrocytes [353], either quantitatively (e.g., too little or too much myelin) or qualitatively (e.g., altered myelin composition) or both [354]. This may lead to altered neuronal conductance that is particularly pronounced in, but clearly not limited to, these newly-born cells.
THE MAM HYPOTHESIS AND NEUROINFLAMMATION
Our hypothesis proposes that higher levels of internalized cholesterol trigger MAM formation and subsequent modulation of metabolic enzymes. In the context of microglia, we note that immune challenges induce the transport of cholesterol from the plasma membrane to the ER [355], resulting in the formation of MAM domains. In turn, MAM formation in microglia induces the recruitment and clustering of enzymes involved in the regulation of bioenergetics (i.e., switching from OxPhos to glycolysis [356, 357]) and lipids (including regulation by TREM2 [triggering receptor expressed on myeloid cells 2], a known AD risk factor that is expressed exclusively in microglia [358]), thereby adapting microglial metabolism and cellular membranes to a pro-inflammatory state [357]. Once the activation subsides, cholesterol removal from the ER dissolves MAM’s structure, leading to a reversal of the changes in mitochondrial metabolism and the resolution of inflammation. From this point of view, the upregulation of MAM domains could explain the pro-inflammatory phenotypes associated with AD and help explain why cholesterol alterations are frequently associated with the emergence of damage-associate microglia (DAM) phenotypes.
THE MAM HYPOTHESIS HAS PREDICTIVE POWER
The hypothesis predicts that since AD is fundamentally a lipid disorder due to altered lipid homeostasis (most notably at the MAM), levels of specific lipids should be altered in predictable ways. To take but one example, MAM-localized ACAT1 [255] has a marked predilection for generating cholesteryl esters containing unsaturated fatty acids, especially those containing oleate (C18:1) [359, 360]. Similarly, MAM-localized long chain fatty acid-CoA ligase 4 (FACL4; gene ACSL4) [361], used in the transfer of fatty acids to phospholipids and sphingolipids, has a preference for C20:4 and C20:5 [362]. On the AβPP processing side, the elevation in C99, and the corollary reduction in Aβ, implies that there should be an AD-specific increase not only in the ratio of Aβ42:Aβ40, but also in the ratio of C99:total Aβ. Thus, alterations in specific lipids and in C99 levels should be pathognomonic of AD.
IMPLICATIONS FOR DIAGNOSIS AND TREATMENT
The specific alterations in both lipid profiles and in the C99:total Aβ ratio predicted by the MAM hypothesis should be detectable in more easily-accessible tissues, such as blood (e.g., in plasma/serum for the lipids and the Aβ; in peripheral blood mononuclear cells for the C99). In fact, analysis of lipids in AD blood has already been reported [363–365] but the specificity and mechanistic interpretation of those “agnostic” shotgun approaches have limited their use as true and robust diagnostics. On the other hand, a mechanism-based diagnostic scheme based on the MAM hypothesis should be both more accurate and more precise, by focusing on specific MAM-mediated lipid species, combined with the predicted elevated C99:Aβ ratio.
From the MAM point of view, it is clear that efforts to treat AD by ameliorating downstream effects (e.g., by modulating calcium levels, increasing mitochondrial bioenergetics, reducing total Aβ, and the like) will probably not work. On the other hand, two different approaches to treatment immediately present themselves: 1) re-normalizing cholesterol homeostasis, and 2) re-normalizing C99 levels. One can imagine techniques to execute both approaches [366], but these are beyond the scope of this review.
AUTHOR CONTRIBUTIONS
Estela Area-Gomez (Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing); Eric A. Schon (Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Resources; Supervision; Validation; Visualization; Writing – original draft; Writing – review & editing)
ACKNOWLEDGMENTS
We thank the many contributors to the development of the MAM hypothesis over the years, including Rishi Agrawal, H. Orhan Akman, Thomas Bird, Istvan Boldogh, Eduardo Bonilla, Gloria Patricia Cardona, Robin Chan, Adrianus de Groof, Gilbert Di Paolo, Karen Duff, Gary Gibson, David Gorin, Cristina Guardia-Laguarta, David Holtzman, Junichi Ikenouchi, Khushbu Kabra, Carla Koehler, Mari Carmen Lara Castillo, Delfina Larrea, Francisco Lopera, Mark Mattson, Mark Mehler, Tamar Michaeli, Jorge Montesinos, Patricia Morcillo, Renu Nandakumar, Marta Pera, Liza Pon, Serge Przedborski, Irina Stavrovskaya, Gary Struhl, Mark Tambini, Kirstin Tamucci, Kurenai Tanji, Masato Umeda, Kevin Velasco, Michael Yaffe, Hua Yang, Wai Haung Yu, Taekyung Yun, and Xiongwei Zhu.
FUNDING
This work was supported by grants from the U.S. National Institutes of Health (to EAG [1R01AG056387] and EAS [1R01NS117538]) and the J. Willard and Alice S. Marriott Foundation (to EAS).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Goedert M , Spillantini MG ((2006) ) A century of Alzheimer’s disease. Science 314: , 777–781. |
[2] | Corder EH , Saunders AM , Strittmatter WJ , Schmechel DE , Gaskell PC , Small GW , Roses AD , Haines JL , Pericak-Vance MA ((1993) ) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261: , 921–923. |
[3] | Agrawal RR , Montesinos J , Larrea D , Area-Gomez E , Pera M ((2020) ) The silence of the fats: A MAM’s story about Alzheimer. Neurobiol Dis 145: , 105062. |
[4] | Passer B , Pellegrini L , Russo C , Siegel RM , Lenardo MJ , Schettini G , Bachmann M , Tabaton M , D’Adamio L ((2000) ) Generation of an apoptotic intracellular peptide by γ-secretase cleavage of Alzheimer’s amyloid β protein precursor. J Alzheimers Dis 2: , 289–301. |
[5] | Flammang B , Pardossi-Piquard R , Sevalle J , Debayle D , Dabert-Gay AS , Thevenet A , Lauritzen I , Checler F ((2012) ) Evidence that the amyloid-β protein precursor intracellular domain, AICD, derives from β-secretase-generated C-terminal fragment. J Alzheimers Dis 30: , 145–153. |
[6] | Hardy J , Allsop D ((1991) ) Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci 12: , 383–388. |
[7] | Selkoe DJ ((1991) ) The molecular pathology of Alzheimer’s disease. Neuron 6: , 487–498. |
[8] | Aizenstein HJ , Nebes RD , Saxton JA , Price JC , Mathis CA , Tsopelas ND , Ziolko SK , James JA , Snitz BE , Houck PR , Bi W , Cohen AD , Lopresti BJ , DeKosky ST , Halligan EM , Klunk WE ((2008) ) Frequent amyloid deposition without significant cognitive impairment among the elderly. Arch Neurol 65: , 1509–1517. |
[9] | Arboleda-Velasquez JF , Lopera F , O’Hare M , Delgado-Tirado S , Marino C , Chmielewska N , Saez-Torres KL , Amarnani D , Schultz AP , Sperling RA , Leyton-Cifuentes D , Chen K , Baena A , Aguillon D , Rios-Romenets S , Giraldo M , Guzman-Velez E , Norton DJ , Pardilla-Delgado E , Artola A , Sanchez JS , Acosta-Uribe J , Lalli M , Kosik KS , Huentelman MJ , Zetterberg H , Blennow K , Reiman RA , Luo J , Chen Y , Thiyyagura P , Su Y , Jun GR , Naymik M , Gai X , Bootwalla M , Ji J , Shen L , Miller JB , Kim LA , Tariot PN , Johnson KA , Reiman EM , Quiroz YT ((2019) ) Resistance to autosomal dominant Alzheimer’s disease in an APOE3 Christchurch homozygote: A case report. Nat Med 25: , 1680–1683. |
[10] | Sengupta U , Nilson AN , Kayed R ((2016) ) The role of amyloid-β oligomers in toxicity, propagation, and immunotherapy. EBioMedicine 6: , 42–49. |
[11] | Reardon S ((2018) ) Frustrated Alzheimer’s researchers seek better lab mice. Nature 563: , 611–612. |
[12] | Espeseth AS , Xu M , Huang Q , Coburn CA , Jones KL , Ferrer M , Zuck PD , Strulovici B , Price EA , Wu G , Wolfe AL , Lineberger JE , Sardana M , Tugusheva K , Pietrak BL , Crouthamel MC , Lai MT , Dodson EC , Bazzo R , Shi XP , Simon AJ , Li Y , Hazuda DJ ((2005) ) Compounds that bind APP and inhibit Aβ processing in vitro suggest a novel approach to Alzheimer disease therapeutics. J Biol Chem 280: , 17792–17797. |
[13] | Das B , Yan R ((2019) ) A close look at BACE1 inhibitors for Alzheimer’s disease treatment. CNS Drugs 33: , 251–263. |
[14] | Yiannopoulou KG , Anastasiou AI , Zachariou V , Pelidou SH ((2019) ) Reasons for failed trials of disease-modifying treatments for Alzheimer disease and their contribution in recent research. Biomedicines 7: , 97. |
[15] | Doody RS , Raman R , Farlow M , Iwatsubo T , Vellas B , Joffe S , Kieburtz K , He F , Sun X , Thomas RG , Aisen PS , Alzheimer’s Disease Cooperative Study Steering Committee , Siemers E , Sethuraman G , Mohs R ((2013) ) A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med 369: , 341–350. |
[16] | Haass C , Selkoe DJ ((1993) ) Cellular processing of β-amyloid precursor protein and the genesis of amyloid β-peptide. Cell 75: , 1039–1042. |
[17] | Kakuda N , Shoji M , Arai H , Furukawa K , Ikeuchi T , Akazawa K , Takami M , Hatsuta H , Murayama S , Hashimoto Y , Miyajima M , Arai H , Nagashima Y , Yamaguchi H , Kuwano R , Nagaike K , Ihara Y , Japanese Alzheimer’s Disease Neuroimaging Initiative ((2012) ) Altered γ-secretase activity in mild cognitive impairment and Alzheimer’s disease. EMBO Mol Med 4: , 344–352. |
[18] | Jayne T , Newman M , Verdile G , Sutherland G , Munch G , Musgrave I , Moussavi Nik SH , Lardelli M ((2016) ) Evidence for and against a pathogenic role of reduced γ-secretase activity in familial Alzheimer’s disease. J Alzheimers Dis 52: , 781–799. |
[19] | McGowan E , Pickford F , Kim J , Onstead L , Eriksen J , Yu C , Skipper L , Murphy MP , Beard J , Das P , Jansen K , DeLucia M , Lin WL , Dolios G , Wang R , Eckman CB , Dickson DW , Hutton M , Hardy J , Golde T ((2005) ) Aβ42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 47: , 191–199. |
[20] | Shimojo M , Sahara N , Murayama M , Ichinose H , Takashima A ((2007) ) Decreased Aβ secretion by cells expressing familial Alzheimer’s disease-linked mutant presenilin 1. Neurosci Res 57: , 446–453. |
[21] | Lauritzen I , Pardossi-Piquard R , Bauer C , Brigham E , Abraham JD , Ranaldi S , Fraser P , St-George-Hyslop P , Le Thuc O , Espin V , Chami L , Dunys J , Checler F ((2012) ) The β-secretase-derived C-terminal fragment of βAPP, C99, but not Aβ, is a key contributor to early intraneuronal lesions in triple-transgenic mouse hippocampus. J Neurosci 32: , 16243–16255. |
[22] | Saito T , Suemoto T , Brouwers N , Sleegers K , Funamoto S , Mihira N , Matsuba Y , Yamada K , Nilsson P , Takano J , Nishimura M , Iwata N , Van Broeckhoven C , Ihara Y , Saido TC ((2011) ) Potent amyloidogenicity and pathogenicity of Aβ43. Nat Neurosci 14: , 1023–1032. |
[23] | Xia D , Kelleher RJ 3rd , Shen J ((2016) ) Loss of Aβ43 production caused by presenilin-1 mutations in the knockin mouse brain. Neuron 90: , 417–422. |
[24] | Wanngren J , Lara P , Ojemalm K , Maioli S , Moradi N , Chen L , Tjernberg LO , Lundkvist J , Nilsson I , Karlstrom H ((2014) ) Changed membrane integration and catalytic site conformation are two mechanisms behind the increased Aβ42/Aβ40 ratio by presenilin 1 familial Alzheimer-linked mutations. FEBS Open Bio 4: , 393–406. |
[25] | Yin YI , Bassit B , Zhu L , Yang X , Wang C , Li YM ((2007) ) γ-Secretase substrate concentration modulates the Aβ42/Aβ40 ratio: Implications for Alzheimer disease. J Biol Chem 282: , 23639–23644. |
[26] | Sun L , Zhou R , Yang G , Shi Y ((2017) ) Analysis of 138 pathogenic mutations in presenilin-1 on the in vitro production of Aβ42 and Aβ40 peptides by γ-secretase. Proc Natl Acad Sci U S A 114: , E476–E485. |
[27] | Fandos N , Perez-Grijalba V , Pesini P , Olmos S , Bossa M , Villemagne VL , Doecke J , Fowler C , Masters CL , Sarasa M , AIBL Research Group ((2017) ) Plasma amyloid β42/40 ratios as biomarkers for amyloid β cerebral deposition in cognitively normal individuals. Alzheimers Dement (Amst) 8: , 179–187. |
[28] | Lardelli M ((2023) ) An alternative view of familial Alzheimer’s disease genetics. J Alzheimers Dis 96: , 13–39. |
[29] | Veugelen S , Saito T , Saido TC , Chavez-Gutierrez L , De Strooper B ((2016) ) Familial Alzheimer’s disease mutations in presenilin generate amyloidogenic Aβ peptide seeds. Neuron 90: , 410–416. |
[30] | Chavez-Gutierrez L , Bammens L , Benilova I , Vandersteen A , Benurwar M , Borgers M , Lismont S , Zhou L , Van Cleynenbreugel S , Esselmann H , Wiltfang J , Serneels L , Karran E , Gijsen H , Schymkowitz J , Rousseau F , Broersen K , De Strooper B ((2012) ) The mechanism of γ-secretase dysfunction in familial Alzheimer disease. EMBO J 31: , 2261–2274. |
[31] | Fernandez MA , Klutkowski JA , Freret T , Wolfe MS ((2014) ) Alzheimer presenilin-1 mutations dramatically reduce trimming of long amyloid β-peptides (Aβ) by γ-secretase to increase 42-to-40-residue Aβ . J Biol Chem 289: , 31043–31052. |
[32] | Shen J , Kelleher RJ 3rd , ((2007) ) The presenilin hypothesis of Alzheimer’s disease: Evidence for a loss-of-function pathogenic mechanism. Proc Natl Acad Sci U S A 104: , 403–409. |
[33] | Di Fede G , Catania M , Morbin M , Rossi G , Suardi S , Mazzoleni G , Merlin M , Giovagnoli AR , Prioni S , Erbetta A , Falcone C , Gobbi M , Colombo L , Bastone A , Beeg M , Manzoni C , Francescucci B , Spagnoli A , Cantu L , Del Favero E , Levy E , Salmona M , Tagliavini F ((2009) ) A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 323: , 1473–1477. |
[34] | Zhou R , Yang G , Shi Y ((2017) ) Dominant negative effect of the loss-of-function γ-secretase mutants on the wild-type enzyme through heterooligomerization. Proc Natl Acad Sci U S A 114: , 12731–12736. |
