
Phosphodiesterase Inhibitors for Alzheimer’s Disease: A Systematic Review of Clinical Trials and Epidemiology with a Mechanistic Rationale
Abstract
Background:
Preclinical studies, clinical trials, and reviews suggest increasing 3’,5’-cyclic adenosine monophosphate (cAMP) and 3’,5’-cyclic guanosine monophosphate (cGMP) with phosphodiesterase inhibitors is disease-modifying in Alzheimer’s disease (AD). cAMP/protein kinase A (PKA) and cGMP/protein kinase G (PKG) signaling are disrupted in AD. cAMP/PKA and cGMP/PKG activate cAMP response element binding protein (CREB). CREB binds mitochondrial and nuclear DNA, inducing synaptogenesis, memory, and neuronal survival gene (e.g., brain-derived neurotrophic factor) and peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1α). cAMP/PKA and cGMP/PKG activate Sirtuin-1, which activates PGC1α. PGC1α induces mitochondrial biogenesis and antioxidant genes (e.g.,Nrf2) and represses BACE1. cAMP and cGMP inhibit BACE1-inducing NFκB and tau-phosphorylating GSK3β.
Objective and Methods:
We review efficacy-testing clinical trials, epidemiology, and meta-analyses to critically investigate whether phosphodiesteraseinhibitors prevent or treat AD.
Results:
Caffeine and cilostazol may lower AD risk. Denbufylline and sildenafil clinical trials are promising but preliminary and inconclusive. PF-04447943 and BI 409,306 are ineffective. Vinpocetine, cilostazol, and nicergoline trials are mixed. Deprenyl/selegiline trials show only short-term benefits. Broad-spectrum phosphodiesterase inhibitor propentofylline has been shown in five phase III trials to improve cognition, dementia severity, activities of daily living, and global assessment in mild-to-moderate AD patients on multiple scales, including the ADAS-Cogand the CIBIC-Plus in an 18-month phase III clinical trial. However, two books claimed based on a MedScape article an 18-month phase III trial failed, so propentofylline was discontinued. Now, propentofylline is used to treat canine cognitive dysfunction, which, like AD, involves age-associated wild-type Aβ deposition.
Conclusion:
Phosphodiesterase inhibitors may prevent and treat AD.
BACKGROUND
Sporadic Alzheimer’s disease (AD) is the most common cause of dementia in the elderly. It involves brain amyloid-β (Aβ) generation, aggregation into oligomers, and deposition into neuritic plaques, tau hyperphosphorylation into neurofibrillary tangles [1, 2], iron and aluminum accumulation [3, 4], infections [5–11], oxidative stress and damage to lipids, RNA, DNA, and proteins [12–16], aberrant calcium and zinc signaling [17–21], mitochondrial dysfunction [22–24],endoplasmic reticulum stress [25–27], lysosomal dysfunction [17, 20, 28–30], defective autophagy [31], neuroinflammation [32–38], neuronal abortive cellcycle re-entry [39–48], insulin and insulin-like growth factor 1 resistance [49–51], synaptic dysfunction [52], and neuron death.
Cyclic nucleotides are endogenous cell signaling molecules that act as second messengers to regulate various physiological processes in the central nervous system. There are multiple cyclic nucleotides that may be involved in the pathogenesis of AD, including Ca2+-mobilizing and transient receptor potential melastatin 2 (TRPM2) channel-openingcyclic adenine diphsophate ribose (cADPR) [53–57] and cytoplasmic DNA-induced cyclic guanosine monophosphate adenosine monophosphate (cyclic GMP-AMP, or cGAMP)[14–16, 58–62]. Of these though, there is the most evidence about the cyclic purine nucleotides 3’,5’-cyclic adenosine monophosphate (cAMP) and 3’,5’-cyclic guanosine monophosphate (cGMP).
Physiologic cAMP and cGMP signaling
Physiologically, cAMP is produced whenGαs-containing complexes activate adenylate cyclases (AC). Adenylate cyclase generates cAMP from adenosine triphosphate (ATP). cAMP activates exchange protein directly activated by cAMP (EPAC) and protein kinase A (PKA). PKA holoenzymes consist of two catalytic and two regulatory subunits. When bound to cAMP, the regulatory subunits release the catalytic PKA subunits to phosphorylate many targets [63].
Regarding cGMP signaling, nitric oxide synthases (NOS) produce nitric oxide (NO).NO activates soluble guanylyl cyclase (sGC) in the cytoplasm. sGC produces cytoplasmic cGMP [64, 65]. In addition, Atrial natriuretic peptide (ANP) and B-type natriuretic peptide (BNP) activate particulate, plasma-membrane localized, receptor guanylyl cyclase-A (GC-A) [66], and C-type natriuretic peptide (CNP) activates the particulate guanylyl cyclase B (GC-B) [67]. Particulate GC-A and GC-B produce cGMP proximal to the plasma membrane [67, 68]. cGMP activates protein kinase G (PKG) [64, 65].
Phosphodiesterases
Phosphodiesterase (PDE) enzymes degrade cyclic purine nucleotides. PDE1, 2, 3, 10, and 11 degrade both cAMP and cGMP [69, 70]. PDE4, 7, and 8 degrade cAMP specifically. PDE5, 6, and 9 degrade cGMP specifically [69, 70]. PDE5 degrades cytoplasmic cGMP produced by NOS/NO/sGC signaling, whereas PDE9 degrades peri-plasma membrane cGMP produced by natriuretic peptide/pGC signaling [71]. Complicating the interpretation of studies about the effects of cGMP, low concentrations of cGMP inhibit PDE3, increasing cAMP levels, whereas moderately high doses of cGMP activate PDE2, decreasing cAMP levels [72].
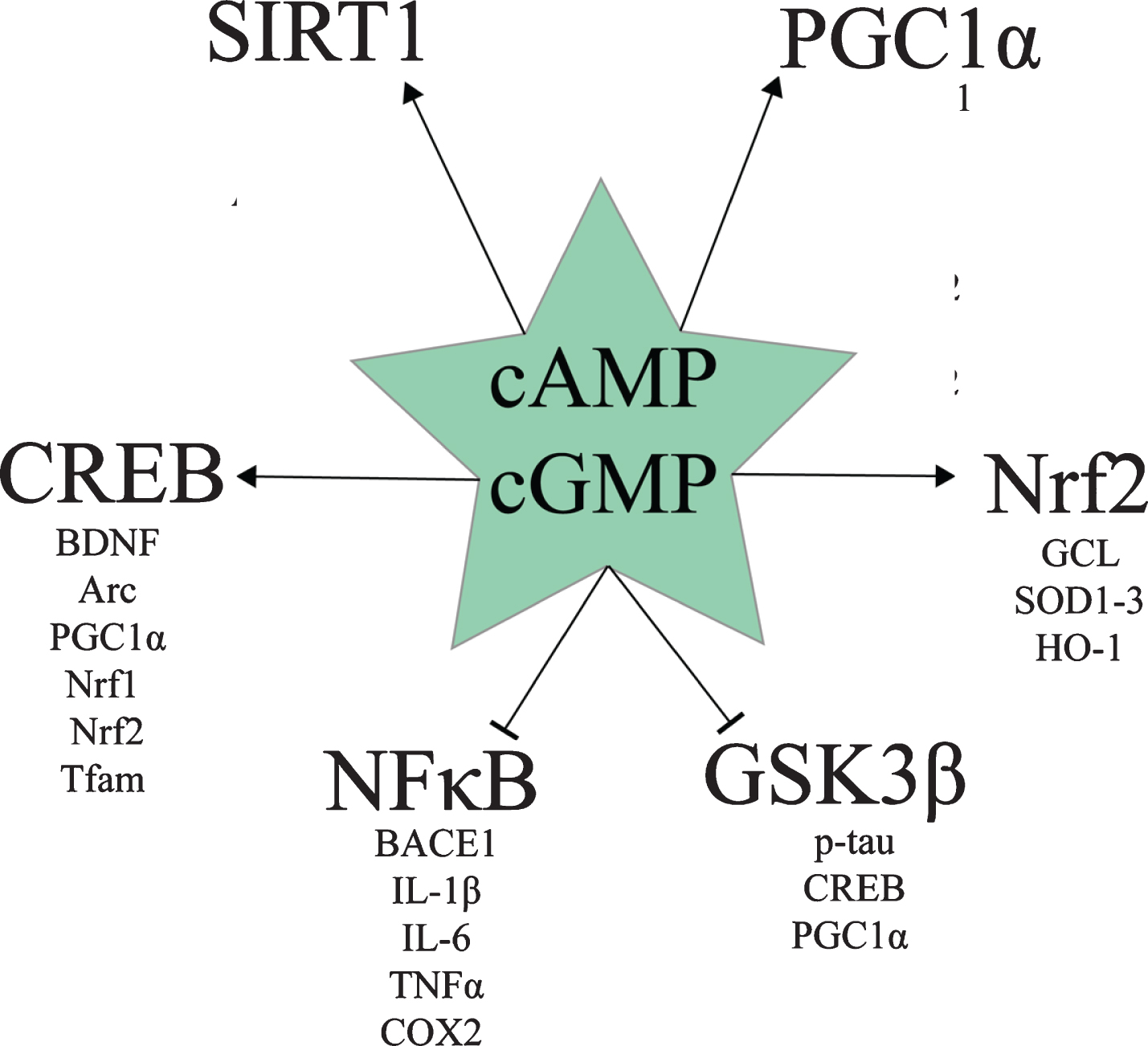
Various drugs and naturally-occurring compounds inhibit one or more PDE enzyme. Based on the various preclinical studies about PDE inhibition for AD, a review by Heckman et al. concluded that PDE2, PDE4, and PDE5 inhibitors appear to hold the most promise for treating AD [73]. Multiple benefits of increasing cAMP and/or cGMP signaling with various PDE inhibitors can be ascribed to the effect of these pathways on cAMP response element binding protein (CREB), Sirtuin 1 (SIRT1), peroxisome proliferator-activated receptor-γ coactivator-1α (PGC1α), nuclear factor erythroid 2-related factor 2 (Nrf2), nuclear factor κ-light-chain-enhancer of activated B cells (NFκB), and glycogen synthase kinase 3β (GSK3β) signaling (Fig. 1). The following section is a discussion of the effects of cAMP and cGMP levels on these molecular pathways.
Fig.1
Downstream targets of cAMP and cGMP signaling relevant to AD.
cAMP, cGMP, and CREB
Both cAMP/PKA and cGMP/PKG signaling activate CREB by phosphorylating it at Ser133 [74–79]. pSer133-CREB protein levels are decreased in the AD hippocampus [80], the AD prefrontal cortex, and AD peripheral blood mononuclear cells [81]. pSr133-CREB activates the transcription of mitochondrial D-loop DNA and nuclear DNA [82, 83]. Activated CREB induces genes involved in synaptogenesis and memory formation [84], such as brain derived neurotrophic factor (BDNF) and Activity-regulated cytoskeleton-associated protein (Arc), and neuronal survival, such as BDNF and Bcl2 [84–90], as well as genes involved in mitochondrial biogenesis and antioxidant defense, such as PGC1α, Nuclear respiratory factor 1 (Nrf1), Nrf2, and mitochondrial transcription factor A (Tfam) [83, 91].
cAMP, cGMP, SIRT1, and PGC1α
Both cAMP/PKA and cGMP/PKG signaling promote mitochondrial biogenesis and antioxidant gene expression via CREB- and SIRT1-mediated PGC1α activation [92–105]. In addition to activating CREB [74–79], which induces PGC1α transcription [83, 91], both cAMP/PKA and cGMP/PKG signaling activate SIRT1 [94–96, 98, 104–110]. SIRT1 exerts multiple beneficial effects in AD, including downregulating pro-amyloidogenic β-secretase (BACE1) and upregulating anti-amyloidogenic A Disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) [106], as well as deacetylating and thereby post-translationally activating PGC1α [97, 111–118].
PGC1α induces the core regulators of mitochondrial biogenesis, including Nrf1, Nrf2, and Tfam [93]. PGC1α induces antioxidant genes when bound to Forkhead box O3 (Foxo3a) and deacetylated by SIRT1, namely manganese superoxide dismutase (MnSOD), catalase, peroxiredoxins 3 and 5, thioredoxin 2, thioredoxin reductase 2, and uncoupling protein 2 (UCP2)[112].Brain PGC1α overexpression has also been shown to upregulate glutathione peroxidase 1 (GPx1), UCP4, and UCP5 [119]. PGC1α also induces genes involved in oxidative respiration [92, 103] and fatty acid β-oxidation [120]. Conversely, in a peroxisome proliferator-activated receptor γ (PPARγ)-dependent mechanism, PGC1α represses the expression of BACE1 [121, 122].
These downstream effects of PGC1α signaling are important for AD treatment because BACE1 is the rate-limiting enzyme in Aβ generation [123], and neural mitochondrial biogenesis and antioxidant enzyme expression are severely disrupted in AD. In the AD patient hippocampus, mitochondrial biogenesis is impaired as shown by decreased number of mitochondria in neurons and lower expression of PGC1α, as well as of downstream Nrf1, Nrf2, Tfam [93], and MnSOD [23, 112]. This deficit in PGC1α signaling in AD appears to begin very early. PGC1α, Nrf1, Nrf2, and Tfam were downregulated at the mRNA level as early as 1 month of age in a transgenic AD mouse model, even before a significant concentration of Aβ oligomers had formed [91]. In rodent brain hypo-perfused like AD and vascular dementia brains are [119, 124–127], PGC1α neural overexpression improved cognitive deficits and the metabolic activity of hypoxic neurons [119], indicating the therapeutic potential of modulating PGC1α signaling pharmacologically.
Therapeutically, PDE inhibitorsprovide a way to modulate this AD deficit in pSer133-CREB and PGC1α signaling. In APPswe M17 cells, cAMP rescued CREB phosphorylation and PGC1α expression in a PKA-dependent manner [93]. Furthermore, cAMP signaling may promote not only synaptogenesis, memory formation, neuron survival, and mitochondrial biogenesis, but also restoration of the physiologic balance between mitochondrial biogenesis and mitophagy. For example, 15 mg/kg/day of the cAMP-elevating PDE7 inhibitor S14 given orally to 6-month-old APP/PS1 mice rescued memory deficits, increased neurogenesis, and rescued mitochondrial mass by increasing mitochondrial biogenesis and decreasing Aβ-induced excessive mitophagy [128]. This balancebetween mitochondrial biogenesis and mitophagy appears to be disrupted in AD patients’ hippocampal pyramidal neurons as shown by the PGC1αdownregulation [93, 129], as well as the finding that many of the few surviving mitochondria accumulate in autophagosomes [22, 130]. Ultimately, in hypoperfused rodent brain, the nonspecific PDE inhibitor propentofylline restores mitochondrial ATP production [131, 132], suggesting that one outcome of modulating CREB and PGC1α signaling with PDE inhibitors is enhanced mitochondrial ATP production.
cAMP, cGMP, and Nrf2
Downstream of CREB and PGC1α, Nrf2 is a transcription factor that induces various genes involved in antioxidant defense, detoxification, autophagy, and cytoprotection, such as glutamate–cysteine ligase (the rate-limiting enzyme in glutathione synthesis), SOD1-3, and heme oxygenase 1 (HO-1) [133–136]. Nrf2 is active when it is localized to the nucleus; however, nuclear Nrf2 levels are decreased in AD patients’ hippocampal cells, indicating decreased Nrf2 activity in AD [137]. Both cAMP and cGMP appear to increase Nrf2 signaling.
cAMP appears to increase Nrf2 signaling in most cell types [138–140]. For example, in mouse and human hepatocytes, cAMP analogue 8-Bromoadenosine-cAMP upregulated Nrf2 target gene expression and antioxidant response element (ARE) transcriptional activity in a PKA-dependent manner [138]. Treatment of keratinocytes and melanocytes with either α-melanocyte stimulating hormone or forskolin (both of which increase cAMP) increased Nrf2 and Nrf2 target gene expression [139]. The PDE4 inhibitor roflumilast has been shown to replenish Nrf2 levels in bronchial epithelial cells infected with respiratory syncytial virus [141, 142]. Therefore, cAMP may increase Nrf2 signaling in neurons and other brain cells [138–140].
cGMP may also activate Nrf2 signaling, as in human bronchial epithelial cells, arsenite activated Nrf2 in a cGMP/PKG-dependent manner [143]. cGMP/PKG signaling has been postulated to be involved in Nrf2 activation in the heart as well [144]. In C6 glioma cells, nitrated cGMP activated Nrf2 [145]. In colon carcinoma cells, nitric oxide activated Nrf2 [146]. This points to context-dependent activation of Nrf2 by cGMP, nitrated cGMP, and NO in diverse cell types. Therefore, both cAMP and cGMP (and thus PDE inhibitors) may promote Nrf2 activity in brain cells.
cAMP, cGMP, and NFκB
Another important therapeutic target in AD is NFκB. NFκB is a family of transcription factor dimers consisting of two of five possible protein subunits: c-Rel, RelA/p65, RelB, p105/NFKB1, and p100/NFKB2. NFκB dimers containing the RelA/p65 subunit induce inflammatory gene expression (e.g., pro-inflammatory cytokines IL-1β, IL-6, and TNFα, chemokines IL-8, RANTES, MIP1, enzymes COX2 and iNOS, adhesion molecules VCAM1 and ICAM1 [147]), as well as the expression of BACE1 [34, 35]. Inflammatory NFκB is activated by oxidative stress [148], diverse pathogen- and damage-associated molecular patterns via toll-like receptors (TLR) [149, 150], fibrillar Aβ via TLR2 [32], pro-inflammatory cytokines (e.g., TNFα via the TNF receptor and IL-1β via the IL-1 receptor) [147], and other immunoreceptors [37, 148], such as the B-cell antigen receptor (BCR) in conjunction with protein tyrosine kinases [151]. Through signaling mediators such as MYD88 [32], TLR, for example, activate inhibitor of NFκB kinase (IKK), which phosphorylates inhibitor of NFκB protein α (IκBα), causing its ubiquitination and degradation, releasing c-Rel, RelA/p65, and RelB containing dimers to translocate to the nucleus and induce gene transcription [151].
cAMP/PKA signaling inhibits NFκB in most but not all cell types and contexts [147, 152]. With relevance to AD, raising cAMP has been shown to inhibit NFκB in microglia stimulated with either TNFα or lipopolysaccharide (LPS) [147, 153, 154]. Disruption of inflammatory NFκB signaling by cAMP has been shown to occur via multiple mechanisms [147, 155]. Specifically, cAMP/PKA-phosphorylated CREB upregulates the IκBα gene [147, 156, 157]. cAMP inhibits IKKβ, decreasing IκB phosphorylation, ubiquitination, and degradation [147, 157]. cAMP inhibits the ubiquitination and degradation of IκBα [147, 151]. Phosphorylated CREB outcompetes NFκB for their common requisite binding partner CREB binding protein (CBP), shifting gene transcription towards CREB and away from NFκB-mediated expression [147]. cAMP induces the exchange of activating p65-p50 NFκB dimers for repressive p50-p50 dimers [147]. cAMP induces the exchange of activating CREB-c-Jun dimers for repressive CREB-ICER dimers in the promoters of certain genes, such as TNFα [147]. cAMP activates the expression of c-Fos, which prevents p65 homodimers from binding to promoters [147].
cGMP inhibits NFκB indirectly via cAMP [155, 158]. cGMP inhibits PDE3, raising cAMP levels. In vascular smooth muscle cells, raising effective cGMP levels with a NO donor or C natriuretic peptide resulted in cGMP-induced PDE3 inhibition, cAMP accumulation, and cAMP/PKA-dependent NFκB inhibition [158].
Therefore, raising cAMP and cGMP levels with PDE inhibitors would likely inhibit NFκB in brain microglia, decreasing neuroinflammation and β-amyloidosis in AD [34, 35, 147, 153, 154, 156–158].
cAMP, cGMP, and GSK3β
GSK3β is the primary kinase of 45 of the 85 phosphorylable Serine and Threonine residues on tau [159–162]. If PKA phosphorylates tau before GSK3β does, then paired helical filament formation is suppressed, whereas if GSK3β phosphorylates tau before PKA does, then paired helical formation is promoted [163], suggesting that high PKA activity and low GSK3β activity may suppress neurofibrillary tangle formation. Furthermore, GSK3β phosphorylates and thereby inhibits CREB and PGC1α [101, 164–169].
Multiple lines of evidence suggest that both cAMP and cGMP inhibit GSK3β. cAMP/PKA suppresses GSK3β activity in most cell types by phosphorylating it at Ser9, including in cortical neurons [170–172], platelets [173], mouse spermatozoa [174], Rat1, NIH 3T3, and HEK293 cells [175], but not in murine melanoma cells [176]. Multiple AD rodent model studies have shown that the cGMP-elevating PDE5 inhibitor sildenafil inhibits GSK3β, suggesting that cGMP typically inhibits GSK3β in the brain [177–181].
Gαs/AC/cAMP/PKA signaling alterations in AD
The beneficial effects of cAMP and cGMP on CREB, PGC1α, Nrf2, NFκB, and GSK3β signaling, however, appear to be lost during the pathogenesis of AD, as preclinical studies and neuropathology evidence suggestthat the AC/cAMP/PKA and NOS/NO/sGC/cGMP pathways may be pathologically disrupted in AD patients’ brains [79, 182–205]. In preclinical models, both BACE1/Aβ and tau pathology disrupt cAMP signaling. Aβ25–35 exposure transiently increases cAMP levels, suppressing neuronal glucose uptake in a cAMP/PKA-dependent manner [206], but long-term Aβ42 exposure decreases cAMP levels [182]. Sublethal doses of Aβ42 (but not Aβ25–35) impair KCl- and N-methyl-D-aspartate-induced CREB phosphorylation [193]. BACE1 overexpression decreases cAMP levels, PKA activity, and CREB phosphorylation independently of Aβ [199]. Human tau overexpression inhibits PKA, decreases phosphorylation of CREB, decreases phosphorylation of glutamate receptor 1 [200] (GluA1, phosphorylation of which increases its membrane localization, facilitating conductivity, long-term potentiation, and memory formation [207]), decreases phosphorylation of tropomyosin receptor kinase B [200] (TrkB, a receptor for BDNF [208]), and downregulates BDNF mRNA and protein levels [200]. Therefore, both Aβ and tau pathology appear to disrupt cAMP/PKA signaling.
