
Multi-Target Neuroprotection of Thiazolidinediones on Alzheimer’s Disease via Neuroinflammation and Ferroptosis
Abstract
Alzheimer’s disease (AD) is the main cause of dementia in older age. The prevalence of AD is growing worldwide, causing a tremendous burden to societies and families. Due to the complexity of its pathogenesis, the current treatment of AD is not satisfactory, and drugs acting on a single target may not prevent AD progression. This review summarizes the multi-target pharmacological effects of thiazolidinediones (TZDs) on AD. TZDs act as peroxisome proliferator-activated receptor gamma (PPARγ) agonists and long-chain acyl-CoA synthetase family member 4 (ACSL4) inhibitors. TZDs ameliorated neuroinflammation and ferroptosis in preclinical models of AD. Here, we discussed recent findings from clinical trials of pioglitazone in the treatment of AD, ischemic stroke, and atherosclerosis. We also dissected the major limitations in the clinical application of pioglitazone and explained the potential benefit of pioglitazone in AD. We recommend the use of pioglitazone to prevent cognitive decline and lower AD risk in a specific group of patients.
INTRODUCTION
Alzheimer’s disease (AD) is the main cause of dementia in older age and its prevalence is growing worldwide. By 2050, the global prevalence of dementia will triple [1]. Currently, pharmaceutical companies are attempting to develop AD drugs mainly based on the amyloid cascade hypothesis. However, many drugs targeting amyloid-β (Aβ) failed to demonstrate clinical efficacy. Aducanumab, an anti-Aβ antibody, was recently granted FDA approval. Although it removes Aβ plaques in patients with AD, it does not sufficiently improve brain function and cognition, nor prevent AD progression [2]. With the development of neuroimaging, neuropathology, and biomarkers, a growing body of evidence suggests that AD is a neurodegenerative disease with complex and multifactorial etiology [3, 4].
Typical pathological changes of AD include diffuse precipitation of extracellular Aβ plaques and intracellular neurofibrillary tangles (NFTs). Aβ peptides are produced by sequential cleavage of Aβ precursor protein (AβPP) by β-site AβPP cleaving enzyme 1 (BACE1) and γ-secretase complex. They are released into extracellular space and aggregated into oligomers and fibrils [5]. The 42-amino acid form (Aβ1–42) of Aβ is its most toxic form, which can directly damage neurons by binding to neuron-specific receptors or altering membrane lipid permeability [6, 7]. NFTs are the abnormal aggregation of hyperphosphorylated tau proteins, which hinder microtubules assembly and impair axonal transport in synapses [8]. In 1991, Braak found that the distribution pattern and packing density of Aβ peptides turned out to be limited for differentiation of neuropathological stages. In contrast, NFTs exhibited a characteristic distribution pattern permitting the differentiation of six stages, which called Braak-like pattern [9, 10]. The spatial and temporal pattern in the appearance of these NFTs correlates with cognitive decline as well as brain atrophy [3, 11, 12].
A growing number of studies have confirmed that neuroinflammation plays an important role in the pathogenesis of AD. The deposition of Aβ peptides drives neuroinflammation by activating microglia. When NLRP3 inflammasome assembles inside of microglia, its activation will promote Aβ deposition and drive tau pathology. Meanwhile, microglia are involved in the processing and spreading of tau [13–15]. Pascoal found microglial activation and tau accumulation colocalize in a Braak-like pattern in the living human brain with positron emission tomography brain imaging, and tau propagation pathways depended on the baseline microglia network rather than the tau network. Their findings support a model where an interaction between Aβ and activated microglia sets the pace for tau spread across Braak stages [16].
Ferroptosis is a newly discovered programmed cell death characterized by iron dyshomeostasis and lipid peroxidation, which was reminiscent of AD. In addition to the accumulation of Aβ plaques, NFTs, and neuroinflammation, AD patients show progressive synaptic loss, and neuronal death, also high oxidative stress that correlates with abnormal levels or overload of brain metals [17]. Cortical iron has been shown to be elevated in AD, the level of cortical iron was strongly associated with cognitive decline in AD [18]. Cortical iron might propel cognitive deterioration by inducing oxidative stress and ferroptosis, suggesting anti-ferroptotic therapies may be efficacious in AD.
The complex pathophysiology of AD necessitates the simultaneous pursuit of multiple targets for successful treatment. However, multi-target drugs generally have poor specificity and diminished efficacy [19]. TZDs are a group of anti-diabetic drugs that improve insulin resistance [20, 21]. TZDs, including troglitazone, rosiglitazone, and pioglitazone, can suppress the inflammatory response and cell ferroptosis with high target specificity. Therefore, in addition to their use in diabetes, researchers administered TZDs in the preclinical and clinical studies of AD. In addition to the pharmacological targets of TZDs, we discuss the neuroprotective effects of TZDs on AD, which is mainly mediated by inhibiting neuroinflammation and ferroptosis.
PHARMACOLOGICAL TARGETS OF TZDS
PPARγ
PPARγ is a member of the peroxisome proliferator-activated receptors (PPARs) family, which consists of three isoforms: PPARα, PPARβ/δ, and PPARγ. PPARγ functions in a ligand-dependent manner [19, 22]. Endogenous polyunsaturated fatty acids (PUFAs) act as the natural ligand of PPARγ, with a low affinity. TZDs are exogenous strong activators of PPARγ due to their high affinity (Fig. 1) [23]. Through transactivation, agonist-bound PPARγ positively regulates cell metabolic homeostasis, including adipogenesis, fat storage, insulin sensitization, and cellular differentiation. Anti-inflammatory properties of PPARγ are regulated by transrepression, not transactivation.
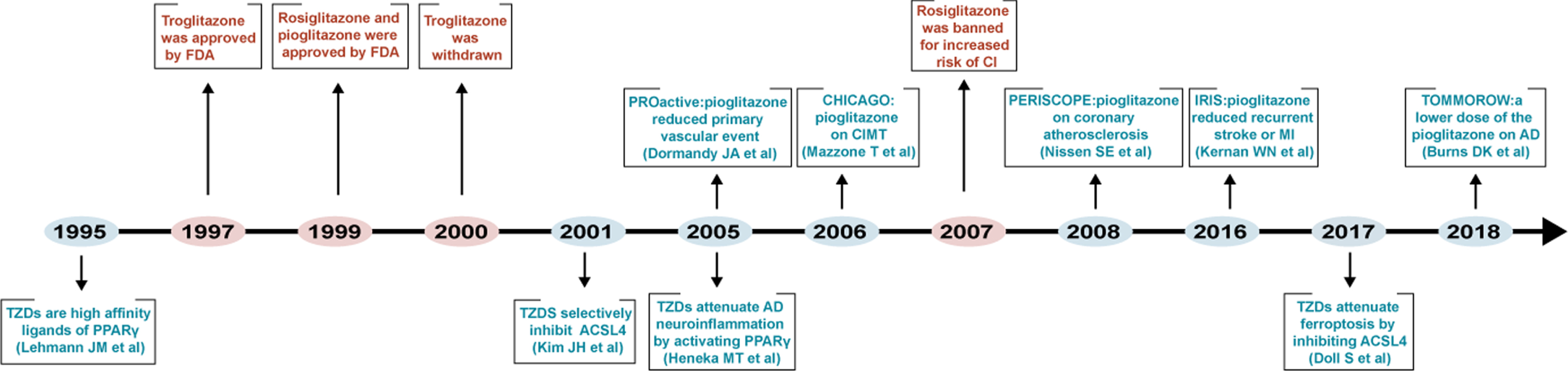
Fig. 1
Timeline diagram depicting mechanism-based studies, clinical application, and clinical trials of TZDs.
Neuroinflammation is the immune response of the central nervous system (CNS) to various harmful stimuli. In AD, neuroinflammation is primarily mediated by immune cells in the CNS. Activated microglia, reactive astrocytes, and increased levels of proinflammatory cytokines are involved in neuroinflammation [24–26]. Agonist-bound PPARγ inhibits neuroinflammation by antagonizing the function of inflammation-related transcription factors, such as nuclear factor-κB (NF-κB), activator protein-1 (AP-1), JAK/STAT, and nuclear factors of activated T-cells (NFAT) [27–29]. Three principal transrepression mechanisms have been proposed. First, activated PPARγ competitively binds most of the coactivators, making them unavailable for other transcription factors. Second, PPARγ directly binds to other transcription factors, preventing them from inducing gene transcription. Third, PPARγ inhibits the activation of mitogen-activated protein kinase (MAPK), thereby inhibiting downstream transcription factors. Among them, NF-κB is the main target for PPARγ, which is involved in the expression of numerous inflammatory factors and pro-inflammatory mediators (Fig. 2) [22].
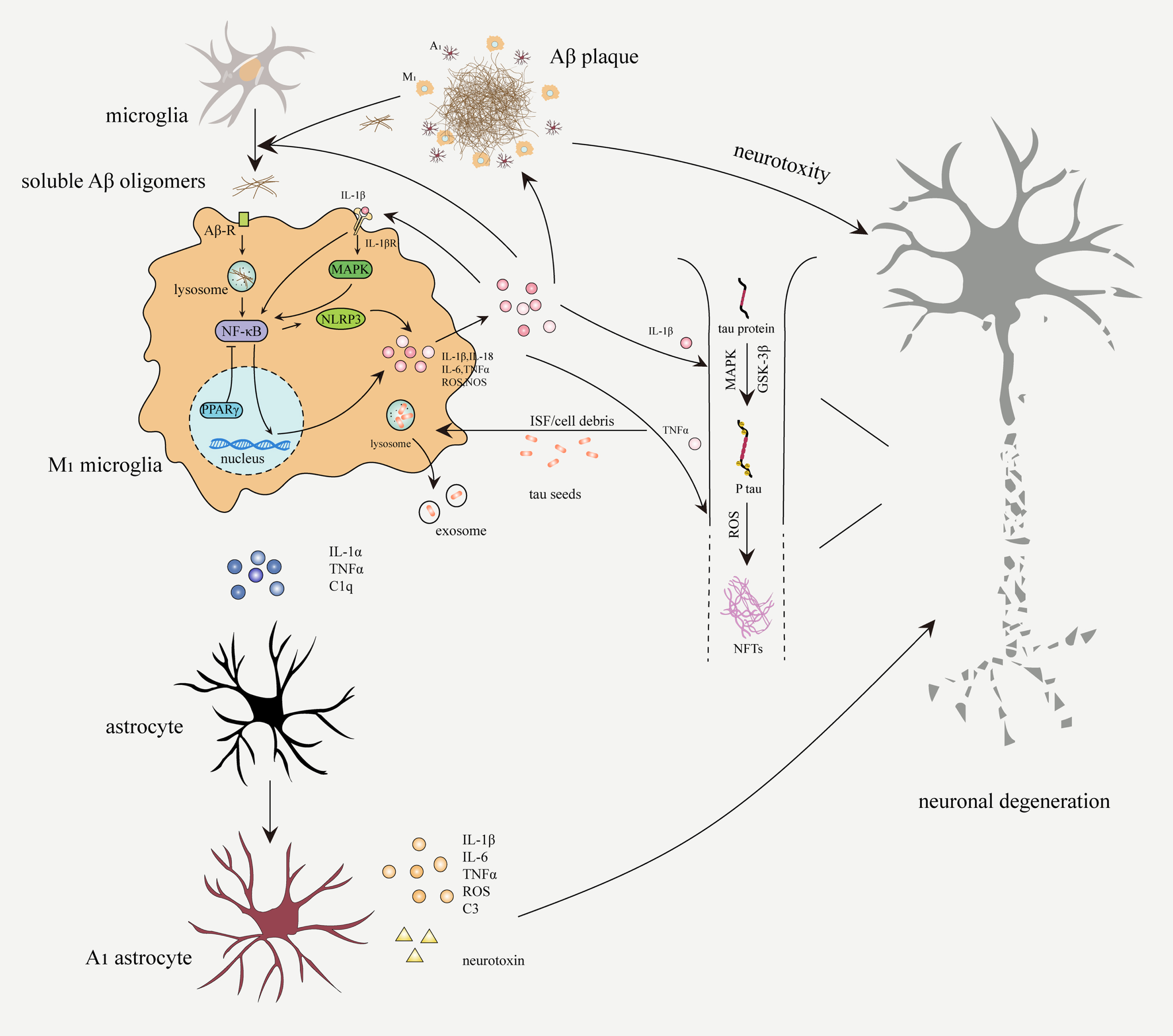
Fig. 2
Neuroinflammation in the pathogenesis of AD. Soluble Aβ oligomers bind to Aβ receptors (Aβ-R) on microglia, which will be subsequently internalized and degraded. Activated microglia secrete proinflammatory factors through NF-κB signaling pathway to amplify microglial activation and phagocytosis. Aβ aggregation due to overproduction or impaired phagocytosis, continuously activates microglia and astrocytes. M1 microglia and A1 astrocytes secrete proinflammatory cytokines (IL-1β, IL-6, IL-18, and TNFα), ROS, complement C3, and neurotoxin, which can accelerate neuronal damage, expand inflammatory response, and promote Aβ deposition and NFTs formation. Activated microglia participate in the spread of pathological tau through the exosome pathway.
