
Protein Quality Control Pathways at the Crossroad of Synucleinopathies
Abstract
The pathophysiology of Parkinson’s disease, dementia with Lewy bodies, multiple system atrophy, and many others converge at alpha-synuclein (α-Syn) aggregation. Although it is still not entirely clear what precise biophysical processes act as triggers, cumulative evidence points towards a crucial role for protein quality control (PQC) systems in modulating α-Syn aggregation and toxicity. These encompass distinct cellular strategies that tightly balance protein production, stability, and degradation, ultimately regulating α-Syn levels. Here, we review the main aspects of α-Syn biology, focusing on the cellular PQC components that are at the heart of recognizing and disposing toxic, aggregate-prone α-Syn assemblies: molecular chaperones and the ubiquitin-proteasome system and autophagy-lysosome pathway, respectively. A deeper understanding of these basic protein homeostasis mechanisms might contribute to the development of new therapeutic strategies envisioning the prevention and/or enhanced degradation of α-Syn aggregates.
INTRODUCTION
Alpha-synuclein (α-Syn) was first identified in human brain extracts more than 25 years ago [1, 2], and since then many physiological roles have been ascribed to this small protein. Although α-Syn has no defined tridimensional structure in aqueous solution [3] and is soluble under most physiological conditions [4], it can adopt beta-strand rich conformations favoring the formation of amyloid fibrils in several neurodegenerative diseases, collectively known as synucleinopathies [5–7]. For instance, α-Syn aggregates are found in distinctive neuronal structures known as Lewy bodies (LBs) and Lewy neurites (LNs) in idiopathic and familial forms of Parkinson’s disease (PD) and dementia with Lewy bodies [8], and in glial cytoplasmatic inclusions in multiple system atrophy [9–12]. However, instead of being able to adopt only one type of structure, recent studies revealed that aggregated α-Syn possess distinct conformations (polymorphs) with unique cytotoxicity profiles [13–17]. This suggests that different synucleinopathies arise from distinct α-Syn polymorphs, as indeed proposed by experiments in animal models [18, 19].
Although the initial events leading to α-Syn aggregation and toxicity in vivo are still poorly understood, several lines of evidence suggest that cellular protein quality control (PQC) pathways play a central role in these processes. Among these are the molecular chaperones and the two main protein degradation pathways, namely the ubiquitin-proteasome system (UPS) and autophagy-lysosome pathway (ALP) [20]. Here, we review basic molecular and cellular principles of α-Syn aggregation and their connection with PQC components, with a special emphasis on the suppression of α-Syn aggregation and/or toxicity by molecular chaperones.
ALPHA-SYNUCLEIN STRUCTURE AND FUNCTION
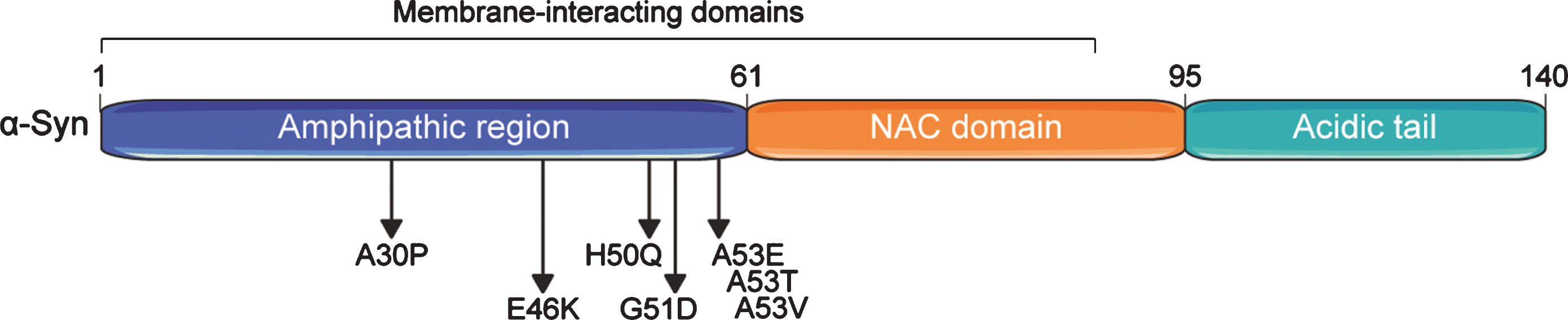
The N-terminal domain of α-Syn contains several motifs with amphipathic properties allowing for interactions with membranes (binding to lipid vesicles) and that can serve in protein-protein interactions [21] (Fig. 1). The central portion (residues 61 to 95) contains the non-amyloid-beta component of Alzheimer’s disease amyloid (NAC) motif [1], which is both sufficient and required for amyloid formation [6, 22–24]. The C-terminal region has an important role in shielding the NAC motif from aggregation [6, 24]. Deletion of only the last 10 amino acids is already sufficient to accelerate α-Syn aggregation in vitro, and this effect is stronger upon larger C-terminal truncations up to amino acids 102–120 [24–26]. α-Syn is subject to several post-translational modifications (PTMs), including N-terminal acetylation, ubiquitylation, SUMOylation, nitration, and phosphorylation [27–32], with diverse consequences for its function and propensity to aggregate (detailed below). Several roles have been ascribed to α-Syn, including facilitating the assembly of N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE)-complexes at presynaptic neuron terminals that mediate release of neurotransmitters [33, 34], and induction of membrane curvatures [35], among many others [36].
Fig. 1
Domain structure of the human alpha-synuclein (α-Syn) protein. α-Syn comprises three basic domains: an N-terminal amphipathic region, a central non-β-amyloid component (NAC) domain, and a C-terminal acidic domain. Seven membrane-interacting amino acid motifs are also present in the first half of the protein. The region preceding the NAC domain concentrates all pathogenic α-Syn mutations identified so far. Numbers on the upper part of the structure refer to amino acid positions.
SNCA MUTATIONS REVEAL UNIQUE FEATURES OF α-Syn TOXICITY AND AGGREGATION
Two types of mutations in the SNCA gene have been linked to autosomal dominant forms of PD, highlighting distinct mechanisms by which α-Syn aggregation can be triggered: (i) increased gene dosage and (ii) point mutations enhancing α-Syn aggregation propensity. The latter, including A30P [37], E46K [38], H50Q [39, 40], G51D [41], and A53T [42], A53V [43], and A53E [44] (see Fig. 1), have been discovered by genetic screens in families with hereditary PD and directly influence α-Syn aggregation to different extents and via discrete pathways [45]. Mutants such as α-SynA53T largely enhance α-Syn aggregation into fibrils [45, 46], most likely by changing the conformational landscape that α-Syn populates towards aggregation-prone conformers, without disrupting vesicular interactions [21]. In contrast, the A30P mutation does not markedly modulate α-Syn aggregation compared to overexpression of α-SynWT in cellular models [46–48]. Instead, it abolishes α-Syn interaction with lipid vesicles both in vitro [21, 49] and in vivo [50], which may lead to a buildup of cytosolic α-Syn levels, and eventually contributes to α-Syn aggregation. This implies that α-Syn aggregation is also extremely dependent on its concentration and can even be triggered by the wild type protein [50, 51]. In fact, familial PD cases caused by duplications or triplications of the SNCA locus have been identified [52–56], with increased gene dosage correlating with earlier age at onset of disease [57].
MODELLING α-Syn AGGREGATION: SEEDED VERSUS NON-SEEDED CONDITIONS
As for any aggregation-prone protein, α-Syn molecules adopt conformations that allow the establishment of non-native interactions between molecules and their coalescence into thermodynamically unstable assemblies [58]. It is upon conformational transition to more regular and complementary interfaces [59] that stable seeds are generated, capable of acting as conformational template of the amyloid state [58]. In contrast, the highly stable preformed α-Syn aggregates commonly used in studies grow by incorporation of α-Syn molecules to their ends, as the binding of additional molecules to fibrillar ends generates an incorporation site for another molecule [58]. The spontaneous aggregation of α-Syn into amyloid fibrils thus is a multi-step process during which various intermediates are generated that provide copious opportunities for PQC interference.
The exogenous provision of preformed fibrils (seeded aggregation) bypasses the initial requirement for seed formation and allows the rapid incorporation of α-Syn monomers to their ends [58], presenting a more limited number of conformational states at which PQC components can interfere. The molecular mechanisms of chaperone modulation of α-Syn aggregation in spontaneous versus seeded aggregation are thus likely to differ significantly. Indeed, some chaperones interfere with unseeded aggregation (e.g., DNAJB6 [60]), whilst others selectively act on the elongation of preformed seeds (e.g., HSPB5 [61]).
The distinction between unseeded and seeded α-Syn aggregation is thus extremely important to our understanding of the α-Syn aggregation process and PQC effects thereon. Cellular studies aimed at investigating PQC components in α-Syn aggregation are most often unable to clearly determine whether unseeded, seeded or both processes are targeted and to what extent.
Non-seeded α-Syn aggregation
It has been surprisingly difficult to consistently model spontaneous, non-seeded α-Syn aggregation in cellular and organismal models. In fact, recent nuclear magnetic resonance data showed that α-Syn at physiological concentrations remains largely in a monomeric, highly dynamic state in cells [4]. Since the crowded cellular environment is expected to facilitate α-Syn aggregation, these data strongly suggest the existence of agents (such as molecular chaperones and protein degradation machineries) that efficiently counteract α-Syn aggregation under normal circumstances.