In neuropathology studies, it appears that AC/cAMP/PKA signaling may be disrupted in AD patients’ brains. To begin with, AC protein and activity levels (especially Gαs-stimulated) have generally been found to be decreased in AD patients’ brains [183–187, 201–205]. Basal, Gαs-stimulated, and forskolin-stimulated AC activity has been found to be decreased in the AD hippocampus [201–203]. However, Gαs-stimulated but not basal or forskolin-stimulated AC activity was found to be decreased in the AD hippocampus, temporal cortex, and angular gyrus in one study [204]. AC-I protein levels were significantly decreased in the AD hippocampus [205]. Parietal cortex membrane AC-I and AC-II protein levels were significantly decreased in AD patients, as was Ca2 + /CaM-stimulated AC activity [183]. Basal AC activity was found to be decreased in the AD frontal cortex, temporal cortex, and angular gyrus [184]. Gαs-stimulated but not basal or forskolin-stimulated AC activity was decreased in the AD superior temporal cortex [185]. Gαs-stimulated but not basal or forskolin-stimulated AC activity was decreased in the AD neocortex [186]. [3H]forskolin binding was decreased in the AD frontal cortex, suggestive of fewer AC catalytic subunits to bind to [187]. These findings overall suggest that a decrease in AC activity may occur in AD brains.
cAMP levels were not altered in the AD patient cerebrospinal fluid (CSF)in two studies [197, 198], but they were elevated in one study in a manner that correlated with tau pathology [209]. Increased cAMP levels were found in AD microvessels [189], and in the cerebral cortical and meningeal vessels in the frontal and temporal cortex and hippocampus in association with vascular Aβ [188]. This suggests that cAMP levels and localization may be subtly dysregulated in AD brains.
Downstream of cAMP, PKA activity appears to be suppressed in AD in most studies [185, 190–192, 194, 200, 210, 211]. [3H]cAMP binding to cytosolic but not particulate PKA was decreased in the AD patient entorhinal cortex and subiculum [190]. Neither soluble nor particulate PKA activity were altered in the AD patient superior temporal cortex in one study [185], nor was PKA activity altered in AD patient cerebral microvessels [210]. In another, however, decreased PKA activity was found in the AD patient temporal cortex [192]. Decreased PKA activity was also observed in the AD patient medial temporal cortex [191]. Regulatory 51 kDa PKA-RIIα and PKA-RIIβ and catalytic PKA-Cβ protein levels were decreased in the AD patient medial temporal cortex [191]. Catalytic PKA-Cα protein levels were decreased in the AD patient frontal cortex [194]. However, a calpain-cleaved 47 kDa fragment of PKA-RIIα was increased in the AD patient medial temporal cortex [191]. In cultured hippocampal neurons, Aβ exposure resulted in PKA inhibition, higher PKA-RIIα protein levels, and less glutamate-induced CREB phosphorylation (the latter of which was reversed by cAMP-elevating PDE4 inhibitor rolipram) [211]. Overexpression of human tau was found to increase nuclear PKA-RIIα proteins [200], suggesting that the concerted action of Ca2+-activated calpain proteases, Aβ, and tau may promote expression of the 47 kDa PKA-RIIα fragment in the nucleus. Silencing PKA-RIIα expression improved tau-induced deficits in PKA activity and CREB phosphorylation [200], suggesting that this nuclear 47 kDa PKA-RIIα upregulation in AD may suppress nuclear PKA activity and CREB phosphorylation. Therefore, overall PKA activity and nuclear PKA activity appear to be suppressed in AD [185, 190–192, 194, 200, 210, 211].
GC/cGMP/PKG signaling alterations in AD
Preclinical and neuropathology data suggest that NOS/NO/sGC/cGMP signaling may be disrupted in AD as well. Aβ inhibits the NOS/NO/sGC/cGMP/CREB pathway required for long-term potentiation in hippocampal slices [79]. NOS activity has been found to be decreased in the AD superior frontal gyrus and hippocampus [196]. The activity of NO-activated sGC—but not basal sGC nor particulate GC (pGC)—was found to be decreased in the AD superior temporal cortex [195].
Upstream of the maintained pGC signaling [195], elevated BNP levels have been associated with cognitive disorders in elderly patients 75–78 years old but not older [212, 213], and multiple studies have found associations between increased BNP levels in blood or plasma andmild cognitive impairment (MCI) occurrence, severity, or risk, conversion of MCI to AD, or dementia [213–223]. For example, one study found plasma BNP levels were associated with diagnosis of MCI or AD as well as CSF Aβ42 levels; furthermore, plasma BNP levels were found to be increased in MCI and AD patients [213, 214]. This suggests that BNP/pGC/cGMP signaling may be either maintained or increased in MCI and AD [195, 212–214].
Overall, cGMP levels were found to be decreased in AD patient CSF [197, 198], which correlated with CSF levels of Aβ42 [197], depression symptoms [198], and cognitive dysfunction as measured by The Mini-Mental Status Exam (MMSE) [197, 198]. This suggests that AD is characterized by a profound and disease severity-correlating decrement in NOS/NO/sGC/cGMP signaling.
Increasing cAMP and cGMP signaling to treat AD
Thus, PDE inhibitors would be predicted to restore disrupted cAMP/PKA, cGMP/PKG, CREB, PGC1α, and Nrf2 signaling, activate SIRT1, repress NFκB-induced BACE1 expression, Aβ generation, and inflammation,inhibit GSK3β-mediated tau phosphorylation,and promote neuron survival, synaptogenesis, mitochondrial biogenesis and survival of mitophagy, ATP generation, antioxidant and detoxification enzyme expression, and memory formation—all of which would be crucial therapeutic benefits in the fight to prevent, halt, and reverse AD etiopathogenesis. In this narrative review, we overview efficacy-testing clinical trials, epidemiological studies, and meta-analyses to investigate whether PDE inhibitors can prevent or treat AD, MCI, or dementia in a disease-modifying fashion.
CLINICAL TRIALS AND EPIDEMIOLOGY OF PDE INHIBITORS
Vinpocetine
Vinpocetine inhibits PDE1, raising cAMP and cGMP [224]. Discovered in the 1970s by C. Szántay and C. Lörincz et al. independently, vinpocetine has been approved in European countries for the treatment of dementia and stroke for over 30 years, and it is available in the United States as a dietary supplement [225]. Preclinical data demonstrate that vinpocetine can rescue cognitive deficits in a rodent AD model [226], increase CREB phosphorylation [227] and BDNF expression [228], downregulate BACE1 [226], upregulate Nrf2 mRNA expression [229], decrease oxidative stress [226, 228–239], mitochondrial dysfunction [240, 241], and apoptosis [230, 239], inhibit GSK3β [226, 228], NFκB [229, 231, 232, 239, 242–250], and the NLPR3 inflammasome [243], decrease levels of pro-inflammatory cytokines IL-1β [226, 229, 231, 232, 236, 237, 243], IL-6 [236, 237, 239, 251], and TNFα [226, 229, 231, 232, 242, 243, 246], and arrest the cell cycle in the G1 phase by downregulating cyclin D1 and upregulating p27(Kip1) [252].
Despite these promising preclinical findings, there is very little evidence about vinpocetine for the treatment of AD specifically, and what little there is is disappointing. In a 1-year-long 1989 open-label pilot trial, 15 patients with AD showed no improvement or delay in decline on the CGI at doses of 30, 45, or 60 mg vinpocetine once daily [253]. In a multicenter, double-blind, placebo-controlled trial, subgroup analysis showed that no significant treatment effect was observed on the CGI, the Short Cognitive Performance Test/Syndrom-Kurztest (SKT), the Brief Cognitive Rating Scale, or the Sandoz Clinical Assessment Geriatric (SCAG) in AD patients [254]. These were the only clinical trials of vinpocetine in AD that could be found at the time of this writing, and both seem to indicate that vinpocetine is ineffective for AD. By contrast, the evidence of vinpocetine for the treatment of all-cause dementia is inconclusive but relatively promising.
In a 2001 meta-analysis of 3 out of 39 studies that met inclusion criteria, 327 patients with dementia or cognitive impairment due to other disorders took vinpocetine or placebo [255]. The vinpocetine treatment group had significantly better scores on the Mini Mental Status Questionnaire (MMSQ), the SCAG, and the CGI. However, the authors noted that vinpocetine is not indicated for the treatment of AD due to the little and inconsistent data available for this indication [255].
In a 2003 Cochrane systematic review and meta-analysis of the three available unconfounded, double-blind, randomized, placebo-controlled clinical trials of vinpocetine for the treatment of 583 patients with dementia, 30 mg and 60 mg vinpocetine were found to produce significant benefit, including on the CGI and on the SKT attention and memory scale at up to and including 13 months. Due to the inconclusive nature of the data, however, the authors concluded vinpocetine is not indicated for dementia treatment [256].
In a 2012 clinical trial, moderately severe MCI patients who took vinpocetine for 18 months showed significant improvements in cognition on the MMSE and the Alzheimer’s Disease Assessment Scale-Cognitive Subscale (ADAS-Cog), daily activity on the Activities of Daily Living, mood on the Hamilton Depression Scale, and overall change in disease status according to patients and investigators on the patient global impression of change and the clinical global impression of change [257].
Compared to placebo, vinpocetine 5 mg twice daily for 3 months in a 2014 pilot study resulted in significantly improved memory and concentration in 56 patients with cognitive impairment due to either dementia of various etiologies or epilepsy [258].
Therefore, the evidence about vinpocetine for AD is very limited, but the two trials available seem to show that vinpocetine is not effective for this indication. However, vinpocetinedoes appear to hold promise for MCI and dementia patients, although further clinical trials with larger patient cohorts are required to further evaluate its efficacy for these indications.
Nicergoline
The ergot derivative nicergoline inhibits Ca2+/calmodulin-dependent PDE1 and cGMP-stimulated PDE2 activity [259] but activates the high Km cAMP-degrading PDE from heart [260]. In addition, it non-competitively inhibits Ca2+/Mg2+-dependent brain adenosine triphosphatase (ATPase). It exerts a complex effect on brain Na+/K+ ATPase whereby it activates the Na+/K+ ATPase at low concentrations but inhibits it at high concentrations [260, 261]. It also acts as a potent α1A adrenergic receptor antagonist [262]. Nicergoline has been available since the 1970s in over 50 countries and has been used to treat a variety of conditions, including AD, vascular dementia, acute and chronic peripheral circulation disorders, and cerebral infarction [263, 264].
In preclinical studies, nicergoline has been shown to restore cognitive deficits in AD mice [265], upregulate BDNF [266], reverse the age-associated cholinergic deficit and enhance K+-induced acetylcholine release [267, 268], activate catalase [269], decrease oxidative stress [265, 269–271], inflammation [265, 266], andapoptosis [265, 266, 272], and decrease levels of IL-1β, IL-6, and TNFα [266].
A 2001 Cochrane systematic review of 14 unconfounded, double-blind, randomized, placebo-controlled trials found that nicergoline provided mild to moderate dementia patients with significant improvements on the SCAG, the MMSE, and the CGI at 6 and 12 months [273]. According to this meta-analysis, there was no significant improvement on the ADAS-Cog at 6 or 12 months in a total of 342 patients, although there was a non-significant trend in favor of nicergoline on this scale [263, 273]. This is important since the ADAS-Cog is used exclusively with AD patients. The claim that differences on the ADAS-Cog were not significant at 6 months are based exclusively on Crook et al. 1996, an unpublished report from Pharmacia and Upjohn [273]. The claim that the change on the ADAS-Cog with nicergoline was not significant at 12 months is based on a 2001 multicenter, randomized, double-blind, placebo-controlled trial in 346 patients with probable mild to moderate AD, in which nicergoline 30 mg twice daily resulted in significantly improved cognition on the ADAS-Cog at 6 months and mean differences in ADAS-Cog scores between treated and control patients that grew increasingly large—including at 12 months [274]. According to the primary source poster abstract by Amaducci et al. 1999: “In the reduced patient cohort treated for 12 months, the ADAS-cog score difference between treatments after 3, 6, 9, 12 months was 0.89, 1.07, 1.45 and 1.64 respectively, increasingly in favour of nicergoline [274].” It was noted that AD patients receiving nicergoline improved over the course of the first 6 months and then deteriorated by month 12 [274]. However, the deterioration on the ADAS-Cog in nicergoline-treated AD patients was less severe than in placebo-treated patients, a benefit that became increasingly apparent up to and including at 12 months [274]. In other words, it appears that nicergoline resulted in a modest but significant improvement in AD symptoms on the ADAS-Cog at 6 monthsand produced no significant improvement at 12 months but rather slowed an increasingly precipitous decline at 12 months [274]. It is unclear why this study was included for the 12-month analysis but not for the 6-month analysis in the Cochrane review, nor is it clear whether this would change the conclusion of the Cochrane meta-analysis that the trend towards improvement on the ADAS-Cog at 6 months was not significant. The finding of significant improvement on the ADAS-Cog at 6 months was formally published after the Cochrane review article was in a journal in 2001 [275]. There was no mention in this formal paper of the trends toward a slowing of decline on the ADAS-Cog at 12 months [274, 275].
Two more recent trials, however, studied smaller groups of patientsand yielded negative results. In a 2017 uncontrolled pilot trial, 16 patients with early AD received 30 mg nicergoline twice daily for 1.5 years on average [276]. No statistically significant differences were noted in the severity of dementia, activities of daily living, cognition, and depressive symptoms between baseline and follow-up. However, nicergoline increased cerebral blood flow in frontal and parietal regions [276].A 2019 study in 22 patients with early AD found that, compared to acetylcholine esterase inhibitors alone, acetylcholine esterase inhibitors plus nicergoline preserved cerebral blood flow to the left temporal pole and the middle cingulate gyrus but did not result in any significant difference in dementia severity [277].
Nicergoline was found by a 2014 systematic review and meta-analysis to have a good safety profile, especially compared to other ergot derivatives, with no more adverse events reported than for placebo [278].
Therefore,it appears that nicergoline may be indicated for dementia patients [273]. There are mixed reports about whether the improvement on the ADAS-Cog at 6 months that nicergoline provided was significant, and there was a non-significant trend toward slower cognitive decline on this scale at 12 months [273–275]. However, two more recent pilot trials had disappointing results [276, 277]. This makes it inconclusive whether nicergoline is useful for the treatment of AD. Further clinical trials of nicergoline in AD involving more patients appear to be warranted.
Deprenyl/selegiline
Deprenyl, also known as selegiline, inhibits the calmodulin-dependent PDE1A2 and monoamine oxidase (MAO) [224, 279]. Discovered in the 1960s by Zoltan Ecseri at Hungarian drug company Chinoin, deprenyl is used clinically to treat Parkinson’s disease, major depressive disorder, and attention-deficit/hyperactivity disorder [280–282]. In preclinical studies, deprenyl has been found to increase CREB phosphorylation [283], activate Nrf2 [284–286] and catalase [283], decrease oxidative stress [284–287], inflammation [286], mitochondrial dysfunction [287], and apoptosis [287], and potentiate UVradiation-induced PARP1 activation yet downregulate PARP1 protein expression [288].
In the latest Cochrane systematic review and meta-analysis from 2003, which included 17 unconfounded, double-blind, randomized, placebo-controlled clinical trials, it was found selegiline significantly improved memory and cognition in AD patients at 4–6 weeks and 8–17 weeks, but not at 21–30, 35–56, or 65–82 weeks, and it improved activities of daily living at 4–6 weeks but not at later time points [289]. These 17 randomized controlled trials (RCTs) found significant improvements in memory on multiple scales, including the MMSE in 6 RCTs, the Randt Memory Index and the Wechsler Memory Scale in 2 RCTs each, and the Buschke Selective Reminding Test, prose recall, the Rey-AVL, and the ADAS-Cog in 1 RCT each. Sideeffects were reported, but very few were severe enough that a patient had to leave the trial, and there was no significant difference between the number of adverse events and trial withdrawals reported with selegiline and placebo. However, meta-analysis of the global rating scales (i.e., the Blessed Dementia Scale, Gottfries-Bråne-Steen scale (GBS), Global Deterioration Scale, and CGI) found no significant difference between selegiline and placebo. Birks and Flicker concluded that, “selegiline for Alzheimer’s disease has proved disappointing ... .there is [] no evidence of a clinically meaningful benefit for Alzheimer’s disease sufferers. This is true irrespective of the outcome measure evaluated, i.e. cognition, emotional state, activities of daily living, and global assessment, whether in the short, or longer term (up to 69 weeks), where this has been assessed [289].” Nevertheless, the authors’ own results do appear to point to modest and short-termyet statistically significant improvements in memory, cognition, and activities of daily living on various scales with selegiline treatment in AD patients, although the short-lived nature of these improvements do appear disappointing.
Cilostazol
PDE3 degrades cAMP and cGMP and is localized primarily to the endoplasmic reticulum membrane [290, 291]. Cilostazol is a PDE3 inhibitor used as an anti-platelet agent. Invented in the late 1980s by Otsuka Pharmaceutical Co., it is used clinically to treat intermittent claudication in peripheral vascular disease and to prevent stroke [292, 293]. Cilostazol has been shown in preclinical studies to rescue cognitive dysfunction and promote soluble Aβ brain efflux in a mouse model of cerebral amyloid angiopathy, decrease oxidative stress and apoptosis, suppress Aβ oligomerization and deposition [294, 295], inhibit GSK3β [168, 173], activate CREB [168, 295–299], induce mitochondrial biogenesis via cAMP/PKA/CREB/PGC1α signaling [298–301], induce Nrf2 and target gene expression [298, 302–305], decrease oxidative stress [306], upregulate SIRT1 protein levels [307], activate adenosine monophosphate-activated protein kinase [308, 309], inhibit NFκB [306–308, 310–312], downregulate BACE1 [307], and decrease levels of IL-1β [305], IL-6 [168, 305], and TNFα [298, 305].
Epidemiologically, taking cilostazol has been associated in a dose-dependent fashion with decreased risk of incident all-cause dementia in patients over 65 with an overall adjusted hazard ratio of 0.75 (meaning that patients over 65 taking cilostazol are 25% less likely to develop dementia compared to those not taking cilostazol), with high-dose cilostazol users having a 0.53 adjusted hazard ratio (meaning that high-dose cilostazol users are 47% less likely to develop dementia)[313]. Subgroup analysis revealed that cilostazol users who had ischemic heart disease or cerebral vascular diseases were significantly protected from dementia (adjusted hazard ratios 0.44 and 0.34, respectively) [313].
A retrospective study found that control patients deteriorated by ∼2 points on the MMSE per year, whereas cilostazol-treated patients had no change in MMSE scores per year, a simsignificant difference [314]. Subgroup analysis revealed that control MCI patients had an annual decrease in MMSE score of 4 points, whereas cilostazol-treated MCI patients had a very small, ∼0.2 point increase in MMSE score per year, a significant difference [314]. The differences between MMSE scores for healthy control patients and dementia patients were not significant, although it is worth mentioning that control dementia patients had an annual decrease in MMSE scores of ∼1.1, whereas cilostazol-treated dementia patients had an annual increase in MMSE scores of ∼0.4 points [314]. In other words, the cilostazol-treated dementia patients appeared to improve slightly more on the MMSE than cilostazol-treated MCI patients (though this trend was non-significant), but the effect of cilostazol in dementia patients may have been found to be non-significant simply because the control dementia patients deteriorated less rapidly than the control MCI patients did [314].
In another retrospective analysis comparing patients with mild dementia who received either donepezil alone or donepezil plus cilostazol, cilostazol users had a significantly slower rate of cognitive decline as measured by the MMSE [315].
In a 12-month case-control study in 60 patients with stable AD taking acetylcholine esterase inhibitors, cilostazol add-on usage was significantly associated with a decreased rate of cognitive decline on the MMSE [316].
In an open-label, uncontrolled pilot trial of 10 patients with AD, improvement in MMSE score was found at 6 months [69, 317]. In a randomized, controlled trial of 20 patients with AD and cerebrovascular disease, compared to taking aspirin or clopidogrel, taking 100 mg/day cilostazol was found to prevent decrements in scores on the Japanese ADAS-Cog, Trail Making Test-A, and the Revised Wechsler Memory Scale (logical memory-I) at 6 months. Only the control patients had decreased cerebral blood flow in the left middle temporal gyrus, whereas the cilostazol-treated patients had significantly increased blood flow in the right anterior cingulate lobe [318].
In a 6-month, randomized, double-blind, controlled trial of 36 patients with mild or moderate AD and white matter lesions, 100 mg cilostazol plus donepezil compared to donepezil alone prevented decreases in glucose metabolism in the parietal and frontal lobes compared to controls and preserved glucose metabolism in the left inferior frontal gyrus [319]. However, no significant differences were observed on the MMSE, the ADAS-Cog, or other outcome measures. That being said, cilostazol-induced improvements in glucose metabolism correlated with better scores on the ADAS-Cog [319]. Further trials of cilostazol are ongoing [69, 320].
Therefore, cilostazol usage may be associated with a decreased risk of dementia [313]. Itmay slow the rate of cognitive decline on the MMSE in MCI patients [314], mild dementia patients [315], and AD patients [69, 316–318]. However, the trial data about cilostazol in AD specifically is very limited and overall disappointing, with efficacy found by two pilot trials (one controlled, the other not) studying 30 total AD patients, but lack of efficacy found by a rigorous clinical trial studying 36 AD patients with white matter lesions [69, 317–319]. Based on these findings, future trials of cilostazol should focus on patients with MCI or early AD, especially those with comorbid cerebrovascular disease.
Denbufylline
Denbufylline inhibits PDE4, raising cAMP [321]. For unclear reasons, there is a dearth of preclinical evidence about denbufylline, although PDE4 inhibition is arguably one of the most promising PDE therapeutic targets for the purpose of cognitive enhancement and AD treatment according to the following preclinical evidence and a recent review of the preclinical evidence about PDE inhibitors for the treatment of AD [73]. For example, treatment with the PDE4 inhibitor rolipram resulted in long-lasting improvements in basal synaptic transmission, long-term potentiation, and working, reference, and associative memory in APP/PS1 transgenic AD mice [322]. In rats exposed to Aβ40 in the CA1 hippocampus, 0.5 mg/kg rolipram rescued memory deficits and CREB phosphorylation [323]. In rats microinfused with Aβ25–35 into the CA1 hippocampus, 0.1, 0.25 and 0.5 mg/kg/day intraperitoneal PDE4-inhibiting rolipram dose-dependently reversed memory deficits [324, 325], restored CREB phosphorylation and Bcl2 expression, decreased p65 NFκB and Bax expression [324], decreased ROS and malondialdehyde levels, rescued glutathione levels and SOD activity, upregulated thioredoxin, and inhibited the inducible iNOS pathway in the hippocampus [325]. In mice injected in the dentate gyrus with aggregated Aβ42, lentiviral RNA silencing of long-form PDE4D rescued Aβ42-induced cAMP decrements and memory deficits on the Morris water maze and the novel object recognition test while upregulating pSer133-CREB and BDNF and downregulating p65 NFκB, IL-1β, and TNFα [182]. In APPswe/PS1dE9 mice, 0.001 mg/kg of the PDE4D isoform-specific inhibitor GEBR-7b for three weeks at 5 months of age improved spatial memory on the object location test at 7 months of age without affecting CREB phosphorylation, BDNF expression, synaptic densities, Aβ levels, tau phosphorylation, or GSK3β activation [326]. Clinical trials have not been performed of any of these preclinically tested PDE4 inhibitors, possibly because the side-effect of emesis has hampered their clinical development [69]. However, these preclinical effects of PDE4 inhibition are likely reasonably representative of denbufylline’s preclinical effects.