[35] | Pera M , Larrea D , Guardia-Laguarta C , Montesinos J , Velasco KR , Agrawal RR , Xu Y , Chan RB , Di Paolo G , Mehler MF , Perumal GS , Macaluso FP , Freyberg ZZ , Acin-Perez R , Enriquez JA , Schon EA , Area-Gomez E ((2017) ) Increased localization of APP-C99 in mitochondria-associated ER membranes causes mitochondrial dysfunction in Alzheimer disease. EMBO J 36: , 3356–3371. |
[36] | Heilig EA , Xia W , Shen J , Kelleher RJ 3rd , ((2010) ) A presenilin-1 mutation identified in familial Alzheimer disease with cotton wool plaques causes a nearly complete loss of γ-secretase activity. J Biol Chem 285: , 22350–22359. |
[37] | Xia D , Watanabe H , Wu B , Lee SH , Li Y , Tsvetkov E , Bolshakov VY , Shen J , Kelleher RJ 3rd , ((2015) ) Presenilin-1 knockin mice reveal loss-of-function mechanism for familial Alzheimer’s disease. Neuron 85: , 967–981. |
[38] | Xu TH , Yan Y , Kang Y , Jiang Y , Melcher K , Xu HE ((2016) ) Alzheimer’s disease-associated mutations increase amyloid precursor protein resistance to γ-secretase cleavage and the Aβ42/Aβ40 ratio. Cell Discov 2: , 16026. |
[39] | Cavanagh C , Colby-Milley J , Bouvier D , Farso M , Chabot JG , Quirion R , Krantic S ((2013) ) βCTF-correlated burst of hippocampal TNFα occurs at a very early, pre-plaque stage in the TgCRND8 mouse model of Alzheimer’s disease. J Alzheimers Dis 36: , 233–238. |
[40] | Kaur G , Pawlik M , Gandy SE , Ehrlich ME , Smiley JF , Levy E ((2017) ) Lysosomal dysfunction in the brain of a mouse model with intraneuronal accumulation of carboxyl terminal fragments of the amyloid precursor protein. Mol Psychiatry 22: , 981–989. |
[41] | Lauritzen I , Pardossi-Piquard R , Bourgeois A , Becot A , Checler F ((2019) ) Does intraneuronal accumulation of carboxyl-terminal fragments of the amyloid precursor protein trigger early neurotoxicity in Alzheimer’s disease?. Curr Alzheimer Res 16: , 453–457. |
[42] | Mondragon-Rodriguez S , Gu N , Manseau F , Williams S ((2018) ) Alzheimer’s transgenic model Is characterized by very early brain network alterations and β-CTF fragment accumulation: Reversal by β-secretase inhibition. Front Cell Neurosci 12: , 121. |
[43] | Jiang Y , Mullaney KA , Peterhoff CM , Che S , Schmidt SD , Boyer-Boiteau A , Ginsberg SD , Cataldo AM , Mathews PM , Nixon RA ((2010) ) Alzheimer’s-related endosome dysfunction in Down syndrome is Aβ-independent but requires APP and is reversed by BACE-1 inhibition. Proc Natl Acad Sci U S A 107: , 1630–1635. |
[44] | Lee KW , Im JY , Song JS , Lee SH , Lee HJ , Ha HY , Koh JY , Gwag BJ , Yang SD , Paik SG , Han PL ((2006) ) Progressive neuronal loss and behavioral impairments of transgenic C57BL/6 inbred mice expressing the carboxy terminus of amyloid precursor protein. Neurobiol Dis 22: , 10–24. |
[45] | Berger-Sweeney J , McPhie DL , Arters JA , Greenan J , Oster-Granite ML , Neve RL ((1999) ) Impairments in learning and memory accompanied by neurodegeneration in mice transgenic for the carboxyl-terminus of the amyloid precursor protein. Brain Res Mol Brain Res 66: , 150–162. |
[46] | Oster-Granite ML , McPhie DL , Greenan J , Neve RL ((1996) ) Age-dependent neuronal and synaptic degeneration in mice transgenic for the C terminus of the amyloid precursor protein. J Neurosci 16: , 6732–6741. |
[47] | Neve RL , Boyce FM , McPhie DL , Greenan J , Oster-Granite ML ((1996) ) Transgenic mice expressing APP-C100 in the brain. Neurobiol Aging 17: , 191–203. |
[48] | Tamayev R , Matsuda S , Arancio O , D’Adamio L ((2012) ) β- but not γ-secretase proteolysis of APP causes synaptic and memory deficits in a mouse model of dementia. EMBO Mol Med 4: , 171–179. |
[49] | Freeman M ((2014) ) The rhomboid-like superfamily: Molecular mechanisms and biological roles. Annu Rev Cell Dev Biol 30: , 235–254. |
[50] | Paschkowsky S , Oestereich F , Munter LM ((2018) ) Embedded in the membrane: How lipids confer activity and specificity to intramembrane proteases. J Membr Biol 251: , 369–378. |
[51] | Hayashi T , Rizzuto R , Hajnoczky G , Su TP ((2009) ) MAM: More than just a housekeeper. Trends Cell Biol 19: , 81–88. |
[52] | Simons K , Sampaio JL ((2011) ) Membrane organization and lipid rafts. Cold Spring Harb Perspect Biol 3: , a004697. |
[53] | Montesinos J , Pera M , Larrea D , Guardia-Laguarta C , Agrawal RR , Velasco KR , Yun TD , Stavrovskaya IG , Xu Y , Koo SY , Snead AM , Sproul AA , Area-Gomez E ((2020) ) The Alzheimer’s disease-associated C99 fragment of APP regulates cellular cholesterol trafficking. EMBO J 39: , e103791. |
[54] | Mignard V , Dubois N , Lanoe D , Joalland MP , Oliver L , Pecqueur C , Heymann D , Paris F , Vallette FM , Lalier L ((2020) ) Sphingolipid distribution at mitochondria-associated membranes (MAMs) upon induction of apoptosis. J Lipid Res 61: , 1025–1037. |
[55] | Risselada HJ , Marrink SJ ((2008) ) The molecular face of lipid rafts in model membranes. Proc Natl Acad Sci U S A 105: , 17367–17372. |
[56] | Simons K , Vaz WL ((2004) ) Model systems, lipid rafts, and cell membranes. Annu Rev Biophys Biomol Struct 33: , 269–295. |
[57] | Janmey PA , Kinnunen PK ((2006) ) Biophysical properties of lipids and dynamic membranes. Trends Cell Biol 16: , 538–546. |
[58] | Urano Y , Hayashi I , Isoo N , Reid PC , Shibasaki Y , Noguchi N , Tomita T , Iwatsubo T , Hamakubo T , Kodama T ((2005) ) Association of active γ-secretase complex with lipid rafts. J Lipid Res 46: , 904–912. |
[59] | Vetrivel KS , Cheng H , Lin W , Sakurai T , Li T , Nukina N , Wong PC , Xu H , Thinakaran G ((2004) ) Association of γ-secretase with lipid rafts in post-Golgi and endosome membranes. J Biol Chem 279: , 44945–44954. |
[60] | Area-Gomez E , de Groof AJ , Boldogh I , Bird TD , Gibson GE , Koehler CM , Yu WH , Duff KE , Yaffe MP , Pon LA , Schon EA ((2009) ) Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am J Pathol 175: , 1810–1816. |
[61] | Newman M , Ebrahimie E , Lardelli M ((2014) ) Using the zebrafish model for Alzheimer’s disease research. Front Genet 5: , 189. |
[62] | Schreiner B , Hedskog L , Wiehager B , Ankarcrona M ((2015) ) Amyloid-β peptides are generated in mitochondria-associated endoplasmic reticulum membranes. J Alzheimers Dis 43: , 369–374. |
[63] | Guardia-Laguarta C , Coma M , Pera M , Clarimon J , Sereno L , Agullo JM , Molina-Porcel L , Gallardo E , Deng A , Berezovska O , Hyman BT , Blesa R , Gomez-Isla T , Lleo A ((2009) ) Mild cholesterol depletion reduces amyloid-β production by impairing APP trafficking to the cell surface. J Neurochem 110: , 220–230. |
[64] | Hur JY , Welander H , Behbahani H , Aoki M , Franberg J , Winblad B , Frykman S , Tjernberg LO ((2008) ) Active γ-secretase is localized to detergent-resistant membranes in human brain. FEBS J 275: , 1174–1187. |
[65] | Wolfe MS ((2020) ) Unraveling the complexity of γ-secretase. Semin Cell Dev Biol 105: , 3–11. |
[66] | Czirr E , Cottrell BA , Leuchtenberger S , Kukar T , Ladd TB , Esselmann H , Paul S , Schubenel R , Torpey JW , Pietrzik CU , Golde TE , Wiltfang J , Baumann K , Koo EH , Weggen S ((2008) ) Independent generation of Aβ42 and Aβ38 peptide species by γ-secretase. J Biol Chem 283: , 17049–17054. |
[67] | Golde TE , Ran Y , Felsenstein KM ((2012) ) Shifting a complex debate on γ-secretase cleavage and Alzheimer’s disease. EMBO J 31: , 2237–2239. |
[68] | Olsson F , Schmidt S , Althoff V , Munter LM , Jin S , Rosqvist S , Lendahl U , Multhaup G , Lundkvist J ((2014) ) Characterization of intermediate steps in amyloid β (Aβ) production under near-native conditions. J Biol Chem 289: , 1540–1550. |
[69] | Perrin F , Papadopoulos N , Suelves N , Opsomer R , Vadukul DM , Vrancx C , Smith SO , Vertommen D , Kienlen-Campard P , Constantinescu SN ((2020) ) Dimeric transmembrane orientations of APP/C99 regulate γ-secretase processing line impacting signaling and oligomerization. iScience 23: , 101887. |
[70] | Qi-Takahara Y , Morishima-Kawashima M , Tanimura Y , Dolios G , Hirotani N , Horikoshi Y , Kametani F , Maeda M , Saido TC , Wang R , Ihara Y ((2005) ) Longer forms of amyloid β prote, Imlications for the mechanism of intramembrane cleavage by γ-secretase. J Neurosci 25: , 436–445 |
[71] | Pantelopulos GA , Straub JE , Thirumalai D , Sugita Y ((2018) ) Structure of APP-C991 - 99 and implications for role of extra-membrane domains in function and oligomerization. Biochim Biophys Acta Biomembr 1860: , 1698–1708. |
[72] | Kaiser HJ , Orlowski A , Rog T , Nyholm TK , Chai W , Feizi T , Lingwood D , Vattulainen I , Simons K ((2011) ) Lateral sorting in model membranes by cholesterol-mediated hydrophobic matching. Proc Natl Acad Sci U S A 108: , 16628–16633. |
[73] | Winkler E , Kamp F , Scheuring J , Ebke A , Fukumori A , Steiner H ((2012) ) Generation of Alzheimer disease-associated amyloid β42/43 peptide by γ-secretase can be inhibited directly by modulation of membrane thickness. J Biol Chem 287: , 21326–21334. |
[74] | Area-Gomez E , Del Carmen Lara Castillo M , Tambini MD , Guardia-Laguarta C , de Groof AJ , Madra M , Ikenouchi J , Umeda M , Bird TD , Sturley SL , Schon EA ((2012) ) Upregulated function of mitochondria-associated ER membranes in Alzheimer disease. EMBO J 31: , 4106–4123. |
[75] | Alonso A , Goni FM ((2018) ) The physical properties of ceramides in membranes. Annu Rev Biophys 47: , 633–654. |
[76] | Kinoshita M , Suzuki KG , Matsumori N , Takada M , Ano H , Morigaki K , Abe M , Makino A , Kobayashi T , Hirosawa KM , Fujiwara TK , Kusumi A , Murata M ((2017) ) Raft-based sphingomyelin interactions revealed by new fluorescent sphingomyelin analogs. J Cell Biol 216: , 1183–1204. |
[77] | Silva LC , Futerman AH , Prieto M ((2009) ) Lipid raft composition modulates sphingomyelinase activity and ceramide-induced membrane physical alterations. Biophys J 96: , 3210–3222. |
[78] | Bretscher MS , Munro S ((1993) ) Cholesterol and the Golgi apparatus. Science 261: , 1280–1281. |
[79] | Hartmann T , Bieger SC , Bruhl B , Tienari PJ , Ida N , Allsop D , Roberts GW , Masters CL , Dotti CG , Unsicker K , Beyreuther K ((1997) ) Distinct sites of intracellular production for Alzheimer’s disease Aβ40/42 amyloid peptides. Nat Med 3: , 1016–1020. |
[80] | Holmes O , Paturi S , Ye W , Wolfe MS , Selkoe DJ ((2012) ) Effects of membrane lipids on the activity and processivity of purified γ-secretase. Biochemistry 51: , 3565–3575. |
[81] | Sobhanifar S , Schneider B , Lohr F , Gottstein D , Ikeya T , Mlynarczyk K , Pulawski W , Ghoshdastider U , Kolinski M , Filipek S , Guntert P , Bernhard F , Dotsch V ((2010) ) Structural investigation of the C-terminal catalytic fragment of presenilin 1. Proc Natl Acad Sci U S A 107: , 9644–9649. |
[82] | Fukumori A , Steiner H ((2016) ) Substrate recruitment of γ-secretase and mechanism of clinical presenilin mutations revealed by photoaffinity mapping. EMBO J 35: , 1628–1643. |
[83] | Chau DM , Crump CJ , Villa JC , Scheinberg DA , Li YM ((2012) ) Familial Alzheimer disease presenilin-1 mutations alter the active site conformation of γ-secretase. J Biol Chem 287: , 17288–17296. |
[84] | Wong E , Liao GP , Chang JC , Xu P , Li YM , Greengard P ((2019) ) GSAP modulates γ-secretase specificity by inducing conformational change in PS1. Proc Natl Acad Sci U S A 116: , 6385–6390. |
[85] | Herl L , Thomas AV , Lill CM , Banks M , Deng A , Jones PB , Spoelgen R , Hyman BT , Berezovska O ((2009) ) Mutations in amyloid precursor protein affect its interactions with presenilin/γ-secretase. Mol Cell Neurosci 41: , 166–174. |
[86] | Uemura K , Lill CM , Li X , Peters JA , Ivanov A , Fan Z , DeStrooper B , Bacskai BJ , Hyman BT , Berezovska O ((2009) ) Allosteric modulation of PS1/γ-secretase conformation correlates with amyloid β42/40 ratio. PLoS One 4: , e7893. |
[87] | Uemura K , Farner KC , Nasser-Ghodsi N , Jones P , Berezovska O ((2011) ) Reciprocal relationship between APP positioning relative to the membrane and PS1 conformation. Mol Neurodegener 6: , 15. |
[88] | Osenkowski P , Ye W , Wang R , Wolfe MS , Selkoe DJ ((2008) ) Direct and potent regulation of γ-secretase by its lipid microenvironment. J Biol Chem 283: , 22529–22540. |
[89] | Schon EA , Area-Gomez E ((2013) ) Mitochondria-associated ER membranes in Alzheimer disease. Mol Cell Neurosci 55: , 26–36. |
[90] | Yan Y , Wang C ((2006) ) Aβ42 is more rigid than Aβ40 at the C terminus: Implications for Aβ aggregation and toxicity. J Mol Biol 364: , 853–862. |
[91] | Zhou L , Brouwers N , Benilova I , Vandersteen A , Mercken M , Van Laere K , Van Damme P , Demedts D , Van Leuven F , Sleegers K , Broersen K , Van Broeckhoven C , Vandenberghe R , De Strooper B ((2011) ) Amyloid precursor protein mutation E682K at the alternative β-secretase cleavage β’-site increases Aβ generation. EMBO Mol Med 3: , 291–302. |
[92] | Li N , Liu K , Qiu Y , Ren Z , Dai R , Deng Y , Qing H ((2016) ) Effect of presenilin mutations on APP cleavage; insights into the pathogenesis of FAD. Front Aging Neurosci 8: , 51. |
[93] | Koo EH , Lansbury PT Jr. , Kelly JW ((1999) ) Amyloid diseases: Abnormal protein aggregation in neurodegeneration. Proc Natl Acad Sci U S A 96: , 9989–9990. |
[94] | Lauritzen I , Becot A , Bourgeois A , Pardossi-Piquard R , Biferi MG , Barkats M , Checler F ((2019) ) Targeting γ-secretase triggers the selective enrichment of oligomeric APP-CTFs in brain extracellular vesicles from Alzheimer cell and mouse models. Transl Neurodegener 8: , 35. |
[95] | Balasubramanian AB , Kawas CH , Peltz CB , Brookmeyer R , Corrada MM ((2012) ) Alzheimer disease pathology and longitudinal cognitive performance in the oldest-old with no dementia. Neurology 79: , 915–921. |
