
The Adult Neurogenesis Theory of Alzheimer’s Disease
Abstract
Alzheimer’s disease starts in neural stem cells (NSCs) in the niches of adult neurogenesis. All primary factors responsible for pathological tau hyperphosphorylation are inherent to adult neurogenesis and migration. However, when amyloid pathology is present, it strongly amplifies tau pathogenesis. Indeed, the progressive accumulation of extracellular amyloid-β deposits in the brain triggers a state of chronic inflammation by microglia. Microglial activation has a significant pro-neurogenic effect that fosters the process of adult neurogenesis and supports neuronal migration. Unfortunately, this “reactive” pro-neurogenic activity ultimately perturbs homeostatic equilibrium in the niches of adult neurogenesis by amplifying tau pathogenesis in AD. This scenario involves NSCs in the subgranular zone of the hippocampal dentate gyrus in late-onset AD (LOAD) and NSCs in the ventricular-subventricular zone along the lateral ventricles in early-onset AD (EOAD), including familial AD (FAD). Neuroblasts carrying the initial seed of tau pathology travel throughout the brain via neuronal migration driven by complex signals and convey the disease from the niches of adult neurogenesis to near (LOAD) or distant (EOAD) brain regions. In these locations, or in close proximity, a focus of degeneration begins to develop. Then, tau pathology spreads from the initial foci to large neuronal networks along neural connections through neuron-to-neuron transmission.
INTRODUCTION
The amyloid hypothesis
The prevailing view in the field is that amyloid-β peptide (Aβ) exhibits a “toxic gain-of-function” when it forms oligomers and aggregates into plaques, directly contributing to the pathogenesis of Alzheimer’s disease (AD) [1, 2]. In particular, the amyloid hypothesis, the prevalent theory of AD pathogenesis, suggests that the accumulation of pathological forms of Aβ is the primary pathological process driven by an imbalance between Aβ production and Aβ clearance [3, 4]. In this pathway, microtubule-associated protein tau pathology with the formation of phospho-tau-immunoreactive neurofibrillary tangles (NFTs) and subsequent neuronal dysfunction and neurodegeneration, perhaps mediated via inflammation, are thought to be the downstream result [4]. The direct influence of Aβ on tau pathogenesis is well documented. For example, injection of Aβ fibrils [5] or Aβ-containing brain extract [6] into mutant tau transgenic mice, crossed between mutant tau and amyloid precursor protein (APP) or 5x familial AD (FAD) transgenic mice, results in exacerbated tau pathology [5–16]. Moreover, “in vitro” [17] and “in vivo” [16] studies have demonstrated that Aβ exerts its detrimental actions by activating a key kinase, glycogen synthase kinase 3β (GSK-3β) [17, 18], implicating this kinase as an important player in the amyloid cascade. Notably, GSK-3β is the primary kinase that phosphorylates tau [18, 19]. In agreement, increased GSK-3β activity has been observed in the brains of AD patients [20]. These data confirm GSK-3β as a cornerstone of AD pathogenesis and support the notion that this kinase represents a crucial molecular link between Aβ and tau [18, 19, 21–24]. Accordingly, human Aβ oligomers induce hyperphosphorylation of tau at AD-relevant epitopes and cause neuritic dystrophy in cultured neurons [25].
The current view
In the current theory, I propose a shift in the paradigm wherein aggregates of the two key players in AD pathogenesis, i.e., Aβ and tau peptide, develop by two different and relatively independent processes. In particular, the central hypothesis is that tau pathogenesis is linked to adult neurogenesis and migration. All elements predisposing to pathological tau hyperphosphorylation are present in the niches of adult neurogenesis. In contrast, as already documented in the literature, metabolism plays a primary role in driving Aβ deposition [26–28]. Despite the fact that the two processes driving Aβ and tau pathogenesis are relatively independent, when Aβ pathology is present, it acts as a strong driving force for tau pathogenesis. Therefore, Aβ pathology also plays a crucial role in AD pathogenesis in the current theory. However, its detrimental effect is explained in quite a different way from the classical amyloid hypothesis. In particular, Aβ not only has a downstream effect on tau pathogenesis, especially when Aβ and tau colocalize, but also has an early indirect effect by influencing the process of adult neurogenesis and migration. In brief, the current theory depicts the following scenario. Progressive accumulation of extracellular Aβ deposits in the brain triggers a state of chronic inflammation by microglia. Microglial activation has a significant pro-neurogenic effect that fosters adult neurogenesis and supports neuronal migration. Unfortunately, this “reactive” pro-neurogenic pathway ultimately perturbs the delicate homeostatic equilibrium in the neurogenic niches by amplifying tau pathogenesis in AD. An imbalance between increased tau phosphorylation, already occurring at a high rate in neural stem cells (NSCs), coupled with less efficient clearance of the byproducts of tau hyperphosphorylation, as well as further increases in hyperphosphorylation during long migrations, could be the primary reasons behind these detrimental effects. This scenario involves NSCs in the subgranular zone (SGZ) of the hippocampal dentate gyrus (DG) in late-onset AD (LOAD) and NSCs in the ventricular-subventricular zone (V-SVZ) along the lateral ventricles in early-onset AD (EOAD), including familial AD (FAD). Neuroblasts carrying the initial seed of tau pathology travel throughout the brain by neuronal migration driven by complex signals, bringing the disease from the niches of adult neurogenesis to near (LOAD) or distant (EOAD) brain regions. At these locations, or in close proximity, a focus of degeneration begins to develop. Then, tau pathology spreads from the initial foci to large neuronal networks along neural connections by neuron-to-neuron transmission.
