
Comprehensive rehabilitative care across the spectrum of amyotrophic lateral sclerosis
Abstract
BACKGROUND: Amyotrophic lateral sclerosis (ALS or Lou Gehrig’s disease) is a neurodegenerative disease that results in progressive muscle weakness and wasting. There is no known cure and the disease is uniformly fatal.
PURPOSE: This review discusses current concepts in ALS care, from breaking the diagnosis to end-of-life care. People with ALS have several multidisciplinary needs due to a complex and dynamic disease process. They benefit from rehabilitation interventions that are individualized and have the goal of optimizing independence, function, and safety. These strategies also help minimize symptomatic burden and maximize quality of life.
CONCLUSION: Patient-centered, multidisciplinary care has a significant impact on the life of people with ALS and is thecurrent standard of care for this patient population.
1Introduction
Amyotrophic lateral sclerosis (ALS, aka Lou Gehrig’s disease) is a daunting diagnosis to receive. The disease is uniformly fatal, with an average life expectancy of only three years after symptom onset (Chio, Logroscino et al., 2009; Miller, Mitchell, & Moore, 2012). The only FDA-approved treatment, riluzole, only confers a modest survival advantage (Miller et al., 2012). The natural history of the disease is characterized by a relentlessly progressive course that is characterized by diffuse weakness and skeletal muscle wasting, and frequently accompanied by dysarthria, dysphagia, and failure of the muscles that support ventilation (Brooks, 1996). However an “atypical” disease presentation may occur, with much slower disease progression occurring in some individuals with about 10% of cases living 10 years or longer (Chio, Logroscino et al., 2009). Thus, the severity of the illness and uncertainties regarding the time course of disability and evolving care burden are a cause of great stress for affected individuals and their families. Receiving a diagnosis of ALS is a life-changing experience for all people involved and health-care providers themselves are not immune from the enormity of the task of caring for these individuals (Bromberg, Schenkenberg, & Brownell, 2011). The clinical care for ALS is optimally centered around “ALS clinics” with disease-focused expertise offering a multi-disciplinary approach that leverages the experience of several health care providers such as neurologists, physiatrists, pulmonologists, physiotherapists, occupational therapists, speech and language pathologists, and assistive technology experts, to name a few (Mayadev, Weiss, Distad, Krivickas, & Carter, 2008). The role of the ALS specialist is multi-faceted and includes ownership of multiple aspects of care as well as delegation to sub-specialists and coordination of their services. This highly integrated, comprehensive care that keeps the person with ALS at the center of attention (Fig. 1) has been shown to result in better quality of life, increased utilization of supportive care services, and possibly prolonged survival (Chio, Bottacchi, Buffa, Mutani, & Mora, 2006; Miller et al., 2009b; Traynor, Alexander, Corr, Frost, & Hardiman, 2003; Van den Berg et al., 2005; Zoccolella et al., 2007). Multidisciplinary care is now considered standard of care and is one of the eleven AAN-endorsed ALS quality measures (Miller et al., 2014). Despite the fatal outcome of the disease process, ALS multidisciplinary care is intrinsically “rehabilitative” where rehabilitation is defined as the process of assisting people to reach their fullest potential despite the presence of a disability. Therefore, in ALS, the goal of the doctor-patient relationship shifts from “cure” to “care”. The disease, while “incurable”, is clearly worth treating, and invites as much creativity as possible to enhance function throughout the course of a disease that is quite dynamic and diverse. This paper will discuss relevant aspects of rehabilitative care across the ALS spectrum, emphasizing issues that frequently arise in caring for this patient population and what the rehabilitation team can offer to assist patients as the disease progresses.
2From symptom onset to diagnosis
2.1Do I have ALS?
The onset of ALS is insidious and may “mimic” a variety of other conditions, often leading to misclassification and delays in diagnosis (Paganoni et al., 2014). The time from symptom onset to confirmed diagnosis ranges from 8 to 15 months (Cellura, Spataro, Taiello, & La Bella, 2012; Chio, 1999; Chio, Mora et al., 2009; Donaghy, Dick, Hardiman, & Patterson, 2008; Househam & Swash, 2000; Mitchell et al., 2010; Paganoni et al., 2014), which represents a significant proportion of total disease duration. This “diagnostic delay” may include a period of time when the diagnosis was suspected (Paganoni et al., 2014), but the physician may have decided to delay discussion while ruling out mimickers or waiting for confirmation from an ALS specialist.
There is no consensus on how or when to deliver the diagnosis of ALS (Miller et al., 2009b). Small studies have highlighted that, unfortunately, many patients and their families are dissatisfied with diagnosis delivery (Borasio, Sloan, & Pongratz, 1998; McCluskey, Casarett, & Siderowf, 2004) and that better training in delivering challenging diagnoses is needed during medical education (Schellenberg, Schofield, Fang, & Johnston, 2014). Importantly, clinical experience supports the view that the way the diagnosis is communicated is the very first step in the rehabilitative process as it will have a major bearing on patient attitudes towards the disease and the medical team’s recommendations (Silani & Borasio, 1999). Establishing a good physician-patient relationship even before the diagnosis is confirmed may help reduce patients’ fears and dispel misconceptions such as “there is nothing that one can do” or the outcome of ALS is “choking to death” (Maessen et al., 2010). The peri-diagnostic time represents a unique opportunity to adjust to the diagnosis in a stepwise fashion, understand the evolving tools for care and research that the medical system offers as well as discover the many opportunities for involvement with patient support groups, advocacy and community resources.
Spending enough time discussing the diagnosis has been identified as a predictor of higher patient and caregiver satisfaction (McCluskey et al., 2004). Patient surveys have also suggested that communication skills such as an empathetic style, delivering the diagnosis when the patient’s support network is present, arranging a short-term follow-up meeting to address additional questions or concerns, and informing patients of sources for help and support including tertiary care centers and clinical trial options are associated with positive patient feedback (Borasio et al., 1998; McCluskey et al., 2004). While diagnosis delivery cannot be standardized, research in cancer has developed various protocols to aid the clinician in this challenging task such as the SPIKES method (Baile et al., 2000). The goals of this strategy are to gather information from the patient to gauge knowledge and expectations, provide intelligible information and support, and develop a plan (Baile et al., 2000). These goals are achieved by using a variety of methods that are applicable to ALS as well (Baile et al., 2000). Examples include choosing an appropriate setting (private room, with significant others present, sitting down with no barriers between clinician and patient), allowing enough time with no interruptions, asking open-ended questions to assess patient’s perceptions (“What have you been told about your medical condition so far?”), providing information in a stepwise fashion using non-technical terms (“Based on the tests we have done, there is a problem with your nerves so that your muscles can’t work properly”; “This condition is called amyotrophic lateral sclerosis”; “There is no cure at the moment although one drug is available to slow down the disease; there is also a lot of research going on at this time to try to find new treatments”), addressing patient’s emotions with empathetic responses (“I know this is not good news and I also wish the news were better”), and forming a strategy to go forward (“We have an experienced team here who works with many people with your same condition”; “We have multiple ideas and strategies to assist you through this”; “Your main problem at this time is ankle weakness and falls, we can help with this”). Finally, variability in disease progression should be discussed as some people with ALS do have a slow progression and physicians’ ability to predict survival times is limited (Chio, Logroscino et al., 2009; Kuffner et al., 2015).
3Early ALS: Exercise and adaptive equipment
Early symptoms of ALS can be quite variable. ALS can first develop in the legs or the arms or the bulbar region, with each region accounting for about a third of presentations. The onset is usually focal. The disease then progresses and spreads to other body regions. Typical presenting symptoms include ankle weakness causing foot drop, falls, difficulties with grip and fine motor skills, and slurred speech.
3.1“Can exercise make me stronger?”
Many people with ALS ask about the role of exercise either because exercise has been an integral component of their pre-morbid lifestyle or because exercise is often thought to be reparative, having positive effects on endurance and strength. When they ask about exercise, most patients refer to aerobic training and strengthening programs.
The evidence regarding the risks and benefits of aerobic/strengthening exercise in ALS is limited to some pre-clinical evidence and two small clinical studies (Dal Bello-Haas & Florence, 2013). In mouse models of ALS, moderate endurance exercise delayed disease onset and increased survival (Carreras et al., 2010; Kirkinezos, Hernandez, Bradley, & Moraes, 2003; Veldink et al., 2003). High-intensity endurance training, on the other hand, was detrimental (Carreras et al., 2010; Mahoney, Rodriguez, Devries, Yasuda, & Tarnopolsky, 2004). Small studies in people with ALS, also supported the safety of moderate-intensity exercise. In a study by Drory and colleagues (Drory, Goltsman, Reznik, Mosek, & Korczyn, 2001), 25 people with ALS were randomized to perform an individualized moderate-intensity daily exercise program “involving most muscle groups of the four limbs and trunk”, as opposed to avoiding any physical activity beyond their usual daily requirements. The goal of the program was to improve muscle endurance by “having the muscles work against only modest loads but undergo significant changes in length” (Drory et al., 2001). The program was well tolerated and was associated with less functional decline on the ALS Functional RatingScale (ALSFRS) and the Ashworth spasticity scale in the exercising group at 3 months following study initiation (Drory et al., 2001). A second randomized,controlled trial of moderate resistance exercise in 27 people with ALS also resulted in better function at 6 months, as measured by total ALSFRS scores and quality of life, without adverse effects (Bello-Haas et al., 2007).
Currently there is an ongoing, larger trial of exercise in ALS to confirm and expand on these findings (NCT01521728). In the meantime, based on the available evidence so far and drawing from the exercise literature in other progressive neuromuscular diseases, we can conclude that moderate-intensity exercise is safe for people with ALS. Overexertion with resulting prolonged post-exercise fatigue, muscle pain, or soreness, however, should be avoided as these symptoms can signal overwork-induced muscle damage (Petrof, 1998). Exercise is unlikely to make muscles significantly stronger in ALS and high-intensity weight training in the gym should be discouraged. On the other hand, gentle, restorative exercise can be used as a tool to avoid further deconditioning and as a means to improve sleep and mood. Aerobic exercise practiced in a community setting (e.g., accessible pool, adaptive golf, chair yoga, tai chi) is generally preferred and can also help promote social interactions. Practical recommendations for exercise in ALS are listed in Table 1. Of note, stretching and range-of-motion exercises should be encouraged starting early in the course of the disease and as part of a gentle, wellness-oriented, physical activity daily routine. Clinical experience suggests that performing simple stretching exercises that target the major joints helps prevent development of painful and function-limiting contractures, especially at the shoulders. Training with a physical therapist who will develop an appropriate exercise routine isrecommended and periodic program modification will be needed as the disease progresses.