To date, most studies investigating de novo, non-seeded, α-Syn aggregation have relied on overexpression of either wild-type (WT) or mutant variants of α-Syn. In one of the earliest models, α-Syn inclusion formation was detected in human neuroglioma H4 cells and mouse primary cortical neurons only upon overexpression of α-Syn constructs (α-SynWT, α-SynA30P, or α-SynA53T) harboring distinct C-terminal tags of variable sizes, which affected proteasomal clearance [47]. Since untagged α-Syn variants remained soluble, the tag potentiated aggregation probably through the exposure of the NAC region. Others have employed the co-expression of α-Syn with distinct aggregation-prone proteins that co-localize with α-Syn in LBs to trigger inclusion formation, such as synphilin-1 [45, 62–65] and tubulin polymerization-promoting protein (TPPP/p25α) [66], but it is not entirely clear whether these are indeed active drivers of α-Syn aggregation. Some studies have also used bimolecular fluorescence complementation assays to assess de novo α-Syn aggregation [45, 67, 68]. In these cases, fluorescence is reconstituted and detected upon co-expression of two α-Syn constructs fused to either the N- or C-terminus halves of a fluorescent protein (for example, the split Venus-system). However, it is still neither clear whether such assemblies are of fibrillar nature, as the interaction of little as two α-Syn molecules is already sufficient to reconstitute fluorescence, nor to what extent the reconstitution of the functional fluorescent protein drives assembly. Nevertheless, some degree of α-Syn fibrillation was detected upon overexpression of distinct split Venus-α-Syn in flies [68]. In any case, true detergent-insoluble α-Syn aggregates are either usually not observed in unseeded α-Syn models, or they comprise only a small fraction of the total α-Syn pool, highlighting the urgent need for better models to document α-Syn aggregation.
Seeded α-Syn aggregation
Seeded aggregation experiments have been instrumental in uncovering many of the basic principles of α-Syn pathology (see for instance [26, 69–71]). Indeed, most of the α-Syn literature relies on experiments in which an exogenous source of α-Syn amyloid fibrils is administered to cells or animals in order to trigger aggregation of the endogenous α-Syn (i.e., the α-Syn pool generated by cells de novo, even if it consists of an artificial transgene). In these cases, exogenous α-Syn fibrils come from either in vitro reactions using recombinant α-Syn [69] or from fibrils isolated from animal models or human postmortem tissue [19, 26, 72]. As stated above, structural differences of α-Syn fibrils may lead to different synucleinopathies [15, 73]. However, it should be noted that there is currently no evidence demonstrating that human pathology starts upon exposure to exogenous α-Syn seeds [36], suggesting that factors such as cellular stress may trigger the formation of the first α-Syn seeds.
α-Syn AGGREGATION AND TOXICITY IN THE CONTEXT OF PQC SYSTEMS
Molecular chaperones
Suppression of α-Syn aggregation by Hsp70 machines
Molecular chaperones are at the heart of several PQC pathways and have been extensively implicated as protective agents against protein aggregation and neurodegeneration [74]. Here, we will primarily focus on the action of Hsp70 machines on α-Syn aggregation and toxicity. The human genome encodes for multiple Hsp70 isoforms and these Hsp70s act with the help of many co-factors a system that we refer to as the Hsp70 machines.
Purified Hsp70s (e.g., HSPA1A or HSPA8) alone can almost completely block α-Syn fibrillation at substoichiometric ratios, generating small aggregates composed of both proteins [25, 75–77]. Interestingly, addition of recombinant Hsp70-interacting protein (Hip) to reactions containing Hsp70 and monomeric α-Syn completely blocked Hsp70 co-aggregation and led to sustained inhibition of α-Syn aggregation in an ATP-dependent manner [78], highlighting the importance of additional co-factors for maximal suppression of α-Syn aggregation by Hsp70 machines (see below). Purified Hsp70s (HSPA1A or HSPA8) have been shown to bind a range of α-Syn assemblies, including monomers [77], pre-fibrillar [76, 78], and fibrillar species [75, 79, 80]. α-Syn amino acid stretches that are bound by Hsp70s span residues 10–45 and 97–102 [77]. Besides the suppression of α-Syn nucleation, Hsp70s also bind to α-Syn seeds [75] and prevent fibril elongation [79, 80]. These latter findings are consistent with a holdase function of Hsp70s against α-Syn fibril elongation, possibly shielding fibrillar ends from further incorporation of α-Syn molecules [79, 80].
In cells, co-expression of Hsp70 decreased the amount of high molecular weight α-Syn species [64, 65], probably by stabilization of α-Syn in assembly-incompetent states [81]. This could account for decreased cytotoxicity of α-Syn upon overexpression of Hsp70 [65, 82]. Indeed, Hsp70 overexpression in primary neurons markedly decreased the size, but not the amount, of secreted α-Syn species [82]. Since Hsp70 was also detected in the culture medium, it was proposed to bind monomeric or low molecular weight pre-fibrillar α-Syn assemblies and prevent further aggregation into mature fibrils [82].
At the organismal level, mice overexpressing both α-Syn and the rat HspA1 showed a 2-fold reduction in 1% Triton-X100-insoluble α-Syn-containing aggregates, compared to animals expressing α-Syn only [65]. In the fruit fly Drosophila melanogaster, selective expression of α-SynWT, α-SynA30P, or α-SynA53T in dopaminergic neurons for 20 days led to a 50% cell loss, but this could be fully rescued by targeted co-expression of the human Hsp70 isoform HSPA1L [83]. Interestingly, despite its cytoprotective effects, HSPA1L did not inhibit α-Syn inclusion formation, but rather co-localized with α-Syn in LB-like structures, suggesting that Hsp70 binding reduced toxic interactions of α-Syn with other biomolecules. Such phenomenon is conserved from flies to humans, with evidence for the accumulation of not only Hsp70, but also its cochaperones Hsp40/DNAJs and Hsp110/NEFs, into LBs and LNs from patients with PD, dementia with Lewy bodies, and other synucleinopathies [63, 83]. Indeed, the titration of Hsp70s out of solution by misfolded α-Syn has been hypothesized to contribute to disease onset due to lowering of the functional pool of Hsp70 available for protein quality control pathways [78, 84].
In vitro, the suppression of α-Syn aggregation by either Hsp70 (HSPA1A) or Hsc70 (HSPA8) does not require ATP/ADP cycling [25, 75, 78, 80], nor co-chaperones, such as DNAJB1 [78]. In fact, DNAJB1, which stimulates Hsp70 cycling, even counteracts such sequestering activity [76]. However, these factors are essential for the proper function of Hsp70 in quality control pathways in vivo [85]. Indeed, overexpression of other members from the family of Hsp70 co-chaperones also successfully prevents α-Syn aggregation and/or toxicity in cell and mouse models of PD. Of special relevance in this context is the large family of Hsp40/DNAJ proteins, which are regarded as the main determinants of specificity of Hsp70 machines, since different DNAJs bind to distinct client proteins and deliver those to Hsp70 [85, 86]. Thus, DNAJs could be exploited to maximize the activity of Hsp70 machines towards specific substrates. For example, besides inhibiting α-Syn aggregation in vitro [76], DNAJB1 almost completely abolished α-Syn inclusion formation in cells overexpressing α-Syn [63]. This has also been shown for DNAJB6 and its close homologue DNAJB8 [51], both of which also strongly suppress the aggregation of other amyloidogenic polypeptides, including expanded polyglutamine-containing proteins [60, 87] and the amyloid-beta protein [88]. Interestingly, α-Syn aggregation was not suppressed by a DNAJB6 mutant that does not interact with Hsp70 [51], strengthening the notion that cooperation between distinct components of Hsp70 machines is essential for optimal function. Despite these examples, little is still known on the contribution of different DNAJs and/or NEFs to the Hsp70-dependent suppression of α-Syn aggregation in vivo. Similar to recently developed in vitro screens for inhibiting tau aggregation [89], or enhancing α-Syn disaggregation (see below) [90], further comparative studies using distinct compositions of Hsp70 machines are urgently required to better understand and manipulate Hsp70 machines in synucleinopathies.
Disaggregation of α-Syn fibrils by Hsp70 machines
The diversity and complexity of Hsp70 machines is also highlighted by studies investigating the potential of these systems to disaggregate pre-existing α-Syn amyloids. For instance, although Hsp70 alone does not modify or disaggregate mature α-Syn fibrils at relevant time-scales in vitro [75, 91], a specific Hsp70/HSPA-Hsp40/DNAJ-Hsp110/NEF combination showed powerful, ATP-dependent disaggregase activity against α-Syn amyloids [90]. Indeed, optimal fragmentation and depolymerization of α-Syn fibrils was detected upon combining Hsc70/HSPA8 with Hdj1/DNAJB1 and the NEF Apg2/HSPA4, but not upon addition of other Hsp70 machine members, such as Hsp70/HSPA1A, DNAJA1, DNAJA2, or BAG1. Moreover, a precise stoichiometry between these components was crucial for productive disaggregation [90, 91], further illustrating the tight balance between specificity and levels of chaperones/co-chaperones for the activity of Hsp70 machines. It is still not known whether Hsp70-mediated disaggregation of α-Syn also occurs in vivo, but it is tempting to speculate that the breakup of fibrils into smaller, more soluble assemblies facilitates their processing by downstream PQC components, as discussed below. However, it is equally possible that disaggregation could be detrimental and facilitate α-Syn seed propagation. Further studies are necessary to clarify these issues.
Clearance of α-Syn assemblies via protein degradation machineries
The two major cellular protein degradation machineries comprise the UPS and ALP, with the latter encompassing both autophagosome-dependent and independent pathways [92]. There is an intricate and tightly regulated crosstalk between proteasomal and lysosomal pathways engaged in the processing of α-Syn, as several studies reported preferential degradation of α-Syn via the UPS or ALP [31, 93–98]. Moreover, α-Syn (WT or distinct mutants) overexpression can impair the activity of both the UPS [99, 100] and distinct components of the ALP [66, 101–104], which would act in a progressive pathogenic feedback loop to accelerate aggregation and toxicity. Whether UPS or ALP lead to the degradation of α-Syn assemblies is still actively debated. Recent findings suggest, however, that the UPS has a more prominent role in degrading smaller α-Syn assemblies at least when protein quality systems are highly active, as is generally the case in young, healthy organisms [99]. Autophagic activity seems to be more required for larger α-Syn assemblies and upon increased α-Syn burden, due to either mutations that lead to α-Syn accumulation or decreased activity of other PQC components, as observed with aging [99]. α-Syn has also been shown to be recognized and degraded by other cellular (extracellular) proteases not directly linked to the UPS and ALP pathways [105, 106]. However, the extent to which such enzymes are required for proper α-Syn turnover and/or inhibition of propagation is still poorly understood.