[96] | Rodrigue KM , Kennedy KM , Park DC ((2009) ) Beta-amyloid deposition and the aging brain. Neuropsychol Rev 19: , 436–450. |
[97] | Pulina MV , Hopkins M , Haroutunian V , Greengard P , Bustos V ((2020) ) C99 selectively accumulates in vulnerable neurons in Alzheimer’s disease. Alzheimers Dement 16: , 273–282. |
[98] | Perez-Tur J , Froelich S , Prihar G , Crook R , Baker M , Duff K , Wragg M , Busfield F , Lendon C , Clark RF , et al. ((1995) ) A mutation in Alzheimer’s disease destroying a splice acceptor site in the presenilin-1 gene. Neuroreport 7: , 297–301. |
[99] | Gomez-Isla T , Growdon WB , McNamara MJ , Nochlin D , Bird TD , Arango JC , Lopera F , Kosik KS , Lantos PL , Cairns NJ , Hyman BT ((1999) ) The impact of different presenilin 1 and presenilin 2 mutations on amyloid deposition, neurofibrillary changes and neuronal loss in the familial Alzheimer’s disease brain: Evidence for otherhenotype-modifying factors. Brain 122: , 1709–1719 |
[100] | Steiner H , Revesz T , Neumann M , Romig H , Grim MG , Pesold B , Kretzschmar HA , Hardy J , Holton JL , Baumeister R , Houlden H , Haass C ((2001) ) A pathogenic presenilin-1 deletion causes abberrant Aβ42 production in the absence of congophilic amyloid plaques. J Biol Chem 276: , 7233–7239. |
[101] | Hunter S , Brayne C ((2017) ) Do anti-amyloid beta protein antibody cross reactivities confound Alzheimer disease research?. J Negat Results Biomed 16: , 1. |
[102] | Grant MKO , Handoko M , Rozga M , Brinkmalm G , Portelius E , Blennow K , Ashe KH , Zahs KR , Liu P ((2019) ) Human cerebrospinal fluid 6E10-immunoreactive protein species contain amyloid precursor protein fragments. PLoS One 14: , e0212815. |
[103] | Song JX , Malampati S , Zeng Y , Durairajan SSK , Yang CB , Tong BC , Iyaswamy A , Shang WB , Sreenivasmurthy SG , Zhu Z , Cheung KH , Lu JH , Tang C , Xu N , Li M ((2020) ) A small molecule transcription factor EB activator ameliorates beta-amyloid precursor protein and Tau pathology in Alzheimer’s disease models. Aging Cell 19: , e13069. |
[104] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[105] | Pettersen JA , Patry DG , St George-Hyslop PH , Curry B ((2011) ) Variant Alzheimer disease with spastic paraparesis: A rare presenilin-1 mutation. Can J Neurol Sci 38: , 659–661. |
[106] | Dermaut B , Kumar-Singh S , Engelborghs S , Theuns J , Rademakers R , Saerens J , Pickut BA , Peeters K , van den Broeck M , Vennekens K , Claes S , Cruts M , Cras P , Martin JJ , Van Broeckhoven C , De Deyn PP ((2004) ) A novel presenilin 1 mutation associated with Pick’s disease but not β-amyloid plaques. Ann Neurol 55: , 617–626. |
[107] | Asher S , Priefer R ((2022) ) Alzheimer’s disease failed clinical trials. Life Sci 306: , 120861. |
[108] | Young-Pearse TL , Chen AC , Chang R , Marquez C , Selkoe DJ ((2008) ) Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev 3: , 15. |
[109] | Zhang T , Chen D , Lee TH ((2019) ) Phosphorylation signaling in APP processing in Alzheimer’s disease. Int J Mol Sci 21: , 209. |
[110] | Manville RW , Abbott GW ((2021) ) The amyloid precursor protein C99 fragment modulates voltage-gated potassium channels. Cell Physiol Biochem 55: , 157–170. |
[111] | Lee SE , Kwon D , Shin N , Kong D , Kim NG , Kim HY , Kim MJ , Choi SW , Kang KS ((2022) ) Accumulation of APP-CTF induces mitophagy dysfunction in the iNSCs model of Alzheimer’s disease. Cell Death Discov 8: , 1. |
[112] | DelBove CE , Strothman CE , Lazarenko RM , Huang H , Sanders CR , Zhang Q ((2019) ) Reciprocal modulation between amyloid precursor protein and synaptic membrane cholesterol revealed by live cell imaging. Neurobiol Dis 127: , 449–461. |
[113] | Yang X , Sun GY , Eckert GP , Lee JC ((2014) ) Cellular membrane fluidity in amyloid precursor protein processing. Mol Neurobiol 50: , 119–129. |
[114] | Zerbinatti CV , Cordy JM , Chen CD , Guillily M , Suon S , Ray WJ , Seabrook GR , Abraham CR , Wolozin B ((2008) ) Oxysterol-binding protein-1 (OSBP1) modulates processing and trafficking of the amyloid precursor protein. Mol Neurodegener 3: , 5. |
[115] | Puglielli L , Tanzi RE , Kovacs DM ((2003) ) Alzheimer’s disease: The cholesterol connection. Nat Neurosci 6: , 345–351. |
[116] | Beel AJ , Mobley CK , Kim HJ , Tian F , Hadziselimovic A , Jap B , Prestegard JH , Sanders CR ((2008) ) Structural studies of the transmembrane C-terminal domain of the amyloid precursor protein (APP): Does APP function as a cholesterol sensor?. Biochemistry 47: , 9428–9446. |
[117] | Barrett PJ , Song Y , Van Horn WD , Hustedt EJ , Schafer JM , Hadziselimovic A , Beel AJ , Sanders CR ((2012) ) The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336: , 1168–1171. |
[118] | Di Scala C , Yahi N , Lelievre C , Garmy N , Chahinian H , Fantini J ((2013) ) Biochemical identification of a linear cholesterol-binding domain within Alzheimer’s β amyloid peptide. ACS Chem Neurosci 4: , 509–517. |
[119] | Yao ZX , Papadopoulos V ((2002) ) Function of β-amyloid in cholesterol transport: A lead to neurotoxicity. FASEB J 16: , 1677–1679. |
[120] | Higashide H , Ishihara S , Nobuhara M , Ihara Y , Funamoto S ((2017) ) Alanine substitutions in the GXXXG motif alter C99 cleavage by γ-secretase but not its dimerization. J Neurochem 140: , 955–962. |
[121] | Song Y , Hustedt EJ , Brandon S , Sanders CR ((2013) ) Competition between homodimerization and cholesterol binding to the C99 domain of the amyloid precursor protein. Biochemistry 52: , 5051–5064. |
[122] | Shi Q , Chen J , Zou X , Tang X ((2022) ) Intracellular cholesterol synthesis and transport. Front Cell Dev Biol 10: , 819281. |
[123] | Ridsdale A , Denis M , Gougeon PY , Ngsee JK , Presley JF , Zha X ((2006) ) Cholesterol is required for efficient endoplasmic reticulum-to-Golgi transport of secretory membrane proteins. Mol Biol Cell 17: , 1593–1605. |
[124] | Luo J , Yang H , Song BL ((2020) ) Mechanisms and regulation of cholesterol homeostasis. Nat Rev Mol Cell Biol 21: , 225–245. |
[125] | Brown MS , Goldstein JL ((1986) ) A receptor-mediated pathway for cholesterol homeostasis. Science 232: , 34–47. |
[126] | Naito T , Ercan B , Krshnan L , Triebl A , Koh DHZ , Wei FY , Tomizawa K , Torta FT , Wenk MR , Saheki Y ((2019) ) Movement of accessible plasma membrane cholesterol by the GRAMD1 lipid transfer protein complex. Elife 8: , e51401. |
[127] | Holtta-Vuori M , Ikonen E ((2006) ) Endosomal cholesterol traffic: Vesicular and non-vesicular mechanisms meet. Biochem Soc Trans 34: , 392–394. |
[128] | Ikonen E , Zhou X ((2021) ) Cholesterol transport between cellular membranes: A balancing act between interconnected lipid fluxes. Dev Cell 56: , 1430–1436. |
[129] | Diaz-Rohrer BB , Levental KR , Simons K , Levental I ((2014) ) Membrane raft association is a determinant of plasma membrane localization. Proc Natl Acad Sci U S A 111: , 8500–8505. |
[130] | Hoglinger D , Burgoyne T , Sanchez-Heras E , Hartwig P , Colaco A , Newton J , Futter CE , Spiegel S , Platt FM , Eden ER ((2019) ) NPC1 regulates ER contacts with endocytic organelles to mediate cholesterol egress. Nat Commun 10: , 4276. |
[131] | Eden ER ((2016) ) The formation and function of ER-endosome membrane contact sites. Biochim Biophys Acta 1861: , 874–879. |
[132] | Chang TY , Chang CC , Ohgami N , Yamauchi Y ((2006) ) Cholesterol sensing, trafficking, and esterification. Annu Rev Cell Dev Biol 22: , 129–157. |
[133] | Kwart D , Gregg A , Scheckel C , Murphy EA , Paquet D , Duffield M , Fak J , Olsen O , Darnell RB , Tessier-Lavigne M ((2019) ) A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP β-CTFs, not Aβ . Neuron 104: , 1022. |
[134] | Lee JH , Yang DS , Goulbourne CN , Im E , Stavrides P , Pensalfini A , Chan H , Bouchet-Marquis C , Bleiwas C , Berg MJ , Huo C , Peddy J , Pawlik M , Levy E , Rao M , Staufenbiel M , Nixon RA ((2022) ) Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat Neurosci 25: , 688–701. |
[135] | Im E , Jiang Y , Stavrides PH , Darji S , Erdjument-Bromage H , Neubert TA , Choi JY , Wegiel J , Lee JH , Nixon RA ((2023) ) Lysosomal dysfunction in Down syndrome and Alzheimer mouse models is caused by v-ATPase inhibition by Tyr682-phosphorylated APP βCTF.eadg. Sci Adv 9: , 1925. |
[136] | Gao S , Casey AE , Sargeant TJ , Makinen VP ((2018) ) Genetic variation within endolysosomal system is associated with late-onset Alzheimer’s disease. Brain 141: , 2711–2720. |
[137] | Jiang Y , Rigoglioso A , Peterhoff CM , Pawlik M , Sato Y , Bleiwas C , Stavrides P , Smiley JF , Ginsberg SD , Mathews PM , Levy E , Nixon RA ((2016) ) Partial BACE1 reduction in a Down syndrome mouse model blocks Alzheimer-related endosomal anomalies and cholinergic neurodegeneration: Role of APP-CTF. Neurobiol Aging 39: , 90–98. |
[138] | La Rosa LR , Perrone L , Nielsen MS , Calissano P , Andersen OM , Matrone C ((2015) ) Y682G mutation of amyloid precursor protein promotes endo-lysosomal dysfunction by disrupting APP-SorLA interaction. Front Cell Neurosci 9: , 109. |
[139] | Simons K , Ehehalt R ((2002) ) Cholesterol, lipid rafts, and disease. J Clin Invest 110: , 597–603. |
[140] | Shibuya Y , Chang CC , Chang TY ((2015) ) ACAT1/SOAT1 as a therapeutic target for Alzheimer’s disease. Future Med Chem 7: , 2451–2467. |
[141] | Choy RW , Cheng Z , Schekman R ((2012) ) Amyloid precursor protein (APP) traffics from the cell surface via endosomes for amyloid β (Aβ) production in the trans-Golgi network. Proc Natl Acad Sci U S A 109: , .E2077–E2082. |
[142] | Chia PZ , Toh WH , Sharples R , Gasnereau I , Hill AF , Gleeson PA ((2013) ) Intracellular itinerary of internalised β-secretase, BACE1, and its potential impact on β-amyloid peptide biogenesis. Traffic 14: , 997–1013. |
[143] | Tan JZA , Fourriere L , Wang J , Perez F , Boncompain G , Gleeson PA ((2020) ) Distinct anterograde trafficking pathways of BACE1 and amyloid precursor protein from the TGN and the regulation of amyloid-β production. Mol Biol Cell 31: , 27–44. |
[144] | Vassar R ((2004) ) BACE1: The β-secretase enzyme in Alzheimer’s disease. J Mol Neurosci 23: , 105–114. |
[145] | Elbaz-Alon Y , Guo Y , Segev N , Harel M , Quinnell DE , Geiger T , Avinoam O , Li D , Nunnari J ((2020) ) PDZD8 interacts with Protrudin and Rab7 at ER-late endosome membrane contact sites associated with mitochondria. Nat Commun 11: , 3645. |
[146] | Friedman JR , Dibenedetto JR , West M , Rowland AA , Voeltz GK ((2013) ) Endoplasmic reticulum-endosome contact increases as endosomes traffic and mature. Mol Biol Cell 24: , 1030–1040. |
[147] | Raiborg C , Wenzel EM , Stenmark H ((2015) ) ER-endosome contact sites: Molecular compositions and functions. EMBO J 34: , 1848–1858. |
[148] | Dong R , Saheki Y , Swarup S , Lucast L , Harper JW , De Camilli P ((2016) ) Endosome-ER contacts control actin nucleation and retromer function through VAP-dependent regulation of PI4P. Cell 166: , 408–423. |
[149] | Mahfoud R , Garmy N , Maresca M , Yahi N , Puigserver A , Fantini J ((2002) ) Identification of a common sphingolipid-binding domain in Alzheimer, prion, and HIV-1 proteins. J Biol Chem 277: , 11292–11296. |
[150] | Hanbouch L , Schaack B , Kasri A , Fontaine G , Gkanatsiou E , Brinkmalm G , Camporesi E , Portelius E , Blennow K , Mourier G , Gilles N , Millan MJ , Marquer C , Zetterberg H , Boussicault L , Potier MC ((2022) ) Specific mutations in the cholesterol-binding site of APP alter its processing and favor the production of shorter, less toxic Aβ peptides. Mol Neurobiol 59: , 7056–7073. |
[151] | Yuyama K , Igarashi Y ((2017) ) Exosomes as carriers of Alzheimer’s amyloid-β . Front Neurosci 11: , 229. |
[152] | Burns MP , Rebeck GW ((2010) ) Intracellular cholesterol homeostasis and amyloid precursor protein processing. Biochim Biophys Acta 1801: , 853–859. |
[153] | Bjorkhem I , Meaney S ((2004) ) Brain cholesterol: Long secret life behind a barrier. Arterioscler Thromb Vasc Biol 24: , 806–815. |
[154] | Kotti TJ , Ramirez DM , Pfeiffer BE , Huber KM , Russell DW ((2006) ) Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc Natl Acad Sci U S A 103: , 3869–3874. |
[155] | Djelti F , Braudeau J , Hudry E , Dhenain M , Varin J , Bieche I , Marquer C , Chali F , Ayciriex S , Auzeil N , Alves S , Langui D , Potier MC , Laprevote O , Vidaud M , Duyckaerts C , Miles R , Aubourg P , Cartier N ((2015) ) CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer’s disease. Brain 138: , 2383–2398. |
[156] | Zhao Y , Hu D , Wang R , Sun X , Ropelewski P , Hubler Z , Lundberg K , Wang Q , Adams DJ , Xu R , Qi X ((2022) ) ATAD3A oligomerization promotes neuropathology and cognitive deficits in Alzheimer’s disease models. Nat Commun 13: , 1121. |
[157] | Issop L , Fan J , Lee S , Rone MB , Basu K , Mui J , Papadopoulos V ((2015) ) Mitochondria-associated membrane formation in hormone-stimulated Leydig cell steroidogenesis: Role of ATAD3. Endocrinology 156: , 334–345. |
[158] | Bryleva EY , Rogers MA , Chang CC , Buen F , Harris BT , Rousselet E , Seidah NG , Oddo S , LaFerla FM , Spencer TA , Hickey WF , Chang TY ((2010) ) ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc Natl Acad Sci U S A 107: , 3081–3086. |
[159] | Puglielli L , Ellis BC , Ingano LA , Kovacs DM ((2004) ) Role of acyl-coenzyme A:cholesterol acyltransferase activity in the processing of the amyloid precursor protein. J Mol Neurosci 24: , 93–96. |
[160] | Puglielli L , Konopka G , Pack-Chung E , Ingano LA , Berezovska O , Hyman BT , Chang TY , Tanzi RE , Kovacs DM ((2001) ) Acyl-coenzyme A:cholesterol acyltransferase modulates the generation of the amyloid β-peptide. Nat Cell Biol 3: , 905–912. |