In a randomized, multicenter, parallel group, double-blind, placebo-controlled clinical trial in 45 patients with mild to moderate AD not taking any other medications, 100 mg of denbufylline given twice daily for 3 months significantly improved scores on the SCAG, the CGI, the Digit Symbol Substitution Test, and the MMSE compared to placebo [327]. It also decreased delta activity and accelerated slow activity on imaging (which are increased in AD patients), indicating an increase in vigilance that correlates with symptomatic improvement [327]. Neither emesis nor any other adverse effects were reported [327].
In a trial in 226 AD patients and 110 patients with vascular or mixed dementia, 25, 50, or 100 mg denbufylline for 4 months significantly improved MMSE scores but only when these patients were considered as a single group [328]. Significantly more all-cause dementia patients who received denbufylline than those who received placebo had improved MMSE scores. No major adverse events were reported [328].
Therefore, the trial data about denbufylline in AD is very limited but somewhat promising. Emesis was not reported in association with this PDE4 inhibitor [327, 328]. Further trials of denbufylline in AD may be warranted.
Sildenafil
PDE5A specifically degrades cytoplasmic cGMP produced by sGC downstream of NO [71]. Sildenafil (also known as Viagra) inhibits PDE5, thereby raising cytoplasmic cGMP levels. Discovered in the late 1980s by Pfizer, sildenafil is used clinically to treat erectile dysfunction and pulmonary arterial hypertension [329, 330]. In preclinical rodent models of AD,sildenafil has been shown to rescue memory [177–180, 331–335], decrease Aβ levels [178, 179, 333–336] and tau hyperphosphorylation [177, 178, 180, 331], inhibit GSK3β [177–180] and JNK [331], decrease IL-1β, IL-6, and TNF-α secretion [335], upregulate pSer133-CREB [180, 333, 335], BDNF [180, 336], Arc [180], and Bcl2 [336], downregulate BACE1 [179], AβPP, caspase-3, and Bax [336], and decrease double-stranded DNA breaks and apoptotic cells [336]. For a recent review of sildenafil for AD, see Sanders 2020 [337].
Single-doses of 50 mg sildenafil have been shown in small groups of AD patients to decrease spontaneous neural activity in the right hippocampus, decrease cerebrovascular reactivity, and increase cerebral blood flow and the cerebral metabolic rate of oxygen [338, 339]. Based on this very limited preliminary evidence, clinical trials of sildenafil in AD patients are warranted.
A dose-limiting effect of sildenafil is that high doses of sildenafil raise cGMP levels to such an extent that cGMP-sensitive PDE2 is activated, degrading cAMP [72]. This effect has been shown to limit the effective dosage of sildenafil in metabolic conditions [72]. Therefore, future clinical trials should test sildenafil combined with a cGMP-stimulated PDE2 inhibitor, such as propentofylline [72, 340].
PF-04447943 and BI 409,306
In a phase II multicenter, double-blind, randomized, placebo-controlled trial, Pfizer’s PDE9A inhibitor PF-04447943 failed to improve scores significantly better than placebo on the ADAS-Cog, the Neuropsychiatric Inventory, or the CGI-Improvement scale [341].
In two multicenter, double-blind, parallel-group, randomized, placebo-controlled phase II studies, MCI and mild to moderate AD patients who took the PDE9 inhibitor BI 409306 developed by Boehringer Ingelheim did not show any significant difference on the Neuropsychological Test Battery total z-score at 12 weeks, nor on the ADAS-Cog11, the Clinical Dementia Rating scale-Sum of Boxes, or the Alzheimer’s Disease Cooperative Study-Activities of Daily Living scale [342].
In preclinical models of AD, PDE9 inhibition has been shown to rescue synaptic plasticity and memory and decrease dendritic spine density degeneration and cytotoxicity [213, 341, 343, 344]. However, PDE9 inhibitors PF-04447943 and BI 409306 have been shown to be ineffective in MCI and AD patients [341, 342]. This might bebecause PDE9A regulates natriuretic peptide-induced pGC-generated peri-plasma membrane cGMP, not NO/sGC-generated cytoplasmic cGMP [71]. As discussed, the NOS/NO/sGC pathway and cytoplasmic cGMP pools are disrupted in AD [195, 196], whereas NP/pGC/cGMP signaling and peri-plasma membrane cGMP are maintained or increased [195, 212–214], which might make increasing peri-plasma membrane cGMP with PDE9inhibitors a less viable therapeutic approach than replenishing cytoplasmic cGMP pools with PDE5 inhibitors [213].
Another consideration is that it is NP-activated pGC, not NO-activated sGC, that produces the peri-cytoplasmic cGMP that activates cyclic-nucleotide gated (CNG) channels [68]. Therefore, PDE9 inhibition might be expected to promote CNG activation, unlike PDE5 inhibition (Fig. 2) [68, 71]. CNG channels are non-specific cation channels that allow Ca2+ ions to influx into the cytoplasm of olfactory sensory neurons [345]. Excessive axonal Ca2+ influx induces Wallerian axonal degeneration, and excessive cytoplasmic Ca2+ influx induces excitotoxicity [346–349]. Olfactory sensory neurons are exposed to multiple sporadic AD risk factors, including combustion-derived carbon nanoparticles and microbial toxins [350–353]. AD pathogenic factors spread between neurons trans-synaptically in extracellular vesicles, such as exosomes [354–358], and tau pathology spreads through the brain in AD in a stereotypical fashion [2]. Olfactory sensory neurons’ axons project into the olfactory bulb [359], a brain region that is critically affected starting from the preclinical stages of AD [350, 351]. Thus, PDE9A inhibition might promote pGC/cGMP-activated CNG-mediated Ca2+ influx into olfactory sensory neurons, axon degeneration, excitotoxicity, and possibly propagation of AD pathologies to the olfactory bulb, possibly counterbalancing its positive effects and providing no net change in AD patients taking PDE9 inhibitors.
Fig.2
The effects of PDE5 versus those of PDE9 on cGMP.
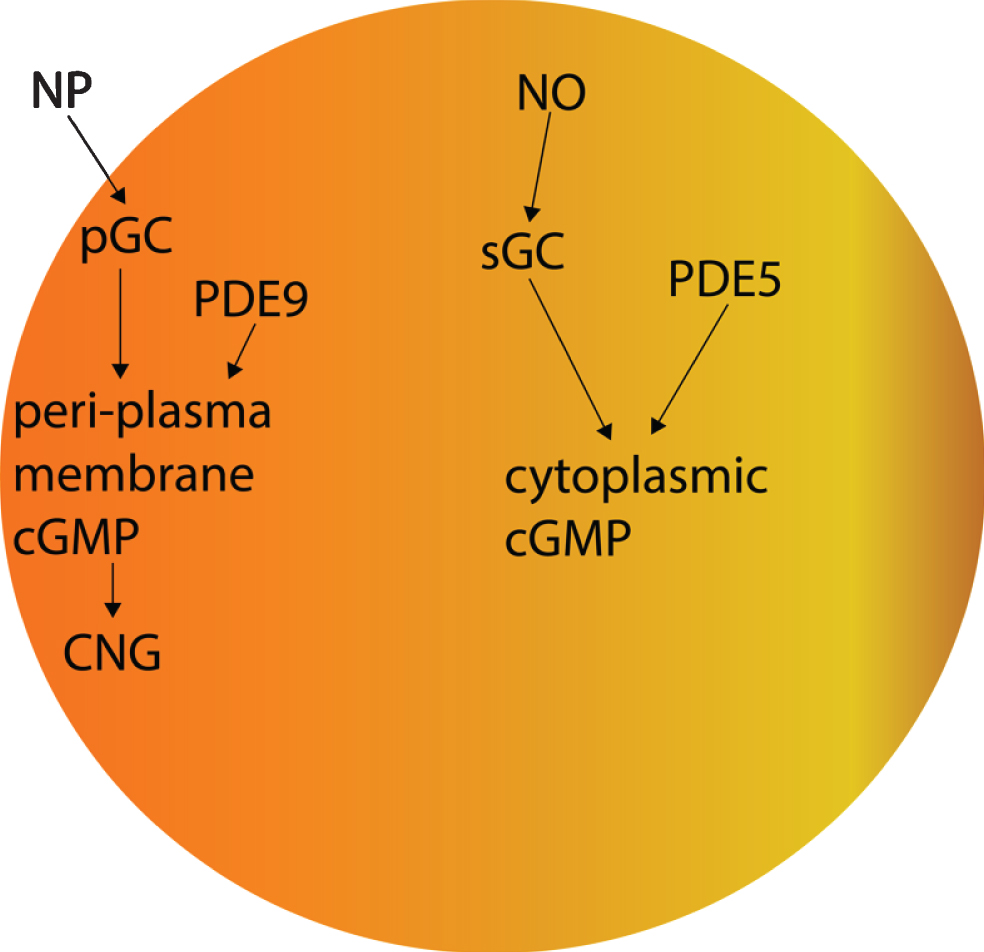
Furthermore, it is unclear whether CNG channel expression is restricted to the AD brain olfactory sensory neurons, since the CNG subunit CNGA1 is significantly upregulated at the mRNA level in AD patients’ entorhinal cortices (Fig. 3) [360], and the cGMP-sensitive CNGA3 subunit is significantly upregulated in AD patients’ hippocampi (Fig. 4) [345, 360]. CNG mRNA was rarely expressed in a non-AD patients’ temporal cortex, but when it was, it was typically expressed by neurons (Figs. 5–10) [360, 361]. This suggests the possibility that PDE9 inhibitors might promote pGC/cGMP/CNG/Ca2+-mediated axonal degeneration and excitotoxicity in the sporadic AD entorhinal cortex and hippocampus.
![CNGA1 upregulated in AD entorhinal cortex, adapted from Xu et al. 2018 with permission [360].](https://content.iospress.com:443/media/adr/2020/4-1/adr-4-1-adr200191/adr-4-adr200191-g003.jpg)
![CNGA3 upregulated in AD hippocampus, adapted from Xu et al. 2018 with permission [360].](https://content.iospress.com:443/media/adr/2020/4-1/adr-4-1-adr200191/adr-4-adr200191-g004.jpg)
Fig.5
Cell type-specific expression of CNGA1 in non-AD temporal cortex, adapted from Xu et al. and Darmanis et al. with permission [360, 361].
![Cell type-specific expression of CNGA1 in non-AD temporal cortex, adapted from Xu et al. and Darmanis et al. with permission [360, 361].](https://content.iospress.com:443/media/adr/2020/4-1/adr-4-1-adr200191/adr-4-adr200191-g005.jpg)
Fig.6
Cell type-specific expression of CNGA2 in non-AD temporal cortex, adapted from Xu et al. and Darmanis et al. with permission [360, 361].
![Cell type-specific expression of CNGA2 in non-AD temporal cortex, adapted from Xu et al. and Darmanis et al. with permission [360, 361].](https://content.iospress.com:443/media/adr/2020/4-1/adr-4-1-adr200191/adr-4-adr200191-g006.jpg)
Fig.7
Cell type-specific expression of CNGA3 in non-AD temporal cortex, adapted from Xu et al. and Darmanis et al. with permission [360, 361].
![Cell type-specific expression of CNGA3 in non-AD temporal cortex, adapted from Xu et al. and Darmanis et al. with permission [360, 361].](https://content.iospress.com:443/media/adr/2020/4-1/adr-4-1-adr200191/adr-4-adr200191-g007.jpg)
Fig.8
Cell type-specific expression of CNGA4 in non-AD temporal cortex, adapted from Xu et al. and Darmanis et al. with permission [360, 361].
![Cell type-specific expression of CNGA4 in non-AD temporal cortex, adapted from Xu et al. and Darmanis et al. with permission [360, 361].](https://content.iospress.com:443/media/adr/2020/4-1/adr-4-1-adr200191/adr-4-adr200191-g008.jpg)
Fig.9
Cell type-specific expression of CNGB1 in non-AD temporal cortex, adapted from Xu et al. and Darmanis et al. with permission [360, 361].
![Cell type-specific expression of CNGB1 in non-AD temporal cortex, adapted from Xu et al. and Darmanis et al. with permission [360, 361].](https://content.iospress.com:443/media/adr/2020/4-1/adr-4-1-adr200191/adr-4-adr200191-g009.jpg)
Fig.10
Cell type-specific expression of CNGB3 in non-AD temporal cortex, adapted from Xu et al. and Darmanis et al. with permission [360, 361].
![Cell type-specific expression of CNGB3 in non-AD temporal cortex, adapted from Xu et al. and Darmanis et al. with permission [360, 361].](https://content.iospress.com:443/media/adr/2020/4-1/adr-4-1-adr200191/adr-4-adr200191-g010.jpg)
Therefore, PDE9 inhibitor might do as much harm as good, equaling out to no net effect in clinical trials. PDE9 inhibitors should not be pursued further for the treatment of AD.
Caffeine
Caffeine is a non-specific/broad-spectrum PDE inhibitor. Like propentofylline, caffeine is a methyl xanthine derivative [362, 363]. In AD transgenic mice, caffeine partially rescued PKA activity and CREB phosphorylation in the striatum but not the frontal cortex [364], decreased plasma, cortical, and hippocampal Aβ levels [365, 366], suppressed BACE1 and PS1 expression [366], and improved working memory [366].
The evidence about whether caffeine lowers the risk of AD, cognitive decline, or dementia is mixed. In a 2015 meta-analysis of 31,479 individuals enrolled in 20 studies, caffeine intake from coffee or tea was not significantly associated with the incidence of cognitive disorders [367]. In a 2016 study (n = 6,467), caffeine intake was associated with a lower risk of incident dementia and cognitive decline in women over the age of 65 [213, 364, 368]. In a 2016 meta-analysis of 29,155 participants enrolled in 11 prospective studies, no association was found between caffeine intake and dementia or cognitive decline, but high caffeine intake was significantly associated with a decreased risk of incident AD [369]. In a 2017 meta-analysis of 34,282 individuals enrolled in 9 prospective cohort studies, a J-shaped association was found whereby 1–2 cups of coffee daily intake was associated with lower risk of cognitive disorders, but the association for 3 cups of coffee or more was not significant [370]. However, a 2018 dose-response meta-analysis of 8 prospective studies found no significant association between caffeine dosage and risk of AD or dementia [371]. Nevertheless, a 2019 study found that compared to drinking less than two cups of coffee a day, lifetime drinking two or more cups of coffee a day was significantly associated with reduced Aβ plaque burden in 411 cognitively-intact older adults [372]. Therefore, caffeine intake may be associated with a decreased risk of dementia, cognitive decline, and/or AD. No clinical trials of caffeine in AD have been conducted.
Propentofylline
Perhaps the most promising PDE inhibitor for the treatment of AD is propentofylline. Propentofylline is a methyl xanthine derivative like caffeine that acts as a relatively potent and nonspecific cAMP/cGMP PDE inhibitor and adenosine reuptake inhibitor [340, 362, 363]. Propentofylline has been shown in preclinical models to suppress Aβ plaque deposition, tau hyperphosphorylation, GSK3β activation [373], microglial ROS generation [374–378], glutamate production, LPS-induced microglial IL-1β and TNFα secretion [375–377], Aβ-induced IL-1β secretion [373], and microglial proliferation [374–377]. It has been shown topromote anti-inflammatory regulatory T cell proliferation [379], restoration of ATP production [131, 132], and cerebral metabolic response to a memory task [380]. It can enhance irradiation-induced G1/S transition block [381]. It has also reversedaluminum-induced brain edema and hypoxia-like metabolic changes [382] and Aβ-induced memory deficits [383], as well as preventednerve cell death induced by either nerve growth factor withdrawal or Aβ [384–386].
Propentofylline has perhaps the most impressive history of phase III clinical trials of any of the PDE inhibitors reviewed in this manuscript.In a 1998 meta-analysis of four phase III, randomized, double-blind, placebo-controlled clinical trials ranging from 6 to 14 months, 300 mg of propentofylline taken thrice daily 1 hour before meals provided consistently significant improvements both in patients with AD and patients with vascular dementia [375]. These benefits persisted even after treatment cessation, suggesting a disease-modifying rather than a purely symptomatic improvement in these dementia patients [375]. Subgroup analysis revealed that, at 6 months, AD patients treated with propentofylline demonstrated significantly improved global function on the GBS, cognition on the MMSE and the SKT, and activities of daily living on the Nürnberger Altersbeobachtungs-skala (NAB questionnaire). At the final visit, treated AD patients exhibited significant improvements in all categories tested, including on the GBS, CGI item II (global improvement), SKT, MMSE, and NAB [375].
A 2003 Cochrane systematic review of unconfounded double-blind, randomized, placebo-controlled clinical trials in dementia patients found that propentofylline treatment resulted insignificantly improved cognition at 3, 6, and 12 months (including on the MMSE at 12 months), global assessment at 3 months, dementia severity at 3, 6, and 12 months (including on the CGI at 12 months), and activities of daily living at 6 and 12 months [376]. However, data from an additional 1200 patients in randomized clinical trials was not shared with the authors by the drug’s manufacturer, Aventis [376]. An update on this systematic literature search performed in 2008 yielded no new clinical trials of propentofylline in dementia patients [376].
In a 1999 18-month-long multinational, randomized, double-blind, placebo-controlled clinical trial in 486 mild to moderate AD patients, 300 mg propentofylline thrice daily 1 hour before meals resulted in significantly improved cognitive performance on the ADAS-Cog and global function on the Clinician’s Interview-Based Impression of Change Plus Caregiver Input (CIBIC-Plus) [376, 387–391]. Benefits persisted 6 months after treatment cessation in the 18-month clinical trial as well, further suggesting a disease-modifying effect [387, 389, 391]. The results of this trial were presented in several poster abstracts [376, 387–391]. This study was listed in the Cochrane review but not included in the meta-analysis due to insufficient raw data from it being available [376].
However, a 2007 book chapter claimed that after two successful phase III clinical trials (there were more than two), an18-month trial “failed to show benefit, and development was discontinued [392].” The author did not cite this claim, making its source unclear. However, a MedScape article published in 2000claimed that the 18-month-long Propentofylline Long-term Use Study (PLUS) showed no significant difference between treatment and placebo in AD patients, leading Aventis to discontinue development of the drug [393]. According to this article, in the late 1990s, the European Agency for the Evaluation of Medicinal Products rejected Aventis’s application to market propentofylline for the treatment of AD and vascular dementia [393], andin 1999, the manufacturer announced a phase III trial of the drug in AD patients to address “regulatory concerns,” but this was later cancelled [393]. The article does not contain any references. A 2008 book reproduced the preceding claimswithout citing its source [394]. Neither of these books nor the MedScape article reference the successful 18-month-long phase III clinical trial presented in the 1999 poster abstracts. It is unclear to this author whether there were two 18-month trials, one successful and one not, or one single trial that this MedScape article and the subsequent two books misreported. Regarding safety, the drug was well tolerated in these studies for periods of up to 18 months [376, 387, 389, 391, 395].
Now, in the UK, propentofylline is used for the treatment of canine cognitive dysfunction [396], which has been proposed as a superior preclinical model of AD compared to rodent models since dogs naturally develop age-associated Aβ plaques with AD-like symptoms [397].
An economic analysis found that adding propentofylline to the standard of care for patients with mild to moderate AD in Sweden would save 3.8–7.6% of the costs associated with caring for this population [398]. A Canadian economic analysis based on clinical trials of 12 months’ duration found that treating dementia patients with propentofylline would improve health outcomes and reduce home care and caregiver costs [399].
Propentofylline is the most effective inhibitor out of four PDE-inhibiting xanthine derivates tested (i.e., pentoxifylline, propentofylline, torbafylline, and albifylline) for all of the PDE isoforms that were measured (i.e., PDE1, PDE2, PDE3, and PDE4), with it being particularly effective at inhibiting cGMP-stimulated PDE2 and PDE4 [340]. This is a remarkable selectivity profile for several reasons.First, this makes propentofylline the only PDE inhibitor reviewed here that inhibits more than one of the most promising to inhibit in AD PDEs (i.e., PDE2 and PDE4) [69, 73, 340]. Second, inhibiting multiple PDEs and boosting both cAMP and cGMP might offer synergistic improvement of CREB, PGC1α, and Nrf2, etc., signaling [72]. Third, high doses of PDE5 inhibitor sildenafil have the side-effect of activating cGMP-sensitive PDE2 [72], resulting in cAMP degradation and potentially disrupting PKA-mediated CREB and PGC1α signaling [72, 82, 93]. This suggests that propentofylline and sildenafil might provide AD and vascular dementia patients with synergistic disease-modifying benefit by inhibiting PDE2, PDE4, and PDE5 simultaneously. Propentofylline, sildenafil, donepezil, and memantine should be compared to donepezil and memantine (standard of care) alone in future preclinical studies and clinical trials for the treatment of AD.
CONCLUSION
Based on the preceding discussion, it can be concluded that modulating cyclic purine nucleotide levels with certain PDE inhibitors can prevent and treat AD, MCI, and dementia in a disease-modifying fashion (see Table 1 for summary of the clinical effectiveness of the PDE inhibitors discussed). Caffeine and cilostazol may be associated with a decreased risk of incident dementia, cognitive decline, and AD [213, 313–316, 364, 368–370, 372], suggesting that PDE inhibitors may help prevent AD. The clinical trials of denbufylline and sildenafil are promising but very preliminary [327, 328, 338, 339], so no conclusions can be drawn from them. Clinical trials of PF-04447943 and BI 409,306 have shown a lack of efficacy [341, 342], which might be because PDE9 inhibition increases peri-plasma membrane cGMP (rather than cytoplasmic cGMP) and allows Ca2+ influx through CNG [68, 71, 213]. The clinical trials of vinpocetine [253, 255–258], cilostazol [69, 317–319], and nicergoline are preliminary and mixed [273–277], with cilostazol and nicergoline likely being the most promising and the worthiest of further study of these three [273–277]. Deprenyl/selegiline clinical trials show short-term but not long-term benefits, making deprenyl somewhat disappointing [289]. By contrast, propentofylline has been shown in five phase III clinical trials to improve cognition, dementia severity, activities of daily living, and global assessment in mild-to-moderate AD patients on multiple scales [375, 376, 387–391], including on the ADAS-Cog and the CIBIC-Plus at 18month in a phase III clinical trial that was presented in several 1999 poster abstracts [376, 387–391]. However, two books published in 2007 and 2008, respectively, claimed based, apparently, on a MedScape article published in 2000 that an 18-month phase IIIb clinical trial failed to show efficacy, so propentofylline was discontinued [392–394]. It is unclear whether there were two 18-month-long phase III trials of propentofylline, or whether the MedScape article was inaccurate. Ultimately, propentofylline may be indicated for the treatment of patients with mild-to-moderate sporadic AD. Regardless, propentofylline is used now to treat canine cognitive dysfunction [396]. Like human AD, canine cognitive dysfunction involves age-associated wild-type Aβ deposition, making it a superior preclinical model of AD compared to rodent models [397]. Ironically, this impliesthe hypothesis that if propentofylline treats canine cognitive dysfunction, then it may treat human sporadic AD.
