
Focal traumatic brain injury induces neuroplastic molecular responses in lumbar spinal cord
Abstract
Background/Objectives:
Motor impairment induced by traumatic brain injury (TBI) may be mediated through changes in spinal molecular systems regulating neuronal plasticity. We assessed whether a focal controlled cortical impact (CCI) TBI in the rat alters expression of the Tgfb1, c-Fos, Bdnf, and Gap43 neuroplasticity genes in lumbar spinal cord.
Approach/Methods:
Adult male Sprague-Dawley rats (n = 8) were subjected to a right-side CCI over the anterior sensorimotor hindlimb representation area or sham-injury (n = 8). Absolute expression levels of Tgfb1, c-Fos, Bdnf, and Gapd43 genes were measured by droplet digital PCR in ipsi-and contralesional, dorsal and ventral quadrants of the L4 and L5 spinal cord. The neuronal activity marker c-Fos was analysed by immunohistochemistry in the dorsal L4 and L5 segments. The contra- vs. ipsilesional expression pattern was examined as the asymmetry index, AI.
Results:
The Tgfb1 mRNA levels were significantly higher in the CCI vs. sham-injured rats, and in the contra- vs. ipsilesional dorsal domains in the CCI group. The number of c-Fos-positive cells was elevated in the L4 and L5 segments; and on the contralesional compared to the ipsilesional side in the CCI group. The c-Fos AI in the dorsal laminae was significantly increased by CCI.
Conclusions:
The results support the hypothesis that focal TBI induces plastic alterations in the lumbar spinal cord that may contribute to either motor recovery or maladaptive motor responses.
1.Introduction
Traumatic brain injury (TBI) is an increasing global health problem and a leading cause of death and morbidity (Werner & Stevens, 2015; Wilson et al., 2017). TBI is a disease process beginning at time of impact, exacerbated by a complicated series of molecular events such as influx of calcium ions, mitochondrial damage, disturbed energy metabolism and extensive damage to the cytoskeleton, that may continue up to years after the initial injury (Brooks & Martin, 2014; Cheng, Kong, Zhang, & Zhang, 2012; Masel & DeWitt, 2010). Following TBI, the endogenous capacity for CNS repair and replacement of lost cells is markedly limited. Clinical recovery depends on CNS plasticity that may vary in type, e.g. be beneficial or maladaptive, and locations, occurring in CNS regions distal to the injury (Tennant, 2014). Based on the assumption that this adaptation machinery was restricted to a few regions of the brain, the vast majority of studies have evaluated cortical mechanisms (Axelson et al., 2013; Combs, Jones, Kozlowski, & Adkins, 2016; Pruitt et al., 2017; Wolpaw, 2007). Knowledge of TBI-induced spinal plasticity is limited, but both early and more recent studies have shown that activity-dependent plasticity may occur in the entire CNS, including the spinal cord, that is plausibly beneficial to CNS recovery (Sist, Fouad, & Winship, 2014; Tennant, 2014; Wolpaw, 2007). Examples of maladaptive post-injury plasticity include the development of spasticity characterized by a velocity-dependent increases in tonic stretch reflexes, which could lead to debilitating motor disturbances for the patient. Although the mechanisms causing post-injury spasticity are multifactorial, numerous contributory changes may appear throughout the injured CNS, including the spinal cord. Such factors include altered neurotransmission and excitability of motoneurons, altered synaptic input and function, and changes in the neuronal network (Bose, Hou, & Thompson, 2015).
Following TBI, descending influences from the injured brain may lead to a rearrangement of locomotor spinal neuronal circuits which may manifest as changes in the expression of c-Fos, a marker of neuronal activity, and other neuroplastic genes including for growth-associated protein (Gap-43), brain-derived neurotrophic factor (Bdnf), and transforming growth factor β (Tgfb1). GAP-43 participates in the developmental regulation of axonal growth and neural network formation (Grasselli & Strata, 2013). The main function of BDNF is to regulate synapses, with structural and functional short-and long-lasting effects (Bramham & Messaoudi, 2005). TGF-β regulates multiple biological processes including inflammation and expression of neuropeptides in the spinal cord (Santibanez, Quintanilla, & Bernabeu, 2011). We here addressed the hypothesis that expression of these genes may be altered in the spinal cord remote from a focal TBI induced using the controlled cortical impact (CCI) TBI model in rats. Analysis of the CCI-induced changes in the contra-and ipsilesional patterns of expression of these genes in the spinal cord could provide further evidence for plastic spinal response and shed light on neural spinal circuits affected by brain trauma (Bose et al., 2015). We here performed analysis of i) c-Fos by immunohistochemistry as a readout of neuronal activation, and ii) the levels of Gap-43, Bdnf and Tgfb1 expression by droplet digital polymerase chain reaction (ddPCR) as indicators of neuroplastic changes in the L4 and L5 spinal segments mediating TBI-induced motor deficits.