3.2“I want to continue to work, take care of my children, go on a trip. What can you do to make this possible?”
As patients develop weakness in the bulbar, cervical, and/or lumbosacral regions, they lose the ability to perform the daily activities they care about. The focus of clinic visits should shift from documenting the inevitable progression of the weakness to pro-active brainstorming with all the members of the multidisciplinary team to find ways to make desired activities possible for as long as possible. Assistive devices and adaptive equipment should be considered and periodically re-evaluated. Some devices may be borrowed from patient support groups as they are expensive (Connolly et al., 2014; Gladman, Dharamshi, & Zinman, 2014) and multiple devices are likely to be needed along the course of disease progression (Bromberg, Brownell, Forshew, & Swenson, 2010). Of note, the introduction of these devices is sometimes viewed by patient’s as a sign of “defeat”. It is important that the rehabilitation team focus on the function-expanding aspect of assistive devices; these are the tools that will ensure that one can continue to enjoy functional independence and safe mobility at home, at work, and in the community for as long as possible.
Lower body weakness leads to a less efficient and more energy-consuming gait pattern (Menotti et al., 2011) as the patient needs to compensate for muscle imbalances to avoid tripping and falling. Proximal leg weakness may compound the problem, making itdifficult to get out of a car or rise from a low surface. Further, muscle imbalance and compensatory gait patterns (e.g., steppage, hip hiking) can trigger musculoskeletal pain such as low back pain and can worsen fatigue. Braces and adaptive equipment are very valuable even early on to help prevent or manage these problems. Braces can be used on an intermittent basis when weakness is mild to help conserve energy (Bean, Walsh, & Frontera, 2001) and assist at times of demanding activities such as walking long distances. Several braces for the lower limbs are available. The most commonly used braces in ALS are ankle-foot-orthoses (AFOs), preferably light-weight and customized. As disease progresses, AFOs may be needed at all times to help reduce the risk of falling.
Adaptive equipment such as swivel cushions and lift chairs can help assist sit-to-stand transitions. Eventually, patients will need mobility aids such as canes, walkers, and wheelchairs. Canes provide the least stability and are appropriate for mild lower extremity weakness only, adding some proprioceptive input. Walkers provide the most support for the ambulant but weak patient, but are often perceived as cumbersome. Crutches have very limited use in ALS, as one needs a high degree of upper body strength to use them. Transitioning to a wheelchair may be challenging both psychologically and logistically. It is important that wheelchairs are presented as a way to allow greater safety and independence such as during holiday trips and when attending social events. Manual and transport wheelchairs are useful in the early stages to help conserve energy especially when traveling long distances. However, power wheelchairs ultimately prove more useful as ALS patients develop upper body weakness that prevents them from propelling manual wheelchairs. Due to difficulties in obtaining insurance reimbursement for more than one wheelchair, it is recommended that ALS patients rent or borrow a manual or transport wheelchair instead of purchasing one. Insurance coverage should be reserved for the expensive (Ward et al., 2010), customized power wheelchairs. Proper seating and positioning in the wheelchair are critical to ensure comfort and prevent secondary problems such as skin breakdown and back pain. Thus, customization of the power wheelchair, teaching pressure relief maneuvers to patients and their caregivers, and providing ongoing monitoring and wheelchair adjustments in a clinic with experience with ALS is crucial for health and comfort.
Adaptive tools are also available to assist upper body weakness (Dal Bello-Haas, Kloos, & Mitsumoto, 1998). Fine motor skills are often affected first and patients complain of difficulties with doorknobs,buttons, zippers, cans, and jars. Occupational therapists (OTs) with experience in ALS may help guide patients to select among several pieces of adaptive equipment (Table 2). Hand splints may also be considered to compensate for intrinsic hand muscle weakness (Ivy, Smith, & Materi, 2014; Tanaka, Saura, Houraiya, & Tanimura, 2009). The goal of these devices is to allow the patient to get dressed and eat independently, read, write and use computers.
4Symptom management as ALS progresses: Medications and assistive technology
4.1“What medications are available to control ALS-related symptoms?”
While we cannot change the ultimate outcome of the disease, there is certainly a lot that can be done to effectively manage ALS-related symptoms and maximize quality of life (Table 3).
4.1.1Pain
The classic tagline is that ALS is a disease characterized by “painless, progressive weakness”. While this is true with respect to the primary disease process, pain can develop as a secondary complication of musculoskeletal dysfunction due to limited mobility, loss of range of motion, and difficulty with positioning in bed or in a wheelchair (Boninger et al., 2003; Gibson & Frank, 2005). Many people with ALS report pain even in the early stages of their disease (Pizzimenti, Aragona, Onesti, & Inghilleri, 2013; Rivera et al., 2013) and pain is associated with reduced quality of life (Pizzimenti et al., 2013). Common sites of pain include the low back, the neck, and the shoulder region (Ho, Ruthazer, & Russell, 2011). Surprisingly, pain in ALS has been poorly studied and is often poorly managed (Wallace et al., 2014). This is very unfortunate as many causes of pain can be prevented and/or managed by simple interventions such as providing adequate lumbar support or adjusting the arm rests of the patient’s wheelchair. For power wheelchair users, power elevating the legs can ease back pain and the tilt-in-space feature helps relieve pain from gluteal pressure. Braces and splints can support joints that are affected by progressive muscle weakness. For example, many people with ALS develop shoulder pain due to loss of strength of the peri-scapular musculature with subsequent loss of range of motion, subluxation, and development of contractures. A regular stretching program, combined with the use of shoulder approximation sleeves, can help prevent shoulder pain. In the legs, discomfort can be compounded by dependent edema which can benefit from leg elevation, massage, and compressionstockings.
Physical medicine modalities can help relieve pain in many patients (Table 3) (Gaujoux-Viala, Dougados, & Gossec, 2009; Gracies, 2001; Green, Buchbinder, & Hetrick, 2003, 2005). Pharmacologic treatment of pain may also be needed using one or more of several agents. The choice of the specific compound is based on clinician experience as there has been no trials of drug therapy for pain in ALS (Brettschneider, Kurent, & Ludolph, 2013). Options include non-steroidal anti-inflammatory medications (NSAIDs), acetaminophen and medications for neuropathic pain. Opioids may be needed, especially in advanced disease (Oliver et al., 2010).
4.1.2Spasticity
Management of spasticity in ALS has been poorly studied; however, several agents are available for use including baclofen, tizanidine, benzodiazepines, and cannabinoids. The choice of one agent vs. another is based on experience, side effect profile, and patient response. Intrathecal baclofen, and botulinum toxin can be considered in primary lateral sclerosis (PLS) and select cases of upper-motor neuron predominant ALS. Of note, reduction of bothersome spasticity needs to be balanced with the fact that some increased tone in certain muscle groups can actually support function (e.g., spasticity in the extensor muscles of the legs can assist standing during transfers and facilitate bed mobility). Non-pharmacologic treatment options for spasticity can be offered as an adjunct, although the effects of such interventions are generally limited and temporary (Table 3) (Gracies, 2001).
4.1.3Cramps
Cramps can be quite bothersome in ALS and yet management of cramps has been understudied in ALS (Baldinger, Katzberg, & Weber, 2012; Miller et al., 2009b). Quinine has been used in the past with some success (El-Tawil et al., 2010), but due to the potential for serious hematologic reactions (Houstoun et al., 2014), quinine treatment for leg cramps has decreased significantly and is not approved by the Food and Drug Administration (FDA) for this use. However, in our experience some people with ALS will drink tonic water to relieve cramps as it contains some quinine. Regardless, concerns about potential quinine-related side effects remain, thus limiting its usefulness. Empirically, many ALS clinicians try one of several medications to address cramps including baclofen, gabapentin and cannabinoids (Weber, Goldman, & Truniger, 2010) with variable success. Additional agents with potential benefit include vitamin B and diltiazem (Katzberg, Khan, & So, 2010) as well as levetiracetam (Bedlack, Pastula, Hawes, & Heydt, 2009). Stretching, gentle exercise (Ribeiro, 2014) and good hydration may also be beneficial. A trial of mexiletine for the treatment of muscle cramps in ALS is ongoing (NCT01811355).
4.1.4Sialorrhea/secretion management
Drooling of saliva occurs in ALS due to oro-pharyngeal weakness that also leads to difficulty expectorating sputum and bronchial secretions. Patients may be bothered by saliva leaking from the corner of the mouth while at the same time they have the feeling that some dry, thick secretions are “stuck in the back of the throat”. This combination of symptoms may be extremely frustrating for patients and may be difficult to manage as finding the right balance between too much drooling and too much “dryness” can be tricky.
Pharmacological agents for sialorrhea management are listed in Table 3 and are available in differentformulations (Banfi et al., 2014). Their use is limited by anticholinergic side effects such as constipation, difficulty urinating, dry eyes, blurred vision, and confusion. Botulinum toxin injection (Anagnostou et al., 2013; Jackson et al., 2009; Stokholm, Bisgard, & Vilholm, 2013) and radiation (Assouline et al., 2014; Kasarskis, Hodskins, & St Clair, 2011) of the salivary glands are alternative strategies that can be considered when adequate symptom control cannot be achieved or is limited by intolerable side effects caused by use of systemic agents. These interventions can be quite effective and have few side effects, although rapid worsening of dysphagia to the point of requiring insertion of a gastrostomy tube is a potential rare complication of botulinum toxin injection.
Treatment of sialorrhea can make clearance of bronchial secretions more difficult by increasing their viscosity. Mucolytics can be used to counteract this problem. In addition, cough assist devices are widely used to promote airway clearance and are recommended treatment when the patient’s peak cough flow is measured at 270 L/min or less during routine pulmonary function testing.
4.1.5Fatigue and sleep
Fatigue is a frequently reported symptom in ALS (McElhiney, Rabkin, Gordon, Goetz, & Mitsumoto, 2009a; Ramirez, Piemonte, Callegaro, & Da Silva, 2008) and is a disabling problem for many. The term “fatigue” refers to both an inability to sustain motor function during exertion and a pervasive tiredness (Gibbons, Thornton, & Young, 2013). Causes of fatigue are multiple, ranging from nocturnal hypoventilation with resulting excessive daytime sleepiness to uncontrolled pain and cramps that interfere with sleep (Lo Coco & La Bella, 2012). Behavioral and medical interventions that may aid in fatigue management are listed in Table 3. Modafinil has been proposed as a pharmacologic treatment for fatigue in ALS based on two small studies (Carter et al., 2005; Rabkin et al., 2009), but cost is a limiting factor and confirmation of efficacy is needed in larger studies.