ACSL4
Acyl-CoA synthetases (ACS) convert fatty acids to their respective acyl-CoA-activated forms, which are involved in energy metabolism and membrane structure. Non-activated free fatty acids are involved in the production of bioactive mediators such as prostaglandins (PGs), leukotrienes (LTs), and thromboxane (TX) [30]. Among the ACS family, long-chain acyl-CoA synthetases (ACSLs) consume a broad range of long-chain fatty acid substrates from 10 to 22 carbons, as their substrate. ACSLs consist of five ACSL isozymes, including ACSL1, ACSL5, ACSL6, ACSL3, and ACSL4. They are classified based on sequence homology and substrate specificities. For instance, ACSL4 especially prefers PUFAs [31]. The study found that TZDs could specifically inhibit ACSL4 in a PPARγ-independent manner (Figs. 1 and 3) [32]. PUFAs refer to long-chain fatty acids with more than two double bonds. PUFAs are susceptible to lipid peroxidation because of weak C–H bonds at the bis-allylic positions [33].
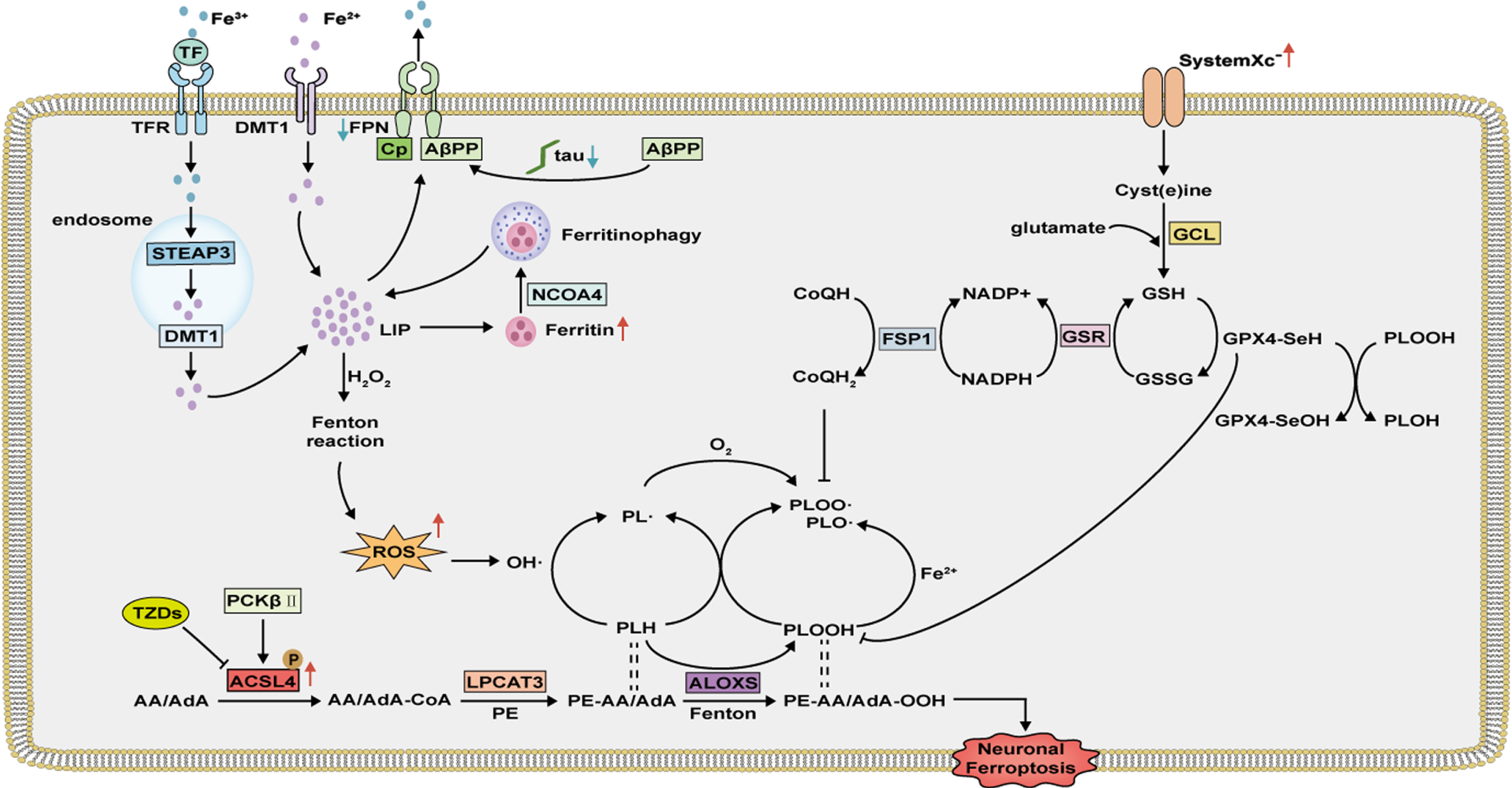
Fig. 3
Neuronal ferroptosis mechanism in AD. Overexpressed ACSL4 is activated and catalyzes AA/AdA to AA/AdA-CoA, which is then esterified to PE by LPCAT3. Decreased iron export and increased expression of ferritin promote neuronal iron accumulation. Fe2 + in LIP, via the Fenton reactions or ALOXs, catalyzes the production of phospholipid hydroperoxides (PLOOH) and phospholipid radicals (PL•, PLO•, and PLOO•), leading to neuronal ferroptosis. Neuronal ferroptosis can be prevented by cyst(e)ine–GSH–GPX4 axis and NADH-FSP1-CoQ10 axis.
Ferroptosis is a recently identified form of non-apoptotic cell death. Ferroptosis is caused by iron-dependent peroxidation of lipids and plays an important role in AD [34, 35]. Membrane phospholipids containing PUFAs (PUFA-PLs) are pivotal for ferroptosis. Blocking the peroxidation site of PUFAs could prevent ferroptosis [36]. ACSL4 is a key enzyme for ferroptosis, activating PUFA to form PUFA-COA, which will be esterified to PUFA-PLs. TZDs, as specific inhibitors of ACSL4, directly inhibit PUFAs incorporation into cellular lipids in a PPARγ-independent manner. 10 uM concentration of troglitazone, rosiglitazone, or pioglitazone could inhibit 85–95% function of ACSL4. Rosiglitazone had the strongest inhibitory effect, comparable to ACSL4 knockout, and effectively prevented ferroptosis [31, 32]. Unlike ACSL4 deletion, deletion of other members of ACSL did not affect ferroptosis [31].
TZDS INHIBIT NEUROINFLAMMATION IN AD
Cell players of neuroinflammation
Microglia
Microglia, the resident phagocytes of the CNS, are widely distributed in the brain. Microglia are the main driver of neuroinflammation. In physiological conditions, microglia play an important role in the maintenance and plasticity of neuronal circuits and contribute to synaptic remodeling. Meanwhile, microglia use constantly motile processes to find pathogens, cellular debris, or misfolded proteins. Once activated by these pathological factors, microglia initiate phagocytosis to maintain CNS homeostasis [6].
The soluble Aβ aggregates in AD bind to cell-surface receptors of microglia (Fig. 2), including CD36, CD47, α6β1-integrin, and toll-like receptors (TLRs). This receptor-ligand interaction activates phagocytic uptake of Aβ by microglia. Internalized Aβ fibers are degraded by lysosomal proteases [37, 38]. Microglial activation simultaneously induces a variety of phenotypes. The classic M1 phenotype of microglia, also known as the proinflammatory phenotype, is characterized by increased secretion of proinflammatory cytokines, such as TNFα, IL-1, IL-6, IL-12, and IL-18. M1 microglia also release reactive oxygen species (ROS) and nitrogen oxide (NO) with less phagocytosis. The classic M2 phenotype of microglia, also known as the anti-inflammatory phenotype, releases anti-inflammatory cytokines, such as IL-4, IL-10, IL-13, and transforming growth factor-β (TGF-β). M2 microglia has outstanding phagocytosis ability, with no toxic products. The M1 and M2 are the extremes of microglial phenotype. There may be a series of transitional phenotypes, which collectively participate in local immune responses in the brain [39, 40]. Activated microglia upregulate NLRP3 inflammasome and release IL-1β and IL-18 through NF-κB signaling pathway [13, 41]. IL-1β is a potent proinflammatory cytokine, which binds to the IL-1β receptor (IL-1R) on neurons and glial cells, activates the classical NF-κB and MAPK signaling pathways, thereby upregulating itself and other proinflammatory cytokines, such as TNF-α and IL-6 (Fig. 2) [42]. In addition, IL-1β induces an M1-like phenotype of microglia to release more proinflammatory factors and ROS [6].
Similar to the effect of PPARγ activation on mononuclear macrophages in peripheral tissues, TZDs inhibit the nuclear translocation of NF-κB by activating PPARγ, and induce microglia transformation from the proinflammatory M1 to the anti-inflammatory M2 phenotype in the CNS [43]. Consistently, pioglitazone decreased the expression of IL-1β, IL-6, and TNF-α, and promoted the expression of IL-4, IL-10, and TGF-β in neuroinflammation [44, 45]. The expression of NLRP3 and IL-1β significantly decreased following treatment with pioglitazone, which protected neurons against microglial NO [46, 47]. Furthermore, pioglitazone upregulated the expression of the microglial cell-surface scavenging receptor, CD36, and enhanced Aβ phagocytosis [48, 49].
Astrocytes
Astrocytes are the most important supporting cells in CNS. They are involved in a variety of physiological functions, including secretion of trophic molecules, regulation of brain microenvironment, maintenance of blood-brain barrier (BBB) integrity, and normal function of synapses [50]. Zamanian et al. used inflammatory and ischemia models to induce two different types of reactive astrocytes and found that inflammation-induced reactive astrocytes upregulated a variety of “harmful” genes, named type A1 astrocytes, while ischemia-induced reactive astrocytes upregulated a variety of “beneficial” genes, named type A2 astrocytes [51].
In neuroinflammation, microglia induce A1 astrocytes by releasing three main cytokines, IL-1α, TNFα, and complement component subunit 1Q (C1q) [52]. A1 astrocytes cannot support neuronal survival, outgrowth, synaptogenesis, and phagocytosis [50]. Similar to M1 microglia, A1 astrocytes upregulate inflammatory markers such as TNFα, IL-1β, IL-6, ROS, and complement component C3 through NF-κB signaling pathway. Complement C3 activates neuronal C3aR to disrupt the morphology and function of dendrites [53]. Moreover, complement C3 can be specifically used as a marker of reactive A1 astrocytes [52]. A1 phenotype simultaneously secretes a neurotoxin that can induce apoptosis in neurons and oligodendrocytes (Fig. 2) [25].
A1 astrocytes reside around Aβ plaques. Rosiglitazone significantly reduced the intensity of glial fibrillary acidic protein (GFAP) staining and the number of A1 astrocytes and promoted the functional recovery of astrocytes [54–56]. Pioglitazone downregulated IL-1β, IL-6, TNF-α, COX2, and iNOX in astrocytes through the PPARγ/NF-KB pathway, thereby alleviating neuroinflammation [57–59].
Neuroinflammation regulate Aβ and tau pathology
In the early stages of AD, microglia release inflammatory cytokines and chemokines to recruit more microglia and astrocytes for phagocytic degradation of Aβ. However, when Aβ accumulation exceeds the phagocytosis capacity of microglia, or when the phagocytosis capacity of microglia is impaired due to the presence of APOE4 or TREM2 variants, Aβ accumulates in the extracellular space and directly damages neurons (Fig. 2) [60, 61]. Normal astrocytes can degrade Aβ by releasing proteases, which play an important role in Aβ clearance. Impaired function and atrophy of reactive astrocytes might contribute to reduced clearance of Aβ [62]. The proinflammatory factors secreted by activated microglia and reactive astrocytes can promote AβPP synthesis and BACE1 expression, thereby enhancing Aβ deposition [63, 64]. Due to the continuous positive feedback loop between Aβ and neuroinflammation, chronic inflammatory response activates microglia and astrocytes in AD [26].
Radiotracer detection method revealed active neuroinflammation in the tau aggregation area [65]. Proinflammatory factors released by activated microglia and reactive astrocytes play an important role in tau pathology. It was demonstrated that tau-hype phosphorylation and aggregation depend on NLRP3 inflammasome in mice treated with intracerebral Aβ [14]. In a mouse model of AD with IL-1β overexpression, it was found that IL-1β promotes tau phosphorylation through p38-MAPK and glycogen synthase kinase 3 (GSK-3β) signaling pathways [66, 67]. TNF-α-treated neurons induce tau aggregation through ROS generation. Similarly, activated microglia co-cultured with neurons induce tau aggregation (Fig. 2) [68]. Current findings support that pathological tau seeds can spread between neurons and induce hyperphosphorylated tau aggregation in normal neurons [69]. Activated microglia remove pathological tau seeds and cell debris in the interstitial fluid. Some will be degraded, and the others will be re-released in exosomes (Fig. 2). Inhibition of exosome synthesis or depletion of microglia can significantly restrain the spread of pathological tau, suggesting that microglia may participate in the progression of pathological tau through exosome release [70].
TZDs improve AD by regulating neuroinflammation
TZDs were first used for AD after discovering that non-steroidal anti-inflammatory drugs (NSAIDs) can activate PPARγ [71]. Epidemiological studies have found that long-term use of NSAIDs can significantly reduce the risk of AD [72]. Subsequently, animal studies confirmed that long-term administration of ibuprofen significantly reduces soluble Aβ1–42 levels and cortical Aβ plaques and prevents microglial activation in 11-month-old Tg2576 mice [73]. PPARγ antagonist also attenuated the protective effect of ibuprofen on Aβ deposition [63]. Given that NSAIDs may inhibit neuroinflammation and disease progression in AD by activating PPARγ, TZDs, as strong activators of PPARγ, are potential candidates for treating AD [74].
