
Intelligent Alzheimer’s Diseases Gene Association Prediction Model Using Deep Regulatory Genomic Neural Networks
Abstract
Alzheimer’s disease (AD) is an illness that affects the nervous system, leading to a loss in cognitive and logical abilities. Gene regulatory expressions, which are the complex language exhibited by DNA, serve several functionalities, including the physical and biological life cycle processes in the human body. The gene expression sequence affects the pathology experienced by an individual, its longevity, and potential for a cure. The transcription factors, from DNA to RNA conversion, and the binding process determine the gene expression, which varies for every human organ and disease. This study proposes Deep convolutional neural network model that reads the gene regulatory expression sequence through various convolutional layers encoded to detect positive spikes in transcription factors. This results in the prediction of disease conversion probability from mild cognitive impairment to AD which is the key-requisite for affected geriatric cohorts.
INTRODUCTION
The leading factor in Alzheimer’s disease (AD) in adults over 65 is a significant concern. “There are currently billion individuals living in the US. By 2050, 14 million Americans will be infected.” This should be revised to: “There are currently billions of individuals living in the US. By 2050, 14 million Americans are projected to be affected.” Although there is currently no cure for AD, researchers and medical professionals are working to alleviate the suffering caused by the disease, comprehend how it works, and ultimately find solutions to halt or slow down its course. It has been demonstrated that synaptic impairment, reduced cerebral glucose metabolism, and cerebral hypoperfusion occur prior to the beginning of amyloid-β (Aβ). Despite its positive effects on lowering brain Aβ deposition, humanized anti-monoclonal antibody Bapineuzumab β did not enhance clinical outcomes in AD patients in clinical trials. Thus, it may be challenging to develop clinical interventions that solely target Aβ.
The proposed study aims to predict the significant motifs that increase the vulnerability of cognitive neurodegenerative diseases like AD. The production and storage of Aβ peptides in the brain are fundamental processes in the beginning stages of AD. However, growing evidence includes the development of ‘Aβ-dependent pathways’ in the disease progression.
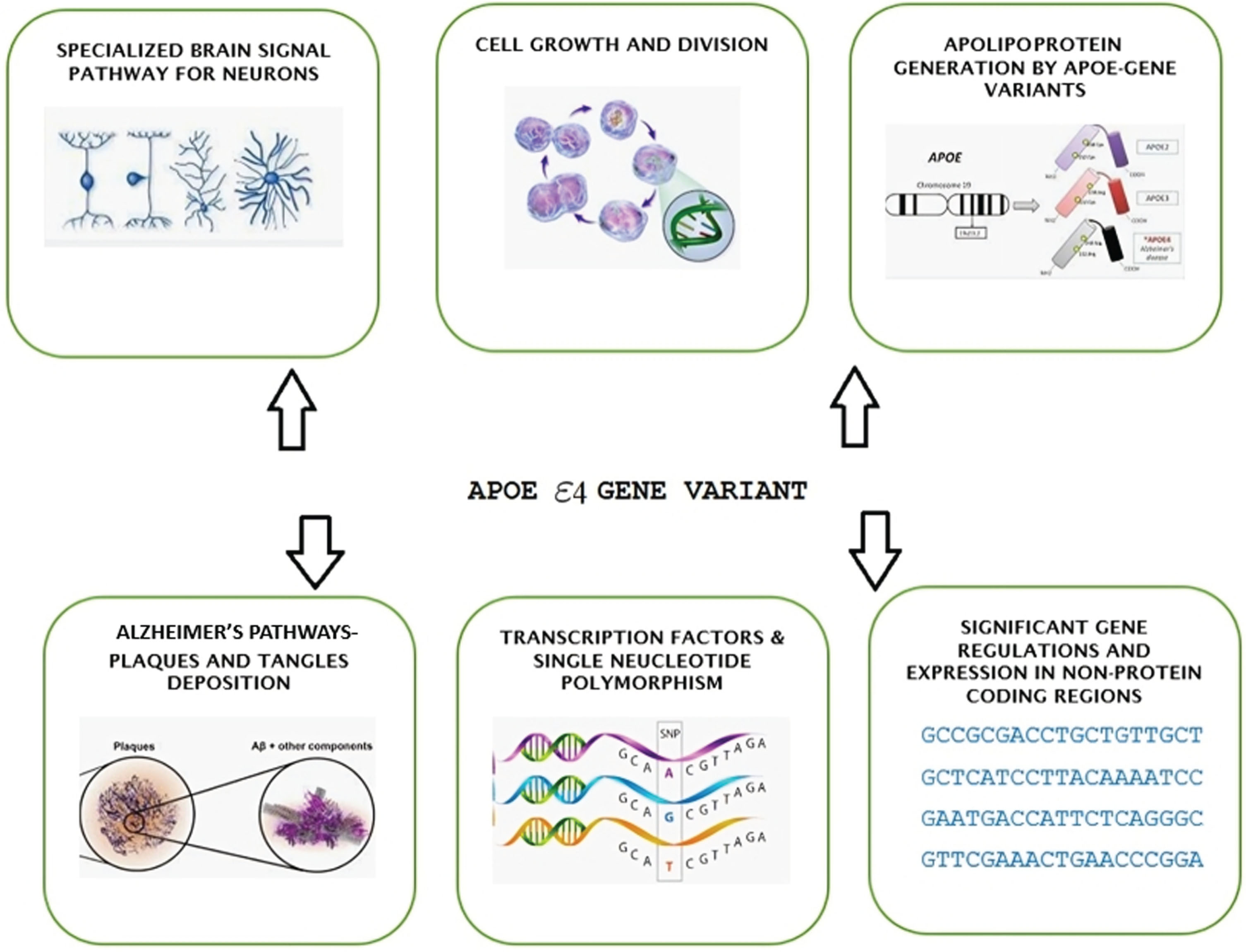
Numerous diversified genome-wide association studies and genome-wide association meta-analyses conducted using the ADNI repository have consistently identified the APOE 4 allele as the most significant genetic risk factor, while the APOE 2 allele has been shown to provide the strongest genetic protective factor [1]. Despite these findings, no specific treatments targeting APOE have been developed thus far. However, in the last five years, our understanding of APOE pathogenesis has expanded beyond its role in amyloid-peptide-centric mechanisms to encompass tau neurofibrillary degeneration, microglia and astrocyte responses, and disruption of the blood-brain barrier.
