
Agent Orange Causes Metabolic Dysfunction and Molecular Pathology Reminiscent of Alzheimer’s Disease
Abstract
Background:
Agent Orange, an herbicide used during the Vietnam War, contains 2,4-dichlorophenoxyacetic acid (2,4-D) and 2,4,5-trichlorophenoxyacetic acid (2,4,5-T). Agent Orange has teratogenic and carcinogenic effects, and population-based studies suggest Agent Orange exposures lead to higher rates of toxic and degenerative pathologies in the peripheral and central nervous system (CNS).
Objective:
This study examines the potential contribution of Agent Orange exposures to neurodegeneration.
Methods:
Human CNS-derived neuroepithelial cells (PNET2) treated with 2,4-D and 2,4,5-T were evaluated for viability, mitochondrial function, and Alzheimer’s disease (AD)-related proteins.
Results:
Treatment with 250μg/ml 2,4-D or 2,4,5-T significantly impaired mitochondrial function, caused degenerative morphological changes, and reduced viability in PNET2 cells. Correspondingly, glyceraldehyde-3-phosphate dehydrogenase expression which is insulin-regulated and marks the integrity of carbohydrate metabolism, was significantly inhibited while 4-hydroxy-2-nonenal, a marker of lipid peroxidation, was increased. Tau neuronal cytoskeletal protein was significantly reduced by 2,4,5-T, and relative tau phosphorylation was progressively elevated by 2,4,5-T followed by 2,4-D treatment relative to control. Amyloid-β protein precursor (AβPP) was increased by 2,4,5-T and 2,4-D, and 2,4,5-T caused a statistical trend (0.05 < p<0.10) increase in Aβ. Finally, altered cholinergic function due to 2,4,5-T and 2,4-D exposures was marked by significantly increased choline acetyltransferase and decreased acetylcholinesterase expression, corresponding with responses in early-stage AD.
Conclusion:
Exposures to Agent Orange herbicidal chemicals rapidly damage CNS neurons, initiating a path toward AD-type neurodegeneration. Additional research is needed to understand the permanency of these neuropathologic processes and the added risks of developing AD in Agent Orange-exposed aging Vietnam Veterans.
INTRODUCTION
Agent Orange, a potent synthetic herbicide, was widely used to defoliate enemy territory during the Vietnam War between 1965 and 1970 [1]. Exposures were highest in Operation Ranch Hand military personnel who carried out much of the spraying from aircraft and in ground troops stationed within 0.5 km of treated zones [1]. Years later, increased rates of severe birth defects, including spina bifida, limb deformities, sensory impairments, and developmental disabilities were observed in the offspring of otherwise young, healthy Vietnamese women who resided in regions that had been sprayed with Agent Orange [2, 3]. Subsequent studies demonstrated higher rates of newly diagnosed carcinomas and soft-tissue sarcomas, as well as cardiovascular disease in military personnel known to have been exposed to Agent Orange [4, 5]. These population-based studies drew attention to the long-term unintended adverse effects of Agent Orange exposures on human health across the lifespan.
Agent Orange is chiefly composed of a 1 : 1 ratio of 2,4-dichlorophenoxyacetic acid (2,4-D) and 2,4,5-trichlorophenoxyacetic acid (2,4,5-T) [1]. However, considerable attention has been paid to the potential role of 2,3,7,8-tetrachloro-p-dioxin (TCDD) as a mediator of human disease because TCDD is highly toxic, and trace quantities contaminated 2,4,5-T during its production [1]. In some respects, that concept may have detracted from larger questions about the teratogenic and mutagenic effects of the herbicide toxins deliberately incorporated into Agent Orange. Given what is already known about the toxic effects of Agent Orange on plant life, a reasonable hypothesis is that its constituents also contribute to mammalian cellular injury and disease. Furthermore, the higher rates of malignancies, cardiovascular diseases, and diabetes mellitus [5–7], years after the exposures suggest that Agent Orange may have indirect or “second-hit” type long-term effects that exacerbate the risk of aging-associated diseases.
In a recent review article, potential links between Agent Orange exposures and the later development of peripheral nervous system (PNS) or central nervous system (CNS) degenerative diseases were discussed [8]. Moreover, in a cohort analysis of Vietnam War veterans included in the Veterans Health Administration database, significantly higher rates and earlier onsets of dementia were correlated with presumed Agent Orange exposures [9]. Since degenerative diseases of the PNS and CNS are often aging-associated and many of them have been linked to Type 2 diabetes mellitus and insulin resistance [10–12], it is of interest to expand our understanding of how Agent Orange exerts its toxic and degenerative effects on human nervous system cells [13–16]. In this regard, previous studies showed that 2,4-D had lipotoxic effects on gangliosides, myelin, and Schwann cells [17–21]. Of note is that Schwann cells generate and maintain myelin, and myelin is needed for efficient neural conductivity and function.
Although dioxins are no longer produced in the United States, and the manufacture of polychlorinated biphenyl-containing products, some of which generate dioxins, was banned by the Environmental Protection Agency in 1979, dioxins are found throughout the world as contaminants in soil, foods including high-fat dairy products, meat, fish, eggs, and shellfish, and at low levels in plants, water and air [22]. Worse yet, Agent Orange pollutants remain in the environment for decades [23]. Besides the increased risk of generating TCDD with the manufacture of 2,4-D and 2,4,5-T, compelling evidence for the harmful effects of 2,4,5-T led to its discontinued registered use in the United States, although information about its global usage remains scant. In contrast, human exposures to 2,4-D broadly impact global populations due to its widespread inclusion in herbicide and pesticide products, including in the United States. To begin addressing questions about neurotoxic and degenerative responses to Agent Orange, we examined the effects of 2,4-D and 2,4,5-T on mitochondrial function, viability, stress, and markers of neurometabolic dysfunction in human PNET2 cells using an established in vitro model. The rationale was that significant impairments related to these indices would provide evidence that Agent Orange exposures establish a deleterious path toward progressive neurodegeneration, including possibly Alzheimer’s disease (AD).
METHODS
Materials
Dulbecco’s Modified Eagle Medium (DMEM), Amplex UltraRed, and 4-methylumbelliferyl phosphate (4-MUP) were obtained from Invitrogen (Carlsbad, CA, USA). The CYQUANT MTT Cell Viability Assay, which is based on the cellular conversion of 3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide (MTT) to an insoluble formazan product, Hoechst (H33342) dye, and Nunc Lab Tek 8-well chamber slides were from Thermo Fisher (Cambridge, MA USA). Alkaline Phosphatase Conjugated to Streptavidin was purchased from Vector Laboratories (Burlingame, CA, USA). Details pertaining to the Agent Orange chemicals are listed in Table 1, and antibodies used in immunoassays are listed in Table 2. All other fine chemicals were purchased from Thermo Fisher (Cambridge, MA USA), Sigma-Aldrich (St. Louis, MO, USA), or CalBiochem (Carlsbad, CA, USA).
Table 1
Agent Orange herbicide chemicals used in vitro
| Compound | Source | Catalog # | M.W. | Solvent | Concentration |
| 2,4-Dichloro-phenoxy Acetic Acid (2,4-D) | Santa Cruz Biotechnologies, Dallas, TX USA | sc-205097 | 221.04 | DMSO | 25–250μg/ml |
| 2,4,5-Trichloro-phenoxyacetic acid (2,4,5-T) | Santa Cruz Biotechnologies, Dallas, TX USA | sc-209335 | 255.48 | DMSO | 25–250μg/ml |
Vehicle control cultures were treated with DMSO.
Table 2
Antibodies used for duplex ELISA studies
| Antibody | Source | Company | Type | Concentration/Dilution | RRID# |
| Glucose-6-Phosphate Dehydrogenase (GAPDH) | Mouse | Santa Cruz, Dallas, TX | Monoclonal | 0.2μg/ml | AB_10847862 |
| Tau | Rabbit | Agilent/Dako, Santa Clara, CA | Polyclonal | 6.2μg/ml | AB_10013724 |
| Phospho-Tau (pTau; pT205-Tau) | Rabbit | Abcam, Waltham, MA | Polyclonal | 0.8μg/ml | AB_304676 |
| Amyloid Precursor Protein (APP) | Rabbit | Cell Signaling, Danvers, MA | Polyclonal | 0.246μg/ml | AB_10694227 |
| Amyloid Precursor Protein-Amyloid Beta (APP-Aβ) | Mouse | Novacastra (LSBio) | Monoclonal | 1 : 250 | LS-C651578-MSS10 |
| Choline Acetyltransferase (ChAT) | Rabbit | Abcam, Waltham, MA | Polyclonal | 1 : 3000 | AB_2244866 |
| Acetylcholinesterase (AChE) | Mouse | Abcam, Waltham, MA | Monoclonal | 0.25μg/ml | AB_303316 |
| 8-Hydroxydeoxyguanosine (8-OHdG) | Mouse | 21st Century Biochemicals, Marlborough, MA | Monoclonal | 0.2μg/ml | [40] |
| 4-Hydroxy-2-nonenal (HNE) | Goat | Abcam, Waltham, MA | Polyclonal | 1.0μg/ml | AB_722493 |
| Large acidic ribosomal protein (RPLPO) | Mouse | Santa Cruz, Dallas TX | Monoclonal | 0.1μg/ml | [39, 41, 42] |
Cell culture model
Human CNS-derived primitive neuroepithelial tumor (PNET2) cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal calf serum, 2 mM glutamine, and 4.5 g/L glucose but without antibiotics. The cells were grown at 37°C in a standard 5% CO2 humidified incubator.
The PNET2 cell line, established in 1993 [24], was found to mimic many properties of immature non-transformed CNS neurons including growth, differentiation and cell motility and adhesion responsiveness to trophic factor stimulation, the expression of complex signal transduction networks, and sensitivity to toxins such as alcohol, oxidative stress, and mediators of neurodegeneration and neurotoxicity [25–30]. Only limited passage pools of PNET2 cells, i.e., 10 or fewer, were used in experiments. In contrast, the use of non-CNS cells including SH-Sy5y and PC12, have not reliably yielded data that could guide subsequent in vivo brain experiments.
Fresh overnight sub-confluent cultures were trypsinized and re-seeded into 96-well Nunc flat-bottom plates at a density of 1.4 x 104 viable cells/well, or 6-well plates at a density of 2.1 x 105 viable cells/well. Cell viability was assessed by Trypan blue exclusion. After 24 h, 16 replicate 96-well cultures were treated with 25μg/ml or 250μg/ml of 2,4-D or 2,4,5-T (Table 1) Agent Orange herbicide chemicals for 48 h. For the 6-well plates, six replicate cultures were treated with 250μg/ml of 2,4-D or 2,4,5-T for 48 h. Since 2,4-D and 2,4,5-T were solubilized in dimethyl sulfoxide (DMSO) and further diluted with serum-free DMEM, vehicle control cultures studied in parallel, were treated with the same content of DMSO diluted in DMEM.