Heneka et al. compared the efficacy of pioglitazone and ibuprofen in 10-month-old AβPP V717I mice. Both treatments significantly reduced the number of activated microglia and reactive astrocytes, downregulated COX2, iNOS, and BACE1, and prevented Aβ1–42 deposition in the hippocampus and cortex (Fig. 1) [74]. Similarly, in mice overexpressing mutant human AβPP, rosiglitazone activated M2 microglia to promote its phagocytic ability, reduce the expression of proinflammatory markers, and remove Aβ plaques and neuropil threads containing phosphorylated tau in the hippocampus [75]. Pioglitazone blocks Aβ-triggered hyperphosphorylated of tau and the subsequent synaptic loss [76]. In addition to improving AD pathology and inflammation in the brain, pioglitazone and rosiglitazone both improved spatial memory impairment in mice induced by Aβ [48, 56].
It has been found that TZDs can also affect AD pathology through PPARγ-dependent non-inflammatory pathways. Researchers identified a PPARγ-responsive element (PPRE) in the BACE1 gene promoter, which controls BACE1 expression. PPARγ could be a repressor of BACE1, since overexpression of PPARγ or treatment with pioglitazone reduces BACE1 expression and intracellular Aβ levels, while PPARγ depletion enhances BACE1 gene promoter activity [77]. Cyclin-dependent kinase 5 (CDK5) is a serine /threonine kinase with many important roles in neuronal development. Enhanced CDK5 activity was observed in mice models of AD, which may be related to Aβ-induced dendritic spine loss. Pioglitazone inhibited CDK5 activity by decreasing p35 protein level, and improved neuronal long-term potentiation deficit [78, 79].
TZDS INHIBIT FERROPTOSIS IN AD
Discovery of ferroptosis and ACSL4
Previous studies have used high-throughput screening to identify a small molecule compound, Erastin, with a selective lethal ability for RAS mutant tumor cells (RAS-selective lethal, RSL) [80]. Erastin initiated non-apoptotic cell death associated with increased levels of intracellular ROS. It is an iron-dependent lethal mechanism that can be prevented by iron chelation [81]. In 2012, Dixon et al. named this specific cell death induced by erastin as ferroptosis. It was found that erastin inhibits cystine/glutamate antiporter systemXc–, which results in cystine uptake disorders and impaired glutathione (GSH) synthesis, and ultimately iron-dependent cell death. Ferrostatin-1 (Fer-1) can effectively inhibit erastin-induced ferroptosis but has no significant effect on cystine and glutathione levels. It mainly reduces the levels of lipid ROS and it is speculated that lipid ROS may be crucial for ferroptosis [34]. Further studies revealed that Fer-1 has a lipophilic N-cyclohexyl, which can protect specific membrane PUFAs against ferroptosis-inducing conditions, such as cystine and glutathione depletion. Fer-1 inhibits oxidative damage of membrane PUFAs and resists cell ferroptosis; thus, lipid peroxidation products are the proximate executioners of ferroptosis [35].
RSL3 can also induce ferroptosis, but unlike erastin, it does not depend on systemXc– pathway [81]. RSL3 does not change GSH level and may directly regulate GSH downstream pathway. One consequence of GSH depletion could be glutathione-dependent peroxidases (GPXs) inactivation. Using GSH as an essential cofactor, GPXs catalyze the reduction of peroxide to water or to the corresponding alcohols. It has been suggested that GPX4 is a central regulator of ferroptosis and the direct target of RSL3. GPX4–/– mice developed acute renal failure and significantly increased cellular lipid peroxidation [36, 82, 83]. This is consistent with previous studies showing that GPX4 specifically reduces lipid peroxidation [84]. Meanwhile, a novel small molecule compound liproxstatin-1 (Lip-1) inhibited RSL3-induced ferroptosis in renal tubular epithelial cells and remarkably prolonged the survival of GPX–/– mice [82]. Like Fer-1, lipophilic free radical-trapping antioxidants, inhibit ferroptosis through a potent reduction of lipid peroxidation [85].
Using a haploid cell line with retroviral-mediated mutations, two specific gene mutations with significant enrichment in ferroptosis-resistant cells, ACSL4 and lysophosphatidylcholine acyltransferase 3 (LPCAT3), were found [86]. Doll et al. applied two approaches: microarray analysis of ferroptosis-resistant cell lines, and a genome-wide CRISPR gRNA library screen. Both approaches identified ACSL4 as a pro-ferroptosis gene and a critical determinant of ferroptosis sensitivity. Acsl4–Gpx4 double-KO cells could be passaged for more than 10 days, which is opposite to previous studies concluding that no cell lines can survive ex vivo without GPX4 [31]. During PUFAs biosynthesis, ACSL4 preferentially catalyzes arachidonic acid (AA)/adrenic acid (AdA) to form acyl-CoA derivative AA/AdA-CoA, which is esterified to phosphatidylethanolamines (PE) by LPCAT3. It has been found that AA/AdA-CoA containing PE is the preferred substrate for lipid peroxidation in ferroptosis. The content of PE-AA/ AdA in ACSL4 knockout cells was significantly lower than that in WT cells. Loss of ACSL4 significantly inhibited RSL3-induced lipid peroxidation PE-AA/AdA-OOH, whereas LPCAT3 knockout only showed a slight protective effect [87].
AD and ferroptosis
As early as 1997, it was reported that oxidative damage mediated by iron accumulation is involved in the pathogenesis of AD [88]. The autopsy indicated that iron distribution in the frontal cortex of AD patients is significantly different from that in aging brains without AD [89]. According to NFTs and Aβ plaque levels of the inferior temporal cortex in the autopsy brain, AD neuropathology was divided into low and high pathology scores. The results showed that in subjects with high pathology scores, the cortical iron level was strongly associated with cognitive decline in AD, but there were no significant correlations with non-AD dementia or with increased AD proteinopathy in the absence of dementia. There is a hypothesis that iron does not merely accumulate with AD pathology but rather potentiates degeneration once the pathology is sufficiently severe [18]. The exact relationship between iron accumulation and the development of AD was not understood until the discovery of ferroptosis, which provided new evidence for the involvement of iron accumulation in the cognitive impairment of AD.
Iron and AD
Ferroptosis is iron-dependent regulatory cell death. Iron chelator deferoxamine can inhibit ferroptosis [34]. Hepatocyte ferroptosis occurred in mice fed with a high-iron diet and in gene knockout-mediated iron overload mouse models [90]. Iron is a highly redox-reactive metal, and the Fenton reaction is involved in lipid peroxidation by producing hydroxyl radicals. In addition, lipoxygenases (LOXs), iron-containing enzymes, selectively catalyze lipid peroxidation by an iron redox reaction [82, 87].
Iron homeostasis is exquisitely controlled in neurons because of their high metabolic activity. By modulating iron uptake, storage, utilization, and efflux, neurons maintain an optimal intracellular iron balance [91]. Iron homeostasis imbalance in AD leads to iron accumulation in the hippocampus and cortex. Ferroportin (Fpn) is the only known protein that exports intracellular iron in mammals [92]. It was found that Fpn expression is significantly lower in the hippocampus of the AD brain than in the normal brain, resulting in decreased iron efflux capacity and increased iron accumulation in neurons [93, 94]. Elevated levels of ferritin were found in AD brain, providing strong evidence for iron accumulation [95]. Fpn conditional knockout mice developed AD-like hippocampal atrophy, memory deficits, and typical ferroptosis characteristics, confirming that iron accumulation is involved in AD pathogenesis through ferroptosis [94]. Fpn is regulated by AβPP in neurons. AβPP possesses ferroxidase activity, oxidizing Fe2 + into Fe3 + and interacting with Fpn to export iron [96]. Tau affects the AβPP translocation to the cell surface. Pathological loss of soluble tau can prevent AβPP trafficking to the cell surface in AD, resulting in reduced iron efflux, which causes iron accumulation and ferroptosis (Fig. 3) [97].
Lipid peroxidation and AD
The core reaction of ferroptosis is lipid peroxidation, and most ferroptosis inhibitors act as lipophilic radical-trapping antioxidants. Lipid peroxidation can occur through non-enzymatic and enzymatic pathways. The non-enzymatic pathway initiates by the Fenton reaction, which utilizes Fe2 + in the labile iron pool (LIP) to react with hydrogen peroxide (H2O2), resulting in the production of hydroxyl radicals (OH•). These radicals remove a bisallylic hydrogen atom from PUFA-PLs to form a carbon-centered phospholipid radical (PL•). Subsequent reactions with molecular oxygen produce phospholipid peroxyl radical (PLOO•), which removes hydrogen from other PUFAs, forming lipid peroxidation PLOOH and the fresh PL•. PLOOH can react with Fe2 + to generate PLO•, which damages the peroxidation chain reaction together with PL•, propagating PLOOH on the lipid membrane (Fig. 3) [98, 99]. The primary executioner of the enzymatic processes are LOXs, which catalyze the dioxygenation of PUFAs to produce lipid peroxidation. LOXs-mediated PUFAs oxidation can exert a lethal effect during certain conditions (GSH depletion) [36].
Lipid peroxidation is common in the brain due to high oxygen utilization and high PUFA levels [100]. The evidence of ferroptosis lipid peroxidation, including structural mitochondrial atrophy and increased levels of lipid peroxidation products 4-hydroxynonenal (4-HNE) and malondialdehyde (MDA), was found in AD brain by histopathology [95]. The ferroptosis inhibitor, Lip-1, can effectively reduce neuronal death in the hippocampal dentate gyrus and improve learning and memory impairment in AD mice [94]. Peroxidation of membrane lipids can disturb cell membrane integrity, decrease membrane flexibility, increase membrane permeability, and finally affect material transport and signal conduction. In addition, lipid peroxidation may also occur in mitochondria, endoplasmic reticulum, and lysosomes, the organelles rich in phospholipid membranes, leading to cell dysfunction and death [101]. PLOOH can be decomposed into toxic derivatives, such as 4-HNEs and MDA, forming covalent electrophilic products with biological macromolecules, such as DNA, lipids, and proteins. They act as the secondary messengers of oxidative stress. They are also severely cytotoxic [33, 102].
Anti-lipid peroxidation system and AD
In physiological conditions, cells constantly produce ROS and lipid peroxidation products, which are removed by the antioxidant system. Excessive lipid peroxidation often leads to ferroptosis. Cyst(e)ine–GSH–GPX4 axis is the main negative regulator of lipid peroxidation. Cystine/glutamate antiporter systemXc– is composed of the light-chain subunit, SLC7A11, and the heavy-chain subunit, SLC3A2. Light-chain subunit, SLC7A11, is responsible for cystine/glutamate transpor. SystemXc– uptakes cystine by exchanging extracellular cystine for intracellular glutamate, and rapidly reduces it to cysteine. Cysteine is involved in GSH synthesis, which is catalyzed by glutamate cysteine ligase. Cystine uptake by systemXc– is the rate-limiting step in GSH synthesis [103]. GSH, the cofactor of GPX4, is a crucial antioxidant, which has two forms: reduced GSH and oxidized GSSG. In physiological conditions, GSH is an electron donor for GPX4. As GPX4 is synthesized, selenium replaces the sulfur of cysteine to form selenocysteine (Sec). Active selenol (-SeH) can be oxidized by PLOOH to selenic acid (-SeOH), and then reduced by GSH, which provides two electrons for oxidized GPX4. Glutathione-disulfide reductase (GSR) produces GSH from GSSG [104]. Meanwhile, PLOOH is reduced to the corresponding phospholipid alcohol (PLOH) (Fig. 3) [104, 105].
Another GPX4-independent pathway is mediated by the NADH-FSP1-CoQ10 axis [106]. The N-terminus of FSP1 contains a canonical myristoylation, which binds to the lipid bilayers. On the cell membrane, FSP1 reduces CoQ10 to CoQ10-H2, which captures lipid free radicals and cooperates with GPX4 to resist lipid peroxidation (Fig. 3) [107].
AD is accompanied by excessive lipid peroxidation. There were lower levels of GSH and GPX4 in AD pathology and animal models, which correlate with cognitive dysfunction [94, 95, 108]. Higher SLC7A11 levels were observed in the AD brain compared with the normal brain, which can be related to compensatory intake of cystine to potentiate the anti-lipid peroxidation system [95]. Mutations in presenilin 1 and 2 cause autosomal dominant familial Alzheimer’s disease (FAD). It was found that PS can regulate selenium uptake to promote GPX4 expression and inhibit ferroptosis. PS mutations can disrupt selenium uptake, suppress GPX4 expression, and increase cell sensitivity to ferroptosis. Therefore, presenilin mutations in FAD may promote neurodegeneration by weakening the anti-lipid peroxidation system [109]. GPX4 conditional knockout mice also showed cognitive impairment and hippocampal neuron loss. On the contrary, AD mice overexpressing GPX4 improved significantly, suggesting that ferroptosis plays an important role in AD [110–112].
TZDs inhibit ACSL4 to protect AD from ferroptosis
Accumulating evidence indicates that ferroptosis is an ACSL4-dependent cell death process. ACSL4 activation is dependent on dimerization and regulated by phosphorylated kinases, which include protein kinase A (PKA) and protein kinase C (PKC), a group of Ser/Thr kinases. When Acsl4 was phosphorylated by PKA, its activity was significantly increased compared to the activity of non-phosphorylated Acsl4. On the contrary, PKC phosphorylation caused a significant reduction in the Acsl4 activity [113]. Zhang identified phosphorylation of Thr328 is critical for the activation of ACSL4. One of the five isoforms of PKC, PKCβII could sense the initial lipid peroxidation, directly phosphorylated and activated ACSL4 at Thr328 to increase PUFA-containing phospholipids and promote ferroptosis [114].
