
Modeling Parkinson’s Disease in C. elegans
Abstract
Parkinson’s disease (PD) is an adult onset neurodegenerative disease that is characterized by selective degeneration of neurons primarily in the substantia nigra. At present, the pathogenesis of PD is incompletely understood and there are no neuroprotective treatments available. Accurate animal models of PD provide the opportunity to elucidate disease mechanisms and identify therapeutic targets. This review focuses on C. elegans models of PD, including both genetic and toxicant models. This microscopic worm offers several advantages for the study of PD including ease of genetic manipulation, ability to complete experiments rapidly, low cost, and ability to perform large scale screens for disease modifiers. A number of C. elegans models of PD have been generated including transgenic worms that express α-synuclein or LRRK2, and worms with deletions in PRKN/pdr-1, PINK1/pink-1, DJ-1/djr-1.1/djr-1.2 and ATP13A2/catp-6. These worms have been shown to exhibit multiple phenotypic deficits including the loss of dopamine neurons, disruption of dopamine-dependent behaviors, increased sensitivity to stress, age-dependent aggregation, and deficits in movement. As a result, these phenotypes can be used as outcome measures to gain insight into disease pathogenesis and to identify disease modifiers. In this way, C. elegans can be used as an experimental tool to elucidate mechanisms involved in PD and to find novel therapeutic targets that can subsequently be validated in other models.
INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disorder affecting more than 10 million patients worldwide [1]. PD is a neurological movement disorder that is characterized by impaired balance, bradykinesia, rigidity, and the presence of resting tremors. In addition to deficits in movement, PD patients can also exhibit non-motor symptoms including depression, apathy, anxiety, dementia, constipation, disrupted sleep, and others. While rare cases of early onset PD have been described, PD is a disease of advanced age. The prevalence of PD is 0.3% among all ages, but increases to more than 3% in individuals over 80 years of age [2].
In the brain, PD patients exhibit progressive degeneration of dopaminergic neurons in the substantia nigra, although many other regions of the brain are also affected. Neuronal loss within the substantia nigra decreases dopamine signaling to the striatum thereby contributing to the motor symptoms of PD. At the cellular level, the disease is characterized by intracellular aggregation of a protein called α-synuclein into Lewy bodies, which are observed in the brains of patients with PD [3]. There are currently no neuroprotective treatments available for PD and the pathogenesis of the disease is incompletely understood.
While PD was traditionally thought of as an entirely idiopathic disorder, work in the past two decades has demonstrated that genetics make a significant contribution to the disease. Currently, it is estimated that 15% of all PD cases are familial. The first gene that was shown to cause monogenic PD is SNCA/PARK1, which encodes α-synuclein [4]. There are now 23 chromosomal regions that are defined as PARK to signify their connection to PD. These regions are numbered chronologically by the order of their discovery, either by linkage analysis of families or by genome wide association studies in large populations. Of the genes definitively linked to heritable PD, mutations in SNCA/PARK1/PARK4 and LRRK2/PARK8 [5, 6] are known to be the cause of autosomal-dominant PD, while mutations in PRKN/PARK2 [7], PINK1/PARK6 [8], DJ-1/PARK7 [9], and ATP13A2/PARK9 [10] are responsible for autosomal-recessive forms of disease. While monogenic forms of PD are relatively rare, the etiology of disease for the most common forms of genetic PD share many similarities with sporadic cases, suggesting that similar cellular pathways are involved in both cases. Thus, with each new gene that is implicated in PD comes the opportunity to learn more about the cellular pathways involved in disease pathogenesis.
C. ELEGANS AS AN ANIMAL MODEL OF PARKINSON’S DISEASE
The discovery of genes that cause monogenic forms of PD allowed for the generation of genetic models of PD in many different species, including C. elegans. Studying the biology of a particular gene implicated in PD in animal models can provide insight into the molecular pathways involved in the human disease. Animal models can also be used to identify biomarkers of disease and to test potential treatments. While models of PD have been generated in multiple species from single cellular yeast to monkeys, this review will focus on C. elegans models of PD.
The nematode C. elegans is a microscopic roundworm that grows to be 1-2 mm in length as adults. After hatching, these animals develop to adulthood in just 2 days under laboratory conditions at 20°C. Once these worms reach adulthood, their average lifespan is 2-3 weeks, making them useful for studies of aging. C. elegans exist primarily as a self-fertilizing hermaphrodite, in which all of the progeny are genetically identical. Males exist as a small fraction of the population (<0.1%) but their numbers can be greatly increased in the laboratory to facilitate genetic crosses. This animal is genetically tractable with robust tools for spatiotemporal control of gene expression and a highly annotated genome. Because C. elegans are transparent, fluorescent proteins can be readily visualized in a live worm to measure levels and location of gene products of interest. These animals have been utilized to address a variety of cellular and genetic questions [11] and specifically to gain insight into neurodegenerative disease [12].
C. elegans have a well-defined, invariant nervous system with exactly 302 neurons in each hermaphrodite out of a total of 959 cells in the organism. Unlike any other organism, all of the connections of all 302 neurons in C. elegans have been mapped using electron micrographs thereby providing the most complete nervous system connectome of any organism [13]. Importantly, these neurons encode complex behaviors, which, in several cases, have been described at the level of a single neuron [14–17]. Such behaviors include chemotaxis, thermotaxis, touch response, mating rituals, social and individual feeding, and scavenging as well as associative and non-associative learning [18–22].
EXPERIMENTAL TOOLS FOR C. ELEGANS
Genetics
One of the greatest advantages of C. elegans as a model organism is the ease of genetic manipulation and the wide array of genetic tools available. Transgenic animals can be easily generated in less than a month through microinjection or microparticle bombardment. Deletion and point mutants are readily available from the Caenorhabditis Genetics Center, which is a central repository for C. elegans mutant strains that currently has over 20,000 strains available. Importantly, generating double mutants to examine the effect of one gene on another is greatly facilitated by the fact that the worms are hermaphrodites because only one cross is required to combine two mutations in one worm, which can then be selfed to generate worms homozygous for both mutations. Once a homozygous double mutant animal is produced, each hermaphrodite will produce up to 300 offspring.
C. elegans have 6 chromosomes and approximately 20,000 genes. The C. elegans genome was the first genome of a multi-cellular organism to be completely sequenced. About 40–50% of protein coding genes have orthologs in humans and it is estimated that 60–80% of human disease causing genes have orthologs in C. elegans [23–25]. Importantly for the study of PD, C. elegans has orthologs to many of the genes implicated in PD including LRRK2/lrk-1, PINK1/pink-1, PRKN/pdr-1, DJ-1/djr-1.1/djr-1.2, and ATP13A2/catp-6.
RNA interference
The precision and adaptability of RNAi in C. elegans makes it a particularly powerful tool. Unlike most animals, in C. elegans RNAi can be administered by simply feeding worms bacteria engineered to express a double stranded RNA targeting a gene of interest. Remarkably, in these animals, RNAi is heritable for up to three or more generations [26, 27]. In C. elegans, there are several methods that can be utilized in order to silence gene expression using RNAi: the dsRNA can be delivered via injection into any tissue [28]; by feeding bacteria expressing the dsRNA [29]; by soaking animals in a solution containing dsRNA [30]; or by production of dsRNA via a transgene for in vivo knock down that can be conditionally controlled [31].
While these methods are sufficient to mediate gene silencing in most tissues, it has been shown that specific tissues, such as neurons, can be less sensitive to RNAi, depending on the method of RNAi delivery [32–34]. Nonetheless, strains with enhanced sensitivity to RNAi, such as rrf-3 or eri-1, have been identified to circumvent this limitation [35, 36]. Alternatively, it has been shown that the transgenic expression of the dsRNA transporter SID-1 in a tissue of interest can sensitize that tissue to RNAi [37]. Tissue-specific knockdown of a gene of interest can be accomplished by expressing a necessary component of the endogenous RNAi machinery in a mutant that is deficient in that component (e.g., the Argonaute protein RDE-1 can be expressed just in the intestine using an intestine specific ges-1 promoter in rde-1 mutant animals to achieve intestine-specific knockdown of a gene of interest when worms are exposed to an RNAi targeting that gene). Using this approach, strains have been constructed in which a gene of interest can be knocked down specifically in GABAergic neurons [38], serotonergic neurons [39], dopaminergic neurons [40], cholinergic neurons [40], glutamatergic neurons [40] or pan-neuronally [37].
Screening for disease modifiers
One of the biggest advantages of using a simple genetic model organism for the study of Parkinson’s disease is the ability to perform large-scale screens. These screens are enabled by the small size, ease of maintenance, affordability, and the large brood size of C. elegans. Screening for disease modifiers can be done using a chemical/compound screen, a classical forward genetic screen [41], or an RNAi interference screen [42]. In fact, RNAi libraries covering nearly the entire genome are available commercially, including the Ahringer library which contains bacterial clones with 17,575 genes [43] and the Vidal RNAi library that includes open reading frame targeting clones of 11,800 genes [44]. A number of groups have performed screens for disease modifiers in C. elegans models of PD using various outcome measures including aggregation and neurodegeneration (Table 1).
Table 1
Summary of screens performed in C. elegans models of Parkinson’s disease
| Type of Screen | Model | Outcome measure | Comments | Reference |
| Compound | MPP+ | Mobility | Proof-of-principle that MPP+ model could be used to screen for modifiers of mobility | Braungart et al., 2004 [100] |
| Compound | 6-OHDA | Neuron loss | D2 receptor agonists bromocriptine and quinpirole are neuroprotective | Marvanova et al., 2007 [101] |
| Compound | Pdat-1:α-syn | Neuron loss | Combined screening with yeast and worms, genes responsible for intracellular trafficking and mitochondrial function were found to be neuroprotective | Su et al., 2010 [140] |
| Genetic | 6-OHDA | Neuron loss | Mutations in dat-1 dopamine transporter gene are neuroprotective | Nass et al., 2005 [141] |
| RNAi | Punc-51:α-syn | Uncoordinated movement, growth retardation | Knockdown of genes in endocytic pathway exacerbate α-syn toxicity | Kuwahara et al., 2008 [70] |
| RNAi | Punc-54:α-syn:YFP | α-synuclein aggregation | Identified genes that increase or decrease aggregation when knocked down | Van Ham et al., 2008 [58] |
| RNAi | Punc-54:α-syn:GFP, Punc-54::tor-2 | α-synuclein aggregation | Homologs of PD-causing genes increase aggregation | Hamamichi et al., 2008 [73] |
| RNAi | Punc-54:α-syn:YFP | Uncoordinated movement | Kynurenine pathway and serotonin production play an important role in regulating protein homeostasis | Van der Goot et al., 2012 [74] |
| RNAi | Punc-54:α-syn:GFP | α-synuclein aggregation | Targeting upregulated genes in Insulin/IGF mutants that reduce α-syn aggregation | Knight et al., 2014 [138] |
| RNAi | Punc-54:α-syn:YFP | α-synuclein aggregation | Knockdown of neuroprotective genes also increased α-syn aggregation as well as causing changed motility, mitochondrial content, and ROS production | Jadiya et al., 2015 [142] |
MPP+, 1-methyl-4-phenylpyridinium; 6-OHDA, 6-hydroxydopamine; RNAi, RNA interference.