[161] | Lange Y , Ye J , Steck TL ((2014) ) Essentially all excess fibroblast cholesterol moves from plasma membranes to intracellular compartments. PLoS One 9: , e98482. |
[162] | Cossec JC , Simon A , Marquer C , Moldrich RX , Leterrier C , Rossier J , Duyckaerts C , Lenkei Z , Potier MC ((2010) ) Clathrin-dependent APP endocytosis and Aβ secretion are highly sensitive to the level of plasma membrane cholesterol. Biochim Biophys Acta 1801: , 846–852. |
[163] | DelBove CE , Deng XZ , Zhang Q ((2018) ) The fate of nascent APP in hippocampal neurons: A live cell imaging study. ACS Chem Neurosci 9: , 2225–2232. |
[164] | Jin C , Wang J , Wang Y , Jia B , Guo X , Yang G , Xu P , Greengard P , Zhou R , Shi Y ((2022) ) Modulation of amyloid precursor protein cleavage by γ-secretase activating protein through phase separation. Proc Natl Acad Sci U S A 119: , e2122292119. |
[165] | Marquer C , Devauges V , Cossec JC , Liot G , Lecart S , Saudou F , Duyckaerts C , Leveque-Fort S , Potier MC ((2011) ) Local cholesterol increase triggers amyloid precursor protein-Bace1 clustering in lipid rafts and rapid endocytosis. FASEB J 25: , 1295–1305. |
[166] | Tambini MD , Pera M , Kanter E , Yang H , Guardia-Laguarta C , Holtzman D , Sulzer D , Area-Gomez E , Schon EA ((2016) ) ApoE4 upregulates the activity of mitochondria-associated ER membranes. EMBO Rep 17: , 27–36. |
[167] | Andrews SJ , Fulton-Howard B , Goate A ((2020) ) Interpretation of risk loci from genome-wide association studies of Alzheimer’s disease. Lancet Neurol 19: , 326–335. |
[168] | Chen X , Hui L , Soliman ML , Geiger JD ((2014) ) Altered cholesterol intracellular trafficking and the development of pathological hallmarks of sporadic AD. J Parkinsons Dis Alzheimers Dis 1: , 8. |
[169] | Heeren J , Grewal T , Laatsch A , Rottke D , Rinninger F , Enrich C , Beisiegel U ((2003) ) Recycling of apoprotein E is associated with cholesterol efflux and high density lipoprotein internalization. J Biol Chem 278: , 14370–14378. |
[170] | Heeren J , Grewal T , Laatsch A , Becker N , Rinninger F , Rye KA , Beisiegel U ((2004) ) Impaired recycling of apolipoprotein E4 is associated with intracellular cholesterol accumulation. J Biol Chem 279: , 55483–55492. |
[171] | Jones L , Holmans PA , Hamshere ML , Harold D , Moskvina V , Ivanov D , Pocklington A , Abraham R , Hollingworth P , Sims R , Gerrish A , Pahwa JS , Jones N , Stretton A , Morgan AR , Lovestone S , Powell J , Proitsi P , Lupton MK , Brayne C , Rubinsztein DC , Gill M , Lawlor B , Lynch A , Morgan K , Brown KS , Passmore PA , Craig D , McGuinness B , Todd S , Holmes C , Mann D , Smith AD , Love S , Kehoe PG , Mead S , Fox N , Rossor M , Collinge J , Maier W , Jessen F , Schurmann B , Heun R , Kolsch H , van den Bussche H , Heuser I , Peters O , Kornhuber J , Wiltfang J , Dichgans M , Frolich L , Hampel H , Hull M , Rujescu D , Goate AM , Kauwe JS , Cruchaga C , Nowotny P , Morris JC , Mayo K , Livingston G , Bass NJ , Gurling H , McQuillin A , Gwilliam R , Deloukas P , Al-Chalabi A , Shaw CE , Singleton AB , Guerreiro R , Muhleisen TW , Nothen MM , Moebus S , Jockel KH , Klopp N , Wichmann HE , Ruther E , Carrasquillo MM , Pankratz VS , Younkin SG , Hardy J , O’Donovan MC , Owen MJ , Williams J ((2010) ) Genetic evidence implicates the immune system and cholesterol metabolism in the aetiology of Alzheimer’s disease. PLoS One 5: , e13950. |
[172] | Fukumoto H , Cheung BS , Hyman BT , Irizarry MC ((2002) ) β-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol 59: , 1381–1389. |
[173] | Holsinger RM , McLean CA , Beyreuther K , Masters CL , Evin G ((2002) ) Increased expression of the amyloid precursor β-secretase in Alzheimer’s disease. Ann Neurol 51: , 783–786. |
[174] | Yang LB , Lindholm K , Yan R , Citron M , Xia W , Yang XL , Beach T , Sue L , Wong P , Price D , Li R , Shen Y ((2003) ) Elevated β-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med 9: , 3–4. |
[175] | Haass C , Lemere CA , Capell A , Citron M , Seubert P , Schenk D , Lannfelt L , Selkoe DJ ((1995) ) The Swedish mutation causes early-onset Alzheimer’s disease by β-secretase cleavage within the secretory pathway. Nat Med 1: , 1291–1296. |
[176] | Busciglio J , Pelsman A , Wong C , Pigino G , Yuan M , Mori H , Yankner BA ((2002) ) Altered metabolism of the amyloid β precursor protein is associated with mitochondrial dysfunction in Down’s syndrome. Neuron 33: , 677–688. |
[177] | Small SA ((2008) ) Retromer sorting: A pathogenic pathway in late-onset Alzheimer disease. Arch Neurol 65: , 323–328. |
[178] | Sullivan CP , Jay AG , Stack EC , Pakaluk M , Wadlinger E , Fine RE , Wells JM , Morin PJ ((2011) ) Retromer disruption promotes amyloidogenic APP processing. Neurobiol Dis 43: , 338–345. |
[179] | Marquer C , Laine J , Dauphinot L , Hanbouch L , Lemercier-Neuillet C , Pierrot N , Bossers K , Le M , Corlier F , Benstaali C , Saudou F , Thinakaran G , Cartier N , Octave JN , Duyckaerts C , Potier MC ((2014) ) Increasing membrane cholesterol of neurons in culture recapitulates Alzheimer’s disease early phenotypes. Mol Neurodegener 9: , 60. |
[180] | Oram JF , Heinecke JW ((2005) ) ATP-binding cassette transporter A1: A cell cholesterol exporter that protects against cardiovascular disease. Physiol Rev 85: , 1343–1372. |
[181] | Nordestgaard LT , Tybjaerg-Hansen A , Nordestgaard BG , Frikke-Schmidt R ((2015) ) Loss-of-function mutation in ABCA1 and risk of Alzheimer’s disease and cerebrovascular disease. Alzheimers Dement 11: , 1430–1438. |
[182] | Vance JE ((2014) ) MAM (mitochondria-associated membranes) in mammalian cells: Lipids and beyond. Biochim Biophys Acta 1841: , 595–609. |
[183] | Friedman JR , Lackner LL , West M , DiBenedetto JR , Nunnari J , Voeltz GK ((2011) ) ER tubules mark sites of mitochondrial division. Science 334: , 358–362. |
[184] | Giacomello M , Pellegrini L ((2016) ) The coming of age of the mitochondria-ER contact: A matter of thickness. Cell Death Differ 23: , 1417–1427. |
[185] | Wu H , Carvalho P , Voeltz GK ((2018) ) Here, there, and everywhere: The importance of ER membrane contact sites. Science 361: , eaan5835. |
[186] | de Brito OM , Scorrano L ((2008) ) Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 456: , 605–610. |
[187] | Stoica R , De Vos KJ , Paillusson S , Mueller S , Sancho RM , Lau KF , Vizcay-Barrena G , Lin WL , Xu YF , Lewis J , Dickson DW , Petrucelli L , Mitchell JC , Shaw CE , Miller CC ((2014) ) ER-mitochondria associations are regulated by the VAPB-PTPIP51 interaction and are disrupted by ALS/FTD-associated TDP-43. Nat Commun 5: , 3996. |
[188] | Thi My Nhung T , Phuoc Long N , Diem Nghi T , Suh Y , Hoang Anh N , Jung CW , Minh Triet H , Jung M , Woo Y , Yoo J , Noh S , Kim SJ , Lee SB , Park S , Thomas G , Simmen T , Mun J , Rhee HW , Kwon SW , Park SK ((2023) ) Genome-wide kinase-MAM interactome screening reveals the role of CK2A1 in MAM Ca(2+) dynamics linked to DEE66. Proc Natl Acad Sci U S A 120: , e2303402120. |
[189] | Yang J , Sun M , Chen R , Ye X , Wu B , Liu Z , Zhang J , Gao X , Cheng R , He C , He J , Wang X , Huang L ((2023) ) Mitochondria-associated membrane protein PACS2 maintains right cardiac function in hypobaric hypoxia. iScience 26: , 106328. |
[190] | Xu P , Chang JC , Zhou X , Wang W , Bamkole M , Wong E , Bettayeb K , Jiang LL , Huang T , Luo W , Xu H , Nairn AC , Flajolet M , Ip NY , Li YM , Greengard P ((2021) ) GSAP regulates lipid homeostasis and mitochondrial function associated with Alzheimer’s disease. J Exp Med 218: , e20202446. |
[191] | Del Prete D , Suski JM , Oules B , Debayle D , Gay AS , Lacas-Gervais S , Bussiere R , Bauer C , Pinton P , Paterlini-Brechot P , Wieckowski MR , Checler F , Chami M ((2017) ) Localization and processing of the Amyloid-β protein precursor in mitochondria-associated membranes. J Alzheimers Dis 55: , 1549–1570. |
[192] | Vance JE ((2020) ) Inter-organelle membrane contact sites: Implications for lipid metabolism. Biol Direct 15: , 24. |
[193] | Doghman-Bouguerra M , Lalli E ((2017) ) The ER-mitochondria couple: In life and death from steroidogenesis to tumorigenesis. Mol Cell Endocrinol 441: , 176–184. |
[194] | Prasad M , Kaur J , Pawlak KJ , Bose M , Whittal RM , Bose HS ((2015) ) Mitochondria-associated endoplasmic reticulum membrane (MAM) regulates steroidogenic activity via steroidogenic acute regulatory protein (StAR)-voltage-dependent anion channel 2 (VDAC2) interaction. J Biol Chem 290: , 2604–2616. |
[195] | Rieusset J ((2018) ) The role of endoplasmic reticulum-mitochondria contact sites in the control of glucose homeostasis: An update. Cell Death Dis 9: , 388. |
[196] | Csordas G , Varnai P , Golenar T , Roy S , Purkins G , Schneider TG , Balla T , Hajnoczky G ((2010) ) Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol Cell 39: , 121–132. |
[197] | Csordas G , Weaver D , Hajnoczky G ((2018) ) Endoplasmic reticulum-mitochondrial contactology: Structure and signaling functions. Trends Cell Biol 28: , 523–540. |
[198] | Yang S , Zhou R , Zhang C , He S , Su Z ((2020) ) Mitochondria-associated endoplasmic reticulum membranes in the pathogenesis of type 2 diabetes mellitus. Front Cell Dev Biol 8: , 571554. |
[199] | Lumsden AL , Rogers JT , Majd S , Newman M , Sutherland GT , Verdile G , Lardelli M ((2018) ) Dysregulation of neuronal iron homeostasis as an alternative unifying effect of mutations causing familial Alzheimer’s disease. Front Neurosci 12: , 533. |
[200] | Xue Y , Schmollinger S , Attar N , Campos OA , Vogelauer M , Carey MF , Merchant SS , Kurdistani SK ((2017) ) Endoplasmic reticulum-mitochondria junction is required for iron homeostasis. J Biol Chem 292: , 13197–13204. |
[201] | van Vliet AR , Agostinis P ((2018) ) Mitochondria-associated membranes and ER stress. Curr Top Microbiol Immunol 414: , 73–102. |
[202] | Yang M , Li C , Yang S , Xiao Y , Xiong X , Chen W , Zhao H , Zhang Q , Han Y , Sun L ((2020) ) Mitochondria-associated ER membranes - the origin site of autophagy. Front Cell Dev Biol 8: , 595. |
[203] | van Vliet AR , Verfaillie T , Agostinis P ((2014) ) New functions of mitochondria associated membranes in cellular signaling. Biochim Biophys Acta 1843: , 2253–2262. |
[204] | Schon EA , Area-Gomez E ((2010) ) Is Alzheimer’s disease a disorder of mitochondria-associated membranes?. J Alzheimers Dis 20: (Suppl 2), S281–292. |
[205] | Supnet C , Bezprozvanny I ((2010) ) Neuronal calcium signaling, mitochondrial dysfunction, and Alzheimer’s disease.. J Alzheimers Dis 20: (Suppl 2), S487–S498. |
[206] | Patergnani S , Suski JM , Agnoletto C , Bononi A , Bonora M , De Marchi E , Giorgi C , Marchi S , Missiroli S , Poletti F , Rimessi A , Duszynski J , Wieckowski MR , Pinton P ((2011) ) Calcium signaling around mitochondria associated membranes (MAMs). Cell Commun Signal 9: , 19. |
[207] | Hayashi T , Su TP ((2007) ) Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2 + signaling and cell survival. Cell 131: , 596–610. |
[208] | Tesei A , Cortesi M , Zamagni A , Arienti C , Pignatta S , Zanoni M , Paolillo M , Curti D , Rui M , Rossi D , Collina S ((2018) ) Sigma receptors as endoplasmic reticulum stress “gatekeepers” and their modulators as emerging new weapons in the fight against cancer. Front Pharmacol 9: , 711. |
[209] | Gong CX , Liu F , Grundke-Iqbal I , Iqbal K ((2006) ) Impaired brain glucose metabolism leads to Alzheimer neurofibrillary degeneration through a decrease in tau O-GlcNAcylation. J Alzheimers Dis 9: , 1–12. |
[210] | Tamucci KA (2022) The role of ER-mitochondria contact sites in the regulation of glucose metabolism: A tale of two mitochondria and its relevance to amyotrophic lateral sclerosis. Doctoral thesis, https://academiccommons.columbia.edu/doi/10.7916/5dg8-9h02, 1-245. |
[211] | Pastorino JG , Hoek JB ((2008) ) Regulation of hexokinase binding to VDAC. J Bioenerg Biomembr 40: , 171–182. |
[212] | Hamasaki M , Furuta N , Matsuda A , Nezu A , Yamamoto A , Fujita N , Oomori H , Noda T , Haraguchi T , Hiraoka Y , Amano A , Yoshimori T ((2013) ) Autophagosomes form at ER-mitochondria contact sites. Nature 495: , 389–393. |
[213] | Swerdlow RH ((2011) ) Brain aging, Alzheimer’s disease, and mitochondria. Biochim Biophys Acta 1812: , 1630–1639. |
[214] | Wang X , Su B , Zheng L , Perry G , Smith MA , Zhu X ((2009) ) The role of abnormal mitochondrial dynamics in the pathogenesis of Alzheimer’s disease. J Neurochem 109: , 153–159. |
[215] | Zhang F , Niu L , Li S , Le W ((2019) ) Pathological impacts of chronic hypoxia on Alzheimer’s disease. ACS Chem Neurosci 10: , 902–909. |
[216] | Sun X , He G , Qing H , Zhou W , Dobie F , Cai F , Staufenbiel M , Huang LE , Song W ((2006) ) Hypoxia facilitates Alzheimer’s disease pathogenesis by up-regulating BACE1 gene expression. Proc Natl Acad Sci U S A 103: , 18727–18732. |
[217] | Wang WX , Prajapati P , Nelson PT , Springer JE ((2020) ) The mitochondria-associated ER membranes are novel subcellular locations enriched for inflammatory-responsive microRNAs. Mol Neurobiol 57: , 2996–3013. |
[218] | Lv Y , Cheng L , Peng F ((2022) ) Compositions and functions of mitochondria-associated endoplasmic reticulum membranes and their contribution to cardioprotection by exercise preconditioning. Front Physiol 13: , 910452. |
[219] | Harned TC , Stan RV , Cao Z , Chakrabarti R , Higgs HN , Chang CCY , Chang TY ((2023) ) Acute ACAT1/SOAT1 blockade increases MAM cholesterol and strengthens ER-mitochondria connectivity. Int J Mol Sci 24: , 5525. |