The effect of ACSL4 on ferroptosis depends on multiple factors. It has been reported that CD8+T cells can promote tumor cells ferroptosis and mediate anti-tumor immunity by releasing IFNγ [115]. Further studies revealed that IFNγ binds to cell surface receptors and activates JAK/STAT1 signaling pathway to regulate the expression of interferon regulatory factor 1 (IRF1). IRF1 upregulates ACSL4 expression by binding to its promoter region, thereby promoting ferroptosis [116]. In addition, it was found that ACSL4 promoter region has abundant GC boxes, which have a high affinity for the transcription factor SP1. Under hypoxic conditions, upregulation or downregulation of SP1 in intestinal adenocarcinoma cells similarly increased or decreased the expression of ACSL4 [117].
It has been found that the expression of ACSL4 and SP1 is significantly higher in AβPP/PS1 mice hippocampal neurons than in wild-type mice, In addition, inhibition of ACSL4 or SP1 effectively prevented Aβ-induced ferroptosis of neurons [118, 119]. TZDs specifically inhibit ACSL4-mediated PUFAs activation and lipid peroxidation. Studies have shown that pioglitazone effectively improves cognitive dysfunction by inhibiting lipid peroxidation [120]. Modulating lipid peroxidation and lipid metabolism disorder alleviated cognitive impairment in AD mice [110, 121]. ACSL4 inhibition by TZDs has also been demonstrated in other diseases. In ischemia-reperfusion injury models, including intestinal, lung, brain, and myocardial ischemia models, TZDs or ACSL4 knockout improved ischemia-reperfusion injury by downregulating abnormally overexpressed ACSL4 expression and inhibiting ferroptosis [117, 122–124]. Similarly, TZDs alleviated acute kidney injury and improved rhabdomyolysis by inhibiting ACSL4 [125, 126]. Given the importance of ferroptosis in AD and its dependence on ACSL4, ACSL4 may be a potential target in AD.
CLINICAL TRIALS OF PIOGLITAZONE IN CNS DISEASES
In 1997, the first TZD, troglitazone, was approved in the United States for the treatment of type 2 diabetes. Subsequently, two other TZDs, rosiglitazone and pioglitazone, were successively introduced into the market in 1999. However, because of hepatotoxicity troglitazone was withdrawn from the market in 2000, and rosiglitazone was banned in Europe and the United States in 2010 due to the high risk of myocardial infarction (Fig. 1). Only pioglitazone is still used in clinics [127]. Therefore, in terms of clinical trials, this review mainly describes the use of pioglitazone in CNS diseases.
AD
In an early prospective randomized controlled study, patients with mild AD and type 2 diabetes were treated with pioglitazone or placebo. Both groups also received donepezil and had good glycemic control. The results showed that pioglitazone improved cognitive function and regional cerebral blood flow in the parietal lobe, but the plasma ratio of Aβ40/42 did not significantly change. This study included only 42 cases [128].
In 2014, Read reported an elderly patient with early AD who took a higher dose (45 mg–60 mg) of pioglitazone. Pioglitazone significantly delayed cognitive decline. During the 4-year observation period, the MMSE score showed cognitive improvement and remained constant while taking 60 mg of pioglitazone. When they attempted to reduce the dose to 30 mg, the MMSE score suddenly dropped and improved after adjusting the dose to 45 mg. These findings suggest that pioglitazone may delay cognitive decline in a dose-dependent manner in the early stages of AD [129].
A prospective cohort study compared the incidence of dementia between non-diabetic subjects, diabetic patients not using pioglitazone, diabetic patients with prescriptions of <2 years of pioglitazone, and diabetics with ≥2 years of pioglitazone. The results showed that diabetic patients without the use of pioglitazone had a 23% increase in dementia risk. The use of pioglitazone was associated with a lower risk of dementia, and this protective effect depended on the duration of use. The risk of dementia among diabetic patients with <2 years of using pioglitazone was comparable to those of non-diabetic subjects. The risk of dementia after long-term use of pioglitazone (≥2 years) was 47% less than that among non-diabetic subjects [130].
In a prospective cohort study with metformin as a first-line glucose-lowering regimen, there was a decreased risk of dementia in those taking pioglitazone compared with those taking other second-line glucose-lowering drugs such as sulfonylureas, acarbose, and insulin [131]. A network meta-analysis measured the effects of 6 hypoglycemic agents on AD and mild cognitive impairment (MCI). It showed that pioglitazone 15–30 mg, intranasal insulin 40 units, and sitagliptin 100 mg all significantly improved cognition compared with placebo. In addition, pioglitazone 15–30 mg demonstrated the greatest efficacy [132].
For measuring the safety and efficacy of pioglitazone among non-diabetic subjects with AD, an 18-month, phase II clinical trial was conducted by Takeda Pharmaceuticals. Twenty-five subjects completed the study and 45 mg/day of pioglitazone was well tolerated, with no significant effect on blood glucose levels, hemoglobin levels, or other blood biochemistry [133]. The TOMMORROW study (Fig. 1) was a randomized controlled study consisting of diabetic subjects ≥65 years of age and without cognitive impairment. Participants at high risk of MCI randomly received low-dose pioglitazone (0.8 mg/day) or placebo. The study started in 2013 and terminated in 2018 after failing to meet the expected efficacy. Low doses of pioglitazone did not delay the onset of MCI nor increase the risk of adverse events [134]. The negative results obtained in the TOMMORROW study were related to the use of a lower dose of pioglitazone. In addition, this study did not measure biomarkers, which might affect the effectiveness analysis of the drug.
Ischemic stroke
Ischemic stroke has a high recurrence risk and is the second most common vascular event after myocardial infarction (MI) [135]. Pioglitazone was found as effective as an intensive lipid-lowering strategy in preventing stroke recurrence [136, 137]. A meta-analysis measured the preventive effect of pioglitazone on secondary stroke. It included three randomized controlled studies and found that the use of pioglitazone in patients with stroke, insulin resistance, prediabetes, and diabetes mellitus was associated with a lower risk of recurrent stroke (decreased by 32%) and future major vascular events (decreased by 25%) [138].
The PROactive study, a randomized controlled trial (Fig. 1), included patients with type 2 diabetes who had evidence of macrovascular disease. They received oral pioglitazone or placebo in addition to their glucose-lowering drugs and other medications. After 34 months, pioglitazone significantly reduced the incidence of the primary composite outcome compared with placebo [139]. The second study was published two years later, which showed that pioglitazone significantly reduced the recurrence rate of stroke and cardiovascular events in diabetic patients with prior stroke [140].
Another randomized double-blind controlled trial, the IRIS study (Fig. 1), included non-diabetic subjects with insulin resistance and recent ischemic stroke or TIA. The primary outcome was recurrent stroke or MI. After 4.8 years, the primary outcome occurred in 9.0% of participants in the pioglitazone group and in 11.8% of participants in the placebo group, and the difference was statistically significant [137]. Multiple secondary analyses of the IRIS study were performed. These analyses found that pioglitazone was effective for the secondary prevention of ischemic stroke or TIA and significantly reduced stroke recurrence [141]. After stratifying patients according to their compliance, the results showed that pioglitazone had a better preventive effect on primary outcomes in subjects with acceptable compliance [142]. Another study stratified patients according to their cardiovascular risk factors and showed that patients at high risk of stroke or MI achieved a greater benefit from pioglitazone [143].
Two retrospective studies confirmed the effect of pioglitazone on the secondary prevention of stroke among diabetic patients with ischemic stroke [144, 145]. Pioglitazone may also have a good preventive effect for primary stroke prevention. Two recent retrospective studies showed that pioglitazone dose-dependently decreased the risk of new-onset ischemic stroke in patients with type 2 diabetes [146, 147].
Carotid atherosclerosis (coronary artery)
Pioglitazone has been found to delay the progression of carotid and coronary atherosclerosis in diabetic patients. As measured by ultrasound, carotid intima-media thickness (CIMT) was highly correlated with future risk of cardiovascular events and was a strong predictor of ischemic stroke and MI. Two randomized controlled studies compared the effects of pioglitazone and glimepiride on CIMT in patients with type 2 diabetes [148, 149]. In the CHICAGO study (Fig. 1), participants were asked to adjust their statin dose to meet American Diabetes Association goals before the study. The results showed in the presence of blood pressure and blood glucose control and adequate use of statins, pioglitazone significantly delayed CIMT progression compared with glimepiride. Another study showed that pioglitazone also decelerated the progression of CIMT in patients with prediabetes [150]. The PERISCOPE study compared pioglitazone with glimepiride on coronary atherosclerosis in patients with diabetes. Intravascular ultrasound was used to assess the variation in plaque size (Fig. 1). With comparable levels of blood glucose, the changes in percent atheroma volume and maximum atheroma thickness from baseline to study completion increased in the glimepiride group and decreased in the pioglitazone group. It shows the benefits of pioglitazone in delaying coronary atherosclerosis in patients with type 2 diabetes and coronary artery disease [151].
Atherosclerosis is an inflammatory disease. Each plaque contains numerous macrophages, which weakens plaque stability. In 18F-fluorodeoxyglucose positron emission tomography (18F-FDG-PET) imaging, FDG uptake was correlated with macrophage abundance in plaques. The application of FDG-PET can help identify the inflammatory activity in atherosclerotic plaques [152]. Compared with glimepiride, pioglitazone could attenuate FDG uptake in carotid and coronary atherosclerotic plaques in patients with impaired glucose tolerance or diabetes [153, 154]. In another study comparing statin alone with the combination of statin and pioglitazone, FDG intake was slightly but not significantly lower in the combination group after 3 months of treatment [155].
The vascular endothelium regulates vascular wall homeostasis by maintaining normal vascular tonicity and modulating platelet aggregation and leukocyte adhesion. Endothelial dysfunction is critical for the development of atherosclerosis. Studies have found that pioglitazone improved endothelial vasodilatation in non-diabetic patients, independent of its effect on insulin sensitivity and inflammation [156, 157].
Pioglitazone also altered the lipid profile in the CHICAGO and PERISCOPE studies, with a significant increase in high-density lipoprotein cholesterol (HDL-C) levels and a significant decrease in triglyceride levels [149, 151]. The favorable effects of pioglitazone on the triglyceride/HDL-C ratio correlated with delayed atheroma progression [158]. The effect of pioglitazone on low-density lipoprotein cholesterol (LDL-C) was reflected in its influence on LDL-C particle size. It was found that pioglitazone can increase LDL-C particle size and reduce particle density, which is associated with slower plaque progression [159]. Small and dense LDL-C particles may promote plaque progression. The measurement of LDL-C particles may be a supplementary risk predictor for people with low LDL-C levels [160].
LIMITATIONS OF CLINICAL APPLICATION OF PIOGLITAZONE
Pioglitazone has shown numerous advantages in the preclinical studies and clinical trials of AD with insulin resistance. The clinical application and clinical trials of pioglitazone are limited in non-diabetic subjects with AD due to side effects. Since its approval in the USA, a potential link between pioglitazone and bladder cancer has been the subject of debate. A re-analysis of the PROactive study showed a significantly increased risk of bladder cancer with pioglitazone [161]. After conducting a review of the safety of pioglitazone, the European Medicines Agency (EMA) decided to maintain the marketing of the drug, whereas the US Food and Drug Administration (FDA) warned about the possible increased risk of bladder cancer. A retrospective cohort study in the UK reached the same conclusion, suggesting that the use of pioglitazone is associated with an increased risk of bladder cancer in patients with type 2 diabetes. The risk of bladder cancer increased with the duration and dose of pioglitazone [162].
On the other hand, pioglitazone may contribute to an increased risk of heart failure by increasing fluid retention. However, the risk of fatal heart failure and all-cause mortality did not increase, and the incidence of cardiovascular events was significantly reduced [163, 164]. In addition, pioglitazone can cause weight gain by redistribution of adipose tissue, and increase the risk of fracture [165, 166]. These adverse events restricted the prescription of pioglitazone, although the overall risk-benefit balance is strongly in favor of prescribing pioglitazone [167].
In patients with diabetes or insulin resistance, the reduced risk of AD with pioglitazone was related not only to its suppressive effects on inflammation and lipid peroxidation but also to its stabilizing effect on glucose metabolism in the brain [134]. The TOMMORROW study was conducted to investigate the efficacy of pioglitazone on non-diabetic patients with AD. In order to achieve good tolerability and safety, a low dose of pioglitazone (0.8 mg/day) was used in the TOMMORROW study. However, low doses of pioglitazone did not delay the onset of MCI in high-risk participants. Previous animal studies suggested that the effect of pioglitazone can be enhanced in a time- and dose-dependent manner [19]. Pioglitazone cannot effectively pass through BBB, and higher doses of pioglitazone are usually used in animal studies to achieve effective concentrations across the BBB. Pioglitazone is an efflux transporter substrate on BBB, preventing pioglitazone from reaching effective concentrations in the brain. The application of lower doses of sustained-release tablets in the TOMMORROW study might keep the transporter in an unsaturated state, and insufficient concentrations of pioglitazone in the brain may be a key factor limiting its efficacy in AD. To address the poor BBB permeability of pioglitazone and its dose-dependent peripheral side effects, intranasal nano lipid carriers of pioglitazone have been formulated for its targeted delivery to the brain [168]. Leriglitazone, which is the active metabolite products of pioglitazone, is under development by Minoryl for the treatment of X-linked adrenoleukodystrophy (X-ALD) and other neurodegenerative diseases due to its adequate BBB penetration, good bioavailability, and safety profile. Compared with pioglitazone, Leriglitazone increase 50% brain exposure ratio in mice [169]. In the ADVANCE study, a randomized, double-blind, phase 2-3 trial of leriglitazone for preventing disease progression in men with adrenomyeloneuropathy. It was found that none of the clinically progressive cerebral adrenoleukodystrophy were occurred in patients receiving leriglitazone [170]. As increasing the effective concentration of TZDs in the brain, there is still a potential for TZDs in treatment of AD.