OUTCOME MEASURES USED TO IDENTIFY DISEASE MODIFIERS
In order to screen for disease modifiers it is necessary to have quantifiable phenotypic deficits that can be used as outcome measures. Although C. elegans is a simple organism, there are a number of assays available to assess the toxicity of PD-related defects including very specific dopamine-dependent behaviors.
Survival of dopamine neurons
Because loss of dopamine neurons is a hallmark of human PD, the primary outcome measure in many C. elegans studies is the survival of dopamine neurons (Fig. 1). While staining for neurons is possible in fixed tissues, due to C. elegan’s transparent cuticle, neuronal survival is normally assessed in live animals by expressing a fluorescent protein specifically in dopamine neurons using the promoter from the dat-1 dopamine transporter gene. In C. elegans hermaphrodites there are 8 dopamine neurons (out of 302 total neurons) including 6 anterior (four CEP neurons, two ADE neurons) and 2 posterior (PDE neurons). Male C. elegans possess 6 additional dopaminergic neurons in the tail ray. In addition to the loss of neuron cell bodies, quantification of more subtle phenotypes is possible. This includes the disappearance of axons, broken neurites, retreat of dendritic terminals, and axonal and dendritic blebbing [45–49].
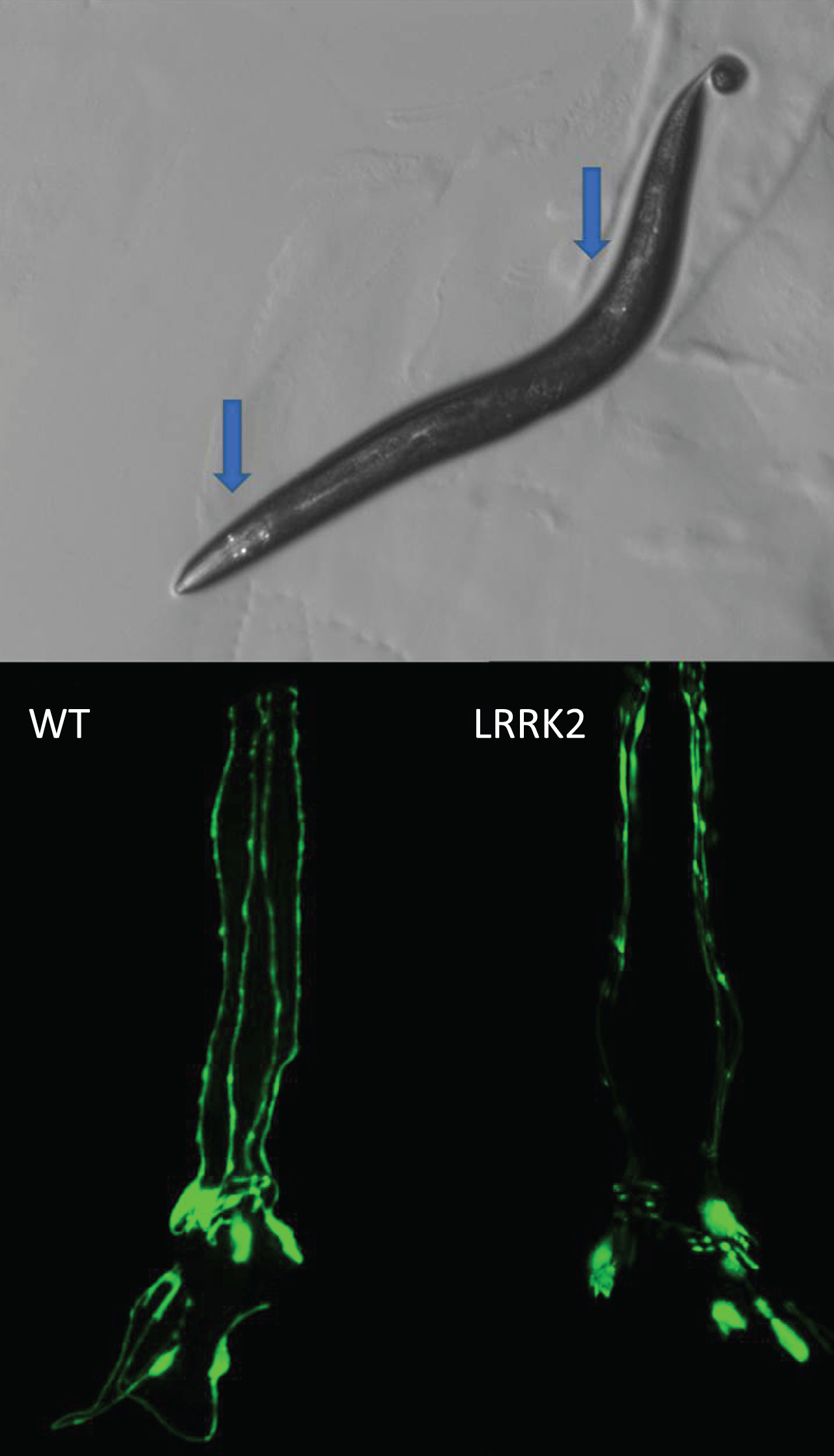
Fig.1
Loss of dopamine neurons. C. elegans hermaphrodites have eight dopamine neurons that can be visualized in live worms by expressing a fluorescent protein such as GFP with a dopamine neuron specific promoter such as dat-1. There are six dopamine neurons in the head and two posterior, as indicated by the blue arrows (top). The progressive loss of dopamine neurons can be monitored throughout the lifespan of the worm. Expression of human mutant LRRK2 with G2019S mutation causes an accelerated loss of dopamine neurons (bottom). Bottom panels show only the head region of the worm with the tip of the nose facing the top of the page.
Dopamine-dependent behaviors
Basal slowing
The dopaminergic circuit has been found to directly affect several behaviors in C. elegans. Basal slowing is a feeding behavior in which the rate of locomotion changes depending upon whether the animal is in the presence or absence of the bacterial lawn that serves as its food source in the laboratory due to a mechanosensory response. Normally animals crawl more slowly in the presence of bacterial food than when there is no food present, but the disruption of dopamine signaling prevents the animal’s ability to slow in the presence of food, rendering a higher crawling speed [22]. The basal slowing ratio can be calculated by determining the difference between crawling speed on and off food, then determining the ratio by dividing by speed off food:
basal slowing = ([rate of movement absence of food – rate of movement presence of food]) / rate of movement presence of food
Ethanol preference
C. elegans can sense environmental chemical cues that cause attractive or repulsive chemotaxis [50]. Chemotaxis assays are assessed by dividing an agar plate into quadrants and seeding two quadrants with the compound in question. Worms are then transferred to the test plate and allowed to freely explore. To assess the effect of the compound, the animals in the seeded and control quadrants are quantified and a preference index (PI) is calculated:
Under laboratory conditions C. elegans avoid ethanol [51, 52] and this is dependent on a functional dopamine signaling system [53]. A similar effect is observed using the compound nonanol, which induces a strong negative chemotaxis response that is dependent on functional dopaminergic circuity [54, 55].
Area-restricted searching (ARS)
ARS is a foraging strategy in which C. elegans prioritize unexplored areas over those that have run out of food. When a worm is searching for food on an empty plate, they will turn frequently at sharp angles, containing their search to a confined area. As this behavior becomes unsuccessful at finding food, the search area is expanded, the number of turns decreases, and the scavenging behavior then consists of wide arcs that increase the efficiency of foraging. Dopamine signaling is involved in the successful switching of strategies [21]. ARS is evaluated by transferring animals to an agar plate without food. Video recordings are then taken 5 minutes after transfer and again after 30 minutes without food. Analysis of the videos then allow for the frequency of turns greater than 90° at each time point.
Swimming induced paralysis (SWIP)
While healthy, well fed animals initially thrash vigorously in liquid, after thrashing for about 6 minutes, C. elegans become paralyzed exhibiting a phenotype called SWIP. Animals with impaired dopaminergic function, such as tyrosine hydroxylase mutants, do not exhibit this behavior [56]. This suggests that endogenous dopamine is responsible for inactivation of the motor circuitry necessary for thrashing behavior. However, while SWIP is readily observed in animals at the L4 stage of development, it becomes less prominent with age.
Accumulation of α-synuclein
As the aggregation of α-synuclein is a key feature of PD, C. elegans models have been developed in which human SNCA is expressed in order to study α-synuclein accumulation. To visualize α-synuclein aggregation in live worms, α-synuclein has been linked to a fluorescent protein (e.g., GFP or YFP) and typically expressed in body wall muscle to facilitate visualization [57, 58] (Fig. 2). While neuronal α-synuclein aggregates can be visualized, due to the small size of nematode neurons, quantifiable intracellular inclusions can only be resolved in fixed tissue [59], which is not suitable large scale or screening analysis.
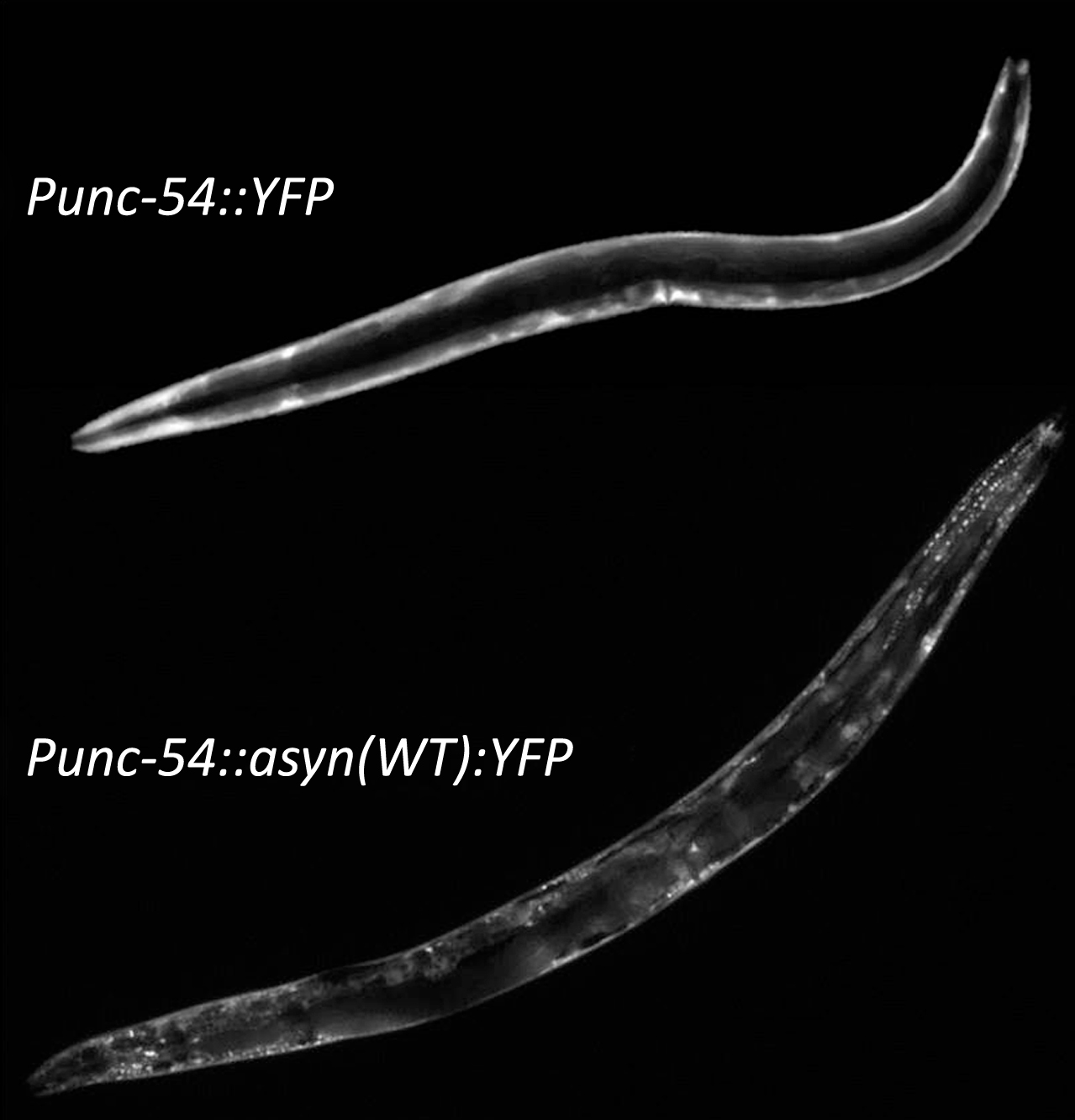
Fig.2
Aggregation of α-synuclein. Although worms do not possess an ortholog of α-synuclein it is possible to study α-synuclein by expressing α-synuclein linked to a fluorescent proteion, such as YFP, with a body wall muscle specific promoter such as unc-54. While expression of YFP alone results in diffuse expression throughout the body wall muscle, the presence of α-synuclein results in the formation of aggregates.