Initial dose-effect studies to select the 2,4-D or 2,4,5-T concentrations for use in our experiments were guided by previous reports in which the effects of these herbicides were evaluated in vitro with a range of cell types [21, 31–33], but probably higher than the concentrations used to spray the herbicides [34]. The doses ultimately selected could not be compared with historical human exposures via Agent Orange, particularly since most of the prior research focused on TCDD. However, the doses employed in our experiments were considerably below the 100 or 200 mg/kg daily in vivo rodent administrations reported previously [18, 19].
Mitochondrial and viability studies
The MTT colorimetric assay was used to measure the effects of 2,4-D and 2,4,5-T treatments on mitochondrial function [35, 36]. Absorbances were measured at 540 nm in a SpectraMax M5 Microplate Reader (Molecular Dynamics, Sunnyvale, CA, USA). After discarding the supernatants and rinsing the wells with phosphate-buffered saline (PBS), the cells were stained with Hoechst H33342 (10μg/ml in PBS) (Thermo Fisher Scientific, Cambridge, MA) for 15 min protected from ambient light with aluminum foil. Fluorescence (Ex360 nm/Em460 nm) intensity corresponding to nuclear staining was measured in a SpectraMax M5 Microplate Reader. Although H33342 labels live and dead cells, the PBS rinses dislodge and remove unfixed non-viable cells, preventing their inclusion in this component of the assay. Using this approach, H33342 fluorescence intensity increases linearly with viable cell culture density in 96-well plates under non-saturated conditions [37]. The calculated MTT/H33342 ratios were used as indices of relative mitochondrial activity/cell viability. These assays were performed in replicates of 16 and repeated twice. and Hoechst H33342 fluorescence was used as an index of cell number/viability.
Protein extraction
Immunoreactivity was measured in protein homogenates to assess the effects of 2,4-D and 2,4,5-T treatments on PNET2 cell expression of standard molecular biomarkers associated with AD neurodegeneration including tau, phosphorylated tau (pT205-Tau), amyloid-β protein precursor (AβPP), amyloid beta fragment of AβPP (APP-Aβ), choline acetyltransferase (ChAT), and acetylcholinesterase (AChE). In addition, 8-hydroxydeoxyguanosine (8-OHdG) and 4-Hydroxy-2-nonenal (HNE) were measured as indices of nucleic acid damage and lipid peroxidation, respectively. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH), an insulin-responsive enzyme [38], served as a marker of metabolic integrity. Large acidic ribosomal protein (RPLPO) served as an internal loading control [39–42]. The antibody descriptions, sources, concentrations, and validation references are provided in Table 2.
Duplex ELISAs
We used duplex ELISAs to measure PNET2 cellular immunoreactivity such that the levels of specific protein expression were normalized to RPLPO measured in the same samples as previously described [43, 44]. In brief, duplex ELISAs were performed in quadruplicate with 50μl pre-diluted protein homogenate aliquots containing 50 ng of protein adsorbed to the bottom flat surfaces of 96-well MaxiSorp plates. Adsorptions were performed overnight at 4°C. After briefly rinsing in phosphate-buffered saline (PBS), pH 7.35, non-specific sites were blocked by a 2-h incubation with Superblock-TBS Blocking Buffer at room temperature and with gentle platform agitation. Adherent, blocked protein samples were then reacted with primary antibodies (0.2–5.0μg/ml) by overnight incubation at 4°C. Immunoreactivity was detected with horseradish peroxidase (HRP)-conjugate secondary antibody and the soluble Amplex UltraRed fluorophore. A SpectraMax M5 microplate reader (Molecular Dynamics, USA) was used to measure fluorescence intensity (Ex530 nm/Em590 nm). Then, RPLPO immunoreactivity, which served as a sample loading control, was measured by incubating the proteins with biotinylated anti-RPLPO followed by streptavidin-conjugated alkaline phosphatase and 4-MUP (Ex360 nm/Em450 nm). The calculated ratios of the target protein to RPLPO fluorescence were used for inter-group statistical comparisons. Six replicate cultures were analyzed per group. PBS or TBS rinses were used between procedural steps. All assays were repeated twice.
Statistical analysis
Inter-group comparisons were made by one-way analysis of variance (ANOVA) with Tukey post hoc multiple comparisons tests (GraphPad Prism 9.4, San Diego, CA). Results were graphed using scatter plots with mean and standard deviation bars, or box plots depicting the means (horizontal bars), 95% Confidence interval limits (upper and lower borders of the boxes), and ranges (upper and lower stems). Significant (p < 0.05) and trend-wise (0.05 < p<0.10) post hoc test differences are shown in the tables and graphs.
RESULTS
Effects of 2,4-D on neuronal mitochondrial function and viability (Table 3 and Fig. 1)
At the 25μg/ml concentration, 2,4-D produced modest increases in mean MTT activity. However, at the 250μg/ml dose, 2,4-D sharply reduced MTT activity relative to control and the 25μg/ml-treated cultures (Fig. 1A). One-way ANOVA demonstrated significant inter-group differences (Table 3A), and the post hoc Tukey test showed that the mean level of MTT activity was significantly lower in PNET2 cultures treated with the 250μg /ml dose of 2,4-D relative to control and the 25μg/ml 2,4-D-treated cultures (p < 0.0001) (Fig. 1A).
Table 3
ANOVA for effects of 2,3-D on neuronal mitochondrial function and viability
| Assay | F-Ratio | p |
| MTT | 38.38 | <0.0001 |
| Viability | 4.77 | 0.0132 |
Inter-group comparisons were made using one-way ANOVA and post-hoc Tukey tests with correction for multiple comparisons (See Fig. 1). F-Ratios correspond to the measured Agent Orange herbicide 2,4-D toxin effects. NS, not significant. DFn, DFd=2, 45 for all assays.
Fig. 1
CNS neurotoxic effects of 2,4-D. Human PNET2 cells seeded into 96-well plates were treated with 0, 25μg/ml, or 250μg/ml of 2,4-D for 48 h. A) Absorbance for MTT activity (540 nM) reflecting mitochondrial function. B) Hoechst H33342 fluorescence (Ex360/Em460) reflecting cell number/viability. N = 16 culture well replicates per dose. Graphs depict individual sample data with means (horizontal bars) and standard deviations (stems). Significant post hoc multiple comparison Tukey test results are shown. MTT = 3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide.
![CNS neurotoxic effects of 2,4-D. Human PNET2 cells seeded into 96-well plates were treated with 0, 25μg/ml, or 250μg/ml of 2,4-D for 48 h. A) Absorbance for MTT activity (540 nM) reflecting mitochondrial function. B) Hoechst H33342 fluorescence (Ex360/Em460) reflecting cell number/viability. N = 16 culture well replicates per dose. Graphs depict individual sample data with means (horizontal bars) and standard deviations (stems). Significant post hoc multiple comparison Tukey test results are shown. MTT = 3-[4,5-dimethylthiazole-2-yl]-2,5-diphenyltetrazolium bromide.](https://content.iospress.com:443/media/adr/2023/7-1/adr-7-1-adr230046/adr-7-adr230046-g001.jpg)
H33342 fluorescence provided an index of adherent cell number and therefore viability since all cultures were seeded identically and non-adherent dead cells in the unfixed cultures were removed by pre-rinsing with PBS. The mean levels of H33342 fluorescence progressively declined with increasing 2,4-D dose. One-way ANOVA revealed significant inter-group differences in Hoechst H33342 fluorescence (Table 3), and the post hoc Tukey test demonstrated that viability was significantly lower in cultures treated with 250μg/ml of 2,4-D relative to vehicle. Moreover, post hoc linear trend analysis showed that the progressive decline in cell viability with increasing 2,4-D dose was significant (R square = 0.175; p = 0.004) (Fig. 1B).
Effects of 2,4,5-T on neuronal mitochondrial function and viability (Table 3 and Fig. 2)
In PNET2 cells treated with 25μg/ml 2,4,5-T, MTT activity was elevated above the levels measured in both control and the 250μg/ml 2,4,5-T-treated cultures, whose mean level of MTT was comparable to control (Fig. 2A). One-way ANOVA demonstrated significant inter-group differences (F = 31.76; p < 0.0001) (Table 4), and the post hoc Tukey tests showed that the differences in mean MTT activity resulting from 25μg/ml versus 0 or the 250μg/ml 2,4,5-T were statistically significant (both p < 0.0001) (Fig. 2A).
Fig. 2
CNS neurotoxic effects of 2,4,5-T. Human PNET2 CNS cells seeded into 96-well plates were treated with 0, 25μg/ml, or 250μg/ml of 2,4,5-T for 48 h. A) Absorbance for MTT activity (540 nM) reflecting mitochondrial function. B) Hoechst H33342 fluorescence (Ex360/Em460) reflecting viable cell number. N = 16 culture well replicates per treatment group. Graphs depict individual sample data with means (horizontal bars) and standard deviations (stems). Significant post hoc multiple comparison Tukey test results are displayed.