Early studies have established a causal link between APOE and amyloid-peptide aggregation and clearance [2]. Since all these disease processes have the potential to lead to cognitive impairment, it becomes essential to capitalize on this newfound knowledge to develop APOE-specific medications. Several treatment strategies have been explored in mice models carrying human APOE alleles.
Early onset AD
This stage of the disease affects those aged 30 to 60 (constituting 5% of all AD cases). It is also known as familial AD, primarily attributed to hereditary factors. The heredity leads to individual genetic mutations and gene regulatory patterns resulting in inappropriate protein synthesis.
Late onset AD
The majority of AD occurs in adults over 60, influenced by genetic, environmental, and lifestyle factors. In-depth pathological diagnosis reveals significant characteristics and unusual morphology in brain regions. This is characterized by the widening of sulci due to cerebral degeneration and significant neuronal loss. Neurotic plaques, biomarkers deposited in neurons, are found around Aβ40/42 types of amyloid-β protein precursor (AβPP) derivative [3]. These are diffuse plaques with Aβ42 predominance. Tau protein types have been identified as the next major factor that increases the deposition of neurofibrillary tangles in cerebral regions.
Genetic factors
AD with an autosomal dominant distribution is linked to single nucleotide polymorphisms (SNPs) in the APP and PS1 genes for AβPP and presenilin, respectively. Mutations in the presenilin (PS2) gene lead to defective protein activity and improper amyloid protein breakdown, producing damaging amyloid plaques, a characteristic of the disease. The precise function of the APOE gene, which encodes a very low-density lipoprotein aiding in removing cholesterol from the bloodstream, is unknown [4]. Varied alleles (2, 3, 4) exhibit different phenotypes. AD risk remains unaffected by SNPs in the gene encoding the microtubule-associated protein tau (MAPT).
Correlation with early onset and elevated tau proteins
Early onset is correlated with elevated tau proteins in the cerebrospinal fluid (CSF). The gene for tumor necrosis factor (TNF), a moderator of the risk for APOE4 carriers as depicted in Fig. 1, is identified as an independent risk factor for the development of the disease.
Fig. 1
Gene variants.
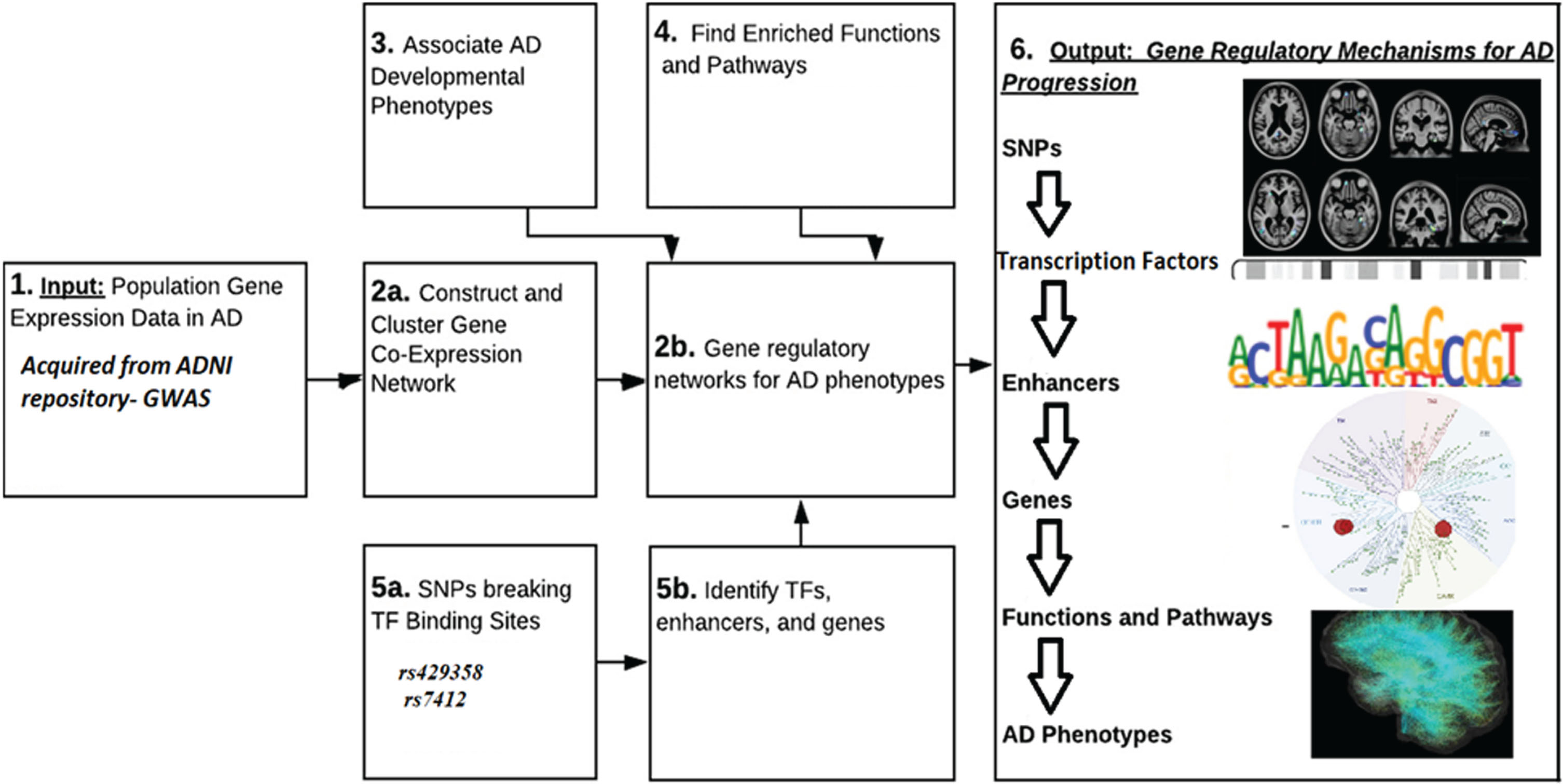
MATERIALS AND METHODS
Genome-wide association studies (GWASs) have found additional gene variants, including CR1, BIN1, CLU, and PICALM, in addition to the APOE gene. These variants are located on human chromosome 19. There are three common gene variants of APOE—ɛ2, ɛ3, and ɛ4—producing various disease phenotypes. These variants are determined by two SNPs observed in every gene expression: rs429358 and rs7412. Rs429358 comprises the common allele morph with T and variant allele morph with C, encoding an amino acid change (Cys130Arg) in exon 4. Rs7412 consists of the common allele morph with C and variant allele morph with T, encoding an amino acid change (Arg176Cys) in exon 4. The majority and increased risk factor are linked to the APOE4 allele, which is independent [5].