Movement: Thrashing and crawling
Because motor deficits are a major component of Parkinson’s disease, multiple assays are utilized to measure movement [49, 60]. The rate of movement in liquid, also known as the thrashing rate, is counted as the number of body bends per unit time and can be assessed manually on individual worms or using video-tracking on entire populations [61]. Crawling speed on solid plates is measured as distance travelled in a given amount on time.
Mitochondrial morphology and function
As deficits in mitochondrial function have been implicated in the pathogenesis of PD, another important outcome measure that is examined in C. elegans models is mitochondrial morphology and function, especially since multiple genes that cause PD are directly involved in mitochondrial function (PRKN/pdr-1, PINK1/pink-1, DJ-1/djr-1.1). Mitochondrial morphology can be visualized in live worms by utilizing transgenic animals that express a fusion protein of a mitochondrially-targeted protein (e.g., TOMM-20) and a fluorescent protein (e.g., GFP or YFP) [62–64] (Fig. 3). Using this technique, disruptions in mitochondrial morphology have been discovered in pdr-1, pink-1 and djr-1.1 mutants [53]. A basic assessment of mitochondrial function can be achieved through measurements of oxygen consumption and ATP levels. Oxygen consumption can be measured in individual or small numbers of samples using a Clark electrode or in multiple samples using a Seahorse Extracellular Flux Analyzer, while ATP can be measured from lysed worms using commercially available lucigenin based quantification kits.
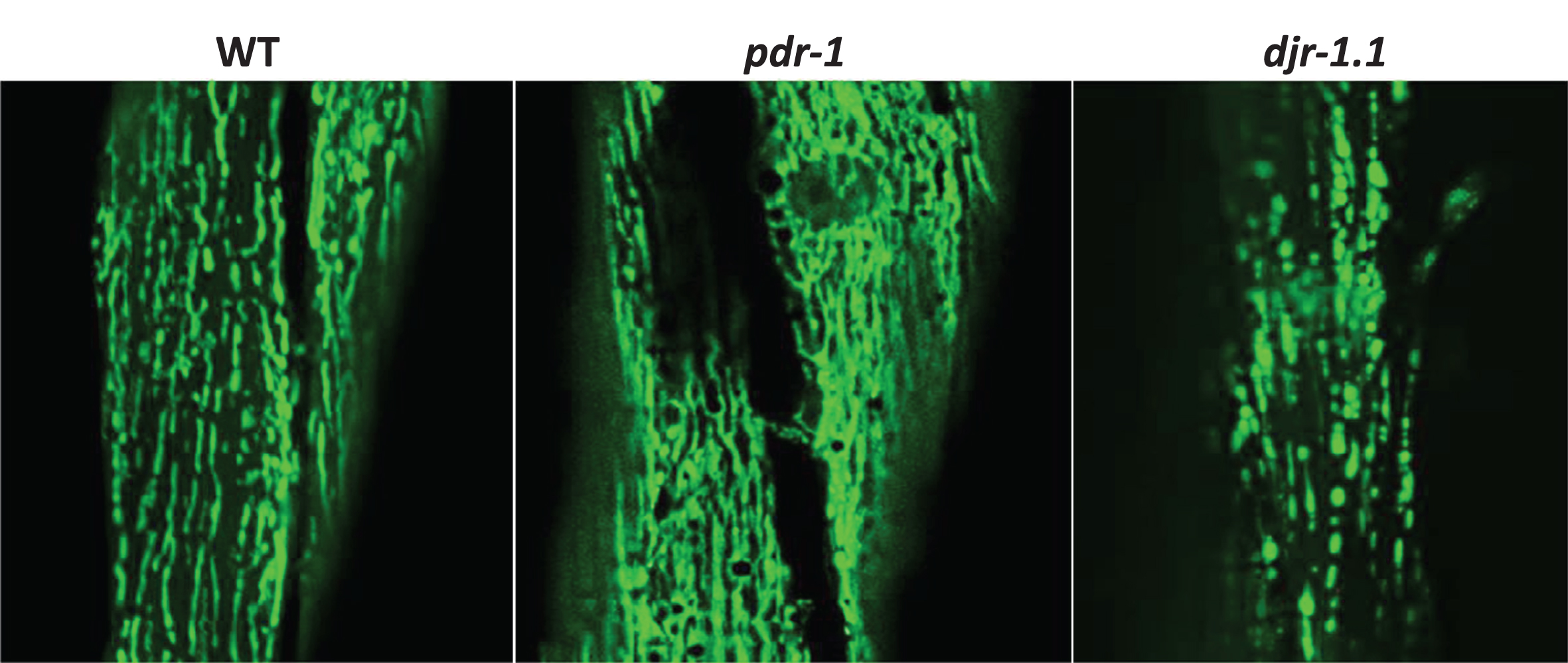
Fig.3
Mitochondrial morphology. The morphology of the mitochondria can be monitored in live worms by expressing a mitochondrially-targeted fluorescent protein, such as GFP, under tissue-specific promoters, such as myo-3 for body wall muscle. In wild-type worms, mitochondria in the body wall muscle exist as parallel tracks of elongated mitochondria. pdr-1 mutants exhibit increased accumulation of mitochondria, while djr-1.1 mutants exhibit mitochondrial fragmentation.
Resistance to stress
Multiple studies have examined resistance to stress in C. elegans models of PD and observed that the genetic defects that cause PD increase susceptibility to various stresses [53, 60, 65]. Interestingly, heat stress, oxidative stress and osmotic stress all act to increase protein aggregation (Cooper and Van Raamsdonk, unpublished data). The connection between increased sensitivity to stress, decreased proteostasis and the aggregation of α-synuclein has yet to be explored.
Overall, there are a number of different outcome measures that can be used to quantify phenotypic deficits in worm models of PD. Of all these measures, we think that loss of dopamine neurons is the most reliable and disease-relevant assay, and this is also the most frequently reported outcome in the literature. Note that it is important to combine this assay with tests of neuronal function to ensure that any increases in neuronal number that are associated with a therapeutic intervention also preserve neuronal function. For this purpose, we have found the basal slowing assay to be the most robust, and this is also the most commonly used assay in published studies. Finally, it should be noted that most of the tests of dopamine-dependent behavior rely on crawling. As a result, it is important to test movement as deficits in movement can be a confounding factor in these assays.
C. ELEGANS MODELS OF PARKINSON’S DISEASE
SNCA
SNCA/PARK1/PARK4, the first gene to be definitively linked to familial PD [4], encodes α-synuclein, a protein involved in synaptic vesicle formation. Mutations in SNCA and duplications or triplications of the gene have been shown to cause autosomal dominant forms of PD [66, 67]. While C. elegans does not have a homolog to SNCA, a number of worm models have been generated by expressing human wild-type or mutant α-synuclein either ubiquitously or in specific tissues (Table 2). Expression of α-synuclein in all neurons or specific populations of neurons causes loss of dopamine neurons, deficits in dopamine dependent behavior, and decreased levels of dopamine [49, 59, 60, 68–72]. In order to study aggregation, α-synuclein has been linked to YFP or GFP and expressed in body wall muscle cells for ease of visualization [58, 73, 74]. In addition to aggregation, these worms show deficits in movement and increased sensitivity to stress (Cooper and Van Raamsdonk, unpublished data). Since worms do not normally express α-synuclein, worm models have also been generated that ubiquitously express wild-type α-synuclein from a single copy transgene (Cooper and Van Raamsdonk, unpublished data).