PTMs also play a role in α-Syn processing and act as important sorting hubs to distinct protein degradation machineries. For instance, the covalent binding of ubiquitin to α-Syn, via either mono- (monoUb) or polyubiquitylation (polyUb) in distinct linkage types, has opposing consequences to the fate of α-Syn. For instance, the co-chaperone CHIP (carboxyl terminus of Hsp70-interacting protein), a ubiquitous E3 Ub-ligase and crucial downstream effector of Hsp70 machineries [85], was shown to promote α-Syn degradation via both the UPS and ALP [107]. Also, while monoubiquitylation by the E3 ubiquitin-ligase SIAH targeted α-Syn to the UPS, removal of the ubiquitin moiety by the deubiquitylase USP9X favored α-Syn degradation via macroautophagy [108]. Yet another ubiquitin-ligase (Nedd4) facilitated the binding of K63-linked polyUb chains to α-Syn and promoted its lysosomal degradation via the ESCRT pathway [109]. Depending on its assembly state, other PTMs such as SUMOylation, phosphorylation, nitration, O-GlcNAcylation, oxidation, and dopamine-modification can also modulate α-Syn processing via downstream degradation pathways [30, 31, 110–114]. In this context, the main findings associated to the partition of α-Syn between the UPS and ALP are discussed below and illustrated in Fig. 2.
Fig. 2
Targeting and processing of alpha-synuclein (α-Syn) by protein quality control (PQC) pathways. Left: in normal conditions, in which the cellular PQC capacity is in balance with the α-Syn burden, soluble as wells as pre-fibrillar α-Syn assemblies (after disassembly) have been shown to be targeted to and degraded by several PQC components. The initial survey of α-Syn species might be performed by molecular chaperones (1), which can facilitate the sorting of α-Syn to distinct degradative routes, such as the ubiquitin-proteasome system (UPS; 2), a ubiquitin-independent proteasomal degradation pathway (3), chaperone-mediated autophagy (CMA; 4), macroautophagy (5), secretion via endosomes (6) [162], and proteolytic digestion by intracellular (7) or extracellular proteases. Right: in aged organisms or pathological conditions, the α-Syn burden surpasses the cellular PQC capacity, leading to α-Syn accumulation and subsequent aggregation. Fibrillar α-Syn assemblies can trap several biomolecules, including molecular chaperones (8), which contributes to chaperone depletion and decreases PQC capacity. Similarly, α-Syn aggregation has been linked to impairment of different steps of macroautophagy (9), CMA (10), and proteasomal degradation (11). In some experimental setups, increased α-Syn levels can also lead to increased autophagic flux and destruction of organelles, such as mitochondria (12). α-Syn species can also be secreted to the extracellular space and taken up by neighboring cells (13), where they seed the aggregation of soluble α-Syn species (14). α-Syn aggregation additionally impairs the intracellular trafficking of other proteins, such as the lysosomal enzyme glucocerebrosidase (GCase; 15). Decreased lysosomal GCase activity, due to either mislocalization of wildtype (wt) GCase or mutant GCase variants (16), leads to accumulation of GCase substrates (such as glycosylceramide; 17), which might potentiate α-Syn aggregation. See main text for further mechanistic details and references. ER: endoplasmic reticulum; Hsc70: heat shock cognate 71 kDa protein; LAMP2a: lysosome-associated membrane protein 2 isoform a; poly-Ub: poly-ubiquitin.
![Targeting and processing of alpha-synuclein (α-Syn) by protein quality control (PQC) pathways. Left: in normal conditions, in which the cellular PQC capacity is in balance with the α-Syn burden, soluble as wells as pre-fibrillar α-Syn assemblies (after disassembly) have been shown to be targeted to and degraded by several PQC components. The initial survey of α-Syn species might be performed by molecular chaperones (1), which can facilitate the sorting of α-Syn to distinct degradative routes, such as the ubiquitin-proteasome system (UPS; 2), a ubiquitin-independent proteasomal degradation pathway (3), chaperone-mediated autophagy (CMA; 4), macroautophagy (5), secretion via endosomes (6) [162], and proteolytic digestion by intracellular (7) or extracellular proteases. Right: in aged organisms or pathological conditions, the α-Syn burden surpasses the cellular PQC capacity, leading to α-Syn accumulation and subsequent aggregation. Fibrillar α-Syn assemblies can trap several biomolecules, including molecular chaperones (8), which contributes to chaperone depletion and decreases PQC capacity. Similarly, α-Syn aggregation has been linked to impairment of different steps of macroautophagy (9), CMA (10), and proteasomal degradation (11). In some experimental setups, increased α-Syn levels can also lead to increased autophagic flux and destruction of organelles, such as mitochondria (12). α-Syn species can also be secreted to the extracellular space and taken up by neighboring cells (13), where they seed the aggregation of soluble α-Syn species (14). α-Syn aggregation additionally impairs the intracellular trafficking of other proteins, such as the lysosomal enzyme glucocerebrosidase (GCase; 15). Decreased lysosomal GCase activity, due to either mislocalization of wildtype (wt) GCase or mutant GCase variants (16), leads to accumulation of GCase substrates (such as glycosylceramide; 17), which might potentiate α-Syn aggregation. See main text for further mechanistic details and references. ER: endoplasmic reticulum; Hsc70: heat shock cognate 71 kDa protein; LAMP2a: lysosome-associated membrane protein 2 isoform a; poly-Ub: poly-ubiquitin.](https://content.iospress.com:443/media/jpd/2020/10-2/jpd-10-2-jpd191790/jpd-10-jpd191790-g002.jpg)
Ubiquitin-proteasome system
In mammalian cells, the central player of the UPS is the 26S proteasome, a large, ATP-dependent multiprotein complex devoted to the selective destruction of target proteins [115]. Evidence for the degradation of α-Syn via proteasomes comes from both in vitro [116] and cellular studies [117–119], with not only monomeric, but maybe also pre-fibrillar α-Syn species (after dissociation) being targeted to this pathway [30, 100]. Several Ub-ligases interact with α-Syn and catalyze the addition of either mono- or polyUb chains with either cytoprotective or toxic consequences depending on the specific experimental setup, presumably due to differential impact on cellular α-Syn half-life [109, 117, 120, 121]. Unmodified α-Syn can also be degraded by proteasomes via an Ub-independent pathway [118], particularly relevant for phosphorylated α-Syn at serine 129 (α-SynpS129) [119]. A mutant mimicking α-SynpS129 (α-SynS129E) was shown to be a poor autophagic substrate [111], re-emphasizing the complementary importance of the different degradation pathways. Several lines of evidence also suggest that an increased α-Syn burden inhibits proteasomal activity, which in turn might lead to a further increase in α-Syn levels, thus establishing a pathogenic feedback loop favoring α-Syn aggregation [100, 104, 122, 123].
Autophagy-lysosome pathway
The numerous reports on α-Syn degradation via the ALP highlight the importance of lysosomal-dependent regulation of α-Syn levels [124]. Not surprisingly, a plethora of therapeutic strategies targeting the ALP have been explored to tackle α-Syn aggregation and toxicity (reviewed in [125]). The ALP comprises catabolic processes that converge at the lysosome, being usually divided in three distinct types: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA) [126]. Macroautophagy relies on the engulfment of substrates within autophagosomes, which are double-layered membrane vesicles that sequester intracellular components and target them to lysosomes for degradation [127]. Most long-lived proteins, protein aggregates and even whole damaged organelles are degraded via macroautophagy [92, 128]. The importance of macroautophagy for normal cellular function is exemplified by experiments in which loss of macroautophagy in neurons led to accumulation of ubiquitylated proteins and inclusion bodies, and neurodegeneration [129]. Moreover, mutations in different autophagy-related genes, such as ATG5, lead to genetic diseases with neurologic phenotypes in humans [130].
Data supporting a role for macroautophagy in the degradation of monomeric and pre-fibrillar α-Syn assemblies come mainly from studies detecting α-Syn buildup upon exposure of cell lines overexpressing either WT or mutant α-Syn variants to the inhibitor of autophagosome formation 3-methyladenine [93, 95, 97, 131]. In vivo, overexpression of beclin-1, which is involved in autophagosome formation via the phosphatidylinositol 3-phosphate kinase complex, rescued neurological deficits of α-Syn transgenic mice [131]. Yet, beclin-1 is involved in other endosomal pathways, not directly linked to macroautophagy [132], which may contribute to the reduction of α-Syn levels and improved performance of animals overexpressing α-Syn [131]. Impairment of lysosomes, toward which all ALP components converge, with bafilomycin A1 also resulted in α-Syn buildup, further supporting a role for the ALP in α-Syn degradation [96, 125, 133, 134]. Nonetheless, whether macroautophagy is capable of degrading aggregated, insoluble α-Syn assemblies, such as those present in LBs, is still debated. For instance, in a cell model of endogenous α-Syn aggregation upon exposure to exogenous α-Syn pre-formed fibrils, α-Syn inclusion resisted lysosomal degradation [134]. In addition, increasing macroautophagy flux upon α-Syn overexpression was also shown to have detrimental effects, ranging from increased degradation of mitochondria (mitophagy) in both cellular [135], and animal models of PD [136] to enhanced secretion of α-Syn assemblies to the extracellular space [66], that may contribute to the spreading of pathogenic α-Syn aggregates. On the other hand, Gao and colleagues (2019) have recently demonstrated enhanced degradation of internalized exogenous α-Syn pre-formed fibrils in neuronal cell lines upon treatment with different autophagy inducers, suggesting that lysosomes might be capable of clearing seeded fibrillar α-Syn [137].