Table 1
| Drug | Phosphodiesterase inhibited | Clinical effectiveness |
| Vinpocetine | PDE1 [224] | Ineffective for the treatment of AD [253, 254]. May hold promise for the treatment of MCI and dementia [255–258]. |
| Nicergoline | PDE1 and cGMP-stimulated PDE2 [259] | May be effective for the treatment of dementia [273]. Inconclusive whether effective for the treatment of AD [273–277]. |
| Deprenyl/selegiline | PDE1A2 [224, 279] | Only short-term improvements in AD [289]. |
| Cilostazol | PDE3 | May be associated with a lower risk of incident dementia [313]. May slow cognitive decline in MCI, mild dementia, and AD patients [69, 314, 315, 316–318]. Inconclusive whether effective for the treatment of AD [69, 317–319]. |
| Denbufylline | PDE4 [321] | Inconclusive whether effective for AD [327, 328]. |
| Sildenafil | PDE5 [71] | No clinical trials yet performed of sildenafil for AD. |
| PF-04447943 and BI 409, 306 | PDE9 | Not effective for AD [341, 342]. |
| Caffeine | Broad-spectrum PDE inhibitor | No clinical trials performed. May decrease risk of dementia, cognitive decline, and AD [213, 364, 367–372]. |
| Propentofylline | Broad-spectrum PDE inhibitor [340] | May be effective and indicated for the treatment of vascular dementia and AD [375, 376, 387–391]. |
Treatment effects of propentofylline persisted months after treatment cessation in multiple trials, suggesting a disease-modifying effect in AD patients [375, 387, 389, 391]. We propose this disease-modifying effect may have been observed because, as a potent and non-specific/broad-spectrum PDE inhibitor [340], propentofylline raises cytoplasmic cAMP and cGMPenough to substantially enhance neural pSer133-CREB, SIRT1, PGC1α, and Nrf2 signaling [74–81, 92–105, 138–140, 143, 144], improving neural synaptogenesis [84–90], memory [84], mitochondrial biogenesis [83, 91, 93, 103], antioxidant [112], detoxification, and survival gene expression [84–90] while also inhibiting BACE1-inducing inflammatory NFκB [34, 35, 147, 153, 154, 156–158] and tau-phosphorylating and CREB-inhibiting GSK3β [159–162, 164–168, 170, 171, 173–175, 177–181].
Interestingly, the only PDE inhibitor that has completed efficacy-testing clinical trials as of 2020 that is known to inhibit more than one of the most promising PDEs to inhibit to treat AD according a review of preclinical studies (i.e., PDE2, PDE4, and PDE5) [73] is propentofylline [340]. Propentofylline is known to inhibit PDE2 and PDE4 [340], two of the three most promising therapeutic targets [73]. It is perhaps unsurprising, then, that propentofylline is also the PDE inhibitor with arguably the most promising clinical trials so far [375, 376, 387–391]. However, it is unknown whether propentofylline inhibits PDE5 as well [340], although it is a non-specific/broad-spectrum PDE inhibitor that raises both cAMP and cGMP, so it might [340]. If it does not, then the PDE5 inhibitor sildenafil might be the ideal therapeutic to co-administer with propentofylline. Furthermore, by potently inhibiting cGMP-stimulated PDE2 [340], propentofylline would be ideal to co-administer with sildenafil to prevent the dose-limiting side-effect of sildenafil of activating cGMP-stimulated PDE2 [72]. Future preclinical studies should study the effect of propentofylline and sildenafil together and apart plus standard of care compared to standard of care alone in AD to determine whether propentofylline and sildenafil combination treatment might provide synergistic benefits. Future clinical trials should investigate whether propentofylline improves AD symptom severity on the ADAS-Cog and overall impression in a third (or second?) 18-month phase III clinical trial in mild-to-moderate AD patients.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Cline EN , Bicca MA , Viola KL , Klein WL ((2018) ) The amyloid-β oligomer hypothesis: Beginning of the third decade. J Alzheimers Dis 64: , S567–S610. |
[2] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[3] | Smith MA , Zhu X , Tabaton M , Liu G , McKeel DW , Cohen ML , Wang X , Siedlak SL , Dwyer BE , Hayashi T , Nakamura M , Nunomura A , Perry G ((2010) ) Increased iron and free radical generation in preclinical Alzheimer disease and mild cognitive impairment. J Alzheimers Dis 19: , 353–372. |
[4] | Yumoto S , Kakimi S , Ishikawa A ((2018) ) Colocalization of aluminum and iron in nuclei of nerve cells in brains of patients with Alzheimer’s disease. J Alzheimers Dis 65: , 1267–1281. |
[5] | Itzhaki RF , Lathe R , Balin BJ , Ball MJ , Bearer EL , Braak H , Bullido MJ , Carter C , Clerici M , Cosby SL , Del Tredici K , Field H , Fulop T , Grassi C , Griffin WST , Haas J , Hudson AP , Kamer AR , Kell DB , Licastro F , Letenneur L , Lövheim H , Mancuso R , Miklossy J , Otth C , Palamara AT , Perry G , Preston C , Pretorius E , Strandberg T , Tabet N , Taylor-Robinson SD , Whittum-Hudson JA , Del K , Kamer AR , Kell DB , Licastro F , Letenneur L , Lövheim H , Mancuso R , Miklossy J , Otth C , Palamara AT , Perry G , Preston C , Pretorius E , Strandberg T , Tabet N , Taylor-Robinson SD , Whittum-Hudson JA ((2017) ) Microbes and Alzheimer’s disease. J Alzheimers Dis 51: , 979–984. |
[6] | Miklossy J , Khalili K , Gern L , Ericson RL , Darekar P , Bolle L , Hurlimann J , Paster BJ ((2004) ) Borrelia burgdorferi persists in the brain in chronic lyme neuroborreliosis and may be associated with Alzheimer disease. J Alzheimers Dis 6: , 639–649; discussion 673-681. |
[7] | Miklossy J ((2011) ) Alzheimer’s disease - a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J Neuroinflammation 8: , 90. |
[8] | Miklossy J ((2015) ) Historic evidence to support a causal relationship between spirochetal infections and Alzheimer’s disease. Front Aging Neurosci 7: , 46. |
[9] | Fülöp T , Itzhaki RF , Balin BJ , Miklossy J , Barron AE ((2018) ) Role of microbes in the development of Alzheimer’s disease: State of the art - an international symposium presented at the 2017 IAGG congress in San Francisco. Front Genet 9: , 1–16. |
[10] | Readhead B , Funk CC , Ehrlich ME , Gandy S , Dudley JT , Readhead B , Funk CC , Richards MA , Shannon P ((2018) ) Multiscale analysis of independent Alzheimer ‘s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron 99: , 64–82.e7. |
[11] | Alonso R , Pisa D , Fernández-Fernández AM , Carrasco L ((2018) ) Infection of fungi and bacteria in brain tissue from elderly persons and patients with Alzheimer’s disease. Front Aging Neurosci 10: , 159. |
[12] | Butterfield DA , Sultana R ((2007) ) Redox proteomics identification of oxidatively modified brain proteins in Alzheimer’s disease and mild cognitive impairment: Insights into the progression of this dementing disorder. J Alzheimers Dis 12: , 61–72. |
[13] | Bradley MAA , Markesbery WRR , Lovell MAA ((2010) ) Increased levels of 4-hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free Radic Biol Med 48: , 1570–1576. |
[14] | Nunomura A , Perry G , Aliev G , Hirai K , Takeda A , Balraj EK , Jones PK , Ghanbari H , Wataya T , Shimohama S , Chiba S , Atwood CS , Petersen RB , Smith MA ((2001) ) Oxidative damage is the earliest event in Alzheimer disease. J Neuropathol Exp Neurol 60: , 759–767. |
[15] | Lovell MA , Soman S , Bradley MA ((2011) ) Oxidatively modified nucleic acids in preclinical Alzheimer’s disease (PCAD) brain. Mech Ageing Dev 132: , 443–448. |
[16] | Wang J , Markesbery WR , Lovell MA ((2006) ) Increased oxidative damage in nuclear and mitochondrial DNA in mild cognitive impairment. J Neurochem 96: , 825–832. |
[17] | Li X , Jiang LH ((2018) ) Multiple molecular mechanisms form a positive feedback loop driving amyloid β42 peptide-induced neurotoxicity via activation of the TRPM2 channel in hippocampal neurons. Cell Death Dis 9: , 1–16. |
[18] | Alawieyah Syed Mortadza S , Sim JA , Neubrand VE , Jiang LH ((2018) ) A critical role of TRPM2 channel in Aβ42-induced microglial activation and generation of tumor necrosis factor-α. Glia 66: , 562–575. |
[19] | Jiang LH , Li X , Syed Mortadza SA , Lovatt M , Yang W ((2018) ) The TRPM2 channel nexus from oxidative damage to Alzheimer’s pathologies: An emerging novel intervention target for age-related dementia. Ageing Res Rev 47: , 67–79. |
[20] | Li X , Yang W , Jiang L-H ((2017) ) Alteration in intracellular Zn2+ homeostasis as a result of TRPM2 channel activation contributes to ROS-induced hippocampal neuronal death. Front Mol Neurosci 10: , 1–10. |
[21] | Gibson G , Cotman C , Lynch G , Blass J ((2017) ) Calcium hypothesis of Alzheimer’s disease and brain aging: A framework for integrating new evidence into a comprehensive theory of pathogenesis. Alzheimers Dement 13: , 178–182. |
[22] | Hirai K , Aliev G , Nunomura A , Fujioka H , Russell RL , Atwood CS , Johnson AB , Kress Y , Vinters H V , Tabaton M , Shimohama S , Cash AD , Siedlak SL , Harris PL , Jones PK , Petersen RB , Perry G , Smith MA ((2001) ) Mitochondrial abnormalities in Alzheimer’s disease. J Neurosci 21: , 3017–3023. |
[23] | Majd S , Power JHT ((2018) ) Oxidative stress and decreased mitochondrial superoxide dismutase 2 and peroxiredoxins 1 and 4 based mechanism of concurrent activation of AMPK and mTOR in Alzheimer’s disease. Curr Alzheimer Res 15: , 1–13. |
[24] | Zhang C , Rissman RA , Feng J ((2015) ) Characterization of ATP alternations in an Alzheimer’s disease transgenic mouse model. J Alzheimers Dis 44: , 375–378. |
[25] | Hoozemans JJMM , Van Haastert ES , Nijholt DATT , Rozemuller AJMM , Eikelenboom P , Scheper W ((2009) ) The unfolded protein response is activated in pretangle neurons in alzheimer’s disease hippocampus. Am J Pathol 174: , 1241–1251. |
[26] | Monte SM de la , Re E , Longato L , Tong M ((2012) ) Dysfunctional pro-ceramide, ER stress, and nsulin/IGF signaling networks with progression of Alzheimer’s disease. J Alzheimers Dis 30: , S217–S229. |
[27] | de la Monte SM ((2012) ) Triangulated mal-signaling in Alzheimer’s disease: Roles of neurotoxic ceramides, ER stress, and insulin resistance reviewed. J Alzheimers Dis 30: , S231–249. |
[28] | Goetzl EJ , Boxer A , Schwartz JB , Abner EL , Petersen RC , Miller BL , Kapogiannis D ((2015) ) Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 85: , 40–47. |
[29] | Colacurcio DJ , Nixon RA ((2016) ) Disorders of lysosomal acidification—The emerging role of v-ATPase in aging and neurodegenerative disease. Ageing Res Rev 32: , 75–88. |
[30] | Ginsberg SD , Alldred MJ , Counts SE , Cataldo AM , Neve RL , Jiang Y , Wuu J , Chao M V. , Mufson EJ , Nixon RA , Che S ((2010) ) Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer’s disease progression. Biol Psychiatry 68: , 885–893. |
[31] | Bordi M , Berg MJ , Mohan PS , Peterhoff CM , Alldred MJ , Che S , Ginsberg SD , Nixon RA ((2016) ) Autophagy flux in CA1 neurons of Alzheimer hippocampus: Increased induction overburdens failing lysosomes to propel neuritic dystrophy. Autophagy 12: , 2467–2483. |
[32] | Rangasamy SB , Jana M , Roy A , Corbett GT , Kundu M , Chandra S , Mondal S , Dasarathi S , Mufson EJ , Mishra RK , Luan C-H , Bennett DA , Pahan K ((2018) ) Selective disruption of TLR2-MyD88 interaction inhibits inflammation and attenuates Alzheimer’s pathology. J Clin Invest 128: , 4297–4312. |
[33] | Liu S , Liu Y , Hao W , Wolf L , Kiliaan AJ , Penke B , Rube CE , Walter J , Heneka MT , Hartmann T , Menger MD , Fassbender K ((2012) ) TLR2 is a primary receptor for Alzheimer’s amyloid peptide to trigger neuroinflammatory activation. J Immunol 188: , 1098–1107. |
[34] | Chami L , Buggia-Prévot V , Duplan E , Del Prete D , Delprete D , Chami M , Peyron J-F , Checler F ((2012) ) Nuclear factor kappa B regulates βAPP and β- and γ-secretases differently at physiological and supraphysiological Aβ concentrations. J Biol Chem 287: , 24573–24584. |
[35] | Chen C-H , Zhou W , Liu S , Deng Y , Cai F , Tone M , Tone Y , Tong Y , Song W ((2012) ) Increased NF-κB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int J Neuropsychopharmacol 15: , 77–90. |
[36] | Cacabelos R , Alvarez XA , Fernández-Novoa L , Franco A , Mangues R , Pellicer A , Nishimura T ((1994) ) Brain interleukin-1 beta in Alzheimer’s disease and vascular dementia. Methods Find Exp Clin Pharmacol 16: , 141–151. |
[37] | Wu D , Zhang X , Zhao M , Zhou A-L ((2015) ) The role of the TLR4/NF-κB signaling pathway in Aβ accumulation in primary hippocampal neurons. Sheng Li Xue Bao 67: , 319–328. |
[38] | Wu Y-Y , Hsu J-L , Wang H-C , Wu S-J , Hong C-J , Cheng IH-J ((2015) ) Alterations of the neuroinflammatory markers IL-6 and TRAIL in Alzheimer’s disease. Dement Geriatr Cogn Dis Extra 5: , 424–434. |
[39] | Westra JW , Barral S , Chun J ((2009) ) A reevaluation of tetraploidy in the Alzheimer’s disease brain. Neurodegener Dis 6: , 221–229. |
[40] | Raina AK , Zhu X , Rottkamp CA , Monteiro M , Takeda A , Smith MA ((2000) ) Cyclin’ toward dementia: Cell cycle abnormalities and abortive oncogenesis in Alzheimer disease. J Neurosci Res 61: , 128–133. |
[41] | Mosch B , Morawski M , Mittag A , Lenz D , Tarnok A , Arendt T ((2007) ) Aneuploidy and DNA replication in the normal human brain and Alzheimer’s disease. J Neurosci 27: , 6859–6867. |
[42] | Frade JM , López-sánchez N ((2010) ) A novel hypothesis for Alzheimer disease based on neuronal tetraploidy induced by p75NTR. Cell Cycle 9: , 1934–1941. |
[43] | Walton CC , Perea G , Frade JM , Barrio-Alonso E , Hernández-Vivanco A , Walton CC , Perea G , Frade JM ((2018) ) Cell cycle reentry triggers hyperploidization and synaptic dysfunction followed by delayed cell death in differentiated cortical neurons. Sci Rep 8: , 1–14. |
[44] | Moh C ((2011) ) Cell cycle deregulation in the neurons of Alzheimer’s disease. Results Probl Cell Differ 53: , 565–576. |
[45] | Zhu X , Raina AK , Perry G , Smith MA ((2004) ) Alzheimer’s disease: The two-hit hypothesis. Lancet Neurol 3: , 219–226. |
[46] | Zhu X , Castellani RJ , Takeda A , Nunomura A , Atwood CS , Perry G , Smith MA ((2001) ) Differential activation of neuronal ERK, JNK / SAPK and p38 in Alzheimer disease: The ‘two hit’ hypothesis. Mech Ageing Dev 123: , 39–46. |
[47] | Fuchsberger T , Lloret A , Viña J ((2017) ) New functions of APC/C ubiquitin ligase in the nervous system and its role in Alzheimers disease. Int J Mol Sci 18: , 1057. |
[48] | Yang Y , Geldmacher DS , Herrup K ((2001) ) DNA replication precedes neuronal cell death in Alzheimer’s disease. J Neurosci 21: , 2661–2668. |
[49] | Talbot K , Wang H , Kazi H , Han L-Y , Bakshi KP , Stucky A , Fuino RL , Kawaguchi KR , Samoyedny AJ , Wilson RS , Arvanitakis Z , Schneider JA , Wolf BA , Bennett DA , Trojanowski JQ , Arnold SE ((2012) ) Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 122: , 1316–1338. |
[50] | Steen E , Terry BM , Rivera EJ , Cannon JL , Neely TR , Tavares R , Xu XJ , Wands JR , de la Monte SM ((2005) ) Impaired insulin and insulin-like growth factor expression and signaling mechanisms in Alzheimer’s disease - is this type 3 diabetes? J Alzheimers Dis 7: , 63–80. |
[51] | de la Monte SM ((2017) ) Insulin resistance and neurodegeneration: Progress towards the development of new therapeutics for Alzheimer’s disease. Drugs 77: , 47–65. |
[52] | Selkoe DJ ((2002) ) Alzheimer’s disease is a synaptic failure. Science 298: , 789–791. |
[53] | Togashi K , Hara Y , Tominaga T , Higashi T , Konishi Y , Mori Y , Tominaga M ((2006) ) TRPM2 activation by cyclic ADP-ribose at body temperature is involved in insulin secretion. EMBO J 25: , 1804–1815. |
[54] | Eraslan E , Tanyeli A , Polat E , Polat E ((2019) ) 8-Br-cADPR, a TRPM2 ion channel antagonist, inhibits renal ischemia-reperfusion injury. J Cell Physiol 234: , 4572–4581. |
[55] | Kolisek M , Beck A , Fleig A , Penner R ((2005) ) Cyclic ADP-ribose and hydrogen peroxide synergize with ADP-ribose in the activation of TRPM2 channels. Mol Cell 18: , 61–69. |
[56] | Choe CU , Lardong K , Gelderblom M , Ludewig P , Leypoldt F , Koch-Nolte F , Gerloff C , Magnus T ((2011) ) CD38 exacerbates focal cytokine production, postischemic inflammation and brain injury after focal cerebral ischemia. PLoS One 6: , e19046. |
[57] | Blacher E , Dadali T , Bespalko A , Haupenthal VJ , Grimm MOW , Hartmann T , Lund FE , Stein R , Levy A ((2015) ) Alzheimer’s disease pathology is attenuated in a CD38-deficient mouse model. Ann Neurol 78: , 88–103. |
[58] | Reinert LS , Lopušná K , Winther H , Sun C , Thomsen MK , Nandakumar R , Mogensen TH , Meyer M , Vægter C , Nyengaard JR , Fitzgerald KA , Paludan SR ((2016) ) Sensing of HSV-1 by the cGAS-STING pathway in microglia orchestrates antiviral defence in the CNS. Nat Commun 7: , 13348. |
[59] | Mathur V , Burai R , Vest RT , Bonanno LN , Lehallier B , Zardeneta ME , Mistry KN , Do D , Marsh SE , Abud EM , Blurton-Jones M , Li L , Lashuel HA , Wyss-Coray T ((2017) ) Activation of the STING-Dependent type I interferon response reduces microglial reactivity and neuroinflammation. Neuron 96: , 1290–1302.e6. |
[60] | Chin AC ((2019) ) Neuroinflammation and the cGAS-STING pathway. J Neurophysiol 121: , 1087–1091. |
[61] | Tetz V , Tetz G ((2016) ) Effect of deoxyribonuclease I treatment for dementia in end-stage Alzheimer’s disease: A case report. J Med Case Rep 10: , 131. |
[62] | Takahashi A , Okada R , Nagao K , Kawamata Y , Hanyu A , Yoshimoto S , Takasugi M , Watanabe S , Kanemaki MT , Obuse C , Hara E ((2017) ) Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat Commun 8: , 1–14. |
[63] | Søberg K , Skålhegg BS ((2018) ) The molecular basis for specificity at the level of the protein kinase a catalytic subunit. Front Endocrinol (Lausanne) 9: , 538. |
[64] | Hasebe M , Yoshino M ((2016) ) Nitric oxide/cGMP/PKG signaling pathway activated by M 1 -type muscarinic acetylcholine receptor cascade inhibits Na+ -activated K+ currents in Kenyon cells. J Neurophysiol 115: , 3174–3185. |
[65] | Denninger JW , Marletta MA ((1999) ) Guanylate cyclase and the.NO/cGMP signaling pathway. Biochim Biophys Acta 1411: , 334–350. |
[66] | Pandey KN ((2011) ) Guanylyl cyclase / atrial natriuretic peptide receptor-A: Role in the pathophysiology of cardiovascular regulation. Can J Physiol Pharmacol 89: , 557–573. |
[67] | Yamamoto K , Kawai M , Yamazaki M , Tachikawa K , Kubota T , Ozono K , Michigami T ((2019) ) CREB activation in hypertrophic chondrocytes is involved in the skeletal overgrowth in epiphyseal chondrodysplasia Miura type caused by activating mutations of natriuretic peptide receptor B. Hum Mol Genet 28: , 1183–1198. |
[68] | Piggott LA , Hassell KA , Berkova Z , Morris AP , Silberbach M , Rich TC ((2006) ) Natriuretic peptides and nitric oxide stimulate cGMP synthesis in different cellular compartments. J Gen Physiol 128: , 3–14. |
[69] | Prickaerts J , Heckman PRA , Blokland A ((2017) ) Investigational phosphodiesterase inhibitors in phase I and phase II clinical trials for Alzheimer’s disease. Expert Opin Investig Drugs 26: , 1033–1048. |