Table 2
Transgenic worm models of Parkinson’s disease
| Expression pattern | Transgene | Phenotypes | Reference |
| α-synuclein models | |||
| Dopamine neurons | Pdat-1::asyn | ↑DA neuron loss, ↓DA, asyn accumulation | Lakso et al., 2003 [49]; Cao et al., 2005 [68]; Kuwahara et al., 2006 [59] |
| Pdat-1::asyn (A53T) | ↑DA neuron loss, ↑DA neurite defects, ↓DA, asyn accumulation, movement deficit, ↓basal slowing, ↓ARS, ↓ethanol avoidance | Lakso et al., 2003 [49]; Kuwahara et al., 2006 [59]; Kuwahara et al., 2008 [70]; Cooper et al., 2015 [60] | |
| Pdat-1::asyn (A30P) | ↑DA neuron loss, ↑DA neurite defects, ↓DA, asyn accumulation, ↓basal slowing | Kuwahara et al., 2006 [59]; Karpinar et al., 2009 [143] | |
| Pdat-1::asyn (A56P) | ↑DA neurite defects, ↓basal slowing | Karpinar et al., 2009 [143] | |
| Motor neurons | Pacr-2::asyn | Movement deficit | Lakso et al., 2003 [49] |
| Pacr-2::asyn (A53T) | Movement deficit | Lakso et al., 2003 [49] | |
| Punc-30::asyn | Movement deficit | Lakso et al., 2003 [49] | |
| Punc-30::asyn (A53T) | Movement deficit | Lakso et al., 2003 [49] | |
| Mechanosensory neurons | Pmec-7::asyn | Impaired touch response | Kuwahara et al., 2008 [70] |
| Pmec-7::asyn (A53T) | Impaired touch response | Kuwahara et al., 2008 [70] | |
| Pan-neuronal | Paex-3::asyn | ↑DA neuron loss, movement deficit | Lakso et al., 2003 [49] |
| Paex-3::asyn (A53T) | ↑DA neuron loss, movement deficit | Lakso et al., 2003 [49] | |
| Punc-51::asyn | Movement deficit, growth defect, impaired touch response | Kuwahara et al., 2008 [70] | |
| Punc-51::asyn (A30P) | Movement deficit, growth defect, impaired touch response | Kuwahara et al., 2008 [70] | |
| Punc-51::asyn (A53T) | Movement deficit, growth defect, impaired touch response | Kuwahara et al., 2008 [70] | |
| Body wall muscle | Punc-54::asyn:GFP | Movement deficit, asyn accumulation, asyn aggregation | Hamamichi et al., 2008 [73] |
| Punc-54::asyn:YFP | Movement deficit, asyn accumulation, asyn aggregation, ↓stress resistance | van Ham et al., 2008 [58]; Cooper and Van Raamsdonk, unpublished data | |
| Ubiquitous | Peft-3::asyn:RFP | asyn accumulation, axon blebbing, ↓basal slowing, ↓stress resistance | Cooper and Van Raamsdonk, unpublished data |
| LRRK2 models | |||
| Dopamine neurons | Pdat-1::LRRK2 | ↑DA neuron loss, ↓DA, movement deficit | Yao et al., 2010 [76]; Cooper et al., 2015 [60] |
| Pdat-1::LRRK2 (G2019S) | ↑DA neuron loss, ↓DA, movement deficit, ↓basal slowing, ↓ARS, ↓ethanol avoidance | Yao et al., 2010 [76]; Liu et al., 2011 [78]; Yao et al., 2013 [77]; Cooper et al., 2015 [60] | |
| Pdat-1::LRRK2 (G2019S/D1994A) | ↑DA neuron loss compared to WT, but ↓DA neuron loss compared to G2019S | Liu et al., 2011 [78] | |
| Pdat-1::LRRK2 (R1441C) | ↑DA neuron loss, ↓DA, movement deficit, ↓basal slowing | Yao et al., 2010 [76]; Yao et al., 2013 [77] | |
| Pan-neuronal | Psnb-1::LRRK2 | ↑DA neuron loss | Saha et al., 2009 [75] |
| Psnb-1::LRRK2 (R1441) | ↑DA neuron loss, ↓DA | Saha et al., 2009 [75] | |
| Psnb-1::LRRK2 (G2019S) | ↑DA neuron loss, ↓DA | Saha et al., 2009 [75] |
DA, dopamine; ARS, area-restricted searching; asyn, α-synuclein.
LRRK2/LRK-1
Mutations in the leucine rich repeat kinase 2 gene LRRK2 have also been shown to cause autosomal dominant PD [6]. lrk-1, the C. elegans homolog of LRRK2, is expressed broadly in these animals, including neurons, in which it associates with the Golgi apparatus [65]. Expression of WT or mutant LRRK2 either pan-neuronally using the synaptobrevin promoter [75] or specifically in dopamine neurons using the dopamine transporter promoter [60, 76, 77] decreases dopamine levels, induces deficits in dopamine-dependent behaviors and causes a progressive loss of dopamine neurons (Table 2). Extrachromosomal arrays expressing LRRK2 in dopamine neurons have also shown that kinase activity of LRRK2(G2019S) is important for the development of age-dependent neurodegeneration [78].
PRKN/PDR-1
Mutations in PRKN/PARK2 cause an autosomal recessive form of PD with an early age of onset [7, 79]. Parkin, which is encoded by pdr-1 in C. elegans, encodes an E3 ubiquitin ligase involved in protein degradation and mitophagy [80]. p dr-1 mutants exhibit a loss of dopamine neurons [81], and deficits in the dopamine-dependent behaviors (Table 3) [53, 82]. In addition, pdr-1 mutants have been shown to have increased sensitivity to various stresses [53, 64, 83]. At a cellular level, an in frame deletion in pdr-1 causes the protein to aggregate and increases sensitivity to proteotoxic stress [71]. Finally, mutations in pdr-1 have been shown to cause the accumulation of dysfunctional mitochondria [53, 64] and deficiencies in oxidative phosphorylation [53, 84], which are associated with activation of the mitochondrial unfolded protein response [53]. Perhaps as a means of jettisoning their accumulating mitochondria, pdr-1 mutant animals also produce more exophers than wild-type worms [85].
Table 3
Genetic loss of function C. elegans models of Parkinson’s disease
| Human gene | C. elegans gene | Phenotypes | Reference |
| PRKN | pdr-1 | ↑DA neuron loss, ↓basal slowing, ↓ethanol avoidance, ↓lifespan, mitochondrial accumulation, ↓stress resistance | Springer et al., 2005 [71]; Ved et al., 2005 [102]; Bornhorst et al., 2014 [81]; Chakraborty et al., 2015 [82]; Kirienko et al., 2015 [83]; Palikaras et al., 2015 [64]; Luz et al., 2015 [84]; Cooper et al., 2017 [53] |
| PINK1 | pink-1 | ↓basal slowing, mitochondrial accumulation, ↓stress resistance | Samann et al., 2009 [65]; Kirienko et al., 2015 [83]; Palikaras et al., 2015 [64]; Luz et al., 2015 [84]; Cooper et al., 2017 [53] |
| DJ-1 | djr-1.1 | ↓stress resistance, mitochondrial fragmentation | Lee et al., 2012 [93]; Cooper et al., 2017 [53] |
| djr-1.2 | ↓stress resistance | Lee et al., 2012 [93] | |
| ATP13A2 | catp-6 | ↑DA neuron loss, ↓basal slowing, movement deficit, ↓stress resistance | Cooper and Van Raamsdonk, unpublished data |
DA, dopamine.
PINK1/PINK-1
Mutations in PINK1/PARK6 cause an autosomal recessive form of PD with early disease onset [8, 86]. PINK1 (PTEN-induced putative kinase 1) is a mitochondrial kinase that acts with Parkin in mitophagy [87, 88] and is also first identified ubiquitin kinase [89]. Mutations in the PINK1 homolog pink-1 result in increased sensitivity to multiple stresses [64, 65, 83]. While pink-1 worms show a significant reduction in basal slowing, these mutants exhibit a wild-type survival of dopamine neurons [53]. The mitochondria of pink-1 worms accumulate with age, show altered morphology and have deficiencies in oxidative phosphorylation [53, 65, 84]. As in pdr-1 mutants, worms with dysfunctional pink-1 also produce significantly more exophers in neurons expressing toxic proteins [85].
DJ-1/DJR-1.1,DJR-1.2
Mutations in the DJ-1/PARK7 gene cause a recessive form of PD with an early age of onset [90]. DJ-1 is a deglycase that has been shown to protect against oxidative stress [91, 92]. There are two orthologs of DJ-1 in C. elegans, djr-1.1, which is expressed primarily in the intestine, and djr-1.2, which is expressed primarily in neurons [93]. Deletion of either ortholog (or both together) does not cause the loss of dopamine neurons or significantly impact dopamine-dependent behaviors [53]. Nonetheless, djr-1.1 mutants show increased sensitivity to oxidative stress, mitochondrial fragmentation and a decreased ability to generate energy [53, 93].
ATP13A2/CATP-6
Mutations in ATP13A2/PARK9 cause an atypical form of early-onset parkinsonism called Kufor-Rakeb Syndrome [10, 94]. This gene encodes a lysosomal P-type ATPase transporter. Loss of ATP13A2 function leads to enhanced oxidative stress, protein misfolding, and aggregation due to deficiencies within the lysosome [94]. Mutations in catp-6 causes a number of deficits including accelerated loss of dopamine neurons, deficits in dopamine-dependent behavior, decreased rate of movement and increased sensitivity to multiple stresses (Cooper and Van Raamsdonk, unpublished data). Levels of CATP-6 have been shown to influence the accumulation of α-synuclein [95].
TOXICANT MODELS
In addition to genetic models, a number of groups have used neurotoxins to induce dopaminergic cell death as a model of PD [45, 96, 97]. Treating worms with MPP+ (1-Methyl-4-phenylpyridinium), the active metabolite of MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine), has been shown to cause deficits in movement and dopamine neuron loss [98–100]. Similarly, treating worms with the dopamine analog 6-hydroxydopamine (6-OHDA), Manganese (Mn), or methylmercury also causes degeneration of dopamine neurons [45, 48, 101]. The insecticide rotenone and the herbicide paraquat have also been used to induce behavioral deficits and neurodegeneration in C. elegans. These compounds cause toxicity by increasing ROS leading to cellular toxicity, damage and disruption of neuronal morphology [45, 46, 97, 105–107].
Overall, there are a large variety of C. elegans models of PD to choose from when designing an experiment. The best model to utilize depends on the experimental question of interest. Of the genetic models, those expressing mutant α-synuclein or LRRK2 in dopamine neurons provide the most robust loss of dopamine neurons and dopamine-dependent behaviors. These models would be most appropriate for the development of neuroprotective strategies. pdr-1, pink-1 and djr-1.1 mutants have subtler phenotypic deficits making them less ideal for therapeutic studies, but can be used to identify factors contributing to disease (e.g., identification of synthetic lethal interactions).
CONSERVATION ACROSS SPECIES
An important premise for studying PD in animal models is that the findings obtained in the animal model are also observed in PD. There have been multiple examples in which findings from C. elegans have been shown to be conserved across species and for the purpose of this review we will highlight only a few of these.
In one study examining the neuroprotective properties of ATP13A2/CATP-6, it was initially shown that the yeast homolog of ATP13A2 could protect against α-synuclein toxicity in a colony growth assay [108]. It was subsequently shown that overexpression of CATP-6 in C. elegans decreases α-synuclein-mediated neuronal loss, while catp-6 RNAi causes increased aggregation of α-synuclein [95]. Finally, this same phenomenon was examined in a mammalian system. In rat primary midbrain neurons, it was shown that expression of ATP13A2 could protect against α-synuclein(A53T) toxicity [95]. Thus, the ability of ATP13A2/CATP-6 to protect against α-synuclein toxicity is conserved from yeast to worm to rodent.
Similarly, another group examined the interaction between glucocerebrosidase (GBA) and α-synuclein. Mutations in GBA1 are the most common genetic risk factor for PD: about 10% of people with PD have mutations in GBA1. Glucocerebrosidase acts to degrade glycolipids and homozygous mutations in GBA1 lead to the development of a lysosomal storage disorder called Gaucher disease. Based on the observation of α-synuclein positive Lewy bodies in patients with Gaucher disease [109], Mazzulli et al. explored the relationship between GBA and α-synuclein. They found that knocking down GBA is primary cortical neurons or human iPS neurons resulted in increased levels of α-synuclein [110]. Similarly, RNAi against gba-2 caused increased aggregation of α-synuclein in C. elegans, while a mouse model of Gaucher disease also showed increased α-synuclein accumulation [110].
It is important to note that the corroboration of findings between organisms is not always observed and validating the results from one species in another can be complicated by the absence of an ortholog and deciding which assay to use. In a recent study seeking to prioritize loss of function variants that were found by whole exome sequencing in 1148 PD patients, only 10 of the 27 genes identified had an ortholog in C. elegans [111]. The effect of 9 of these genes on developmental lethality and survival was tested and it was found that 3 of the genes exhibited a detrimental effect. Interestingly, two of those genes, DIS3 and KALRN, were known to also reduce viability in Drosophila [112, 113] and rodents [114, 115]. The fact that 6 of the 9 genes did not reduce survival does not exclude the possibility that these genes contribute to PD, since most worm models of PD exhibit normal longevity. It is possible that knocking down these genes would impact dopamine neuron survival or dopamine-dependent behavior but these outcomes were not assessed.