Therefore, the new core statement of the current theory is that AD starts in NSCs in the niches of adult neurogenesis. Interestingly, recent findings suggest that the current paradigm and the classic amyloid hypothesis might not be incompatible. In particular, some authors found clear evidence of intracellular oligomers of Aβ generated in NSCs within the SGZ niche at a very early stage in a transgenic AD mouse model [29]. In the current theory, this finding could be interpreted as proof that Aβ pathology not only indirectly influences tau pathogenesis by fostering neurogenesis and migration but also directly contributes to pathological tau hyperphosphorylation within these niches, most likely by activating GSK-3β. This view also reinforces the role of amyloid pathology as a leading factor in the current model, bridging the gap between the two theories.
In the first section of this manuscript, I propose the core hypothesis of tau pathogenesis in AD as linked to adult neurogenesis and migration. In the second section, I present the indirect microglia-mediated Aβ-tau interaction. In the third section, I consider the scaling of molecular pathology to the macroscopic brain. In the Discussion section, I put the new theory into context and consider the potential merits and limitations of the current proposal.
TAU PATHOGENESIS IS LINKED TO ADULT NEUROGENESIS AND MIGRATION
Tau isoform and phosphorylation during postnatal and adult neurogenesis
The tau isoform featured by the presence of three-repeat microtubule-binding domains (3R-tau) predominates at early developmental stages [30, 31]. The 3R-tau isoform exhibits a lower affinity for microtubules than the mature brain tau isoform [32], so it confers lower stability to the cytoskeleton and allows the morphological differentiation and migration of developing neurons. In contrast, 4R-tau is the predominant isoform at mature developmental stages [30, 31]. It binds microtubules with a greater affinity and displaces the previously bound 3R-tau from microtubules [30], guaranteeing the stability of the cytoskeleton required to maintain neuronal integrity. In addition to the presence or absence of exon 10 shaping the 4R- or 3R-tau isoform, tau phosphorylation is developmentally regulated: it is higher in fetal neurons and decreases with age during development [33–35]. As phosphorylation decreases the affinity of tau protein for microtubules [36], hyperphosphorylation of fetal tau [33, 37] contributes to maintaining a dynamic microtubule network as required by the outgrowth of axons during embryogenic neurogenesis [38].
In the adult human brain, both 3R- and 4R-tau are present, although in newborn neurons in the niches of adult neurogenesis, 3R-tau is the primary isoform [32, 39]. In particular, it has been demonstrated that 3R-tau is transiently expressed during the maturation of NSCs in the hippocampal SGZ [32, 38, 40, 41]. For instance, in rodents, individual new subgranular neurons exhibit the highest expression of 3R-tau when cells are 2 weeks old [40], and expression of this molecule is maintained until 4 weeks, a time point at which 3R-tau is replaced by 4R-tau [32]. Moreover, high tau phosphorylation in fetal epitopes is related to adult neurogenesis in both the V-SVZ and SGZ [39, 42], although fetal tau phosphorylation can be found in the adult brain in additional areas [35]. Transient expression of the 3R-tau isoform and fetal tau hyperphosphorylation in adult neurogenesis are not unexpected, considering that new neurons require a high degree of plasticity to migrate, differentiate, project axons, and integrate into the cell layer, and both the 3R-tau isoform and high phosphorylation guarantee a dynamic microtubule network [38, 39]. Furthermore, abnormally hyperphosphorylated tau in AD constitutes paired helical filaments (PHFs) of NFTs [43–47]. Interestingly, the 3R-tau isoform is said to facilitate PHFs, such as those seen in classical AD NFTs [48]. Additionally, several sites of hyperphosphorylation of the fetal 3R-tau during development were the same as those in the AD brain [33, 37, 49, 50]. Additionally, as already reported, GSK-3β is the first identified tau kinase [51, 52] that plays a key role in AD-like tau hyperphosphorylation [18, 19, 21, 53–55]. Interestingly, during development, expression of GSK-3β reaches its highest level in the late embryonic/early postnatal period, markedly decreasing with maturation into adulthood [56, 57]. More importantly, activated GSK-3β is believed to be the primary tau kinase in newborn neurons during adult neurogenesis [39, 58].