Different from macroautophagy, CMA encompasses the selective targeting of substrates to lysosomes via Hsc70 (HSPA8) and its co-chaperones, to specifically recognize cargo proteins with a KFERQ-like pentapetide motif, and lysosomal-associated membrane protein 2a (LAMP2a)-mediated substrate translocation across lysosomal membranes [138, 139]. Several lines of evidence support the involvement of CMA in the processing of α-Syn [125]. In an in vitro lysosomal reconstitution assay, α-SynWT was selectively targeted to lysosomes by LAMP2a, and mutations within a KFERQ-like motif in α-Syn C-terminus abolished this activity [94]. In cultured cells overexpressing α-Syn, macroautophagy inhibition led to higher α-Syn clearance via CMA [95, 140], while α-Syn protein levels were increased upon specific knockdown of LAMP2a [141], or HSPA8 [141, 142]. Compared to healthy controls, lower LAMP2a protein levels were detected in brains from early-stage PD patients, accompanied by a buildup of α-Syn and other known CMA substrates, such as myocyte-specific enhancer factor 2D (MEF2D) and nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor alpha (IκBα) [143]. The importance of CMA in processing α-Syn monomers and dimers, but not pre-fibrillar assemblies [111], is somewhat diminished by the finding that α-Syn steady-state levels were unchanged in Lamp2 knockout mice [144]. This however may be due to developmental adaptations in other PQC components, such as the UPS, as outlined above, thus masking the influence of CMA. Indeed, in vivo downregulation of Lamp2a in rats resulted in accumulation of ubiquitin-positive α-Syn inclusions in the substantia nigra followed by loss of dopaminergic neurons [145]. Additional evidence for CMA involvement in α-Syn degradation comes from observations that distinct PTMs, including oxidation, nitration, and modification by oxidized dopamine, impair α-Syn degradation via CMA, resulting in its buildup [111]. Importantly, similar to the rare α-SynA30P and α-SynA53T mutations [94], dopamine-modified α-Syn (present in sporadic PD cases) also interferes with the processing of other CMA substrates [111], further contributing to the imbalance of protein homeostasis.
Upon convergence of distinct ALP routes at lysosomes, soluble α-Syn assemblies can be degraded by acidic proteases, such as cathepsin D [146–148]. Another lysosomal enzyme that has attracted much attention in synucleinopathies is glucocerebrosidase (GCase). While homozygous mutations in the GCase-encoding gene GBA1 cause Gaucher’s disease [149], heterozygous mutations are a well-established risk factor for developing PD [150]. Indeed, α-Syn buildup is observed in several models of GCase deficiency. This occurs upon pharmacological inhibition of GCase activity in cultured cells [151, 152] and also in GBA1 mutant backgrounds, both in mouse models overexpressing α-Syn [153–155] and in PD patient iPS-derived dopaminergic neurons [156, 157]. α-Syn buildup impairs GCase trafficking and targeting to lysosomes [158, 159]. Conversely, rescue of GCase activity in mice overexpressing α-SynA53T reduced α-Syn levels and toxicity [155], establishing a pathogenic feedback loop that promotes loss of GCase function, and α-Syn accumulation, aggregation and, potentially, cell-to-cell transmission of α-Syn seeds [160, 161]. Altogether, these results suggest that the upregulation of autophagy without a simultaneous improvement of lysosomal capacity might not be a true therapeutic strategy in synucleinopathies.
CONCLUDING REMARKS
The topics discussed here paint a complex picture of cellular strategies engaged in the tight regulation of α-Syn protein levels, which ultimately determine its aggregation propensity and associated toxicity. Even though there are still some fundamental gaps in our understanding of α-Syn biology, it has become increasingly clear that the activity of dedicated PQC components, such as molecular chaperones, the UPS, and ALP is a crucial line of defense against α-Syn-mediated pathology. Failure of these systems (e.g., due to cellular stress, genetic predisposition, or aging) will influence α-Syn levels and solubility, eventually leading to disease. However, it is tempting to envision that novel therapeutic strategies to prevent, slow down and/or halt progression of synucleinopathies will emerge based on our understanding of protein homeostasis in general and in particular in components that prevent initiation of α-Syn protein aggregation or help clearing them before they affect neuronal health and synaptic integrity.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
This is an EU Joint Program – Neurodegenerative Disease Research (JPND) project. The project is supported through the following funding organizations under the aegis of JPND – www.jpnd.eu. France, National Research Agency (ANR); Germany, Federal Ministry of Education and Research (BMBF); Netherlands, Netherlands Organization for Scientific Research (ZonMw); Sweden, Swedish Research Council (VR). We would also like to acknowledge Prof. Bernd Bukau for his support and the Deutsche Forschungsgemeinschaft (SFB1036), AmPro program of the Helmholtz Society and the Landesstiftung Baden-Württemberg.
REFERENCES
[1] | Uéda K , Fukushima H , Masliah E , Xia Y , Iwai A , Yoshimoto M , Otero DA , Kondo J , Ihara Y , Saitoh T ((1993) ) Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 90: , 11282–11286. |
[2] | Jakes R , Spillantini MG , Goedert M ((1994) ) Identification of two distinct synucleins from human brain. FEBS Lett 345: , 27–32. |
[3] | Weinreb PH , Zhen W , Poon AW , Conway KA , Lansbury PT ((1996) ) NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 35: , 13709–13715. |
[4] | Theillet F-X , Binolfi A , Bekei B , Martorana A , Rose HM , Stuiver M , Verzini S , Lorenz D , van Rossum M , Goldfarb D , Selenko P ((2016) ) Structural disorder of monomeric α-synuclein persists in mammalian cells. Nature 530: , 45–50. |
[5] | Vilar M , Chou H-T , Lührs T , Maji SK , Riek-Loher D , Verel R , Manning G , Stahlberg H , Riek R ((2008) ) The fold of alpha-synuclein fibrils. Proc Natl Acad Sci U S A 105: , 8637–8642. |
[6] | Rodriguez JA , Ivanova MI , Sawaya MR , Cascio D , Reyes FE , Shi D , Sangwan S , Guenther EL , Johnson LM , Zhang M , Jiang L , Arbing MA , Nannenga BL , Hattne J , Whitelegge J , Brewster AS , Messerschmidt M , Boutet S , Sauter NK , Gonen T , Eisenberg DS ((2015) ) Structure of the toxic core of α-synuclein from invisible crystals. Nature 525: , 486–490. |
[7] | Goedert M , Jakes R , Spillantini MG ((2017) ) The synucleinopathies: Twenty years on. J Parkinsons Dis 7: , S51–S69. |
[8] | Spillantini MG , Schmidt ML , Lee VM , Trojanowski JQ , Jakes R , Goedert M ((1997) ) Alpha-synuclein in Lewy bodies. Nature 388: , 839–840. |
[9] | Wakabayashi K , Yoshimoto M , Tsuji S , Takahashi H ((1998) ) α-Synuclein immunoreactivity in glial cytoplasmic inclusions in multiple system atrophy. Neurosci Lett 249: , 180–182. |
[10] | Tu PH , Galvin JE , Baba M , Giasson B , Tomita T , Leight S , Nakajo S , Iwatsubo T , Trojanowski JQ , Lee VMY ((1998) ) Glial cytoplasmic inclusions in white matter oligodendrocytes of multiple system atrophy brains contain insoluble α-synuclein. Ann Neurol 44: , 415–422. |
[11] | Arima K , Uáda K , Sunohara N , Arakawa K , Hirai S , Nakamura M , Tonozuka-Uehara H , Kawai M ((1998) ) NACP/α-synuclein immunoreactivity in fibrillary components of neuronal and oligodendroglial cytoplasmic inclusions in the pontine nuclei in multiple system atrophy. Acta Neuropathol 96: , 439–444. |
[12] | Spillantini MG , Crowther RA , Jakes R , Cairns NJ , Lantos PL , Goedert M ((1998) ) Filamentous α-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci Lett 251: , 205–208. |
[13] | Bousset L , Pieri L , Ruiz-Arlandis G , Gath J , Jensen PH , Habenstein B , Madiona K , Olieric V , Böckmann A , Meier BH , Melki R ((2013) ) Structural and functional characterization of two alpha-synuclein strains. Nat Commun 4: , 2575. |
[14] | Li B , Ge P , Murray KA , Sheth P , Zhang M , Nair G , Sawaya MR , Shin WS , Boyer DR , Ye S , Eisenberg DS , Zhou ZH , Jiang L ((2018) ) Cryo-EM of full-length α-synuclein reveals fibril polymorphs with a common structural kernel. Nat Commun 9: , 3609. |
[15] | Peelaerts W , Bousset L , Van der Perren A , Moskalyuk A , Pulizzi R , Giugliano M , Van den Haute C , Melki R , Baekelandt V ((2015) ) α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522: , 340–344. |