CONCLUSION
The growing incidence of AD has brought a tremendous burden to societies and families. People are paying more attention to AD worldwide. Many large pharmaceutical companies are developing drugs for AD; however, there has been no breakthrough in improving the disease outcome. Most of these drugs target Aβ. Although the pathogenesis of AD is still controversial, it can be known that AD is an intricate and multi-factorial pathological process. Drugs that act on a single target or pathway may not be effective in halting AD progression. TZDs, as PPARγ agonists and ACSL4 inhibitors, play a dual role by their potent anti-inflammatory and anti-ferroptosis properties. In the preclinical studies of AD, TZDs simultaneously regulated various pathological pathways, effectively inhibited chronic inflammation, and protected against neuronal loss.
In clinical trials, as a treatment for type 2 diabetes, TZDs effectively controlled blood glucose, improved insulin resistance, and reduced the risk of AD in patients with diabetes or insulin resistance. Low-dose pioglitazone failed to show efficacy in non-diabetic patients with AD. It may be necessary to reduce drug side effects by optimizing drug carriers and improving drug targeting.
AD is the leading cause of dementia, followed by vascular dementia. Autopsy studies found that mixed dementia mediated by these two causes also accounted for a high proportion, which should not be ignored in the treatment of patients who have non-categorized dementia. Clinical trials of pioglitazone have shown its efficacy in the prevention and treatment of arteriosclerosis and ischemic stroke, making it, especially a suitable candidate for mixed dementia.
Currently, among TZDs, only pioglitazone is still used in clinical practice, but many clinicians are discouraged by the increased risk of obesity, fractures, heart failure, and bladder cancer. Although the results of clinical trials are confusing, the use of pioglitazone for preventing cognitive decline is still a possible long-term option. A careful selection of patients is helpful. Patients with prediabetes or insulin resistance, those with increased risk of myocardial infarction and ischemic stroke, mixed dementia, low risk of heart failure and bladder cancer, can obtain the utmost benefit from pioglitazone.
ACKNOWLEDGMENTS
We appreciated EditSprings (https://www.editsprings.com/) for the expert linguistic services provided.
FUNDING
This work was supported by the International Cooperation Program of Science and Technology Development Plan in Jilin Province (Grant No. 20210402020GH).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Scheltens P , De Strooper B , Kivipelto M , Holstege H , Chételat G , Teunissen CE , Cummings J , van der Flier WM ((2021) ) Alzheimer’s disease. Lancet 397: , 1577–1590. |
[2] | Karran E , De Strooper B ((2022) ) The amyloid hypothesis in Alzheimer disease: New insights from new therapeutics. Nat Rev Drug Discov 21: , 306–318. |
[3] | Braak H , Thal DR , Ghebremedhin E , Del Tredici K ((2011) ) Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol 70: , 960–969. |
[4] | Ossenkoppele R , van der Kant R , Hansson O ((2022) ) Tau biomarkers in Alzheimer’s disease: Towards implementation in clinical practice and trials. Lancet Neurol 21: , 726–734. |
[5] | Long JM , Holtzman DM ((2019) ) Alzheimer disease: An update on pathobiology and treatment strategies. Cell 179: , 312–339. |
[6] | Heneka MT , Carson MJ , El Khoury J , Landreth GE , Brosseron F , Feinstein DL , Jacobs AH , Wyss-Coray T , Vitorica J , Ransohoff RM , Herrup K , Frautschy SA , Finsen B , Brown GC , Verkhratsky A , Yamanaka K , Koistinaho J , Latz E , Halle A , Petzold GC , Town T , Morgan D , Shinohara ML , Perry VH , Holmes C , Bazan NG , Brooks DJ , Hunot S , Joseph B , Deigendesch N , Garaschuk O , Boddeke E , Dinarello CA , Breitner JC , Cole GM , Golenbock DT , Kummer MP ((2015) ) Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14: , 388–405. |
[7] | Joshi P , Turola E , Ruiz A , Bergami A , Libera DD , Benussi L , Giussani P , Magnani G , Comi G , Legname G , Ghidoni R , Furlan R , Matteoli M , Verderio C ((2014) ) Microglia convert aggregated amyloid-beta into neurotoxic forms through the shedding of microvesicles. Cell Death Differ 21: , 582–593. |
[8] | Leyns CEG , Holtzman DM ((2017) ) Glial contributions to neurodegeneration in tauopathies. Mol Neurodegener 12: , 50. |
[9] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[10] | Braak H , Braak E ((1997) ) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18: , 351–357. |
[11] | Kulkarni B , Cruz-Martins N , Kumar D ((2022) ) Microglia in Alzheimer’s disease: An unprecedented opportunity as prospective drug target. Mol Neurobiol 59: , 2678–2693. |
[12] | Pontecorvo MJ , Devous MD Sr. , Navitsky M , Lu M , Salloway S , Schaerf FW , Jennings D , Arora AK , McGeehan A , Lim NC , Xiong H , Joshi AD , Siderowf A , Mintun MA , 18F-AV-1451-A05 investigators ((2017) ) Relationships between flortaucipir PET tau binding and amyloid burden, clinical diagnosis, age and cognition. Brain 140: , 748–763. |
[13] | Heneka MT , Kummer MP , Stutz A , Delekate A , Schwartz S , Vieira-Saecker A , Griep A , Axt D , Remus A , Tzeng TC , Gelpi E , Halle A , Korte M , Latz E , Golenbock DT ((2013) ) NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493: , 674–678. |
[14] | Ising C , Venegas C , Zhang S , Scheiblich H , Schmidt SV , Vieira-Saecker A , Schwartz S , Albasset S , McManus RM , Tejera D , Griep A , Santarelli F , Brosseron F , Opitz S , Stunden J , Merten M , Kayed R , Golenbock DT , Blum D , Latz E , Buée L , Heneka MT ((2019) ) NLRP3 inflammasome activation drives tau pathology. Nature 575: , 669–673. |
[15] | Hopp SC , Lin Y , Oakley D , Roe AD , DeVos SL , Hanlon D , Hyman BT ((2018) ) The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J Neuroinflammation 15: , 269. |
[16] | Pascoal TA , Benedet AL , Ashton NJ , Kang MS , Therriault J , Chamoun M , Savard M , Lussier FZ , Tissot C , Karikari TK , Ottoy J , Mathotaarachchi S , Stevenson J , Massarweh G , Schöll M , de Leon MJ , Soucy JP , Edison P , Blennow K , Zetterberg H , Gauthier S , Rosa-Neto P ((2021) ) Microglial activation and tau propagate jointly across Braak stages. Nat Med 27: , 1592–1599. |
[17] | Plascencia-Villa G , Perry G ((2021) ) Preventive and therapeutic strategies in Alzheimer’s disease: Focus on oxidative stress, redox metals, and ferroptosis. Antioxid Redox Signal 34: , 591–610. |
[18] | Ayton S , Wang Y , Diouf I , Schneider JA , Brockman J , Morris MC , Bush AI ((2020) ) Brain iron is associated with accelerated cognitive decline in people with Alzheimer pathology. Mol Psychiatry 25: , 2932–2941. |
[19] | Saunders AM , Burns DK , Gottschalk WK ((2021) ) Reassessment of pioglitazone for Alzheimer’s disease. Front Neurosci 15: , 666958. |
[20] | Nolan JJ , Ludvik B , Beerdsen P , Joyce M , Olefsky J ((1994) ) Improvement in glucose tolerance and insulin resistance in obese subjects treated with troglitazone. N Engl J Med 331: , 1188–1193. |
[21] | Miyazaki Y , Mahankali A , Matsuda M , Mahankali S , Hardies J , Cusi K , Mandarino LJ , DeFronzo RA ((2002) ) Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab 87: , 2784–2791. |
[22] | Daynes RA , Jones DC ((2002) ) Emerging roles of PPARs in inflammation and immunity. Nat Rev Immunol 2: , 748–759. |
[23] | Lehmann JM , Moore LB , Smith-Oliver TA , Wilkison WO , Willson TM , Kliewer SA ((1995) ) An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor gamma (PPAR gamma). J Biol Chem 270: , 12953–12956. |
[24] | Lian H , Litvinchuk A , Chiang AC , Aithmitti N , Jankowsky JL , Zheng H ((2016) ) Astrocyte-microglia cross talk through complement activation modulates amyloid pathology in mouse models of Alzheimer’s disease. J Neurosci 36: , 577–589. |
[25] | Liddelow SA , Barres BA ((2017) ) Reactive astrocytes: Production, function, and therapeutic potential. Immunity 46: , 957–967. |
[26] | Cai Y , Liu J , Wang B , Sun M , Yang H ((2022) ) Microglia in the neuroinflammatory pathogenesis of Alzheimer’s disease and related therapeutic targets. Front Immunol 13: , 856376. |
[27] | Yang XY , Wang LH , Chen TS , Hodge DR , Resau JH , DaSilva L , Farrar WL ((2000) ) Activation of human T lymphocytes is inhibited by peroxisome proliferator-activated receptor gamma (PPAR gamma) agonists - PPAR gamma co-association with transcription factor NFAT. J Biol Chem 275: , 4541–4544. |
[28] | Shi L , Lin Q , Li X , Nie Y , Sun S , Deng X , Wang L , Lu J , Tang Y , Luo F ((2017) ) Alliin, a garlic organosulfur compound, ameliorates gut inflammation through MAPK-NF-kappa B/AP-1/STAT-1 inactivation and PPAR-7 activation. Mol Nutr Food Res 61: , 1601013. |
[29] | Khera R , Mehan S , Kumar S , Sethi P , Bhalla S , Prajapati A ((2022) ) Role of JAK-STAT and PPAR-gamma signalling modulators in the prevention of autism and neurological dysfunctions. Mol Neurobiol 59: , 3888–3912. |
[30] | Kuwata H , Hara S ((2019) ) Role of acyl-CoA synthetase ACSL4 in arachidonic acid metabolism. Prostaglandins Other Lipid Mediat 144: , 106363. |
[31] | Doll S , Proneth B , Tyurina YY , Panzilius E , Kobayashi S , IngoId I , Irmler M , Beckers J , Aichler M , Walch A , Prokisch H , Trumbach D , Mao G , Qu F , Bayir H , Fullekrug J , Scheel CH , Wurst W , Schick JA , Kagan VE , Angeli JPF , Conrad M ((2017) ) ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13: , 91–98. |
[32] | Kim JH , Lewin TM , Coleman RA ((2001) ) Expression and characterization of recombinant rat acyl-CoA synthetases 1, 4, and 5 - Selective inhibition by triacsin C and thiazolidinediones. J Biol Chem 276: , 24667–24673. |
[33] | Yan HF , Zou T , Tuo QZ , Xu S , Li H , Belaidi AA , Lei P ((2021) ) Ferroptosis: Mechanisms and links with diseases. Signal Transduct Target Ther 6: , 49. |
[34] | Dixon SJ , Lemberg KM , Lamprecht MR , Skouta R , Zaitsev EM , Gleason CE , Patel DN , Bauer AJ , Cantley AM , Yang WS , Morrison B III , Stockwell BR ((2012) ) Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149: , 1060–1072. |
[35] | Skouta R , Dixon SJ , Wang J , Dunn DE , Orman M , Shimada K , Rosenberg PA , Lo DC , Weinberg JM , Linkermann A , Stockwell BR ((2014) ) Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 136: , 4551–4556. |
[36] | Yang WS , Kim KJ , Gaschler MM , Patel M , Shchepinov MS , Stockwell BR ((2016) ) Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113: , E4966–E4975. |
[37] | Bamberger ME , Harris ME , McDonald DR , Husemann J , Landreth GE ((2003) ) A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci 23: , 2665–2674. |
[38] | Brown MR , Radford SE , Hewitt EW ((2020) ) Modulation of β-amyloid fibril formation in Alzheimer’s disease by microglia and infection. Front Mol Neurosci 13: , 609073. |
[39] | Varnum MM , Ikezu T ((2012) ) The classification of microglial activation phenotypes on neurodegeneration and regeneration in Alzheimer’s disease brain. Arch Immunol Ther Exp (Warsz) 60: , 251–266. |
[40] | Onyango IG , Jauregui GV , Čarná M , Bennett JP Jr. , Stokin GB ((2021) ) Neuroinflammation in Alzheimer’s disease. Biomedicines 9: , 524. |
[41] | Hanslik KL , Ulland TK ((2020) ) The role of microglia and the Nlrp3 inflammasome in Alzheimer’s disease. Front Neurol 11: , 570711. |
[42] | Shaftel SS , Griffin WS , O’Banion MK ((2008) ) The role of interleukin-1 in neuroinflammation and Alzheimer disease: An evolving perspective. J Neuroinflammation 5: , 7. |