4.2“The worst part of ALS is that I lost my ability to communicate”
Dysarthria and the prospect of losing the ability to communicate are understandably frightening symptoms for people with ALS. Reduced speaking rate often precedes loss of intelligibility (Yunusova et al., 2012; Yunusova et al., 2010). Unfortunately, oral motorexercises are not likely to help and, in fact, many people with ALS report worsening of their speech the longer they talk. Speech therapy, however, can teach people to develop compensatory techniques such as minimizing the distance to the listeners, speaking face-to-face in a well lit room, overarticulating, and spelling out words. Taking rest breaks before speaking engagements can help to maximize speaking endurance.
As dysarthria progresses, augmentative and alternative communication (AAC) devices such as text-to-speech devices and computer/tablet applications for speech production are needed (Brownlee & Palovcak, 2007). AAC use is associated with improved quality of life and mood in people with severe dysarthria/anarthria (Caligari, Godi, Guglielmetti, Franchignoni, & Nardone, 2013; Korner et al., 2013). Message (voice) banking allows people to record their own voice and play it back on speech devices. These devices can be operated manually, by eye gaze (Caligari et al., 2013; Spataro, Ciriacono, Manno, & La Bella, 2014), or by head movement tracking technology, allowing communication to occur even in the later stages of the disease (Caligari et al., 2013). Even for patients who use AAC effectively, however, it important to develop personalized communication strategies with the caregivers such as a system for quickly confirming understanding or asking for help. Finally, brain-computer interfaces (BCIs) are an active area of research. BCIs transform brain signals (Collinger et al., 2013; Hochberg et al., 2012; Sellers, Ryan, & Hauser, 2014; Simeral, Kim, Black, Donoghue, & Hochberg, 2011) into commands that activate and move a cursor or a switch and are currently being evaluated for use in ALS (Huggins, Wren, & Gruis, 2011; McCane et al., 2014; Nijboer et al., 2008).
4.3“Why do I cough when I eat?”
Coughing at mealtimes, frequent throat clearing, wet vocal quality, feeling that food gets “stuck”, and choking are all symptoms of oropharyngeal and tongue weakness leading to dysphagia. Dysphagia can lead to aspiration and malnutrition, a negative prognostic factor in ALS (Desport et al., 1999; Paganoni, Deng, Jaffa, Cudkowicz, & Wills, 2011). As development of dysphagia is expected in ALS, bedside assessment of swallowing by a speech and language pathologist (SLP) is generally enough, although Modified Barium Swallow (MBS) may be considered to help educate patients on compensatory swallowing strategies and behavioral changes (e.g., head tilt, chin tuck,alternating bites of solid food with sips of liquid). Patients also need to be trained on diet modifications that can promote safe swallowing such as favoring soft, moist foods over dry, crumbly, or chewy foods. Neuromuscular dysphagia generally begins with difficulty managing liquids. Therefore, even in the early stages of the disease switching to thicker liquids (fruit nectar, smoothies) or addition of commercially available thickeners may be needed.
When oral intake becomes inadequate, too effortful or fatiguing, and/or compromises safety, alternative routes for nutrition may be considered to stabilize body weight (Miller et al., 2009a) and, possibly, prolong survival, although the effect on survival is still debated (Atassi, Cudkowicz, & Schoenfeld, 2011; Miller et al., 2009a). These methods include percutaneous endoscopic gastrostomy (PEG) and radiologically inserted gastrostomy (RIG), with the latter being safer in ALS (Allen et al., 2013). The risk of gastrostomy tube placement increases when forced vital capacity falls below 50% of predicted (Miller et al., 2009a). Therefore, early intervention is practiced by many although the best timing for performing the procedure is unknown (Miller et al., 2009a).
4.4“I cannot lie flat”
Failure of the muscles that support ventilation is the leading cause of death in ALS. Diaphragm weakness first manifests as nocturnal hypoventilation and may lead to interrupted sleep, increased anxiety, early morning headaches, and excessive daytime fatigue. As disease progresses, patients develop orthopnea with inability to lie flat, dyspnea on exertion and eventually shortness of breath when sitting. Weak cough and difficulty clearing secretions are associated symptoms. Patients may also develop a soft voice as sufficient respiratory support is needed to speak loudly.
Most patients with ALS remain asymptomatic until their vital capacity (VC) is less than 50% of predicted. VC correlates with survival (Traynor, Zhang, Shefner, Schoenfeld, & Cudkowicz, 2004), is employed in clinical trials as an outcome measure and is routinely monitored using portable spirometers in most ALS clinics (Miller et al., 2009a). Additional and probably more sensitive methods to monitor diaphragm weakness include nocturnal oximetry, supine VC, maximal inspiratory pressure (MIP), and sniff nasal pressure (SNP) (Miller et al., 2009a). These methods areavailable in many centers although VC remains the most commonly used measure.
When patients develop symptoms or their VC falls below 50% , non-invasive positive pressure ventilation (NIPPV) is offered to support ventilation, initially at night, and then during daytime naps or longer as needed (Miller et al., 2009a). Discussion about the natural course of breathing difficulties in ALS should be initiated early after diagnosis, so that patients can learn about the benefits and limitations of all forms of assistive ventilation and make informed choices. A randomized controlled study from 2006 showed that NIV was effective at prolonging survival in people with ALS, with a median survival benefit of 205 days (Bourke et al., 2006). This benefit, however, was not seen in patients with poor bulbar function (Bourke et al., 2006). NIV also improved quality of life and sleep-related symptoms, even when bulbar dysfunction was present (Bourke et al., 2006). Based on this trial, and a few other subsequent observational studies, NIV is now considered part of the standard of care for ALS, although its use may be limited by patient acceptance and compliance (Miller et al., 2009a). Diaphragm pacing is a newer, alternative means of supporting ventilation. Its effectiveness in prolonging survival in ALS, however, is unclear at this time (Gonzalez-Bermejo et al., 2012; Mahajan, Bach, Saporito, & Perez, 2012; Onders, Elmo, Kaplan, Katirji, & Schilz, 2014) and is the subject of ongoing research (McDermott et al., 2012). Additional preventive interventions include influenza and pneumococcal vaccination that are recommended for all people with ALS. Supplemental oxygen is rarely used unless there is concurrent pulmonary disease as it may suppress respiratory drive and lead to carbon dioxide retention. Finally, some people with ALS choose tracheostomy and invasive mechanical ventilation to prolong life (Dreyer, Lorenzen, Schou, & Felding, 2014). This practice is frequent in some countries (Fini et al., 2014; Tagami et al., 2014), but chosen by only a minority in the United States (J. Rabkin et al., 2013).
5Psychological health and quality of life
People with amyotrophic lateral sclerosis (ALS) face enormous physical and emotional challenges. It is therefore expected for ALS to have a psychological impact on patients both at the time of diagnosis and throughout the course of the disease (Atassi et al., 2011; Cupp et al., 2011; Felgoise et al., 2010; Kurt, Nijboer, Matuz, & Kubler, 2007; McElhiney, Rabkin, Gordon, Goetz, & Mitsumoto, 2009b; Rabkin et al., 2005; Tedman, Young, & Williams, 1997; Wicks et al., 2007). Interestingly, prior studies have shown that psychological distress is not directly related to functional impairment (Atassi, Cook et al., 2011; Cupp et al., 2011; McElhiney et al., 2009b; Rabkin, Wagner, & Del Bene, 2000). It has been speculated that in ALS the predicted functional losses lead the patient to change his or her health expectations (Cupp et al., 2011). Changes in expectations may be accompanied by a response shift so that the factors that contribute most to psychological health shift from those that are dependent on physical function to those that are not (social, spiritual, and existential factors) (Cupp et al., 2011; Lule et al., 2009). Perceived social support is a strong predictor of psychosocial adjustment (Lule et al., 2009).
Still, mildly-to-severely depressed mood is present in 10– 56% of people with ALS, with prevalence varying greatly depending on the scale used to define depression (Atassi, Cook et al., 2011; Cupp et al., 2011; Felgoise et al., 2010; Kurt et al., 2007; McElhiney et al., 2009b; Rabkin et al., 2005; Tedman et al., 1997; Wicks et al., 2007). When present, depression can be effectively treated with a combination of pharmacological treatments and cognitive-behavioral interventions (Table 3). An additional, disabling symptom is pseudobulbar affect (PBA), characterized by excessive laughing or crying incongruous to circumstances. This problem affects 20% – 50% of people with ALS and responds well to the combination of dextromethorphan and quinidine (Brooks et al., 2004; Pioro et al., 2010; Yang & Deeks, 2015). Although PBA is not a mood disorder, antidepressants are also frequently employed and can be effective. Interestingly, wish to die does not correlate with depression (Albert et al., 2005; Rabkin et al., 2014), pain (Maessen et al., 2010), or inadequate care (Maessen et al., 2010). Rather, wish to die is linked to hopelessness, fear of suffocation, and fear of being a burden (Albert et al., 2005; Lule et al., 2014; Maessen et al., 2009, 2010).
Multiple studies have shown that quality of life (QOL) is maintained as ALS progresses (Gauthier et al., 2007; Rousseau, Pietra, Blaya, & Catala, 2011; Rousseau, Pietra, Nadji, & Billette de Villemeur, 2013) and that QOL does not directly correlate with strength and physical function (Chio et al., 2004; Grehl, Rupp, Budde, Tegenthoff, & Fangerau, 2011; Simmons, Bremer, Robbins, Walsh, & Fischer, 2000). Rather, quality of life depends primarily on psychological and existential factors (Bremer, Simone, Walsh, Simmons, & Felgoise, 2004; Calvo et al., 2011; Chio et al., 2004; Simmons et al., 2000). Interestingly, the QOL of people with ALS is often underestimated by caregivers and healthcare professionals (Lule et al., 2013; Trail, Nelson, Van, Appel, & Lai, 2003).