In order to develop metabolic pathway AD therapies, it is crucial to comprehend how these Aβ-independent factors lead to the disease. Recent AD genetic wide studies show that the biomarker indicating pathology in the positive spike model reveals APOE2 carriers exist 1 year longer than APOE4 carriers and live 1.2 years less than those with the APOE 3/3 genotype. Further research is required; however, given that APOE4 has been shown to both increase the incidence of AD and accelerate the age at which it manifests, APOE’s possible impacts on neuronal death or chromosomal length may also affect AD risk.
Hazard ratio (HR)
A measure of how much the risk of the event (AD onset) changes for a one-unit change in the gene expression. HR > 1 indicates increased risk, HR < 1 indicates decreased risk [6].
95% CI for HR
The 95% confidence interval for the hazard ratio provides a range of values within which we can be 95% confident that the true hazard ratio lies.
p-value
The p-value assesses the statistical significance of the association. A p-value less than a chosen significance level (e.g., 0.05) suggests a significant association.
Survival curve
Links to Motif classification survival curves or other relevant plots illustrating the survival differences based on gene expression levels.
Risk connected to the APOE4 variation
Each copy of the APOE4 mutation raises the risk of getting AD, and having only one copy increases the risk by nearly two times. The chances are boosted by around 11 times when there are 2 alleles. Many individuals who carry the APOE4 variation never acquire AD [7]. More than half of AD patients have zero copies of chromosome 4. Retaining an APOE4 copy is less important than genetic history. As people age, there are more diagnoses; however, as a person ages, the residual risk goes down. The impact of APOE4 on cultures outside of Europe is not widely known.
Reinforcement learning is made accessible to computational biologists working on genomics problems as well as computational intelligence experts interested in applications in genomics. Software for model construction, model interpretation, and benchmarking DNA sequence simulations are all easily accessible through the DragoNN Collections of classes. A command-line interface enables modeling and interpretation on user-defined data, while web-based lessons utilizing the Jupyter framework offer interactive model editing and visualization for inexperienced users.