ENVIRONMENT FACTORS CONTRIBUTING TO PD AND GENE-ENVIRONMENT INTERACTIONS
While monogenetic forms of PD facilitate studying the molecular mechanisms underlying the disease, most cases of PD likely result from a complex interaction of genetic and environmental risk factors [116]. Work in C. elegans has been able to connect exposure to environmental toxins such as pesticides, Mn or methylmercury to the degeneration of dopamine neurons [102–104]. Interestingly, it has also been shown that specific bacterial metabolites can also contribute to neurotoxicity in dopamine neurons [117–119]. The availability of genetic models of PD permits studying gene-environment interactions in C. elegans. For example, it was shown that genes implicated in PD (SNCA, pdr-1, djr-1.1) cause increased susceptibility to mitochondrial complex I inhibitors, such as rotenone [102]. Similarly, others have examined the effect of PD-causing mutations on Mn toxicity [81, 120] and showed that exposure to pesticides can exacerbate α-synuclein aggregation [121].
DRUG SCREENING AND DISCOVERY
C. elegans models of PD can also be used as a tool for drug discovery. Compounds can be added to the solid agar plates on which the worms are maintained, or worms can be grown in liquid culture and the compounds added directly to the culture medium. While worms have a thick cuticle that can limit the ability of compounds to enter the worm, genetically modified strains are available with increased drug permeability [122]. Previous work has examined the beneficial effect of specific compounds in genetic and toxicant models of PD, including acetaminophen [123] and valproic acid [124]. In some cases, the compounds investigated are specific to certain genotypes, such as testing kinase inhibitors in LRRK2 mutants [77, 78]. In addition, multiple studies have performed targeted drug screens most commonly for compounds that protect against neurotoxin induced deficits [100–102].
RECENT ADVANCES IN PARKINSON’S DISEASE RESEARCH IN C. ELEGANS
C. elegans models to dissect the mechanism of cell-to-cell transfer of α-synuclein
Recent work has suggested that the cell-to-cell transfer of α-synuclein may be a key step in the pathogenesis of PD [125]. While the transfer of α-synuclein has been demonstrated in multiple model systems [126, 127] and importantly in neurons grafted into PD patients [128, 129], the mechanisms involved have yet to be elucidated. To explore the underlying mechanisms of α-synuclein transfer, two groups have developed worm models using a bimolecular fluorescence (BiFC) approach. In this approach, two parts of a fluorescent protein (e.g., GFP) are fused to α-synuclein such that they will only emit fluorescence if two molecules of α-synuclein interact thereby bringing the two parts of the fluorescent protein close enough together to act as one protein [130]. In order to detect neuron-to-neuron transfer of α-synuclein, the two different BiFC-α-synuclein fusion proteins were expressed in separate populations of neurons, which are synaptically connected [131]. The observation of fluorescence in these worms indicated that the BiFC-α-synuclein molecules move from one cell to another. Having shown that neuron-to-neuron transfer occurs in C. elegans, this model can now be used to screen for modifiers of α-synuclein transfer. As a proof of principle, it was shown that genes involved in autophagy, endocytosis and exocytosis all modulate α-synuclein transfer [131]. Interestingly, silencing C. elegans orthologs of PD-related genes PRKN, PINK1, DJ-1, ATP13A2, VPS-35, and LRRK2 using RNAi increased α-synuclein accumulation in these animals [131]. Another study that used a different BiFC strain to visualize α-synuclein transfer between neuron and muscle demonstrated that transfer of α-synuclein increased with age and could be decreased by genes that have been shown to delay aging [132].
Targeting aging pathways is protective in C. elegans models of Parkinson’s disease
While it has long been known that aging is the greatest risk factor for the development of PD [133, 134], the role of aging in PD is still poorly defined. The large number of similarities that exist between the normal aging process and the development of PD indicates a strong association [135]. However, in order to demonstrate a causative role of aging in the pathogenesis of PD, it is necessary to experimentally modulate aging and show an effect on PD. Because C. elegans has been used extensively to study the genetics of aging, it provides the ideal organism to study the relationship between aging and PD. To test this idea, two groups examined the effect of modulating molecular pathways that have been shown to extend longevity in C. elegans models of PD. Both groups chose to test the effect of decreasing insulin-IGF1 signaling, which has been shown to double the lifespan of a worm [136]. It was found that delaying aging by decreasing insulin-IGF1 signaling decreased aggregation and protected against dopamine neuronal loss in multiple different worm models of PD [60, 137]. Decreasing insulin-IGF1 signaling also reduced the cell-to-cell propagation of α-synuclein [132]. Importantly, a beneficial effect of delaying aging was also observed in a Drosophila model indicating conservation across species [137]. Since decreasing insulin-IGF1 signaling has also been shown to increase lifespan in mice [138], and genetic variants in this pathway are associated with longevity in humans [139], targeting this pathway may provide an effective treatment for patients with PD.
CONCLUSIONS
C. elegans provides a number of advantages as an animal model for the study of PD. The most significant advantages include the wide array of genetic tools available and the ability to screen for disease modifiers in a rapid and cost effective manner. On the other hand, the main limitations of using C. elegans for the study of PD include the lack of a homolog to α-synuclein, the challenge of performing molecular biology specifically on dopamine neurons (which make up only 8 of 959 cells), and the fact that neuronal connectivity differs from humans. With these limitations in mind, the model system has enabled numerous significant contributions to our understanding of PD. For example, although C. elegans lack an ortholog to α-synuclein, this has not precluded using this model to identify numerous modifiers of α-synuclein aggregation and toxicity, which are shared with mammals. As with all model systems, in using C. elegans for PD research, it is important to take advantage of its strengths while using other models to complement its limitations.
CONFLICT OF INTEREST
The authors have no conflict of interests to declare.
ACKNOWLEDGMENTS
This work was supported by the National Institute on Aging (NIA) by grant R21 AG058241 (PI: Van Raamsdonk), the National Institutes of General Medical Sciences by grant R01 GM121756 (PI: Van Raamsdonk) and the Van Andel Research Institute (VARI). JC was supported by the Van Andel Institute Graduate School (VAIGS).
REFERENCES
[1] | Poewe W , Seppi K , Tanner CM , Halliday GM , Brundin P , Volkmann J , Schrag AE , Lang AE ((2017) ) Parkinson disease. Nat Rev Dis Primers 3: , 17013. |
[2] | Pringsheim T , Jette N , Frolkis A , Steeves TD ((2014) ) The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov Disord 29: , 1583–1590. |
[3] | Spillantini MG , Crowther RA , Jakes R , Hasegawa M , Goedert M ((1998) ) Alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci U S A 95: , 6469–6473. |
[4] | Polymeropoulos MH , Lavedan C , Leroy E , Ide SE , Dehejia A , Dutra A , Pike B , Root H , Rubenstein J , Boyer R , Stenroos ES , Chandrasekharappa S , Athanassiadou A , Papapetropoulos T , Johnson WG , Lazzarini AM , Duvoisin RC , Di Iorio G , Golbe LI , Nussbaum RL ((1997) ) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276: , 2045–2047. |
[5] | Paisan-Ruiz C , Jain S , Evans EW , Gilks WP , Simon J , van der Brug M , Lopez de Munain A , Aparicio S , Gil AM , Khan N , Johnson J , Martinez JR , Nicholl D , Carrera IM , Pena AS , de Silva R , Lees A , Marti-Masso JF , Perez-Tur J , Wood NW , Singleton AB ((2004) ) Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 44: , 595–600. |
[6] | Zimprich A , Biskup S , Leitner P , Lichtner P , Farrer M , Lincoln S , Kachergus J , Hulihan M , Uitti RJ , Calne DB , Stoessl AJ , Pfeiffer RF , Patenge N , Carbajal IC , Vieregge P , Asmus F , Muller-Myhsok B , Dickson DW , Meitinger T , Strom TM , Wszolek ZK , Gasser T ((2004) ) Mutations in LRRK2 cause autosomal-dominant parkinsonism with pleomorphic pathology. Neuron 44: , 601–607. |
[7] | Kitada T , Asakawa S , Hattori N , Matsumine H , Yamamura Y , Minoshima S , Yokochi M , Mizuno Y , Shimizu N ((1998) ) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: , 605–608. |
[8] | Valente EM , Abou-Sleiman PM , Caputo V , Muqit MM , Harvey K , Gispert S , Ali Z , Del Turco D , Bentivoglio AR , Healy DG , Albanese A , Nussbaum R , Gonzalez-Maldonado R , Deller T , Salvi S , Cortelli P , Gilks WP , Latchman DS , Harvey RJ , Dallapiccola B , Auburger G , Wood NW ((2004) ) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304: , 1158–1160. |
[9] | Bonifati V , Rizzu P , van Baren MJ , Schaap O , Breedveld GJ , Krieger E , Dekker MC , Squitieri F , Ibanez P , Joosse M , van Dongen JW , Vanacore N , van Swieten JC , Brice A , Meco G , van Duijn CM , Oostra BA , Heutink P ((2003) ) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299: , 256–259. |
[10] | Ramirez A , Heimbach A , Grundemann J , Stiller B , Hampshire D , Cid LP , Goebel I , Mubaidin AF , Wriekat AL , Roeper J , Al-Din A , Hillmer AM , Karsak M , Liss B , Woods CG , Behrens MI , Kubisch C ((2006) ) Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat Genet 38: , 1184–1191. |