[16] | Guerrero-Ferreira R , Taylor NM , Mona D , Ringler P , Lauer ME , Riek R , Britschgi M , Stahlberg H ((2018) ) Cryo-EM structure of alpha-synuclein fibrils. Elife 7: , e36402. |
[17] | Guerrero-Ferreira R , Taylor NMI , Arteni A-A , Kumari P , Mona D , Ringler P , Britschgi M , Lauer ME , Verasdock J , Riek R , Melki R , Meier BH , Böckmann A , Bousset L , Stahlberg H ((2019) ) Two new polymorphic structures of alpha-synuclein solved by cryo-electron microscopy. bioRxiv, doi: 10.1101/654582v2 |
[18] | Recasens A , Dehay B , Bové J , Carballo-Carbajal I , Dovero S , Pérez-Villalba A , Fernagut PO , Blesa J , Parent A , Perier C , Fariñas I , Obeso JA , Bezard E , Vila M ((2014) ) Lewy body extracts from Parkinson disease brains trigger α-synuclein pathology and neurodegeneration in mice and monkeys. Ann Neurol 75: , 351–362. |
[19] | Watts JC , Giles K , Oehler A , Middleton L , Dexter DT , Gentleman SM , DeArmond SJ , Prusiner SB ((2013) ) Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A 110: , 19555–19560. |
[20] | Balchin D , Hayer-Hartl M , Hartl FU ((2016) ) In vivo aspects of protein folding and quality control. Science 353: , aac4354. |
[21] | Jensen PH , Nielsen MS , Jakes R , Dotti CG , Goedert M ((1998) ) Binding of α-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J Biol Chem 273: , 26292–26294. |
[22] | Iwai A , Yoshimoto M , Masliah E , Saitoh T ((1995) ) Non-aβ component of Alzheimer’s disease amyloid (NAC) is amyloidogenic. Biochemistry 34: , 10139–10145. |
[23] | Giasson BI , Murray IVJ , Trojanowski JQ , Lee VMY ((2001) ) A Hydrophobic Stretch of 12 amino acid residues in the middle of α-synuclein is essential for filament assembly. J Biol Chem 276: , 2380–2386. |
[24] | Murray IVJ , Giasson BI , Quinn SM , Koppaka V , Axelsen PH , Ischiropoulos H , Trojanowski JQ , Lee VM-Y ((2003) ) Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 42: , 8530–8540. |
[25] | Luk KC , Mills IP , Trojanowski JQ , Lee VM-Y ((2008) ) Interactions between Hsp70 and the hydrophobic core of alpha-synuclein inhibit fibril assembly. Biochemistry 47: , 12614–12625. |
[26] | Luk KC , Kehm VM , Zhang B , O’Brien P , Trojanowski JQ , Lee VMY ((2012) ) Intracerebral inoculation of pathological α-synuclein initiates a rapidly progressive neurodegenerative α-synucleinopathy in mice. J Exp Med 209: , 975–986. |
[27] | Bhattacharjee P , Öhrfelt A , Lashley T , Blennow K , Brinkmalm A , Zetterberg H ((2019) ) Mass spectrometric analysis of Lewy body-enriched α-synuclein in Parkinson’s disease. J Proteome Res 18: , 2109–2120. |
[28] | Kellie JF , Higgs RE , Ryder JW , Major A , Beach TG , Adler CH , Merchant K , Knierman MD ((2014) ) Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci Rep 4: , 5797. |
[29] | Rott R , Szargel R , Shani V , Hamza H , Savyon M , Abd Elghani F , Bandopadhyay R , Engelender S ((2017) ) SUMOylation and ubiquitination reciprocally regulate α-synuclein degradation and pathological aggregation. Proc Natl Acad Sci U S A 114: , 13176–13181. |
[30] | Shahpasandzadeh H , Popova B , Kleinknecht A , Fraser PE , Outeiro TF , Braus GH ((2014) ) Interplay between sumoylation and phosphorylation for protection against α-synuclein inclusions. J Biol Chem 289: , 31224–31240. |
[31] | Paxinou E , Chen Q , Weisse M , Giasson BI , Norris EH , Rueter SM , Trojanowski JQ , Lee VM-Y , Ischiropoulos H ((2001) ) Induction of alpha-synuclein aggregation by intracellular nitrative insult. J Neurosci 21: , 8053–8061. |
[32] | Anderson JP , Walker DE , Goldstein JM , De Laat R , Banducci K , Caccavello RJ , Barbour R , Huang J , Kling K , Lee M , Diep L , Keim PS , Shen X , Chataway T , Schlossmacher MG , Seubert P , Schenk D , Sinha S , Gai WP , Chilcote TJ ((2006) ) Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic lewy body disease. J Biol Chem 281: , 29739–29752. |
[33] | Burré J , Sharma M , Tsetsenis T , Buchman V , Etherton MR , Südhof TC ((2010) ) Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 329: , 1663–1667. |
[34] | Burré J , Sharma M , Südhof TC ((2014) ) α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A 111: , E4274–4283. |
[35] | Varkey J , Isas JM , Mizuno N , Jensen MB , Bhatia VK , Jao CC , Petrlova J , Voss JC , Stamou DG , Steven AC , Langen R ((2010) ) Membrane curvature induction and tubulation are common features of synucleins and apolipoproteins. J Biol Chem 285: , 32486–32493. |
[36] | Peelaerts W , Bousset L , Baekelandt V , Melki R ((2018) ) α-Synuclein strains and seeding in Parkinson’s disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: Similarities and differences. Cell Tissue Res 373: , 195–212. |
[37] | Krüger R , Kuhn W , Müller T , Woitalla D , Graeber M , Kösel S , Przuntek H , Epplen JT , Schöls L , Riess O ((1998) ) Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat Genet 18: , 106–108. |
[38] | Lezcano E , Vidal L , Ser T del , Tortosa EG , Llorens V , Atarés B , Hoenicka J , Ampuero I , Rodriguez O , Muñoz DG , Zarranz JJ , Gómez-Esteban JC , Yebenes de JG , Alegre J , Ros R ((2004) ) The new mutation, E46K, of alpha-synuclein causes parkinson and Lewy body dementia. Ann Neurol 55: , 164–173. |
[39] | Proukakis C , Dudzik CG , Brier T , MacKay DS , Cooper JM , Millhauser GL , Houlden H , Schapira AH ((2013) ) A novel α-synuclein missense mutation in Parkinson disease. Neurology 80: , 1062–1064. |
[40] | Appel-Cresswell S , Vilarino-Guell C , Encarnacion M , Sherman H , Yu I , Shah B , Weir D , Thompson C , Szu-Tu C , Trinh J , Aasly JO , Rajput A , Rajput AH , Jon Stoessl A , Farrer MJ ((2013) ) Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov Disord 28: , 811–813. |
[41] | Lesage S , Anheim M , Letournel F , Bousset L , Honoré A , Rozas N , Pieri L , Madiona K , Dürr A , Melki R , Verny C , Brice A , French Parkinson’s Disease Genetics Study Group ((2013) ) G51D α-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann Neurol 73: , 459–471. |
[42] | Polymeropoulos MH , Lavedan C , Hollmann M , Glutamate I , Silva AJ , Stevens CF , Tonegawa S , Wang Y , Mayford M , Kandel ER , Dell TJO , Patton BL , Malloy SS , Kennedy MB , Biol M , Polymeropoulos MH , Lavedan C , Leroy E , Ide SE , Dehejia A , Dutra A , Pike B , Root H , Rubenstein J , Boyer R , Stenroos ES , Chandrasekharappa S , Athanassiadou A , Papapetropoulos T , Johnson WG , Lazzarini AM , Duvoisin RC , Iorio G Di , Golbe LI , Nussbaum RL ((1997) ) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276: , 2045–2048. |
[43] | Yoshino H , Hirano M , Stoessl AJ , Imamichi Y , Ikeda A , Li Y , Funayama M , Yamada I , Nakamura Y , Sossi V , Farrer MJ , Nishioka K , Hattori N ((2017) ) Homozygous alpha-synuclein p.A53V in familial Parkinson’s disease. Neurobiol Aging 57: , 248.e7–248.e12. |
[44] | Picillo M , Lizarraga KJ , Friesen EL , Chau H , Zhang M , Sato C , Rooke G , Munhoz RP , Rogaeva E , Fraser PE , Kalia SK , Kalia LV ((2018) ) Parkinsonism due to A53E α-synuclein gene mutation: Clinical, genetic, epigenetic, and biochemical features. Mov Disord 33: , 1950–1955. |
[45] | Lázaro DF , Rodrigues EF , Langohr R , Shahpasandzadeh H , Ribeiro T , Guerreiro P , Gerhardt E , Kröhnert K , Klucken J , Pereira MD , Popova B , Kruse N , Mollenhauer B , Rizzoli SO , Braus GH , Danzer KM , Outeiro TF ((2014) ) Systematic comparison of the effects of alpha-synuclein mutations on its oligomerization and aggregation. PLoS Genet 10: , e1004741. |
[46] | Conway KA , Harper JD , Lansbury PT ((1998) ) Accelerated in vitro fibril formation by a mutant α-synuclein linked to early-onset Parkinson disease. Nat Med 4: , 1318–1320. |
[47] | McLean PJ , Kawamata H , Hyman BT ((2001) ) Alpha-synuclein-enhanced green fluorescent protein fusion proteins form proteasome sensitive inclusions in primary neurons. Neuroscience 104: , 901–912. |
[48] | Conway KA , Lee SJ , Rochet JC , Ding TT , Williamson RE , Lansbury PT ((2000) ) Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc Natl Acad Sci U S 97: , 571–576. |
[49] | Samuel F , Flavin WP , Iqbal S , Pacelli C , Renganathan SDS , Trudeau LE , Campbell EM , Fraser PE , Tandon A ((2016) ) Effects of serine 129 phosphorylation on α-synuclein aggregation, membrane association, and internalization. J Biol Chem 291: , 4374–4385. |
[50] | Outeiro TF , Lindquist S ((2003) ) Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 302: , 1772–1775. |
[51] | Aprile FA , Källstig E , Limorenko G , Vendruscolo M , Ron D , Hansen C ((2017) ) The molecular chaperones DNAJB6 and Hsp70 cooperate to suppress α-synuclein aggregation. Sci Rep 7: , 1–11. |