Table 1
Risk connected genetic parameters
| Parameter | Description |
| GWAS Variants | CR1, BIN1, CLU, PICALM, and APOE gene variants located on human chromosome 19. |
| APOE Gene Variants | APOE gene has three common variants: ɛ2, ɛ3, and ɛ4. Determined by SNPs rs429358 and rs7412. |
| rs429358 | SNP with common allele morph T and variant allele morph C, encoding amino acid change (Cys130Arg) in exon 4. |
| rs7412 | SNP with common allele morph C and variant allele morph T, encoding amino acid change (Arg176Cys) in exon 4. |
| APOE4 Allele | APOE4 allele linked to increased risk of Alzheimer’s disease (AD). Each copy raises risk; having one copy increases risk nearly two times, and having two alleles increases risk by around 11 times. |
| AD Biomarker Study | APOE2 carriers live 1 year longer than APOE4 carriers and 1.2 years less than APOE3/3 carriers. |
| APOE4 and AD Risk | APOE4 increases incidence of AD and accelerates age at manifestation. Impact on neuronal death and chromosomal length may affect AD risk. |
| Genetic History | APOE4 carriers may not necessarily develop AD. More than half of AD patients have zero copies of APOE4. Retaining an APOE4 copy is less important than genetic history. |
| Age and AD Risk | Risk increases with age, but residual risk decreases as a person ages. The impact of APOE4 on cultures outside of Europe is not widely known. |
| Reinforcement Learning | Made accessible to computational biologists and computational intelligence experts for genomics applications. DragoNN Collections provide tools for model construction, interpretation, and DNA sequence simulations. |
| Software Tools | DragoNN Collections include a command-line interface for modeling and interpretation, as well as web-based lessons with Jupyter framework for interactive model editing and visualization. |
| APOE Isoform Effects | Discussion covers APOE isoform independent effects on cholesterol metabolism, glucose metabolism, mitochondrial functions, cerebrovascular system, and inflammation, all implicated in AD etiology and cognitive functions. |
| Sex-Dependent Effects | Unclear if APOE isoforms affect specific processes due to irreversible linkage. Changed metabolic pathways may contribute to sex-dependent differences in APOE4-related AD risk, as sex significantly affects metabolic homeostasis. |
| AD Pathways Impact | APOE isoforms impact cholesterol metabolism, glucose metabolism, mitochondrial functions, cerebrovascular system, and inflammation. Risk factors also affect Aβ production/degradation and Aβ-independent AD pathways. |
| Molecular Activities | γ-Secretase activities increase Aβ generation by raising neuronal cholesterol. Increased insulin levels hinder Aβ clearance in type 2 diabetes. Abnormalities in cerebrovascular cells aggravate Aβ deposition in the brain. |