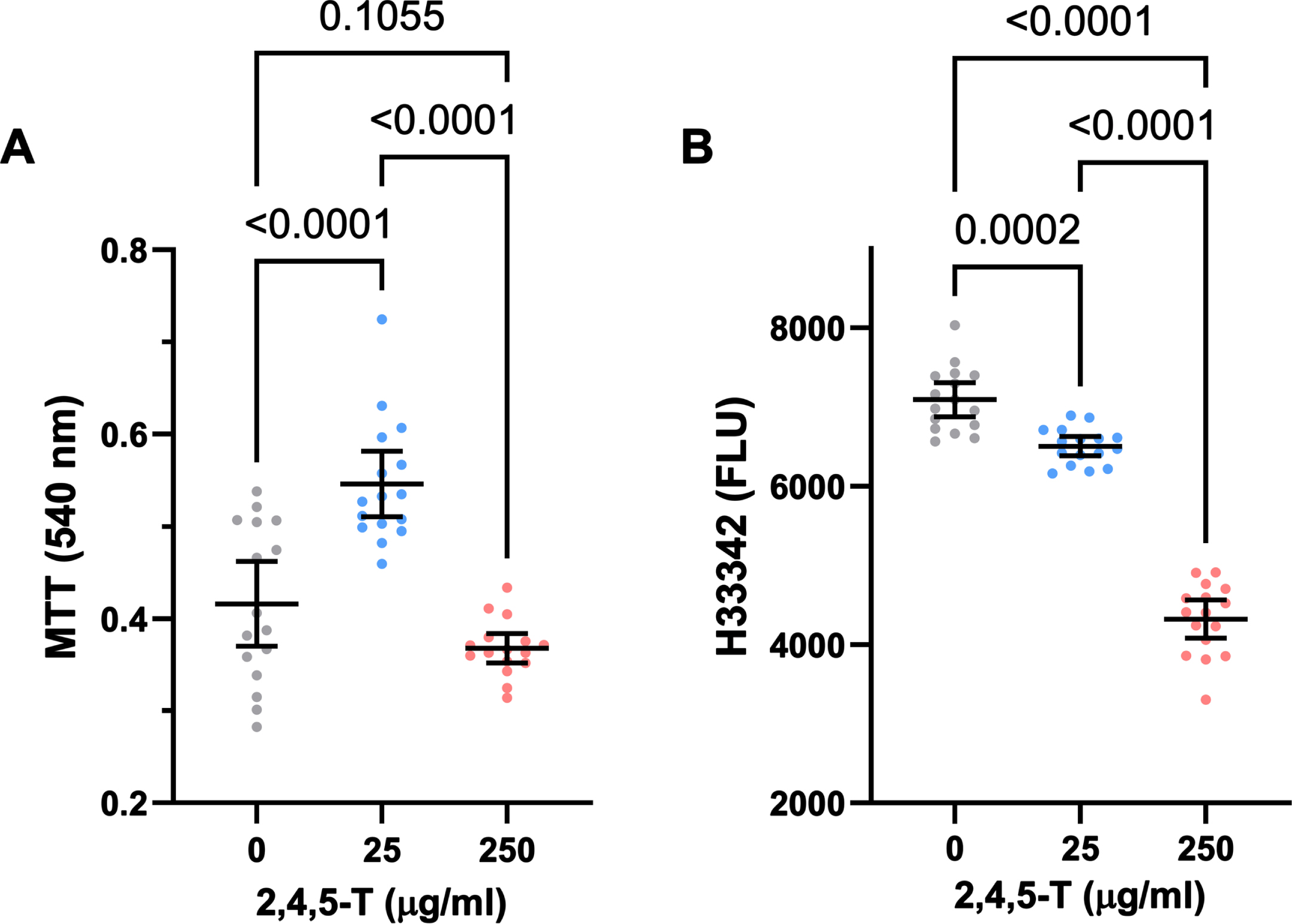
Treatment of PNET2 cultures with 2,4,5-T caused dose-dependent declines in Hoechst H33342 fluorescence (Fig. 2B), reflecting cell loss. While exposure to 25μg/ml of 2,4,5-T led to modest but significant reductions in cell number, treatment with the 250μg/ml dose caused a precipitous decline in PNET2 cell viability. One-way ANOVA demonstrated significant inter-group differences in mean H33342 fluorescence (F = 243.4; p < 0.0001) (Table 4), and post hoc Tukey tests revealed significant reductions in H33342 in cells treated with 25μg/ml of 2,4,5-T relative to control (p < 0.0001) and significantly lower levels of H33342 fluorescence in PNET2 cultures treated with 250μg/ml of 2,4,5-T relative to both control and the 25μg/ml of 2,4,5-T-treated cells (both p < 0.0001). Furthermore, post hoc linear trend analysis showed that the progressive decline in cell viability with increasing 2,4,5-T dose was significant (R square = 0.824; p < 0.0001).
Table 4
ANOVA for effects of 2,4,5-T on neuronal mitochondrial function and viability
| Assay | F-Ratio | p |
| MTT | 31.76 | <0.0001 |
| Viability | 243.4 | <0.0001 |
Inter-group comparisons were made using one-way ANOVA and post-hoc Tukey tests with correction for multiple comparisons (See Fig. 1). F-Ratios correspond to the measured Agent Orange herbicide 2,4-D toxin effects. NS, not significant. DFn, DFd=2, 45 for all assays.
Cytopathological features of 2,4-D and 2,4,5-T exposed PNET2 cells
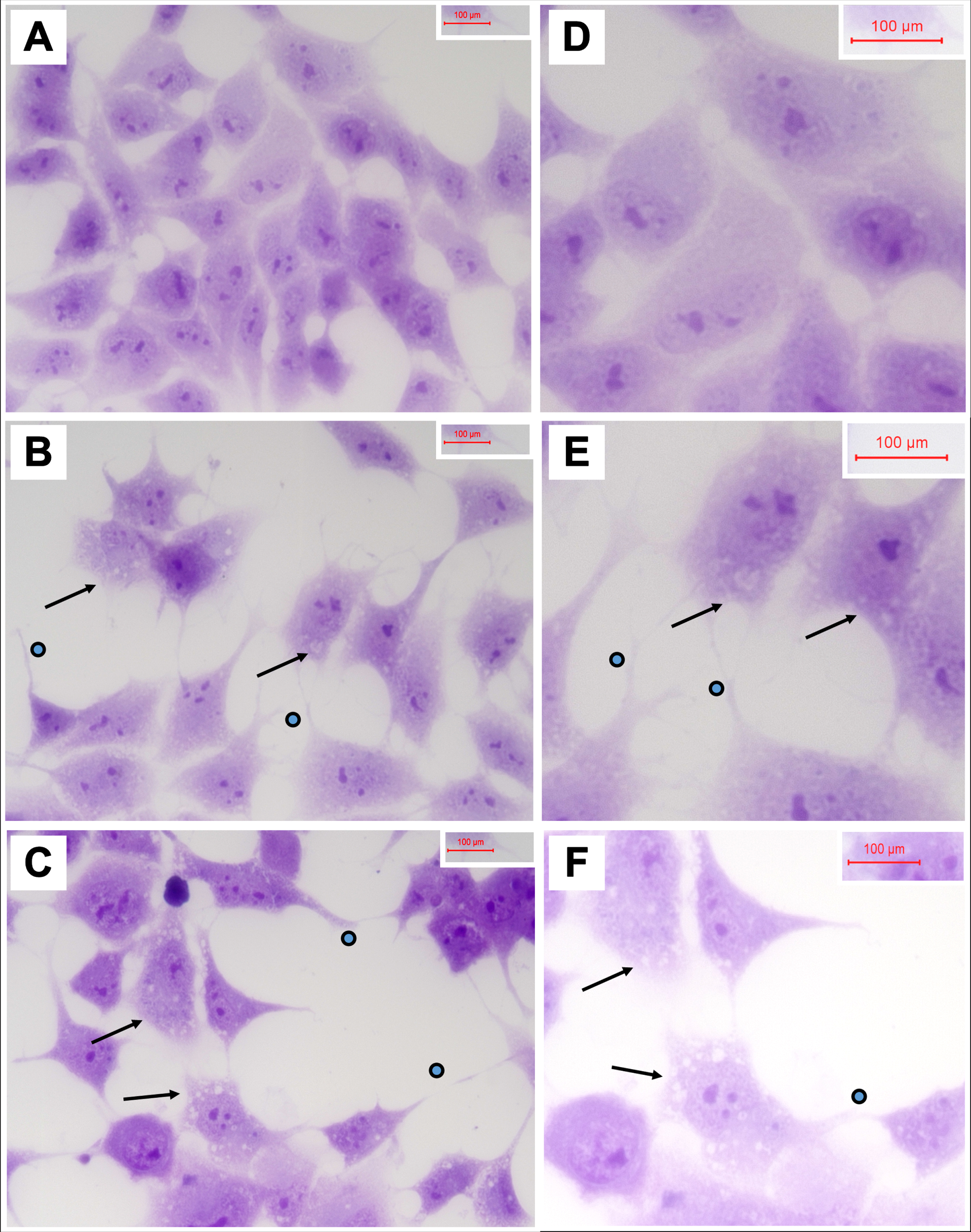
Cells seeded on 8-well chamber slides and treated with vehicle, 250μg/ml 2,4-D, or 250μg/ml 2,4,5-T for 48 h were fixed in 10% neutral buffered formalin, stained with Hematoxylin, and mounted under coverglass for microscopic examination. Relative to vehicle, 2,4-D and 2,4,5-T caused cytoplasmic vacuolation, shrinkage of cell bodies, and conspicuous cell process extension (sprouting) (Fig. 3). In addition, the cell densities were reduced, corresponding with the H33342 results. Cell loss, sprouting and cell body shrinkage appeared more pronounced in the 2,4,5-T than 2,4-D treated cultures.
Fig. 3
Degenerative cytomorphological effects of 2,4-D and 2,4,5-T. PNET2 cells seeded into 8-well chamber slides were treated with A, D) vehicle, B, E) 250μg/ml 2,4-D, or C, F) 250μg/ml 2,4,5-T for 48 h. Cells were stained with Hematoxylin after 10% formalin fixation. Note lower cell densities, cytoplasmic vacuolations (arrows), and irregular cell process extensions (dots) in the 2,4-D and 2,4,5-T treated cultures. Photographs were taken at A-C) 400x and D-F) 600x. Final image magnification scale bars are displayed.
Neuronal function markers
Duplex ELISAs were used to compare the effects of 250μg/ml 2,4-D and 2,4,5-T on GAPDH, 8-OHdG, HNE, Tau, pTau, AβPP, Aβ, ChAT, and AChE immunoreactivity in human PNET2 cells. The data were analyzed by one-way ANOVA (Table 5). The post hoc Tukey test results (corrected for repeated measures) are depicted graphically in Figs. 4–7.
Table 5
Biomarkers of neurotoxicity and neurodegeneration
| Biomarker | F-Ratio | p |
| GAPDH | 37.12 | <0.0001 |
| 8-hydroxydeoxyguanosine | 0.059 | N.S. |
| 4-hydroxy-2-nonenal | 5.067 | 0.021 |
| Amyloid Precursor Protein | 5.819 | 0.0135 |
| Amyloid Precursor Protein-Aβ | 1.722 | N.S. |
| Tau | 3.517 | 0.056 |
| pTau | 7.642 | 0.005 |
| pTau/Tau | 3.580 | 0.054 |
| Choline acetyltransferase | 8.228 | 0.004 |
| Acetylcholinesterase | 28.14 | <0.0001 |
Biomarker immunoreactivity was measured in PNET2 cell homogenates by Duplex ELISA. were configured to quantify immunoreactivity in target proteins using specific unlabeled monoclonal antibodies coupled with horseradish peroxidase-conjugated secondary antibody and the Amplex UltraRed soluble fluorophore. To correct for modest differences in sample loading, results were normalized to large acidic ribosomal protein (RPLPO) with biotinylated antibody, Streptavidin-conjugated alkaline phosphatase and the 4-Methylumbelliferyl phosphate (4-MUP) substrate. Fluorescence intensities (Amplex Red: Ex560 nm/Em590 nm; 4-MUP: Ex360/Em450) were measured in a SpectraMax M5. Inter-group comparisons were made using one-way ANOVA and post-hoc Tukey tests with correction for multiple comparisons (See Figs. 3-5). F-Ratios correspond to measured Agent Orange herbicide toxin effects. NS, not significant. DFn, DFd=1, 40 for all assays.