[52] | Singleton AB , Farrer M , Johnson J , Singleton A , Hague S , Kachergus J , Hulihan M , Peuralinna T , Dutra A , Nussbaum R , Lincoln S , Crawley A , Hanson M , Maraganore D , Adler C , Cookson MR , Muenter M , Baptista M , Miller D , Blancato J , Hardy J , Gwinn-Hardy K ((2003) ) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302: , 841. |
[53] | Nishioka K , Ross OA , Ishii K , Kachergus JM , Ishiwata K , Kitagawa M , Kono S , Obi T , Mizoguchi K , Inoue Y , Imai H , Takanashi M , Mizuno Y , Farrer MJ , Hattori N ((2009) ) Expanding the clinical phenotype of SNCA duplication carriers. Mov Disord 24: , 1811–1819. |
[54] | Ibáñez P , Bonnet A-M , Débarges B , Lohmann E , Tison F , Pollak P , Agid Y , Dürr A , Brice A , A Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364: , 1169–1171. |
[55] | Konno T , Ross OA , Puschmann A , Dickson DW , Wszolek ZK ((2016) ) Autosomal dominant Parkinson’s disease caused by SNCA duplications. Parkinsonism Relat Disord 22: Suppl 1, S1–S6. |
[56] | Fuchs J , Nilsson C , Kachergus J , Munz M , Larsson E-M , Schüle B , Langston JW , Middleton FA , Ross OA , Hulihan M , Gasser T , Farrer MJ ((2007) ) Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology 68: , 916–922. |
[57] | Book A , Guella I , Candido T , Brice A , Hattori N , Jeon B , Farrer MJ , SNCA Multiplication Investigators of the GEoPD Consortium ((2018) ) A meta-analysis of α-synuclein multiplication in familial parkinsonism. Front Neurol 9: , 1021. |
[58] | Oosawa F , Asakura S ((1975) ) Thermodynamics of the Polymerization of Protein, Academic Press, London. |
[59] | Cremades N , Cohen SIA , Deas E , Abramov AY , Chen AY , Orte A , Sandal M , Clarke RW , Dunne P , Aprile FA , Bertoncini CW , Wood NW , Knowles TPJ , Dobson CM , Klenerman D ((2012) ) Direct observation of the interconversion of normal and toxic forms of α-synuclein. Cell 149: , 1048–1059. |
[60] | Kakkar V , Månsson C , de Mattos EP , Bergink S , van der Zwaag M , van Waarde MAWH , Kloosterhuis NJ , Melki R , van Cruchten RTP , Al-Karadaghi S , Arosio P , Dobson CM , Knowles TPJ , Bates GP , van Deursen JM , Linse S , van de Sluis B , Emanuelsson C , Kampinga HH ((2016) ) The S/T-rich motif in the DNAJB6 chaperone delays polyglutamine aggregation and the onset of disease in a mouse model. Mol Cell 62: , 272–283. |
[61] | Waudby CA , Knowles TPJ , Devlin GL , Skepper JN , Ecroyd H , Carver JA , Welland ME , Christodoulou J , Dobson CM , Meehan S ((2010) ) The interaction of alphaB-crystallin with mature alpha-synuclein amyloid fibrils inhibits their elongation. Biophys J 98: , 843–851. |
[62] | Shin Y , Klucken J , Patterson C , Hyman BT , McLean PJ ((2005) ) The Co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates α-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem 280: , 23727–23734. |
[63] | McLean PJ , Kawamata H , Shariff S , Hewett J , Sharma N , Ueda K , Breakefield XO , Hyman BT ((2002) ) TorsinA and heat shock proteins act as molecular chaperones: Suppression of alpha-synuclein aggregation. J Neurochem 83: , 846–854. |
[64] | Klucken J , Shin Y , Hyman BT , McLean PJ ((2004) ) A single amino acid substitution differentiates Hsp70-dependent effects on α-synuclein degradation and toxicity. Biochem Biophys Res Commun 325: , 367–373. |
[65] | Klucken J , Shin Y , Masliah E , Hyman BT , McLean PJ ((2004) ) Hsp70 reduces alpha-synuclein aggregation and toxicity. J Biol Chem 279: , 25497–25502. |
[66] | Ejlerskov P , Rasmussen I , Nielsen TT , Bergström AL , Tohyama Y , Jensen PH , Vilhardt F ((2013) ) Tubulin polymerization-promoting protein (TPPP/p25α) promotes unconventional secretion of α-synuclein through exophagy by impairing autophagosome-lysosome fusion. J Biol Chem 288: , 17313–17335. |
[67] | Outeiro TF , Putcha P , Tetzlaff JE , Spoelgen R , Koker M , Carvalho F , Hyman BT , McLean PJ ((2008) ) Formation of toxic oligomeric α-synuclein species in living cells. PLoS One 3: , e1867. |
[68] | Prasad V , Wasser Y , Hans F , Goswami A , Katona I , Outeiro TF , Kahle PJ , Schulz JB , Voigt A ((2018) ) Monitoring α-synuclein multimerization in vivo. FASEB J 33: , 2116–2131. |
[69] | Luk KC , Kehm V , Carroll J , Zhang B , Brien PO , Trojanowski JQ , Lee VM ((2012) ) Pathological α-synuclein transmission in nontransgenic mice. Science 949: , 949–953. |
[70] | Hansen C , Angot E , Bergström A , Steiner JA , Pieri L , Paul G , Outeiro TF , Melki R , Kallunki P , Fog K ((2011) ) a-synuclein propagates from mouse brain to grafted dopmainergic neurons and seeds aggregation in cultured human cells. J Clin Invest 121: , 715–725. |
[71] | Volpicelli-Daley LA , Luk KC , Patel TP , Tanik SA , Riddle DM , Stieber A , Meaney DF , Trojanowski JQ , Lee VM-Y ((2011) ) Exogenous α-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 72: , 57–71. |
[72] | Bernis ME , Babila JT , Breid S , Wüsten KA , Wüllner U , Tamgüney G ((2015) ) Prion-like propagation of human brain-derived alpha-synuclein in transgenic mice expressing human wild-type alpha-synuclein. Acta Neuropathol Commun 3: , 75. |
[73] | Prusiner SB , Woerman AL , Mordes DA , Watts JC , Rampersaud R , Berry DB , Patel S , Oehler A , Lowe JK , Kravitz SN , Geschwind DH , Glidden DV , Halliday GM , Middleton LT , Gentleman SM , Grinberg LT , Giles K ((2015) ) Evidence for α-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Nat. Acad Sci U S A 112: , E5308–E5317. |
[74] | Kampinga HH , Bergink S ((2016) ) Heat shock proteins as potential targets for protective strategies in neurodegeneration. Lancet Neurol 15: , 748–759. |
[75] | Huang C , Cheng H , Hao S , Zhou H , Zhang X , Gao J , Sun Q-H , Hu H , Wang C-C ((2006) ) Heat shock protein 70 inhibits alpha-synuclein fibril formation via interactions with diverse intermediates. J Mol Biol 364: , 323–336. |
[76] | Dedmon MM , Christodoulou J , Wilson MR , Dobson CM ((2005) ) Heat shock protein 70 inhibits α-synuclein fibril formation via preferential binding to prefibrillar species. J Biol Chem 280: , 14733–14740. |
[77] | Redeker V , Pemberton S , Bienvenut W , Bousset L , Melki R ((2012) ) Identification of protein interfaces between α-synuclein, the principal component of Lewy bodies in Parkinson disease, and the molecular chaperones human Hsc70 and the yeast Ssa1p. J Biol Chem 287: , 32630–32639. |
[78] | Roodveldt C , Bertoncini CW , Andersson A , van der Goot AT , Hsu S-T , Fernández-Montesinos R , de Jong J , van Ham TJ , Nollen EA , Pozo D , Christodoulou J , Dobson CM ((2009) ) Chaperone proteostasis in Parkinson’s disease: Stabilization of the Hsp70/alpha-synuclein complex by Hip. EMBO J 28: , 3758–3770. |
[79] | Aprile FA , Arosio P , Fusco G , Chen SW , Kumita JR , Dhulesia A , Tortora P , Knowles TPJ , Vendruscolo M , Dobson CM , Cremades N ((2017) ) Inhibition of α-synuclein fibril elongation by Hsp70 is governed by a kinetic binding competition between α-synuclein species. Biochemistry 56: , 1177–1180. |
[80] | Pemberton S , Madiona K , Pieri L , Kabani M , Bousset L , Melki R ((2011) ) Hsc70 protein interaction with soluble and fibrillar α-synuclein. J Biol Chem 286: , 34690–34699. |
[81] | Klucken J , Outeiro TF , Nguyen P , McLean PJ , Hyman BT ((2006) ) Detection of novel intracellular α-synuclein oligomeric species by fluorescence lifetime imaging. FASEB J 20: , 2050–2057. |
[82] | Danzer KM , Ruf WP , Putcha P , Joyner D , Hashimoto T , Glabe C , Hyman BT , McLean PJ ((2010) ) Heat-shock protein 70 modulates toxic extracellular α-synuclein oligomers and rescues trans-synaptic toxicity. FASEB J 25: , 326–336. |
[83] | Auluck PK , Chan HYE , Trojanowski JQ , Lee VMY , Bonini NM ((2002) ) Chaperone suppression of alpha-synuclein toxicity in a Drosophila model for Parkinson’s disease. Science 295: , 865–868. |
[84] | Hinault MP , Cuendet AFH , Mattoo RUH , Mensi M , Dietler G , Lashuel HA , Goloubinoff P ((2010) ) Stable α-synuclein oligomers strongly inhibit chaperone activity of the Hsp70 system by weak interactions with J-domain co-chaperones. J Biol Chem 285: , 38173–38182. |
[85] | Kampinga HH , Craig EA ((2010) ) The HSP70 chaperone machinery: J proteins as drivers of functional specificity. Nat Rev Mol Cell Biol 11: , 579–592. |
[86] | Craig EA , Marszalek J ((2017) ) How do J-proteins get Hsp70 to do so many different things? Trends Biochem Sci 42: , 355–368. |
[87] | Hageman J , Rujano MA , van Waarde MAWH , Kakkar V , Dirks RP , Govorukhina N , Oosterveld-Hut HMJ , Lubsen NH , Kampinga HH ((2010) ) A DNAJB chaperone subfamily with HDAC-dependent activities suppresses toxic protein aggregation. Mol Cell 37: , 355–369. |