[70] | Bollen E , Prickaerts J ((2012) ) Phosphodiesterases in neurodegenerative disorders. IUBMB Life 64: , 965–970. |
[71] | Lee DI , Zhu G , Sasaki T , Cho GS , Hamdani N , Holewinski R , Jo SH , Danner T , Zhang M , Rainer PP , Bedja D , Kirk JA , Ranek MJ , Dostmann WR , Kwon C , Margulies KB , Van Eyk JE , Paulus WJ , Takimoto E , Kass DA ((2015) ) Phosphodiesterase 9A controls nitric-oxide-independent cGMP and hypertrophic heart disease. Nature 519: , 472–476. |
[72] | Banerjee J , Bruckbauer A , Thorpe T , Zemel MB ((2019) ) Biphasic effect of sildenafil on energy sensing is mediated by phosphodiesterases 2 and 3 in adipocytes and hepatocytes. Int J Mol Sci 20: , 2992. |
[73] | Heckman PRA , Blokland A , Prickaerts J ((2017) ) From age-related cognitive decline to alzheimer’s disease: A translational overview of the potential role for phosphodiesterases. Adv Neurobiol 17: , 135–168. |
[74] | Lu Y-F , Hawkins RD ((2002) ) Ryanodine receptors contribute to cGMP-induced late-phase LTP and CREB phosphorylation in the hippocampus. J Neurophysiol 88: , 1270–1278. |
[75] | Chen Y , Zhuang S , Cassenaer S , Casteel DE , Gudi T , Boss GR , Pilz RB ((2003) ) Synergism between calcium and cyclic GMP in cyclic AMP response element-dependent transcriptional regulation requires cooperation between CREB and C/EBP-beta. Mol Cell Biol 23: , 4066–4082. |
[76] | Fiorito J , Deng S , Landry DW , Arancio O ((2018) ) Targeting the NO/cGMP/CREB phosphorylation signaling pathway in Alzheimer’s disease. In Neurochemical Basis of Brain Function and Dysfunction, HeinbockelT, CsokaAB, eds. IntechOpen, pp. 1–24. |
[77] | Ciani E , Guidi S , Bartesaghi R , Contestabile A ((2002) ) Nitric oxide regulates cGMP-dependent cAMP-responsive element binding protein phosphorylation and Bcl-2 expression in cerebellar neurons: Implication for a survival role of nitric oxide. J Neurochem 82: , 1282–1289. |
[78] | Nagai-Kusuhara A , Nakamura M , Mukuno H , Kanamori A , Negi A , Seigel GM ((2007) ) cAMP-responsive element binding protein mediates a cGMP/protein kinase G-dependent anti-apoptotic signal induced by nitric oxide in retinal neuro-glial progenitor cells. Exp Eye Res 84: , 152–162. |
[79] | Puzzo D , Vitolo O , Trinchese F , Jacob JP , Palmeri A , Arancio O ((2005) ) Amyloid-β peptide inhibits activation of the nitric oxide/cGMP/cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. J Neurosci 25: , 6887–6897. |
[80] | Yamamoto-Sasaki M , Ozawa H , Saito T , Rösler M , Riederer P ((1999) ) Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Res 824: , 300–303. |
[81] | Bartolotti N , Bennett DA , Lazarov O ((2016) ) Reduced pCREB in Alzheimer’s disease prefrontal cortex is reflected in peripheral blood mononuclear cells. Mol Psychiatry 21: , 1158–1166. |
[82] | Ryu H , Lee J , Impey S , Ratan RR , Ferrante RJ ((2005) ) Antioxidants modulate mitochondrial PKA and increase CREB binding to D-loop DNA of the mitochondrial genome in neurons. Proc Natl Acad Sci U S A 102: , 13915–13920. |
[83] | De Rasmo D , Signorile A , Papa F , Roca E , Papa S ((2010) ) cAMP/Ca2+ response element-binding protein plays a central role in the biogenesis of respiratory chain proteins in mammalian cells. IUBMB Life 62: , 447–452. |
[84] | Yiu AP , Rashid AJ , Josselyn SA ((2011) ) Increasing CREB function in the CA1 region of dorsal hippocampus rescues the spatial memory deficits in a mouse model of Alzheimer’s disease. Neuropsychopharmacology 36: , 2169–2186. |
[85] | Yang J-L , Lin Y-T , Chuang P-C , Bohr VA , Mattson MP ((2014) ) BDNF and exercise enhance neuronal DNA repair by stimulating CREB-mediated production of apurinic/apyrimidinic endonuclease 1. Neuromolecular Med 16: , 161–174. |
[86] | Xue W , Wang W , Gong T , Zhang H , Tao W , Xue L , Sun Y , Wang F , Chen G ((2016) ) PKA-CREB-BDNF signaling regulated long lasting antidepressant activities of Yueju but not ketamine. Sci Rep 6: , 1–9. |
[87] | Luo Y , Kuang S , Li H , Ran D , Yang J ((2017) ) cAMP/PKA-CREB-BDNF signaling pathway in hippocampus mediates cyclooxygenase 2-induced learning/memory deficits of rats subjected to chronic unpredictable mild stress. Oncotarget 8: , 35558–35572. |
[88] | Lesiak A , Pelz C , Ando H , Zhu M , Davare M , Lambert TJ , Hansen KF , Obrietan K , Appleyard SM , Impey S , Wayman GA ((2013) ) A genome-wide screen of CREB occupancy identifies the RhoA inhibitors Par6C and Rnd3 as regulators of BDNF-induced synaptogenesis. PLoS One 8: , e64658. |
[89] | Wilson BE , Mochon E , Boxer LM ((1996) ) Induction of bcl-2 expression by phosphorylated CREB proteins during B-cell activation and rescue from apoptosis. Mol Cell Biol 16: , 5546–5556. |
[90] | Riccio A , Ahn S , Davenport CM , Blendy JA , Ginty DD ((1999) ) Mediation by a CREB family transcription factor of NGF-dependent survival of sympathetic neurons. Science 286: , 2358–2361. |
[91] | Singulani MP , Pereira CPM , Ferreira AFF , Garcia PC , Ferrari GD , Alberici LC , de Britto LRG ((2020) ) Impairment of PGC-1α-mediated mitochondrial biogenesis precedes mitochondrial dysfunction and Alzheimer’s pathology in the 3xTg mouse model of Alzheimer’s disease. Exp Gerontol 133: , 110882. |
[92] | Fernandez-Marcos PJ , Auwerx J ((2011) ) Regulation of PGC-1α, a nodal regulator of mitochondrial biogenesis. Am J Clin Nutr 93: , 884S-90. |
[93] | Sheng B , Wang X , Su B , Lee HG , Casadesus G , Perry G , Zhu X ((2012) ) Impaired mitochondrial biogenesis contributes to mitochondrial dysfunction in Alzheimer’s disease. J Neurochem 120: , 419–429. |
[94] | Ayrapetyan T ((2018) ) Defective NO/cGMP/PKG Signaling Downregulates Sirt1 Expression in Aged Osteoblasts (thesis). Advisor: Renate B. Pilz, Immo Scheffler. |
[95] | Jia H , Guo Z , Yao Y ((2019) ) PDE5 inhibitor protects the mitochondrial function of hypoxic myocardial cells. Exp Ther Med 17: , 199–204. |
[96] | Li N , Yuan Y , Li S , Zeng C , Yu W , Shen M , Zhang R , Li C , Zhang Y , Wang H ((2016) ) Pde5 inhibitors protect against post-infarction heart failure. Front Biosci (Landmark Ed) 21: , 1194–1210. |
[97] | Nemoto S , Fergusson MM , Finkel T ((2005) ) SIRT1 functionally interacts with the metabolic regulator and transcriptional coactivator PGC-1α. J Biol Chem 280: , 16456–16460. |
[98] | Gerhart-Hines Z , Dominy JE , Blättler SM , Jedrychowski MP , Banks AS , Lim JH , Chim H , Gygi SP , Puigserver P ((2011) ) The cAMP/PKA pathway rapidly activates SIRT1 to promote fatty acid oxidation independently of changes in NAD (+). Mol Cell 44: , 851–863. |
[99] | Whitaker RM , Wills LP , Stallons LJ , Schnellmann RG ((2013) ) cGMP-selective phosphodiesterase inhibitors stimulate mitochondrial biogenesis and promote recovery from acute kidney injury. J Pharmacol Exp Ther 347: , 626–634. |
[100] | Nisoli E , Clementi E , Paolucci C , Cozzi V , Tonello C , Sciorati C , Bracale R , Valerio A , Francolini M , Moncada S , Carruba MO ((2003) ) Mitochondrial biogenesis in mammals: The role of endogenous nitric oxide. Science 299: , 896–899. |
[101] | Gureev AP , Shaforostova EA , Popov VN ((2019) ) Regulation of mitochondrial biogenesis as a way for active longevity: Interaction between the Nrf2 and PGC-1α signaling pathways. Front Genet 10: , 435. |
[102] | Gutsaeva DR , Carraway MS , Suliman HB , Demchenko IT , Shitara H , Yonekawa H , Piantadosi CA ((2008) ) Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J Neurosci 28: , 2015–2024. |
[103] | Nisoli E , Falcone S , Tonello C , Cozzi V , Palomba L , Fiorani M , Pisconti A , Brunelli S , Cardile A , Francolini M , Cantoni O , Carruba MO , Moncada S , Clementi E ((2004) ) Mitochondrial biogenesis by NO yields functionally active mitochondria in mammals. Proc Natl Acad Sci U S A 101: , 16507–16512. |
[104] | Shalwala M , Zhu S-G , Das A , Salloum FN , Xi L , Kukreja RC ((2014) ) Sirtuin 1 (SIRT1) activation mediates sildenafil induced delayed cardioprotection against ischemia-reperfusion injury in mice. PLoS One 9: , e86977. |
[105] | Fiore D , Gianfrilli D , Giannetta E , Galea N , Panio G , di Dato C , Pofi R , Pozza C , Sbardella E , Carbone I , Naro F , Lenzi A , Venneri MA , Isidori AM ((2016) ) PDE5 inhibition ameliorates visceral adiposity targeting the miR-22/SIRT1 pathway: Evidence from the CECSID trial. J Clin Endocrinol Metab 101: , 1525–1534. |
[106] | Zhang Z , Shen Q , Wu X , Zhang D , Xing D ((2020) ) Activation of PKA/SIRT1 signaling pathway by photobiomodulation therapy reduces Aβ levels in Alzheimer’s disease models. Aging Cell 19: , e13054. |
[107] | Nisoli E , Tonello C , Cardile A , Cozzi V , Bracale R , Tedesco L , Falcone S , Valerio A , Cantoni O , Clementi E , Moncada S , Carruba MO ((2005) ) Calorie restriction promotes mitochondrial biogenesis by inducing the expression of eNOS. Science 310: , 314–317. |
[108] | Kitada M , Ogura Y , Koya D ((2016) ) The protective role of Sirt1 in vascular tissue: Its relationship to vascular aging and atherosclerosis. Aging (Albany NY) 8: , 2290–2307. |
[109] | Shao H , Xue Q , Zhang F , Luo Y , Zhu H , Zhang X , Zhang H , Ding W , Yu B ((2014) ) Spinal SIRT1 activation attenuates neuropathic pain in mice. PLoS One 9: , e100938. |
[110] | Bermúdez-Ocaña DY , Ambriz-Tututi M , Pérez-Severiano F , Granados-Soto V ((2006) ) Pharmacological evidence for the participation of NO-cyclic GMP-PKG-K+ channel pathway in the antiallodynic action of resveratrol. Pharmacol Biochem Behav 84: , 535–542. |
[111] | Rodgers JT , Lerin C , Haas W , Gygi SP , Spiegelman BM , Puigserver P ((2005) ) Nutrient control of glucose homeostasis through a complex of PGC-1α and SIRT1. Nature 434: , 113–118. |
[112] | Olmos Y , Sánchez-Gómez FJ , Wild B , García-Quintans N , Cabezudo S , Lamas S , Monsalve M ((2013) ) SirT1 regulation of antioxidant genes is dependent on the formation of a FoxO3a/PGC-1α complex. Antioxidants Redox Signal 19: , 1507–1521. |
[113] | Lagouge M , Argmann C , Gerhart-Hines Z , Meziane H , Lerin C , Daussin F , Messadeq N , Milne J , Lambert P , Elliott P , Geny B , Laakso M , Puigserver P , Auwerx J ((2006) ) Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1α . Cell 127: , 1109–1122. |
[114] | Gurd BJ ((2011) ) Deacetylation of PGC-1a by SIRT1: Importance for skeletal muscle function and exercise-induced mitochondrial biogenesis. Appl Physiol Nutr Metab 36: , 589–597. |
[115] | Gerhart-Hines Z , Rodgers JT , Bare O , Lerin C , Kim SH , Mostoslavsky R , Alt FW , Wu Z , Puigserver P ((2007) ) Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1α . EMBO J 26: , 1913–1923. |
[116] | Dominy JE , Lee Y , Gerhart-Hines Z , Puigserver P ((2010) ) Nutrient-dependent regulation of PGC-1α’s acetylation state and metabolic function through the enzymatic activities of Sirt1/GCN5. Biochim Biophys Acta 1804: , 1676–1683. |
[117] | Amat R , Planavila A , Chen SL , Iglesias R , Giralt M , Villarroya F ((2009) ) SIRT1 controls the transcription of the PGC-1a gene in skeletal muscle through PGC-1a auto-regulatory loop and interaction with MyoD. J Biol Chem 284: , 21872–21880. |
[118] | Tang BL ((2016) ) Sirt1 and the mitochondria. Mol Cells 39: , 87–95. |
[119] | Han B , Jiang W , Liu H , Wang J , Zheng K , Cui P , Feng Y , Dang C , Bu Y , Wang QM , Ju Z , Hao J ((2020) ) Upregulation of neuronal PGC-1α ameliorates cognitive impairment induced by chronic cerebral hypoperfusion. Theranostics 10: , 2832–2848. |
[120] | Wang H , Peiris TH , Mowery A , Lay J Le , Gao Y , Greenbaum LE ((2008) ) CCAAT/enhancer binding protein-β is a transcriptional regulator of peroxisome-proliferator-activated receptor-γ coactivator-1α in the regenerating liver. Mol Endocrinol 22: , 1596–1605. |
[121] | Wang R , Li JJ , Diao S , Kwak YD , Liu L , Zhi L , Büeler H , Bhat NR , Williams RW , Park EA , Liao FF ((2013) ) Metabolic stress modulates Alzheimer’s β-secretase gene transcription via SIRT1-PPARγ-PGC-1 in neurons. Cell Metab 17: , 685–694. |
[122] | Kwak Y-D , Wang R , Li J , Zhang Y-W , Xu H , Liao F-F ((2011) ) Differential regulation of BACE1 expression by oxidative and nitrosative signals. Mol Neurodegener 6: , 17. |
[123] | Yan R , Vassar R ((2014) ) Targeting the β secretase BACE1 for Alzheimer’s disease therapy. Lancet Neurol 13: , 319–329. |
[124] | Alsop DC , Casement M , De Bazelaire C , Fong T , Press DZ ((2008) ) Hippocampal hyperperfusion in Alzheimer’s disease. Neuroimage 42: , 1267–1274. |
[125] | Ishii K , Kitagaki H , Kono M , Mori E ((1996) ) Decreased medial temporal oxygen metabolism in Alzheimer’s disease shown by PET. J Nucl Med 37: , 1159–1165. |
[126] | Hays CC , Zlatar ZZ , Wierenga CE ((2016) ) The utility of cerebral blood flow as a biomarker of preclinical Alzheimer’s disease. Cell Mol Neurobiol 36: , 167–179. |
[127] | Ishii K , Sasaki M , Yamaji S , Sakamoto S , Kitagaki H , Mori E ((1998) ) Paradoxical hippocampus perfusion in mild-to-moderate Alzheimer’s disease. J Nucl Med 39: , 293–298. |
[128] | Bartolome F , De La Cueva M , Pascual C , Antequera D , Fernandez T , Gil C , Martinez A , Carro E ((2018) ) Amyloid β-induced impairments on mitochondrial dynamics, hippocampal neurogenesis, and memory are restored by phosphodiesterase 7 inhibition. Alzheimers Res Ther 10: , 24. |
[129] | Qin W , Haroutunian V , Katsel P , Cardozo CP , Ho L , Buxbaum JD , Pasinetti GM ((2009) ) PGC-1α expression decreases in the Alzheimer disease brain as a function of dementia. Arch Neurol 66: , 352–361. |
[130] | Moreira PI , Siedlak SL , Wang X , Santos MS , Oliveira CR , Tabaton M , Nunomura A , Szweda LI , Aliev G , Smith MA , Zhu X , Perry G ((2007) ) Autophagocytosis of mitochondria is prominent in Alzheimer disease. J Neuropathol Exp Neurol 66: , 525–532. |
[131] | Plaschke K , Martin E , Bardenheuer HJ ((1998) ) Effect of propentofylline on hippocampal brain energy state and amyloid precursor protein concentration in a rat model of cerebral hypoperfusion. J Neural Transm 105: , 1065–1077. |
[132] | Turcani M , Pavlik A , Turcani P ((2003) ) Propentofylline rapidly normalizes mitochondrial respiration in a gerbil low flow unilateral forebrain ischemia. Neurosci Lett 349: , 87–90. |
[133] | Mani M , Khaghani S , Mohammadi TG , Zamani Z , Azadmanesh K , Meshkani R , Pasalar P , Mostafavi E ((2013) ) Activation of Nrf2-antioxidant response element mediated glutamate cysteine ligase expression in hepatoma cell line by homocysteine. Hepat Mon 13: , e8394. |
[134] | Pajares M , Jiménez-Moreno N , García-Yagüe ÁJ , Escoll M , de Ceballos ML , Van Leuven F , Rábano A , Yamamoto M , Rojo AI , Cuadrado A ((2016) ) Transcription factor NFE2L2/NRF2 is a regulator of macroautophagy genes. Autophagy 12: , 1902–1916. |
[135] | Dong J , Sulik KK , Chen SY ((2008) ) Nrf2-mediated transcriptional induction of antioxidant response in mouse exposed to ehanol in vivo: Implications for the prevention of fetal alcohol spectrum disorders. Antioxid Redox Signal 10: , 2023. |
[136] | Espada S , Ortega F , Molina-Jijón E , Rojo AI , Pérez-Sen R , Pedraza-Chaverri J , Miras-Portugal MT , Cuadrado A ((2010) ) The purinergic P2Y13 receptor activates the Nrf2/HO-1 axis and protects against oxidative stress-induced neuronal death. Free Radic Biol Med 49: , 416–426. |
[137] | Ramsey CP , Glass CA , Montgomery MB , Lindl KA , Ritson GP , Chia LA , Hamilton RL , Chu CT , Jordan-Sciutto KL ((2007) ) Expression of Nrf2 in neurodegenerative diseases [nuclear Nrf2 decreased in AD]. J Neuropathol Exp Neurol 66: , 75–85. |
[138] | Kulkarni SR , Donepudi AC , Xu J , Wei W , Cheng QC , Driscoll M V , Johnson DA , Johnson JA , Li X , Slitt AL ((2014) ) Fasting induces nuclear factor E2-related factor 2 and ATP-binding cassette transporters via protein kinase a and sirtuin-1 in mouse and human. Antioxidants Redox Signal 20: , 15–30. |
[139] | Kokot A , Metze D , Mouchet N , Galibert MD , Schiller M , Luger TA , Böhm M ((2009) ) α-melanocyte-stimulating hormone counteracts the suppressive effect of UVB on Nrf2 and Nrf-dependent gene expression in human skin. Endocrinology 150: , 3197–3206. |
[140] | Ziady AG , Sokolow A , Shank S , Corey D , Myers R , Plafker S , Kelley TJ ((2012) ) Interaction with CREB binding protein modulates the activities of Nrf2 and NF-κB in cystic fibrosis airway epithelial cells. Am J Physiol Lung Cell Mol Physiol 302: , L1221–L1231. |
[141] | Mata M , Martinez I , Melero JA , Tenor H , Cortijo J ((2013) ) Roflumilast inhibits respiratory syncytial virus infection in human differentiated bronchial epithelial cells. PLoS One 8: , e69670. |
[142] | Lee C ((2018) ) Therapeutic modulation of virus-induced oxidative stress via the Nrf2-dependent antioxidative pathway. Oxid Med Cell Longev 2018: , 26. |
[143] | Chen C , Jiang X , Gu S , Lai Y , Liu Y , Zhang Z ((2017) ) Protection of Nrf2 against arsenite-induced oxidative damage is regulated by the cyclic guanosine monophosphate-protein kinase G signaling pathway. Environ Toxicol 32: , 2004–2020. |
[144] | Pall ML ((2015) ) A probable protective mechanism for NO/cGMP/G-kinase activity is to raise Nrf2 activity. J Clin Exp Cardiol 6: , 157. |
[145] | Fujii S , Sawa T , Ihara H , Tong KI , Ida T , Okamoto T , Ahtesham AK , Ishima Y , Motohashi H , Yamamoto M , Akaike T ((2010) ) The critical role of nitric oxide signaling, via protein S-guanylation and nitrated cyclic GMP, in the antioxidant adaptive response. J Biol Chem 285: , 23970–23984. |
[146] | Li CQ , Min YK , Godoy LC , Thiantanawat A , Trudel LJ , Wogan GN ((2009) ) Nitric oxide activation of Keap1/Nrf2 signaling in human colon carcinoma cells. Proc Natl Acad Sci U S A 106: , 14547–14551. |
[147] | Gerlo S , Kooijman R , Beck IM , Kolmus K , Spooren A , Haegeman G ((2011) ) Cyclic AMP: A selective modulator of NF-κB action. Cell Mol Life Sci 68: , 3823–3841. |
[148] | Oliveira-Marques V , Marinho HS , Cyrne L , Antunes F ((2009) ) Role of hydrogen peroxide in NF-κB activation: From inducer to modulator. Antioxidants Redox Signal 11: , 2223–2243. |
[149] | Calvo-Rodríguez M , de la Fuente C , García-Durillo M , García-Rodríguez C , Villalobos C , Núñez L ((2017) ) Aging and amyloid β oligomers enhance TLR4 expression, LPS-induced Ca2+ responses, and neuron cell death in cultured rat hippocampal neurons. J Neuroinflammation 14: , 1–13. |
[150] | Zhan X , Stamova B , Sharp FR ((2018) ) Lipopolysaccharide associates with amyloid plaques, neurons and oligodendrocytes in Alzheimer’s disease brain: A review. Front Aging Neurosci 10: , 1–14. |
[151] | Minguet S , Huber M , Rosenkranz L , Schamel WWA , Reth M , Brummer T ((2005) ) Adenosine and cAMP are potent inhibitors of the NF-κB pathway downstream of immunoreceptors. Eur J Immunol 35: , 31–41. |
[152] | Hervé R , Schmitz T , Evain-Brion D , Cabrol D , Leroy M-J , Méhats C ((2008) ) The PDE4 inhibitor rolipram prevents NF-κB binding activity and proinflammatory cytokine release in human chorionic cells. J Immunol 181: , 2196–2202. |