| Immunological Impact | Glial cell-mediated Aβ clearance and AβPP processing are impacted by immunological responses through inflammasomes and cytokines, playing a crucial role in Aβ deposition. |
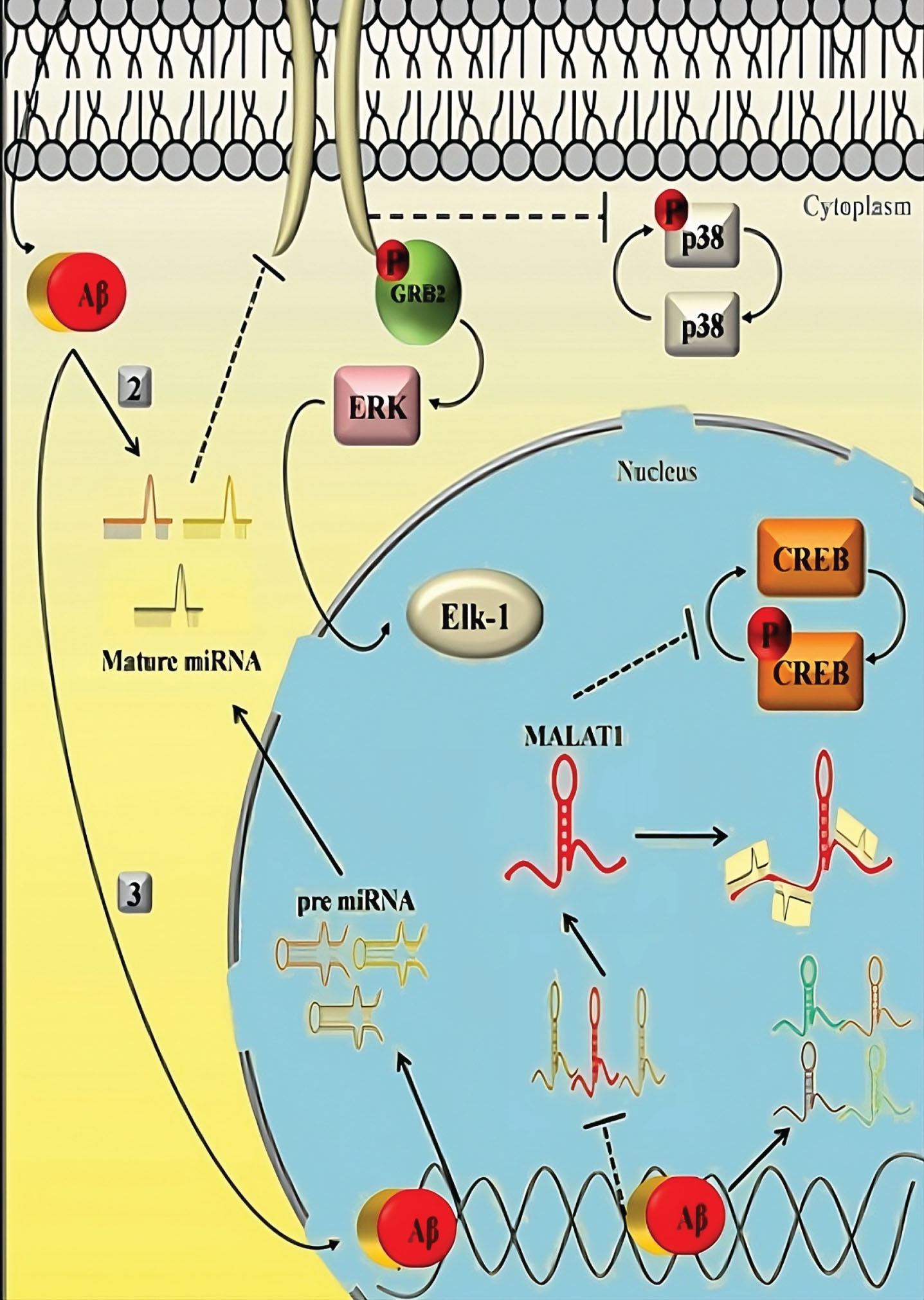
The discussion has covered APOE isoform independent effects on cholesterol metabolism, glucose metabolism, mitochondrial functions, cerebrovascular system, and inflammation, which are significant pathways as shown in Fig. 2, implicated in the etiology of AD and cognitive functions. It is still unclear if APOE isoforms affect any particular processes because of their irreversible linkage. The changed metabolic pathways may potentially be the cause of the sex-dependent differences in APOE4-related risk of developing AD, as sex significantly affects metabolic homeostasis. Most intriguingly, these risk factors have a significant impact on Aβ production and/or degradation and Aβ-independent AD pathways (Fig. 2).
Fig. 2
Aβ-independent AD pathways.
For instance, higher β- and γ-secretase activities speed up the generation of Aβ by increasing neuronal cholesterol. Increased levels of insulin hinder Aβ clearance in type 2 diabetes by competing with Aβ for an important Aβ-degrading enzyme called insulin-degrading enzyme. Since the cerebrovascular system is essential for regulating brain Aβ clearance, abnormalities of cerebrovascular cells, particularly vascular mural cells, lead to an aggravation of Aβ deposition as senile plaques in the brain parenchyma and cerebral amyloid. Antipathy is observed with the cerebral vascular system. Additionally, glial cell-mediated Aβ clearance and AβPP processing are both significantly impacted by immunological responses via inflammasomes and cytokines, which play a crucial role in Aβ deposition.
RESULTS
Pathogenic pathways for AD are distinct for each APOE isoform, involving both Aβ-dependent and independent processes [8]. It is crucial to note that abnormalities in these metabolic pathways worsen Aβ buildup, potentially setting off a vicious cycle in AD, as increased Aβ likely disrupts these pathways in turn. Epidemiological research indicates that factors such as age, sex, and lifestyle (including rest, fitness, and education) have a significant impact on APOE-related AD etiology. APOE probably increases the risk of AD through these intricate predictions and experiments in an isoform-dependent manner (E4 > E3 > E2).
In contrast to “simple” negative sets, training the models genome-wide by segmenting the genome into small (200 bp), overlapping (stride = 50) segments (Fig. 3) leads to improved generalization performance on the test set. For example, it is found that training on the entire genome performed better on test sets for the SPI1 and CTCF tasks than using shuffled reference negatives.
Fig. 3
Motif scores of Alzheimer’s disease associated genes.