[88] | Månsson C , Arosio P , Hussein R , Kampinga HH , Hashem RM , Boelens WC , Dobson CM , Knowles TPJ , Linse S , Emanuelsson C ((2014) ) Interaction of the molecular chaperone DNAJB6 with growing amyloid-beta 42 (Aβ42) aggregates leads to sub-stoichiometric inhibition of amyloid formation. J Biol Chem 289: , 31066–31076. |
[89] | Mok S-A , Condello C , Freilich R , Gillies A , Arhar T , Oroz J , Kadavath H , Julien O , Assimon VA , Rauch JN , Dunyak BM , Lee J , Tsai FTF , Wilson MR , Zweckstetter M , Dickey CA , Gestwicki JE ((2018) ) Mapping interactions with the chaperone network reveals factors that protect against tau aggregation. Nat Struct Mol Biol 25: , 384–393. |
[90] | Gao X , Carroni M , Nussbaum-Krammer C , Mogk A , Nillegoda NB , Szlachcic A , Guilbride DL , Saibil HR , Mayer MP , Bukau B ((2015) ) Human Hsp70 disaggregase reverses Parkinson’s-linked α-synuclein amyloid fibrils. Mol Cell 59: , 781–793. |
[91] | Duennwald ML , Echeverria A , Shorter J ((2012) ) Small heat shock proteins potentiate amyloid dissolution by protein disaggregases from yeast and humans. PLoS Biol 10: , e1001346. |
[92] | Dikic I ((2017) ) Proteasomal and autophagic degradation systems. Annu Rev Biochem 86: , 193–224. |
[93] | Webb JL , Ravikumar B , Atkins J , Skepper JN , Rubinsztein DC ((2003) ) α-synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278: , 25009–25013. |
[94] | Cuervo AM , Stefanis L , Fredenburg R , Lansbury PT , Sulzer D ((2004) ) Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 305: , 1292–1295. |
[95] | Vogiatzi T , Xilouri M , Vekrellis K , Stefanis L ((2008) ) Wild type α-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem 283: , 23542–23556. |
[96] | Lee H-J , Khoshaghideh F , Patel S , Lee SJ ((2004) ) Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J Neurosci 24: , 1888–1896. |
[97] | Yu WH , Dorado B , Figueroa HY , Wang L , Planel E , Cookson MR , Clark LN , Duff KE ((2009) ) Metabolic activity determines efficacy of macroautophagic clearance of pathological oligomeric α-synuclein. Am J Pathol 175: , 736–747. |
[98] | Bennett MC , Bishop JF , Leng Y , Chock PB , Chase TN , Mouradian MM ((1999) ) Degradation of alpha-synuclein by proteasome. J Biol Chem 274: , 33855–33858. |
[99] | Ebrahimi-Fakhari D , Cantuti-Castelvetri I , Fan Z , Rockenstein E , Masliah E , Hyman BT , McLean PJ , Unni VK ((2011) ) Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J Neurosci 31: , 14508–14520. |
[100] | Emmanouilidou E , Stefanis L , Vekrellis K ((2010) ) Cell-produced α-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol Aging 31: , 953–968. |
[101] | Yan JQ , Yuan YH , Gao YN , Huang JY , Ma KL , Gao Y , Zhang WQ , Guo XF , Chen NH ((2014) ) Overexpression of human E46K mutant α-Synuclein impairs macroautophagy via inactivation of JNK1-Bcl-2 pathway. Mol Neurobiol 50: , 685–701. |
[102] | Winslow AR , Chen CW , Corrochano S , Acevedo-Arozena A , Gordon DE , Peden AA , Lichtenberg M , Menzies FM , Ravikumar B , Imarisio S , Brown S , O’Kane CJ , Rubinsztein DC ((2010) ) α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J Cell Biol 190: , 1023–1037. |
[103] | Song JX , Lu JH , Liu LF , Chen LL , Durairajan SSK , Yue Z , Zhang HQ , Li M ((2014) ) HMGB1 is involved in autophagy inhibition caused by SNCA/a-synuclein overexpression: A process modulated by the natural autophagy inducer corynoxine B. Autophagy 10: , 144–154. |
[104] | Stefanis L , Larsen KE , Rideout HJ , Sulzer D , Greene LA ((2001) ) Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J Neurosci 21: , 9549–9560. |
[105] | Tatebe H , Watanabe Y , Kasai T , Mizuno T , Nakagawa M , Tanaka M , Tokuda T ((2010) ) Extracellular neurosin degrades α-synuclein in cultured cells. Neurosci Res 67: , 341–346. |
[106] | Sung JY , Park SM , Lee C-H , Um JW , Lee HJ , Kim J , Oh YJ , Lee S-T , Paik SR , Chung KC ((2005) ) Proteolytic cleavage of extracellular secreted α-synuclein via matrix metalloproteinases. J Biol Chem 280: , 25216–25224. |
[107] | Shin Y , Klucken J , Patterson C , Hyman BT , McLean PJ ((2005) ) The Co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates α-synuclein degradation decisions between proteasomal and lysosomal pathways. J Biol Chem 280: , 23727–23734. |
[108] | Rott R , Szargel R , Haskin J , Bandopadhyay R , Lees AJ , Shani V , Engelender S ((2011) ) α-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc Natl Acad Sci U S A 108: , 18666–18671. |
[109] | Tofaris GK , Kim HT , Hourez R , Jung J-W , Kim KP , Goldberg AL ((2011) ) Ubiquitin ligase Nedd4 promotes α-synuclein degradation by the endosomal-lysosomal pathway. Proc Natl Acad Sci U S A 108: , 17004–17009. |
[110] | Levine PM , Galesic A , Balana AT , Mahul-Mellier A-L , Navarro MX , De Leon CA , Lashuel HA , Pratt MR ((2019) ) α-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proc Natl Acad Sci U S A 116: , 1511–1519. |
[111] | Martinez-Vicente M , Talloczy Z , Kaushik S , Massey AC , Mazzulli J , Mosharov EV , Hodara R , Fredenburg R , Wu D , Follenzi A , Dauer W , Przedborski S , Ischiropoulos H , Lansbury PT , Sulzer D , Cuervo AM ((2008) ) Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J Clin Invest 118: , 777–788. |
[112] | Tenreiro S , Reimão-Pinto MM , Antas P , Rino J , Wawrzycka D , Macedo D , Rosado-Ramos R , Amen T , Waiss M , Magalhães F , Gomes A , Santos CN , Kaganovich D , Outeiro TF ((2014) ) Phosphorylation modulates clearance of alpha-synuclein inclusions in a yeast model of Parkinson’s disease. PLoS Genet 10: , e1004302. |
[113] | Oueslati A , Schneider BL , Aebischer P , Lashuel HA ((2013) ) Polo-like kinase 2 regulates selective autophagic alpha-synuclein clearance and suppresses its toxicity in vivo. Proc Natl Acad Sci U S A 110: , E3945–E3954. |
[114] | Dahmene M , Bérard M , Oueslati A ((2017) ) Dissecting the molecular pathway involved in PLK2 kinase-mediated α-synuclein-selective autophagic degradation. J Biol Chem 292: , 3919–3928. |
[115] | Collins GA , Goldberg AL ((2017) ) The logic of the 26S proteasome. Cell 169: , 792–806. |
[116] | Liu CW , Corboy MJ , DeMartino GN , Thomas PJ ((2003) ) Endoproteolytic activity of the proteasome. Science 299: , 408–411. |
[117] | Shimura H , Schlossmacher MG , Hattori N , Frosch MP , Trockenbacher A , Schneider R , Mizuno Y , Kosik KS , Selkoe DJ ((2001) ) Ubiquitination of a new form of alpha-synuclein by parkin from human brain: Implications for Parkinson’s disease. Science 293: , 263–269. |
[118] | Tofaris GK , Layfield R , Spillantini MG ((2001) ) α-Synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett 509: , 22–26. |
[119] | Machiya Y , Hara S , Arawaka S , Fukushima S , Sato H , Sakamoto M , Koyama S , Kato T ((2010) ) Phosphorylated α-Synuclein at Ser-129 is targeted to the proteasome pathway in a ubiquitin-independent manner. J Biol Chem 285: , 40732–40744. |
[120] | Rott R , Szargel R , Haskin J , Shani V , Shainskaya A , Manov I , Liani E , Avraham E , Engelender S ((2008) ) Monoubiquitylation of α-Synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J Biol Chem 283: , 3316–3328. |
[121] | Lee JT , Wheeler TC , Li L , Chin LS ((2008) ) Ubiquitination of α-synuclein by Siah-1 promotes α-synuclein aggregation and apoptotic cell death. Hum Mol Genet 17: , 906–917. |
[122] | Petrucelli L , O’Farrell C , Lockhart PJ , Baptista M , Kehoe K , Vink L , Choi P , Wolozin B , Farrer M , Hardy J , Cookson MR ((2002) ) Parkin protects against the toxicity associated with mutant alpha-synuclein: Proteasome dysfunction selectively affects catecholaminergic neurons. Neuron 36: , 1007–1019. |
[123] | Tanaka Y , Engelender S , Igarashi S , Rao RK , Wanner T , Tanzi RE , Sawa A , L Dawson V , Dawson TM , Ross CA ((2001) ) Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum Mol Genet 10: , 919–926. |
[124] | Vidyadhara DJ , Lee JE , Chandra SS ((2019) ) Role of the endolysosomal system in Parkinson’s disease. J Neurochem 150: , 487–506. |
[125] | Xilouri M , Brekk OR , Stefanis L ((2016) ) Autophagy and alpha-synucle, Relevance toarkinson’s disease and related synucleopathies. Mov Disord 31: , 178–192. |
[126] | Kaur J , Debnath J ((2015) ) Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 16: , 461–472. |
[127] | Lilienbaum A ((2013) ) Relationship between the proteasomal system and autophagy. Int J Biochem Mol Biol 4: , 1–26. |
[128] | Sontag EM , Samant RS , Frydman J ((2017) ) Mechanisms and functions of spatial protein quality control. Annu Rev Biochem 86: , 97–122. |