6Advanced disease and end-of-life care
Early involvement of palliative care as part of the multidisciplinary team ensures an integrated approach across the spectrum of the illness that includes multi-modal symptom management, advanced planning of end-of-life care, and timely utilization of hospice (Bede et al., 2011; Blackhall, 2012; Karam, Paganoni, Joyce, Carter, & Bedlack, 2014). Patients with advanced disease and their caregivers face increasing physical, financial, and emotional demands (Chio, Gauthier, Calvo, Ghiglione, & Mutani, 2005; Connolly et al., 2014; Gauthier et al., 2007; Gladman et al., 2014; Lillo, Mioshi, & Hodges, 2012; Qutub, Lacomis, Albert, & Feingold, 2014). Adequate social support is associated with reduced burden (Pagnini et al., 2010; Qutub et al., 2014) and continued engagement with the multidisciplinary team assists patients and their caregivers in coping with worsening functional status. Home visits and telemedicine visits reduce the need for logistically difficult trips to the ALS clinic (Vitacca et al., 2010). The main focus of palliative care for people with advanced ALS is proactive management of symptoms such as pain and shortness of breath, which can be effectively managed with a variety of medications including opioids and benzodiazepines (Oliver et al., 2010), coordination of home care services, and caregiver support.
7Research and future directions
Basic ALS research has seen several breakthrough discoveries in recent years including a novel understanding of the genetic complexities and molecular mechanisms of the disease (Renton, Chio, & Traynor, 2014; Turner et al., 2013). While the lack of disease-modifying treatments at this time is upsetting, one should keep in mind that the pace of discovery in ALS has greatly accelerated over the last 20 years (Fig. 2). Little progress was made between the original description of ALS in the 19th century and 1993, the year that the first ALS-causing gene was discovered. On the other hand, over the last two decades, there has been an exploding interest in ALS genetics, pre-clinicalmodeling, and pathophysiology. Hope for ALS is fostered by a vibrant patient community with high involvement with research (Chad et al., 2013) and fundraising (Vaidya, 2014) as well as by a collaborative clinical research community that has developed infrastructure resources, shared metrics, and a roadmap to optimize translational research (Atassi et al., 2013; Beghi et al., 2011; Cudkowicz et al., 2010; Otto et al., 2012; Sherman et al., 2013). Admittedly, the recent exciting basic science discoveries have yet to translate into effective treatments for patients. However, they have already led to a large pipeline of potential therapies that await testing in clinical trials. The hope is that this momentum will soon translate into new treatments for ALS.
Acknowledgments
This manuscript is dedicated to the memory of Lisa Krivickas, M.D., exceptional mentor, ALS clinician and researcher whose work made an invaluable contribution to the field of ALS rehabilitation.
REFERENCE
1 | Albert SM, Rabkin JG, Del Bene ML, Tider T, O’Sullivan I, Rowland LP(2005) Wish todie in end-stage ALS. [Research Support, N.I.H., Extramural Research Support, U.S. Gov’t, P.H.S.]Neurology65: 16874 |
3 | Allen JA, Chen R, Ajroud-Driss S, Sufit RL, Heller S, Siddique T(2013) Gastrostomy tubeplacement by endoscopy versus radiologic methods in patients with ALS: A retrospective study of complications andoutcome. [Comparative Study]Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration14: 4308314 |
4 | Anagnostou E, Rentzos M, Alexakis T, Zouvelou V, Zambelis T, Evdokimidis I(2013) Volume matters: The influence of different botulinum toxin-A dilutions for sialorrhea in amyotrophic lateral sclerosisMuscle & Nerve47: 2276278 |
5 | Assouline A, Levy A, Abdelnour-Mallet M, Gonzalez-Bermejo J, Lenglet T, Le Forestier N(2014) Radiation therapy for hypersalivation: A prospective study in amyotrophic lateral sclerosis patients.[Research Support, Non-U.S. Gov’t]International Journal of Radiation Oncology, Biology, Physics88: 3589595 |
6 | Atassi N, Cook A, Pineda CM, Yerramilli-Rao P, Pulley D, Cudkowicz M(2011) Depression in amyotrophic lateralsclerosisAmyotroph Lateral Scler12: 2109112 |
7 | Atassi N, Cudkowicz ME, Schoenfeld DA(2011) Advanced statistical methods to study the effects of gastric tubeand non-invasive ventilation on functional decline and survival inamyotrophic lateral sclerosis. [Research Support, N.I.H.,Extramural Research Support, Non-U.S. Gov’t]Amyotrophiclateral Sclerosis: Official Publication of the World Federation of Neurology Research Group on Motor Neuron Diseases12: 4272277 |
9 | Atassi N, Yerramilli-Rao P, Szymonifka J, Yu H, Kearney M, Grasso D(2013) Analysis ofstart-up, retention, and adherence in ALS clinical trialsNeurology81: 1513501355 |
10 | Baile WF, Buckman R, Lenzi R, Glober G, Beale EA, Kudelka AP(2000) SPIKES-A six-stepprotocol for delivering bad news: Application to the patient with cancerThe Oncologist5: 4302311 |
11 | Baldinger R, Katzberg HD, Weber M(2012) Treatment for cramps in amyotrophic lateral sclerosis/motor neuron disease. [Meta-Analysis Research Support, Non-U.S. Gov’t Review]The Cochrane Database of Systematic Reviews4: CD004157 |
14 | Banfi P, Ticozzi N, Lax A, Guidugli GA, Nicolini A, Silani V(2014) A Review of Options for Treating Sialorrhea in Amyotrophic Lateral Sclerosis. [Review]Respiratory Care |
15 | Bean J, Walsh A, Frontera W(2001) Brace modification improves aerobic performance inCharcot-Marie-Tooth disease: A single-subject design. [Case Reports]American Journal of Physical Medicine & Rehabilitation/Association of Academic Physiatrists80: 8578582 |
16 | Bede P, Oliver D, Stodart J, van den Berg L, Simmons Z, D OB(2011) Palliative care inamyotrophic lateral sclerosis: A review of current international guidelines and initiativesJ NeurolNeurosurg Psychiatry82: 4413418 |
17 | Bedlack RS, Pastula DM, Hawes J, Heydt D(2009) Open-label pilot trial of levetiracetam for cramps and spasticity in patients with motor neuron disease. [Clinical Trial Research Support, Non-U.S. Gov’t]Amyotrophic Lateral Sclerosis: Official Publication of the World Federation of Neurology Research Group on Motor Neuron Diseases10: 4210215 |
19 | Beghi E, Chio A, Couratier P, Esteban J, Hardiman O, Logroscino G(2011) The epidemiologyand treatment of ALS: Focus on the heterogeneity of the disease and critical appraisal of therapeutic trialsAmyotroph Lateral Scler12: 1110 |
20 | Bello-Haas VD, Florence JM, Kloos AD, Scheirbecker J, Lopate G, Hayes SM(2007) Arandomized controlled trial of resistance exercise in individuals with ALSNeurology68: 2320032007 |
21 | Blackhall LJ(2012) Amyotrophic lateral sclerosis and palliative care: Where we are, and the road aheadMuscle Nerve45: 3311318 |
22 | Boninger ML, Cooper RA, Fitzgerald SG, Lin J, Cooper R, Dicianno B(2003) Investigating neck pain in wheelchair users. [Research Support, U.S. Gov’t, Non-P.H.S.]Journal of Physical Medicine & Rehabilitation/Association of Academic Physiatrists82: 3197202 |
23 | Borasio GD, Sloan R, Pongratz DE(1998) Breaking the news in amyotrophic lateral sclerosisJournal of the Neurological Sciences160 Suppl 1S127S133 |
24 | Bourke SC, Tomlinson M, Williams TL, Bullock RE, Shaw PJ, Gibson GJ(2006) Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: A randomised controlled trial. [Randomized Controlled Trial Research Support, Non-U.S. Gov’t]The Lancet Neurology5: 2140147 |
26 | Bremer BA, Simone AL, Walsh S, Simmons Z, Felgoise SH(2004) Factors supporting quality of life over time for individuals with amyotrophic lateral sclerosis: The role of positive self-perception and religiosityAnn Behav Med28: 2119125 |
27 | Brettschneider J, Kurent J, Ludolph A(2013) Drug therapy for pain in amyotrophic lateral sclerosis or motor neuron diseaseCochrane Database Syst Rev6: CD005226 |
28 | Bromberg MB, Brownell AA, Forshew DA, Swenson M(2010) A timeline for predicting durablemedical equipment needs and interventions for amyotrophic lateral sclerosis patients. [Research Support, Non-U.S.Gov’t]Amyotrophic Lateral Sclerosis: Official Publication of the World Federation of Neurology ResearchGroup on Motor Neuron Diseases11: 1-2110115 |
29 | Bromberg MB, Schenkenberg T, Brownell AA(2011) A survey of stress among amyotrophic lateralsclerosis care providers. [Research Support, Non-U.S. Gov’t]Amyotrophic Lateral Sclerosis: Official Publication of the World Federation of Neurology Research Group on Motor Neuron Diseases12: 3162167 |
30 | Brooks BR(1996) Natural history of ALS: Symptoms, strength, pulmonary function, and disability. [ResearchSupport, Non-U.S. Gov’t Research Support, U.S. Gov’t, Non-P.H.S. Research Support, U.S. Gov’t, P.H.S. Review]Neurology47: 4 Suppl 2S71S78discussion S81-S72. |
34 | Brooks BR, Thisted RA, Appel SH, Bradley WG, Olney RK, Berg JE(2004) Treatmentof pseudobulbar affect in ALS with dextromethorphan/quinidine: A randomized trial. [Clinical Trial MulticenterStudy Randomized Controlled Trial Research Support, Non-U.S. Gov’t]Neurology63: 813641370 |