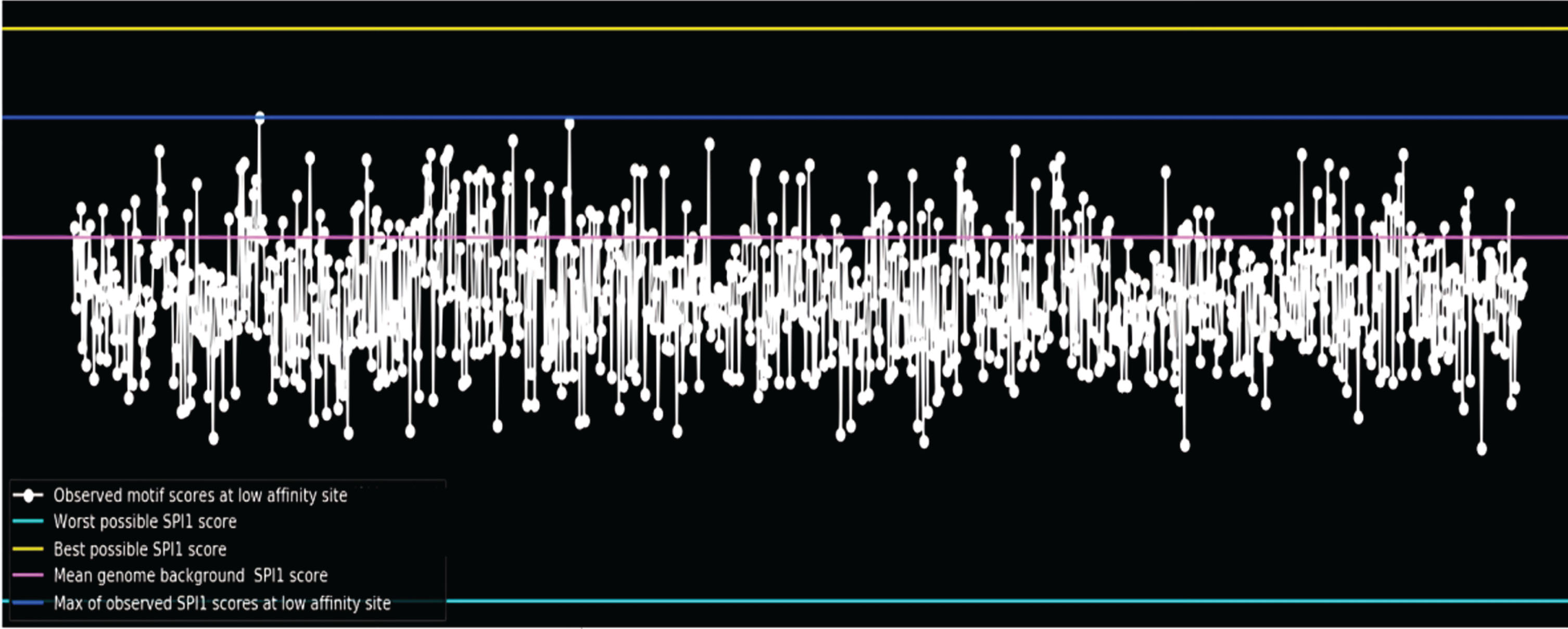
To aid model training, it is required to up-sample positive cases in each batch due to the large class imbalance in in vivo data [9]. The model correctly learned the motif in the CTCF and SPI1 (Fig. 4) datasets, according to interpretation with DeepLIFT, which shows that up-sampling positives to make up 30% of each batch worked effectively. We ensure whether the model still able to learn well at up-sample ratio = 0.1? Upsample ratio = 0.5: Does it Improve Learning? Similar tasks, such as CTCF/ZNF143/SIX5, can perform better when performed simultaneously. However, multitasking does not enhance performance on various jobs (i.e., the SPI1 motif does not resemble the other three motifs, so multitasking does not improve performance for the SPI1 task).
Fig. 4
Gene regulatory mechanism.
In a Deep Regulatory CNN (DRCNN) model, a locally connected linear unit may constitute a Position-Specific Scoring Matrix (PSM) for the GWAS studies obtained from ADNI. The PSM across the sequence is multiplied, the PSM scores are under threshold, and the maximum value is taken to create the weighted sequence of PSM range [10].
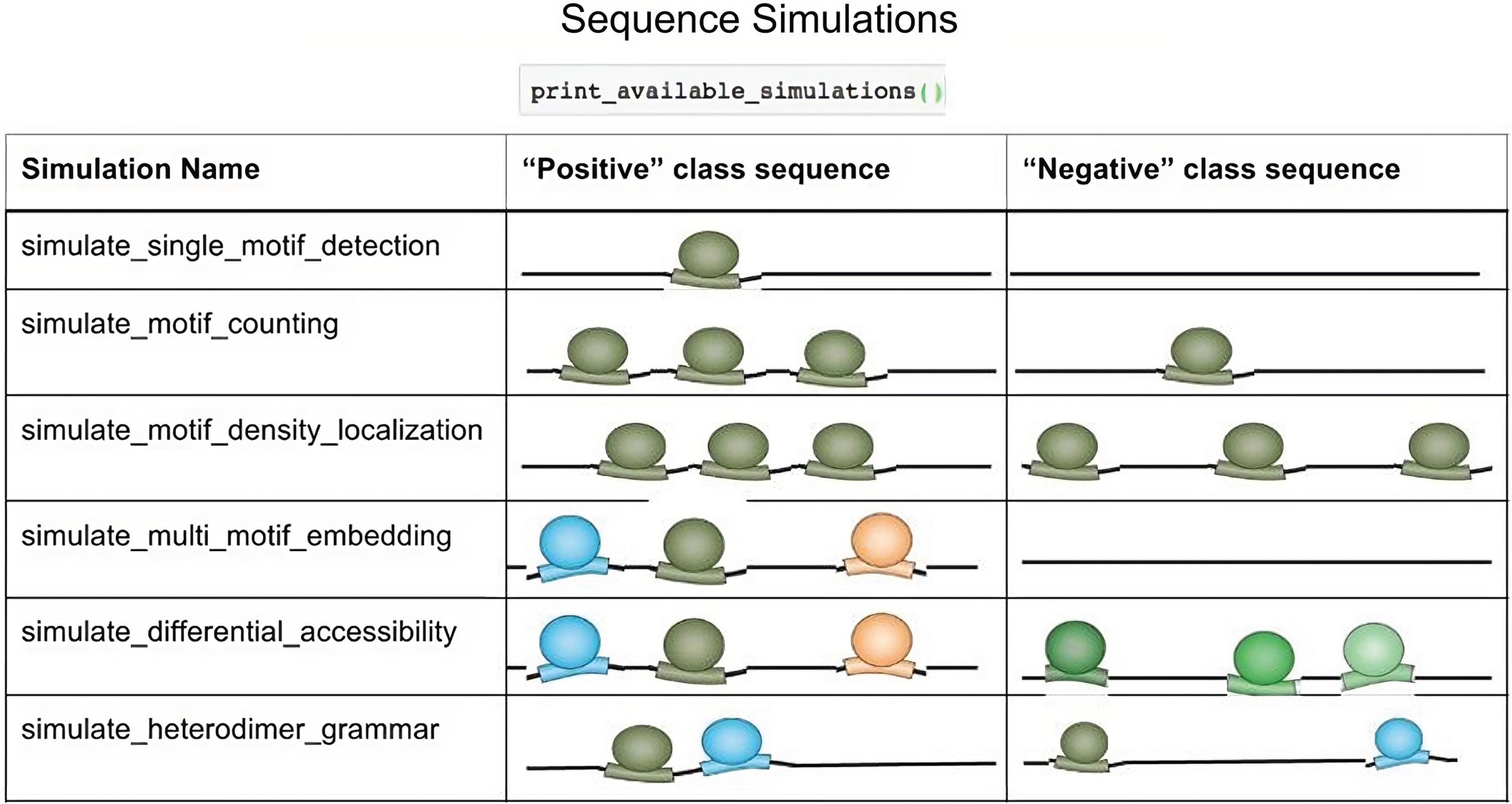
The Deep Genome Regulatory Neural Network constitutes a CNN model, which is a collection of locally connected linear modules performing the functions of convolution. At each layer, a convolutional filter is applied, evaluating the PSM scores followed by ReLU thresholding and max-pooling. To create and train the DRCNN model, the TensorFlow library is combined at the backend with Keras and include deep learning modules. DRCNN may visualize a variety of genome sequence features (Fig. 5) in a compositional manner by using many convolutional layers and filters [11]. We simulate a set of significant sequences with several motif occurrences in the middle and a set of non-significant sequences with numerous motif occurrences dispersed throughout the sequence. The parameters for the TAL1 gene expression motif density localization is defined for a sequence around 1,000 bp long. They include a 1/4 GC fraction and 2–5 instances of the motif in the middle 150 bp of the sequence for deriving significant regions. A total of 3,000 significant and 3,000 non-significant sequences are simulated.
Fig. 5
Significant sequence simulations.
DISCUSSION