[43] | Mandrekar-Colucci S , Karlo JC , Landreth GE ((2012) ) Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J Neurosci 32: , 10117–10128. |
[44] | Zhao Q , Wu X , Yan S , Xie X , Fan Y , Zhang J , Peng C , You Z ((2016) ) The antidepressant-like effects of pioglitazone in a chronic mild stress mouse model are associated with PPARγ-mediated alteration of microglial activation phenotypes. J Neuroinflammation 13: , 259. |
[45] | Han Y , Wang J , Zhao Q , Xie X , Song R , Xiao Y , Kang X , Zhang L , Zhang Y , Peng C , You Z ((2020) ) Pioglitazone alleviates maternal sleep deprivation-induced cognitive deficits in male rat offspring by enhancing microglia-mediated neurogenesis. Brain Behav Immun 87: , 568–578. |
[46] | Yi HJ , Lee JE , Lee DH , Kim YI , Cho CB , Kim IS , Sung JH , Yang SH ((2019) ) The role of NLRP3 in traumatic brain injury and its regulation by pioglitazone. J Neurosurg 133: , 1083–1091. |
[47] | Gray E , Ginty M , Kemp K , Scolding N , Wilkins A ((2012) ) The PPAR-γ agonist pioglitazone protects cortical neurons from inflammatory mediators via improvement in peroxisomal function. J Neuroinflammation 9: , 63. |
[48] | Yamanaka M , Ishikawa T , Griep A , Axt D , Kummer MP , Heneka MT ((2012) ) PPARγ/RXRα-induced and CD36-mediated microglial amyloid-β phagocytosis results in cognitive improvement in amyloid precursor protein/presenilin 1 mice. J Neurosci 32: , 17321–17331. |
[49] | Odegaard JI , Ricardo-Gonzalez RR , Goforth MH , Morel CR , Subramanian V , Mukundan L , Red Eagle A , Vats D , Brombacher F , Ferrante AW , Chawla A ((2007) ) Macrophage-specific PPARgamma controls alternative activation and improves insulin resistance. Nature 447: , 1116–1120. |
[50] | Ding ZB , Song LJ , Wang Q , Kumar G , Yan YQ , Ma CG ((2021) ) Astrocytes: A double-edged sword in neurodegenerative diseases. Neural Regen Res 16: , 1702–1710. |
[51] | Zamanian JL , Xu L , Foo LC , Nouri N , Zhou L , Giffard RG , Barres BA ((2012) ) Genomic analysis of reactive astrogliosis. J Neurosci 32: , 6391–6410. |
[52] | Liddelow SA , Guttenplan KA , Clarke LE , Bennett FC , Bohlen CJ , Schirmer L , Bennett ML , Münch AE , Chung WS , Peterson TC , Wilton DK , Frouin A , Napier BA , Panicker N , Kumar M , Buckwalter MS , Rowitch DH , Dawson VL , Dawson TM , Stevens B , Barres BA ((2017) ) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541: , 481–487. |
[53] | Lian H , Yang L , Cole A , Sun L , Chiang AC , Fowler SW , Shim DJ , Rodriguez-Rivera J , Taglialatela G , Jankowsky JL , Lu HC , Zheng H ((2015) ) NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 85: , 101–115. |
[54] | Nicolakakis N , Aboulkassim T , Ongali B , Lecrux C , Fernandes P , Rosa-Neto P , Tong XK , Hamel E ((2008) ) Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J Neurosci 28: , 9287–9296. |
[55] | Zhao Z , Zhang L , Guo XD , Cao LL , Xue TF , Zhao XJ , Yang DD , Yang J , Ji J , Huang JY , Sun XL ((2017) ) Rosiglitazone exerts an anti-depressive effect in unpredictable chronic mild-stress-induced depressive mice by maintaining essential neuron autophagy and inhibiting excessive astrocytic apoptosis. Front Mol Neurosci 10: , 293. |
[56] | Toledo EM , Inestrosa NC ((2010) ) Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1 Delta E9 mouse model of Alzheimer’s disease. Mol Psychiatry 15: , 272–285. |
[57] | Dehmer T , Heneka MT , Sastre M , Dichgans J , Schulz JB ((2004) ) Protection by pioglitazone in the MPTP model of Parkinson’s disease correlates with I kappa B alpha induction and block of NF kappa B and iNOS activation. J Neurochem 88: , 494–501. |
[58] | Tureyen K , Kapadia R , Bowen KK , Satriotomo I , Liang J , Feinstein DL , Vemuganti R ((2007) ) Peroxisome proliferator-activated receptor-gamma agonists induce neuroprotection following transient focal ischemia in normotensive, normoglycemic as well as hypertensive and type-2 diabetic rodents. J Neurochem 101: , 41–56. |
[59] | Chistyakov DV , Azbukina NV , Lopachev AV , Kulichenkova KN , Astakhova AA , Sergeeva MG ((2018) ) Rosiglitazone as a modulator of TLR4 and TLR3 signaling pathways in rat primary neurons and astrocytes. Int J Mol Sci 19: , 113. |
[60] | Lin YT , Seo J , Gao F , Feldman HM , Wen HL , Penney J , Cam HP , Gjoneska E , Raja WK , Cheng J , Rueda R , Kritskiy O , Abdurrob F , Peng Z , Milo B , Yu CJ , Elmsaouri S , Dey D , Ko T , Yankner BA , Tsai LH ((2018) ) APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s disease phenotypes in human iPSC-derived brain cell types. Neuron 98: , 1141–1154.e7. |
[61] | Prokop S , Miller KR , Labra SR , Pitkin RM , Hoxha Kh , Narasimhan S , Changolkar L , Rosenbloom A , Lee VMY , Trojanowski JQ ((2019) ) Impact of TREM2 risk variants on brain region-specific immune activation and plaque microenvironment in Alzheimer’s disease patient brain samples. Acta Neuropathol 138: , 613–630. |
[62] | Pihlaja R , Koistinaho J , Kauppinen R , Sandholm J , Tanila H , Koistinaho M ((2011) ) Multiple cellular and molecular mechanisms are involved in human Aβ clearance by transplanted adult astrocytes. Glia 59: , 1643–1657. |
[63] | Sastre M , Dewachter I , Landreth GE , Willson TM , Klockgether T , van Leuven F , Heneka MT ((2003) ) Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci 23: , 9796–9804. |
[64] | Sutinen EM , Pirttilä T , Anderson G , Salminen A , Ojala JO ((2012) ) Pro-inflammatory interleukin-18 increases Alzheimer’s disease-associated amyloid-β production in human neuron-like cells. J Neuroinflammation 9: , 199. |
[65] | Maeda J , Zhang MR , Okauchi T , Ji B , Ono M , Hattori S , Kumata K , Iwata N , Saido TC , Trojanowski JQ , Lee VM , Staufenbiel M , Tomiyama T , Mori H , Fukumura T , Suhara T , Higuchi M ((2011) ) In vivo positron emission tomographic imaging of glial responses to amyloid-beta and tau pathologies in mouse models of Alzheimer’s disease and related disorders. J Neurosci 31: , 4720–4730. |
[66] | Li Y , Liu L , Barger SW , Griffin WS ((2003) ) Interleukin-1 mediates pathological effects of microglia on tau phosphorylation and on synaptophysin synthesis in cortical neurons through a p38-MAPK pathway. J Neurosci 23: , 1605–1611. |
[67] | Ghosh S , Wu MD , Shaftel SS , Kyrkanides S , LaFerla FM , Olschowka JA , O’Banion MK ((2013) ) Sustained interleukin-1β overexpression exacerbates tau pathology despite reduced amyloid burden in an Alzheimer’s mouse model. J Neurosci 33: , 5053–5064. |
[68] | Gorlovoy P , Larionov S , Pham TT , Neumann H ((2009) ) Accumulation of tau induced in neurites by microglial proinflammatory mediators. FASEB J 23: , 2502–2513. |
[69] | Guo JL , Lee VM ((2011) ) Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem 286: , 15317–15331. |
[70] | Asai H , Ikezu S , Tsunoda S , Medalla M , Luebke J , Haydar T , Wolozin B , Butovsky O , Kügler S , Ikezu T ((2015) ) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18: , 1584–1593. |
[71] | Lehmann JM , Lenhard JM , Oliver BB , Ringold GM , Kliewer SA ((1997) ) Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem 272: , 3406–3410. |
[72] | in t’ Veld BA , Ruitenberg A , Hofman A , Launer LJ , van Duijn CM , Stijnen T , Breteler MM , Stricker BH ((2001) ) Nonsteroidal antiinflammatory drugs and the risk of Alzheimer’s disease. N Engl J Med 345: , 1515–1521. |
[73] | Yan Q , Zhang J , Liu H , Babu-Khan S , Vassar R , Biere AL , Citron M , Landreth G ((2003) ) Anti-inflammatory drug therapy alters beta-amyloid processing and deposition in an animal model of Alzheimer’s disease. J Neurosci 23: , 7504–7509. |
[74] | Heneka MT , Sastre M , Dumitrescu-Ozimek L , Hanke A , Dewachter I , Kuiperi C , O’Banion K , Klockgether T , Van Leuven F , Landreth GE ((2005) ) Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1-42 levels in APPV717I transgenic mice. Brain 128: , 1442–1453. |
[75] | Escribano L , Simon A-M , Gimeno E , Cuadrado-Tejedor M , Lopez de Maturana R , Garcia-Osta A , Ricobaraza A , Perez-Mediavilla A , Del Rio J , Frechilla D ((2010) ) Rosiglitazone rescues memory impairment in Alzheimer’s transgenic mice: Mechanisms involving a reduced amyloid and tau pathology. Neuropsychopharmacology 35: , 1593–1604. |
[76] | Moosecker S , Gomes P , Dioli C , Yu S , Sotiropoulos I , Almeida OFX ((2019) ) Activated PPARγ abrogates misprocessing of amyloid precursor protein, tau missorting and synaptotoxicity. Front Cell Neurosci 13: , 239. |
[77] | Sastre M , Dewachter I , Rossner S , Bogdanovic N , Rosen E , Borghgraef P , Evert BO , Dumitrescu-Ozimek L , Thal DR , Landreth G , Walter J , Klockgether T , van Leuven F , Heneka MT ((2006) ) Nonsteroidal anti-inflammatory drugs repress beta-secretase gene promoter activity by the activation of PPAR gamma. Proc Natl Acad Sci U S A 103: , 443–448. |
[78] | Chen J , Li S , Sun W , Li J ((2015) ) Anti-diabetes drug pioglitazone ameliorates synaptic defects in AD transgenic mice by inhibiting cyclin-dependent kinase5 activity. PLoS One 10: , e0123864. |
[79] | Quan Q , Qian Y , Li X , Li M ((2019) ) Pioglitazone reduces β amyloid levels via inhibition of PPARγ phosphorylation in a neuronal model of Alzheimer’s disease. Front Aging Neurosci 11: , 178. |
[80] | Dolma S , Lessnick SL , Hahn WC , Stockwell BR ((2003) ) Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 3: , 285–296. |
[81] | Yang WS , Stockwell BR ((2008) ) Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 15: , 234–245. |
[82] | Angeli JPF , Schneider M , Proneth B , Tyurina YY , Tyurin VA , Hammond VJ , Herbach N , Aichler M , Walch A , Eggenhofer E , Basavarajappa D , Radmark O , Kobayashi S , Seibt T , Beck H , Neff F , Esposito I , Wanke R , Foerster H , Yefremova O , Heinrichmeyer M , Bornkamm GW , Geissler EK , Thomas SB , Stockwell BR , O’Donnell VB , Kagan VE , Schick JA , Conrad M ((2014) ) Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16: , 1180-U120. |
[83] | Yang WS , SriRamaratnam R , Welsch ME , Shimada K , Skouta R , Viswanathan VS , Cheah JH , Clemons PA , Shamji AF , Clish CB , Brown LM , Girotti AW , Cornish VW , Schreiber SL , Stockwell BR ((2014) ) Regulation of ferroptotic cancer cell death by GPX4. Cell 156: , 317–331. |
[84] | Imai H , Nakagawa Y ((2003) ) Biological significance of phospholipid hydroperoxide glutathione peroxidase (PHGPx, GPx4) in mammalian cells. Free Radic Biol Med 34: , 145–169. |
[85] | Zilka O , Shah R , Li B , Angeli JPF , Griesser M , Conrad M , Pratt DA ((2017) ) On the mechanism of cytoprotection by ferrostatin-1 and liproxstatin-1 and the role of lipid peroxidation in ferroptotic cell death. ACS Cent Sci 3: , 232–243. |
[86] | Dixon SJ , Winter GE , Musavi LS , Lee ED , Snijder B , Rebsamen M , Superti-Furga G , Stockwell BR ((2015) ) Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol 10: , 1604–1609. |
[87] | Kagan VE , Mao GW , Qu F , Angeli JPF , Doll S , St Croix C , Dar HH , Liu B , Tyurin VA , Ritov VB , Kapralov AA , Amoscato AA , Jiang J , Anthonymuthu T , Mohammadyani D , Yang Q , Proneth B , Klein-Seetharaman J , Watkins S , Bahar H , Greenberger J , Mallampalli RK , Stockwell BR , Tyurina YY , Conrad M , Bayir H ((2017) ) Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13: , 81–90. |
[88] | Smith MA , Harris PL , Sayre LM , Perry G ((1997) ) Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc Natl Acad Sci U S A 94: , 9866–9868. |