Fig. 4
Metabolic and oxidative stress indices modulated by Agent Orange herbicide toxins. Human CNS-derived PNET2 cells were treated for 48 h with Vehicle, 250μg/ml 2,4,5-T, or 250μg/ml of 2,4-D. (A) GAPDH, (B) 8-OHdG, and (C) HNE immunoreactivity were measured by duplex ELISA with results normalized to RPLPO. Each group included 6 replicate cultures. Results were analyzed by one-way ANOVA with post hoc Tukey tests (repeated measures; See Table 5). Significant p-values are indicated within the graphs. 2,4,5-T, 2,4,5-trichlorophenoxyacetic acid; 2,4-D, 2,4-dichlorophenoxyacetic acid; GAPDH, glyceraldehyde 3-phosphate dehydrogenase; 8-OHdG, 8-hydroxydeoxyguanosine; HNE, 4-hydroxy-2- nonenal; RPLPO, Large acidic ribosomal protein.
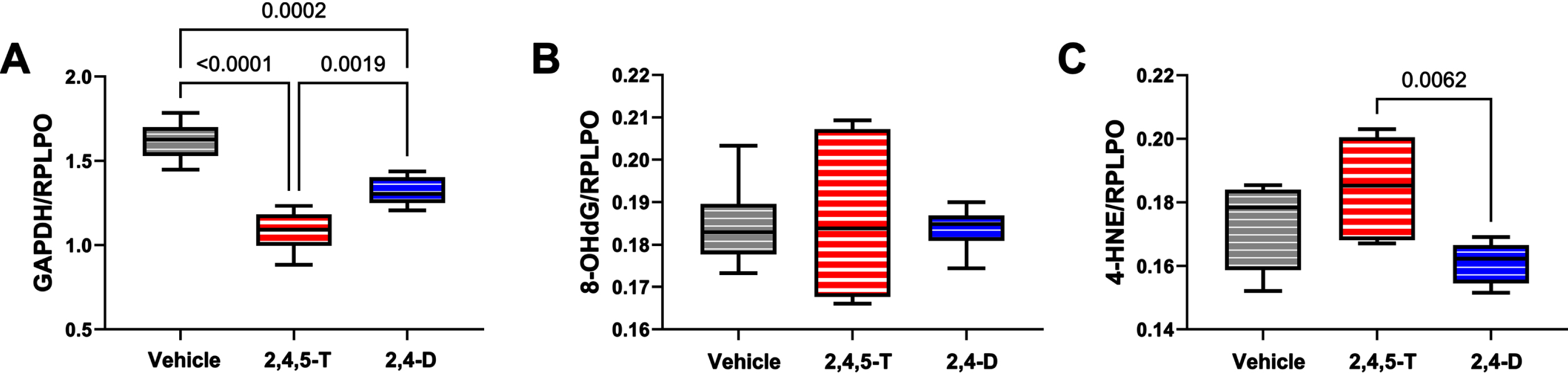
Fig. 5
Effects of Agent Orange herbicidal toxin exposures on AβPP and AβPP-Aβ. Human PNET2 cells treated for 48 h with Vehicle, 250μg/ml 2,4,5-T or 250μg/ml 2,4-D were analyzed for (A) AβPP, (B) Aβ immunoreactivity by duplex ELISA with results normalized to large acidic ribosomal protein (RPLPO). Each group included 6 replicate cultures. Results were analyzed by one-way ANOVA (Table 5). Significant (p < 0.05) and statistical trend (0.05 < p<0.10) results of repeated measures post hoc Tukey tests are shown. 2,4,5-T, 2,4,5-trichlorophenoxyacetic acid; 2,4-D, 2,4-dichlorophenoxyacetic acid.
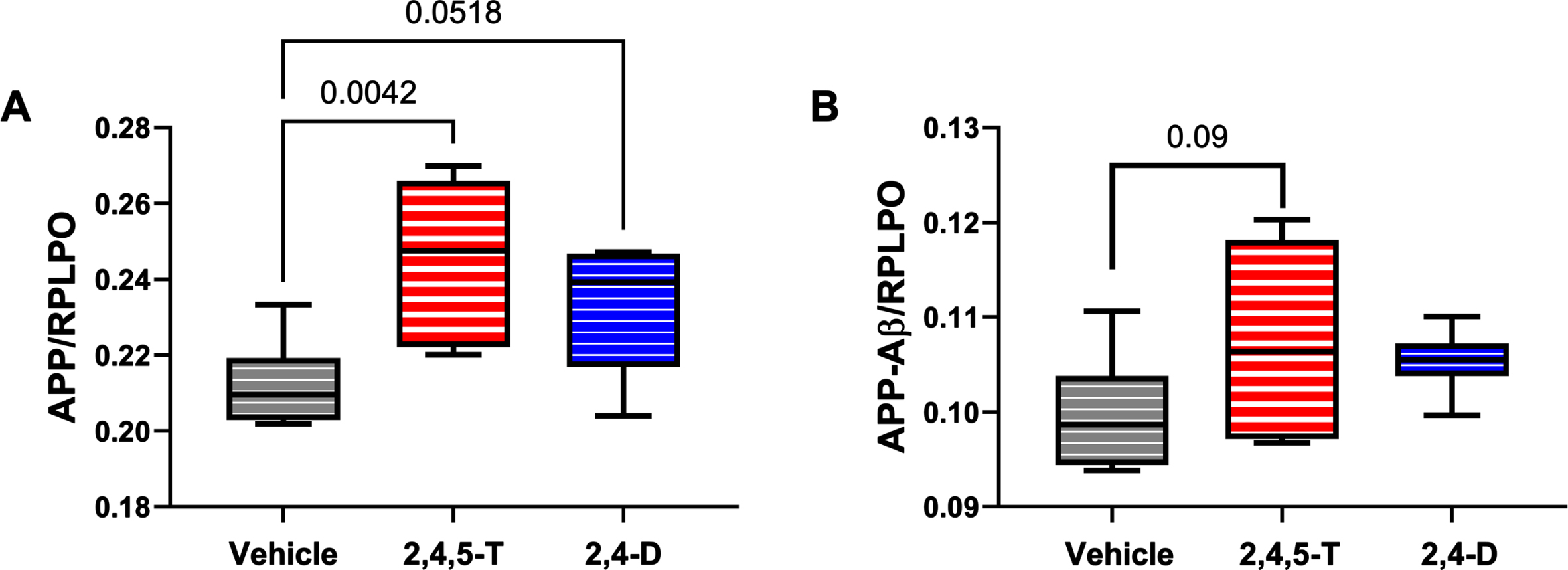
Fig. 6
Effects of Agent Orange herbicidal toxin exposures on tau expression and pTau. Human PNET2 cells treated for 48 h with Vehicle, 250μg/ml 2,4,5-T or 250μg/ml 2,4-D were analyzed for (A) tau and (B) pTau (pT205-Tau) immunoreactivity by duplex ELISA with results normalized to large acidic ribosomal protein (RPLPO). C) The calculated pTau/tau ratios assessed effects on the levels of relative tau phosphorylation. Each group included 6 replicate cultures. Results were analyzed by one-way ANOVA (Table 5). Post hoc significant (p < 0.05) or statistical trend (0.05 < p<0.10) differences by Tukey tests (repeated measures) are displayed. 2,4,5-T, 2,4,5-trichlorophenoxyacetic acid; 2,4-D, 2,4-dichlorophenoxyacetic acid.
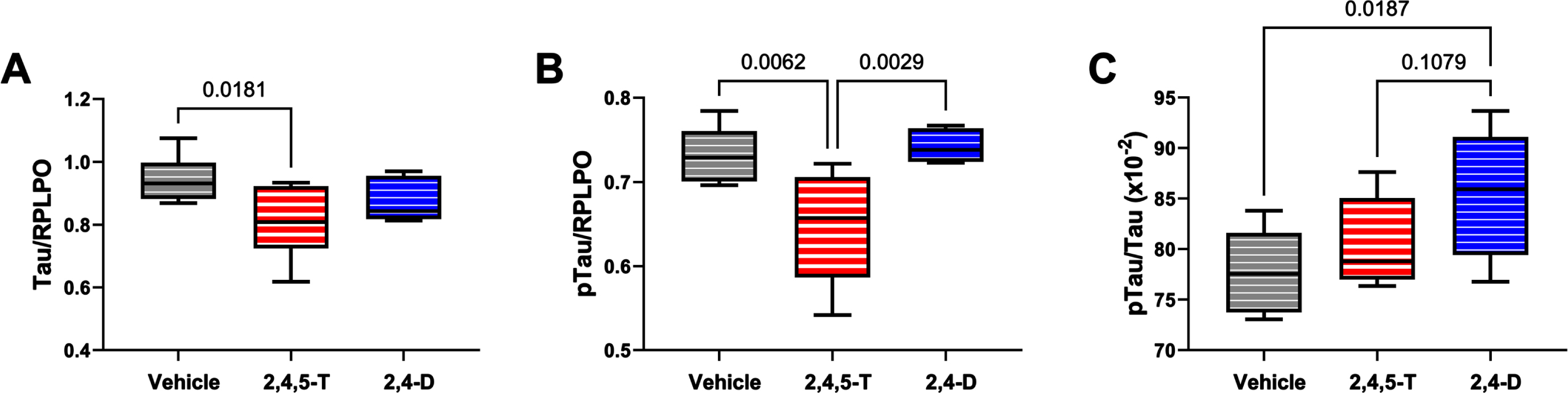
Fig. 7
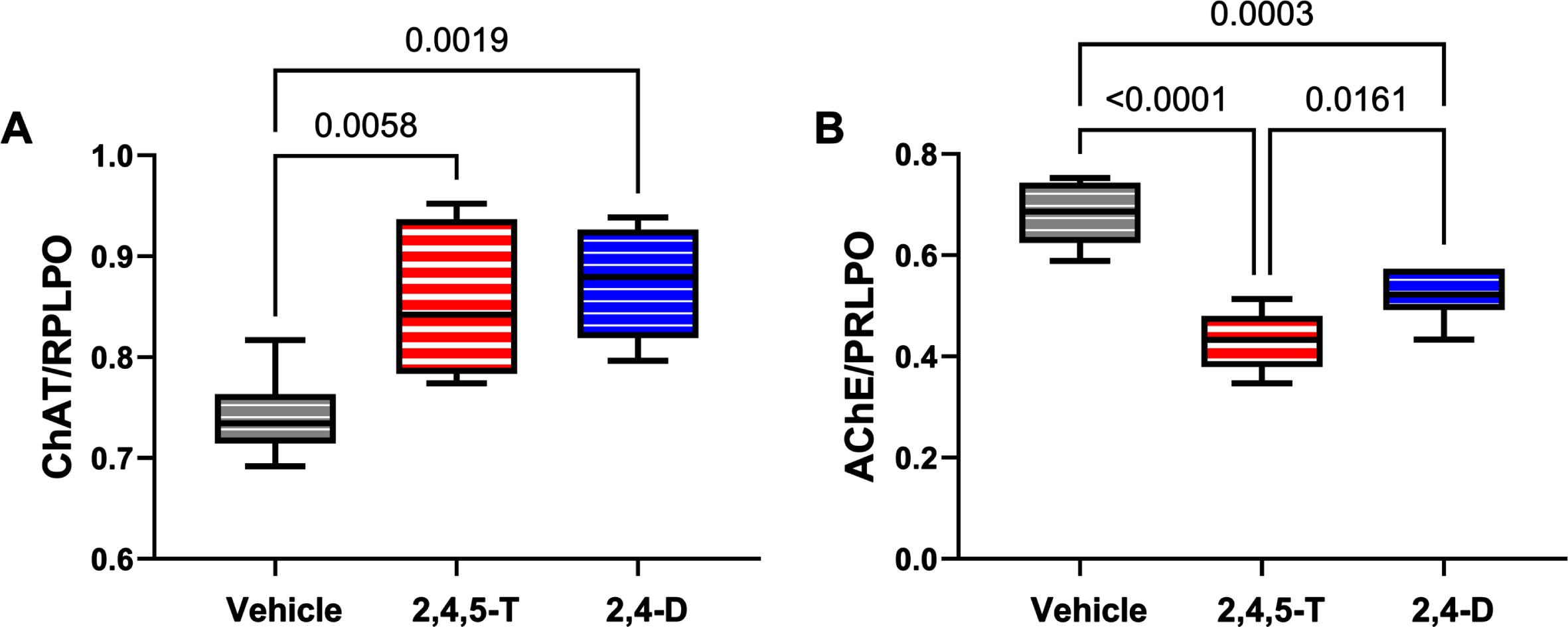
Effects of Agent Orange herbicidal toxin exposures on cholinergic enzyme protein expression. Human PNET2 cells treated for 48 h with Vehicle, 250μg/ml 2,4,5-T or 250μg/ml 2,4-D were analyzed for (A) ChAT and (B) AChE immunoreactivity by duplex ELISA with results normalized to large acidic ribosomal protein (RPLPO). Results were analyzed by one-way ANOVA (Table 5). Significant differences (p < 0.05) by post hoc Tukey tests are indicated. Each group included 6 replicate cultures. 2,4,5-T, 2,4,5-trichlorophenoxyacetic acid; 2,4-D, 2,4-dichlorophenoxyacetic acid; ChAT, choline acetyltransferase; AChE, acetylcholinesterase.