38 | Brownlee A, Palovcak M(2007) The role of augmentative communication devices in the medical management of ALS. [Review]NeuroRehabilitation22: 6445450 |
39 | Caligari M, Godi M, Guglielmetti S, Franchignoni F, Nardone A(2013) Eye tracking communication devices in amyotrophic lateral sclerosis: Impact on disability and quality of life. [Research Support, Non-U.S. Gov’t]Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration14: 7-8546552 |
40 | Calvo A, Moglia C, Ilardi A, Cammarosano S, Gallo S, Canosa A(2011) Religiousness ispositively associated with quality of life of ALS caregivers. [Research Support, Non-U.S. Gov’t]Amyotrophic Lateral Sclerosis: Official Publication of the World Federation of Neurology Research Group onMotor Neuron Diseases12: 3168171 |
41 | Carreras I, Yuruker S, Aytan N, Hossain L, Choi JK, Jenkins BG(2010) Moderate exercisedelays the motor performance decline in a transgenic model of ALSBrain Res1313: 192201 |
42 | Carter GT, Weiss MD, Lou JS, Jensen MP, Abresch RT, Martin TK(2005) Modafinilto treat fatigue in amyotrophic lateral sclerosis: An open label pilot study. [Clinical Trial Controlled ClinicalTrial Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]Palliative Care22: 15559 |
46 | Cellura E, Spataro R, Taiello AC, La BellaV(2012) Factors affecting the diagnostic delay in amyotrophic lateral sclerosisClin Neurol Neurosurg114: 6550554 |
47 | Chad DA, Bidichandani S, Bruijn L, Capra JD, Dickie B, Ferguson J(2013) Fundingagencies and disease organizations: Resources and recommendations to facilitate ALS clinical research. [Review]Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration14 suppl 16266 |
48 | Chio A(1999) ISIS Survey: An international study on thediagnostic process and its implications in amyotrophic lateral sclerosisJ Neurol246 Suppl 3III1III5 |
49 | Chio A, Bottacchi E, Buffa C, Mutani R, Mora G(2006) Positive effects of tertiary centres for amyotrophic lateral sclerosis on outcome and use of hospital facilities. [Multicenter Study Research Support, Non-U.S. Gov’t]Journal of Neurology, Neurosurgery, and Psychiatry77: 8948950 |
51 | Chio A, Gauthier A, Calvo A, Ghiglione P, Mutani R(2005) Caregiver burden and patients’ perception of being a burden in ALSNeurology64: 1017801782 |
52 | Chio A, Gauthier A, Montuschi A, Calvo A, Di Vito N, Ghiglione P(2004) A cross sectionalstudy on determinants of quality of life in ALSJ Neurol Neurosurg Psychiatry75: 1115971601 |
53 | Chio A, Logroscino G, Hardiman O, Swingler R, Mitchell D, Beghi E(2009) Prognostic factorsin ALS: A critical review. [Review]Amyotrophic Lateral Sclerosis: Official Publication of the World Federation of Neurology Research Group on Motor Neuron Diseases10: 5-6310323 |
54 | Chio A, Mora G, Calvo A, Mazzini L, Bottacchi E, Mutani R(2009) Epidemiology of ALS in Italy: A 10-year prospective population-based studyNeurology72: 8725731 |
55 | Collinger JL, Wodlinger B, Downey JE, Wang W, Tyler-Kabara EC, Weber DJ(2013) High-performance neuroprosthetic control by an individual with tetraplegiaLancet381: 9866557564 |
56 | Connolly S, Heslin C, Mays I, Corr B, Normand C, Hardiman O(2014) Health and social care costs of managing amyotrophic lateral sclerosis (ALS): An Irish perspectiveAmyotrophic Lateral Sclerosis & Frontotemporal Degeneration15 |
57 | Cudkowicz ME, Katz J, Moore DH, O’Neill G, Glass JD, Mitsumoto H(2010) Toward moreefficient clinical trials for amyotrophic lateral sclerosisAmyotroph Lateral Scler11: 3259265 |
58 | Cupp J, Simmons Z, Berg A, Felgoise SH, Walsh SM, Stephens HE(2011) Psychological health in patients with ALS is maintained as physical function declinesAmyotroph Lateral Scler12: 4290296 |
59 | Dal Bello-Haas V, Florence JM(2013) Therapeutic exercise for people with amyotrophic lateral sclerosis or motor neuron diseaseCochrane Database Syst Rev5: CD005229 |
60 | Dal Bello-Haas V, Kloos AD, Mitsumoto H(1998) Physical therapy for a patient through six stages of amyotrophic lateral sclerosisPhys Ther78: 1213121324 |
61 | Desport JC, Preux PM, Truong TC, Vallat JM, Sautereau D, Couratier P(1999) Nutritional status is a prognostic factor for survival in ALS patientsNeurology53: 510591063 |
62 | Donaghy C, Dick A, Hardiman O, Patterson V(2008) Timeliness of diagnosis in motor neurone disease: A population-based studyUlster Med J77: 11821 |
63 | Dreyer P, Lorenzen CK, Schou L, Felding M(2014) Survival in ALS with home mechanical ventilation non-invasively and invasively: A 15-year cohort study in west Denmark. [Research Support, Non-U.S. Gov’t]Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration15: 1-26267 |
64 | Drory VE, Goltsman E, Reznik JG, Mosek A, Korczyn AD(2001) The value of muscle exercise in patients with amyotrophic lateral sclerosisJ Neurol Sci191: 1-2133137 |
65 | El-Tawil S, Al MusaT, Valli H, Lunn MP, El-Tawil T, Weber M(2010) Quinine for muscle cramps.[Meta-Analysis Review]The Cochrane Database of Systematic Reviews12: CD005044 |
67 | Felgoise SH, Chakraborty BH, Bond E, Rodriguez J, Bremer BA, Walsh SM(2010) Psychological morbidity in ALS: The importance of psychological assessment beyond depression aloneAmyotroph Lateral Scler11: 4351358 |
68 | Fini N, Georgoulopoulou E, Vinceti M, Monelli M, Pinelli G, Vacondio P(2014) Noninvasiveand invasive ventilation and enteral nutrition for ALS in ItalyMuscle & Nerve50: 4508516 |
69 | Gaujoux-Viala C, Dougados M, Gossec L(2009) Efficacy and safety of steroid injections for shoulder and elbow tendonitis: A meta-analysis of randomised controlled trialsAnn Rheum Dis68: 1218431849 |
70 | Gauthier A, Vignola A, Calvo A, Cavallo E, Moglia C, Sellitti L(2007) A longitudinal studyon quality of life and depression in ALS patient-caregiver couples. [Comparative Study Research Support, Non-U.S.Gov’t]Neurology68: 12923926 |
72 | Gibbons CJ, Thornton EW, Young CA(2013) The patient experience of fatigue in motor neurone diseaseFrontiers in Psychology4: 788 |
73 | Gibson J, Frank A(2005) Pain experienced by electric-powered chair users: A pilot exploration using paindrawings. [Research Support, Non-U.S. Gov’t]Physiotherapy Research International: The Journal forResearchers and Clinicians in Physical Therapy10: 2110115 |
74 | Gladman M, Dharamshi C, Zinman L(2014) Economic burden of amyotrophic lateral sclerosis: A Canadian study of out-of-pocket expenses. [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration15: 5-6426432 |
76 | Gonzalez-Bermejo J, Morelot-Panzini C, Salachas F, Redolfi S, Straus C, Becquemin MH(2012) Diaphragm pacing improves sleep in patients with amyotrophic lateral sclerosis. [Research Support,Non-U.S. Gov’t]Amyotrophic lateral sclerosis: Official Publication of the World Federation of NeurologyResearch Group on Motor Neuron Diseases13: 14454 |
77 | Gracies JM(2001) Physical modalities other than stretch in spastic hypertonia., viPhys Med Rehabil Clin N Am12: 4769792 |
78 | Green S, Buchbinder R, Hetrick S(2003) Physiotherapy interventions for shoulder painCochrane Database Syst Rev2: CD004258 |
79 | Green S, Buchbinder R, Hetrick S(2005) Acupuncture for shoulder painCochrane Database SystRev2: CD005319 |
80 | Grehl T, Rupp M, Budde P, Tegenthoff M, Fangerau H(2011) Depression and QOL in patients with ALS:How do self-ratings and ratings by relatives differ? [Comparative Study]Quality of Life Research: AnInternational Journal of Quality of Life Aspects of Treatment, Care and Rehabilitation20: 4569574 |
81 | Ho DT, Ruthazer R, Russell JA(2011) Shoulder pain in amyotrophic lateral sclerosisJ ClinNeuromuscul Dis13: 15355 |
82 | Hochberg LR, Bacher D, Jarosiewicz B, Masse NY, Simeral JD, Vogel J(2012) Reach andgrasp by people with tetraplegia using a neurally controlled robotic armNature485: 7398372375 |
83 | Househam E, Swash M(2000) Diagnostic delay in amyotrophic lateral sclerosis: What scope for improvement?J Neurol Sci180: 1-27681 |
84 | Houstoun M, Reichman ME, Graham DJ, Nambiar S, Shamsuddin H, Jones SC(2014) Use of anactive surveillance system by the FDA to observe patterns of quinine sulfate use and adverse hematologic outcomesin CMS Medicare data. [Research Support, Non-U.S. Gov’t]Pharmacoepidemiology and Drug Safety23: 9911917 |
85 | Huggins JE, Wren PA, Gruis KL(2011) What would brain-computer interface users want? Opinions andpriorities of potential users with amyotrophic lateral sclerosis. [Comparative Study]Amyotrophic LateralSclerosis: Official Publication of the World Federation of Neurology Research Group on Motor Neuron Diseases12: 5318324 |
86 | Ivy CC, Smith SM, Materi MM(2014) Upper extremity orthoses use in amyotrophic lateral sclerosis/motor neuron disease: Three case reportsHand9: 4543550 |
87 | Jackson CE, Gronseth G, Rosenfeld J, Barohn RJ, Dubinsky R, Simpson CB(2009) Randomized double-blind study of botulinum toxin type B for sialorrhea in ALS patients. [Randomized ControlledTrial Research Support, Non-U.S. Gov’t]Nerve39: 2137143 |