[11] | Corsi AK , Wightman B , Chalfie M ((2015) ) A transparent window into biology: A primer on Caenorhabditis elegans. Genetics 200: , 387–407. |
[12] | Dexter PM , Caldwell KA , Caldwell GA ((2012) ) A predictable worm: Application of Caenorhabditis elegans for mechanistic investigation of movement disorders. Neurotherapeutics 9: , 393–404. |
[13] | White JG , Southgate E , Thomson JN , Brenner S ((1986) ) The structure of the nervous-system of the nematode Caenorhabditis-elegans. Philos Trans R Soc Lond B Biol Sci 314: , 1–340. |
[14] | Xue D , Finney M , Ruvkun G , Chalfie M ((1992) ) Regulation of the Mec-3-gene by the C-elegans homeoproteins Unc-86 and Mec-3. EMBO J 11: , 4969–4979. |
[15] | Gonzalez-Hunt CP , Leung MCK , Bodhicharla RK , McKeever MG , Arrant AE , Margillo KM , Ryde IT , Cyr DD , Kosmaczewski SG , Hammarlund M , Meyer JN ((2014) ) Exposure to mitochondrial genotoxins and dopaminergic neurodegeneration in Caenorhabditis elegans. Plos One 9: , e114459. |
[16] | Lee H , Kim SA , Coakley S , Mugno P , Hammarlund M , Hilliard MA , Lu H ((2014) ) A multi-channel device for high-density target-selective stimulation and long-term monitoring of cells and subcellular features in C. elegans. Lab on a Chip 14: , 4513–4522. |
[17] | Patel MR , Lehrman EK , Poon VY , Crump JG , Zhen M , Bargmann CI , Shen K ((2006) ) Hierarchical assembly of presynaptic components in defined C. elegans synapses. Nat Neurosci 9: , 1488–1498. |
[18] | Markey KM , Glendinning KJ , Morgan JAW , Hart CA , Winstanley C ((2006) ) Caenorhabditis elegans killing assay as an infection model to study the role of type III secretion in Burkholderia cenocepacia. J Med Microbiol 55: , 967–969. |
[19] | Bae Y , Knobel K , Qin H , Hu J , Rosenbaum J , Barr M ((2006) ) Mechanisms of polycystin ciliary localization in C-elegans. Mol Biol Cell 17: . |
[20] | Shaham S ((2015) ) Glial Development and Function in the Nervous System of Caenorhabditis elegans. Cold Spring Harb Perspect Biol 7: , a020578. |
[21] | Hills T , Brockie PJ , Maricq AV ((2004) ) Dopamine and glutamate control area-restricted search behavior in Caenorhabditis elegans. J Neurosci 24: , 1217–1225. |
[22] | Sawin ER , Ranganathan R , Horvitz HR ((2000) ) C-elegans locomotory rate is modulated by the environment through a dopaminergic pathway and by experience through a serotonergic pathway. Neuron 26: , 619–631. |
[23] | Shaye DD , Greenwald I ((2011) ) OrthoList: A compendium of C. elegans genes with human orthologs. Plos One 6: , e20085. |
[24] | Kaletta T , Hengartner MO ((2006) ) Finding function in novel targets: C-elegans as a model organism. Nat Rev Drug Discov 5: , 387–398. |
[25] | Culetto E , Sattelle DB ((2000) ) A role for Caenorhabditis elegans in understanding the function and interactions of human disease genes. Hum Mol Gen 9: , 869–877. |
[26] | Fire A , Xu SQ , Montgomery MK , Kostas SA , Driver SE , Mello CC ((1998) ) Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 391: , 806–811. |
[27] | Montgomery MK , Xu SQ , Fire A ((1998) ) RNA as a target of double-stranded RNA-mediated genetic interference in Caenorhabditis elegans. Proc Natl Acad Sci U S A 95: , 15502–15507. |
[28] | Kelly WG , Fire A ((1998) ) Chromatin silencing and the maintenance of a functional germline in Caenorhabditis elegans. Development 125: , 2451–2456. |
[29] | Timmons L , Court DL , Fire A ((2001) ) Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263: , 103–112. |
[30] | Tabara H , Grishok A , Mello CC ((1998) ) RNAi in C-elegans: Soaking in the genome sequence. Science 282: , 430–431. |
[31] | Tavernarakis N , Wang SL , Dorovkov M , Ryazanov A , Driscoll M ((2000) ) Heritable and inducible genetic interference by double-stranded RNA encoded by transgenes. Nat Genet 24: , 180–183. |
[32] | Hunter CP , Winston WM , Molodowitch C , Feinberg EH , Shih J , Sutherlin M , Wright AJ , Fitzgerald MC ((2006) ) Systemic RNAi in Caenorhabditis elegans. Cold Spring Harb Symp Quant Biol 71: , 95–100. |
[33] | Winston WM , Molodowitch C , Hunter CP ((2002) ) Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science 295: , 2456–2459. |
[34] | Timmons L , Tabara H , Mello CC , Fire AZ ((2003) ) Inducible systemic RNA silencing in Caenorhabditis elegans. Mol Biol Cell 14: , 2972–2983. |
[35] | Simmer F , Tijsterman M , Parrish S , Koushika SP , Nonet ML , Fire A , Ahringer J , Plasterk RH ((2002) ) Loss of the putative RNA-directed RNA polymerase RRF-3 makes C. elegans hypersensitive to RNAi. Curr Biol 12: , 1317–1319. |
[36] | Kennedy S , Wang D , Ruvkun G ((2004) ) A conserved siRNA-degrading RNase negatively regulates RNA interference in C. elegans. Nature 427: , 645–649. |
[37] | Calixto A , Chelur D , Topalidou I , Chen X , Chalfie M ((2010) ) Enhanced neuronal RNAi in C. elegans using SID-1. Nat Methods 7: , 554–559. |
[38] | McIntire SL , Reimer RJ , Schuske K , Edwards RH , Jorgensen EM ((1997) ) Identification and characterization of the vesicular GABA transporter. Nature 389: , 870–876. |
[39] | Harris G , Korchnak A , Summers P , Hapiak V , Law WJ , Stein AM , Komuniecki P , Komuniecki R ((2011) ) Dissecting the serotonergic food signal stimulating sensory-mediated aversive behavior in C. elegans. Plos One 6: , e21897. |
[40] | Firnhaber C , Hammarlund M ((2013) ) Neuron-specific feeding RNAi in C. elegans and its use in a screen for essential genes required for GABA neuron function. Plos Genet 9: , e1003921. |
[41] | Jorgensen EM , Mango SE ((2002) ) The art and design of genetic screens: Caenorhabditis elegans. Nat Rev Genet 3: , 356–369. |
[42] | Boutros M , Ahringer J ((2008) ) The art and design of genetic screens: RNA interference. Nat Rev Genet 9: , 554–566. |
[43] | Kamath RS , Martinez-Campos M , Zipperlen P , Fraser AG , Ahringer J ((2001) ) Effectiveness of specific RNA-mediated interference through ingested double-stranded RNA in Caenorhabditis elegans. Genome Biol 2: , RESEARCH0002. |
[44] | Rual JF , Ceron J , Koreth J , Hao T , Nicot AS , Hirozane-Kishikawa T , Vandenhaute J , Orkin SH , Hill DE , van den Heuvel S , Vidal M ((2004) ) Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res 14: , 2162–2168. |
[45] | Nass R , Hall DH , Miller DM , Blakely RD ((2002) ) Neurotoxin-induced degeneration of dopamine neurons in Caenorhabditis elegans. Proc Natl Acad Sci U S A 99: , 3264–3269. |
[46] | Pu P , Le W ((2008) ) Dopamine neuron degeneration induced by MPP+ is independent of CED-4 pathway in Caenorhabditis elegans. Cell Res 18: , 978–981. |
[47] | Ali SJ , Rajini PS ((2012) ) Elicitation of dopaminergic features of Parkinson’s disease in C. elegans by monocrotophos, an organophosphorous insecticide. CNS Neurol Disord Drug Targets 11: , 993–1000. |
[48] | Masoudi N , Ibanez-Cruceyra P , Offenburger SL , Holmes A , Gartner A ((2014) ) Tetraspanin (TSP-17) protects dopaminergic neurons against 6-OHDA-induced neurodegeneration in C. elegans. PLoS Genet 10: , e1004767. |
[49] | Lakso M , Vartiainen S , Moilanen AM , Sirvio J , Thomas JH , Nass R , Blakely RD , Wong G ((2003) ) Dopaminergic neuronal loss and motor deficits in Caenorhabditis elegans overexpressing human alpha-synuclein. J Neurochem 86: , 165–172. |
[50] | Bargmann CI ((2006) ) Chemosensation in C. elegans. WormBook 1–29. |
[51] | Davies AG , McIntire SL ((2004) ) Using C. elegans to screen for targets of ethanol and behavior-altering drugs. Biol Proced Online 6: , 113–119. |
[52] | Lee J , Jee C , McIntire SL ((2009) ) Ethanol preference in C. elegans. Genes Brain Behav 8: , 578–585. |
[53] | Cooper JF , Machiela E , Dues DJ , Spielbauer KK , Senchuk MM , Van Raamsdonk JM ((2017) ) Activation of the mitochondrial unfolded protein response promotes longevity and dopamine neuron survival in Parkinson’s disease models. Sci Rep 7: , 16441. |
[54] | Kimura KD , Fujita K , Katsura I ((2010) ) Enhancement of odor avoidance regulated by dopamine signaling in Caenorhabditis elegans. J Neurosci 30: , 16365–16375. |
[55] | Sashidhara KV , Modukuri RK , Jadiya P , Dodda RP , Kumar M , Sridhar B , Kumar V , Haque R , Siddiqi MI , Nazir A ((2014) ) Benzofuran-chalcone hybrids as potential multifunctional agents against Alzheimer’s disease: Synthesis and in vivo studies with transgenic Caenorhabditis elegans. ChemMedChem 9: , 2671–2684. |
[56] | McDonald PW , Hardie SL , Jessen TN , Carvelli L , Matthies DS , Blakely RD ((2007) ) Vigorous motor activity in Caenorhabditis elegans requires efficient clearance of dopamine mediated by synaptic localization of the dopamine transporter DAT-1. J Neurosci 27: , 14216–14227. |
[57] | Bodhicharla R , Nagarajan A , Winter J , Adenle A , Nazir A , Brady D , Vere K , Richens J , O’Shea P , Bell DR , de Pomerai D ((2012) ) Effects of alpha-synuclein overexpression in transgenic Caenorhabditis elegans strains. CNS Neurol Disord Drug Targets 11: , 965–975. |
[58] | van Ham TJ , Thijssen KL , Breitling R , Hofstra RM , Plasterk RH , Nollen EA ((2008) ) C. elegans model identifies genetic modifiers of alpha-synuclein inclusion formation during aging. PLoS Genet 4: , e1000027. |
[59] | Kuwahara T , Koyama A , Gengyo-Ando K , Masuda M , Kowa H , Tsunoda M , Mitani S , Iwatsubo T ((2006) ) Familial Parkinson mutant alpha-synuclein causes dopamine neuron dysfunction in transgenic Caenorhabditis elegans. J Biol Chem 281: , 334–340. |
[60] | Cooper JF , Dues DJ , Spielbauer KK , Machiela E , Senchuk MM , Van Raamsdonk JM ((2015) ) Delaying aging is neuroprotective in Parkinson’s disease: A genetic analysis in C. elegans models. NPJ Parkinsons Dis 1: , 15022. |
[61] | Swierczek NA , Giles AC , Rankin CH , Kerr RA ((2011) ) High-throughput behavioral analysis in C. elegans. Nat Methods 8: , 592-U112. |
[62] | Labrousse AM , Zappaterra MD , Rube DA , van der Bliek AM ((1999) ) C-elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell 4: , 815–826. |