GAPDH: This insulin-regulated enzyme has a critical role in carbohydrate metabolism [38]. Reduced expression or enzyme activity is a feature of CNS neurotoxic or neurodegenerative pathology [45, 46]. One-way ANOVA demonstrated significant inter-group differences in GAPDH expression (p < 0.0001). Post hoc Tukey tests demonstrated significant reductions in GAPDH in both 2,4,5-T, and 2,4-D treated PNET2 cells, and greater reductions associated with 2,4,5-T than 2,4-D (Fig. 4A).
8-OHdG: This biomarker of oxidative stress [47] is associated with RNA [48], mitochondrial DNA [49], and nuclear DNA [50] damage. Following short-term exposures, no significant effects of 2,4,5-T or 2,4-D were observed with respect to 8-OHdG immunoreactivity (Fig. 4B).
HNE: This established biomarker of lipid peroxidation that reflects membrane damage [51] and is increased in various neurodegenerative or neurotoxic disease states [29, 52, 53], was significantly affected by Agent Orange herbicide toxin exposures as demonstrated by one-way ANOVA (p = 0.021). Post-hoc Tukey tests demonstrated the highest levels of HNE in cells exposed to 2,4,5-T with a significant difference from the 2,4-D-exposed cultures (p = 0.0062) (Fig. 4C).
A βPP: Amyloid precursor protein is abundantly expressed in the brain [54], and its levels increase with sporadic AD [55]. Proteolytic cleavage of AβPP by beta secretase followed by gamma secretase [54] generates C-terminal 40 or 42 amino acid length Aβ peptides that can aggregate and are thought to impact the development of AD [54]. Duplex ELISAs demonstrated significantly altered expression of AβPP (p = 0.01) associated with Agent Orange herbicide toxin exposure. Post hoc tests showed significantly elevated levels of AβPP in cells treated with 2,4,5-T (p = 0.004) and statistical trend increases following 2,4-D exposure (p = 0.05) (Fig. 5A).
A β: The 40 or 42-amino acid long C-terminal peptides, once accumulated, have the propensity to aggregate or form oligomers [54]. Aβ deposition is pro-inflammatory, and increased levels of Aβ oligomers are neurotoxic [54]. The high levels of Aβ associated with AD neurodegeneration led to the amyloid hypothesis and subsequent development of treatments designed to clear Aβ from the brain in hope of disease remediation. The ANOVA test did not detect significant inter-group differences. However, the graph and post hoc test revealed somewhat higher levels of Aβ in the Agent Orange herbicide toxin-exposed groups with a statistical trend difference between 2,4,5-T and vehicle (p = 0.09) (Fig. 5B).
Tau: This neuronal microtubule binding protein functions in controlling the assembly and stability of microtubules and impacts axonal transport and growth cone development [56]. Tau is targeted in a broad array of neurodegenerative diseases, including AD. Neurotoxic injury and oxidative stress have been associated with tau protein fragmentation leading to its reduced levels with attendant cytoskeletal collapse and synaptic disconnection [57]. One-way ANOVA demonstrated a statistical trend effect of Agent Orange herbicide toxin exposure, and significant post hoc reduction in tau protein in cells exposed to 2,4,5-T versus Vehicle (Fig. 6A). The differences between 2,4-D and vehicle or 2,4,5-T were not significant.
pTau: The pTau antibody detected the Thr205 phosphorylated epitope of tau, which is one of the many sites contributing to tau hyperphosphorylation in AD [58]. In addition to neurodegeneration, tau phosphorylation occurs following hypoxic-ischemic injury or oxidative stress and, like AD, it is largely mediated by glycogen synthase kinase-3β (GSK-3β) activation [59]. Under physiological conditions, insulin and insulin-like growth factor type 1 (IGF-1) modulate tau phosphorylation via inhibition of GSK-3β [60], but with AD and other forms of neurodegeneration, insulin and IGF-1 signaling are impaired, resulting in the activation of GSK-3β and other kinases that mediate tau hyperphosphorylation on Ser and Thr residues [58].
Consequences of aberrant phosphorylation include tau protein aggregation with formation of paired-helical filament structural lesions characteristic of AD pathology [58]. However, pTau levels are also linked to the abundance of tau protein such that reductions in tau could lead to reductions in the measured pTau, despite elevated levels of GSK-3β activity. In the present study, we observed significant effects of Agent Orange herbicide toxin exposures (p = 0.005) by one-way ANOVA, and significant 2,4,5-T-induced reductions in pTau relative to Vehicle and 2,4-D by post hoc Tukey tests (Fig. 6B). Note the parallel sharp reductions in tau and pTau in 2,4,5-T-treated PNET2 cells. On the other hand, the calculated pTau/tau ratio, reflecting relative tau phosphorylation, correcting for altered tau protein levels, revealed a different trend characterized by progressive elevations in relative tau phosphorylation from Vehicle to 2,4,5-T to 2,4-D with a significant difference between 2,4-D and Vehicle (control) (Fig. 6C).
ChAT: Choline acetyltransferase is critical for the synthesis of acetylcholine, a neurotransmitter that gets reduced in AD and other neurodegenerative diseases [61]. ANOVA demonstrated significant effects of Agent Orange herbicide toxin exposure (p = 0.004), and the graph and post hoc test revealed significantly elevated levels of ChAT in cultures exposed to 2,4,5-T or 2,4-D (Fig. 7A).
AChE: Acetylcholinesterase is the enzyme responsible for the breakdown of acetylcholine which is needed to terminate cholinergic neurotransmission [62]. AChE expression is significantly reduced in AD [63–65], yet one of the standard-of-care treatments is acetylcholinesterase inhibitor medication that has been purposed to preserve already reduced levels of acetylcholine [62, 64, 66, 67]. ANOVA demonstrated significant effects of Agent Orange herbicide toxin exposure on AChE expression (p < 0.0001), and the graph and post hoc test revealed significantly reduced levels of AChE in cultures exposed to 2,4,5-T or 2,4-D relative to vehicle (Fig. 7B).
DISCUSSION
This study examined the direct neurotoxic and early neurodegenerative effects of the two main constituents of Agent Orange, 2,4-D and 2,4,5-T, in the absence of the proposed disease-promoting contaminant, TCDD. Importantly, both agents independently impaired CNS-derived PNET2 cell function, but in different manners. The 2,4-D caused a significant reduction in MTT activity only at the higher dose, reflecting mitochondrial dysfunction, but also caused a dose-dependent decline in cell number/viability. Therefore, cell death occurred at the lower concentration of 2,4-D without compromise in mitochondrial function (MTT activity). In contrast, at the higher dose of 2,4-D, the precipitous decline in MTT vis-à-vis gradual decline in cell viability suggests that mitochondrial dysfunction was a major adverse effect that contributed to PNET2 cell loss.
Treatment with 2,4,5-T had strikingly disparate dose-dependent effects on mitochondrial function such that the lower concentration increased whilst the higher concentration decreased MTT activity. One potential explanation for these seemingly paradoxical responses is that at the lower dose, increased MTT activity reflected over-activation of mitochondrial function related to stress, whereas the apparent normalization of MTT at the higher dose corresponded to mitochondrial dysfunction possibly due to uncoupling. This interpretation falls in line with the H33342 fluorescence results which showed PNET2 cell loss vis-à-vis increased MTT activity at the 25μg/ml dose, and further striking PNET2 cell loss with “normalization” of MTT activity at the 250μg/ml dose. Similar phenomena have been observed following ethanol and nitrosamine treatment of CNS-derived PNET2 cells [26, 46, 68, 69].
The most significant finding in this study was that both components of Agent Orange had adverse effects on PNET2 cell mitochondrial function and viability, except that the 2,4-D impacted mitochondria more than viability while 2,4,5-T adversely affected both mitochondria and viability. In essence, the results suggest that impaired CNS-derived PNET2 cell functions caused by either 2,4-D or 2,4,5-T exposure were likely mitochondrial-mediated and that although both compounds caused PNET2 cell death, 2,4,5-T is the more potent neurotoxin. Importantly, the combined effects of 2,4-D and 2,4,5-T resulting from Agent Orange exposures may have additive neurotoxic effects leading to substantial impairments in energy metabolism and cell survival.
To further examine potential mechanisms of increased cell loss and mitochondrial dysfunction, the cells were analyzed for GAPDH, 8-OHdG, and HNE immunoreactivity. The significant reductions in GAPDH reflect the inhibitory effects of 2,4,5-T and 2,4-D on energy metabolism. Importantly, GAPDH is an insulin-responsive gene [38], and Agent Orange exposures correlate with higher rates of diabetes mellitus [8]. The findings with respect to GAPDH may reflect early impairments in insulin-stimulated GAPDH linked to deficits in energy metabolism via carbohydrate pathways that are critical for neuronal function [55, 70]. The findings with respect to 8-OHdG provide no evidence that the short-term exposures to Agent Orange herbicidal toxins increased guanine oxidation leading to DNA or RNA adduct formation, suggesting the mitochondrial dysfunction marked by reductions in MTT was functional rather than mediated by nucleic acid structural damage. However, with prolonged exposures, mitochondrial DNA damage would likely ensue due to superimposed oxidative stress. HNE is a major α,β-unsaturated aldehyde product of n-6 fatty acid oxidation and lipid peroxidation end-product that functions as a second messenger of oxidative/electrophilic stress [71]. HNE modulates cell survival/death via ER stress induction and promotes cell death via apoptosis [71]. The 2,4,5-T-induced statistical trend increase in HNE suggests a role for lipid peroxidation as a mediator of oxidative stress and impaired PNET2 cell survival.