2.Results
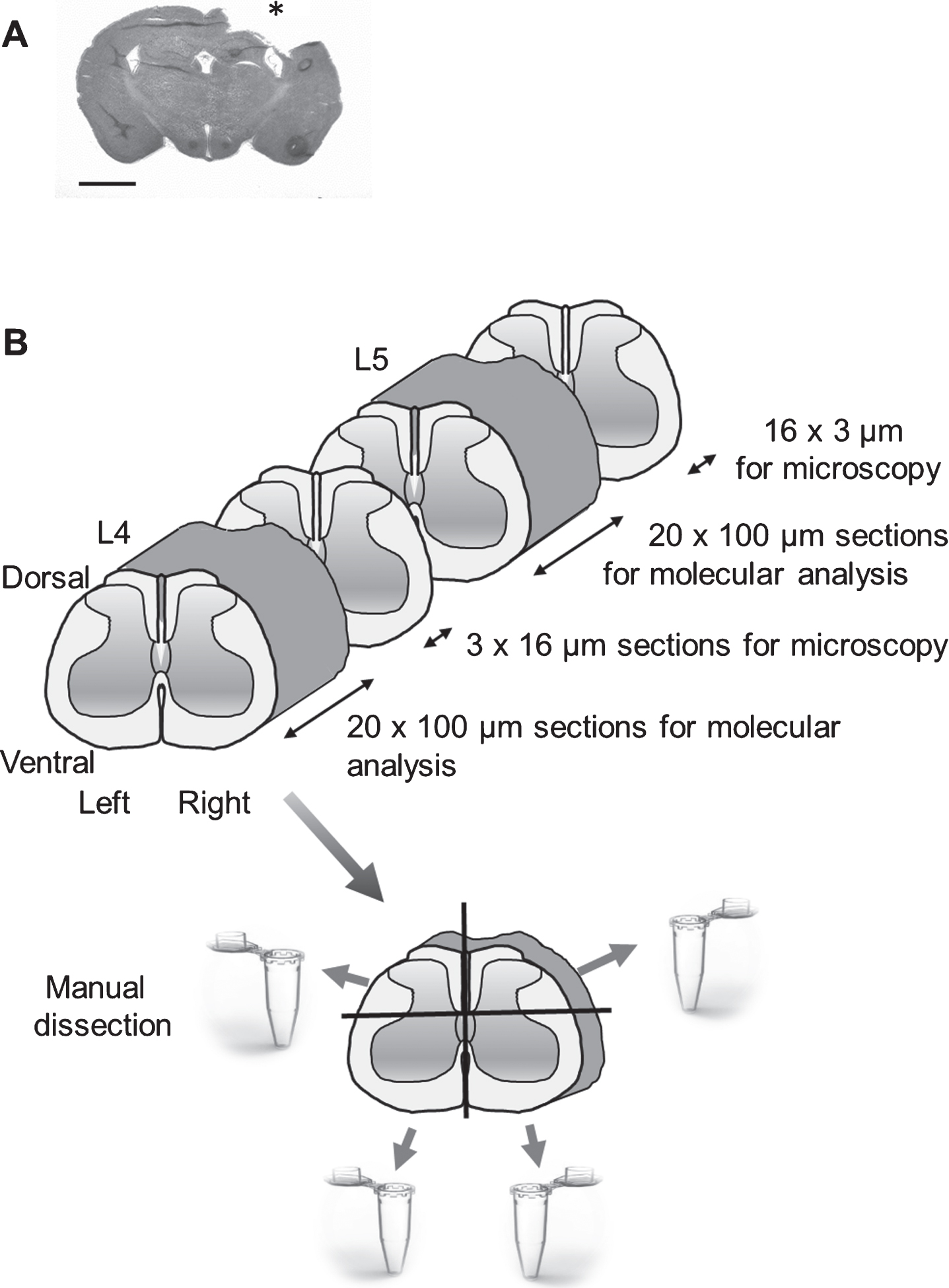
There was no mortality from the surgery, and no animal was excluded from the study. In CCI-injured animals, there was a minor weight loss during the first post-injury days that then recovered. In all brain-injured animals, a focal cortical brain injury was observed (Fig. 1A).
Fig.1
(A) A coronal section of a rat brain following controlled cortical impact (CCI) traumatic brain injury (hematoxylin/eosin (H&E) staining); Scale bar, 2.5 mm. Impact area indicated by *, (B). Schematic overview of the experimental design. The new method for dissection of four tissue quadrants (ipsi-and contralesional, dorsal and ventral) from the rat spinal cord is shown. The frozen tissue block was cut into transverse 100μm sections. After collecting six 100μm sections, a 16μm cryosection was used to control for anatomical localization using H&E staining. From each L4 and L5 level, approximately 13 100μm sections were prepared. Each of these sections was cut along the central axial line (at the laminae VI level), dividing the dorsal and ventral parts to domains of approximately equal size; this operation was followed by an incision along the central sagittal line resulting in separation of the left and right spinal cord halves.
2.1.Effects of CCI on expression of plastic genes in contra-and ipsilesional domains of lumbar spinal cord
Pilot analysis by ddPCR and RNAscope revealed stochastic variability in gene expression between the left and right-side of adjacent thin coronal spinal sections. This micro-heterogeneity in the left-right expression pattern apparently arises due to differences in proportion of inter-and motoneurons and glia among the sides and among the sections. To surpass this heterogeneity, we pooled segments dissected from a number of coronal tissue sections of the L4 and L5 segments.
Following ddPCR (see Fig. 1B for schematic overview), absolute expression levels of the Tgfb1, c-Fos, Bdnf, and Gap43 genes were quantified in dorsal and ventral ipsi-and contralesional domains of L4 and L5 segments of lumbar spinal cord. Two-way ANOVA was performed with operation type (CCI and sham injury) and measurement side (contra-and ipsilesional) as independent factors separately for each gene and for the dorsal and ventral domains of the L4 and L5 lumbar segments.
Significant measurement side and injury type effects of CCI on Tgfb1 expression were revealed in the dorsal domains of the L4 (side: F1,12 = 21.5, p = 0.0006; group: F1,12 = 31.6, p = 0.0001) and L5- (side: F1,12 = 5.0, p = 0.046) segments. In the L4 segment post hoc analysis by Tukey HSD showed a significantly higher Tgfb1 expression in the contralesional domain in the CCI group compared to the ipsilesional side (Left CCI vs Right CCI; L4: p = 0.002; L5: p = 0.015) (Fig. 2A). At the same time a significantly higher Tgfb1 expression in the left dorsal domain in the CCI group was found when compared to the sham-injured group (L4: p = 0.0008; L5: p = 0.025) (Fig. 2A). No significant differences between the injury (CCI and sham) types were evident for the ventral domains (two-way ANOVA for L4: measurement side: F1,10 = 0.5, p = 0.51; injury type: F1,10 = 0.8, p = 0.40; for L5: measurement side: F1,12 = 3.5, p = 0.087; injury type: F1,12 = 0.1, p = 0.720; Fig. 2B). No significant effects were revealed for Bdnf, c-Fos and Gap43 expression levels in dorsal and ventral parts of the lumbar spinal cord (data not shown).
Fig.2
Tgfb1 expression in dorsal (A) and ventral (B) parts of lumbar spinal cord (L4 and L5 levels) measured by droplet digital PCR following controlled cortical impact (CCI) and sham injury. Two-way ANOVA analysis revealed significant injury type (sham and CCI) and measurement side (left and right) effects on Tgfb1 expression in L4 level (measurement side and injury type: p < 0.001) and L5 level (measurement side: p < 0.05) in the dorsal part of the spinal cord. L, left; R, right: n = 4 rats per group. *, p < 0.05, **, p < 0.01, ***, p < 0.001, post hoc analysis Tukey HSD.