[89] | van Duijn S , Bulk M , van Duinen SG , Nabuurs RJA , van Buchem MA , van der Weerd L , Natté R ((2017) ) Cortical iron reflects severity of Alzheimer’s disease. J Alzheimers Dis 60: , 1533–1545. |
[90] | Wang H , An P , Xie E , Wu Q , Fang X , Gao H , Zhang Z , Li Y , Wang X , Zhang J , Li G , Yang L , Liu W , Min J , Wang F ((2017) ) Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 66: , 449–465. |
[91] | Zhang D-L , Ghosh MC , Rouault TA ((2014) ) The physiological functions of on regulatory proteins in iron homeostasis - an update. Front Pharmacol 5: , 124. |
[92] | Wang F , Wang J , Shen Y , Li H , Rausch W-D , Huang X ((2022) ) Iron dyshomeostasis and ferroptosis: A new Alzheimer’s disease hypothesis? Front Aging Neurosci 14: , 830569. |
[93] | Raha AA , Vaishnav RA , Friedland RP , Bomford A , Raha-Chowdhury R ((2013) ) The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in Alzheimer’s disease. Acta Neuropathol Commun 1: , 55. |
[94] | Bao W-D , Pang P , Zhou X-T , Hu F , Xiong W , Chen K , Wang J , Wang F , Xie D , Hu Y-Z , Han Z-T , Zhang H-H , Wang W-X , Nelson PT , Chen J-G , Lu Y , Man H-Y , Liu D , Zhu L-Q ((2021) ) Loss of ferroportin induces memory impairment by promoting ferroptosis in Alzheimer’s disease. Cell Death Differ 28: , 1548–1562. |
[95] | Ashraf A , Jeandriens J , Parkes HG , So PW ((2020) ) Iron dyshomeostasis, lipid peroxidation and perturbed expression of cystine/glutamate antiporter in Alzheimer’s disease: Evidence of ferroptosis. Redox Biol 32: , 101494. |
[96] | Duce JA , Tsatsanis A , Cater MA , James SA , Robb E , Wikhe K , Leong SL , Perez K , Johanssen T , Greenough MA , Cho HH , Galatis D , Moir RD , Masters CL , McLean C , Tanzi RE , Cappai R , Barnham KJ , Ciccotosto GD , Rogers JT , Bush AI ((2010) ) Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer’s disease. Cell 142: , 857–867. |
[97] | Lei P , Ayton S , Finkelstein DI , Spoerri L , Ciccotosto GD , Wright DK , Wong BXW , Adlard PA , Cherny RA , Lam LQ , Roberts BR , Volitakis I , Egan GF , McLean CA , Cappai R , Duce JA , Bush AI ((2012) ) Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat Med 18: , 291–295. |
[98] | Feng H , Stockwell BR ((2018) ) Unsolved mysteries: How does lipid peroxidation cause ferroptosis? PLoS Biol 16: , e2006203. |
[99] | Jiang X , Stockwell BR , Conrad M ((2021) ) Ferroptosis: Mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 22: , 266–282. |
[100] | Sultana R , Perluigi M , Butterfield DA ((2006) ) Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer’s disease: Insights into mechanism of neurodegeneration from redox proteomics. Antioxid Redox Signal 8: , 2021–2037. |
[101] | Catala A , Diaz M ((2016) ) Editorial: Impact of lipid peroxidation on the physiology and pathophysiology of cell membranes. Front Physiol 7: , 423. |
[102] | Barrera G , Pizzimenti S , Daga M , Dianzani C , Arcaro A , Cetrangolo GP , Giordano G , Cucci MA , Graf M , Gentile F ((2018) ) Lipid peroxidation-derived aldehydes, 4-hydroxynonenal and malondialdehyde in aging-related disorders. Antioxidants 7: , 102. |
[103] | Dixon SJ , Patel D , Welsch M , Skouta R , Lee E , Hayano M , Thomas AG , Gleason C , Tatonetti N , Slusher BS , Stockwell BR ((2014) ) Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3: , e02523. |
[104] | Tang D , Chen X , Kang R , Kroemer G ((2021) ) Ferroptosis: Molecular mechanisms and health implications. Cell Res 31: , 107–125. |
[105] | Ingold I , Berndt C , Schmitt S , Doll S , Poschmann G , Buday K , Roveri A , Peng X , Freitas FP , Seibt T , Mehr L , Aichler M , Walch A , Lamp D , Jastroch M , Miyamoto S , Wurst W , Ursini F , Amer ESJ , Fradejas-Villar N , Schweizer U , Zischka H , Angeli JPF , Conrad M ((2018) ) Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell 172: , 409-422. e21. |
[106] | Doll S , Freitas FP , Shah R , Aldrovandi M , da Silva MC , Ingold I , Grocin AG , da Silva TNX , Panzilius E , Scheel CH , Mourao A , Buday K , Sato M , Wanninger J , Vignane T , Mohana V , Rehberg M , Flatley A , Schepers A , Kurz A , White D , Sauer M , Sattler M , Tate EW , Schmitz W , Schulze A , O’Donnell V , Proneth B , Popowicz GM , Pratt DA , Angeli JPF , Conrad M ((2019) ) FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575: , 693–698. |
[107] | Bersuker K , Hendricks JM , Li Z , Magtanong L , Ford B , Tang PH , Roberts MA , Tong B , Maimone TJ , Zoncu R , Bassik MC , Nomura DK , Dixon SJ , Olzmann JA ((2019) ) The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575: , 688–692. |
[108] | Mandal PK , Saharan S , Tripathi M , Murari G ((2015) ) Brain glutathione levels–a novel biomarker for mild cognitive impairment and Alzheimer’s disease. Biol Psychiatry 78: , 702–710. |
[109] | Greenough MA , Lane DJR , Balez R , Anastacio HTD , Zeng Z , Ganio K , McDevitt CA , Acevedo K , Belaidi AA , Koistinaho J , Ooi L , Ayton S , Bush AI ((2022) ) Selective ferroptosis vulnerability due to familial Alzheimer’s disease presenilin mutations. Cell Death Differ 29: , 2123–2136. |
[110] | Hambright WS , Fonseca RS , Chen L , Na R , Ran Q ((2017) ) Ablation of ferroptosis regulator glutathione peroxidase 4 in forebrain neurons promotes cognitive impairment and neurodegeneration. Redox Biol 12: , 8–17. |
[111] | Seiler A , Schneider M , Foerster H , Roth S , Wirth EK , Culmsee C , Plesnila N , Kremmer E , Radmark O , Wurst W , Bornkamm GW , Schweizer U , Conrad M ((2008) ) Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-Mediated cell death. Cell Metab 8: , 237–248. |
[112] | Chen L , Dar NJ , Na R , McLane KD , Yoo K , Han X , Ran Q ((2022) ) Enhanced defense against ferroptosis ameliorates cognitive impairment and reduces neurodegeneration in 5xFAD mice. Free Radic Biol Med 180: , 1–12. |
[113] | Emilia Smith M , Saraceno GE , Capani F , Castilla R ((2013) ) Long-chain acyl-CoA synthetase 4 is regulated by phosphorylation. Biochem Biophys Res Commun 430: , 272–277. |
[114] | Zhang HL , Hu BX , Li ZL , Du T , Shan JL , Ye ZP , Peng XD , Li X , Huang Y , Zhu XY , Chen YH , Feng GK , Yang D , Deng R , Zhu XF ((2022) ) PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat Cell Biol 24: , 88–98. |
[115] | Wang W , Green M , Choi JE , Gijon M , Kennedy PD , Johnson JK , Liao P , Lang X , Kryczek I , Sell A , Xia H , Zhou J , Li G , Li J , Li W , Wei S , Vatan L , Zhang H , Szeliga W , Gu W , Liu R , Lawrence TS , Lamb C , Tanno Y , Cieslik M , Stone E , Georgiou G , Chan TA , Chinnaiyan A , Zou W ((2019) ) CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 569: , 270–274. |
[116] | Liao P , Wang W , Wang W , Kryczek I , Li X , Bian Y , Sell A , Wei S , Grove S , Johnson JK , Kennedy PD , Gijon M , Shah YM , Zou W ((2022) ) CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell 40: , 365-378. e6. |
[117] | Li Y , Feng D , Wang Z , Zhao Y , Sun R , Tian D , Liu D , Zhang F , Ning S , Yao J , Tian X ((2019) ) Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ 26: , 2284–2299. |
[118] | Gao Y , Li J , Wu Q , Wang S , Yang S , Li X , Chen N , Li L , Zhang L ((2021) ) Tetrahydroxy stilbene glycoside ameliorates Alzheimer’s disease in APP/ PS1 mice via glutathione peroxidase related ferroptosis. Int Immunopharmacol 99: , 108002. |
[119] | Zhu ZY , Liu YD , Gong Y , Jin W , Topchiy E , Turdi S , Gao YF , Culver B , Wang SY , Ge W , Zha WL , Ren J , Pei ZH , Qin X ((2022) ) Mitochondrial aldehyde dehydrogenase (ALDH2) rescues cardiac contractile dysfunction in an APP/PS1 murine model of Alzheimer’s disease via inhibition of ACSL4-dependent ferroptosis. Acta Pharmacol Sin 43: , 39–49. |
[120] | Alzoubi KH , Khabour OF , Alfaqih M , Tashtoush M , Al-Azzam SI , Mhaidat NM , Alrabadi N ((2022) ) The protective effectsof pioglitazone against cognitive impairment caused by L-methionine administration in a rat model. CNSNeurol Disord Drug Targets 21: , 77–84. |
[121] | Ates G , Goldberg J , Currais A , Maher P ((2020) ) CMS121, a fatty acid synthase inhibitor, protects against excess lipid peroxidation and inflammation and alleviates cognitive loss in a transgenic mouse model of Alzheimer’s disease. Redox Biol 36: , 101648. |
[122] | Xu Y , Li X , Cheng Y , Yang M , Wang R ((2020) ) Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J 34: , 16262–16275. |
[123] | Cui Y , Zhang Y , Zhao X , Shao L , Liu G , Sun C , Xu R , Zhang Z ((2021) ) ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav Immun 93: , 312–321. |
[124] | Fan Z , Cai L , Wang S , Wang J , Chen B ((2021) ) Baicalin prevents myocardial ischemia/reperfusion injury through inhibiting ACSL4 mediated ferroptosis. Front Pharmacol 12: , 628988. |
[125] | He S , Li R , Peng Y , Wang Z , Huang J , Meng H , Min J , Wang F , Ma Q ((2022) ) ACSL4 contributes to ferroptosis-mediated rhabdomyolysis in exertional heat stroke. J Cachexia Sarcopenia Muscle 13: , 1717–1730. |
[126] | Wang Y , Zhang M , Bi R , Su Y , Quan F , Lin Y , Yue C , Cui X , Zhao Q , Liu S , Yang Y , Zhang D , Cao Q , Gao X ((2022) ) ACSL4 deficiency confers protection against ferroptosis-mediated acute kidney injury. Redox Biol 51: , 102262. |
[127] | Papaetis GS ((2022) ) Pioglitazone, bladder cancer, and the presumption of innocence. Curr Drug Saf 17: , 294–318. |
[128] | Sato T , Hanyu H , Hirao K , Kanetaka H , Sakurai H , Iwamoto T ((2011) ) Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging 32: , 1626–1633. |
[129] | Read S , Wu P , Biscow M ((2014) ) sustained 4-year cognitive and functional response in early Alzheimer’s disease with pioglitazone. J Am Geriatr Soc 62: , 584–586. |
[130] | Heneka MT , Fink A , Doblhammer G ((2015) ) Effect of pioglitazone medication on the incidence of dementia. Ann Neurol 78: , 284–294. |
[131] | Lu C-H , Yang C-Y , Li C-Y , Hsieh C-Y , Ou H-T ((2018) ) Lower risk of dementia with pioglitazone, compared with other second-line treatments, in metformin-based dual therapy: A population-based longitudinal study. Diabetologia 61: , 562–573. |
[132] | Cao B , Rosenblat JD , Brietzke E , Park C , Lee Y , Musial N , Pan Z , Mansur RB , McIntyre RS ((2018) ) Comparative efficacy and acceptability of antidiabetic agents for Alzheimer’s disease and mild cognitive impairment: A systematic review and network meta-analysis. Diabetes Obes Metab 20: , 2467–2471. |
[133] | Geldmacher DS , Fritsch T , McClendon MJ , Landreth G ((2011) ) A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch Neurol 68: , 45–50. |
[134] | Burns DK , Alexander RC , Welsh-Bohmer KA , Culp M , Chiang C , O’Neil J , Evans RM , Harrigan P , Plassman BL , Burke JR , Wu J , Lutz MW , Haneline S , Schwarz AJ , Schneider LS , Yaffe K , Saunders AM , Ratti E , TOMMORROW study investigators ((2021) ) Safety and efficacy of pioglitazone for the delay of cognitive impairment in people at risk of Alzheimer’s disease (TOMMORROW): A prognostic biomarker study and a phase 3, randomised, double-blind, placebo-controlled trial. Lancet Neurol 20: , 537–547. |
[135] | Dawson J ((2018) ) Pioglitazone use after stroke: Story of hearts, minds, and bones. Circulation 138: , 1221–1223. |
[136] | Amarenco P , Bogousslavsky J , Callahan A III , Goldstein LB , Hennerici M , Rudolph AE , Sillesen H , Simunovic L , Szarek M , Welch KMA , Zivin JA , Stroke Prevention by Aggressive Reduction in Cholesterol Levels (SPARCL) Investigators ((2006) ) High-dose atorvastatin after stroke or transient ischemic attack. N Engl J Med 355: , 549–559. |
[137] | Viscoli CM , Kernan WN , Young LH ((2016) ) Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med 375: , 704. |