AβPP has important physiological roles in cell signaling through Notch pathways which mediate a broad range of neuronal functions including plasticity, cell survival, and growth [72]. Previous studies showed elevated levels of AβPP in brains with sporadic AD [65, 73], suggesting that the upregulated expression and protein processing contribute to the accumulation of Aβ cleavage products. This concept is important for explaining any associated increases in Aβ unrelated to germline mutations or the APOE4 genotype. Earlier studies also showed that AβPP expression and Aβ immunoreactivity increase with oxidative stress [57]. Similarly, in the present study, we found that AβPP and Aβ immunoreactivities were highest in 2,4,5-T-exposed cells which also had the lowest levels of GAPDH and MTT, and the highest levels of HNE. The responses in 2,4-D-treated cells were intermediate between Vehicle and 2,4,5-T. Together, these findings support the concept that Agent Orange herbicide toxins exert oxidative neuronal injury accompanied by AβPP and Aβ-related molecular pathologies that have relevance to AD-type neurodegeneration.
The neuronal cytoskeleton is largely composed of the microtubule-associated binding protein, tau, which has critical roles in maintaining the cytoarchitecture and inter-neuronal connections [56]. Oxidative injury disrupts the neuronal cytoskeleton and renders it vulnerable to degradation. Further oxidative injury destabilizes tau, leading to growth cone collapse and synaptic disconnection [74, 75]. In AD, stress-associated increases in GSK-3β [59], cyclin dependent kinase 5 (Cdk5) [59, 76], and calmodulin-dependent protein kinase (CaMKII) [75] activities contribute to tau hyper-phosphorylation on Ser and Thr residues, rendering tau susceptible to misfolding, aggregation, and intracellular accumulation. The studies herein showed that both tau and pTau were significantly reduced in 2,4,5-T-treated cells relative to Vehicle. Most likely, the reductions in tau protein were mediated by oxidative stress and corresponding reductions in pTau were due to the sharp declines in cellular tau. In support of this notion, the calculated pTau/Tau ratios showed that the Agent Orange herbicide toxin exposures enhanced relative tau phosphorylation, and significantly elevated the levels in PNET2 cell cultures treated with 2,4-D. The trend reversals for relative phosphorylation compared with tau and pTau immunoreactivities likely reflects enhanced activity of kinases already known to promote tau phosphorylation under stress conditions. The finding that pTau/Tau was significantly elevated in 2,4-D-treated cultures which exhibited a moderate but non-significant reduction in tau yet pTau levels that were similar to control, supports the argument that Agent Orange herbicide toxin exposures can promote stress-related tau phosphorylation, similar to the findings in AD.
ChAT is enzymatically responsible for the biosynthesis of acetylcholine and a specific indicator of cholinergic neuron functioning [77]. Previous studies linked reductions in ChAT to AD-associated cognitive impairment [61], and dysregulated insulin signaling in the brain and cultured neuronal cells [73, 78–80]. In contrast, very early stages of neurodegeneration that are associated with mild cognitive impairment paradoxically exhibit markedly increased ChAT activity [81], possibly suggesting a compensatory response to evolving failure of cholinergic pathways. The significantly elevated levels of ChAT associated with 2,4,5-T and 2,4-D correspond to increased cellular efforts to generate acetylcholine in Agent Orange Herbicide toxin-stressed cells and appear to be reminiscent of the very early stages of AD.
AChE, a serine hydrolase, has a key role in hydrolytic metabolism [82, 83]. However, apart from its enzymatic functions, AChE has roles in mediating axonal growth, synaptogenesis, neuronal migration and cell adhesion [83]. Previous studies demonstrated CNS cholinergic dysfunction marked by increased activity surrounding senile plaques in AD [82], but reduced expression or activity in regions that were unrelated to plaques in AD, as well as in other neurodegenerative diseases [63] or following chronic ethanol exposure [84–86]. Inhibition of AChE may have neurotoxic effects due to prolonged stimulation. Of interest is that AChE is a specific molecular target of organophosphate and carbamate pesticides which inhibit enzyme activity [83]. The finding of Agent Orange herbicide toxin-mediated inhibition of AChE is likely significant in relation to dysregulation of cholinergic activities as well as enzyme activity-independent neuronal functions needed for plasticity and repair.
Conclusions
The findings suggest that the primary herbicidal constituents of Agent Orange are sufficient to damage CNS-derived PNET2 cells, and likely cause harm to the immature human brain [26]. The teratogenic effects of Agent Orange on the human fetal CNS could have been mediated by the neurotoxic effects of Agent Orange, in the absence of TCDD trace contamination. Our studies complement prior publications that also examined the toxic effects of 2,4-D or 2,4,5-T using in vitro systems [21, 31, 33, 34, 87], and support the concept that apart from TCDD, the intended herbicide exposures have deleterious effects on health. The findings herein are also relevant to earlier in vitro and in vivo studies demonstrating neurotoxic effects of 2,4-D and 2,4,5-T on myelin-producing cells [18, 21], neuronal cells [20], and cognitive-behavioral functions [19].
The results suggest that Agent Orange damages CNS neuronal cells in ways that typically occur with neurodegeneration [88–90], which could potentially account for the suspected increased risk of neurodegenerative diseases among Vietnam Veterans. Of note in these regards is that mitochondrial dysfunction impairs cellular energy metabolism, which is one of the core pathophysiological processes in both diabetes mellitus and neurodegeneration. The potential contributions of Agent Orange exposure to brain atrophy [91] and neurodegenerative diseases have been considered, and epidemiologic [8, 92] or biomarker [93] links have been demonstrated in retrospective studies.
Although the doses of 2,4-D and 2,4,5-T used correspond with previous studies [21, 31–33], they were likely higher than the levels included in Agent Orange herbicide sprays [34]. However, the chronicity and repeated nature of exposures, long intervals of post-exposure follow-up, and persistence of chemicals in the environment and body could significantly impact the development and nature of diseases linked to Agent Orange. Of further importance is the growing appreciation of the broad toxic and carcinogenic effects of 2,4-D which has taken years to prove and still the evidence has not achieved a level that government agencies would deem sufficient to ban its use [94]. Currently, the widespread, uncontrolled use of 2,4-D in herbicide and pesticide products is such that one in three Americans has biomarkers of exposure [95]. Therefore, despite the study design’s limitations including the inability to draw direct dose-effect comparisons with human exposures, the results support the notion that both 2,4-D and 2,4-T exert alarming adverse effects on CNS-derived neuroepithelial cell function, resulting in abnormalities reminiscent of AD. Additional studies are needed to further characterize the time course and full spectrum of Agent Orange herbicide toxin-mediated neurodegeneration in brain tissue, particularly in relation to insulin resistance, which is a feature of diabetes mellitus and AD-type neurodegeneration [10, 11].
ACKNOWLEDGMENTS
This research was by supported by R01AA011431 (SMD) and R01AA028408 (SMD) from the National Institute on Alcohol Abuse and Alcoholism at the National Institutes of Health, and the Karen T. Romer Undergraduate Teaching & Research Award (UTRA) from Brown University (AG).
FUNDING
This work was supported by grants from the National Institutes of Health, National Institutes on Alcohol Abuse and Alcoholism (AA-011431, AA-028408), and a Brown University Undergraduate Teaching and Research Award.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
The data supporting the findings of this study are available on request from the corresponding author.