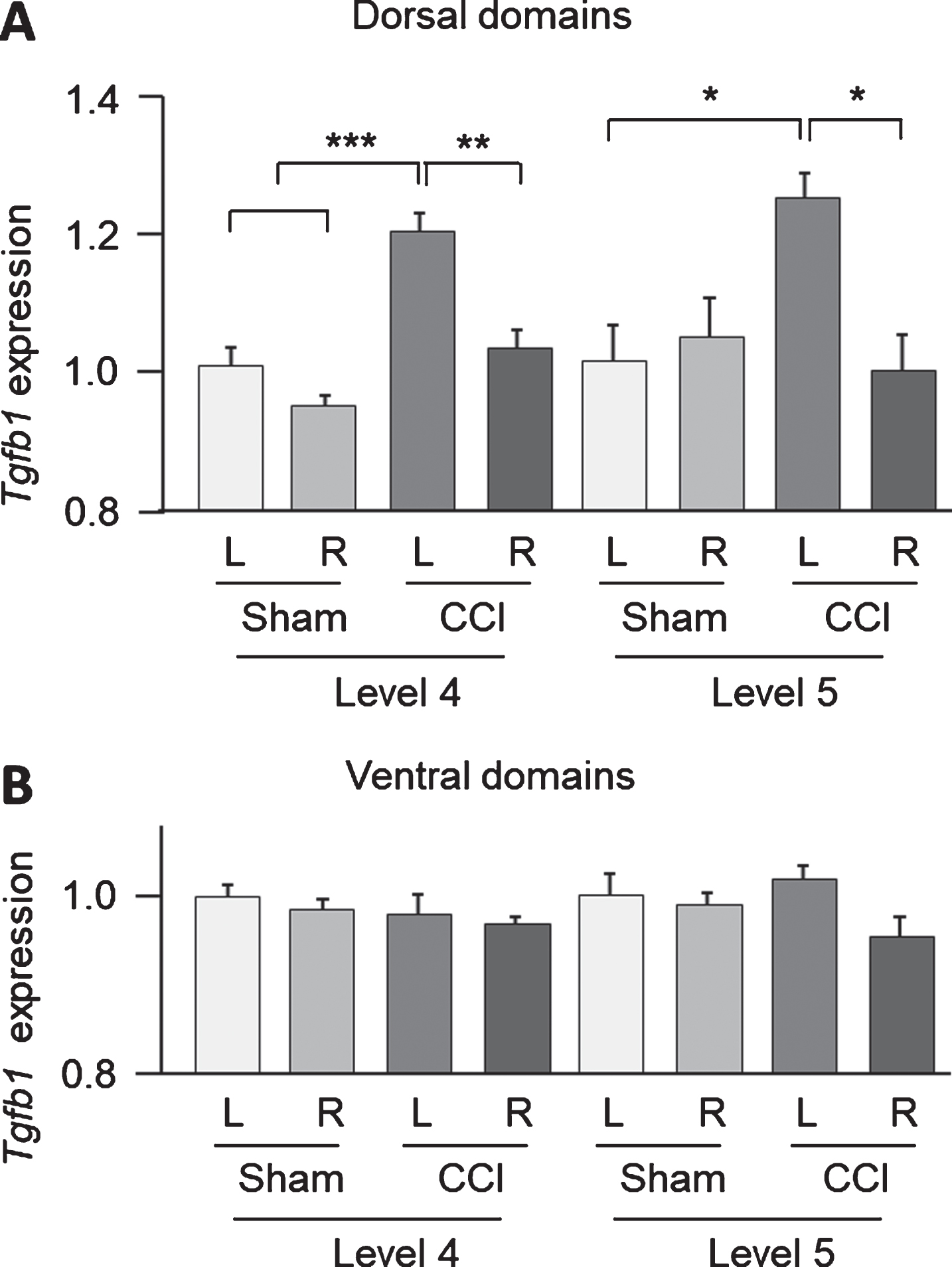
In conclusion, strong Tgfb1 expression upregulation was indicated in contralesional domain in the CCI group.
2.2.CCI effects on a number of c-Fos-positive neurons in the lumbar spinal cord
The c-Fos expressing neurons were immunolabelled to characterize the CCI induced changes in anatomical patterns of neuronal activation in the lumbar spinal cord (Fig. 3). Two groups of c-Fos-positive cells were distinguished in the dorsal domains. One group was located in the laminae I–III and enriched in lamina III. The second group showed diffuse distribution across the laminas V and VI. These groups were analysed separately as ROI 1 and ROI 2. Virtually no c-Fos staining was evident in other parts of spinal cord.
Fig.3
c-Fos-immunoreactive neurons in the dorsal lumbar spinal cord after right side traumatic brain injury using the controlled cortical impact (CCI) model. Photomicrographs of c-Fos expression in sham-injured (A) and CCI (B) animals. Scale bars, 50μm. Bars show the number of c-Fos-positive cells±SEM in the ROI 1 (C) and ROI 2 (E), measured in L4 and L5 levels (n = 4 rats per group). Asymmetry index (AI) for c-Fos expression in ROI 1 (D) and ROI 2 (F) are shown. *, p < 0.05, **, p < 0.01 compared with the sham injured group. The regions of interest (ROI) for IHC analysis: ROI 1 (laminae I–III) and ROI 2 (laminae IV–VI).