[220] | Pettegrew JW , Panchalingam K , Hamilton RL , McClure RJ ((2001) ) Brain membrane phospholipid alterations in Alzheimer’s disease. Neurochem Res 26: , 771–782. |
[221] | Wells K , Farooqui AA , Liss L , Horrocks LA ((1995) ) Neural membrane phospholipids in Alzheimer disease. Neurochem Res 20: , 1329–1333. |
[222] | Stone SJ , Vance JE ((2000) ) Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J Biol Chem 275: , 34534–34540. |
[223] | Monteiro-Cardoso VF , Rochin L , Arora A , Houcine A , Jaaskelainen E , Kivela AM , Sauvanet C , Le Bars R , Marien E , Dehairs J , Neveu J , El Khallouki N , Santonico E , Swinnen JV , Tareste D , Olkkonen VM , Giordano F ((2022) ) ORP5/8 and MIB/MICOS link ER-mitochondria and intra-mitochondrial contacts for non-vesicular transport of phosphatidylserine. Cell Rep 40: , 111364. |
[224] | Voelker DR ((1989) ) Phosphatidylserine translocation to the mitochondrion is an ATP-dependent process in permeabilized animal cells. Proc Natl Acad Sci U S A 86: , 9921–9925. |
[225] | Zborowski J , Dygas A , Wojtczak L ((1983) ) Phosphatidylserine decarboxylase is located on the external side of the inner mitochondrial membrane. FEBS Lett 157: , 179–182. |
[226] | Vance JE ((2003) ) Molecular and cell biology of phosphatidylserine and phosphatidylethanolamine metabolism. Prog Nucleic Acid Res Mol Biol 75: , 69–111. |
[227] | Schumacher MM , Choi JY , Voelker DR ((2002) ) Phosphatidylserine transport to the mitochondria is regulated by ubiquitination. J Biol Chem 277: , 51033–51042. |
[228] | Mielke MM , Lyketsos CG ((2010) ) Alterations of the sphingolipid pathway in Alzheimer’s disease: New biomarkers and treatment targets?. Neuromolecular Med 12: , 331–340. |
[229] | van Echten-Deckert G , Walter J ((2012) ) Sphingolipids: Critical players in Alzheimer’s disease. Prog Lipid Res 51: , 378–393. |
[230] | Cutler RG , Kelly J , Storie K , Pedersen WA , Tammara A , Hatanpaa K , Troncoso JC , Mattson MP ((2004) ) Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proc Natl Acad Sci U S A 101: , 2070–2075. |
[231] | Filippov V , Song MA , Zhang K , Vinters HV , Tung S , Kirsch WM , Yang J , Duerksen-Hughes PJ ((2012) ) Increased ceramide in brains with Alzheimer’s and other neurodegenerative diseases. J Alzheimers Dis 29: , 537–547. |
[232] | Han X , Holtzman DM , McKeel DW Jr. , Kelley J , Morris JC ((2002) ) Substantial sulfatide deficiency and ceramide elevation in very early Alzheimer’s disease: Potential role in disease pathogenesis. J Neurochem 82: , 809–818. |
[233] | Hartmann T , Kuchenbecker J , Grimm MO ((2007) ) Alzheimer’s disease: The lipid connection. J Neurochem 103: (Suppl 1), 159–170. |
[234] | Grimm MO , Grosgen S , Rothhaar TL , Burg VK , Hundsdorfer B , Haupenthal VJ , Friess P , Muller U , Fassbender K , Riemenschneider M , Grimm HS , Hartmann T ((2011) ) Intracellular APP domain regulates serine-palmitoyl-CoA transferase expression and is affected in Alzheimer’s disease. Int J Alzheimers Dis 2011: , 695413. |
[235] | He X , Huang Y , Li B , Gong CX , Schuchman EH ((2010) ) Deregulation of sphingolipid metabolism in Alzheimer’s disease. Neurobiol Aging 31: , 398–408. |
[236] | Ardail D , Popa I , Bodennec J , Louisot P , Schmitt D , Portoukalian J ((2003) ) The mitochondria-associated endoplasmic-reticulum subcompartment (MAM fraction) of rat liver contains highly active sphingolipid-specific glycosyltransferases. Biochem J 371: , 1013–1019. |
[237] | Wu BX , Rajagopalan V , Roddy PL , Clarke CJ , Hannun YA ((2010) ) Identification and characterization of murine mitochondria-associated neutral sphingomyelinase (MA-nSMase), the mammalian sphingomyelin phosphodiesterase 5. J Biol Chem 285: , 17993–178002. |
[238] | Rubanov LI , Zaraisky AG , Shilovsky GA , Seliverstov AV , Zverkov OA , Lyubetsky VA ((2019) ) Screening for mouse genes lost in mammals with long lifespans. BioData Min 12: , 20. |
[239] | Kong JN , Zhu Z , Itokazu Y , Wang G , Dinkins MB , Zhong L , Lin HP , Elsherbini A , Leanhart S , Jiang X , Qin H , Zhi W , Spassieva SD , Bieberich E ((2018) ) Novel function of ceramide for regulation of mitochondrial ATP release in astrocytes. J Lipid Res 59: , 488–506. |
[240] | London M , London E ((2004) ) Ceramide selectively displaces cholesterol from ordered lipid domains (rafts): Implications for lipid raft structure and function. J Biol Chem 279: , 9997–10004. |
[241] | Yu C , Alterman M , Dobrowsky RT ((2005) ) Ceramide displaces cholesterol from lipid rafts and decreases the association of the cholesterol binding protein caveolin-1. J Lipid Res 46: , 1678–1691. |
[242] | Dinkla S , Wessels K , Verdurmen WP , Tomelleri C , Cluitmans JC , Fransen J , Fuchs B , Schiller J , Joosten I , Brock R , Bosman GJ ((2012) ) Functional consequences of sphingomyelinase-induced changes in erythrocyte membrane structure. Cell Death Dis 3: , e410. |
[243] | Takasugi N , Sasaki T , Shinohara M , Iwatsubo T , Tomita T ((2015) ) Synthetic ceramide analogues increase amyloid-β 42 production by modulating γ-secretase activity. Biochem Biophys Res Commun 457: , 194–199. |
[244] | Gudz TI , Tserng KY , Hoppel CL ((1997) ) Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J Biol Chem 272: , 24154–24158. |
[245] | Kennedy MA , Moffat TC , Gable K , Ganesan S , Niewola-Staszkowska K , Johnston A , Nislow C , Giaever G , Harris LJ , Loewith R , Zaremberg V , Harper ME , Dunn T , Bennett SAL , Baetz K ((2016) ) A signaling lipid associated with Alzheimer’s disease promotes mitochondrial dysfunction. Sci Rep 6: , 19332. |
[246] | Kogot-Levin A , Saada A ((2014) ) Ceramide and the mitochondrial respiratory chain. Biochimie 100: , 88–94. |
[247] | Nelson MB , Swensen AC , Winden DR , Bodine JS , Bikman BT , Reynolds PR ((2015) ) Cardiomyocyte mitochondrial respiration is reduced by receptor for advanced glycation end-product signaling in a ceramide-dependent manner. Am J Physiol Heart Circ Physiol 309: , H63–H69. |
[248] | Smith ME , Tippetts TS , Brassfield ES , Tucker BJ , Ockey A , Swensen AC , Anthonymuthu TS , Washburn TD , Kane DA , Prince JT , Bikman BT ((2013) ) Mitochondrial fission mediates ceramide-induced metabolic disruption in skeletal muscle. Biochem J 456: , 427–439. |
[249] | Stefani M , Liguri G ((2009) ) Cholesterol in Alzheimer’s disease: Unresolved questions. Curr Alz Res 6: , 15–29. |
[250] | Di Paolo G , Kim TW ((2011) ) Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat Rev Neurosci 12: , 284–296. |
[251] | Maulik M , Westaway D , Jhamandas JH , Kar S ((2013) ) Role of cholesterol in APP metabolism and its significance in Alzheimer’s disease pathogenesis. Mol Neurobiol 47: , 37–63. |
[252] | Cossec JC , Marquer C , Panchal M , Lazar AN , Duyckaerts C , Potier MC ((2010) ) Cholesterol changes in Alzheimer’s disease: Methods of analysis and impact on the formation of enlarged endosomes. Biochim Biophys Acta 1801: , 839–845. |
[253] | Ganji R , Paulo JA , Xi Y , Kline I , Zhu J , Clemen CS , Weihl CC , Purdy JG , Gygi SP , Raman M ((2023) ) The p97-UBXD8 complex regulates ER-mitochondria contact sites by altering membrane lipid saturation and composition. Nat Commun 14: , 638. |
[254] | Pierrot N , Tyteca D , D’Auria L , Dewachter I , Gailly P , Hendrickx A , Tasiaux B , Haylani LE , Muls N , N’Kuli F , Laquerriere A , Demoulin JB , Campion D , Brion JP , Courtoy PJ , Kienlen-Campard P , Octave JN ((2013) ) Amyloid precursor protein controls cholesterol turnover needed for neuronal activity. EMBO Mol Med 5: , 608–625. |
[255] | Rusinol AE , Cui Z , Chen MH , Vance JE ((1994) ) A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J Biol Chem 269: , 27494–27502. |
[256] | Das A , Brown MS , Anderson DD , Goldstein JL , Radhakrishnan A ((2014) ) Three pools of plasma membrane cholesterol and their relation to cholesterol homeostasis. Elife 3: , e02882. |
[257] | Pani A , Mandas A , Diaz G , Abete C , Cocco PL , Angius F , Brundu A , Mucaka N , Pais ME , Saba A , Barberini L , Zaru C , Palmas M , Putzu PF , Mocali A , Paoletti F , La Colla P , Dessi S ((2009) ) Accumulation of neutral lipids in peripheral blood mononuclear cells as a distinctive trait of Alzheimer patients and asymptomatic subjects at risk of disease. BMC Med 7: , 66–77. |
[258] | Gómez-Ramos P , Asunción Morán M ((2007) ) Ultrastructural localization of intraneuronal Aβ-peptide in Alzheimer disease brains. J Alzheimers Dis 11: , 53–59. |
[259] | Huttunen HJ , Peach C , Bhattacharyya R , Barren C , Pettingell W , Hutter-Paier B , Windisch M , Berezovska O , Kovacs DM ((2009) ) Inhibition of acyl-coenzyme A: Cholesterol acyl transferase modulates amyloid precursor protein trafficking in the early secretory pathway. FASEB J 23: , 3819–3828. |
[260] | Huttunen HJ , Puglielli L , Ellis BC , MacKenzie Ingano LA , Kovacs DM ((2009) ) Novel N-terminal cleavage of APP precludes Aβ generation in ACAT-defective AC29 cells. J Mol Neurosci 37: , 6–15. |
[261] | van der Kant R , Langness VF , Herrera CM , Williams DA , Fong LK , Leestemaker Y , Steenvoorden E , Rynearson KD , Brouwers JF , Helms JB , Ovaa H , Giera M , Wagner SL , Bang AG , Goldstein LSB ((2019) ) Cholesterol metabolism is a druggable axis that independently regulates tau and amyloid-β in iPSC-derived Alzheimer’s disease neurons. Cell Stem Cell 24: , 369–375 e369. |
[262] | Bittman R , Kasireddy CR , Mattjus P , Slotte JP ((1994) ) Interaction of cholesterol with sphingomyelin in monolayers and vesicles. Biochemistry 33: , 11776–11781. |
[263] | Endapally S , Frias D , Grzemska M , Gay A , Tomchick DR , Radhakrishnan A ((2019) ) Molecular discrimination between two conformations of sphingomyelin in plasma membranes. Cell 176: , 1040–1053 e1017. |
[264] | Gulati S , Liu Y , Munkacsi AB , Wilcox L , Sturley SL ((2010) ) Sterols and sphingolipids: Dynamic duo or partners in crime?. Prog Lipid Res 49: , 353–365. |
[265] | Scheek S , Brown MS , Goldstein JL ((1997) ) Sphingomyelin depletion in cultured cells blocks proteolysis of sterol regulatory element binding proteins at site 1. Proc Natl Acad Sci U S A 94: , 11179–11183. |
[266] | Dai J , Alwarawrah M , Huang J ((2010) ) Instability of cholesterol clusters in lipid bilayers and the cholesterol’s umbrella effect. J Phys Chem B 114: , 840–848. |
[267] | Fantini J , Barrantes FJ ((2013) ) How cholesterol interacts with membrane proteins: An exploration of cholesterol-binding sites including CRAC, CARC, and tilted domains. Front Physiol 4: , 31. |
[268] | Cordy JM , Hooper NM , Turner AJ ((2006) ) The involvement of lipid rafts in Alzheimer’s disease. Mol Membr Biol 23: , 111–122. |
[269] | Hare J ((2010) ) Trafficking of amyloid c-precursor protein products C83 and C99 on the endocytic pathway. Biochem Biophys Res Commun 401: , 219–224. |
[270] | Grimm MO , Grimm HS , Patzold AJ , Zinser EG , Halonen R , Duering M , Tschape JA , De Strooper B , Muller U , Shen J , Hartmann T ((2005) ) Regulation of cholesterol and sphingomyelin metabolism by amyloid-β and presenilin. Nat Cell Biol 7: , 1118–1123. |
[271] | Panchal M , Gaudin M , Lazar AN , Salvati E , Rivals I , Ayciriex S , Dauphinot L , Dargere D , Auzeil N , Masserini M , Laprevote O , Duyckaerts C ((2014) ) Ceramides and sphingomyelinases in senile plaques. Neurobiol Dis 65: , 193–201. |
[272] | Ferrari A , He C , Kennelly JP , Sandhu J , Xiao X , Chi X , Jiang H , Young SG , Tontonoz P ((2020) ) Aster proteins regulate the accessible cholesterol pool in the plasma membrane. Mol Cell Biol 40: , e00255–00220. |
[273] | Capitini C , Bigi A , Parenti N , Emanuele M , Bianchi N , Cascella R , Cecchi C , Maggi L , Annunziato F , Pavone FS , Calamai M ((2023) ) APP and Bace1: Differential effect of cholesterol enrichment on processing and plasma membrane mobility. iScience 26: , 106611. |
[274] | Evin G , Zhu A , Holsinger RM , Masters CL , Li QX ((2003) ) Proteolytic processing of the Alzheimer’s disease amyloid precursor protein in brain and platelets. J Neurosci Res 74: , 386–392. |
[275] | Nicsanu R , Cervellati C , Benussi L , Squitti R , Zanardini R , Rosta V , Trentini A , Ferrari C , Saraceno C , Longobardi A , Bellini S , Binetti G , Zanetti O , Zuliani G , Ghidoni R ((2022) ) Increased serum beta-secretase 1 activity is an early marker of Alzheimer’s disease. J Alzheimers Dis 87: , 433–441. |
[276] | Chaudhuri A , Chattopadhyay A ((2011) ) Transbilayer organization of membrane cholesterol at low concentrations: Implications in health and disease. Biochim Biophys Acta 1808: , 19–25. |
[277] | Pappolla MA , Bryant-Thomas TK , Herbert D , Pacheco J , Fabra Garcia M , Manjon M , Girones X , Henry TL , Matsubara E , Zambon D , Wolozin B , Sano M , Cruz-Sanchez FF , Thal LJ , Petanceska SS , Refolo LM ((2003) ) Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology 61: , 199–205. |
[278] | Sandhu J , Li S , Fairall L , Pfisterer SG , Gurnett JE , Xiao X , Weston TA , Vashi D , Ferrari A , Orozco JL , Hartman CL , Strugatsky D , Lee SD , He C , Hong C , Jiang H , Bentolila LA , Gatta AT , Levine TP , Ferng A , Lee R , Ford DA , Young SG , Ikonen E , Schwabe JWR , Tontonoz P ((2018) ) Aster proteins facilitate nonvesicular plasma membrane to ER cholesterol transport in mammalian cells.514-529 e. Cell 175: , 514–529 e520. |
[279] | Xiao X , Kennelly JP , Ferrari A , Clifford BL , Whang E , Gao Y , Qian K , Sandhu J , Jarrett KE , Brearley-Sholto MC , Nguyen A , Nagari RT , Lee MS , Zhang S , Weston TA , Young SG , Bensinger SJ , Villanueva CJ , de Aguiar Vallim TQ , Tontonoz P ((2023) ) Hepatic nonvesicular cholesterol transport is critical for systemic lipid homeostasis. Nat Metab 5: , 165–181. |