[153] | Delgado M ((2002) ) Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit CBP-NF-kappaB interaction in activated microglia. Biochem Biophys Res Commun 297: , 1181–1185. |
[154] | Woo MS , Jang PG , Park JS , Kim WK , Joh TH , Kim HS ((2003) ) Selective modulation of lipopolysaccharide-stimulated cytokine expression and mitogen-activated protein kinase pathways by dibutyryl-cAMP in BV2 microglial cells. Brain Res Mol Brain Res 113: , 86–96. |
[155] | Peixoto CA , Nunes AKS , Rapôso C ((2017) ) The role of NO/cGMP signaling on neuroinflammation: A new therapeutic opportunity. In Mechanisms of Neuroinflammation, Aranda AbreuGE, ed. InTech, pp. 167–208. |
[156] | Kamthong PJ , Wu MC ((2001) ) Inhibitor of nuclear factor-κB induction by cAMP antagonizes interleukin-1-induced human macrophage-colony-stimulating-factor expression. Biochem J 356: , 525–530. |
[157] | Chong YH , Shin SA , Lee HJ , Kang JHL , Suh Y-H ((2002) ) Molecular mechanisms underlying cyclic AMP inhibition of macrophage dependent TNF-alpha production and neurotoxicity in response to amyloidogenic C-terminal fragment of Alzheimer’s amyloid precursor protein. J Neuroimmunol 133: , 160–174. |
[158] | Aizawa T , Wei H , Miano JM , Abe J , Berk BC , Yan C ((2003) ) Role of phosphodiesterase 3 in NO/cGMP-mediated antiinflammatory effects in vascular smooth muscle cells. Circ Res 93: , 406–413. |
[159] | Chiarini A , Armato U , Gardenal E , Gui L , Dal Prà I ((2017) ) Amyloid β-exposed human astrocytes overproduce phospho-Tau and overrelease it within exosomes, effects suppressed by calcilytic NPS 2143—further implications for Alzheimer’s therapy. Front Neurosci 11: , 217. |
[160] | Sergeant N , Bretteville A , Hamdane M , Caillet-Boudin ML , Grognet P , Bombois S , Blum D , Delacourte A , Pasquier F , Vanmechelen E , Schraen-Maschke S , Buée L ((2008) ) Biochemistry of Tau in Alzheimer’s disease and related neurological disorders. Expert Rev Proteomics 5: , 207–224. |
[161] | Tavares IA , Touma D , Lynham S , Troakes C , Schober M , Causevic M , Garg R , Noble W , Killick R , Bodi I , Hanger DP , Morris JDH ((2013) ) Prostate-derived sterile 20-like kinases (PSKs/TAOKs) phosphorylate Tau protein and are activated in tangle-bearing neurons in alzheimer disease. J Biol Chem 288: , 15418–15429. |
[162] | Buée L , Bussière T , Buée-Scherrer V , Delacourte A , Hof PR ((2000) ) Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res Rev 33: , 95–130. |
[163] | Zheng-Fischhöfer Q , Biernat J , Mandelkow EM , Illenberger S , Godemann R , Mandelkow E ((1998) ) Sequential phosphorylation of Tau by glycogen synthase kinase-3β and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur J Biochem 252: , 542–552. |
[164] | Bullock BP , Habener JF ((1998) ) Phosphorylation of the cAMP response element binding protein CREB by cAMP-dependent protein kinase A and glycogen synthase kinase-3 alters DNA- binding affinity, conformation, and increases net charge. Biochemistry 37: , 3795–3809. |
[165] | Tullai JW , Sepulveda S , Brennan JF , Moss ME , Naya FJ , Cooper GM ((2013) ) Inhibition of GSK-3 activates CREB transcriptional targets during induction of cardiomyocyte hypertrophy. FASEB J 27: (1 Suppl). |
[166] | Tullai JW , Chen J , Schaffer ME , Kamenetsky E , Kasif S , Cooper GM ((2007) ) Glycogen synthase kinase-3 represses cyclic AMP response element-binding protein (CREB)-targeted immediate early genes in quiescent cells. J Biol Chem 282: , 9482–9491. |
[167] | Grimes CA , Jope RS ((2001) ) Creb DNA binding activity is inhibited by glycogen synthase kinase-3β and facilitated by lithium. J Neurochem 78: , 1219–1232. |
[168] | El-Abhar H , Abd El Fattah MA , Wadie W , El-Tanbouly DM ((2018) ) Cilostazol disrupts TLR-4, Akt/GSK-3β/CREB, and IL-6/JAK-2/STAT-3/SOCS-3 crosstalk in a rat model of Huntington’s disease. PLoS One 13: , e0203837. |
[169] | Anderson RM , Barger JL , Edwards MG , Braun KH , O’connor CE , Prolla TA , Weindruch R ((2008) ) Dynamic regulation of PGC-1α localization and turnover implicates mitochondrial adaptation in calorie restriction and the stress response. Aging Cell 7: , 101–111. |
[170] | Shelly M , Cancedda L , Lim BK , Popescu AT , Cheng P lin , Gao H , Poo M ming ((2011) ) Semaphorin3A regulates neuronal polarization by suppressing axon formation and promoting dendrite growth. Neuron 71: , 433–446. |
[171] | Kim YT , Hur E-M , Snider WD , Zhou F-Q ((2011) ) Role of GSK3 signaling in neuronal morphogenesis. Front Mol Neurosci 4: , 48. |
[172] | Perez-Gonzalez R , Pascual C , Antequera D , Bolos M , Redondo M , Perez DI , Pérez-Grijalba V , Krzyzanowska A , Sarasa M , Gil C , Ferrer I , Martinez A , Carro E ((2013) ) Phosphodiesterase 7 inhibitor reduced cognitive impairment and pathological hallmarks in a mouse model of Alzheimer’s disease. Neurobiol Aging 34: , 2133–2145. |
[173] | Hayashi H , Sudo T ((2009) ) Effects of the cAMP-elevating agents cilostamide, cilostazol and forskolin on the phosphorylation of Akt and GSK-3β in platelets. Thromb Haemost 102: , 327–335. |
[174] | Dey S , Goswami S , Eisa A , Bhattacharjee R , Brothag C , Kline D , Vijayaraghavan S ((2018) ) Cyclic AMP and glycogen synthase kinase 3 form a regulatory loop in spermatozoa. J Cell Physiol 233: , 7239–7252. |
[175] | Fang X , Yu SX , Lu Y , Bast RC , Woodgett JR , Mills GB ((2000) ) Phosphorylation and inactivation of glycogen synthase kinase 3 by protein kinase A. Proc Natl Acad Sci U S A 97: , 11960–11965. |
[176] | Khaled M , Larribere L , Bille K , Aberdam E , Ortonne JP , Ballotti R , Bertolotto C ((2002) ) Glycogen synthase kinase 3β is activated by cAMP and plays an active role in the regulation of melanogenesis. J Biol Chem 277: , 33690–33697. |
[177] | García-Barroso C , Ricobaraza A , Pascual-Lucas M , Unceta N , Rico AJ , Goicolea MA , Sallés J , Lanciego JL , Oyarzabal J , Franco R , Cuadrado-Tejedor M , García-Osta A ((2013) ) Tadalafil crosses the blood-brain barrier and reverses cognitive dysfunction in a mouse model of AD. Neuropharmacology 64: , 114–123. |
[178] | Zhu L , Yang JY , Xue X , Dong YX , Miao FR , Wang YF , Xue H , Wu CF ((2015) ) A novel phosphodiesterase-5 Inhibitor: Yonkenafil modulates neurogenesis, gliosis to improve cognitive function and ameliorates amyloid burden in an APP/PS1 transgenic mice model. Mech Ageing Dev 150: , 34–45. |
[179] | Orejana L , Barros-Miñones L , Jordan J , Cedazo-Minguez A , Tordera RM , Aguirre N , Puerta E ((2015) ) Sildenafil decreases BACE1 and cathepsin B levels and reduces APP amyloidogenic processing in the SAMP8 mouse. J Gerontol Ser A Biol Sci Med Sci 70: , 675–685. |
[180] | Cuadrado-Tejedor M , Hervias I , Ricobaraza A , Puerta E , Pérez-Roldán JM , García-Barroso C , Franco R , Aguirre N , García-Osta A ((2011) ) Sildenafil restores cognitive function without affecting β-amyloid burden in a mouse model of Alzheimer’s disease. Br J Pharmacol 164: , 2029–2041. |
[181] | García-Osta A , Cuadrado-Tejedor M , García-Barroso C , Oyarzábal J , Franco R ((2012) ) Phosphodiesterases as therapeutic targets for Alzheimer’s disease. ACS Chem Neurosci 3: , 832–844. |
[182] | Zhang C , Cheng Y , Wang H , Wang C , Wilson SP , Xu J , Zhanga HT ((2014) ) RNA interference-mediated knockdown of long-form phosphodiesterase-4D (PDE4D) enzyme reverses amyloid-β42-induced memory deficits in mice. J Alzheimers Dis 38: , 269–280. |
[183] | Yamamoto M , Ozawa H , Saito T , Hatta S , Riederer P , Takahata N ((1997) ) Ca2+/CaM-sensitive adenylyl cyclase activity is decreased in the Alzheimer’s brain: Possible relation to type I adenylyl cyclase. J Neural Transm 104: , 721–732. |
[184] | Cowburn RF , O’Neill C , Ravid R , Winblad B , Fowler CJ ((1992) ) Preservation of Gi-protein inhibited adenylyl cyclase activity in the brains of patients with Alzheimer’s disease. Neurosci Lett 141: , 16–20. |
[185] | Bonkale WL , Fastbom J , Wiehager B , Ravid R , Winblad B , Cowburn RF ((1996) ) Impaired G-protein-stimulated adenylyl cyclase activity in Alzheimer’s disease brain is not accompanied by reduced cyclic-AMP-dependent protein kinase A activity. Brain Res 737: , 155–161. |
[186] | Cowburn RF , O’Neill C , Ravid R , Alafuzoff I , Winblad B , Fowler CJ ((1992) ) Adenylyl cyclase activity in postmortem human brain: Evidence of altered Grotein mediation in Alzheimer’s disease. J Neurochem 58: , 1409–1419. |
[187] | Dewar D , Horsburgh K , Graham DI , Brooks DN , McCulloch J ((1990) ) Selective alterations of high affinity [3H]forskolin binding sites in Alzheimer’s disease: A quantitative autoradiographic study. Brain Res 511: , 241–248. |
[188] | Martínez M , Hernández AI , Hernanz A ((2001) ) Increased cAMP immunostaining in cerebral vessels in Alzheimer’s disease. Brain Res 922: , 148–152. |
[189] | Grammas P , Roher AE , Ball MJ ((1994) ) Increased accumulation of cAMP in cerebral microvessels in Alzheimer’s disease. Neurobiol Aging 15: , 113–116. |
[190] | Bonkale WL , Cowburn RF , Ohm TG , Bogdanovic N , Fastbom J ((1999) ) A quantitative autoradiographic study of [3H]cAMP binding to cytosolic and particulate protein kinase A in post-mortem brain staged for Alzheimer’s disease neurofibrillary changes and amyloid deposits. Brain Res 818: , 383–396. |
[191] | Liang Z , Liu F , Grundke-Iqbal I , Iqbal K , Gong CX ((2007) ) Down-regulation of cAMP-dependent protein kinase by over-activated calpain in Alzheimer disease brain. J Neurochem 103: , 2462–2470. |
[192] | Kim SH , Nairn AC , Cairns N , Lubec G ((2001) ) Decreased levels of ARPP-19 and PKA in brains of Down syndrome and Alzheimer’s disease. J Neural Transm Suppl, pp. 263–272. |
[193] | Tong L , Thornton PL , Balazs R , Cotman CW ((2001) ) β-Amyloid-(1-42) impairs activity-dependent cAMP-response element-binding protein signaling in neurons at Concentrations in which cell survival is not compromised. J Biol Chem 276: , 17301–17306. |
[194] | Shi J , Qian W , Yin X , Iqbal K , Grundke-Iqbal I , Gu X , Ding F , Gong CX , Liu F ((2011) ) Cyclic AMP-dependent protein kinase regulates the alternative splicing of tau exon 10: A mechanism involved in tau pathology of Alzheimer disease. J Biol Chem 286: , 14639–14648. |
[195] | Bonkale WL , Winblad B , Ravid R , Cowburn RF ((1995) ) Reduced nitric oxide responsive soluble guanylyl cyclase activity in the superior temporal cortex of patients with Alzheimer’s disease. Neurosci Lett 187: , 5–8. |
[196] | Liu P , Fleete MS , Jing Y , Collie ND , Curtis MA , Waldvogel HJ , Faull RLM , Abraham WC , Zhang H ((2014) ) Altered arginine metabolism in Alzheimer’s disease brains. Neurobiol Aging 35: , 1992–2003. |
[197] | Ugarte A , Gil-Bea F , García-Barroso C , Cedazo-Minguez Á , Ramírez MJ , Franco R , García-Osta A , Oyarzabal J , Cuadrado-Tejedor M ((2015) ) Decreased levels of guanosine 3’, 5’-monophosphate (cGMP) in cerebrospinal fluid (CSF) are associated with cognitive decline and amyloid pathology in Alzheimer’s disease. Neuropathol Appl Neurobiol 41: , 471–482. |
[198] | Hesse R , Lausser L , Gummert P , Schmid F , Wahler A , Schnack C , Kroker KS , Otto M , Tumani H , Kestler HA , Rosenbrock H , von Arnim CAF ((2017) ) Reduced cGMP levels in CSF of AD patients correlate with severity of dementia and current depression. Alzheimers Res Ther 9: , 17. |
[199] | Chen Y , Huang X , Zhang Y wu , Rockenstein E , Bu G , Golde TE , Masliah E , Xu H ((2012) ) Alzheimer’s β-secretase (BACE1) regulates the cAMP/PKA/CREB pathway independently of β-amyloid. J Neurosci 32: , 11390–11395. |
[200] | Ye J , Yin Y , Liu H , Fang L , Tao X , Wei L , Zuo Y , Yin Y , Ke D , Wang JZ ((2020) ) Tau inhibits PKA by nuclear proteasome-dependent PKAR2α elevation with suppressed CREB/GluA1 phosphorylation. Aging Cell 19: , e13055. |
[201] | Ohm TG , Bohl J , Lemmer B ((1989) ) Reduced cAMP-signal transduction in postmortem hippocampus of demented old people. Prog Clin Biol Res 317: , 501–509. |
[202] | Schnecko A , Witte K , Bohl J , Ohm T , Lemmer B ((1994) ) Adenylyl cyclase activity in Alzheimer’s disease brain: Stimulatory and inhibitory signal transductionathways are differently affected. Brain Res 644: , 291–296. |
[203] | Ohm TG , Bohl J , Lemmer B ((1991) ) Reduced basal and stimulated (isoprenaline, Gpp(NH)p, forskolin) adenylate cyclase activity in Alzheimer’s disease correlated with histopathological changes. Brain Res 540: , 229–236. |
[204] | O’Neill C , Wiehager B , Fowler CJ , Ravid R , Winblad B , Cowburn RF ((1994) ) Regionally selective alterations in G protein subunit levels in the Alzheimer’s disease brain. Brain Res 636: , 193–201. |
[205] | Yamamoto M , Götz ME , Ozawa H , Luckhaus C , Saito T , Rösler M , Riederer P ((2000) ) Hippocampal level of neural specific adenylyl cyclase type I is decreased in Alzheimer’s disease. Biochim Biophys Acta 1535: , 60–68. |
[206] | Prapong T , Uemura E , Hsu WH ((2001) ) G protein and cAMP-dependent protein kinase mediate amyloid β-peptide inhibition of neuronal glucose uptake. Exp Neurol 167: , 59–64. |
[207] | Song Q , Zheng HW , Li XH , Huganir RL , Kuner T , Zhuo M , Chen T ((2017) ) Selective phosphorylation of AMPA receptor contributes to the network of long-term potentiation in the anterior cingulate cortex. J Neurosci 37: , 8534–8548. |
[208] | Mizuno M , Yamada K , He J , Nakajima A , Nabeshima T ((2003) ) Involvement of BDNF receptor TrkB in spatial memory formation. Learn Mem 10: , 108–115. |
[209] | Martínez M , Fernández E , Frank A , Guaza C , de la Fuente M , Hernanz A ((1999) ) Increased cerebrospinal fluid cAMP levels in Alzheimer’s disease. Brain Res 846: , 265–267. |
[210] | Cashman RE , Grammas P ((1995) ) cAMP-dependent protein kinase in cerebral microvessels in aging and Alzheimer disease. Mol Chem Neuropathol 26: , 247–258. |
[211] | Vitolo OV , Sant’angelo A , Costanzo V , Battaglia F , Arancio O , Shelanski M ((2002) ) Amyloid beta-peptide inhibition of the PKA/CREB pathway and long-term potentiation: Reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci U S A 99: , 13217–13221. |
[212] | Hiltunen M , Kerola T , Kettunen R , Hartikainen S , Sulkava R , Vuolteenaho O , Nieminen T ((2013) ) The prognostic capacity of B-type natriuretic peptide on cognitive disorder varies by age. Ann Med 45: , 74–78. |
[213] | Kelly MP ((2018) ) Cyclic nucleotide signaling changes associated with normal aging and age-related diseases of the brain. Cell Signal 42: , 281–291. |
[214] | Hu WT , Holtzman DM , Fagan AM , Shaw LM , Perrin R , Arnold SE , Grossman M , Xiong C , Craig-Schapiro R , Clark CM , Pickering E , Kuhn M , Chen Y , Van Deerlin VM , McCluskey L , Elman L , Karlawish J , Chen-Plotkin A , Hurtig HI , Siderowf A , Swenson F , Lee VMY , Morris JC , Trojanowski JQ , Soares H ((2012) ) Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology 79: , 897–905. |
[215] | Marksteiner J , Imarhiagbe D , Defrancesco M , Deisenhammer EA , Kemmler G , Humpel C ((2013) ) Analysis of 27 vascular-related proteins reveals that NT-proBNP is a potential biomarker for Alzheimer’s disease and mild cognitive impairment: A pilot-study. Exp Gerontol 50: , 114–121. |
[216] | Naito J , Naka Y , Watanabe H ((2009) ) Clinical impression of brain natriuretic peptide levels in demented patients without cardiovascular disease. Geriatr Gerontol Int 9: , 242–245. |
[217] | Mirza SS , De Bruijn RFAG , Koudstaal PJ , Van Den Meiracker AH , Franco OH , Hofman A , Tiemeier H , Ikram MA ((2016) ) The N-terminal pro B-type natriuretic peptide, and risk of dementia and cognitive decline: A 10-year follow-up Study in the general population. J Neurol Neurosurg Psychiatry 87: , 356–362. |
[218] | Kondziella D , Göthlin M , Fu M , Zetterberg H , Wallin A ((2009) ) B-type natriuretic peptide plasma levels are elevated in subcortical vascular dementia. Neuroreport 20: , 825–827. |
[219] | Kara K , Mahabadi AA , Weimar C , Winkler A , Neumann T , Kälsch H , Dragano N , Moebus S , Erbel R , Jöckel KH , Jokisch M ((2016) ) N-terminal pro-B type natriuretic peptide is associated with mild cognitive impairment in the general population. J Alzheimers Dis 55: , 359–369. |
[220] | Hilal S , Chai YL , Ikram MK , Elangovan S , Yeow TB , Xin X , Chong JY , Venketasubramanian N , Richards AM , Chong JPC , Lai MKP , Chen C ((2015) ) Markers of cardiac dysfunction in cognitive impairment and dementia. Medicine (Baltimore) 94: , e297. |
[221] | Tynkkynen J , Laatikainen T , Salomaa V , Havulinna AS , Blankenberg S , Zeller T , Hernesniemi JA ((2015) ) NT-proBNP and the risk of dementia: A prospective cohort study with 14 years of follow-up. J Alzheimers Dis 44: , 1007–1013. |
[222] | Tynkkynen J , Hernesniemi JA , Laatikainen T , Havulinna AS , Salo P , Blankenberg S , Zeller T , Salomaa V ((2017) ) High-sensitivity cardiac troponin I and NT-proBNP as predictors of incident dementia and Alzheimer’s disease: The FINRISK Study. J Neurol 264: , 503–511. |
[223] | Soares HD , Potter WZ , Pickering E , Kuhn M , Immermann FW , Shera DM , Ferm M , Dean RA , Simon AJ , Swenson F , Siuciak JA , Kaplow J , Thambisetty M , Zagouras P , Koroshetz WJ , Wan HI , Trojanowski JQ , Shaw LM ((2012) ) Plasma biomarkers associated with the apolipoprotein E genotype and alzheimer disease. Arch Neurol 69: , 1310–1317. |
[224] | Medina AE ((2011) ) Therapeutic utility of phosphodiesterase type I inhibitors in neurological conditions. Front Neurosci 5: , 1–5. |
[225] | Zhang Y shuai , Li J dong , Yan C ((2018) ) An update on vinpocetine: New discoveries and clinical implications. Eur J Pharmacol 819: , 30–34. |
[226] | Ali AA , Ahmed HI , Khaleel SA , Abu-Elfotuh K ((2019) ) Vinpocetine mitigates aluminum-induced cognitive impairment in socially isolated rats. Physiol Behav 208: , 112571. |
[227] | Krahe TE , Wang W , Medina AE ((2009) ) Phosphodiesterase inhibition increases CREB phosphorylation and restores orientation selectivity in a model of fetal alcohol spectrum disorders. PLoS One 4: , e6643. |
[228] | Ahmed HI , Abdel-Sattar SA , Zaky HS ((2019) ) Vinpocetine halts ketamine-induced schizophrenia-like deficits in rats: Impact on BDNF and GSK-3β/β-catenin pathway. Naunyn Schmiedebergs Arch Pharmacol 391: , 1327–1338. |
[229] | Lourenco-Gonzalez Y , Fattori V , Domiciano TP , Rossaneis AC , Borghi SM , Zaninelli TH , Bernardy CCF , Alves-Filho JC , Cunha TM , Cunha FQ , Casagrande R , Verri WA ((2019) ) Repurposing of the nootropic drug vinpocetine as an analgesic and anti-inflammatory agent: Evidence in a mouse model of superoxide anion-triggered inflammation. Mediators Inflamm 2019: , 6481812. |
[230] | Abdel-Salam OME , Hamdy SM , Seadawy SAM , Galal AF , Abouelfadl DM , Atrees SS ((2016) ) Effect of piracetam, vincamine, vinpocetine, and donepezil on oxidative stress and neurodegeneration induced by aluminum chloride in rats. Comp Clin Path 25: , 305–318. |