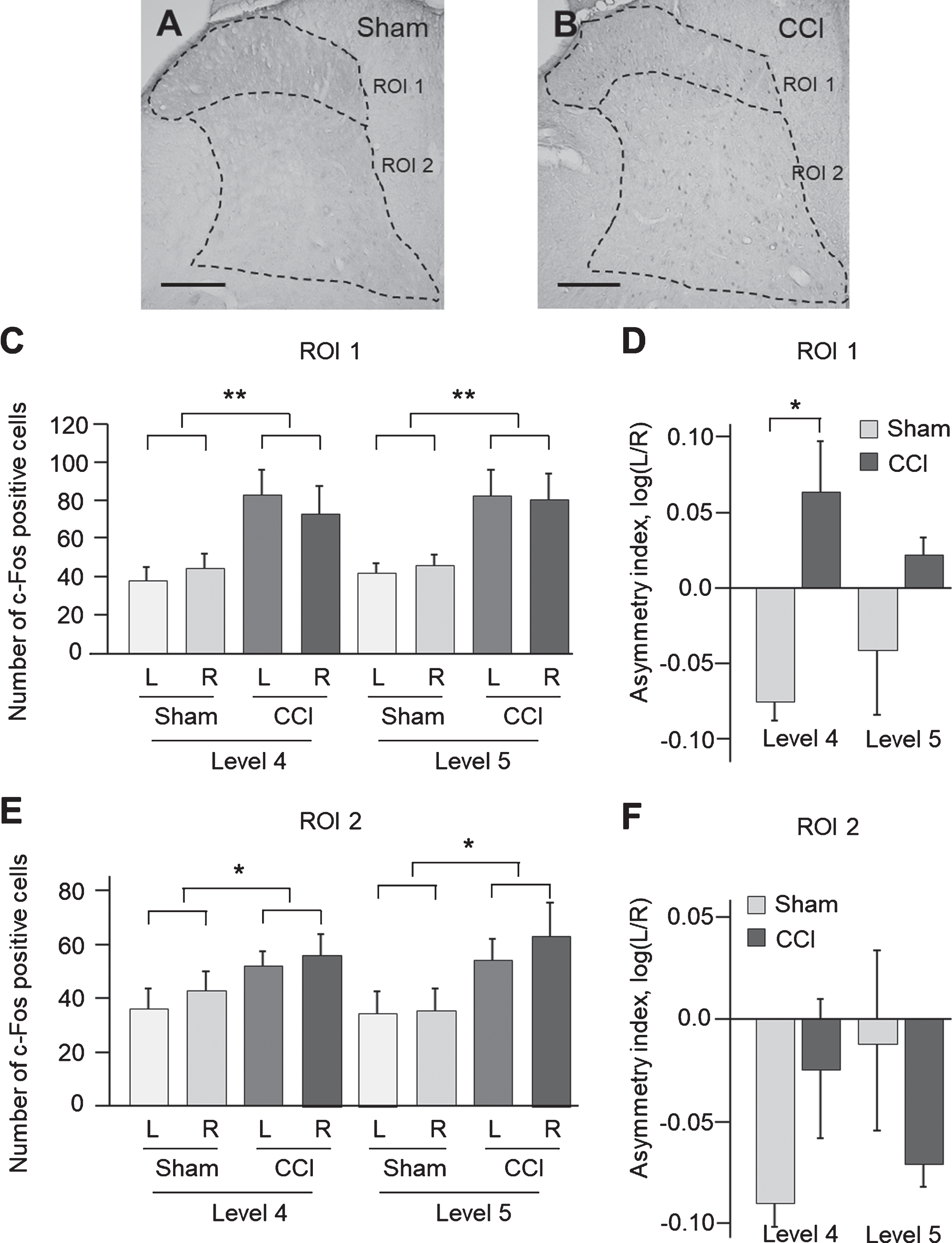
The CCI effects on a number of c-Fos-positive cells in ROI 1 and ROI 2 of the L4 and L5 segments were analysed by two-way ANOVA with injury type (CCI vs sham injury) and measurement side (ipsi vs. contralesional side) as independent factors (Fig. 3, A–F). In the L4 segment, a significant CCI effect was revealed in ROI 1 (F1,13 = 11.7, p = 0.005) and ROI 2 (F1,13 = 5.2, p = 0.04). Post hoc Tukey HSD test showed a significant increase in the number of c-Fos positive cells in CCI rats compared to sham-injured controls (ROI 1: p = 0.005, 1.9-fold; ROI 2: p = 0.04, 1.4-fold) (Fig. 3C, E). In the L5 segment, a significant CCI effect was revealed in ROI 1 (F1,13 = 13.7, p = 0.003) and ROI 2 (F1,13 = 7.2, p = 0.018). Post hoc Tukey HSD test showed a significant increase in the number of c-Fos-positive cells in CCI rats when compared to sham-injured controls (ROI 1: p = 0.003, 1.8-fold; ROI 2: p = 0.02, 1.7-fold) (Fig. 3C, E).
To investigate whether the c-Fos response to focal TBI differs between the ipsi-and contralesional sides the asymmetry index (AI) was analysed. The AI was defined as log (L/R), where L and R were a number of c-Fos-positive cells in the left (contralesional) and right (ipsilesional) halves of ROI 1 and ROI 2. The AI was calculated separately for the L4 and L5 spinal segments. Two-way ANOVA for the AI with injury type (CCI vs. sham) and lumbar segment (L4 vs. L5) as independent factors performed separately for ROI 1 and ROI 2 revealed significant effect of CCI on the AI in ROI 1 (F13,4 = 11.1, p = 0.005; Fig. 3D) but not in ROI 2 (Fig. 3F). Post hoc analysis Tukey HSD showed a significant increase of the AI in the CCI group compared to sham-injured group in ROI 1 for the L4 (p = 0.03) (Fig. 3D) but not L5 segment. Changes in the AI demonstrated higher activation of the contra-vs. ipsilesional c-Fos expressing neural circuits in ROI 1 (Laminae I-III) in the L4 lumbar segment after the unilateral CCI.
3.Discussion
We used a well-established model of focal traumatic brain injury (TBI) and observed significant changes in molecular markers with implication for regeneration and plasticity mechanisms in the lumbar spinal cord, remote from the brain injury site. Elevated c-Fos signals were found in the dorsal horn of the lumbar spinal cord in rats subjected to focal TBI. In each L4 and L5 segments and in each region of interest, the number of c-Fos-positive cells was increased by TBI. Moreover, focal TBI resulted in a higher number of c-Fos-positive cells on the side contralateral to the brain injury. In addition, the absolute copy number of Tgfb1, a marker of activation of neuroprotective functions (Dobolyi, Vincze, Pal, & Lovas, 2012), was significantly upregulated on the contralesional side in the CCI group.
The results of the c-Fos analysis support the hypothesis that the focal TBI induced spinal plastic response remote from a brain injury (Tennant, 2014; Wolpaw, 2007). The elevated c-Fos signals in the brain-injured animals implicate that cortical descending fibres mediate transmission of signals from the injured cortex to the lumbar spinal cord (Hunt, Pini, & Evan, 1987; Kovacs, 1998). Temporal analysis of c-Fos induction by noxious stimuli in rat spinal cord (Draisci & Iadarola, 1989) showed that the c-Fos protein concentration gradually returned to basal conditions at 8–24 hours after the stimulation. In our experiments, c-Fos expression was found to be elevated in the CCI rats in 3 days post-injury. The lasting activation of spinal neurons in the spinal cord of brain-injured animals may indicate a development of spinal circuit plasticity. Descending CST axons arising from sensory cortical areas mainly terminate in laminae III and IV, the corresponding to the ventral part of ROI 1 and dorsal part of ROI 2, and descending CST fibres from traditional motor cortex have a more ventral termination into laminae V and VI, which form the ventral part of our ROI 2 (Brown, 1982; Sist et al., 2014). Thus, activation of neurons in both ROI 1 and 2 in our experiments produced by sensorimotor cortex injury is in consistence with descending corticospinal tract (CST) dorsal horn innervation, supporting the notion of cortically generated stimuli.
In addition to a general elevation of c-Fos signals in the spinal cords of brain-injured rats, a significant distribution difference was also detected between the treatment groups. In brain-injured rats the activated spinal neurons were concentrated to the contralesional side with a more prominent elevation in the L4 segment. Our data suggest that c-Fos-positive neurons may represent the part of neuronal circuits activated by cortical stimuli induced by the focal TBI.