[280] | Nagashima S , Tabara LC , Tilokani L , Paupe V , Anand H , Pogson JH , Zunino R , McBride HM , Prudent J ((2020) ) Golgi-derived PI(4)P-containing vesicles drive late steps of mitochondrial division. Science 367: , 1366–1371. |
[281] | Galmes R , Houcine A , van Vliet AR , Agostinis P , Jackson CL , Giordano F ((2016) ) ORP5/ORP8 localize to endoplasmic reticulum-mitochondria contacts and are involved in mitochondrial function. EMBO Rep 17: , 800–810. |
[282] | Andersen JP , Zhang J , Sun H , Liu X , Liu J , Nie J , Shi Y ((2020) ) Aster-B coordinates with Arf1 to regulate mitochondrial cholesterol transport. Mol Metab 42: , 101055. |
[283] | Trinh MN , Brown MS , Goldstein JL , Han J , Vale G , McDonald JG , Seemann J , Mendell JT , Lu F ((2020) ) Last step in the path of LDL cholesterol from lysosome to plasma membrane to ER is governed by phosphatidylserine. Proc Natl Acad Sci U S A 117: , 18521–18529. |
[284] | Trinh MN , Brown MS , Seemann J , Vale G , McDonald JG , Goldstein JL , Lu F ((2022) ) Interplay between Asters/GRAMD1s and phosphatidylserine in intermembrane transport of LDL cholesterol. Proc Natl Acad Sci U S A 119: , e2120411119. |
[285] | Giordano F ((2018) ) Non-vesicular lipid trafficking at the endoplasmic reticulum-mitochondria interface. Biochem Soc Trans 46: , 437–452. |
[286] | Weggen S , Beher D ((2012) ) Molecular consequences of amyloid precursor protein and presenilin mutations causing autosomal-dominant Alzheimer’s disease. Alzheimers Res Ther 4: , 9. |
[287] | Hudry E , Van Dam D , Kulik W , De Deyn PP , Stet FS , Ahouansou O , Benraiss A , Delacourte A , Bougneres P , Aubourg P , Cartier N ((2010) ) Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer’s disease. Mol Ther 18: , 44–53. |
[288] | Lutjohann D , Papassotiropoulos A , Bjorkhem I , Locatelli S , Bagli M , Oehring RD , Schlegel U , Jessen F , Rao ML , von Bergmann K , Heun R ((2000) ) Plasma 24S-hydroxycholesterol (cerebrosterol) is increased in Alzheimer and vascular demented patients. J Lipid Res 41: , 195–198. |
[289] | Papassotiropoulos A , Lutjohann D , Bagli M , Locatelli S , Jessen F , Rao ML , Maier W , Bjorkhem I , von Bergmann K , Heun R ((2000) ) Plasma 24S-hydroxycholesterol: A peripheral indicator of neuronal degeneration and potential state marker for Alzheimer’s disease. Neuroreport 11: , 1959–1962. |
[290] | Feringa FM , van der Kant R ((2021) ) Cholesterol and Alzheimer’s disease; from risk genes to pathological effects. Front Aging Neurosci 13: , 690372. |
[291] | Acharya TK , Kumar A , Kumar S , Goswami C ((2022) ) TRPV4 interacts with MFN2 and facilitates endoplasmic reticulum-mitochondrial contact points for Ca2 +-buffering. Life Sci 310: , 121112. |
[292] | Du K , Dou Y , Chen K , Zhao Y ((2023) ) Activation of TRPV4 induces intraneuronal tau hyperphosphorylation via cholesterol accumulation. Exp Neurol 364: , 114392. |
[293] | Bai X , Wu J , Zhang M , Xu Y , Duan L , Yao K , Zhang J , Bo J , Zhao Y , Xu G , Zu H ((2021) ) DHCR24 knock-down induced Tau hyperphosphorylation at Thr181, Ser199, Thr231, Ser262, Ser396 epitopes and inhibition of autophagy by overactivation of GSK3β/mTOR signaling. Front Aging Neurosci 13: , 513605. |
[294] | Marchi C , Adorni MP , Caffarra P , Ronda N , Spallazzi M , Barocco F , Galimberti D , Bernini F , Zimetti F ((2019) ) ABCA1- and ABCG1-mediated cholesterol efflux capacity of cerebrospinal fluid is impaired in Alzheimer’s disease. J Lipid Res 60: , 1449–1456. |
[295] | Shibuya Y , Niu Z , Bryleva EY , Harris BT , Murphy SR , Kheirollah A , Bowen ZD , Chang CCY , Chang TY ((2015) ) Acyl-coenzyme A:cholesterol acyltransferase 1 blockage enhances autophagy in the neurons of triple transgenic Alzheimer’s disease mouse and reduces human P301L-tau content at the presymptomatic stage. Neurobiol Aging 36: , 2248–2259. |
[296] | Szabo L , Cummins N , Paganetti P , Odermatt A , Papassotiropoulos A , Karch C , Gotz J , Eckert A , Grimm A ((2023) ) ER-mitochondria contacts and cholesterol metabolism are disrupted by disease-associated tau protein. EMBO Rep 24: , e57499. |
[297] | Israel MA , Yuan SH , Bardy C , Reyna SM , Mu Y , Herrera C , Hefferan MP , Van Gorp S , Nazor KL , Boscolo FS , Carson CT , Laurent LC , Marsala M , Gage FH , Remes AM , Koo EH , Goldstein LS ((2012) ) Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482: , 216–220. |
[298] | Moore S , Evans LD , Andersson T , Portelius E , Smith J , Dias TB , Saurat N , McGlade A , Kirwan P , Blennow K , Hardy J , Zetterberg H , Livesey FJ ((2015) ) APP metabolism regulates tau proteostasis in human cerebral cortex neurons. Cell Rep 11: , 689–696. |
[299] | Area-Gomez E , Schon EA ((2016) ) Mitochondria-associated ER membranes and Alzheimer disease. Curr Opin Genet Dev 38: , 90–96. |
[300] | Area-Gomez E , Schon EA ((2017) ) Alzheimer Disease. Adv Exp Med Biol 997: , 149–156. |
[301] | Pera M , Montesinos J , Larrea D , Agrawal RR , Velasco KR , Stavrovskaya IG , Yun TD , Area-Gomez E ((2020) ) MAM and C99, key players in the pathogenesis of Alzheimer’s disease. Int Rev Neurobiol 154: , 235–278. |
[302] | Smirnov DS , Galasko D , Hiniker A , Edland SD , Salmon DP ((2021) ) Age-at-onset and APOE-related heterogeneity in pathologically confirmed sporadic Alzheimer disease. Neurology 96: , e2272–e2283. |
[303] | Rudge JD ((2022) ) A new hypothesis for Alzheimer’s disease: The lipid invasion model. J Alzheimers Dis Rep 6: , 129–161. |
[304] | Chew H , Solomon VA , Fonteh AN ((2020) ) Involvement of lipids in Alzheimer’s disease pathology and potential therapies. Front Physiol 11: , 598. |
[305] | Castello MA , Soriano S ((2013) ) Rational heterodoxy: Cholesterol reformation of the amyloid doctrine. Ageing Res Rev 12: , 282–288. |
[306] | Barber CN , Raben DM ((2019) ) Lipid metabolism crosstalk in the brain : Glia and neurons. Front Cell Neurosci 13: , 212 |
[307] | Montesinos J , Guardia-Laguarta C , Area-Gomez E ((2020) ) The fat brain. Curr Opin Clin Nutr Metab Care 23: , 68–75. |
[308] | Borroni V , Barrantes FJ ((2011) ) Cholesterol modulates the rate and mechanism of acetylcholine receptor internalization. J Biol Chem 286: , 17122–17132. |
[309] | Duarte A , Poderoso C , Cooke M , Soria G , Cornejo Maciel F , Gottifredi V , Podesta EJ ((2012) ) Mitochondrial fusion is essential for steroid biosynthesis. PLoS One 7: , e45829. |
[310] | Essayan-Perez S , Sudhof TC ((2023) ) Neuronal γ-secretase regulates lipid metabolism, linking cholesterol to synaptic dysfunction in Alzheimer’s disease. Neuron 111: , 3176–3194 e3177.. |
[311] | Rossignol M , Uso T , Thomas P ((1985) ) Relationship between fluidity and ionic permeability of bilayers from natural mixtures of phospholipids. J Membr Biol 87: , 269–275. |
[312] | Montani L ((2021) ) Lipids in regulating oligodendrocyte structure and function. Semin Cell Dev Biol 112: , 114–122. |
[313] | Ginzburg L , Kacher Y , Futerman AH ((2004) ) The pathogenesis of glycosphingolipid storage disorders. Semin Cell Dev Biol 15: , 417–431. |
[314] | Ferretti MT , Iulita MF , Cavedo E , Chiesa PA , Schumacher Dimech A , Santuccione Chadha A , Baracchi F , Girouard H , Misoch S , Giacobini E , Depypere H , Hampel H ; Women’s Brain Project and the Alzheimer Precision Medicine Initiative ((2018) ) Sex differences in Alzheimer disease - the gateway to precision medicine. Nat Rev Neurol 14: , 457–469. |
[315] | Cheng YJ , Lin CH , Lane HY ((2021) ) From menopause to neurodegeneration-molecular basis and potential therapy. Int J Mol Sci 22: , 8654. |
[316] | Ferrini RL , Barrett-Connor E ((1998) ) Sex hormones and age: A cross-sectional study of testosterone and estradiol and their bioavailable fractions in community-dwelling men. Am J Epidemiol 147: , 750–754. |
[317] | Bose HS , Bose M , Whittal RM ((2023) ) Tom40 in cholesterol transport. iScience 26: , 106386. |
[318] | Larsen MC , Lee J , Jorgensen JS , Jefcoate CR ((2020) ) STARD1 functions in mitochondrial cholesterol metabolism and nascent HDL formation. gene expression and molecular mRNA imaging show novel splicing and a 1:1 mitochondrial association. Front Endocrinol (Lausanne) 11: , 559674. |
[319] | Rogers MA , Liu J , Kushnir MM , Bryleva E , Rockwood AL , Meikle AW , Shapiro D , Vaisman BL , Remaley AT , Chang CCY , Chang TY ((2012) ) Cellular pregnenolone esterification by acyl-CoA:cholesterol acyltransferase. J Biol Chem 287: , 17483–17492. |
[320] | Cui J , Ait-Ghezala G , Sambamurti K , Gao F , Shen Y , Li R ((2022) ) Sex-specific regulation of β-secretase: A novel estrogen response element (ERE)-dependent mechanism in Alzheimer’s disease. J Neurosci 42: , 1154–1165. |
[321] | Sambamurti K , Kinsey R , Maloney B , Ge YW , Lahiri DK ((2004) ) Gene structure and organization of the human β-secretase (BACE) promoter. FASEB J 18: , 1034–1036. |
[322] | Fukumoto H , Rosene DL , Moss MB , Raju S , Hyman BT , Irizarry MC ((2004) ) β-secretase activity increases with aging in human, monkey, and mouse brain. Am J Pathol 164: , 719–725. |
[323] | Shi W , Wu H , Liu S , Wu Z , Wu H , Liu J , Hou Y ((2021) ) Progesterone suppresses cholesterol esterification in APP/PS1 mice and a cell model of Alzheimer’s disease. Brain Res Bull 173: , 162–173. |
[324] | Mazzone T , Krishna M , Lange Y ((1995) ) Progesterone blocks intracellular translocation of free cholesterol derived from cholesteryl ester in macrophages. J Lipid Res 36: , 544–551. |
[325] | Cui J , Shen Y , Li R ((2013) ) Estrogen synthesis and signaling pathways during aging: From periphery to brain. Trends Mol Med 19: , 197–209. |
[326] | Guennoun R ((2020) ) Progesterone in the brain : Hormone, neurosteroid and neurorotectant. Int J Mol Sci 21: , 5271 |
[327] | Kim YJ , Soto M , Branigan GL , Rodgers K , Brinton RD ((2021) ) Association between menopausal hormone therapy and risk of neurodegenerative diseases: Implications for precision hormone therapy. Alzheimers Dement 7: , e12174. |
[328] | Sung YF , Tsai CT , Kuo CY , Lee JT , Chou CH , Chen YC , Chou YC , Sun CA ((2022) ) Use of hormone replacement therapy and risk of dementia: A nationwide cohort study.. Neurology 99: , e1835–e1842. |
[329] | Bekris LM , Yu CE , Bird TD , Tsuang DW ((2010) ) Genetics of Alzheimer disease. J Geriatr Psychiatry Neurol 23: , 213–227. |
[330] | Olmastroni E , Molari G , De Beni N , Colpani O , Galimberti F , Gazzotti M , Zambon A , Catapano AL , Casula M ((2022) ) Statin use and risk of dementia or Alzheimer’s disease: A systematic review and meta-analysis of observational studies. Eur J Prev Cardiol 29: , 804–814. |
[331] | Trompet S , van Vliet P , de Craen AJ , Jolles J , Buckley BM , Murphy MB , Ford I , Macfarlane PW , Sattar N , Packard CJ , Stott DJ , Shepherd J , Bollen EL , Blauw GJ , Jukema JW , Westendorp RG ((2010) ) Pravastatin and cognitive function in the elderly. Results of the PROSPER study. J Neurol 257: , 85–90. |
[332] | Lai MT , Chen E , Crouthamel MC , DiMuzio-Mower J , Xu M , Huang Q , Price E , Register RB , Shi XP , Donoviel DB , Bernstein A , Hazuda D , Gardell SJ , Li YM ((2003) ) Presenilin-1 and presenilin-2 exhibit distinct yet overlapping γ-secretase activities. J Biol Chem 278: , 22475–22481. |
[333] | Filadi R , Greotti E , Turacchio G , Luini A , Pozzan T , Pizzo P ((2016) ) Presenilin 2 modulates endoplasmic reticulum-mitochondria coupling by tuning the antagonistic effect of mitofusin 2. Cell Rep 15: , 2226–2238. |
[334] | Rossini M , Garcia-Casas P , Filadi R , Pizzo P ((2021) ) Loosening ER-mitochondria coupling by the expression of the presenilin 2 loop domain. Cells 10: , 1968. |
[335] | Naon D , Hernandez-Alvarez MI , Shinjo S , Wieczor M , Ivanova S , Martins de Brito O , Quintana A , Hidalgo J , Palacin M , Aparicio P , Castellanos J , Lores L , Sebastian D , Fernandez-Veledo S , Vendrell J , Joven J , Orozco M , Zorzano A , Scorrano L ((2023) ) Splice variants of mitofusin 2 shape the endoplasmic reticulum and tether it to mitochondria. Science 380: , eadh9351. |
[336] | Nguyen HN , Son DJ , Lee JW , Hwang DY , Kim YK , Cho JS , Lee US , Yoo HS , Moon DC , Oh KW , Hong JT ((2006) ) Mutant presenilin 2 causes abnormality in the brain lipid profile in the development of Alzheimer’s disease. Arch Pharm Res 29: , 884–889. |
[337] | Bassendine MF , Taylor-Robinson SD , Fertleman M , Khan M , Neely D ((2020) ) Is Alzheimer’s disease a liver disease of the brain?. J Alzheimers Dis 75: , 1–14. |
[338] | Mandas A , Abete C , Putzu PF , la Colla P , Dessi S , Pani A ((2012) ) Changes in cholesterol metabolism-related gene expression in peripheral blood mononuclear cells from Alzheimer patients. Lipids Health Dis 11: , 39. |
[339] | Liu CC , Zhao J , Fu Y , Inoue Y , Ren Y , Chen Y , Doss SV , Shue F , Jeevaratnam S , Bastea L , Wang N , Martens YA , Qiao W , Wang M , Zhao N , Jia L , Yamazaki Y , Yamazaki A , Rosenberg CL , Wang Z , Kong D , Li Z , Kuchenbecker LA , Trottier ZA , Felton L , Rogers J , Quicksall ZS , Linares C , Knight J , Chen Y , Kurti A , Kanekiyo T , Fryer JD , Asmann YW , Storz P , Wang X , Peng J , Zhang B , Kim BYS , Bu G ((2022) ) Peripheral apoE4 enhances Alzheimer’s pathology and impairs cognition by compromising cerebrovascular function. Nat Neurosci 25: , 1020–1033. |
[340] | Anastasia I , Ilacqua N , Raimondi A , Lemieux P , Ghandehari-Alavijeh R , Faure G , Mekhedov SL , Williams KJ , Caicci F , Valle G , Giacomello M , Quiroga AD , Lehner R , Miksis MJ , Toth K , de Aguiar Vallim TQ , Koonin EV , Scorrano L , Pellegrini L ((2021) ) Mitochondria-rough-ER contacts in the liver regulate systemic lipid homeostasis. Cell Rep 34: , 108873. |