89 | Karam CY, Paganoni S, Joyce N, Carter GT, Bedlack R(2014) Palliative Care Issues inAmyotrophic Lateral Sclerosis: An Evidenced-Based ReviewThe American Journal of Hospice & Palliative Care |
90 | Kasarskis EJ, Hodskins J, St Clair WH(2011) Unilateral parotid electron beam radiotherapy as palliative treatment for sialorrhea in amyotrophic lateral sclerosis. [Comparative Study]Journal of the Neurological Sciences308: 1-2155157 |
91 | Katzberg HD, Khan AH, So YT(2010) Assessment: Symptomatic treatment for muscle cramps (an evidence-based review): Report of the therapeutics and technology assessment subcommittee of the American academy of neurology. [Research Support, Non-U.S. Gov’t Review]Neurology74: 8691696 |
93 | Kirkinezos IG, Hernandez D, Bradley WG, Moraes CT(2003) Regular exercise is beneficial to a mouse model of amyotrophic lateral sclerosisAnn Neurol53: 6804807 |
94 | Korner S, Sieniawski M, Kollewe K, Rath KJ, Krampfl K, Zapf A(2013) Speech therapy andcommunication device: Impact on quality of life and mood in patients with amyotrophic lateral sclerosis.[Clinical Trial]Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration14: 12025 |
95 | Kuffner R, Zach N, Norel R, Hawe J, Schoenfeld D, Wang L(2015) Crowdsourced analysis ofclinical trial data to predict amyotrophic lateral sclerosis progressionNature Biotechnology33: 15157 |
96 | Kurt A, Nijboer F, Matuz T, Kubler A(2007) Depression and anxiety in individuals with amyotrophic lateral sclerosis: Epidemiology and managementCNS Drugs21: 4279291 |
97 | Lillo P, Mioshi E, Hodges JR(2012) Caregiver burden in amyotrophic lateral sclerosis is more dependent on patients’ behavioral changes than physical disability: A comparative study. [Research Support, Non-U.S. Gov’t]BMC Neurology12: 156 |
98 | Lo Coco D, La Bella V(2012) Fatigue, sleep, and nocturnal complaints in patients with amyotrophiclateral sclerosisEuropean Journal of Neurology: The Official Journal of the European Federation ofNeurological Societies19: 5760763 |
99 | Lule D, Ehlich B, Lang D, Sorg S, Heimrath J, Kubler A(2013) Quality of life in fataldisease: The flawed judgement of the social environment. [Research Support, Non-U.S. Gov’t]Journal of Neurology260: 1128362843 |
100 | Lule D, Nonnenmacher S, Sorg S, Heimrath J, Hautzinger M, Meyer T(2014) Live and let die:Existential decision processes in a fatal disease. [Research Support, Non-U.S. Gov’t]Journal ofNeurology261: 3518525 |
101 | Lule D, Zickler C, Hacker S, Bruno MA, Demertzi A, Pellas F(2009) Life can be worth living in locked-in syndrome. [Research Support, Non-U.S. Gov’t Review]Progress in Brain Research177: 339351 |
103 | Maessen M, Veldink JH, Onwuteaka-Philipsen BD, de Vries JM, Wokke JH, van der Wal G(2009) Trends and determinants of end-of-life practices in ALS in the Netherlands. [Research Support, Non-U.S.Gov’t]Neurology73: 12954961 |
104 | Maessen M, Veldink JH, van den Berg LH, Schouten HJ, van der Wal G, Onwuteaka-Philipsen BD(2010) Requests for euthanasia: Origin of suffering in ALS, heart failure, and cancer patientsJournal of Neurology257: 711921198 |
105 | Mahajan KR, Bach JR, Saporito L, Perez N(2012) Diaphragm pacing and noninvasive respiratory management of amyotrophic lateral sclerosis/motor neuron diseaseMuscle & Nerve46: 6851855 |
106 | Mahoney DJ, Rodriguez C, Devries M, Yasuda N, Tarnopolsky MA(2004) Effects of high-intensity endurance exercise training in the GA mouse model of amyotrophic lateral sclerosisMuscle Nerve29: 5656662 |
107 | Mayadev AS, Weiss MD, Distad BJ, Krivickas LS, Carter GT(2008) Theamyotrophic lateral sclerosis center: A model of multidisciplinary management. [Review]Physical Medicineand Rehabilitation Clinics of North America19: 3619631xi |
108 | McCane LM, Sellers EW, McFarland DJ, Mak JN, Carmack CS, Zeitlin D(2014) Brain-computer interface (BCI) evaluation in people with amyotrophic lateral sclerosis. [Research Support,N.I.H., Extramural Research Support, Non-U.S. Gov’t]Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration15: 3-4207215 |
110 | McCluskey L, Casarett D, Siderowf A(2004) Breaking the news: A survey of ALS patients and theircaregivers. [Comparative Study]Amyotrophic lateral sclerosis and other motor neuron disorders: Officialpublication of the World Federation of Neurology, Research Group on Motor Neuron Diseases5: 3131135 |
111 | McDermott CJ, Maguire C, Cooper CL, Ackroyd R, Baird WO, Baudouin S(2012) Protocolfor diaphragm pacing in patients with respiratory muscle weakness due to motor neurone disease (DiPALS): Arandomised controlled trial. [Randomized Controlled Trial Research Support, Non-U.S. Gov’t]BMC Neurology12: 74 |
113 | McElhiney MC, Rabkin JG, Gordon PH, Goetz R, Mitsumoto H(2009a) Prevalence of fatigue and depression in ALS patients and change over timeJournal of Neurology, Neurosurgery, and Psychiatry80: 1011461149 |
114 | McElhiney MC, Rabkin JG, Gordon PH, Goetz R, Mitsumoto H(2009b) Prevalence of fatigue and depression in ALS patients and change over timeJ Neurol Neurosurg Psychiatry80: 1011461149 |
115 | Menotti F, Felici F, Damiani A, Mangiola F, Vannicelli R, Macaluso A(2011) Charcot-Marie-Tooth 1A patients with low level of impairment have a higher energy cost of walking than healthy individualsNeuromuscular Disorders: NMD21: 15257 |
116 | Miller RG, Brooks BR, Swain-Eng RJ, Basner RC, Carter GT, Casey P(2014) Qualityimprovement in neurology: Amyotrophic lateral sclerosis quality measures. Report of the Quality Measurement andReporting Subcommittee of the American Academy of NeurologyAmyotrophic Lateral Sclerosis & Frontotemporal Degeneration15: 3-4165168 |
117 | Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W(2009a) Practice parameter update: The care of the patient with amyotrophic lateral sclerosis: Drug, nutritional, andrespiratory therapies (an evidence-based review): Report of the Quality Standards Subcommittee of the AmericanAcademy of NeurologyNeurology73: 1512181226 |
118 | Miller RG, Jackson CE, Kasarskis EJ, England JD, Forshew D, Johnston W(2009b) Practice parameter update: The care of the patient with amyotrophic lateral sclerosis: Multidisciplinary care,symptom management, and cognitive/behavioral impairment (an evidence-based review): Report of the QualityStandards Subcommittee of the American Academy of Neurology. [Practice Guideline Research Support, N.I.H.,Extramural Research Support, Non-U.S. Gov’t Review]Neurology73: 1512271233 |
122 | Miller RG, Mitchell JD, Moore DH(2012) Riluzole for amyotrophic lateral sclerosis (ALS)/motor neuron disease (MND). [Meta-Analysis Research Support, Non-U.S. Gov’t Review]The Cochrane Database of Systematic Reviews3: CD001447 |
125 | Mitchell JD, Callagher P, Gardham J, Mitchell C, Dixon M, Addison-Jones R(2010) Timelinesin the diagnostic evaluation of people with suspected amyotrophic lateral sclerosis (ALS)/motor neuron disease(MND)–a 20-year review: Can we do better?Amyotroph Lateral Scler11: 6537541 |
126 | Nijboer F, Sellers EW, Mellinger J, Jordan MA, Matuz T, Furdea A(2008) A P300-based brain-computer interface for people with amyotrophic lateral sclerosis. [Research Support, N.I.H., ExtramuralResearch Support, Non-U.S. Gov’t]Clinical Neurophysiology: Official Journal of the International Federation of Clinical Neurophysiology119: 819091916 |
128 | Oliver DJ, Campbell C, O’Brien T, Sloan R, Sykes N, Tallon C(2010) Medication in the lastdays of life for motor neuron disease/amyotrophic lateral sclerosisAmyotrophic Lateral Sclerosis: OfficialPublication of the World Federation of Neurology Research Group on Motor Neuron Diseases11: 6562564 |
129 | Onders RP, Elmo M, Kaplan C, Katirji B, Schilz R(2014) Final analysis of the pilot trial of diaphragm pacing in amyotrophic lateral sclerosis with long-term follow-up: Diaphragm pacing positively affects diaphragm respiration.; discussionAmerican Journal of Surgery207: 3397 |
130 | Otto M, Bowser R, Turner M, Berry J, Brettschneider J, Connor J(2012) Roadmap and standardoperating procedures for biobanking and discovery of neurochemical markers in ALSAmyotroph Lateral Scler13: 1110 |
131 | Paganoni S, Deng J, Jaffa M, Cudkowicz ME, Wills AM(2011) Body mass index, not dyslipidemia,is an independent predictor of survival in amyotrophic lateral sclerosisMuscle Nerve44: 12024 |
132 | Paganoni S, Macklin EA, Lee A, Murphy A, Chang J, Zipf A(2014) Diagnostic timelines anddelays in diagnosing amyotrophic lateral sclerosis (ALS). [Research Support, Non-U.S. Gov’t]Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration15: 5-6453456 |
133 | Pagnini F, Rossi G, Lunetta C, Banfi P, Castelnuovo G, Corbo M(2010) Burden, depression,and anxiety in caregivers of people with amyotrophic lateral sclerosis. [ResearchSupport, Non-U.S. Gov’t]Psychology, Health & Medicine15: 6685693 |
134 | Petrof BJ(1998) The molecular basis of activity-induced muscle injury in Duchenne muscular dystrophy. [Research Support, Non-U.S. Gov’t Review]Molecular and Cellular Biochemistry179: 1-2111123 |
136 | Pioro EP, Brooks BR, Cummings J, Schiffer R, Thisted RA, Wynn D(2010) Dextromethorphan plus ultra low-dose quinidine reduces pseudobulbar affect. [Randomized Controlled Trial ResearchSupport, Non-U.S. Gov’t]Annals of Neurology68: 5693702 |