Activation of microglia is a hallmark of brain pathology. The inflammatory response is mediated by activated microglia, the resident immune cells of the CNS, which normally respond to neuronal damage. TGF-βs were reported to inhibit microglial cells, and thereby exert an anti-inflammatory action. Since microglial cells are a major source of TGF-β1 in the CNS (Kiefer, Streit, Toyka, Kreutzberg, & Hartung, 1995), they might exert an auto-inhibitory control on microglial cells (Chio, Lin, Chang, & Lin, 2016). Furthermore, activated TGF-β signalling alters the microenvironment around inhibitory interneurons, and may cause deficits in neuronal inhibition following brain injuries (Kim et al., 2017). A role for TGF-β in neuroprotection and astrogliosis was also suggested (Wang et al., 2015). TGF-β1 also has a capability of promoting axonal regeneration after brain lesion (Bae et al., 2011) and regulate neuroinflammation as well apoptosis after mild traumatic brain injury (Patel, Prasad, Kuwar, Haldar, & Abdul-Muneer, 2017). In further studies, the contribution of the strong contralesional Tgfb1 upregulation observed in the spinal cord following CCI on cell death, the inflammatory response or the development of spasticity should be addressed.
Neurons exhibit a remarkable plasticity both during neural development and during the subsequent remodeling of synaptic connectivity (Grasselli & Strata, 2013). GAP-43 is a protein induced during periods of axonal extension and highly enriched on the inner surface of the growth cone membrane. Consistent with its role as a “plasticity protein,” there is evidence that GAP-43 can directly alter cell shape and neurite extension and induction of synaptogenesis and altered excitability of spinal motor neurons has been exhibited after strength training (Adkins, Boychuk, Remple, & Kleim, 2006). Thus, this motivated us to evaluate changes in GAP43 expression in the lumbar spinal cord. Using the present experimental set-up, no GAP43 alterations were found. Although these findings argue against a role for this factor in post-TBI lumbar plasticity, we cannot exclude that GAP43 expression is changed at earlier or later time points, not evaluated in the present report.
Brain derived neurotrophic factor (BDNF) may promote neuronal survival and axonal sprouting in the spinal cord (Hayashi, Ueyama, Nemoto, Tamaki, & Senba, 2000; Vavrek, Girgis, Tetzlaff, Hiebert, & Fouad, 2006). Since it was upregulated in the spinal cord at three days following stroke, it was suggested as an initiator of structural plasticity (Bose et al., 2013). Treatment blocking early onset of spasticity was accompanied by marked up-regulation of BDNF expression in spinal cord tissue. Finally, in fluid percussion TBI in rats reduced levels of BDNF in the spinal cord was observed in the lumbar spinal cord at one week post-injury (Ying, Feng, Agrawal, Zhuang, & Gomez-Pinilla, 2012). Here, we did not observe a changed expression of BDNF expression which may be related to the chosen TBI model as well as the evaluation time point, and should be addressed in further studies. Of note, the absence of changed expression of three analyzed genes at a single time point does not exclude the possibility that the expression of these genes are altered by CCI at other post-injury time points.
TBI may result in many changes in the spinal cord, including fewer spinal cord motor neurons (Wright et al., 2017), and a reduction in intermediate reticular fibers (Bose et al., 2013). Unilateral brain injury may also induce a “pathological spinal memory” (Wolpaw, 2007) or maladaptive spinal plasticity (Tennant, 2014; Wolpaw, 2007). Although deficit in motor function is a common consequence of traumatic brain injury (TBI), not much is known about the influence of brain injury on motor centers in the spinal cord. Our current results emphasize TBI-induced effects on CNS regions, distant from the brain lesion. We evaluated spinal cord changes at three days post-injury, since the neuroplastic changes develop rapidly and are initiated during the first post-injury hours and days (Singer, Jegasothy, Singer, Allison, & Dunne, 2004). Furthermore, neuroplasticity may peak during the initial post-injury period due to an increased structural plasticity and growth factor expression occurring in the distinct time window (Sist et al., 2014).
Our study is limited by analysis of a single time point; acute changes and those emerging at later post-injury time period were not examined. Motor deficits in the CCI model are well established and last from the first day up to several weeks post-injury (Marklund, 2016). At the time point and injury severity chosen, rats subjected to CCI show robust motor impairment. Yet, our results demonstrate that alterations in expression of molecular regulators/markers of spinal plasticity occur in the lumbar spinal cord remote from the focal TBI. These molecular results corroborate with the concept of diaschisis coined by von Monakow already in 1914 to describe the neurophysiological changes developing distantly from a focal brain lesion (for references, see Carrera & Tononi, 2014). This concept postulates that distant changes varying in their characteristics and location nonetheless represent remote alterations induced by focal and non-focal brain lesions. Altogether, the identified changes in gene expression may contribute to sensorimotor recovery and/or development of post-TBI maladaptive spinal plasticity.
4.Experimental procedure
4.1.Animals
Male 325–375 g Sprague-Dawley rats (n = 16; Scanbur BK AB, Sweden), were used in this experiment. Animals were housed four to a cage with a 12:12 light/dark cycle with food and water ad libitum. Eight rats were used for IHC staining and eight rats were used for the ddPCR quantification. The experiment was approved by the Uppsala animal ethics committee (C165-14 and C166-14) and conducted in accordance with the guidelines of Swedish legislation on animal experimentation (Animal Welfare act SDS1998:56) and European Union Legislation (Convention ETS123 and Directive 86/609/EEC).
4.2.Surgery
The rats were anaesthetized using isoflurane (4% in air) and then moved to a stereotaxic frame where anaesthesia was maintained through a nose cone delivering 1.2% isoflurane in a mixture of 70% nitrous oxide and 30% oxygen.
TBI was induced using the controlled cortical impact (CCI) method, resulting in a focal contusion injury and forelimb motor impairments (Combs et al., 2016; Jefferson et al., 2016; Jones et al., 2012). The CCI has a high reproducibility and accuracy, is considered a clinically-relevant TBI model, and induces brain tissue injury of small variability (Osier, Korpon, & Dixon, 2015). CCI and sham groups received a 4 mm-diameter craniotomy centred on 0.5 mm anterior and 3.5 mm lateral to the bregma, directly over the anterior hindlimb representation area of the right hemisphere. Sham-injured animals underwent the same procedures including craniotomy, except that the impact was omitted (Marklund, 2016). The brain injury was delivered by a small-bore electromagnetically controlled device (Benchmark Stereotaxic Impactor; Cortech Holdings, St. Louis, MO, USA). The electromagnetic controlled probe tip was angled 18° away from vertical, placing the flat impactor tip (4.0 mm in diameter) perpendicular to the surface of the brain. The tip was connected to a sensor that signals when the tip comes into contact with dura. The impactor then penetrated the exposed brain at 2.4 m/s at a depth of 1.0 mm below the cortical surface with a dwell time of 100 ms. After the injury, the bone flap was put back to cover exposed cortex and was held in place using tissue glue (Histoacryl; Braun Surgical, Barcelona, Spain).