The basic DragoNN model comprises of one convolutional layer with 15 convolutional filters and max-pooling of around 40 width units [12]. The model’s inputs are 1500-character input motif regulatory regions and a 15-character filter. On the input profile, neurons serve as informal alignments of data. The model scans the whole input stream in search of a certain pattern represented by the filter’s weights. Convolution filter weights around 15 specify the dimension of the filter weights. The maximum value of a significant motif in sliding windows of size 40 is calculated using the maximum pool of width. The pooling layer is included because TF motifs often only occur in a small number of positions in DNA sequences. Using the pooling layer, we may scale down the size of the resulting sequence by applying the collection of GWAS datasets. Thus, training the DRCNN for about 200 self-iterations with pre-defined stopping conditions for every loss in validation data, and the performance is not showing improvement in every three iterations. The model runs through the training data completely for each epoch and modifies its weights on each gene motif to minimize the loss, which measures the performance and error in each DRCNN layer. The model’s performance measurements on the validation data were saved after each epoch [13].
In the significant regions example, the motif scan produces a cluster of three high-scoring motif alignment positions at a predetermined distance from the gene expression’s center region. In the non-significant region, the spacing between the high-scoring motif alignments is random. If the randomly spaced motifs happen to have a spacing close to the significant example, we practice to provide another index value to select a different region. Only by visualizing the significant sequence, motifs are not entirely predicted and evaluated.
Transcription factors (TF) might be able to bind to numerous comparable but distinct sequences. Some of the motif’s bases could be more significant than others. The preference the TF has for each potential base at each position inside the motif is frequently represented as a Position-Specific Scoring Matrix. That, however, presumes that each place inside the theme is autonomous, which is not necessarily the case. Even the length of a motif might change at times. Even the DNA on each side of the motif can affect binding; the bases within the motif are predominantly responsible for it. Other DNA characteristics may also be significant. The physical environment affects numerous TFs [14]. The TF is affected and decided by several factors, including the physical composition of the DNA and how firmly the double helix is twisted. Methylation of the DNA may also affect TF binding. It is studied that the majority of the DNA in eukaryotes is tightly coiled around histones. Only the unwinding sections are accessible to TFs.