[63] | Morsci NS , Hall DH , Driscoll M , Sheng ZH ((2016) ) Age-related phasic patterns of mitochondrial maintenance in adult Caenorhabditis elegans neurons. J Neurosci 36: , 1373–1385. |
[64] | Palikaras K , Lionaki E , Tavernarakis N ((2015) ) Coordination of mitophagy and mitochondrial biogenesis during ageing in C-elegans. Nature 521: , 525-U241. |
[65] | Samann J , Hegermann J , von Gromoff E , Eimer S , Baumeister R , Schmidt E ((2009) ) Caenorhabditits elegans LRK-1 and PINK-1 act antagonistically in stress response and neurite outgrowth. J Biol Chem 284: , 16482–16491. |
[66] | Singleton AB , Farrer M , Johnson J , Singleton A , Hague S , Kachergus J , Hulihan M , Peuralinna T , Dutra A , Nussbaum R , Lincoln S , Crawley A , Hanson M , Maraganore D , Adler C , Cookson MR , Muenter M , Baptista M , Miller D , Blancato J , Hardy J , Gwinn-Hardy K ((2003) ) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302: , 841. |
[67] | Soldner F , Stelzer Y , Shivalila CS , Abraham BJ , Latourelle JC , Barrasa MI , Goldmann J , Myers RH , Young RA , Jaenisch R ((2016) ) Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature 533: , 95–99. |
[68] | Cao SS , Gelwix CC , Caldwell KA , Caldwell GA ((2005) ) Torsin-mediated protection from cellular stress in the dopaminergic neurons of Caenorhabditis elegans. J Neurosci 25: , 3801–3812. |
[69] | Buttner S , Broeskamp F , Sommer C , Markaki M , Habernig L , Alavian-Ghavanini A , Carmona-Gutierrez D , Eisenberg T , Michael E , Kroemer G , Tavernarakis N , Sigrist SJ , Madeo F ((2014) ) Spermidine protects against alpha-synuclein neurotoxicity. Cell Cycle 13: , 3903–3908. |
[70] | Kuwahara T , Koyama A , Koyama S , Yoshina S , Ren CH , Kato T , Mitani S , Iwatsubo T ((2008) ) A systematic RNAi screen reveals involvement of endocytic pathway in neuronal dysfunction in alpha-synuclein transgenic C. elegans. Hum Mol Genet 17: , 2997–3009. |
[71] | Springer W , Hoppe T , Schmidt E , Baumeister R ((2005) ) A Caenorhabditis elegans Parkin mutant with altered solubility couples alpha-synuclein aggregation to proteotoxic stress. Hum Mol Genet 14: , 3407–3423. |
[72] | Kuwahara T , Tonegawa R , Ito G , Mitani S , Iwatsubo T ((2012) ) Phosphorylation of alpha-synuclein protein at Ser-129 reduces neuronal dysfunction by lowering its membrane binding property in Caenorhabditis elegans. J Biol Chem 287: , 7098–7109. |
[73] | Hamamichi S , Rivas RN , Knight AL , Cao S , Caldwell KA , Caldwell GA ((2008) ) Hypothesis-based RNAi screening identifies neuroprotective genes in a Parkinson’s disease model. Proc Natl Acad Sci U S A 105: , 728–733. |
[74] | van der Goot AT , Zhu W , Vazquez-Manrique RP , Seinstra RI , Dettmer K , Michels H , Farina F , Krijnen J , Melki R , Buijsman RC , Ruiz Silva M , Thijssen KL , Kema IP , Neri C , Oefner PJ , Nollen EA ((2012) ) Delaying aging and the aging-associated decline in protein homeostasis by inhibition of tryptophan degradation. Proc Natl Acad Sci U S A 109: , 14912–14917. |
[75] | Saha S , Guillily MD , Ferree A , Lanceta J , Chan D , Ghosh J , Hsu CH , Segal L , Raghavan K , Matsumoto K , Hisamoto N , Kuwahara T , Iwatsubo T , Moore L , Goldstein L , Cookson M , Wolozin B ((2009) ) LRRK2 modulates vulnerability to mitochondrial dysfunction in Caenorhabditis elegans. J Neurosci 29: , 9210–9218. |
[76] | Yao C , El Khoury R , Wang W , Byrd TA , Pehek EA , Thacker C , Zhu X , Smith MA , Wilson-Delfosse AL , Chen SG ((2010) ) LRRK2-mediated neurodegeneration and dysfunction of dopaminergic neurons in a Caenorhabditis elegans model of Parkinson’s disease. Neurobiol Dis 40: , 73–81. |
[77] | Yao C , Johnson WM , Gao Y , Wang W , Zhang J , Deak M , Alessi DR , Zhu X , Mieyal JJ , Roder H , Wilson-Delfosse AL , Chen SG ((2013) ) Kinase inhibitors arrest neurodegeneration in cell and C. elegans models of LRRK2 toxicity. Hum Mol Genet 22: , 328–344. |
[78] | Liu ZH , Hamamichi S , Lee BD , Yang DJ , Ray A , Caldwell GA , Caldwell KA , Dawson TM , Smith WW , Dawson VL ((2011) ) Inhibitors of LRRK2 kinase attenuate neurodegeneration and Parkinson-like phenotypes in Caenorhabditis elegans and Drosophila Parkinson’s disease models. Hum Mol Genet 20: , 3933–3942. |
[79] | Farrer M , Chan P , Chen R , Tan L , Lincoln S , Hernandez D , Forno L , Gwinn-Hardy K , Petrucelli L , Hussey J , Singleton A , Tanner C , Hardy J , Langston JW ((2001) ) Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol 50: , 293–300. |
[80] | Shimura H , Hattori N , Kubo S , Mizuno Y , Asakawa S , Minoshima S , Shimizu N , Iwai K , Chiba T , Tanaka K , Suzuki T ((2000) ) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25: , 302–305. |
[81] | Bornhorst J , Chakraborty S , Meyer S , Lohren H , Brinkhaus SG , Knight AL , Caldwell KA , Caldwell GA , Karst U , Schwerdtle T , Bowman A , Aschner M ((2014) ) The effects of pdr1, djr1.1 and pink1 loss in manganese-induced toxicity and the role of alpha-synuclein in C. elegans. Metallomics 6: , 476–490. |
[82] | Chakraborty S , Chen P , Bornhorst J , Schwerdtle T , Schumacher F , Kleuser B , Bowman AB , Aschner M ((2015) ) Loss of pdr-1/parkin influences Mn homeostasis through altered ferroportin expression in C. elegans. Metallomics 7: , 847–856. |
[83] | Kirienko NV , Ausubel FM , Ruvkun G ((2015) ) Mitophagy confers resistance to siderophore-mediated killing by Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 112: , 1821–1826. |
[84] | Luz AL , Rooney JP , Kubik LL , Gonzalez CP , Song DH , Meyer JN ((2015) ) Mitochondrial morphology and fundamental parameters of the mitochondrial respiratory chain are altered in Caenorhabditis elegans strains deficient in mitochondrial dynamics and homeostasis processes. PLoS One 10: , e0130940. |
[85] | Melentijevic I , Toth ML , Arnold ML , Guasp RJ , Harinath G , Nguyen KC , Taub D , Parker JA , Neri C , Gabel CV , Hall DH , Driscoll M ((2017) ) C-elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 542: , 367–371. |
[86] | Vives-Bauza C , Zhou C , Huang Y , Cui M , de Vries RL , Kim J , May J , Tocilescu MA , Liu W , Ko HS , Magrane J , Moore DJ , Dawson VL , Grailhe R , Dawson TM , Li C , Tieu K , Przedborski S ((2010) ) PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci U S A 107: , 378–383. |
[87] | Deas E , Wood NW , Plun-Favreau H ((2011) ) Mitophagy and Parkinson’s disease: The PINK1-parkin link. Biochim Biophys Acta 1813: , 623–633. |
[88] | Narendra DP , Jin SM , Tanaka A , Suen DF , Gautier CA , Shen J , Cookson MR , Youle RJ ((2010) ) PINK1 is selectively stabilized on impaired mitochondria to activate parkin. Plos Biol 8: , e1000298. |
[89] | Koyano F , Okatsu K , Kosako H , Tamura Y , Go E , Kimura M , Kimura Y , Tsuchiya H , Yoshihara H , Hirokawa T , Endo T , Fon EA , Trempe JF , Saeki Y , Tanaka K , Matsuda N ((2014) ) Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: , 162–166. |
[90] | Bonifati V , Rizzu P , Squitieri F , Krieger E , Vanacore N , van Swieten JC , Brice A , van Duijn CM , Oostra B , Meco G , Heutink P ((2003) ) DJ-1 (PARK7), a novel gene for autosomal recessive, early onset parkinsonism. Neurol Sci 24: , 159–160. |
[91] | Zhang L , Shimoji M , Thomas B , Moore DJ , Yu SW , Marupudi NI , Torp R , Torgner IA , Ottersen OP , Dawson TM , Dawson VL ((2005) ) Mitochondrial localization of the Parkinson’s disease related protein DJ-1: Implications for pathogenesis. Hum Mol Genet 14: , 2063–2073. |
[92] | Fitzgerald JC , Plun-Favreau H ((2008) ) Emerging pathways in genetic Parkinson’s disease: Autosomal-recessive genes in Parkinson’s disease - a common pathway? FEBS J 275: , 5758–5766. |
[93] | Lee JY , Song J , Kwon K , Jang S , Kim C , Baek K , Kim J , Park C ((2012) ) Human DJ-1 and its homologs are novel glyoxalases. Hum Mol Genet 21: , 3215–3225. |
[94] | Dehay B , Ramirez A , Martinez-Vicente M , Perier C , Canron MH , Doudnikoff E , Vital A , Vila M , Klein C , Bezard E ((2012) ) Loss of P-type ATPase ATP13A2/PARK9 function induces general lysosomal deficiency and leads to Parkinson disease neurodegeneration. Proc Natl Acad Sci U S A 109: , 9611–9616. |
[95] | Gitler AD , Chesi A , Geddie ML , Strathearn KE , Hamamichi S , Hill KJ , Caldwell KA , Caldwell GA , Cooper AA , Rochet JC , Lindquist S ((2009) ) Alpha-synuclein is part of a diverse and highly conserved interaction network that includes PARK9 and manganese toxicity. Nat Genet 41: , 308–315. |
[96] | Nass R , Blakely RD ((2003) ) The Caenorhabditis elegans dopaminergic system: Opportunities for insights into dopamine transport and neurodegeneration. Annu Rev Pharmacol Toxicol 43: , 521–544. |
[97] | Zhou S , Wang Z , Klaunig JE ((2013) ) Caenorhabditis elegans neuron degeneration and mitochondrial suppression caused by selected environmental chemicals. Int J Biochem Mol Biol 4: , 191–200. |
[98] | Hwang O ((2013) ) Role of oxidative stress in Parkinson’s disease. Exp Neurobiol 22: , 11–17. |
[99] | Wang YM , Pu P , Le WD ((2007) ) ATP depletion is the major cause of MPP+ induced dopamine neuronal death and worm lethality in alpha-synuclein transgenic C. elegans. Neurosci Bull 23: , 329–335. |
[100] | Braungart E , Gerlach M , Riederer P , Baumeister R , Hoener MC ((2004) ) Caenorhabditis elegans MPP+ model of Parkinson’s disease for high-throughput drug screenings. Neurodegener Dis 1: , 175–183. |
[101] | Marvanova M , Nichols CD ((2007) ) Identification of neuroprotective compounds of caenorhabditis elegans dopaminergic neurons against 6-OHDA. J Mol Neurosci 31: , 127–137. |
[102] | Ved R , Saha S , Westlund B , Perier C , Burnam L , Sluder A , Hoener M , Rodrigues CM , Alfonso A , Steer C , Liu L , Przedborski S , Wolozin B ((2005) ) Similar patterns of mitochondrial vulnerability and rescue induced by genetic modification of alpha-synuclein, parkin, and DJ-1 in Caenorhabditis elegans. J Biol Chem 280: , 42655–42668. |