4.3.Tissue dissection for ddPCR analysis
Rats were sacrificed by decapitation on the third day after CCI and sham injury. The spinal cord was dissected by cutting the vertebral column at the base of the skull and just above the hip bone. A syringe was inserted at the lower opening, and the spinal cord was flushed out with ice cold saline onto a glass plate placed on ice. The lumbar enlargement region was immediately isolated, embedded in cryo-embedding media (Tissue-Tek, Sakura Finetek, Netherlands), frozen on dry ice and kept at – 80°C until dissection. The frozen tissue block was cut into transverse 100μm sections at – 22°C (Cryostar NX 70, Thermo Fisher Scientific, MiCROM international Gmbh). Each time after collecting from 5 to 6 100μm sections, a 16μm cryosection was obtained and used to control for anatomic localization by staining with hematoxylin and eosin. A section position was verified using the spinal cord atlas. From each L4 and L5 levels from 12 to 15 100-μm sections were prepared. Each of these sections was cut along the central axial line (at the laminae VI level) that divided the dorsal and ventral parts to domains of approximately equal size; this operation was followed by cut along the central sagittal line resulting in separation of the left and right halves. The dissection was performed by a surgical scalpel blade (Swann-Morton Ltd, England; No. 20) at – 22°C in the cryochamber. Cryo-embedding media was removed from each domain. The obtained dorsal left and right side tissue fragments and the ventral left and right side tissue fragments dissected from each L4 and L5 segments were separately pooled into four RNase-and DNase-free microcentrifuge tubes and stored at – 80°C.
4.4.RNA purification and cDNA synthesis
Total RNA was purified by using RNeasy Lipid Tissue Mini Kit (Qiagen, Valencia, CA, USA) from each quadrant and level of lumbar spinal cord separately. RNA concentrations were measured with Nanodrop (Nanodrop Technologies, Wilmington, DE, USA). RNA (30 ng) was reverse-transcribed to cDNA with cDNA iScript kit (Bio-Rad, Hercules, CA, USA) according to manufacturer’s protocol. cDNA samples were aliquoted and stored at – 20°C.
4.5.Droplet digital PCR (ddPCR)
The ddPCR assay was described elsewhere (McDermott et al., 2013). Reaction mixture contained droplet digital PCR supermix for probes (Bio-Rad, Hercules, CA, USA), PrimerPCR Probe Assay (Bio-Rad, Hercules, CA, USA; Table 1), and cDNA corresponding to 4 ng transcribed RNA. The mixture was supplemented with droplet generation oil (Bio-Rad, Hercules, CA, USA) and partitioned into 14,000–19,000 droplets in QX200 droplet generator (Bio-Rad, Hercules, CA, USA). PCR was performed in a T100 Thermal Cycler (Bio-Rad, Hercules, CA, USA) under the following conditions: 10 min at 95°C, 40 cycles for 30 s at 94°C followed by incubation for 60 s at 60°C and for 10 min at 98°C. Fluorescence intensity of the droplets was measured by using the QX200 droplet reader (Bio-Rad, Hercules, CA, USA). Data analysis was performed with Quanta Soft droplet reader software (Bio-Rad, Hercules, CA, USA). Positive and negative droplet populations were automatically detected. Absolute levels of Tgfb1, Bdnf, and Gapd43 mRNAs were quantified in four domains (left dorsal, right dorsal, left ventral, and right ventral; n = 4 rats) by ddPCR. Absolute mRNA amount was calculated by using Poisson statistics (Hindson et al., 2013), and background was corrected on the basis of the no template control data. Absolute transcript levels were expressed as RNA copy numbers per nanogram of total RNA. The expression data were normalised to geometric mean of expression of beta-actin (Actb) and glyceraldehyde 3-phosphate dehydrogenase (Gapdh) housekeeping genes. The expression level of Tgfb1 in the left L4 and L5 segments in sham-injured group was taken as a unit and the CCI data was scaled accordingly.
Table 1
Primer PCR probe assays used for droplet digital PCR (Bio-Rad, Hercules, CA, USA)
| Gene Name | Assay ID |
| Tgfb1 | qRnoCIP0031022 |
| c-Fos | Rn02396759 |
| Bdnf | Rn02531967_s1 |
| Gap43 | Rn01474579_m1 |
| Actb | Rn00667869_m1 |
| Gapdh | Rn01775763_g1 |
4.6.Tissue perfusion and fixation
Three days after the surgery, rats were deeply anesthetized with sodium pentobarbital (50 mg/kg body weight, i.p.), perfused transcardially with saline followed by freshly prepared 4% paraformaldehyde in 0.1 M phosphate-buffered saline (pH 7.4) (Kasukurthi et al., 2009). The complete spinal cord was removed, post fixed overnight at 4°C and then transferred to 20% glycerol in PBS for 24 h at 4°C. Tissues were frozen on dry ice and stored at – 80°C. Spinal cords were then sectioned into 40μm coronal sections (Cryostar NX 70, Thermo Fisher Scientific, MiCROM international Gmbh) and stored in PBS at 4°C.
4.7.Immunohistochemistry
Anti-cFos polyclonal antibodies (Merck Millipore, Darmstadt, Germany) were used on free-floating sections at a 1:20 000 dilution. Specificity of signal was verified by lack of staining in no-primary antibodies and no-secondary antibodies experiments. Sections were washed in PBS for 30 minutes followed by 15 min incubation in PBS with 0.3% H2O2 and four more 5 minute washes in PBS Triton X-100. Sections were then blocked in normal goat serum in PBS with Triton X-100 for 1 hours, and exposed to the primary antibodies (1:20 000) for 16 hours at room temperature. Slices were washed in PBS-Triton X three times for 10 minutes each and incubated for 1 hour at RT in biotinylated secondary antibodies at 1:500 dilutions. Following the incubation, sections were washed three times in PBS Triton X-100 for 10 minutes each. Sections were then incubated in avidin-biotin complex reagent using a kit (ABC Vectastain ®, Burlingame, CA) for 45 minutes at RT, washed 3 times (5 minutes each) in PBS Triton X-100, and nickel-enhanced diaminobenzadine (DAB) (Sigma-Aldrich, MO, USA) added to visualize primary antibody staining. Sections were washed once in water, mounted, dried and dehydrated by immersing in 70% ethanol for 10 minutes, 95% ethanol for 10 minutes, 100% ethanol for 10 minutes, and xylenes for 12 minutes. After dehydration, the mounted slides were cover slipped and photomicrographs of DAB grains taken using a Leica DSM microscope. Grains were counted using Image-Pro Plus 5.0 software program (Media Cybernetics Inc., Silver Spring, MD).