Alternative dependencies include monitoring the other binding components, which also perform crucial functions [15]. TFs frequently occupy interactions with each single binding component, which can have an impact on gene regulations and expression. For instance, a TF may build a complex with another component by binding to it, and that complex may subsequently bind to a different DNA motif than the TF alone.
Conclusion
TF binding is a highly unpredictable process, and the DNA language is as complex as natural language processing. Therefore, a DRCNN solution would be appropriate for predicting significant gene expression in AD using only the sequence data, immediately building a model for TF binding. Thus, the work implemented the prediction of significant motifs that increase the vulnerability of cognitive neurodegenerative disease stages. The production and storage of Aβ peptides in the brain are fundamental processes that are the growing evidence in the development of ‘Aβ-dependent pathways’ in the disease progression. The DRCNN model uses convolutional filters and locally connected linear units to provide localized pattern recognition, making it possible to identify complex DNA motifs. The model incorporates adaptive parameters for motif density localization and offers a thorough display of genomic characteristics, enhanced by PSM. The model learns more via the use of simulated sequences, and it is compatible with TensorFlow and Keras, which makes training it more effective. Nevertheless, drawbacks include motif scanning in important regions being static, dependence on a single convolutional layer, and possible sensitivity to motif width assumptions. Challenges include sensitivity to starting and halting conditions and presumptions about autonomous positions in the PSM.
AUTHOR CONTRIBUTIONS
Rohini M (Conceptualization; Data curation; Investigation; Methodology, Writing - Original draft); Oswalt Manoj S (Formal analysis; Validation); Surendran D (Writing – review & editing).
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are openly available in https://adni.loni.usc.edu/data-samples/data-types/genetic-data/.
REFERENCES
[1] | Dubois B , Feldman HH , Jacova C , Hampel H , Molinuevo JL , Blennow K , DeKosky ST , Gauthier S , Selkoe D , Bateman R , Cappa S ((2014) ) Advancing research diagnostic criteria for Alzheimer’s disease: The IWG-2 criteria. Lancet Neurol 13: , 614–629. |
[2] | Ha J , Park S ((2022) ) NCMD: Node2vec-based neural collaborative filtering for predicting miRNA-disease association. IEEE/ACM Trans Comput Biol Bioinform 20: , 1257–1268. |
[3] | Ha J ((2022) ) MDMF: Predicting miRNA–disease association based on matrix factorization with disease similarity constraint. J Pers Med 12: , 885. |
[4] | Ha J , Park C , Park C , Park S ((2020) ) Improved prediction of miRNA-disease associations based on matrix completion with network regularization. Cells 9: , 881. |
[5] | Jeong S ((2017) ) Molecular and cellular basis of neurodegeneration in Alzheimer’s disease. Mol Cells 40: , 613. |
[6] | Ha J ((2023) ), SMAP: Similarity-based matrix factorization framework for inferring miRNA-disease association. Knowl Based Syst 263: , 110295. |
[7] | Drenos F , Kirkwood TB ((2010) ) Selection on alleles affecting human longevity and late-life disease: The example of apolipoprotein E. PLoS One 5: , e10022. |
[8] | Corder EH , Saunders AM , Strittmatter WJ , Schmechel DE , Gaskell PC , Small G , Roses AD , Haines JL , Pericak-Vance MA ((1993) ) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261: , 921–923. |
[9] | Grimm MO , Zimmer VC , Lehmann J , Grimm HS , Hartmann T ((2013) ) The impact of cholesterol, DHA, and sphingolipids on Alzheimer’s disease. Biomed Res Int 2013: , 814390. |
[10] | Huang K , Lin Y , Yang L , Wang Y , Cai S , Pang L , Wu X , Huang L , Alzheimer’s Disease Neuroimaging Initiative ((2020) ) A multipredictor model to predict the conversion of mild cognitive impairment to Alzheimer’s disease by using a predictive nomogram. Neuropsychopharmacology 45: , 358–366. |
[11] | Huddar MG , Sannakki SS , Rajpurohit VS ((2020) ) Attention-based word-level contextual feature extraction and cross-modality fusion for sentiment analysis and emotion classification. Int J Intell Eng Inform 8: , 1–8. |
[12] | Jha D , Alam S , Pyun JY , Lee KH , Kwon GR ((2018) ) Alzheimer’s disease detection using extreme learning machine, complex dual tree wavelet principal coefficients and linear discriminant analysis. J Med Imaging Health Inform 8: , 881–890. |
[13] | Jo T , Nho K , Saykin AJ ((2019) ) Deep learning in Alzheimer’s disease: Diagnostic classification and prognostic prediction using neuroimaging data. Front Aging Neurosci 11: , 220. |
[14] | Kaur S , Chahal KK ((2020) ) Hybrid ANFIS-genetic algorithm based forecasting model for predicting cholera-waterborne disease. Int J Intell Eng Inform 8: , 374–393. |
[15] | Morris JC , Storandt M , Miller JP , McKeel DW , Price JL , Rubin EH , Berg L ((2001) ) Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol 58: , 397–405. |