[129] | Hara T , Nakamura K , Matsui M , Yamamoto A , Nakahara Y , Suzuki-Migishima R , Yokoyama M , Mishima K , Saito I , Okano H , Mizushima N ((2006) ) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441: , 885–889. |
[130] | [130] Kim M , Sandford E , Gatica D , Qiu Y , Liu X , Zheng Y , Schulman BA , Xu J , Semple I , Ro SH , Kim B , Mavioglu RN , Tolun A , Jipa A , Takats S , Karpati M , Li JZ , Yapici Z , Juhasz G , Lee JH , Klionsky DJ , Burmeister M ((2016) ) Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. Elife 5: , 1–18. |
[131] | Spencer B , Potkar R , Trejo M , Rockenstein E , Patrick C , Gindi R , Adame A , Wyss-Coray T , Masliah E ((2009) ) Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in alpha-synuclein models of Parkinson’s and Lewy body diseases. J Neurosci 29: , 13578–13588. |
[132] | Levine B , Liu R , Dong X , Zhong Q ((2015) ) Beclin orthologs: Integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol 25: , 533–544. |
[133] | Klucken J , Poehler AM , Ebrahimi-Fakhari D , Schneider J , Nuber S , Rockenstein E , Schlötzer-Schrehardt U , Hyman BT , McLean PJ , Masliah E , Winkler J ((2012) ) Alpha-synuclein aggregation involves a bafilomycin A1-sensitive autophagy pathway. Autophagy 8: , 754–766. |
[134] | Tanik SA , Schultheiss CE , Volpicelli-Daley LA , Brunden KR , Lee VMY ((2013) ) Lewy body-like α-synuclein aggregates resist degradation and impair macroautophagy. J Biol Chem 288: , 15194–15210. |
[135] | Choubey V , Safiulina D , Vaarmann A , Cagalinec M , Wareski P , Kuum M , Zharkovsky A , Kaasik A ((2011) ) Mutant A53T α-Synuclein induces neuronal death by increasing mitochondrial autophagy. J Biol Chem 286: , 10814–10824. |
[136] | Chen L , Xie Z , Turkson S , Zhuang X ((2015) ) A53T Human α-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J Neurosci 35: , 890–905. |
[137] | Gao J , Perera G , Bhadbhade M , Halliday GM , Dzamko N ((2019) ) Autophagy activation promotes clearance of α-synuclein inclusions in fibril-seeded human neural cells. J Biol Chem 294: , 14241–14256. |
[138] | Chiang HL , Terlecky SR , Plant CP , Dice JF ((1989) ) A role for a 70-kilodalton heat shock protein in lysosomal degradation of intracellular proteins. Science 246: , 382–385. |
[139] | Cuervo AM , Dice JF ((1996) ) A receptor for the selective uptake and degradation of proteins by lysosomes. Science 273: , 501–503. |
[140] | Alvarez-Erviti L , Rodriguez-Oroz MC , Cooper JM , Caballero C , Ferrer I , Obeso JA , Schapira AHV ((2010) ) Chaperone-mediated autophagy markers in Parkinson disease brains. Arch Neurol 67: , 1464–1472. |
[141] | Alvarez-Erviti L , Seow Y , Schapira AHV , Rodriguez-Oroz MC , Obeso JA , Cooper JM ((2013) ) Influence of microRNA deregulation on chaperone-mediated autophagy and α-synuclein pathology in parkinson’s disease. Cell Death Dis 4: , e545–e548. |
[142] | Li G , Yang H , Zhu D , Huang H , Liu G , Lun P ((2014) ) Targeted suppression of chaperone-mediated autophagy by miR-320a promotes α-synuclein aggregation. Int J Mol Sci 15: , 15845–15857. |
[143] | Murphy KE , Gysbers AM , Abbott SK , Spiro AS , Furuta A , Cooper A , Garner B , Kabuta T , Halliday GM ((2015) ) Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov Disord 30: , 1639–1647. |
[144] | Rothaug M , Stroobants S , Schweizer M , Peters J , Zunke F , Allerding M , D’Hooge R , Saftig P , Blanz J ((2015) ) LAMP-2 deficiency leads to hippocampal dysfunction but normal clearance of neuronal substrates of chaperone-mediated autophagy in a mouse model for Danon disease. Acta Neuropathol Commun 3: , 6. |
[145] | Xilouri M , Brekk OR , Polissidis A , Chrysanthou-Piterou M , Kloukina I , Stefanis L ((2016) ) Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 12: , 2230–2247. |
[146] | Sevlever D , Jiang P , Yen S-HCHC ((2008) ) Cathepsin D is the main lysosomal enzyme involved in the degradation of α-synuclein and generation of its carboxy-terminally truncated species. Biochemistry 47: , 9678–9687. |
[147] | Qiao L , Hamamichi S , Caldwell KA , Caldwell GA , Yacoubian TA , Wilson S , Xie ZL , Speake LD , Parks R , Crabtree D , Liang Q , Crimmins S , Schneider L , Uchiyama Y , Iwatsubo T , Zhou Y , Peng L , Lu YM , Standaert DG , Walls KC , Shacka JJ , Roth KA , Zhang J ((2008) ) Lysosomal enzyme cathepsin D protects against alpha-synuclein aggregation and toxicity. Mol Brain 1: , 17. |
[148] | Cullen V , Lindfors M , Ng J , Paetau A , Swinton E , Kolodziej P , Boston H , Saftig P , Woulfe J , Feany MB , Myllykangas L , Schlossmacher MG , Tyynelä J ((2009) ) Cathepsin D expression level affects alpha-synuclein processing, aggregation, and toxicity in vivo. Mol Brain 2: , 1–17. |
[149] | Pastores GM , Hughes DA ((2000) ) Gaucher disease. In GeneReviews® [Internet], AdamMP, ArdingerHH, PagonRA, WallaceSE, eds. University of Washington, Seattle, WA. |
[150] | Sidransky E , Nalls MA , Aasly JO , Aharon-Peretz J , Annesi G , Barbosa ER , Bar-Shira A , Berg D , Bras J , Brice A , Chen C-M , Clark LN , Condroyer C , De Marco E V , Dürr A , Eblan MJ , Fahn S , Farrer MJ , Fung H-C , Gan-Or Z , Gasser T , Gershoni-Baruch R , Giladi N , Griffith A , Gurevich T , Januario C , Kropp P , Lang AE , Lee-Chen G-J , Lesage S , Marder K , Mata IF , Mirelman A , Mitsui J , Mizuta I , Nicoletti G , Oliveira C , Ottman R , Orr-Urtreger A , Pereira LV , Quattrone A , Rogaeva E , Rolfs A , Rosenbaum H , Rozenberg R , Samii A , Samaddar T , Schulte C , Sharma M , Singleton A , Spitz M , Tan E-K , Tayebi N , Toda T , Troiano AR , Tsuji S , Wittstock M , Wolfsberg TG , Wu Y-R , Zabetian CP , Zhao Y , Ziegler SG ((2009) ) Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med 361: , 1651–1661. |
[151] | Manning-Boǧ AB , Schüle B , Langston JW ((2009) ) Alpha-synuclein-glucocerebrosidase interactions in pharmacological Gaucher models: A biological link between Gaucher disease and parkinsonism. Neurotoxicology 30: , 1127–1132. |
[152] | Cleeter MWJ , Chau KY , Gluck C , Mehta A , Hughes DA , Duchen M , Wood NW , Hardy J , Mark Cooper J , Schapira AH ((2013) ) Glucocerebrosidase inhibition causes mitochondrial dysfunction and free radical damage. Neurochem Int 62: , 1–7. |
[153] | Xu YH , Sun Y , Ran H , Quinn B , Witte D , Grabowski GA ((2011) ) Accumulation and distribution of β-synuclein and ubiquitin in the CNS of Gaucher disease mouse models. Mol Genet Metab 102: , 436–447. |
[154] | Fishbein I , Kuo YM , Giasson BI , Nussbaum RL ((2014) ) Augmentation of phenotype in a transgenic Parkinson mouse heterozygous for a Gaucher mutation. Brain 137: , 3235–3247. |
[155] | Sardi SP , Clarke J , Viel C , Chan M , Tamsett TJ , Treleaven CM , Bu J , Sweet L , Passini MA , Dodge JC , Yu WH , Sidman RL , Cheng SH , Shihabuddin LS ((2013) ) Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc Natl Acad Sci U S A 110: , 3537–3542. |
[156] | Woodard CM , Campos BA , Kuo SH , Nirenberg MJ , Nestor MW , Zimmer M , Mosharov EV , Sulzer D , Zhou H , Paull D , Clark L , Schadt EE , Sardi SP , Rubin L , Eggan K , Brock M , Lipnick S , Rao M , Chang S , Li A , Noggle SA ((2014) ) IPSC-derived dopamine neurons reveal differences between monozygotic twins discordant for parkinson’s disease. Cell Rep 9: , 1173–1182. |
[157] | Schöndorf DC , Aureli M , McAllister FE , Hindley CJ , Mayer F , Schmid B , Sardi SP , Valsecchi M , Hoffmann S , Schwarz LK , Hedrich U , Berg D , Shihabuddin LS , Hu J , Pruszak J , Gygi SP , Sonnino S , Gasser T , Deleidi M ((2014) ) IPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat Commun 5: , 4028. |
[158] | Mazzulli JR , Xu YH , Sun Y , Knight AL , McLean PJ , Caldwell GA , Sidransky E , Grabowski GA , Krainc D ((2011) ) Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146: , 37–52. |
[159] | Gegg ME , Burke D , Heales SJR , Cooper JM , Hardy J , Wood NW , Schapira AHV ((2012) ) Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol 72: , 455–463. |
[160] | Bae EJ , Yang NY , Song M , Lee CS , Lee JS , Jung BC , Lee HJ , Kim S , Masliah E , Sardi SP , Lee SJ ((2014) ) Glucocerebrosidase depletion enhances cell-to-cell transmission of α-synuclein. Nat Commun 5: , 1–11. |
[161] | Bae EJ , Yang NY , Lee C , Lee HJ , Kim S , Sardi SP , Lee SJ ((2015) ) Loss of glucocerebrosidase 1 activity causes lysosomal dysfunction and α-synuclein aggregation. Exp Mol Med 47: , e153–e158. |
[162] | Wentink A , Nussbaum-Krammer C , Bukau B ((2019) ) Modulation of amyloid states by molecular chaperones. Cold Spring Harb Perspect Biol 11: , a033969. |