4.8.Quantification of immunolabeling
For quantification of c-Fos-positive cells six sections were taken from each L4 and L5 spinal segment of each CCI (n = 4) and sham-injured (n = 4) rat. c-Fos labelling was analysed by The number of c-Fos-positive cells was manually counted blinded to each experimental group in the laminas I–III (region of interest 1, ROI 1), and the laminas IV–VI (ROI 2). Neurons were considered to be c-Fos-positive if they showed nuclear localization of the protein identified as round structures. The number of c-Fos-positive cells (with labelled nuclei) but not intensity of the staining was taken into account. Results were expressed as the mean number of c-Fos-positive neurons per ROI.
4.9.Statistical analysis
Normality of data distribution was tested using the Kolmogorov-Smirnov test, and homogeneity of variances by Levene’s test. Analysis was performed using two-way analysis of variance (ANOVA) followed by a post hoc Tukey HSD test. Asymmetry index (AI) was defined asx [log (L/R)], where L and R were the expression levels in the left (contralesional) side and right (ipsilesional) side dissected spinal domains, respectively (Watanabe et al., 2015). A significance level of P < 0.05 was accepted as statistically significant. Data was presented as the mean value±standard error of the mean (SEM).
Competing interest
No conflict of interest.
Acknowledgments
This work was supported by Local ALF funds from Uppsala University hospital (to NM); research grants from the Swedish Science Research Council (VR; to GB); the Swedish Institute (Visby Program; to OK and GB); P.O. Zetterling and K. and O.F. Hedström foundations (to OK).
References
1 | Adkins, D.L. , Boychuk, J. , Remple, M.S. , & Kleim, J.A. ((2006) ). Motor training induces experience-specific patterns of plasticity across motor cortex and spinal cord. Journal of Applied Physiology, 101: (6), 1776–1782. |
2 | Axelson, H.W. , Winkler, T. , Flygt, J. , Djupsjo, A. , Hanell, A. , & Marklund, N. ((2013) ). Plasticity of the contralateral motor cortex following focal traumatic brain injury in the rat. Restorative Neurology and Neuroscience, 37: (1), 73–85. |
3 | Bae, J.J. , Xiang, Y.Y. , Martinez-Canabal, A. , Frankland, P.W. , Yang, B.B. , & Lu, W.Y. ((2011) ). Increased transforming growth factor-betal modulates glutamate receptor expression in the hippocampus. International Journal of Physiology, Pathophysiology and Pharmacology, 3: (1), 9–20. |
4 | Bose, P. , Hou, J. , Nelson, R. , Nissim, N. , Parmer, R. , Keener, J. ,... & Thompson, F.J. ((2013) ). Effects of acute intrathecal baclofen in an animal model of TBI-induced spasticity, cognitive, and balance disabilities. Journal of Neurotrauma, 30: (13), 1177–1191. |
5 | Bose, P. , Hou, J. , & Thompson, F.J. ((2015) ). Traumatic Brain Injury (TBI)-Induced Spasticity: Neurobiology, Treatment, and Rehabilitation. In KobeissyF.H. (Ed.), Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton (FL): CRC Press/Taylor & Francis; Chapter 14. |
6 | Bramham, C.R. , & Messaoudi, E. ((2005) ). BDNF function in adult synaptic plasticity: The synaptic consolidation hypothesis. Progress in Neurobiology, 76: (2), 99–125. |
7 | Brooks, G.A. , & Martin, N.A. ((2014) ). Cerebral metabolismfollow-ing traumatic brain injury: New discoveries with implications for treatment. Frontiers in Neuroscience, 8: , 408. |
8 | Brown, A.G. ((1982) ). The dorsal horn of the spinal cord. Quarterly Journal of Experimental Physiology, 67: (2), 193–212. |
9 | Carrera, E. , & Tononi, G. ((2014) ). Diaschisis: Past, present and future. Brain, 137: , 2408–2422. |
10 | Cheng, G. , Kong, R.H. , Zhang, L.M. , & Zhang, J.N. ((2012) ). Mitochondria in traumatic brain injury and mitochondrial-targeted multipotential therapeutic strategies. British Journal of Pharmacology, 167: (4), 699–719. |
11 | Chio, C.C. , Lin, M.T. , Chang, C.P. , & Lin, H.J. ((2016) ). A positive correlation exists between neurotrauma and TGF-beta1-containing microglia in rats. European Journal of Clinical Investigation, 46: (12), 1063–1069. |
12 | Combs, H.L. , Jones, T.A. , Kozlowski, D.A. , & Adkins, D.L. ((2016) ). Combinatorial Motor Training Results in Functional Reorganization of Remaining Motor Cortex after Controlled Cortical Impact in Rats. Journal ofNeurotrauma, 33: (8), 741–747. |
13 | Dobolyi, A. , Vincze, C. , Pal, G. , & Lovas, G. ((2012) ). Theneuropro-tective functions of transforming growth factor beta proteins. International Journal of Molecular Sciences, 13: (7), 8219–8258. |
14 | Draisci, G. , & Iadarola, M.J. ((1989) ). Temporal analysis of increases in c-fos, preprodynorphin and preproenkephalin mRNAs in rat spinal cord. Brain Research: Molecular Brain Research, 6: (1), 31–37. |
15 | Grasselli, G. , & Strata, P. ((2013) ). Structural plasticity of climbing fibers and the growth-associated protein GAP-43. Frontiers in Neural Circuits, 7: , 25. |
16 | Hayashi, M. , Ueyama, T. , Nemoto, K. , Tamaki, T. , & Senba, E. ((2000) ). Sequential mRNA expression for immediate early genes, cytokines, and neurotrophins in spinal cord injury. Journal of Neurotrauma, 17: (3), 203–218. |
17 | Hindson, C.M. , Chevillet, J.R. , Briggs, H.A. , Gallichotte, E.N. , Ruf, I.K. , Hindson, B.J. ,... & Tewari, M. ((2013) ). Absolute quantification by droplet digital PCR versus analog real-time PCR. Nature Methods, 70: (10), 1003–1005. |
18 | Hunt, S.P. , Pini, A. , & Evan, G. ((1987) ). Induction of c-fos-like protein in spinal cord neurons following sensory stimulation. Nature, 328: (6131), 632–634. |