138 | Pizzimenti A, Aragona M, Onesti E, Inghilleri M(2013) Depression, pain and quality of life in patients with amyotrophic lateral sclerosis: A cross-sectional studyFunctional Neurology28: 2115119 |
139 | Qutub K, Lacomis D, Albert SM, Feingold E(2014) Life factors affecting depression and burden in amyotrophic lateral sclerosis caregiversAmyotrophic Lateral Sclerosis & Frontotemporal Degeneration15: 3-4292297 |
140 | Rabkin J, Ogino M, Goetz R, McElhiney M, Marziliano A, Imai T(2013) Tracheostomy withinvasive ventilation for ALS patients: Neurologists’ roles in the US and Japan. [Comparative Study ResearchSupport, Non-U.S. Gov’t]Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration14: 2116123 |
142 | Rabkin JG, Albert SM, Del Bene ML, O’Sullivan I, Tider T, Rowland LP(2005) Prevalence of depressive disorders and change over time in late-stage ALSNeurology65: 16267 |
143 | Rabkin JG, Goetz R, Factor-Litvak P, Hupf J, McElhiney M, Singleton J(2014) Depressionand wish to die in a multicenter cohort of ALS patientsAmyotrophic lateral sclerosis & Frontotemporal Degeneration19 |
144 | Rabkin JG, Gordon PH, McElhiney M, Rabkin R, Chew S, Mitsumoto H(2009) Modafinil treatment of fatigue in patients with ALS: A placebo-controlled study. [Randomized Controlled Trial Research Support, Non-U.S. Gov’t]Muscle & nerve39: 3297303 |
146 | Rabkin JG, Wagner GJ, Del Bene M(2000) Resilience and distress among amyotrophic lateral sclerosis patients and caregiversPsychosom Med62: 2271279 |
147 | Ramirez C, Piemonte ME, Callegaro D, Da Silva HC(2008) Fatigue in amyotrophic lateral sclerosis:Frequency and associated factorsAmyotrophic lateral sclerosis: Official publication of the World Federation of Neurology Research Group on Motor Neuron Diseases9: 27580 |
148 | Renton AE, Chio A, Traynor BJ(2014) State of play in amyotrophic lateral sclerosis genetics.[Research Support, N.I.H., Extramural Research Support, N.I.H., Intramural Review]Nature Neuroscience17: 11723 |
151 | Ribeiro S(2014) Iyengar yoga therapy as an intervention for cramp management in individuals with amyotrophic lateral sclerosis: Three case reports. [Case Reports]Journal of Alternative and Complementary Medicine20: 4322326 |
152 | Rivera I, Ajroud-Driss S, Casey P, Heller S, Allen J, Siddique T(2013) Prevalence and characteristics ofpain in early and late stages of ALSAmyotroph Lateral SclerFrontotemporal Degener14: 5-6369372 |
153 | Rousseau MC, Pietra S, Blaya J, Catala A(2011) Quality of life of ALS and LIS patients with and without invasivemechanical ventilationJournal of Neurology258: 1018011804 |
154 | Rousseau MC, Pietra S, Nadji M, Billette de Villemeur T(2013) Evaluation of quality of life in completelocked-insyndrome patientsJournal of Palliative Medicine16: 1114551458 |
155 | Schellenberg KL, Schofield SJ, Fang S, Johnston WS(2014) Breaking bad news in amyotrophic lateral sclerosis: The need for medical education. [Research Support, Non-U.S. Gov’t]Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration15: 1-24754 |
156 | Sellers EW, Ryan DB, Hauser CK(2014) Noninvasive brain-computer interface enables communication after brainstem stroke. [Research Support, N.I.H., Extramural]. reScience Translational Medicine6: 257257re257 |
157 | Sherman AV, Gubitz AK, Al-Chalabi A, Bedlack R, Berry J, Conwit R(2013) Infrastructureresources for clinical research in amyotrophic lateral sclerosisSuppl5361 |
158 | Silani V, Borasio GD(1999) Honesty and hope: Announcement of diagnosis in ALS. [Review]Neurology53: 8 Suppl 5S37S39discussion S40-S32. |
159 | Simeral JD, Kim SP, Black MJ, Donoghue JP, Hochberg LR(2011) Neural control of cursor trajectory and click by a human with tetraplegia days after implant of an intracortical microelectrode arrayJ Neural Eng8: 2025027 |
160 | Simmons Z, Bremer BA, Robbins RA, Walsh SM, Fischer S(2000) Quality of life in ALS depends on factors other than strength and physical functionNeurology55: 3388392 |
161 | Spataro R, Ciriacono M, Manno C, La BellaV(2014) The eye-tracking computer device for communication in amyotrophic lateral sclerosis. [Research Support, Non-U.S. Gov’t]Acta Neurologica Scandinavica130: 14045 |
162 | Stokholm MG, Bisgard C, Vilholm OJ(2013) Safety and administration of treatment with botulinum neurotoxin for sialorrhoea in ALS patients: Review of the literature and a proposal for tailored treatment. [Research Support, Non-U.S. Gov’t Review]Amyotrophic Lateral Sclerosis & Frontotemporal Degeneration14: 7-8516520 |
164 | Tagami M, Kimura F, Nakajima H, Ishida S, Fujiwara SY(2014) Tracheostomy and invasiveventilation in Japanese ALS patients: Decision-making and survival analysis: 1990-2010Journal of the Neurological Sciences344: 1-2158164 |
165 | Tanaka K, Saura R, Houraiya K, Tanimura H(2009) A simple and useful hand orthosis for patients with amyotrophic lateral sclerosis: A simple web spacer for thumb opposition weakness. [Case Reports]Disability and Rehabilitation Assistive Technology4: 5364366 |
166 | Tedman BM, Young CA, Williams IR(1997) Assessment of depression in patients with motor neurondisease and other neurologically disabling illnessJ Neurol Sci152 Suppl 1S75S79 |
167 | Trail M, Nelson ND, Van JN, Appel SH, Lai EC(2003) A study comparing patients with amyotrophic lateral sclerosis and their caregivers on measures of quality of life, depression, and their attitudes toward treatment options. [Comparative Study]Journal of the Neurological Sciences209: 1-27985 |
168 | Traynor BJ, Alexander M, Corr B, Frost E, Hardiman O(2003) Effect of a multidisciplinary amyotrophic lateral sclerosis (ALS) clinic on ALS survival: A population based studyJournal of Neurology, Neurosurgery, and Psychiatry74: 912581261 |
169 | Traynor BJ, Zhang H, Shefner JM, Schoenfeld D, Cudkowicz ME(2004) Functional outcome measures as clinical trial endpoints in ALS. [Clinical Trial Randomized Controlled Trial Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t Research Support, U.S. Gov’t, P.H.S.]Neurology63: 1019331935 |
174 | Turner MR, Hardiman O, Benatar M, Brooks BR, Chio A, de Carvalho M(2013) Controversiesand priorities in amyotrophic lateral sclerosis. [Research Support, N.I.H., Intramural Research Support, Non-U.S.Gov’t Review]Lancet Neurology12: 3310322 |
177 | Vaidya M(2014) Ice bucket challenge cash may help derisk ALS drug research. [News]Nature Medicine20: 101080 |
178 | Van den Berg JP, Kalmijn S, Lindeman E, Veldink JH, de Visser M, Van der Graaff MM(2005) Multidisciplinary ALS care improves quality of life in patients with ALS. [Research Support, Non-U.S.Gov’t]Neurology65: 812641267 |
179 | Veldink JH, Bar PR, Joosten EA, Otten M, Wokke JH, van den Berg LH(2003) Sexual differences in onset of disease and response to exercise in a transgenic model of ALSNeuromuscul Disord13: 9737743 |
180 | Vitacca M, Comini L, Tentorio M, Assoni G, Trainini D, Fiorenza D(2010) A pilot trial oftelemedicine-assisted, integrated care for patients with advanced amyotrophic lateral sclerosis and theircaregiversJ Telemed Telecare16: 28388 |
181 | Wallace VC, Ellis CM, Burman R, Knights C, Shaw CE, Al-Chalabi A(2014) The evaluation ofpain in amyotrophic lateral sclerosis: A case controlled observational studyAmyotrophic Lateral Sclerosis & Frontotemporal Degeneration15: 7-8520527 |
182 | Ward AL, Sanjak M, Duffy K, Bravver E, Williams N, Nichols M(2010) Power wheelchairprescription, utilization, satisfaction, and cost for patients with amyotrophic lateral sclerosis: Preliminarydata for evidence-based guidelinesArchives of Physical Medicine and Rehabilitation91: 2268272 |
183 | Weber M, Goldman B, Truniger S(2010) Tetrahydrocannabinol (THC) for cramps in amyotrophic lateralsclerosis: A randomised, double-blind crossover trial. [Randomized Controlled Trial Research Support, Non-U.S.Gov’t]Journal of Neurology, Neurosurgery, and Psychiatry81: 1011351140 |
184 | Wicks P, Abrahams S, Masi D, Hejda-Forde S, Leigh PN, Goldstein LH(2007) Prevalence of depression in a 12-month consecutive sample of patients with ALSEur J Neurol14: 99931001 |
185 | Yang LP, Deeks ED(2015) Dextromethorphan/Quinidine: A review of its use in adults with pseudobulbar affectDrugs75: 18390 |
186 | Yunusova Y, Green JR, Greenwood L, Wang J, Pattee GL, Zinman L(2012) Tongue movements and their acoustic consequences in amyotrophic lateral sclerosisFolia Phoniatr Logop64: 294102 |
187 | Yunusova Y, Green JR, Lindstrom MJ, Ball LJ, Pattee GL, Zinman L(2010) Kinematics of disease progression in bulbar ALSJ Commun Disord43: 1620 |
188 | Zoccolella S, Beghi E, Palagano G, Fraddosio A, Guerra V, Lepore V(2007) ALSmultidisciplinary clinic and survival. Results from a population-based study in Southern Italy. [MulticenterStudy]Journal of Neurology254: 811071112 |
Figures and Tables
Fig.1
A model of person-centered multidisciplinary network of care. Abbreviations: MD: medical doctor; PT: physical therapist; OT: occupational therapist; RT: respiratory therapist; SLP: speech and language pathologist.