[138] | Lee M , Saver JL , Liao H-W , Lin C-H , Ovbiagele B ((2017) ) Pioglitazone for secondary stroke prevention: A systematic review and meta-analysis. Stroke 48: , 388–393. |
[139] | Dormandy JA , Charbonnel B , Eckland DJA , Erdmann E , Massi-Benedetti M , Kmoules IK , Skene AM , Tan MH , Lefebvre PJ , Murray GD , Standl E , Wilcox RG , Wlhelmsen L , Betteridge J , Birkeland K , Golay A , Heine RJ , Koranyi L , Laakso M , Mokan M , Norkus A , Pirags V , Podar T , Scheen A , Scherbaum W , Schernthaner G , Schmitz O , Skrha J , Smith U , Taton J , PROactive Investigators ((2005) ) Secondary prevention of macrovascular events in patients with type 2 diabetes in the PROactive Study (PROspective pioglitAzone Clinical Trial In macroVascular Events): A randomised controlled trial. Lancet 366: , 1279–1289. |
[140] | Wilcox R , Bousser M-G , Betteridge DJ , Schernthaner G , Pirags V , Kupfer S , Dormandy J , PROactive Investigators ((2007) ) Effects of pioglitazone in patients with type 2 diabetes with or without previous stroke - Results from PROactive (PROspective pioglitAzone Clinical Trial in macroVascular Events 04). Stroke 38: , 865–873. |
[141] | Yaghi S , Furie KL , Viscoli CM , Kamel H , Gorman M , Dearborn J , Young LH , Inzucchi SE , Lovejoy AM , Kasner SE , Conwit R , Kernan WN ; IRIS Trial Investigators ((2018) ) Pioglitazone prevents stroke in patients with a recent transient ischemic attack or ischemic stroke: A planned secondary analysis of the IRIS Trial (Insulin Resistance Intervention After Stroke). Circulation 137: , 455–463. |
[142] | Spence JD , Viscoli CM , Inzucchi SE , Dearborn-Tomazos J , Ford GA , Gorman M , Furie KL , Lovejoy AM , Young LH , Kernan WN , et al. ((2019) ) Pioglitazone therapy in patients with stroke and prediabetes aanalysis of the IRIS randomized clinical trial. JAMA Neurol 76: , 526–535. |
[143] | Kernan WN , Viscoli CM , Dearborn JL , Kent DM , Conwit R , Fayad P , Furie KL , Gorman M , Guarino PD , Inzucchi SE , Stuart A , Young LH , Insulin Resistance Intervention After Stroke (IRIS) Trial Investigators ((2017) ) Targeting pioglitazone hydrochloride therapy after stroke or transient ischemic attack according to pretreatment risk for stroke ormyocardial infarction. JAMA Neurol 74: , 1319–1327. |
[144] | Morgan CL , Inzucchi SE , Puelles J , Jenkins-Jones S , Currie CJ ((2018) ) Impact of treatment with pioglitazone on stroke outcomes: A real-world database analysis. Diabetes Obes Metab 20: , 2140–2147. |
[145] | Woo M-H , Lee HS , Kim J ((2019) ) Effect of pioglitazone in acute ischemic stroke patients with diabetes mellitus: A nested case-control study. Cardiovasc Diabetol 18: , 67. |
[146] | Hung Y-C , Chiu L-T , Huang H-Y , Bau D-T ((2020) ) Pioglitazone for primary stroke prevention in Asian patients with type 2 diabetes and cardiovascular risk factors: A retrospective study. Cardiovasc Diabetol 19: , 94. |
[147] | Ha J , Choi D-W , Kim KY , Nam CM , Kim E ((2021) ) Pioglitazone use associated with reduced risk of the first attack of ischemic stroke in patients with newly onset type 2 diabetes: A nationwide nested case-control study. Cardiovasc Diabetol 20: , 152. |
[148] | Langenfeld MR , Forst T , Hohberg C , Kann P , Lubben G , Konrad T , Fullert SD , Sachara C , Pfutzner A ((2005) ) Pioglitazone decreases carotid intima-media thickness independently of glycemic control in patients with type 2 diabetes mellitus - Results from a controlled randomized study. Circulation 111: , 2525–2531. |
[149] | Mazzone T , Meyer PM , Feinstein SB , Davidson MH , Kondos GT , D’Agostino RB Sr , Perez A , Provost JC , Haffner SM ((2006) ) Effect of pioglitazone compared with glimepiride on carotid intima-media thickness in type 2 diabetes: A randomized trial. JAMA 296: , 2572–2581. |
[150] | Saremi A , Schwenke DC , Buchanan TA , Hodis HN , Mack WJ , Banerji M , Bray GA , Clement SC , Henry RR , Kitabchi AE , Mudaliar S , Ratner RE , Stentz FB , Musi N , Tripathy D , DeFronzo RA , Reaven PD ((2013) ) Pioglitazone slows progression of atherosclerosis in prediabetes independent of changes in cardiovascular risk factors. Arterioscler Thromb Vasc Biol 33: , 393–399. |
[151] | Nissen SE , Nicholls SJ , Wolski K , Nesto R , Kupfer S , Perez A , Jure H , De Larochellière R , Staniloae CS , Mavromatis K , Saw J , Hu B , Lincoff AM , Tuzcu EM ((2008) ) Comparison of pioglitazone vs glimepiride on progression of coronary atherosclerosis in patients with type 2 diabetes: The PERISCOPE randomized controlled trial. JAMA 299: , 1561–1573. |
[152] | Tawakol A , Migrino RQ , Bashian GG , Bedri S , Vermylen D , Cury RC , Yates D , LaMuraglia GM , Furie K , Houser S , Gewirtz H , Muller JE , Brady TJ , Fischman AJ ((2006) ) In vivo 18F-fluorodeoxyglucose positron emission tomography imaging provides a noninvasive measure of carotid plaque inflammation in patients. J Am Coll Cardiol 48: , 1818–1824. |
[153] | Mizoguchi M , Tahara N , Tahara A , Nitta Y , Kodama N , Oba T , Mawatari K , Yasukawa H , Kaida H , Ishibashi M , Hayabuchi N , Harada H , Ikeda H , Yamagishi S , Imaizumi T ((2011) ) Pioglitazone attenuates atherosclerotic plaque inflammation in patients with impaired glucose tolerance or diabetes a prospective, randomized, comparator-controlled study using serial FDG PET/CT imaging study of carotid artery and ascending aorta. JACC Cardiovasc Imaging 4: , 1110–1118. |
[154] | Nitta Y , Tahara N , Tahara A , Honda A , Kodama N , Mizoguchi M , Kaida H , Ishibashi M , Hayabuchi N , Ikeda H , Yamagishi S , Imaizumi T ((2013) ) Pioglitazone decreases coronary artery inflammation in impaired glucose tolerance and diabetes mellitus: Evaluation by FDG-PET/CT imaging. JACC Cardiovasc Imaging 6: , 1172–1182. |
[155] | Choo EH , Han EJ , Kim CJ , Kim SH , O JH , Chang K , Seung KB ((2018) ) Effect of pioglitazone in combination with moderate dose statin on atherosclerotic inflammation: Randomized controlled clinical trial using serial FDG-PET/CT. Korean Circ J 48: , 591–601. |
[156] | Campia U , Matuskey LA , Panza JA ((2006) ) Peroxisome proliferator-activated receptor-gamma activation with pioglitazone improves endothelium-dependent dilation in nondiabetic patients with major cardiovascular risk factors. Circulation 113: , 867–875. |
[157] | Sakatani Y , Miyoshi T , Oe H , Noda Y , Ohno Y , Nakamura K , Saito Y , Osawa K , Morita H , Kohno K , Ito H ((2014) ) Pioglitazone prevents the endothelial dysfunction induced by ischemia and reperfusion in healthy subjects. J Cardiovasc Pharmacol 64: , 326–331. |
[158] | Nicholls SJ , Tuzcu EM , Wolski K , Bayturan O , Lavoie A , Uno K , Kupfer S , Perez A , Nesto R , Nissen SE ((2011) ) Lowering the triglyceride/high-density lipoprotein cholesterol ratio is associated with the beneficial impact of pioglitazone on progression of coronary atherosclerosis in diabetic patients: Insights from the PERISCOPE (Pioglitazone Effect on Regression of Intravascular Sonographic Coronary Obstruction Prospective Evaluation) study. J Am Coll Cardiol 57: , 153–159. |
[159] | Mani P , Uno K , St John J , Kupfer S , Perez A , Tuzcu EM , Hazen SL , Nissen SE , Nicholls SJ ((2015) ) Favorable impact on LDL particle size in response to treatment with pioglitazone is associated with less progression of coronary atherosclerosis in patients with type 2 diabetes. J Am Coll Cardiol 66: , 328–329. |
[160] | Goldberg RB , Kendall DM , Deeg MA , Buse JB , Zagar AJ , Pinaire JA , Tan MH , Khan MA , Perez AT , Jacober SJ ((2005) ) A comparison of lipid and glycemic effects of pioglitazone and rosiglitazone in patients with type 2 diabetes and dyslipidemia. Diabetes Care 28: , 1547–1554. |
[161] | Hillaire-Buys D , Faillie JL , Montastruc JL ((2011) ) Pioglitazone and bladder cancer. Lancet 378: , 1543–1544; author reply 1544-1545. |
[162] | Azoulay L , Yin H , Filion KB , Assayag J , Majdan A , Pollak MN , Suissa S ((2012) ) The use of pioglitazone and the risk of bladder cancer in people with type 2 diabetes: Nested case-control study. BMJ 344: , e3645. |
[163] | Erdmann E , Dormandy JA , Charbonnel B , Massi-Benedetti M , Moules IK , Skene AM ((2007) ) The effect of pioglitazone on recurrent myocardial infarction in 2,445 patients with type 2 diabetes and previous myocardial infarction: Results from the PROactive (PROactive 05) Study. J Am Coll Cardiol 49: , 1772–1780. |
[164] | Erdmann E , Charbonnel B , Wilcox RG , Skene AM , Massi-Benedetti M , Yates J , Tan M , Spanheimer R , Standl E , Dormandy JA ((2007) ) Pioglitazone use and heart failure in patients with type 2 diabetes and preexisting cardiovascular disease: Data from the PROactive study (PROactive 08). Diabetes Care 30: , 2773–2778. |
[165] | White U , Fitch MD , Beyl RA , Hellerstein MK , Ravussin E ((2021) ) Adipose depot-specific effects of 16 weeks of pioglitazone on in vivo adipogenesis in women with obesity: A randomised controlled trial. Diabetologia 64: , 159–167. |
[166] | Vaccaro O , Masulli M , Nicolucci A , Bonora E , Del Prato S , Maggioni AP , Rivellese AA , Squatrito S , Giorda CB , Sesti G , Mocarelli P , Lucisano G , Sacco M , Signorini S , Cappellini F , Perriello G , Babini AC , Lapolla A , Gregori G , Giordano C , Corsi L , Buzzetti R , Clemente G , Di Cianni G , Iannarelli R , Cordera R , La Macchia O , Zamboni C , Scaranna C , Boemi M , Iovine C , Lauro D , Leotta S , Dall’Aglio E , Cannarsa E , Tonutti L , Pugliese G , Bossi AC , Anichini R , Dotta F , Di Benedetto A , Citro G , Antenucci D , Ricci L , Giorgino F , Santini C , Gnasso A , De Cosmo S , Zavaroni D , Vedovato M , Consoli A , Calabrese M , di Bartolo P , Fornengo P , Riccardi G ((2017) ) Effects on the incidence of cardiovascular events of the addition of pioglitazone versus sulfonylureas in patients with type 2 diabetes inadequately controlled with metformin (TOSCA.IT): A randomised, multicentre trial. Lancet Diabetes Endocrinol 5: , 887–897. |
[167] | Kernan WN , Viscoli CM , Furie KL , Young LH , Inzucchi SE , Gorman M , Guarino PD , Lovejoy AM , Peduzzi PN , Conwit R , Brass LM , Schwartz GG , Adams HP Jr. , Berger L , Carolei A , Clark W , Coull B , Ford GA , Kleindorfer D , O’Leary JR , Parsons MW , Ringleb P , Sen S , Spence JD , Tanne D , Wang D , Winder TR ((2016) ) Pioglitazone after ischemic stroke or transient ischemic attack. N Engl J Med 374: , 1321–1331. |
[168] | Jojo GM , Kuppusamy G ((2019) ) Scope of new formulation approaches in the repurposing of pioglitazone for the management of Alzheimer’s disease. J Clin Pharm Ther 44: , 337–348. |
[169] | Rodríguez-Pascau L , Vilalta A , Cerrada M , Traver E , Forss-Petter S , Weinhofer I , Bauer J , Kemp S , Pina G , Pascual S , Meya U , Musolino PL , Berger J , Martinell M , Pizcueta P ((2021) ) The brain penetrant PPARγ agonist leriglitazone restores multiple altered pathways in models of X-linked adrenoleukodystrophy. Sci Transl Med 13: , eabc0555. |
[170] | Köhler W , Engelen M , Eichler F , Lachmann R , Fatemi A , Sampson J , Salsano E , Gamez J , Molnar MJ , Pascual S , Rovira M , Vilà A , Pina G , Martín-Ugarte I , Mantilla A , Pizcueta P , Rodríguez-Pascau L , Traver E , Vilalta A , Pascual M , Martinell M , Meya U , Mochel F ((2023) ) Safety and efficacy of leriglitazone for preventing disease progression in men with adrenomyeloneuropathy (ADVANCE): A randomised, double-blind, multi-centre, placebo-controlled phase 2-3 trial. Lancet Neurol 22: , 127–136. |