[341] | Ilacqua N , Anastasia I , Aloshyn D , Ghandehari-Alavijeh R , Peluso EA , Brearley-Sholto MC , Pellegrini LV , Raimondi A , de Aguiar Vallim TQ , Pellegrini L ((2022) ) Expression of Synj2bp in mouse liver regulates the extent of wrappER-mitochondria contact to maintain hepatic lipid homeostasis. Biol Direct 17: , 37. |
[342] | Ilacqua N , Anastasia I , Raimondi A , Lemieux P , de Aguiar Vallim TQ , Toth K , Koonin EV , Pellegrini L ((2022) ) A three-organelle complex made by wrappER contacts with peroxisomes and mitochondria responds to liver lipid flux changes. J Cell Sci 135: , jcs259091. |
[343] | Xu J , Patassini S , Rustogi N , Riba-Garcia I , Hale BD , Phillips AM , Waldvogel H , Haines R , Bradbury P , Stevens A , Faull RLM , Dowsey AW , Cooper GJS , Unwin RD ((2019) ) Regional protein expression in human Alzheimer’s brain correlates with disease severity. Commun Biol 2: , 43. |
[344] | Liu L , Drouet V , Wu JW , Witter MP , Small SA , Clelland C , Duff K ((2012) ) Trans-synaptic spread of tau pathology in vivo. PLoS One 7: , e31302. |
[345] | Pooler AM , Polydoro M , Wegmann S , Nicholls SB , Spires-Jones TL , Hyman BT ((2013) ) Propagation of tau pathology in Alzheimer’s disease: Identification of novel therapeutic targets. Alzheimers Res Ther 5: , 49. |
[346] | Christen-Zaech S , Kraftsik R , Pillevuit O , Kiraly M , Martins R , Khalili K , Miklossy J ((2003) ) Early olfactory involvement in Alzheimer’s disease. Can J Neurol Sci 30: , 20–25. |
[347] | Jacobs DM , Sano M , Dooneief G , Marder K , Bell KL , Stern Y ((1995) ) Neuropsychological detection and characterization of preclinical Alzheimer’s disease. Neurology 45: , 957–962. |
[348] | Maldonado-Soto AR , Oakley DH , Wichterle H , Stein J , Doetsch FK , Henderson CE ((2014) ) Stem cells in the nervous system. Am J Phys Med Rehabil 93: , S132–S144. |
[349] | Kempermann G , Gage FH , Aigner L , Song H , Curtis MA , Thuret S , Kuhn HG , Jessberger S , Frankland PW , Cameron HA , Gould E , Hen R , Abrous DN , Toni N , Schinder AF , Zhao X , Lucassen PJ , Frisen J ((2018) ) Human adult neurogenesis: Evidence and remaining questions. Cell Stem Cell 23: , 25–30. |
[350] | Hussain G , Wang J , Rasul A , Anwar H , Imran A , Qasim M , Zafar S , Kamran SKS , Razzaq A , Aziz N , Ahmad W , Shabbir A , Iqbal J , Baig SM , Sun T ((2019) ) Role of cholesterol and sphingolipids in brain development and neurological diseases. Lipids Health Dis 18: , 26. |
[351] | Hubler Z , Allimuthu D , Bederman I , Elitt MS , Madhavan M , Allan KC , Shick HE , Garrison E , M TK , Factor DC , Nevin ZS , Sax JL , Thompson MA , Fedorov Y , Jin J , Wilson WK , Giera M , Bracher F , Miller RH , Tesar PJ , Adams DJ ((2018) ) Accumulation of 8,9-unsaturated sterols drives oligodendrocyte formation and remyelination. Nature 560: , 372–376. |
[352] | Jure I , De Nicola AF , Labombarda F ((2019) ) Progesterone effects on the oligodendrocyte linage: All roads lead to the progesterone receptor. Neural Regen Res 14: , 2029–2034. |
[353] | Bartzokis G ((2004) ) Age-related myelin breakdown: A developmental model of cognitive decline and Alzheimer’s disease.. Neurobiol Aging 25: ,5–18; author reply 49-62. |
[354] | Blanchard JW , Akay LA , Davila-Velderrain J , von Maydell D , Mathys H , Davidson SM , Effenberger A , Chen CY , Maner-Smith K , Hajjar I , Ortlund EA , Bula M , Agbas E , Ng A , Jiang X , Kahn M , Blanco-Duque C , Lavoie N , Liu L , Reyes R , Lin YT , Ko T , R’Bibo L , Ralvenius WT , Bennett DA , Cam HP , Kellis M , Tsai LH ((2022) ) APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 611: , 769–779. |
[355] | Munoz Herrera OM , Zivkovic AM ((2022) ) Microglia and cholesterol handling: Implications for Alzheimer’s disease. Biomedicines 10: , 3105. |
[356] | Fairley LH , Lai KO , Wong JH , Chong WJ , Vincent AS , D’Agostino G , Wu X , Naik RR , Jayaraman A , Langley SR , Ruedl C , Barron AM ((2023) ) Mitochondrial control of microglial phagocytosis by the translocator protein and hexokinase 2 in Alzheimer’s disease. Proc Natl Acad Sci U S A 120: , e2209177120. |
[357] | Lauro C , Limatola C ((2020) ) Metabolic eprograming of microglia in the regulation of the innate inflammatory response. Front Immunol 11: , 493. |
[358] | Yamamoto S , Masuda T ((2023) ) Lipid in microglial biology - from material to mediator. Inflamm Regen 43: , 38. |
[359] | Chang CC , Miyazaki A , Dong R , Kheirollah A , Yu C , Geng Y , Higgs HN , Chang TY ((2010) ) Purification of recombinant acyl-coenzyme A:cholesterol acyltransferase 1 (ACAT1) from H293 cells and binding studies between the enzyme and substrates using difference intrinsic fluorescence spectroscopy. Biochemistry 49: , 9957–9963. |
[360] | Seo T , Oelkers PM , Giattina MR , Worgall TS , Sturley SL , Deckelbaum RJ ((2001) ) Differential modulation of ACAT1 and ACAT2 transcription and activity by long chain free fatty acids in cultured cells. Biochemistry 40: , 4756–4762. |
[361] | Radif Y , Ndiaye H , Kalantzi V , Jacobs R , Hall A , Minogue S , Waugh MG ((2018) ) The endogenous subcellular localisations of the long chain fatty acid-activating enzymes ACSL3 and ACSL4 in sma and breast cancer cells. Mol Cell Biochem 448: , 275–286 arco. |
[362] | Golej DL , Askari B , Kramer F , Barnhart S , Vivekanandan-Giri A , Pennathur S , Bornfeldt KE ((2011) ) Long-chain acyl-CoA synthetase 4 modulates prostaglandin E(2) release from human arterial smooth muscle cells. J Lipid Res 52: , 782–793. |
[363] | Han X , Rozen S , Boyle SH , Hellegers C , Cheng H , Burke JR , Welsh-Bohmer KA , Doraiswamy PM , Kaddurah-Daouk R ((2011) ) Metabolomics in early Alzheimer’s disease: Identification of altered plasma sphingolipidome using shotgun lipidomics. PLoS One 6: , e21643. |
[364] | Mapstone M , Cheema AK , Fiandaca MS , Zhong X , Mhyre TR , MacArthur LH , Hall WJ , Fisher SG , Peterson DR , Haley JM , Nazar MD , Rich SA , Berlau DJ , Peltz CB , Tan MT , Kawas CH , Federoff HJ ((2014) ) Plasma phospholipids identify antecedent memory impairment in older adults. Nat Med 20: , 415–418. |
[365] | Olazaran J , Gil-de-Gomez L , Rodriguez-Martin A , Valenti-Soler M , Frades-Payo B , Marin-Munoz J , Antunez C , Frank-Garcia A , Acedo-Jimenez C , Morlan-Gracia L , Petidier-Torregrossa R , Guisasola MC , Bermejo-Pareja F , Sanchez-Ferro A , Perez-Martinez DA , Manzano-Palomo S , Farquhar R , Rabano A , Calero M ((2015) ) A blood-based, 7-metabolite signature for the early diagnosis of Alzheimer’s disease. J Alzheimers Dis 45: , 1157–1173. |
[366] | Schon EA , Area-Gomez E (2022) Reduction of ERMAM-localized APP-C99 and methods of treating Alzheimer‘s disease. US Patent 11,266,626. |
[367] | Strittmatter WJ , Saunders AM , Schmechel D , Pericak-Vance M , Enghild J , Salvesen GS , Roses AD ((1993) ) Apolipoprotein E: High-avidity binding to β-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci U S A 90: , 1977–1981. |
[368] | Sims R , van der Lee SJ , Naj AC , Bellenguez C , Badarinarayan N , Jakobsdottir J , Kunkle BW , Boland A , Raybould R , Bis JC , et al. ((2017) ) Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 49: , 1373–1384. |
[369] | Smith DC , Karahan H , Sagara Wijeratne HR , Al-Amin M , McCord B , Moon Y , Kim J ((2022) ) Deletion of the Alzheimer’s disease risk gene locus results in obesity and systemic metabolic disruption in mice. Front Aging Neurosci 14: , 1035572. |
[370] | Vardarajan BN , Ghani M , Kahn A , Sheikh S , Sato C , Barral S , Lee JH , Cheng R , Reitz C , Lantigua R , Reyes-Dumeyer D , Medrano M , Jimenez-Velazquez IZ , Rogaeva E , St George-Hyslop P , Mayeux R ((2015) ) Rare coding mutations identified by sequencing of Alzheimer disease genome-wide association studies loci. Ann Neurol 78: , 487–498. |
[371] | Keenan BT , Shulman JM , Chibnik LB , Raj T , Tran D , Sabuncu MR , Alzheimer’s Disease Neuroimaging Initiative, Allen AN , Corneveaux JJ , Hardy JA , Huentelman MJ , Lemere CA , Myers AJ , Nicholson-Weller A , Reiman EM , Evans DA , Bennett DA , De Jager PL ((2012) ) A coding variant in CR1 interacts with APOE-epsilon4 to influence cognitive decline. Hum Mol Genet 21: , 2377–2388. |
[372] | Bellenguez C , Kucukali F , Jansen IE , Kleineidam L , Moreno-Grau S , Amin N , Naj AC , Campos-Martin R , Grenier-Boley B , Andrade V , et al. ((2022) ) New insights into the genetic etiology of Alzheimer’s disease and related dementias. Nat Genet 54: , 412–436. |
[373] | Sahid MNA , Liu S , Kiyoi T , Maeyama K ((2017) ) Inhibition of the mevalonate pathway by simvastatin interferes with mast cell degranulation by disrupting the interaction between Rab27a and double C2 alpha proteins. Eur J Pharmacol 814: , 255–263. |
[374] | Yamauchi Y , Iwamoto N , Rogers MA , Abe-Dohmae S , Fujimoto T , Chang CC , Ishigami M , Kishimoto T , Kobayashi T , Ueda K , Furukawa K , Chang TY , Yokoyama S ((2015) ) Deficiency in the lipid exporter ABCA1 impairs retrograde sterol movement and disrupts sterol sensing at the endoplasmic reticulum. J Biol Chem 290: , 23464–23477. |
[375] | Dib S , Pahnke J , Gosselet F ((2021) ) Role of ABCA7 in human health and in Alzheimer’s disease. Int J Mol Sci 22: , 4603. |
[376] | Kielar D , Kaminski WE , Liebisch G , Piehler A , Wenzel JJ , Mohle C , Heimerl S , Langmann T , Friedrich SO , Bottcher A , Barlage S , Drobnik W , Schmitz G ((2003) ) Adenosine triphosphate binding cassette (ABC) transporters are expressed and regulated during terminal keratinocyte differentiation: A potential role for ABCA7 in epidermal lipid reorganization. J Invest Dermatol 121: , 465–474. |
[377] | Holstege H , Hulsman M , Charbonnier C , Grenier-Boley B , Quenez O , Grozeva D , van Rooij JGJ , Sims R , Ahmad S , Amin N , Norsworthy PJ , Dols-Icardo O , Hummerich H , Kawalia A , Amouyel P , Beecham GW , Berr C , Bis JC , Boland A , Bossu P , Bouwman F , Bras J , Campion D , Cochran JN , Daniele A , Dartigues JF , Debette S , Deleuze JF , Denning N , DeStefano AL , Farrer LA , Fernandez MV , Fox NC , Galimberti D , Genin E , Gille JJP , Le Guen Y , Guerreiro R , Haines JL , Holmes C , Ikram MA , Ikram MK , Jansen IE , Kraaij R , Lathrop M , Lemstra AW , Lleo A , Luckcuck L , Mannens M , Marshall R , Martin ER , Masullo C , Mayeux R , Mecocci P , Meggy A , Mol MO , Morgan K , Myers RM , Nacmias B , Naj AC , Napolioni V , Pasquier F , Pastor P , Pericak-Vance MA , Raybould R , Redon R , Reinders MJT , Richard AC , Riedel-Heller SG , Rivadeneira F , Rousseau S , Ryan NS , Saad S , Sanchez-Juan P , Schellenberg GD , Scheltens P , Schott JM , Seripa D , Seshadri S , Sie D , Sistermans EA , Sorbi S , van Spaendonk R , Spalletta G , Tesi N , Tijms B , Uitterlinden AG , van der Lee SJ , Visser PJ , Wagner M , Wallon D , Wang LS , Zarea A , Clarimon J , van Swieten JC , Greicius MD , Yokoyama JS , Cruchaga C , Hardy J , Ramirez A , Mead S , van der Flier WM , van Duijn CM , Williams J , Nicolas G , Bellenguez C , Lambert JC ((2022) ) Exome sequencing identifies rare damaging variants in ATP8B4 and ABCA1 as risk factors for Alzheimer’s disease. Nat Genet 54: , 1786–1794. |
[378] | Lyssenko NN , Miteva Y , Gilroy S , Hanna-Rose W , Schlegel RA ((2008) ) An unexpectedly high degree of specialization and a widespread involvement in sterol metabolism among the C. elegans putative aminophospholipid translocases. BMC Dev Biol 8: , 96. |
[379] | Lindquist SG , Holm IE , Schwartz M , Law I , Stokholm J , Batbayli M , Waldemar G , Nielsen JE ((2008) ) Alzheimer disease-like clinical phenotype in a family with FTDP-17 caused by a MAPT R406W mutation. Eur J Neurol 15: , 377–385. |
[380] | Logue MW , Lancour D , Farrell J , Simkina I , Fallin MD , Lunetta KL , Farrer LA ((2018) ) Targeted sequencing of Alzheimer disease genes in African Americans implicates novel risk variants. Front Neurosci 12: , 592. |
[381] | Ikezu T , Chen C , DeLeo AM , Zeldich E , Fallin MD , Kanaan NM , Lunetta KL , Abraham CR , Logue MW , Farrer LA ((2018) ) Tau phosphorylation is impacted by rare AKAP9 mutations associated with Alzheimer disease in African Americans. J Neuroimmune Pharmacol 13: , 254–264. |
[382] | Crameri A , Biondi E , Kuehnle K , Lutjohann D , Thelen KM , Perga S , Dotti CG , Nitsch RM , Ledesma MD , Mohajeri MH ((2006) ) The role of seladin-1/DHCR24 in cholesterol biosynthesis, APP processing and Aβ generation. EMBO J 25: , 432–443. |
[383] | Kim M , Suh J , Romano D , Truong MH , Mullin K , Hooli B , Norton D , Tesco G , Elliott K , Wagner SL , Moir RD , Becker KD , Tanzi RE ((2009) ) Potential late-onset Alzheimer’s disease-associated mutations in the ADAM10 gene attenuate α-secretase activity. Hum Mol Genet 18: , 3987–3996. |
[384] | Suh J , Choi SH , Romano DM , Gannon MA , Lesinski AN , Kim DY , Tanzi RE ((2013) ) ADAM10 missense mutations potentiate β-amyloid accumulation by impairing prodomain chaperone function. Neuron 80: , 385–401. |
[385] | Ubelmann F , Burrinha T , Salavessa L , Gomes R , Ferreira C , Moreno N , Guimas Almeida C ((2017) ) Bin1 and CD2AP polarise the endocytic generation of beta-amyloid. EMBO Rep 18: , 102–122. |
[386] | Lomoio S , Willen R , Kim W , Ho KZ , Robinson EK , Prokopenko D , Kennedy ME , Tanzi RE , Tesco G ((2020) ) Gga3 deletion and a GGA3 rare variant associated with late onset Alzheimer’s disease trigger BACE1 accumulation in axonal swellings. Sci Transl Med 12: , eaba1871. |
[387] | Patel D , Mez J , Vardarajan BN , Staley L , Chung J , Zhang X , Farrell JJ , Rynkiewicz MJ , Cannon-Albright LA , Teerlink CC , Stevens J , Corcoran C , Gonzalez Murcia JD , Lopez OL , Mayeux R , Haines JL , Pericak-Vance MA , Schellenberg G , Kauwe JSK , Lunetta KL , Farrer LA , Alzheimer’s Disease Sequencing Project ((2019) ) Association of rare coding mutations with Alzheimer disease and other dementias among adults of European ancestry. JAMA Netw Open 2: , e191350. |