REFERENCES
[1] | Institute of Medicine (US) Committee to Review the Health Effects in Vietnam Veterans of Exposure to Herbicides ((1994) ) Veterans and Agent Orange: Health Effects of Herbicides Used in Vietnam. The National Academies Press, Washington, DC. |
[2] | Le TN , Johansson A ((2001) ) Impact of chemical warfare with agent orange on women’s reproductive lives in Vietnam: A pilot study. Reprod Health Matters 9: , 156–164. |
[3] | Ngo AD , Taylor R , Roberts CL , Nguyen TV ((2006) ) Association between Agent Orange and birth defects: Systematic review and meta-analysis. Int J Epidemiol 35: , 1220–1230. |
[4] | Stellman SD , Stellman JM , Sommer JF Jr. ((1988) ) Health and reproductive outcomes among American Legionnaires in relation to combat and herbicide exposure in Vietnam. Environ Res 47: , 150–174. |
[5] | Wolfe WH , Michalek JE , Miner JC , Rahe A , Silva J , Thomas WF , Grubbs WD , Lustik MB , Karrison TG , Roegner RH , et al. ((1990) ) Health status of Air Force veterans occupationally exposed to herbicides in Vietnam. I. Physical health. JAMA 264: , 1824–1831. |
[6] | Henriksen GL , Ketchum NS , Michalek JE , Swaby JA ((1997) ) Serum dioxin and diabetes mellitus in veterans of Operation Ranch Hand. Epidemiology 8: , 252–258. |
[7] | Michalek JE , Ketchum NS , Akhtar FZ ((1998) ) Postservice mortality of US Air Force veterans occupationally exposed to herbicides in Vietnam: 15-year follow-up. Am J Epidemiol 148: , 786–792. |
[8] | de la Monte SM , Goel A ((2022) ) Agent Orange reviewed: Potential role in peripheral neuropathy and neurodegeneration. J Mil Veterans Health 30: , 17–26. |
[9] | Martinez S , Yaffe K , Li Y , Byers AL , Peltz CB , Barnes DE ((2021) ) Agent Orange exposure and dementia diagnosis in US Veterans of the Vietnam Era. JAMA Neurol 78: , 473–477. |
[10] | de la Monte SM ((2017) ) Insulin resistance and neurodegeneration: Progress towards the development of new therapeutics for Alzheimer’s disease. Drugs 77: , 47–65. |
[11] | de la Monte SM ((2019) ) The Full spectrum of Alzheimer’s disease is rooted in metabolic derangements that drive type 3 diabetes. Adv Exp Med Biol 1128: , 45–83. |
[12] | de la Monte SM , Tong M , Lawton M , Longato L ((2009) ) Nitrosamine exposure exacerbates high fat diet-mediated type 2 diabetes mellitus, non-alcoholic steatohepatitis, and neurodegeneration with cognitive impairment. Mol Neurodegener 4: , 54. |
[13] | Kim JS , Lim HS , Cho SI , Cheong HK , Lim MK ((2003) ) Impact of Agent Orange exposure among Korean Vietnam veterans. Ind Health 41: , 149–157. |
[14] | Michalek JE , Akhtar FZ , Arezzo JC , Garabrant DH , Albers JW ((2001) ) Serum dioxin and peripheral neuropathy in veterans of Operation Ranch Hand. Neurotoxicology 22: , 479–490. |
[15] | Yi SW , Hong JS , Ohrr H , Yi JJ ((2014) ) Agent Orange exposure and disease prevalence in Korean Vietnam veterans: The Korean veterans health study. Environ Res 133: , 56–65. |
[16] | Yi SW , Ohrr H , Hong JS , Yi JJ ((2013) ) Agent Orange exposure and prevalence of self-reported diseases in Korean Vietnam veterans. J Prev Med Public Health 46: , 213–225. |
[17] | Bortolozzi AA , Duffard RO , Evangelista de Duffard AM ((1999) ) Behavioral alterations induced in rats by a pre- and postnatal exposure to 2,4-dichlorophenoxyacetic acid. Neurotoxicol Teratol 21: , 451–465. |
[18] | Duffard R , Garcia G , Rosso S , Bortolozzi A , Madariaga M , di Paolo O , Evangelista de Duffard AM ((1996) ) Central nervous system myelin deficit in rats exposed to 2,4-dichlorophenoxyacetic acid throughout lactation. Neurotoxicol Teratol 18: , 691–696. |
[19] | Oliveira GH , Palermo-Neto J ((1993) ) Effects of 2,4-dichlorophenoxyacetic acid (2,4-D) on open-field behaviour and nerocheumical parameters of rats. Pharmacol Toxicol 73: , 79–85. |
[20] | Rosso SB , Di Paolo OA , Evangelista de Duffard AM , Duffard R ((1997) ) Effects of 2,4-dichlorophenoxyacetic acid on central nervous system of developmental rats. Associated changes in ganglioside pattern. Brain Res 769: , 163–167. |
[21] | Sharifi Pasandi M , Hosseini Shirazi F , Gholami MR , Salehi H , Najafzadeh N , Mazani M , Ghasemi Hamidabadi H , Niapour A ((2017) ) Epi/perineural and Schwann cells as well as perineural sheath integrity are affected following 2,4-D exposure. Neurotox Res 32: , 624–638. |
[22] | Hites RA ((2011) ) Dioxins: An overview and history. Environ Sci Technol 45: , 16–20. |
[23] | Olson KR , Morton LW ((2019) ) Long-term fate of Agent Orange and dioxin TCDD contaminated soils and sediments in Vietnam hotspots. Open J Soil Sci 09: , 1–34. |
[24] | The I , Murthy AE , Hannigan GE , Jacoby LB , Menon AG , Gusella JF , Bernards A ((1993) ) Neurofibromatosis type 1 gene mutations in neuroblastoma. Nat Genet 3: , 62–66. |
[25] | Carter JJ , Tong M , Silbermann E , Lahousse SA , Ding FF , Longato L , Roper N , Wands JR , de la Monte SM ((2008) ) Ethanol impaired neuronal migration is associated with reduced aspartyl-asparaginyl-beta-hydroxylase expression. Acta Neuropathol 116: , 303–315. |
[26] | de la Monte SM , Ganju N , Banerjee K , Brown NV , Luong T , Wands JR ((2000) ) Partial rescue of ethanol-induced neuronal apoptosis by growth factor activation of phosphoinositol-3-kinase. Alcohol Clin Exp Res 24: , 716–726. |
[27] | Lawton M , Tong M , Gundogan F , Wands JR , de la Monte SM ((2010) ) Aspartyl-(asparaginyl) beta-hydroxylase, hypoxia-inducible factor-alpha and Notch cross-talk in regulating neuronal motility. Oxid Med Cell Longev 3: , 347–356. |
[28] | de La Monte SM , Wands JR ((2001) ) Mitochondrial DNA damage and impaired mitochondrial function contribute to apoptosis of insulin-stimulated ethanol-exposed neuronal cells. Alcohol Clin Exp Res 25: , 898–906. |
[29] | Tong M , Dong M , de la Monte SM ((2009) ) Brain insulin-like growth factor and neurotrophin resistance in Parkinson’s disease and dementia with Lewy bodies: Potential role of manganese neurotoxicity. J Alzheimers Dis 16: , 585–599. |
[30] | Tong M , Gao JS , Borgas D , de la Monte SM ((2013) ) Phosphorylation modulates aspartyl-(asparaginyl)-beta hydroxylase protein expression, catalytic activity and migration in human immature neuronal cerebellar cells. Cell Biol (Henderson, NV) 6: , 133. |
[31] | Soloneski S , Gonzalez NV , Reigosa MA , Larramendy ML ((2007) ) Herbicide 2,4-dichlorophenoxyacetic acid (2,4-D)-induced cytogenetic damage in human lymphocytes in vitro presence of erythrocytes. Cell Biol Int 31: , 1316–1322. |
[32] | Bertheussen K , Yousef MI , Figenschau Y ((1997) ) A new sensitive cell culture test for the assessment of pesticide toxicity. J Environ Sci Health B 32: , 195–211. |
[33] | Gonzalez M , Soloneski S , Reigosa MA , Larramendy ML ((2005) ) Genotoxicity of the herbicide 2,4-dichlorophenoxyacetic and a commercial formulation, 2,4-dichlorophenoxyacetic acid dimethylamine salt. I. Evaluation of DNA damage and cytogenetic endpoints in Chinese Hamster ovary (CHO) cells. Toxicol In Vitro 19: , 289–297. |
[34] | Basrur SV , Fletcher RA , Basrur PK ((1976) ) In vitro effects of 2,4-dichlorophenoxy acetic acid (2,4-D) on bovine cells. Can J Comp Med 40: , 408–415. |
[35] | Morgan DM ((1998) ) Tetrazolium (MTT) assay for cellular viability and activity. Methods Mol Biol 79: , 179–183. |
[36] | Ghasemi M , Turnbull T , Sebastian S , Kempson I ((2021) ) The MTT assay: Utility, limitations, pitfalls, and interpretation in bulk and single-cell analysis. Int J Mol Sci 22: , 12827. |
[37] | de la Monte SM , Ganju N , Wands JR ((1999) ) Microtiter immunocytochemical ELISA assay. Biotechniques 26: , 1073–1076,1078. |
[38] | Alexander BM , Dugast I , Ercolani L , Kong XF , Giere L , Nasrin N ((1992) ) Multiple insulin-responsive elements regulate transcription of the GAPDH gene. Adv Enzyme Regul 32: , 149–159. |
[39] | DaDalt AA , Bonham CA , Lotze GP , Luiso AA , Vacratsis PO ((2022) ) Src-mediated phosphorylation of the ribosome biogenesis factor hYVH1 affects its localization, promoting partitioning to the 60S ribosomal subunit. J Biol Chem 298: , 102679. |
[40] | de la Monte SM , Tong M , Carlson RI , Carter JJ , Longato L , Silbermann E , Wands JR ((2009) ) Ethanol inhibition of aspartyl-asparaginyl-beta-hydroxylase in fetal alcohol spectrum disorder: Potential link to the impairments in central nervous system neuronal migration. Alcohol 43: , 225–240. |
[41] | Liu HT , Zou YX , Zhu WJ , Sen L , Zhang GH , Ma RR , Guo XY , Gao P ((2022) ) lncRNA THAP7-AS1, transcriptionally activated by SP1 and post-transcriptionally stabilized by METTL3-mediated m6A modification, exerts oncogenic properties by improving CUL4B entry into the nucleus. Cell Death Differ 29: , 627–641. |
[42] | Xie JJ , Jiang YY , Jiang Y , Li CQ , Lim MC , An O , Mayakonda A , Ding LW , Long L , Sun C , Lin LH , Chen L , Wu JY , Wu ZY , Cao Q , Fang WK , Yang W , Soukiasian H , Meltzer SJ , Yang H , Fullwood M , Xu LY , Li EM , Lin DC , Koeffler HP ((2018) ) Super-enhancer-driven long non-coding RNA LINC01503, regulated by TP63, is over-expressed and oncogenic in squamous cell carcinoma. Gastroenterology 154: , 2137–2151e2131. |
[43] | Andreani T , Tong M , Gundogan F , Silbermann E , de la Monte SM ((2016) ) Differential effects of 3rd trimester-equivalent binge ethanol and tobacco-specific nitrosamine ketone exposures on brain insulin signaling in adolescence. J Diabetes Relat Disord 1: , 105–114. |
[44] | Deochand C , Tong M , Agarwal AR , Cadenas E , de la Monte SM ((2015) ) Tobacco smoke exposure impairs brain insulin/IGF signaling: Potential co-factor role in neurodegeneration. J Alzheimers Dis 50: , 373–386. |
[45] | de la Monte SM , Tong M , Bowling N , Moskal P ((2011) ) si-RNA inhibition of brain insulin or insulin-like growth factor receptors causes developmental cerebellar abnormalities: Relevance to fetal alcohol spectrum disorder. Mol Brain 4: , 13. |
[46] | de la Monte SM , Wands JR ((2002) ) Chronic gestational exposure to ethanol impairs insulin-stimulated survival and mitochondrial function in cerebellar neurons. Cell Mol Life Sci 59: , 882–893. |
[47] | Omari Shekaftik S , Nasirzadeh N ((2021) ) 8-Hydroxy-2’-deoxyguanosine (8-OHdG) as a biomarker of oxidative DNA damage induced by occupational exposure to nanomaterials: A systematic review. Nanotoxicology 15: , 850–864. |