[231] | Ruiz-Miyazawa KW , Zarpelon AC , Pinho-Ribeiro FA , Pavão-de-Souza GF , Casagrande R , Verri WA ((2015) ) Vinpocetine reduces carrageenan-induced inflammatory hyperalgesia in mice by inhibiting oxidative stress, cytokine production and NF-κB activation in the paw and spinal cord. PLoS One 10: , e0118942. |
[232] | Ruiz-Miyazawa KW , Pinho-Ribeiro FA , Zarpelon AC , Staurengo-Ferrari L , Silva RL , Alves-Filho JC , Cunha TM , Cunha FQ , Casagrande R , Verri WA ((2015) ) Vinpocetine reduces lipopolysaccharide-induced inflammatory pain and neutrophil recruitment in mice by targeting oxidative stress, cytokines and NF-κB. Chem Biol Interact 237: , 9–17. |
[233] | Ali AA , Abo El-Ella DM , El-Emam SZ , Shahat AS , El-Sayed RM ((2019) ) Physical & mental activities enhance the neuroprotective effect of vinpocetine & coenzyme Q10 combination against Alzheimer & bone remodeling in rats. Life Sci 229: , 21–35. |
[234] | Gupta S , Sharma B ((2014) ) Protective effects of phosphodiesterase-1 (PDE1) and ATP sensitive potassium (KATP) channel modulators against 3-nitropropionic acid induced behavioral and biochemical toxicities in experimental Huntingtons disease. Eur J Pharmacol 732: , 111–122. |
[235] | Nadeem RI , Ahmed HI , El-Sayeh BM ((2018) ) Protective effect of vinpocetine against neurotoxicity of manganese in adult male rats. Naunyn Schmiedebergs Arch Pharmacol 391: , 729–742. |
[236] | Hu C , Li L ((2017) ) Pre-conditions for eliminating mitochondrial dysfunction and maintaining liver function after hepatic ischaemia reperfusion. J Cell Mol Med 21: , 1719–1731. |
[237] | Zaki HF , Abdelsalam RM ((2013) ) Vinpocetine protects liver against ischemia-reperfusion injury. Can J Physiol Pharmacol 91: , 1064–1070. |
[238] | Jha SK , Jha NK , Kumar D , Ambasta RK , Kumar P ((2017) ) Linking mitochondrial dysfunction, metabolic syndrome and stress signaling in neurodegeneration. Biochim Biophys Acta Mol Basis Dis 1863: , 1132–1146. |
[239] | Fattori V , Borghi SM , Guazelli CFS , Giroldo AC , Crespigio J , Bussmann AJC , Coelho-Silva L , Ludwig NG , Mazzuco TL , Casagrande R , Verri WA ((2017) ) Vinpocetine reduces diclofenac-induced acute kidney injury through inhibition of oxidative stress, apoptosis, cytokine production, and NF-κB activation in mice. Pharmacol Res 120: , 10–22. |
[240] | Svab G , Doczi J , Gerencser AA , Ambrus A , Gallyas F , Sümegi B , Tretter L ((2019) ) The mitochondrial targets of neuroprotective drug vinpocetine on primary neuron cultures, brain capillary endothelial cells, synaptosomes, and brain mitochondria. Neurochem Res 44: , 2435–2447. |
[241] | Tárnok K , Kiss E , Luiten PGM , Nyakas C , Tihanyi K , Schlett K , Eisel ULM ((2008) ) Effects of Vinpocetine on mitochondrial function and neuroprotection in primary cortical neurons. Neurochem Int 53: , 289–295. |
[242] | Wang H , Zhang K , Zhao L , Tang J , Gao L , Wei Z ((2014) ) Anti-inflammatory effects of vinpocetine on the functional expression of nuclear factor-kappa B and tumor necrosis factor-alpha in a rat model of cerebral ischemia-reperfusion injury. Neurosci Lett 566: , 247–251. |
[243] | Liu RT , Wang A , To E , Gao J , Cao S , Cui JZ , Matsubara JA ((2014) ) Vinpocetine inhibits amyloid-beta induced activation of NF-κB, NLRP3 inflammasome and cytokine production in retinal pigment epithelial cells. Exp Eye Res 127: , 49–58. |
[244] | Zhang L , Yang L ((2014) ) Anti-inflammatory effects of Vinpocetine in atherosclerosis and ischemic stroke: A review of the literature. Molecules 20: , 335–347. |
[245] | Jeon KI , Xu X , Aizawa T , Lim JH , Jono H , Kwon DS , Abe JI , Berk BC , Li JD , Yan C ((2010) ) Vinpocetine inhibits NF-κB-dependent inflammation via an IKK-dependent but PDE-independent mechanism. Proc Natl Acad Sci U S A 107: , 9795–9800. |
[246] | Essam RM , Ahmed LA , Abdelsalam RM , El-Khatib AS ((2019) ) Phosphodiestrase-1 and 4 inhibitors ameliorate liver fibrosis in rats: Modulation of cAMP/CREB/TLR4 inflammatory and fibrogenic pathways. Life Sci 222: , 245–254. |
[247] | Zhu M , Liu H , Sun K , Liu J , Mou Y , Qi D , Zhou C , Abudunaibi M , Tasiken B , Li J , Cheng H , Huang H ((2020) ) Vinpocetine inhibits RANKL-induced osteoclastogenesis and attenuates ovariectomy-induced bone loss. Biomed Pharmacother 123: , 109769. |
[248] | Zhuang J , Peng W , Li H , Lu Y , Ke W , Li Y , Fan F , Xu Y ((2013) ) ASSA13-03-13 Inhibitory effects of Vinpocetine on the progression of atherosclerosis are mediated by Akt/NF-kB dependent mechanisms. Heart 99: , A18.1-A18. |
[249] | Zhang F , Yan C , Wei C , Yao Y , Ma X , Gong Z , Liu S , Zang D , Chen J , Shi FD , Hao J ((2018) ) Vinpocetine inhibits NF-κB-dependent inflammation in acute ischemic stroke patients. Transl Stroke Res 9: , 174–184. |
[250] | Colombo BB , Fattori V , Guazelli CFS , Zaninelli TH , Carvalho TT , Ferraz CR , Bussmann AJC , Ruiz-Miyazawa KW , Baracat MM , Casagrande R , Verri WA ((2018) ) Vinpocetine ameliorates acetic acid-induced colitis by inhibiting NF-κB activation in mice. Inflammation 41: , 1276–1289. |
[251] | Kim NJ , Baek JH , Lee JA , Kim HN , Song JK , Chun KH ((2019) ) A PDE1 inhibitor reduces adipogenesis in mice via regulation of lipolysis and adipogenic cell signaling. Exp Mol Med 51: , 1–15. |
[252] | Cai Y , Knight WE , Guo S , Li JD , Knight PA , Yan C ((2012) ) Vinpocetine suppresses pathological vascular remodeling by inhibiting vascular smooth muscle cell proliferation and migration. J Pharmacol Exp Ther 343: , 479–488. |
[253] | Thal LJ , Salmon DP , Lasker B , Bower D , Klauber MR ((1989) ) The safety and lack of efficacy of Vinpocetine in Alzheimer’s disease. J Am Geriatr Soc 37: , 515–520. |
[254] | Wolters EC , Scheltens P , Zwart J , Persijn L , Moll C , van Genuchten H , Lowenthal A , Sennef C ((1992) ) A double blind, placebo and piracetam controlled, multicenter trial of vinpocetine in dementia of Alzheimer’s type and vascular dementia. Neurobiol Aging 13: , s127. |
[255] | Wollschlaeger B ((2001) ) Efficacy of Vinpocetine in the management of cognitive impairment and memory loss. JANA 4: , 25–30. |
[256] | Szatmari SZ , Whitehouse PJ ((2003) ) Vinpocetine for cognitive impairment and dementia. Cochrane Database Syst Rev, CD003119. |
[257] | Valikovics A , Csányi A , Németh L ((2012) ) [Study of the effects of vinpocetin on cognitive functions]. Ideggyogy Sz 65: , 115–120. |
[258] | Ogunrin A ((2014) ) Effect of vinpocetine (cognitolTM) on cognitive performances of a Nigerian population. Ann Med Health Sci Res 4: , 654. |
[259] | Schaeffer P , Luginer C , Follenius-Wund A , Gerard D , Stoclet JC ((1987) ) Comparative effects of calmodulin inhibitors on calmodulin’s hydrophobic sites and on the activation of cyclic nucleotide phosphodiesterase by calmodulin. Biochem Pharmacol 36: , 1989–1996. |
[260] | Leskovac V ((1980) ) Modulatory actions of nicergoline in the central nervous system. In Synaptic Constituents in Health and Disease, BrzinM, SketD, BachelardH, eds. Elsevier, p. 609. |
[261] | Brzin M , Sket D , Bachelard H ((1980) ), Synaptic Constituents in Health and Disease: Proceedings of the Third Meeting of the European Society for Neurochemistry, Bled, August 31st to September 5th, 1980. |
[262] | Alvarez-Guerra M , Bertholom N , Garay RP ((1999) ) Selective blockade by nicergoline of vascular responses elicited by stimulation of alpha(1A)-adrenoceptor subtype in the rat. Fundam Clin Pharmacol 13: , 50–58. |
[263] | Winblad B , Fioravanti M , Dolezal T , Logina I , Milanov IG , Popescu DC , Solomon A ((2008) ) Therapeutic use of nicergoline. Clin Drug Investig 28: , 533–552. |
[264] | Saletu B , Garg A , Shoeb A ((2014) ) Safety of nicergoline as an agent for management of cognitive function disorders. Biomed Res Int 2014: , 610103. |
[265] | Zang G , Fang L , Chen L , Wang C ((2018) ) Ameliorative effect of nicergoline on cognitive function through the PI3K/AKT signaling pathway in mouse models of Alzheimer’s disease. Mol Med Rep 17: , 7293–7300. |
[266] | Mizuno T , Kuno R , Nitta A , Nabeshima T , Zhang G , Kawanokuchi J , Wang J , Jin S , Takeuchi H , Suzumura A ((2005) ) Protective effects of nicergoline against neuronal cell death induced by activated microglia and astrocytes. Brain Res 1066: , 78–85. |
[267] | Carfagna N , Di Clemente A , Cavanus S , Damiani D , Gerna M , Salmoiraghi P , Cattaneo B , Post C ((1995) ) Modulation of hippocampal ACh release by chronic nicergoline treatment in freely moving young and aged rats. Neurosci Lett 197: , 195–198. |
[268] | McArthur RA , Carfagna N , Banfi L , Cavanus S , Cervini MA , Fariello R , Post C ((1997) ) Effects of nicergoline on age-related decrements in radial maze performance and acetylcholine levels. Brain Res Bull 43: , 305–311. |
[269] | Iwata E , Miyazaki I , Asanuma M , Iida A , Ogawa N ((1998) ) Protective effects of nicergoline against hydrogen peroxide toxicity in rat neuronal cell line. Neurosci Lett 251: , 49–52. |
[270] | Yoshida T , Tanaka M , Okamoto K ((2001) ) Inhibitory effect of nicergoline on superoxide generation by activated rat microglias measured using a simple chemiluminescence method. Neurosci Lett 297: , 5–8. |
[271] | Tanaka M , Yoshida T , Okamoto K , Hirai S ((1998) ) Antioxidant properties of nicergoline; inhibition of brain auto- oxidation and superoxide production of neutrophils in rats. Neurosci Lett 248: , 68–72. |
[272] | Caraci F , Chisari M , Frasca G , Canonico PL , Battaglia A , Calafiore M , Battaglia G , Bosco P , Nicoletti F , Copani A , Sortino MA ((2005) ) Nicergoline, a drug used for age-dependent cognitive impairment, protects cultured neurons against β-amyloid toxicity. Brain Res 1047: , 30–37. |
[273] | Fioravanti M , Flicker L ((2001) ) Nicergoline for dementia and other age associated forms of cognitive impairment. Cochrane Database Syst Rev 2001: , CD003159. |
[274] | Amaducci L , Maurer K , Winblad B , Dom R , Bullock R , Bonura L , Battaglia A , Musch B ((1999) ) A long-term, double-blind, placebo-controlled efficacy and safety study of nicergoline in patients with mild to moderate Alzheimer’s disease. Eur Neuropsychopharmacol 9: , 323–324. |
[275] | Winblad B , Bonura ML , Rossini BM , Battaglia A ((2001) ) Nicergoline in the treatment of mild-to-moderate Alzheimer’s disease: A European multicentre trial. Clin Drug Investig 21: , 621–632. |
[276] | Im JJ , Jeong HS , Park JS , Yang Y , Na SH , Oh JK , Chung YA , Song IU ((2017) ) Changes in regional cerebral perfusion after nicergoline treatment in early Alzheimer’s disease: A pilot study. Dement Neurocognitive Disord 16: , 104. |
[277] | Im JJ , Jeong H , Oh JK , Chung YA , Song IU , Lee KS ((2019) ) Effects of nicergoline treatment on regional cerebral blood flow in early Alzheimer’s disease. Int J Imaging Syst Technol 29: , 323–328. |
[278] | Fioravanti M , Nakashima T , Xu J , Garg A ((2014) ) A systematic review and meta-analysis assessing adverse event profile and tolerability of nicergoline. BMJ Open 4: , e005090. |
[279] | Kakkar R , Raju RVS , Rajput AH , Sharma RK ((1996) ) Inhibition of bovine brain calmodulin-dependent cyclic nucleotide phosphodiesterase isozymes by deprenyl. Life Sci 59: , PL337–41. |
[280] | Magyar K ((2011) ) The pharmacology of selegiline. In Monoamine Oxidases and their Inhibitors, YoudimM, RiedererP, eds. Academic Press, p. 65. |
[281] | Moore JJ , Saadabadi A ((2020) ) Selegiline. In StatPearls [Internet]. StatPearls Publishing, Treasure Island, FL. |
[282] | Selegiline, Drugs.com, https://www.drugs.com/international/selegiline.html, Accessed 12 May 2020. |
[283] | Priyanka HP , Bala P , Ankisettipalle S , Thyagarajan S ((2013) ) Bacopa monnieri and l-deprenyl differentially enhance the activities of antioxidant enzymes and the expression of tyrosine hydroxylase and nerve growth factor via ERK 1/2 and NF-κB pathways in the spleen of female Wistar rats. Neurochem Res 38: , 141–152. |
[284] | Nakaso K , Nakamura C , Sato H , Imamura K , Takeshima T , Nakashima K ((2006) ) Novel cytoprotective mechanism of anti-parkinsonian drug deprenyl: PI3K and Nrf2-derived induction of antioxidative proteins. Biochem Biophys Res Commun 339: , 915–922. |
[285] | Xiao H , Lv F , Xu W , Zhang L , Jing P , Cao X ((2011) ) Deprenyl prevents MPP+-induced oxidative damage in PC12 cells by the upregulation of Nrf2-mediated NQO1 expression through the activation of PI3K/Akt and Erk. Toxicology 290: , 286–294. |
[286] | Cui Y , Liu KWK , Liang Y , Ip MSM , Mak JCW ((2017) ) Inhibition of monoamine oxidase-B by selegiline reduces cigarette smoke-induced oxidative stress and inflammation in airway epithelial cells. Toxicol Lett 268: , 44–50. |
[287] | Naoi M , Maruyama W , Shamoto-Nagai M ((2020) ) Rasagiline and selegiline modulate mitochondrial homeostasis, intervene apoptosis system and mitigate α-synuclein cytotoxicity in disease-modifying therapy for Parkinson’s disease. J Neural Transm 127: , 131–147. |
[288] | Brabeck C , Pfeiffer R , Leake A , Beneke S , Meyer R , Bürkle A ((2003) ) L-selegiline potentiates the cellular poly(ADP-ribosyl)ation response to ionizing radiation. J Pharmacol Exp Ther 306: , 973–979. |
[289] | Birks J , Flicker L ((2003) ) Selegiline for Alzheimer’s disease. Cochrane Database Syst Rev, CD000442. |
[290] | Shakur Y , Takeda K , Kenan Y , Yu ZX , Rena G , Brandt D , Houslay MD , Degerman E , Ferrans VJ , Manganiello VC ((2000) ) Membrane localization of cyclic nucleotide phosphodiesterase 3 (PDE3). Two n-terminal domains are required for the efficient targeting to, and association of, PDE3 with endoplasmic reticulum. J Biol Chem 275: , 38749–38761. |
[291] | Consortium TU ((2017) ) UniProt: The universal protein knowledgebase. Nucleic Acids Res 45: , D158–D169. |
[292] | Cilostazol Monograph for Professionals, Drugs.com, https://www.drugs.com/monograph/cilostazol.html, Accessed 12 May 2020. |
[293] | Nishi T , Kimura Y , Nakagawa K ((2000) ) [Research and development of cilostazol: An antiplatelet agent]. Yakugaku Zasshi 120: , 1247–1260. |
[294] | Ono K , Tsuji M ((2019) ) Pharmacological potential of cilostazol for Alzheimer’s disease. Front Pharmacol 10: , 559. |
[295] | Watanabe T , Zhang N , Liu M , Tanaka R , Mizuno Y , Urabe T ((2006) ) Cilostazol protects against brain white matter damage and cognitive impairment in a rat model of chronic cerebral hypoperfusion. Stroke 37: , 1539–1545. |
[296] | Lee JH , Shin HK , Park SY , Kim CD , Lee WS , Hong KW ((2009) ) Cilostazol preserves CA1 hippocampus and enhances generation of immature neuroblasts in dentate gyrus after transient forebrain ischemia in rats. Exp Neurol 215: , 87–94. |
[297] | Chen WJ , Chen YH , Lin KH , Ting CH , Yeh YH ((2011) ) Cilostazol promotes vascular smooth muscles cell differentiation through the cAMP response element-binding protein-dependent pathway. Arterioscler Thromb Vasc Biol 31: , 2106–2113. |
[298] | Joe Y , Zheng M , Kim HJ , Uddin MJ , Kim SK , Chen Y , Park J , Cho GJ , Ryter SW , Chung HT ((2015) ) Cilostazol attenuates murine hepatic ischemia and reperfusion injury via heme oxygenase-dependent activation of mitochondrial biogenesis. Am J Physiol Gastrointest Liver Physiol 309: , G21–29. |
[299] | Wang B , Zhu L , Sui S , Sun C , Jiang H , Ren D ((2014) ) Cilostazol induces mitochondrial fatty acid β-oxidation in C2C12 myotubes. Biochem Biophys Res Commun 447: , 441–445. |
[300] | Zuo L , Li Q , Sun B , Xu Z , Ge Z ((2013) ) Cilostazol promotes mitochondrial biogenesis in human umbilical vein endothelial cells through activating the expression of PGC-1α . Biochem Biophys Res Commun 433: , 52–57. |
[301] | Sheu JJ , Lin KC , Tsai CY , Tsai TH , Leu S , Yen CH , Chen YL , Chang HW , Sun CK , Chua S , Yang JL , Yip HK ((2012) ) Combination of cilostazol and clopidogrel attenuates Rat critical limb ischemia. J Transl Med 10: , 164. |
[302] | Hafez HM , Ibrahim MA , Zedan MZ , Hassan M , Hassanein H ((2019) ) Nephroprotective effect of cilostazol and verapamil against thioacetamide-induced toxicity in rats may involve Nrf2/HO-1/NQO-1 signaling pathway. Toxicol Mech Methods 29: , 146–152. |
[303] | Hassan M , Ibrahim MA , Hafez HM , Mohamed MZ , Zenhom NM , Abd Elghany HM ((2019) ) Role of Nrf2/HO-1 and PI3K/Akt genes in the hepatoprotective effect of cilostazol. Curr Clin Pharmacol 14: , 61–67. |
[304] | Abuelezz SA , Hendawy N ((2018) ) Insights into the potential antidepressant mechanisms of cilostazol in chronically restraint rats. Behav Pharmacol 29: , 28–40. |
[305] | Park SY , Lee SW , Shin HK , Chung WT , Lee WS , Rhim BY , Hong KW , Kim CD ((2010) ) Cilostazol enhances apoptosis of synovial cells from rheumatoid arthritis patients with inhibition of cytokine formation via Nrf2-linked heme oxygenase 1 induction. Arthritis Rheum 62: , 732–741. |
[306] | Su SC , Hung YJ , Huang CL , Shieh YS , Chien CY , Chiang CF , Liu JS , Lu CH , Hsieh CH , Lin CM , Lee CH ((2019) ) Cilostazol inhibits hyperglucose-induced vascular smooth muscle cell dysfunction by modulating the RAGE/ERK/NF-κB signaling pathways. J Biomed Sci 26: , 68. |
[307] | Park SY , Kim HY , Park HJ , Shin HK , Hong KW , Kim CD ((2016) ) Concurrent treatment with taxifolin and cilostazol on the lowering of β-Amyloid accumulation and neurotoxicity via the suppression of P-JAK2/P-STAT3/NF-κB/BACE1 signaling pathways. PLoS One 11: , e0168286. |
[308] | da Motta NAV , de Brito FCF ((2016) ) Cilostazol exerts antiplatelet and anti-inflammatory effects through AMPK activation and NF-kB inhibition on hypercholesterolemic rats. Fundam Clin Pharmacol 30: , 327–337. |
[309] | Aoki C , Hattori Y , Tomizawa A , Jojima T , Kasai K ((2010) ) Anti-inflammatory role of cilostazol in vascular smooth muscle cells in vitro and in vivo. J Atheroscler Thromb 17: , 503–509. |
[310] | Sakamoto T , Ohashi W , Tomita K , Hattori K , Matsuda N , Hattori Y ((2018) ) Anti-inflammatory properties of cilostazol: Its interruption of DNA binding activity of NF-κB from the Toll-like receptor signaling pathways. Int Immunopharmacol 62: , 120–131. |
[311] | Otsuki M , Saito H , Xu X , Sumitani S , Kouhara H , Kurabayashi M , Kasayama S ((2001) ) Cilostazol represses vascular cell adhesion molecule-1 gene transcription via inhibiting NF-κB binding to its recognition sequence. Atherosclerosis 158: , 121–128. |
[312] | Kim HN , Gil CH , Kim YR , Shin HK , Choi BT ((2016) ) Anti-photoaging properties of the phosphodiesterase 3 inhibitor cilostazol in ultraviolet B-irradiated hairless mice. Sci Rep 6: , 1–10. |
[313] | Tai SY , Chien CY , Chang YH , Yang YH ((2017) ) Cilostazol use is associated with reduced risk of dementia: A nationwide cohort study. Neurotherapeutics 14: , 784–791. |
[314] | Taguchi A , Takata Y , Ihara M , Kasahara Y , Tsuji M , Nishino M , Stern D , Okada M ((2013) ) Cilostazol improves cognitive function in patients with mild cognitive impairment: A retrospective analysis. Psychogeriatrics 13: , 164–169. |
[315] | Ihara M , Nishino M , Taguchi A , Yamamoto Y , Hattori Y , Saito S , Takahashi Y , Tsuji M , Kasahara Y , Takata Y , Okada M ((2014) ) Cilostazol add-on therapy in patients with mild dementia receiving donepezil: A retrospective study. PLoS One 9: , e89516. |