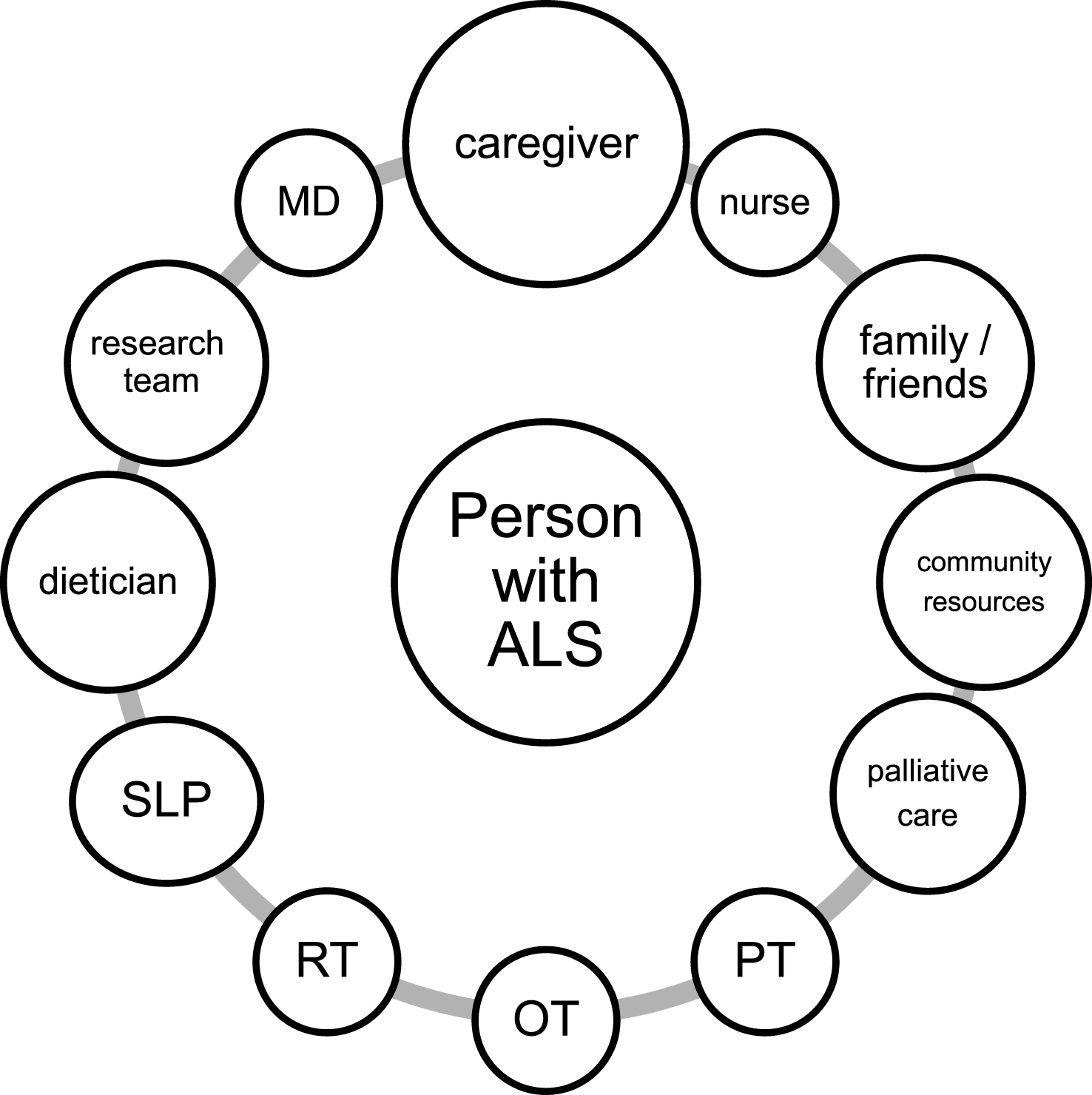
Fig.2
Timeline of ALS research. Abbreviations: C9orf72: chromosome 9 open reading frame 72; iPSC: induced pluripotent stem cells; SOD1: copper zinc superoxide dismutase 1; TDP-43: TAR DNA-binding protein 43.
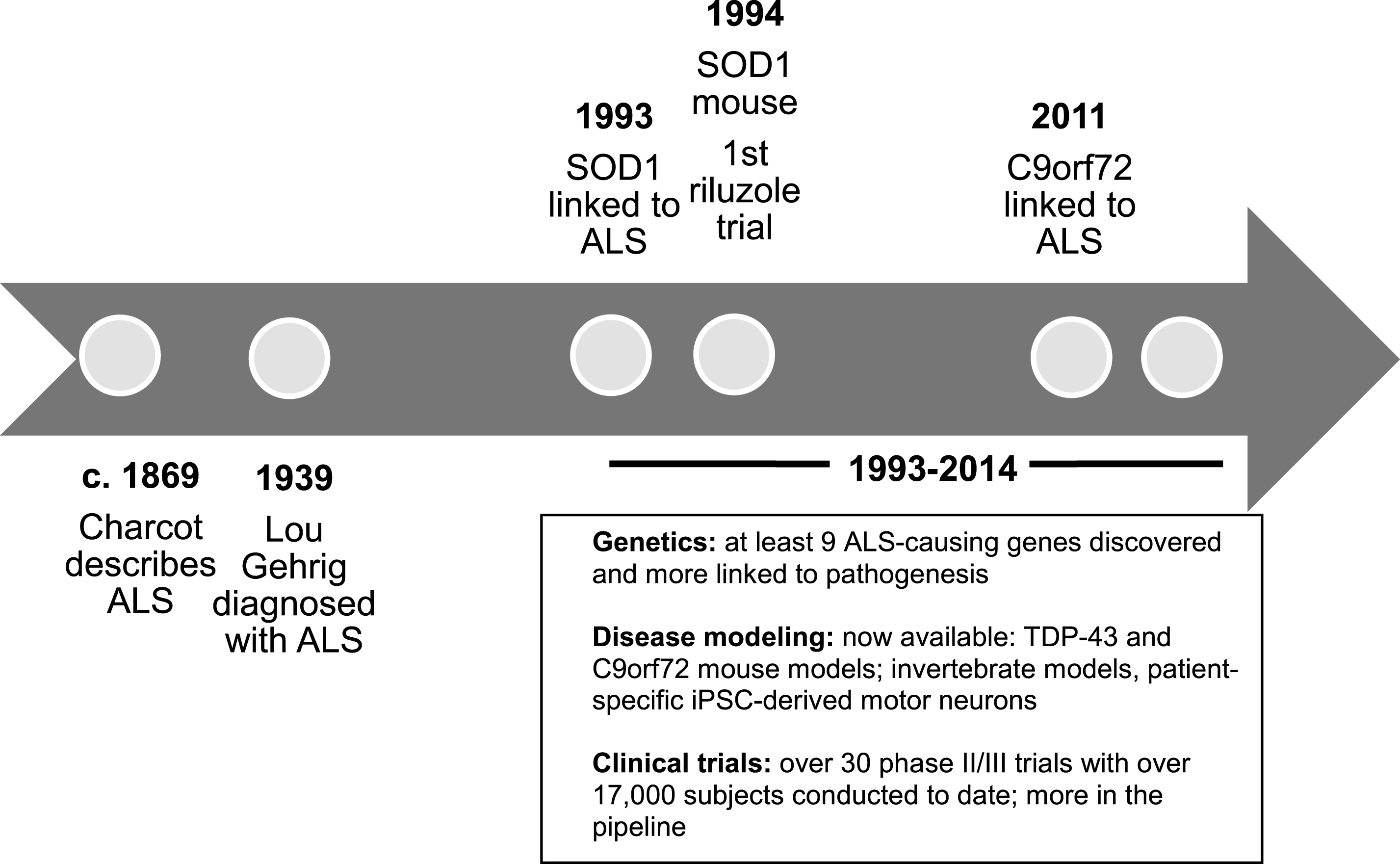
Table 1
Practical, empiric recommendations for exercise in ALS
| Type of exercise | Potential benefits | Practical considerations |
| Flexibility | Prevention and management of contractures; might also help reduce pain and spasticity | Start early in the disease course and incorporate in gentle daily routine with caregiver participation as needed |
| Strengthening | Potential role in maintaining muscle strength | Do not exercise muscles that do not have antigravity strength |
| Avoid high-resistance exercise | ||
| Avoid eccentric exercise | ||
| Progress as tolerated (“start low, go slow”) | ||
| Aerobic | Potential role in reducing deconditioning and improving mood, sleep, spasticity and quality of life | Perform at a moderate, sub-maximum level |
| If the patient cannot talk comfortably during exercise, the program is too vigorous | ||
| Progress as tolerated (“start low, go slow”) | ||
| Consider community-based programs that encourage social interaction and participation such as adaptive sports program (e.g., adaptive golf) |
Table 2
Adaptive equipment for ADLs/IADLs
| Activity | Example |
| Meal preparation and self-feeding | Large-handled utensils or foam cylinders placed around utensils, rocker knives, universal cuffs for holding utensils, long straws, scoop dishes, nonskid pads, mobile arm supports |
| Dressing | Button hooks, zipper pulls, Velcro fasteners, elastic shoelaces, reachers, dressing sticks, long-handled shoe horns |
| Grooming and personal hygiene | Strap-fitted hairbrush, long-handled comb and sponge, foam cylinders placed around the handle of bath tools to facilitate grip |
| Reading and writing | Page-turning devices, large-handled pens or foam cylinders placed around pens to improve grip, hand splints |
Abbreviations: ADLs: activities of daily living; IADLs: instrumental activities of daily living.
Table 3
Treatable ALS-related symptoms
| Symptom | Treatment options |
| Pain | Physical medicine and rehabilitation strategies: optimization of transfers, seating and positioning; bracing; physical therapy; stretching and gentle exercise; massage; TENS; ice/heat; ultrasound; inontophoresis; acupuncture; trigger point injections; joint injections (with lidocaine and steroids). Note that these strategies may be enough to effectively manage pain in many patients with none to minimal side effects. |
| Medications: topical analgesics, acetaminophen, NSAIDs, gabapentin, pregabalin, opioids. | |
| Spasticity | Medications: baclofen, tizanidine, benzodiazepines, cannabinoids (ITB and botulinum toxin in PLS and select cases of UMN-predominant ALS). |
| Adjunctive treatments: as above in the pain section, although the effect on spasticity is limited and temporary. | |
| Cramps | Medications: baclofen, gabapentin, cannabinoids. |
| Adjunctive treatments: stretching, gentle exercise, hydration (tonic water). | |
| Sialorrhea | Medications: glycopyrrolate (may be used SC or IM in advanced disease), amitriptyline, atropine drops (SL), scopolamine transdermal patch, hyoscyamine. |
| Other: botulinum toxin or radiation to salivary glands. | |
| Difficulty with secretion clearance | Most effective: cough-assist device. |
| Adjuncts: guaifenesin, N-acetylcysteine nebulizing treatment, good hydration. | |
| Fatigue | Screen for nocturnal hypoventilation and initiate NIV as needed. |
| Screen for co-morbidities such as anemia and malnutrition and correct if present. | |
| Treat pain, cramps and spasticity that may interfere with sleep. | |
| Review medication list for side effects. | |
| Use braces and assistive devices to help conserve energy. Use energy conservation techniques such as pacing and taking rest breaks. | |
| ? modafinil. | |
| Depression | SSRIs; SNRIs; bupropion; tricyclics. |
| Cognitive-behavioral therapy. |
Abbreviations: ALS: amyotrophic lateral sclerosis; IM: intramuscular; ITB: intratechal baclofen; NIV: non-invasive ventilation; NSAIDs: non-steroidal anti-inflammatory drugs; PLS: primary lateral sclerosis; SC: subcutaneous; SL: sublingual; SSRIs: selective serotonin reuptake inhibitors; SNRIs: serotonin norepinephrine reuptake inhibitors; TENS: transcutaneous electrical nerve stimulation; UMN: upper motor neuron.