19 | Jefferson, S.C. , Clayton, E.R. , Donlan, N.A. , Kozlowski, D.A. , Jones, T.A. , & Adkins, D.L. ((2016) ). Cortical Stimulation Concurrent With Skilled Motor Training Improves Forelimb Function and Enhances Motor Cortical Reorganization Following Controlled Cortical Impact. Neurorehabilitation and Neural Repair, 30: (2), 155–158. |
20 | Jones, T.A. , Liput, D.J. , Maresh, E.L. , Donlan, N. , Parikh, T.J. , Marlowe, D. , & Kozlowski, D.A. ((2012) ). Use-dependent dendritic regrowth is limited after unilateral controlled cortical impact to the forelimb sensorimotor cortex. Journal of Neurotrauma, 29: (7), 1455–1468. |
21 | Kiefer, R. , Streit, W.J. , Toyka, K.V. , Kreutzberg, G.W. , & Hartung, H.P. ((1995) ). Transforming growth factor-beta 1: A lesion-associated cytokine of the nervous system. International Journal of Developmental Neuroscience, 73: (3-4), 331–339. |
22 | Kim, S.Y. , Senatorov, V.V., Jr. , Morrissey, C.S. , Lippmann, K. , Vazquez, O. , Milikovsky, D.Z. ,... & Kaufer, D. ((2017) ). TGF-beta signaling is associated with changes in inflammatory gene expression and perineuronal net degradation around inhibitory neurons following various neurological insults. Scientific Reports, 7: (1), 7711. |
23 | Kovacs, K.J. ((1998) ). c-Fos as a transcription factor: A stressful (re)view from a functional map. Neurochemistry International, 33: (4), 287–297. |
24 | Marklund, N. ((2016) ). Rodent Models of Traumatic Brain Injury: Methods and Challenges. Methods in Molecular Biology, 1462: , 29–46. |
25 | Masel, B.E. , & DeWitt, D.S. ((2010) ). Traumatic brain injury: A disease process, not an event. Journal ofNeurotrauma, 27: (8), 1529–1540. |
26 | McDermott, G.P. , Do, D. , Litterst, C.M. , Maar, D. , Hindson, C.M. , Steenblock, E.R. ,...& Lowe, A.J. ((2013) ). Multiplexed target detection using DNA-binding dye chemistry in droplet digital PCR. Analytical Chemistry, 85: (23), 11619–11627. |
27 | Osier, N.D. , Korpon, J.R. , & Dixon, C.E. ((2015) ). Controlled Cortical Impact Model. KobeissyF.H. (Ed.), Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects. Boca Raton (FL): CRC Press/Taylor & Francis; Chapter 16. |
28 | Patel, R.K. , Prasad, N. , Kuwar, R. , Haldar, D. , & Abdul-Muneer, P.M. ((2017) ). Transforming growth factor-beta 1 signaling regulates neuroinflammation and apoptosis in mild traumatic brain injury. Brain, Behavior, and Immunity, 64: , 244–258. |
29 | Pruitt, D.T. , Danaphongse, T.T. , Schmid, A.N. , Morrison, R.A. , Kilgard, M.P. , Rennaker, R.L., 2nd , & Hays, S.A. ((2017) ). Traumatic Brain Injury Occludes Training-Dependent Cortical Reorganization in the Contralesional Hemisphere. Journal of Neurotrauma, 34: (17), 2495–2503. |
30 | Santibanez, J.F. , Quintanilla, M. , & Bernabeu, C. ((2011) ). TGF-beta/TGF-beta receptor system and its role in physiological and pathological conditions. Clinical Science, 727: (6), 233–251. |
31 | Singer, B.J. , Jegasothy, G.M. , Singer, K.P. , Allison, G.T. , & Dunne, J.W. ((2004) ). Incidence of ankle contracture after moderate to severe acquired brain injury. Archives of Physical Medicine and Rehabilitation, 85: (9), 1465–1469. |
32 | Sist, B. , Fouad, K. , & Winship, I.R. ((2014) ). Plasticity beyond peri-infarct cortex: Spinal up regulation of structural plasticity, neurotrophins, and inflammatory cytokines during recovery from cortical stroke. Experimental Neurology, 252: , 47–56. |
33 | Tennant, K.A. ((2014) ). Thinking outside the brain: Structural plasticity in the spinal cord promotes recovery from cortical stroke. Experimental Neurology, 254: , 195–199. |
34 | Vavrek, R. , Girgis, J. , Tetzlaff, W. , Hiebert, G.W. , & Fouad, K. ((2006) ). BDNF promotes connections of corticospinal neurons onto spared descending interneurons in spinal cord injured rats. Brain, 129: (Pt 6), 1534–1545. |
35 | Wang, X.Y. , Ba, Y.C. , Xiong, L.L. , Li, X.L. , Zou, Y. , Zhu, Y.C. , … & Li, J.T. ((2015) ). Endogenous TGFbeta1 Plays a Crucial Role in Functional Recovery After Traumatic Brain Injury Associated with Smad3 Signal in Rats. Neurochemical Research, 40: (8), 1671–1680. |
36 | Watanabe, H. , Fitting, S. , Hussain, M.Z. , Kononenko, O. , Iatsyshyna, A. , Yoshitake, T. , ... & Bakalkin, G. ((2015) ). Asymmetry of the endogenous opioid system in the human anterior cingulate: A putative molecular basis for lateralization of emotions and pain. Cerebral Cortex, 25: (1), 97–108. |
37 | Werner, J.K. , & Stevens, R.D. ((2015) ). Traumatic brain injury: Recent advances in plasticity and regeneration. CurrentOpin-ion in Neurology, 28: (6), 565–573. |
38 | Wilson, L. , Stewart, W. , Dams-O'Connor, K. , Diaz-Arrastia, R. , Horton, L. , Menon, D.K. , & Polinder, S. ((2017) ). The chronic and evolving neurological consequences of traumatic brain injury. The Lancet Neurology, 16: (10), 813–825. |
39 | Wolpaw, J.R. ((2007) ). Spinal cord plasticity in acquisition and maintenance of motor skills. Acta Physiologica, 189: (2), 155–169. |
40 | Wright, D.K. , Liu, S. , van der Poel, C. , McDonald, S.J. , Brady, R.D. , Taylor, L. ,.... & Shultz, S.R. ((2017) ). Traumatic Brain Injury Results in Cellular, Structural and Functional Changes Resembling Motor Neuron Disease. Cerebral Cortex, 27: (9), 4503–4515. |
41 | Ying, Z. , Feng, C. , Agrawal, R. , Zhuang, Y. , & Gomez-Pinilla, F. ((2012) ). Dietary omega-3 deficiency from gestation increases spinal cord vulnerability to traumatic brain injury-induced damage. PLoS One, 7: (12), e52998. |