[316] | Tai SY , Chen CH , Chien CY , Yang YH ((2017) ) Cilostazol as an add-on therapy for patients with Alzheimer’s disease in Taiwan: A case control study. BMC Neurol 17: , 40. |
[317] | Arai H , Takahashi T ((2009) ) A combination therapy of Donepezil and Cilostazol for patients with moderate Alzheimer disease: Pilot follow-up study. Am J Geriatr Psychiatry 17: , 353–354. |
[318] | Sakurai H , Hanyu H , Sato T , Kume K , Hirao K , Kanetaka H , Iwamoto T ((2013) ) Effects of cilostazol on cognition and regional cerebral blood flow in patients with Alzheimer’s disease and cerebrovascular disease: A pilot study. Geriatr Gerontol Int 13: , 90–97. |
[319] | Lee JY , Lee H , Yoo H Bin , Choi JS , Jung HY , Yoon EJ , Kim H , Jung YH , Lee HY , Kim YK ((2019) ) Efficacy of cilostazol administration in Alzheimer’s disease patients with white matter lesions: A positron-emission tomography study. Neurotherapeutics 16: , 394–403. |
[320] | Saito S , Kojima S , Oishi N , Kakuta R , Maki T , Yasuno F , Nagatsuka K , Yamamoto H , Fukuyama H , Fukushima M , Ihara M ((2016) ) A multicenter, randomized, placebo-controlled trial for cilostazol in patients with mild cognitive impairment: The COMCID study protocol. Alzheimers Dement (N Y) 2: , 250–257. |
[321] | Rickards KJ , Page CP , Lees P , Gettinby G , Cunningham FM ((2003) ) In vitro and ex vivo effects of the phosphodiesterase 4 inhibitor, rolipram, on thromboxane production in equine blood. J Vet Pharmacol Ther 26: , 123–130. |
[322] | Gong B , Vitolo OV , Trinchese F , Liu S , Shelanski M , Arancio O ((2004) ) Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest 114: , 1624–1634. |
[323] | Cheng YF , Wang C , Lin HB , Li YF , Huang Y , Xu JP , Zhang HT ((2010) ) Inhibition of phosphodiesterase-4 reverses memory deficits produced by Aβ25-35 or Aβ1-40 peptide in rats. Psychopharmacology (Berl) 212: , 181–191. |
[324] | Wang C , Yang XM , Zhuo YY , Zhou H , Lin HB , Cheng YF , Xu JP , Zhang HT ((2012) ) The phosphodiesterase-4 inhibitor rolipram reverses Aβ-induced cognitive impairment and neuroinflammatory and apoptotic responses in rats. Int J Neuropsychopharmacol 15: , 749–766. |
[325] | Zhuo Y , Guo H , Cheng Y , Wang C , Wang C , Wu J , Zou Z , Gan D , Li Y , Xu J ((2016) ) Inhibition of phosphodiesterase-4 reverses the cognitive dysfunction and oxidative stress induced by Aβ25–35 in rats. Metab Brain Dis 31: , 779–791. |
[326] | Sierksma ASR , Van Den Hove DLA , Pfau F , Philippens M , Bruno O , Fedele E , Ricciarelli R , Steinbusch HWM , Vanmierlo T , Prickaerts J ((2014) ) Improvement of spatial memory function in APPswe/PS1dE9 mice after chronic inhibition of phosphodiesterase type 4D. Neuropharmacology 77: , 120–130. |
[327] | Saletu B , Anderer P , Fischhof PK , Lorenz H , Barousch R , Böhmer F ((1992) ) EEG Mapping and psychopharmacological studies with denbufylline in SDAT and MID. Biol Psychiatry 32: , 668–681. |
[328] | Treves TA , Korczyn AD ((1999) ) Denbufylline in dementia: A double-blind controlled study. Dement Geriatr Cogn Disord 10: , 505–510. |
[329] | Goldstein I , Burnett AL , Rosen RC , Park PW , Stecher VJ ((2019) ) The serendipitous story of sildenafil: An unexpected oral therapy for erectile dysfunction. Sex Med Rev 7: , 115–128. |
[330] | Sildenafil Citrate Monograph for Professionals, Drugs.com, https://www.drugs.com/monograph/sildenafil-citrate.html, Accessed 12 May 2020. |
[331] | Orejana L , Barros-Miñones L , Aguirre N , Puerta E ((2013) ) Implication of JNK pathway on tau pathology and cognitive decline in a senescence-accelerated mouse model. Exp Gerontol 48: , 565–571. |
[332] | Devan BD , Pistell PJ , Duffy KB , Kelley-Bell B , Spangler EL , Ingram DK ((2014) ) Phosphodiesterase inhibition facilitates cognitive restoration in rodent models of age-related memory decline. Neurorehabilitation 34: , 101–111. |
[333] | Puzzo D , Staniszewski A , XianDeng S , Privitera L , Leznik E , Liu S , Zhang H , Feng Y , Palmeri A , Landry DW , Arancio O ((2009) ) Phosphodiesterase 5 inhibition improves synaptic function, memory, and amyloid-β load in an Alzheimer’s disease mouse model. J Neurosci 29: , 8075–8086. |
[334] | Jin F , Gong QH , Xu YS , Wang LN , Jin H , Li F , Li LS , Ma YM , Shi JS ((2014) ) Icariin, a phoshphodiesterase-5 inhibitor, improves learning and memory in APP/PS1 transgenic mice by stimulation of NO/cGMP signalling. Int J Neuropsychopharmacol 17: , 871–881. |
[335] | Zhang J , Guo J , Zhao X , Chen Z , Wang G , Liu A , Wang Q , Zhou W , Xu Y , Wang C ((2013) ) Phosphodiesterase-5 inhibitor sildenafil prevents neuroinflammation, lowers beta-amyloid levels and improves cognitive performance in APP/PS1 transgenic mice. Behav Brain Res 250: , 230–237. |
[336] | Puzzo D , Loreto C , Giunta S , Musumeci G , Frasca G , Podda MV , Arancio O , Palmeri A ((2014) ) Effect of phosphodiesterase-5 inhibition on apoptosis and beta amyloid load in aged mice. Neurobiol Aging 35: , 520–531. |
[337] | Sanders O ((2020) ) Sildenafil for the treatment of Alzheimer’s disease: A systematic review. J Alzheimers Dis Rep 4: , 91–106. |
[338] | Sheng M , Lu H , Liu P , Li Y , Ravi H , Peng SL , Diaz-Arrastia R , Devous MD , Womack KB ((2017) ) Sildenafil improves vascular and metabolic function in patients with Alzheimer’s disease. J Alzheimers Dis 60: , 1351–1364. |
[339] | Samudra N , Motes M , Lu H , Sheng M , Diaz-Arrastia R , Devous M , Hart J , Womack KB ((2019) ) A pilot study of changes in medial temporal lobe fractional amplitude of low frequency fluctuations after sildenafil administration in patients with Alzheimer’s disease. J Alzheimers Dis 70: , 163–170. |
[340] | Meskini N , Némoz G , Okyayuz-Baklouti I , Lagarde M , Prigent AF ((1994) ) Phosphodiesterase inhibitory profile of some related xanthine derivatives pharmacologically active on the peripheral microcirculation. Biochem Pharmacol 47: , 781–788. |
[341] | Schwam E , Nicholas T , Chew R , Billing C , Davidson W , Ambrose D , Altstiel L ((2014) ) A multicenter, double-blind, placebo-controlled trial of the PDE9A inhibitor, PF-04447943, in Alzheimer’s disease. Curr Alzheimer Res 11: , 413–421. |
[342] | Frölich L , Wunderlich G , Thamer C , Roehrle M , Garcia M , Dubois B ((2019) ) Evaluation of the efficacy, safety and tolerability of orally administered BI 409306, a novel phosphodiesterase type 9 inhibitor, in two randomised controlled phase II studies in patients with prodromal and mild Alzheimer’s disease. Alzheimers Res Ther 11: , 18. |
[343] | Li J , Liu CN , Wei N , Li XD , Liu YY , Yang R , Jia YJ ((2016) ) Protective effects of BAY 73-6691, a selective inhibitor of phosphodiesterase 9, on amyloid-β peptides-induced oxidative stress in in-vivo and in-vitro models of Alzheimer’s disease. Brain Res 1642: , 327–335. |
[344] | Kroker KS , Mathis C , Marti A , Cassel JC , Rosenbrock H , Dorner-Ciossek C ((2014) ) PDE9A inhibition rescues amyloid beta-induced deficits in synaptic plasticity and cognition. Neurobiol Aging 35: , 2072–2078. |
[345] | Kaupp UB , Seifert R ((2002) ) Cyclic nucleotide-gated ion channels. Physiol Rev 82: , 769–824. |
[346] | Di Stefano M , Loreto A , Orsomando G , Mori V , Zamporlini F , Hulse RP , Webster J , Donaldson LF , Gering M , Raffaelli N , Coleman MP , Gilley J , Conforti L ((2017) ) NMN deamidase delays wallerian degeneration and rescues axonal defects caused by NMNAT2 deficiency in vivo. Curr Biol 27: , 784–794. |
[347] | Loreto A , Di Stefano M , Gering M , Conforti L ((2015) ) Wallerian degeneration is executed by an NMN-SARM1-dependent late Ca(2+) influx but only modestly influenced by mitochondria. Cell Rep 13: , 2539–2552. |
[348] | Duan Y , Gross RA , Sheu S-S ((2007) ) Ca2+-dependent generation of mitochondrial reactive oxygen species serves as a signal for poly(ADP-ribose) polymerase-1 activation during glutamate excitotoxicity. J Physiol 585: , 741–758. |
[349] | Olloquequi J , Cornejo-Córdova E , Verdaguer E , Soriano FX , Binvignat O , Auladell C , Camins A ((2018) ) Excitotoxicity in the pathogenesis of neurological and psychiatric disorders: Therapeutic implications. J Psychopharmacol 32: , 265–275. |
[350] | Lachén-Montes M , González-Morales A , Zelaya MV , Pérez-Valderrama E , Ausín K , Ferrer I , Fernández-Irigoyen J , Santamaría E ((2017) ) Olfactory bulb neuroproteomics reveals a chronological perturbation of survival routes and a disruption of prohibitin complex during Alzheimer’s disease progression. Sci Rep 7: , 1–15. |
[351] | Calderón-Garcidueñas L , González-Maciel A , Kulesza RJ , González-González LO , Reynoso-Robles R , Mukherjee PS , Torres-Jardón R ((2019) ) Air pollution, combustion and friction derived nanoparticles, and Alzheimer’s disease in urban children and young adults. J Alzheimers Dis 70: , 343–360. |
[352] | Bredesen DE ((2016) ) Inhalational Alzheimer’s disease: An unrecognized-and treatable- epidemic. Aging (Albany NY) 8: , 304–313. |
[353] | González-Maciel A , Reynoso-Robles R , Torres-Jardón R , Mukherjee PS , Calderón-Garcidueñas L ((2017) ) Combustion-derived nanoparticles in key brain target cells and organelles in young urbanites: Culprit hidden in plain sight in Alzheimer’s disease development. J Alzheimers Dis 59: , 189–208. |
[354] | Liu L , Drouet V , Wu JW , Witter MP , Small SA , Clelland C , Duff K ((2012) ) Trans-synaptic spread of Tau pathology in vino. PLoS One 7: , e31302. |
[355] | Wang Y , Balaji V , Kaniyappan S , Krüger L , Irsen S , Tepper K , Chandupatla R , Maetzler W , Schneider A , Mandelkow E-ME , Mandelkow E-ME ((2017) ) The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener 12: , 5. |
[356] | Dinkins MB , Wang G , Bieberich E ((2017) ) Sphingolipid-enriched extracellular vesicles and Alzheimer’s disease: A decade of research. J Alzheimers Dis 60: , 757–768. |
[357] | Malm T , Loppi S , Kanninen KM ((2016) ) Exosomes in Alzheimer’s disease. Neurochem Int 97: , 193–199. |
[358] | Sardar Sinha M , Ansell-Schultz A , Civitelli L , Hildesjö C , Larsson M , Lannfelt L , Ingelsson M , Hallbeck M ((2018) ) Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol 136: , 41–56. |
[359] | Nagayama S , Homma R , Imamura F ((2014) ) Neuronal organization of olfactory bulb circuits. Front Neural Circuits 8: , 98. |
[360] | Xu M , Zhang DF , Luo R , Wu Y , Zhou H , Kong LL , Bi R , Yao YG ((2018) ) A systematic integrated analysis of brain expression profiles reveals YAP1 and other prioritized hub genes as important upstream regulators in Alzheimer’s disease. Alzheimers Dement 14: , 215–229. |
[361] | Darmanis S , Sloan SA , Zhang Y , Enge M , Caneda C , Shuer LM , Hayden Gephart MG , Barres BA , Quake SR ((2015) ) A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci U S A 112: , 7285–7290. |
[362] | Singh N , Shreshtha AK , Thakur MS , Patra S ((2018) ) Xanthine scaffold: Scope and potential in drug development. Heliyon 4: , e00829. |
[363] | Grome JJ , Hofmann W , Gojowczyk G , Stefanovich V ((1996) ) Effects of a xanthine derivative, propentofylline, on local cerebral blood flow and glucose utilization in the rat. Brain Res 740: , 41–46. |
[364] | Zeitlin R , Patel S , Burgess S , Arendash GW , Echeverria V ((2011) ) Caffeine induces beneficial changes in PKA signaling and JNK and ERK activities in the striatum and cortex of Alzheimer’s transgenic mice. Brain Res 1417: , 127–136. |
[365] | Cao C , Cirrito JR , Lin X , Wang L , Verges DK , Dickson A , Mamcarz M , Zhang C , Mori T , Arendash GW , Holtzman DM , Potter H ((2009) ) Caffeine suppresses amyloid-β levels in plasma and brain of alzheimer’s disease transgenic mice. J Alzheimers Dis 17: , 681–697. |
[366] | Arendash GW , Mori T , Cao C , Mamcarz M , Runfeldt M , Dickson A , Rezai-Zadeh K , Tan J , Citron BA , Lin X , Echeverria V , Potter H ((2009) ) Caffeine reverses cognitive impairment and decreases brain amyloid-β levels in aged alzheimer’s disease mice. J Alzheimers Dis 17: , 661–680. |
[367] | Kim YS , Kwak SM , Myung SK ((2015) ) Caffeine intake from coffee or tea and cognitive disorders: A meta-analysis of observational studies. Neuroepidemiology 44: , 51–63. |
[368] | Driscoll I , Shumaker SA , Snively BM , Margolis KL , Manson JE , Vitolins MZ , Rossom RC , Espeland MA ((2016) ) Relationships between caffeine intake and risk for probable dementia or global cognitive impairment: The women’s health initiative memory study. J Gerontol A Biol Sci Med Sci 71: , 1596–1602. |
[369] | Liu QP , Wu YF , Cheng HY , Xia T , Ding H , Wang H , Wang ZM , Xu Y ((2016) ) Habitual coffee consumption and risk of cognitive decline/dementia: A systematic review and meta-analysis of prospective cohort studies. Nutrition 32: , 628–636. |
[370] | Wu L , Sun D , He Y ((2017) ) Coffee intake and the incident risk of cognitive disorders: A dose-response meta-analysis of nine prospective cohort studies. Clin Nutr 36: , 730–736. |
[371] | Larsson SC , Orsini N ((2018) ) Coffee consumption and risk of dementia and alzheimer’s disease: A dose-response meta-analysis of prospective studies. Nutrients 10: , E1501. |
[372] | Kim JW , Byun MS , Yi D , Lee JH , Jeon SY , Jung G , Lee HN , Sohn BK , Lee JY , Kim YK , Shin SA , Sohn CH , Lee DY ((2019) ) Coffee intake and decreased amyloid pathology in human brain. Transl Psychiatry 9: , 270. |
[373] | Chauhan NB , Siegel GJ , Feinstein DL ((2005) ) Propentofylline attenuates tau hyperphosphorylation in Alzheimer’s Swedish mutant model Tg2576. Neuropharmacology 48: , 93–104. |
[374] | Banati RB , Schubert P , Rothe G , Gehrmann J , Rudolphi K , Valet G , Kreutzberg GW ((1994) ) Modulation of intracellular formation of reactive oxygen intermediates in peritoneal macrophages and microglia/brain macrophages by propentofylline. J Cereb Blood Flow Metab 14: , 145–149. |
[375] | Rother M , Erkinjuntti T , Roessner M , Kittner B , Marcusson J , Karlsson I ((1998) ) Propentofylline in the treatment of Alzheimer’s disease and vascular dementia: A review of phase III trials. Dement Geriatr Cogn Disord 9: , 36–43. |
[376] | Frampton MA , Harvey RJ , Kirchner V ((2003) ) Propentofylline for dementia. Cochrane Database Syst Rev, CD002853. |
[377] | Si Q , Nakamura Y , Ogata T , Kataoka K , Schubert P ((1998) ) Differential regulation of microglial activation by propentofylline via cAMP signaling. Brain Res 812: , 97–104. |
[378] | McRae A , Schubert P , Ogata T , Nakamura Y , Rudolphi K ((1996) ) Postischemic glial responses and amyloid accumulation are modified by propentofylline: Neuroprotective pharmacon for Alzheimer’s disease? Neurobiol Aging 17: , S152. |
[379] | Szczypka M , Lis M , Suszko-Pawłowska A , Pawlak A , Sysak A , Obmińska-Mrukowicz B ((2017) ) Propentofylline, phosphodiesterase and adenosine reuptake inhibitor modulates lymphocyte subsets and lymphocyte activity after in-vivo administration in non-immunized and SRBC-immunized mice. J Pharm Pharmacol 69: , 1166–1177. |
[380] | Mielke R , Ghaemi M , Kessler J , Kittner B , Szelies B , Herholz K , Heiss WD ((1998) ) Propentofylline enhances cerebral metabolic response to auditory memory stimulation in Alzheimer’s disease. J Neurol Sci 154: , 76–82. |
[381] | Bohm L , Theron T , Binder A ((2000) ) Influence of pentoxifylline, A-802710, propentofylline and A-802715 (Hoechst) on the expression of cell cycle blocks and S-phase content after irradiation damage. Biochim Biophys Acta 1499: , 1–10. |
[382] | Stefanovich V , Joó F ((1990) ) Effect of propentofylline on the biochemical lesion of the rat brain in aluminium-induced neurotoxicity. Metab Brain Dis 5: , 7–17. |
[383] | Yamada K , Tanaka T , Senzaki K , Kameyama T , Nabeshima T ((1998) ) Propentofylline improves learning and memory deficits in rats induced by β-amyloid protein-(1-40). Eur J Pharmacol 349: , 15–22. |
[384] | Wirtz-Brugger F , Giovanni A ((2000) ) Guanosine 3’,5’-cyclic monophosphate mediated inhibition of cell death induced by nerve growth factor withdrawal and β-amyloid: Protective effects of Propentofylline. Neuroscience 99: , 737–750. |
[385] | Koriyama Y , Yamazaki M , Chiba K , Mohri T ((2000) ) Evaluation of neurotoxicity of Alzheimer’s amyloid-beta protein (beta-42) in cultured hippocampal cells and its prevention by propentofylline. Jpn J Pharmacol 82: , 301–306. |
[386] | Koriyama Y , Chiba K , Mohri T ((2003) ) Propentofylline protects β-amyloid protein-induced apoptosis in cultured rat hippocampal neurons. Eur J Pharmacol 458: , 235–241. |
[387] | Rother M ((1999) ) A 72-week, placebo-controlled study assessing propentofylline’s safety, efficacy, and impact on disease progression in patients with Alzheimer’s disease. Eur Neuropsychopharmacol 9: , 319. |
[388] | Kittner B , De Deyn PP , Erkinjuntti T ((2000) ) Investigating the natural course and treatment of vascular dementia and Alzheimer’s disease. Parallel study populations in two randomized, placebo-controlled trials. Ann N Y Acad Sci 903: , 535–541. |
[389] | Kittner B ((1999) ) Using a combined randomized start/withdrawal design to assess propentofylline’s effects on disease progression in Alzheimer’s disease and vascular dementia: Results of clinical studies. Eur Neuropsychopharmacol 9: , 320. |
[390] | Rother M ((1999) ) Long-term effects of propentofylline in patients with Alzheimer’s disease: Safety, efficacy, and impact on disease progession. J Am Geriatr Soc 47: , s2. |
[391] | Rother M ((1999) ) Propentofylline versus placebo in patients with Alzheimer’s disease: A 72-Week study examining safety, efficacy, and impact on disease progression. Neurology 52: , 172. |
[392] | Bozyczko-Coyne D , Williams M ((2007) ) 6.08 - Neurodegeneration. In Comprehensive medicinal chemistry II, pp. 193–228. |
[393] | Aventis Pharma Discontinues Development of Propentofylline. MedScape, Accessed 1 March 2020. |
[394] | Jones R ((2008) ) Antidementia drugs. In Drug treatment in dementia, p. 60. |
[395] | Rother M , Kittner B , Rudolphi K , Rößner M , Labs KH ((1996) ) HWA 285 (propentofylline) - A new compound for the treatment of both vascular dementia and dementia of the Alzheimer type. Ann N Y Acad Sci 777: , 404–409. |
[396] | Can Dogs Get Dementia? Discover CCD, Vet’s Kitchen Blog https://www.vetknowhow.co.uk/blog/can-dogs-get-dementia, Accessed 1 March 2020. |
[397] | Mihevc SP , Majdic G ((2019) ) Canine cognitive dysfunction and Alzheimer’s disease-two facets of the same disease? Front Neurosci 13: , 604. |
[398] | Wimo A , Witthaus E , Rother M , Winblad B ((1998) ) Economic impact of introducing propentofylline for the treatment of dementia in Sweden. Clin Ther 20: , 552–566. |
[399] | Bachynsky J , McCracken P , Lier D , Alloul K , Jacobs P ((2000) ) Propentofylline treatment for Alzheimer disease and vascular dementia: An economic evaluation based on functional abilities. Alzheimer Dis Assoc Disord 14: , 102–111. |