[48] | Cattley RC , Glover SE ((1993) ) Elevated 8-hydroxydeoxyguanosine in hepatic DNA of rats following exposure to peroxisome proliferators: Relationship to carcinogenesis and nuclear localization. Carcinogenesis 14: , 2495–2499. |
[49] | Torres-Gonzalez M , Gawlowski T , Kocalis H , Scott BT , Dillmann WH ((2014) ) Mitochondrial 8-oxoguanine glycosylase decreases mitochondrial fragmentation and improves mitochondrial function in H9C2 cells under oxidative stress conditions. Am J Physiol Cell Physiol 306: , C221–229. |
[50] | Qing X , Shi D , Lv X , Wang B , Chen S , Shao Z ((2019) ) Prognostic significance of 8-hydroxy-2’-deoxyguanosine in solid tumors: A meta-analysis. BMC Cancer 19: , 997. |
[51] | Ayala A , Munoz MF , Arguelles S ((2014) ) Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxid Med Cell Longev 2014: , 360438. |
[52] | Moon KH , Tajuddin N , Brown J, 3rd , Neafsey EJ , Kim HY , Collins MA ((2014) ) Phospholipase A2, oxidative stress, and neurodegeneration in binge ethanol-treated organotypic slice cultures of developing rat brain. Alcohol Clin Exp Res 38: , 161–169. |
[53] | Liou CJ , Tong M , Vonsattel JP , de la Monte SM ((2019) ) Altered brain expression of insulin and insulin-like growth factors in frontotemporal lobar degeneration: Another degenerative disease linked to dysregulation of insulin metabolic pathways. ASN Neuro 11: , 1759091419839515. |
[54] | Evin G , Weidemann A ((2002) ) Biogenesis and metabolism of Alzheimer’s disease Abeta amyloid peptides. Peptides 23: , 1285–1297. |
[55] | de la Monte SM ((2012) ) Contributions of brain insulin resistance and deficiency in amyloid-related neurodegeneration in Alzheimer’s disease. Drugs 72: , 49–66. |
[56] | Hamm M , Bailey R , Shaw G , Yen SH , Lewis J , Giasson BI ((2015) ) Physiologically relevant factors influence tau phosphorylation by leucine-rich repeat kinase 2. J Neurosci Res 93: , 1567–1580. |
[57] | Chen GJ , Xu J , Lahousse SA , Caggiano NL , de la Monte SM ((2003) ) Transient hypoxia causes Alzheimer-type molecular and biochemical abnormalities in cortical neurons: Potential strategies for neuroprotection. J Alzheimers Dis 5: , 209–228. |
[58] | Miao J , Shi R , Li L , Chen F , Zhou Y , Tung YC , Hu W , Gong CX , Iqbal K , Liu F ((2019) ) Pathological tau from Alzheimer’s brain induces site-specific hyperphosphorylation and SDS- and reducing agent-resistant aggregation of tau in vivo. Front Aging Neurosci 11: , 34. |
[59] | Lovestone S , Reynolds CH , Latimer D , Davis DR , Anderton BH , Gallo JM , Hanger D , Mulot S , Marquardt B , Stabel S , Woodgett J.R. , Miller CCJ ((1994) ) Alzheimer’s disease-like phosphorylation of the microtubule-associated protein tau by glycogen synthase kinase-3 in transfected mammalian cells. Curr Biol 4: , 1077–1086. |
[60] | Hong M , Lee VM ((1997) ) Insulin and insulin-like growth factor-1 regulate tau phosphorylation in cultured human neurons. J Biol Chem 272: , 19547–19553. |
[61] | Ikonomovic MD , Mufson EJ , Wuu J , Bennett DA , DeKosky ST ((2005) ) Reduction of choline acetyltransferase activity in primary visual cortex in mild to moderate Alzheimer’s disease. Arch Neurol 62: , 425–430. |
[62] | Trang A , Khandhar PB ((2023) ) Physiology, acetylcholinesterase. In StatPearls, StatPearls Publishing, Treasure Island (FL), pp. 1–4. |
[63] | Kish SJ , Schut L , Simmons J , Gilbert J , Chang LJ , Rebbetoy M ((1988) ) Brain acetylcholinesterase activity is markedly reduced indominantly-inherited olivopontocerebellar atrophy. J Neurol Neurosurg Psychiatry 51: , 544–548. |
[64] | Meena VK , Chaturvedi S , Sharma RK , Mishra AK , Hazari PP ((2019) ) Potent acetylcholinesterase selective and reversible homodimeric agent based on tacrine for theranostics. Mol Pharm 16: , 2296–2308. |
[65] | Steen E , Terry BM , Rivera EJ , Cannon JL , Neely TR , Tavares R , Xu XJ , Wands JR , de la Monte SM ((2005) ) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease–is this type 3 diabetes? J Alzheimers Dis 7: , 63–80. |
[66] | Hull M , Berger M , Heneka M ((2006) ) Disease-modifying therapies in Alzheimer’s disease: How far have we come? Drugs 66: , 2075–2093. |
[67] | Davis RE , Doyle PD , Carroll RT , Emmerling MR , Jaen J ((1995) ) Cholinergic therapies for Alzheimer’s disease. Palliative or disease altering? Arzneimittelforschung 45: , 425–431. |
[68] | Chu J , Tong M , de la Monte SM ((2007) ) Chronic ethanol exposure causes mitochondrial dysfunction and oxidative stress in immature central nervous system neurons. Acta Neuropathol 113: , 659–673. |
[69] | de la Monte SM , Longato L , Tong M , Wands JR ((2009) ) Insulin resistance and neurodegeneration: Roles of obesity, type 2 diabetes mellitus and non-alcoholic steatohepatitis. Curr Opin Investig Drugs 10: , 1049–1060. |
[70] | Bukke VN , Villani R , Archana M , Wawrzyniak A , Balawender K , Orkisz S , Ferraro L , Serviddio G , Cassano T ((2020) ) The glucose metabolic pathway as a potential target for therapeutics: Crucial role of glycosylation in Alzheimer’s disease. Int J Mol Sci 21: , 7739. |
[71] | Dalleau S , Baradat M , Gueraud F , Huc L ((2013) ) Cell death and diseases related to oxidative stress: 4-hydroxynonenal (HNE) in the balance. Cell Death Differ 20: , 1615–1630. |
[72] | Luu L , Ciccotosto GD , Cappai R ((2021) ) The Alzheimer’s disease amyloid precursor protein and its neuritogenic actions. Curr Alzheimer Res 18: , 772–786. |
[73] | Rivera EJ , Goldin A , Fulmer N , Tavares R , Wands JR , de la Monte SM ((2005) ) Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: Link to brain reductions in acetylcholine. J Alzheimers Dis 8: , 247–268. |
[74] | Mandelkow EM , Stamer K , Vogel R , Thies E , Mandelkow E ((2003) ) Clogging of axons by tau, inhibition of axonal traffic and starvation of synapses. Neurobiol Aging 24: , 1079–1085. |
[75] | Kamat PK , Kalani A , Rai S , Swarnkar S , Tota S , Nath C , Tyagi N ((2016) ) Mechanism of oxidative stress and synapse dysfunction in the pathogenesis of Alzheimer’s disease: Understanding the therapeutics strategies. Mol Neurobiol 53: , 648–661. |
[76] | Pei JJ , Grundke-Iqbal I , Iqbal K , Bogdanovic N , Winblad B , Cowburn RF ((1998) ) Accumulation of cyclin-dependent kinase 5 (cdk5) in neurons with early stages of Alzheimer’s disease neurofibrillary degeneration. Brain Res 797: , 267–277. |
[77] | Oda Y ((1999) ) Choline acetyltransferase: The structure, distribution and pathologic changes in the central nervous system. Pathol Int 49: , 921–937. |
[78] | Cohen AC , Tong M , Wands JR , de la Monte SM ((2007) ) Insulin and insulin-like growth factor resistance with neurodegeneration in an adult chronic ethanol exposure model. Alcohol Clin Exp Res 31: , 1558–1573. |
[79] | Le T , Tong M , Nguyen V , de la Monte SM ((2013) ) PPAR agonist rescue of ethanol-impaired brain insulin signaling: Cerebellar slice culture model. J Drug Alcohol Res 2: , 235611. |
[80] | Soscia SJ , Tong M , Xu XJ , Cohen AC , Chu J , Wands JR , de la Monte SM ((2006) ) Chronic gestational exposure to ethanol causes insulin and IGF resistance and impairs acetylcholine homeostasis in the brain. Cell Mol Life Sci 63: , 2039–2056. |
[81] | DeKosky ST , Ikonomovic MD , Styren SD , Beckett L , Wisniewski S , Bennett DA , Cochran EJ , Kordower JH , Mufson EJ ((2002) ) Upregulation of choline acetyltransferase activity in hippocampus and frontal cortex of elderly subjects with mild cognitive impairment. Ann Neurol 51: , 145–155. |
[82] | Garcia-Ayllon MS , Small DH , Avila J , Saez-Valero J ((2011) ) Revisiting the role of acetylcholinesterase in Alzheimer’s disease: Cross-talk with P-tau and beta-amyloid. Front Mol Neurosci 4: , 22. |
[83] | Lionetto MG , Caricato R , Calisi A , Giordano ME , Schettino T ((2013) ) Acetylcholinesterase as a biomarker in environmental and occupational medicine: New insights and future perspectives. Biomed Res Int 2013: , 321213. |
[84] | Pires RG , Pereira SR , Oliveira-Silva IF , Franco GC , Ribeiro AM ((2005) ) Cholinergic parameters and the retrieval of learned and re-learned spatial information: A study using a model of Wernicke-Korsakoff Syndrome. Behav Brain Res 162: , 11–21. |
[85] | Tong M , Yu R , Deochand C , de la Monte SM ((2015) ) Differential contributions of alcohol and the nicotine-derived nitrosamine ketone (NNK) to insulin and insulin-like growth factor resistance in the adolescent rat brain. Alcohol Alcohol 50: , 670–679. |
[86] | Jamal M , Ameno K , Miki T , Wang W , Kumihashi M , Isse T , Kawamoto T , Kitagawa K , Nakayama K , Ijiri I , Kinoshita H ((2009) ) Cholinergic alterations following alcohol exposure in the frontal cortex of Aldh2-deficient mice models. Brain Res 1295: , 37–43. |
[87] | Walker EM Jr. , Gadsden RH , Atkins LM , Gale GR ((1972) ) Some effects of 2,4-D and 2,4,5-T on Ehrlich ascites tumor cells in vivo and in vitro. IMS Ind Med Surg 41: , 22–27. |
[88] | de la Monte SM , Wands JR ((2006) ) Molecular indices of oxidative stress and mitochondrial dysfunction occur early and often progress with severity of Alzheimer’s disease. J Alzheimers Dis 9: , 167–181. |
[89] | Friese MA , Schattling B , Fugger L ((2014) ) Mechanisms of neurodegeneration and axonal dysfunction in multiple sclerosis. Nat Rev Neurol 10: , 225–238. |
[90] | Yan MH , Wang X , Zhu X ((2013) ) Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic Biol Med 62: , 90–101. |
[91] | Lee HA , Kyeong S , Kim DH ((2022) ) Long-term effects of defoliant exposure on brain atrophy progression in humans. Neurotoxicology 92: , 25–32. |
[92] | Yang Y , Cheon M , Kwak YT ((2016) ) Is Parkinson’s disease with history of Agent Orange exposure different from idiopathic Parkinson’s disease? Dement Neurocogn Disord 15: , 75–81. |
[93] | Yang Y , Giau VV , An SSA , Kim S ((2018) ) Plasma oligomeric beta amyloid in Alzheimer’s disease with history of Agent Orange exposure. Dement Neurocogn Disord 17: , 41–49. |
[94] | Solomon G, Agent Orange in Your Backyard: The Harmful Pesticide 2,4-D, https://www.theatlantic.com/health/archive/2012/02/agent-orange-in-your-backyard-the-harmful-pesticide-2-4-d/253506. |
[95] | Freisthler MS , Robbins CR , Benbrook CM , Young HA , Haas DM , Winchester PD , Perry MJ ((2022) ) Association between increasing agricultural use of 2,4-D and population biomarkers of exposure: Findings from the National Health and Nutrition Examination Survey, 2001– 2014. Environ Health 21: , 23. |