[388] | Konishi J , Kawaguchi KS , Vo H , Haruki N , Gonzalez A , Carbone DP , Dang TP ((2007) ) γ-Secretase inhibitor prevents Notch3 activation and reduces proliferation in human lung cancers. Cancer Res 67: , 8051–8057. |
[389] | Jakobsdottir J , van der Lee SJ , Bis JC , Chouraki V , Li-Kroeger D , Yamamoto S , Grove ML , Naj A , Vronskaya M , Salazar JL , DeStefano AL , Brody JA , Smith AV , Amin N , Sims R , Ibrahim-Verbaas CA , Choi SH , Satizabal CL , Lopez OL , Beiser A , Ikram MA , Garcia ME , Hayward C , Varga TV , Ripatti S , Franks PW , Hallmans G , Rolandsson O , Jansson JH , Porteous DJ , Salomaa V , Eiriksdottir G , Rice KM , Bellen HJ , Levy D , Uitterlinden AG , Emilsson V , Rotter JI , Aspelund T ; Cohorts for Heart and Aging Research in Genomic Epidemiology consortium ; Alzheimer’s Disease Genetic Consortium; Genetic and Environmental Risk in Alzheimer’s Disease consortium; Donnell CJ , Fitzpatrick AL , Launer LJ , Hofman A , Wang LS , Williams J , Schellenberg GD , Boerwinkle E , Psaty BM , Seshadri S , Shulman JM , Gudnason V , van Duijn CM ((2016) ) Rare functional variant in TM2D3 is associated with late-onset Alzheimer’s disease. PLoS Genet 12: , e1006327. |
[390] | Salazar JL , Yang SA , Lin YQ , Li-Kroeger D , Marcogliese PC , Deal SL , Neely GG , Yamamoto S ((2021) ) TM2D genes regulate Notch signaling and neuronal function in. Drosophila. PLoS Genet 17: , e1009962. |
[391] | Asanomi Y , Shigemizu D , Akiyama S , Miyashita A , Mitsumori R , Hara N , Ikeuchi T , Niida S , Ozaki K ((2022) ) A functional variant of SHARPIN confers increased risk of late-onset Alzheimer’s disease. J Hum Genet 67: , 203–208. |
[392] | Tamiya H , Urushihara N , Shizuma K , Ogawa H , Nakai S , Wakamatsu T , Takenaka S , Kakunaga S ((2023) ) SHARPIN enhances ferroptosis in synovial sarcoma cells via NF-kB- and PRMT5-mediated PGC1α reduction. Cancers (Basel) 15: , 3484. |
[393] | Zeng C , Lin J , Zhang K , Ou H , Shen K , Liu Q , Wei Z , Dong X , Zeng X , Zeng L , Wang W , Yao J ((2022) ) SHARPIN promotes cell proliferation of cholangiocarcinoma and inhibits ferroptosis via p53/SLC7A11/GPX4 signaling. Cancer Sci 113: , 3766–3775. |
[394] | Chen Y , Li S , Yin M , Li Y , Chen C , Zhang J , Sun K , Kong X , Chen Z , Qian J ((2023) ) Isorhapontigenin attenuates cardiac microvascular injury in diabetes via the inhibition of mitochondria-associated ferroptosis through PRDX2-MFN2-ACSL4 pathways. Diabetes 72: , 389–404. |
[395] | Ponneri Babuharisankar A , Kuo CL , Chou HY , Tangeda V , Fan CC , Chen CH , Kao YH , Lee AY ((2023) ) Mitochondrial Lon-induced mitophagy benefits hypoxic resistance via Ca2 +-dependent FUNDC1 phosphorylation at the ER-mitochondria interface. Cell Death Dis 14: , 199. |
[396] | Kamboh MI ((2022) ) Genomics and functional genomics of Alzheimer’s disease. Neurotherapeutics 19: , 152–172. |
[397] | Sato K , Tanabe C , Yonemura Y , Watahiki H , Zhao Y , Yagishita S , Ebina M , Suo S , Futai E , Murata M , Ishiura S ((2012) ) Localization of mature neprilysin in lipid rafts. J Neurosci Res 90: , 870–877. |
[398] | El-Amouri SS , Zhu H , Yu J , Marr R , Verma IM , Kindy MS ((2008) ) Neprilysin : An enzyme candidate to slow therogression of Alzheimer’s disease,Am J Pathol 172: , 1342–1354 |
[399] | Pardossi-Piquard R , Petit A , Kawarai T , Sunyach C , Alves da Costa C , Vincent B , Ring S , D’Adamio L , Shen J , Muller U , St George Hyslop P , Checler F ((2005) ) Presenilin-dependent transcriptional control of the Aβ-degrading enzyme neprilysin by intracellular domains of βAPP and APLP. Neuron 46: , 541–554. |
[400] | Wetzel-Smith MK , Hunkapiller J , Bhangale TR , Srinivasan K , Maloney JA , Atwal JK , Sa SM , Yaylaoglu MB , Foreman O , Ortmann W , Rathore N , Hansen DV , Tessier-Lavigne M , Alzheimer’s Disease Genetics Consortium , Mayeux R , Pericak-Vance M , Haines J , Farrer LA , Schellenberg GD , Goate A , Behrens TW , Cruchaga C , Watts RJ , Graham RR ((2014) ) A rare mutation in UNC5C predisposes to late-onset Alzheimer’s disease and increases neuronal cell death. Nat Med 20: , 1452–1457. |
[401] | Hernaiz-Llorens M , Rosello-Busquets C , Durisic N , Filip A , Ulloa F , Martinez-Marmol R , Soriano E ((2021) ) Growth cone repulsion to Netrin-1 depends on lipid raft microdomains enriched in UNC5 receptors. Cell Mol Life Sci 78: , 2797–2820. |
[402] | Zhao Y , Li X , Huang T , Jiang LL , Tan Z , Zhang M , Cheng IH , Wang X , Bu G , Zhang YW , Wang Q , Xu H ((2017) ) Intracellular trafficking of TREM2 is regulated by presenilin 1. Exp Mol Med 49: , e405. |
[403] | Wang Y , Cella M , Mallinson K , Ulrich JD , Young KL , Robinette ML , Gilfillan S , Krishnan GM , Sudhakar S , Zinselmeyer BH , Holtzman DM , Cirrito JR , Colonna M ((2015) ) TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell 160: , 1061–1071. |
[404] | Nugent AA , Lin K , van Lengerich B , Lianoglou S , Przybyla L , Davis SS , Llapashtica C , Wang J , Kim DJ , Xia D , Lucas A , Baskaran S , Haddick PCG , Lenser M , Earr TK , Shi J , Dugas JC , Andreone BJ , Logan T , Solanoy HO , Chen H , Srivastava A , Poda SB , Sanchez PE , Watts RJ , Sandmann T , Astarita G , Lewcock JW , Monroe KM , Di Paolo G ((2020) ) TREM2 regulates microglial cholesterol metabolism upon chronic phagocytic challenge. Neuron 105: , 837–854. |
[405] | Malik M , Simpson JF , Parikh I , Wilfred BR , Fardo DW , Nelson PT , Estus S ((2013) ) CD33 Alzheimer’s risk-altering polymorphism, CD33 expression, and exon 2 splicing. J Neurosci 33: , 13320–13325. |
[406] | Chan G , White CC , Winn PA , Cimpean M , Replogle JM , Glick LR , Cuerdon NE , Ryan KJ , Johnson KA , Schneider JA , Bennett DA , Chibnik LB , Sperling RA , Bradshaw EM , De Jager PL ((2015) ) CD33 modulates TREM2: Convergence of Alzheimer loci. Nat Neurosci 18: , 1556–1558. |
[407] | Deming Y , Filipello F , Cignarella F , Cantoni C , Hsu S , Mikesell R , Li Z , Del-Aguila JL , Dube U , Farias FG , Bradley J , Budde J , Ibanez L , Fernandez MV , Blennow K , Zetterberg H , Heslegrave A , Johansson PM , Svensson J , Nellgard B , Lleo A , Alcolea D , Clarimon J , Rami L , Molinuevo JL , Suarez-Calvet M , Morenas-Rodriguez E , Kleinberger G , Ewers M , Harari O , Haass C , Brett TJ , Benitez BA , Karch CM , Piccio L , Cruchaga C ((2019) ) The MS4A gene cluster is a key modulator of soluble TREM2 and Alzheimer’s disease risk. Sci Transl Med 11: , eaau2291. |
[408] | Hartl D , May P , Gu W , Mayhaus M , Pichler S , Spaniol C , Glaab E , Bobbili DR , Antony P , Koegelsberger S , Kurz A , Grimmer T , Morgan K , Vardarajan BN , Reitz C , Hardy J , Bras J , Guerreiro R , Balling R , Schneider JG , Riemenschneider M , AESG ((2020) ) A rare loss-of-function variant of ADAM17 is associated with late-onset familial Alzheimer disease. Mol Psychiatry 25: , 629–639. |
[409] | Feuerbach D , Schindler P , Barske C , Joller S , Beng-Louka E , Worringer KA , Kommineni S , Kaykas A , Ho DJ , Ye C , Welzenbach K , Elain G , Klein L , Brzak I , Mir AK , Farady CJ , Aichholz R , Popp S , George N , Neumann U ((2017) ) ADAM17 is the main sheddase for the generation of human triggering receptor expressed in myeloid cells (hTREM2) ectodomain and cleaves TREM2 after Histidine 157. Neurosci Lett 660: , 109–114. |
[410] | Li X , Ma Y , Wei X , Li Y , Wu H , Zhuang J , Zhao Z ((2014) ) Clusterin in Alzheimer’s disease: A player in the biological behavior of amyloid-beta. Neurosci Bull 30: , 162–168. |
[411] | Leeb C , Eresheim C , Nimpf J ((2014) ) Clusterin is a ligand for apolipoprotein E receptor 2 (ApoER2) and very low density lipoprotein receptor (VLDLR) and signals via the Reelin-signaling pathway. J Biol Chem 289: , 4161–4172. |
[412] | Gelissen IC , Hochgrebe T , Wilson MR , Easterbrook-Smith SB , Jessup W , Dean RT , Brown AJ ((1998) ) Apolipoprotein J (clusterin) induces cholesterol export from macrophage-foam cells: A potential anti-atherogenic function? Biochem J 331: (Pt 1), 231–237. |
[413] | Magno L , Lessard CB , Martins M , Lang V , Cruz P , Asi Y , Katan M , Bilsland J , Lashley T , Chakrabarty P , Golde TE , Whiting PJ ((2019) ) Alzheimer’s disease phospholipase C-gamma-2 (PLCG2) protective variant is a functional hypermorph. Alzheimers Res Ther 11: , 16. |
[414] | Cruchaga C , Karch CM , Jin SC , Benitez BA , Cai Y , Guerreiro R , Harari O , Norton J , Budde J , Bertelsen S , Jeng AT , Cooper B , Skorupa T , Carrell D , Levitch D , Hsu S , Choi J , Ryten M , U K Brain Expression Consortium (UKBEC), Sassi C , Bras J , Gibbs RJ , Hernandez DG , Lupton MK , Powell J , Forabosco P , Ridge PG , Corcoran CD , Tschanz JT , Norton MC , Munger RG , Schmutz C , Leary M , Demirci FY , Bamne MN , Wang X , Lopez OL , Ganguli M , Medway C , Turton J , Lord J , Braae A , Barber I , Brown K , The Alzheimer’s Research UK (ARUK) Consortium , Pastor P , Lorenzo-Betancor O , Brkanac Z , Scott E , Topol E , Morgan K , Rogaeva E , Singleton A , Hardy J , Kamboh MI , George-Hyslop PS , Cairns N , Morris JC , Kauwe JSK , Goate AM ((2014) ) Rare coding variants in the phospholipase D3 gene confer risk for Alzheimer’s disease. Nature 505: , 550–554. |
[415] | Van Acker ZP , Perdok A , Hellemans R , North K , Vorsters I , Cappel C , Dehairs J , Swinnen JV , Sannerud R , Bretou M , Damme M , Annaert W ((2023) ) Phospholipase D3 degrades mitochondrial DNA to regulate nucleotide signaling and APP metabolism. Nat Commun 14: , 2847. |
[416] | Brouwers N , Sleegers K , Engelborghs S , Maurer-Stroh S , Gijselinck I , van der Zee J , Pickut BA , Van den Broeck M , Mattheijssens M , Peeters K , Schymkowitz J , Rousseau F , Martin JJ , Cruts M , De Deyn PP , Van Broeckhoven C ((2008) ) Genetic variability in progranulin contributes to risk for clinically diagnosed Alzheimer disease. Neurology 71: , 656–664. |
[417] | Evers BM , Rodriguez-Navas C , Tesla RJ , Prange-Kiel J , Wasser CR , Yoo KS , McDonald J , Cenik B , Ravenscroft TA , Plattner F , Rademakers R , Yu G , White CL 3rd , Herz J ((2017) ) Lipidomic and transcriptomic basis of lysosomal dysfunction in progranulin deficiency. Cell Rep 20: , 2565–2574. |
[418] | Verheijen J , Van den Bossche T , van der Zee J , Engelborghs S , Sanchez-Valle R , Llado A , Graff C , Thonberg H , Pastor P , Ortega-Cubero S , Pastor MA , Benussi L , Ghidoni R , Binetti G , Clarimon J , Lleo A , Fortea J , de Mendonca A , Martins M , Grau-Rivera O , Gelpi E , Bettens K , Mateiu L , Dillen L , Cras P , De Deyn PP , Van Broeckhoven C , Sleegers K ((2016) ) A comprehensive study of the genetic impact of rare variants in SORL1 in European early-onset Alzheimer’s disease. Acta Neuropathol 132: , 213–224. |
[419] | Fjorback AW , Andersen OM ((2012) ) SorLA is a molecular link for retromer-dependent sorting of the amyloid precursor protein. Commun Integr Biol 5: , 616–619. |
[420] | Spoelgen R , von Arnim CA , Thomas AV , Peltan ID , Koker M , Deng A , Irizarry MC , Andersen OM , Willnow TE , Hyman BT ((2006) ) Interaction of the cytosolic domains of sorLA/LR11 with the amyloid precursor protein (APP) and β-secretase β-site APP-cleaving enzyme. J Neurosci 26: , 418–428. |
[421] | Bohm C , Seibel NM , Henkel B , Steiner H , Haass C , Hampe W ((2006) ) SorLA signaling by regulated intramembrane proteolysis. J Biol Chem 281: , 14547–14553. |
[422] | Tian Y , Chang JC , Fan EY , Flajolet M , Greengard P ((2013) ) Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc Natl Acad Sci U S A 110: , 17071–17076. |
[423] | Tian Y , Chang JC , Greengard P , Flajolet M ((2014) ) The convergence of endosomal and autophagosomal pathways: Implications for APP-CTF degradation. Autophagy 10: , 694–696. |
[424] | Thomas RS , Henson A , Gerrish A , Jones L , Williams J , Kidd EJ ((2016) ) Decreasing the expression of PICALM reduces endocytosis and the activity of β-secretase: Implications for Alzheimer’s disease. BMC Neurosci 17: , 50. |
[425] | Mercer JL , Argus JP , Crabtree DM , Keenan MM , Wilks MQ , Chi JT , Bensinger SJ , Lavau CP , Wechsler DS ((2015) ) Modulation of PICALM levels perturbs cellular cholesterol homeostasis. PLoS One 10: , e0129776. |
[426] | Kizil C , Sariya S , Kim YA , Rajabli F , Martin E , Reyes-Dumeyer D , Vardarajan B , Maldonado A , Haines JL , Mayeux R , Jimenez-Velazquez IZ , Santa-Maria I , Tosto G ((2022) ) Admixture mapping of Alzheimer’s disease in Caribbean Hispanics identifies a new locus on 22q13.1. Mol Psychiatry 27: , 2813–2820. |
[427] | Seo J , Byun MS , Yi D , Lee JH , Jeon SY , Shin SA , Kim YK , Kang KM , Sohn CH , Jung G , Park JC , Han SH , Byun J , Mook-Jung I , Lee DY , Choi M , KBASE Research Group ((2020) ) Genetic associations of pathology influence Alzheimer’s disease susceptibility. Alzheimers Res Ther 12: , 156. |
[428] | Rathore N , Ramani SR , Pantua H , Payandeh J , Bhangale T , Wuster A , Kapoor M , Sun Y , Kapadia SB , Gonzalez L , Zarrin AA , Goate A , Hansen DV , Behrens TW , Graham RR ((2018) ) Paired Immunoglobulin-like type 2 receptor alpha G78R variant alters ligand binding and confers protection to Alzheimer’s disease. PLoS Genet 14: , e1007427. |
[429] | N’Songo A , Carrasquillo MM , Wang X , Burgess JD , Nguyen T , Asmann YW , Serie DJ , Younkin SG , Allen M , Pedraza O , Duara R , Greig Custo MT , Graff-Radford NR , Ertekin-Taner N ((2017) ) African American exome sequencing identifies potential risk variants at Alzheimer disease loci. Neurol Genet 3: , e141. |