[103] | Settivari R , Levora J , Nass R ((2009) ) The divalent metal transporter homologues SMF-1/2 mediate dopamine neuron sensitivity in caenorhabditis elegans models of manganism and parkinson disease. J Biol Chem 284: , 35758–35768. |
[104] | VanDuyn N , Settivari R , Wong G , Nass R ((2010) ) SKN-1/Nrf2 inhibits dopamine neuron degeneration in a Caenorhabditis elegans model of methylmercury toxicity. Toxicol Sci 118: , 613–624. |
[105] | Miller RL , Sun GY , Sun AY ((2007) ) Cytotoxicity of paraquat in microglial cells: Involvement of PKCdelta- and ERK1/2-dependent NADPH oxidase. Brain Res 1167: , 129–139. |
[106] | Tanner CM , Kamel F , Ross GW , Hoppin JA , Goldman SM , Korell M , Marras C , Bhudhikanok GS , Kasten M , Chade A , Comyns K , Meng C , Priestley B , Fernandez HH , Cambi F , Umbach DM , Blair A , Sandler DP , Langston JW ((2010) ) Rotenone, paraquat and Parkinson’s disease (PD). Ann Neurol 68: , S17–S17. |
[107] | Garcia-Garcia A , Anandhan A , Burns M , Chen H , Zhou Y , Franco R ((2013) ) Impairment of Atg5-dependent autophagic flux promotes paraquat- and MPP(+)-induced apoptosis but not rotenone or 6-hydroxydopamine toxicity. Toxicol Sci 136: , 166–182. |
[108] | Cooper AA , Gitler AD , Cashikar A , Haynes CM , Hill KJ , Bhullar B , Liu KN , Xu KX , Strathearn KE , Liu F , Cao SS , Caldwell KA , Caldwell GA , Marsischky G , Kolodner RD , LaBaer J , Rochet JC , Bonini NM , Lindquist S ((2006) ) alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 313: , 324–328. |
[109] | Wong K , Sidransky E , Verma A , Mixon T , Sandberg GD , Wakefield LK , Morrison A , Lwin A , Colegial C , Allman JM , Schiffmann R ((2004) ) Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab 82: , 192–207. |
[110] | Mazzulli JR , Xu YH , Sun Y , Knight AL , McLean PJ , Caldwell GA , Sidransky E , Grabowski GA , Krainc D ((2011) ) Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146: , 37–52. |
[111] | Jansen IE , Ye H , Heetveld S , Lechler MC , Michels H , Seinstra RI , Lubbe SJ , Drouet V , Lesage S , Majounie E , Gibbs JR , Nalls MA , Ryten M , Botia JA , Vandrovcova J , Simon-Sanchez J , Castillo-Lizardo M , Rizzu P , Blauwendraat C , Chouhan AK , Li Y , Yogi P , Amin N , van Duijn CM , International Parkinson’s Disease Genetics Consortium, Morris HR , Brice A , Singleton AB , David DC , Nollen EA , Jain S , Shulman JM , Heutink P ((2017) ) Discovery and functional prioritization of Parkinson’s disease candidate genes from large-scale whole exome sequencing. Genome Biol 18: , 22. |
[112] | Newsome TP , Asling B , Dickson BJ ((2000) ) Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaics. Development 127: , 851–860. |
[113] | Neumuller RA , Richter C , Fischer A , Novatchkova M , Neumuller KG , Knoblich JA ((2011) ) Genome-wide analysis of self-renewal in Drosophila neural stem cells by transgenic RNAi. Cell Stem Cell 8: , 580–593. |
[114] | Ma XM , Kiraly DD , Gaier ED , Wang Y , Kim EJ , Levine ES , Eipper BA , Mains RE ((2008) ) Kalirin-7 is required for synaptic structure and function. J Neurosci 28: , 12368–12382. |
[115] | Mandela P , Yankova M , Conti LH , Ma XM , Grady J , Eipper BA , Mains RE ((2012) ) Kalrn plays key roles within and outside of the nervous system. BMC Neurosci 13: , 136. |
[116] | Goldman SM ((2014) ) Environmental toxins and Parkinson’s disease. Annu Rev Pharmacol Toxicol 54: , 141–164. |
[117] | Caldwell KA , Tucci ML , Armagost J , Hodges TW , Chen J , Memon SB , Blalock JE , DeLeon SM , Findlay RH , Ruan QM , Webber PJ , Standaert DG , Olson JB , Caldwell GA ((2009) ) Investigating bacterial sources of toxicity as an environmental contributor to dopaminergic neurodegeneration. Plos One 4: , e7227. |
[118] | Ray A , Martinez BA , Berkowitz LA , Caldwell GA , Caldwell KA ((2014) ) Mitochondrial dysfunction, oxidative stress, and neurodegeneration elicited by a bacterial metabolite in a C. elegans Parkinson’s model. Cell Death Dis 5: , e984. |
[119] | Martinez BA , Kim H , Ray A , Caldwell GA , Caldwell KA ((2015) ) A bacterial metabolite induces glutathione-tractable proteostatic damage, proteasomal disturbances, and PINK1-dependent autophagy in C. elegans.. Cell Death Dis 6: , e1908. |
[120] | Chen P , DeWitt MR , Bornhorst J , Soares FA , Mukhopadhyay S , Bowman AB , Aschner M ((2015) ) Age- and manganese-dependent modulation of dopaminergic phenotypes in a C. elegans DJ-1 genetic model of Parkinson’s disease. Metallomics 7: , 289–298. |
[121] | Jadiya P , Nazir A ((2012) ) Environmental toxicants as extrinsic epigenetic factors for parkinsonism: Studies employing transgenic C. elegans model. CNS Neurol Disord Drug Targets 11: , 976–983. |
[122] | Xiong H , Pears C , Woollard A ((2017) ) An enhanced C. elegans based platform for toxicity assessment. Sci Rep 7: , 9839. |
[123] | Locke CJ , Fox SA , Caldwell GA , Caldwell KA ((2008) ) Acetaminophen attenuates dopamine neuron degeneration in animal models of Parkinson’s disease. Neurosci Lett 439: , 129–133. |
[124] | Kautu BB , Carrasquilla A , Hicks ML , Caldwell KA , Caldwell GA ((2013) ) Valproic acid ameliorates C. elegans dopaminergic neurodegeneration with implications for ERK-MAPK signaling. Neurosci Lett 541: , 116–119. |
[125] | Desplats P , Lee HJ , Bae EJ , Patrick C , Rockenstein E , Crews L , Spencer B , Masliah E , Lee SJ ((2009) ) Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A 106: , 13010–13015. |
[126] | Reyes JF , Olsson TT , Lamberts JT , Devine MJ , Kunath T , Brundin P ((2015) ) A cell culture model for monitoring alpha-synuclein cell-to-cell transfer. Neurobiol Dis 77: , 266–275. |
[127] | Rey NL , Petit GH , Bousset L , Melki R , Brundin P ((2013) ) Transfer of human alpha-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol 126: , 555–573. |
[128] | Li JY , Englund E , Holton JL , Soulet D , Hagell P , Lees AJ , Lashley T , Quinn NP , Rehncrona S , Bjorklund A , Widner H , Revesz T , Lindvall O , Brundin P ((2008) ) Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat Med 14: , 501–503. |
[129] | Kordower JH , Chu Y , Hauser RA , Freeman TB , Olanow CW ((2008) ) Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat Med 14: , 504–506. |
[130] | Outeiro TF , Putcha P , Tetzlaff JE , Spoelgen R , Koker M , Carvalho F , Hyman BT , McLean PJ ((2008) ) Formation of toxic oligomeric alpha-synuclein species in living cells. PLoS One 3: , e1867. |
[131] | Tyson T , Senchuk M , Cooper JF , George S , Van Raamsdonk JM , Brundin P ((2017) ) Novel animal model defines genetic contributions for neuron-to-neuron transfer of alpha-synuclein. Sci Rep 7: , 7506. |
[132] | Kim DK , Lim HS , Kawasaki I , Shim YH , Vaikath NN , El-Agnaf OM , Lee HJ , Lee SJ ((2016) ) Anti-aging treatments slow propagation of synucleinopathy by restoring lysosomal function. Autophagy 12: , 1849–1863. |
[133] | Driver JA , Logroscino G , Gaziano JM , Kurth T ((2009) ) Incidence and remaining lifetime risk of Parkinson disease in advanced age. Neurology 72: , 432–438. |
[134] | Collier TJ , Kanaan NM , Kordower JH ((2011) ) Ageing as a primary risk factor for Parkinson’s disease: Evidence from studies of non-human primates. Nat Rev Neurosci 12: , 359–366. |
[135] | Rodriguez M , Rodriguez-Sabate C , Morales I , Sanchez A , Sabate M ((2015) ) Parkinson’s disease as a result of aging. Aging Cell 14: , 293–308. |
[136] | Kenyon C , Chang J , Gensch E , Rudner A , Tabtiang R ((1993) ) A C. elegans mutant that lives twice as long as wild type. Nature 366: , 461–464. |
[137] | Knight AL , Yan XH , Hamamichi S , Ajjuri RR , Mazzulli JR , Zhang MW , Daigle JG , Zhang SY , Borom AR , Roberts LR , Lee SK , DeLeon SM , Viollet-Djelassi C , Krainc D , O’Donnell JM , Caldwell KA , Caldwell GA ((2014) ) The glycolytic enzyme, GPI, is a functionally conserved modifier of dopaminergic neurodegeneration in Parkinson’s dodels. Cell Metab 20: , 145–157. |
[138] | Holzenberger M , Dupont J , Ducos B , Leneuve P , Geloen A , Even PC , Cervera P , Le Bouc Y ((2003) ) IGF-1 receptor regulates lifespan and resistance to oxidative stress in mice. Nature 421: , 182–187. |
[139] | Suh Y , Atzmon G , Cho MO , Hwang D , Liu B , Leahy DJ , Barzilai N , Cohen P ((2008) ) Functionally significant insulin-like growth factor I receptor mutations in centenarians. Proc Natl Acad Sci U S A 105: , 3438–3442. |
[140] | Su LJ , Auluck PK , Outeiro TF , Yeger-Lotem E , Kritzer JA , Tardiff DF , Strathearn KE , Liu F , Cao S , Hamamichi S , Hill KJ , Caldwell KA , Bell GW , Fraenkel E , Cooper AA , Caldwell GA , McCaffery JM , Rochet JC , Lindquist S ((2010) ) Compounds from an unbiased chemical screen reverse both ER-to-Golgi trafficking defects and mitochondrial dysfunction in Parkinson’s disease models. Dis Model Mech 3: , 194–208. |
[141] | Nass R , Hahn MK , Jessen T , McDonald PW , Carvelli L , Blakely RD ((2005) ) A genetic screen in Caenorhabditis elegans for dopamine neuron insensitivity to 6-hydroxydopamine identifies dopamine transporter mutants impacting transporter biosynthesis and trafficking. J Neurochem 94: , 774–785. |
[142] | Jadiya P , Fatima S , Baghel T , Mir SS , Nazir A ((2016) ) A systematic RNAi screen of neuroprotective genes identifies novel modulators of alpha-synuclein-associated effects in transgenic Caenorhabditis elegans. Mol Neurobiol 53: , 6288–6300. |
[143] | Karpinar DP , Balija MB , Kugler S , Opazo F , Rezaei-Ghaleh N , Wender N , Kim HY , Taschenberger G , Falkenburger BH , Heise H , Kumar A , Riedel D , Fichtner L , Voigt A , Braus GH , Giller K , Becker S , Herzig A , Baldus M , Jackle H , Eimer S , Schulz JB , Griesinger C , Zweckstetter Mr ((2009) ) Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. EMBO J 28: , 3256–3268. |