In summary, tau isoform and phosphorylation in NSCs during postnatal, as well as adult, neurogenesis seem equivalent to those predisposing to the typical tau alterations observed in AD. In this regard, it is worth noting that the primary cause of the tau functional change and NFT formation in AD is believed to be abnormal hyperphosphorylation [59–67]. In addition, abnormal tau hyperphosphorylation seems to reflect exaggerated physiological phosphorylation rather than disorganized phosphorylation at random sites [68, 69]. Therefore, tau pathogenesis in AD seems to depend exclusively on the extent of phosphorylation and the combination of multiple specific phosphorylation sites [38, 70, 71].
Differences between adult and postnatal neurogenesis associated with tau pathogenesis
Interestingly, although 3R-tau is said to facilitate PHFs, such as those seen in classical AD NFTs [48], and several sites of high phosphorylation of the fetal 3R-tau are the same as those in the AD brain [33, 37, 49, 50], during development, fetal tau remains functional and does not polymerize into NFTs. At this point, I speculate that a further crucial factor responsible for tau pathogenesis in AD could be found among those aspects that distinguish adult and postnatal neurogenesis. In this regard, it is worth noting that although postnatal and adult neurogenesis share some niches and signals, there are some important differences between the two.
The primary difference is that the neurogenic niches are surrounded by different environments. In particular, in the large, evolutionarily developed brain of adult mammals, neuroblasts originating in the neurogenic niches must migrate long distances through a complex and generally inhibitory environment [72] made up of neuronal, glial, and vascular networks to reach their destination [73]. Considering that tau hyperphosphorylation in neuroblasts contributes to maintaining a dynamic microtubule network that is amenable to migration, demanding and long migration through the inhibitory environment of the adult brain could have the detrimental effect of further increasing tau hyperphosphorylation. I believe this factor could be crucial to tau pathogenesis, especially in EOAD. Indeed, in the current theory, EOAD pathogenesis is linked to adult neurogenesis, especially in the V-SVZ. Here, migrating neuroblasts carrying the seeds of tau pathology deviate from the conventional rostral migration stream (RMS) to the olfactory bulb (OB) and take different and long migration paths toward various regions of the cortex, driven by complex signals released, in particular, by activated microglia.
I believe a further difference between adult and postnatal neurogenesis relevant to the current theory is related to clearance activity in the niches. In this respect, rapidly accumulating data suggest that autophagy fulfils some roles in NSC function [74, 75]. Specifically, autophagy may serve both a surveillance role by ensuring the quality of NSCs by degrading and eliminating intracellular components and aggregates and an elimination role by ensuring the removal of defective or damaged NSCs through its cell death promotion abilities [74, 75]. Consequently, reduced autophagy during aging compared to the postnatal period may contribute to the accumulation of hyperphosphorylated tau aggregates in NSCs. This event would be a crucial factor in AD pathogenesis, especially in LOAD. Indeed, in the current theory, LOAD is primarily linked to neurogenesis in the hippocampal SGZ, where neural precursors migrate very briefly to the granular layer of the DG. Accordingly, an increase in tau hyperphosphorylation through migration would be a less relevant factor in LOAD pathogenesis than in EOAD pathogenesis.
In summary, in the niches of adult neurogenesis, all conditions predispose patients to pathological tau hyperphosphorylation and accumulation of tau aggregates. When microglia activated by Aβ deposition foster neurogenesis in the niches and support long migrations, the situation is taken to extremes.
THE MICROGLIA-MEDIATED Aβ-TAU INTERACTION
According to the current theory, the slow progressive accumulation of Aβ deposits in the brain provokes microglial activation. Activated microglia foster the process of neurogenesis in niches and support neuronal migration. This “reactive” increase in neurogenesis and migration amplifies tau pathogenesis in AD, and available data in the literature seem to support this scenario.
Microglial activation in AD
Microglial activation in AD is well documented [76]. In the early AD brain, microglia are found in high densities surrounding Aβ plaques [77]. Both postmortem [78, 79] and “in vivo” clinical studies using PET ligands that bind to activated microglia [80, 81] have consistently confirmed the finding that microglia colocalize with amyloid plaques in AD. In particular, senile plaques are infiltrated by astrocytes and microglia in and around their central amyloid core [82, 83]. From this evidence, it has been proposed that Aβ plaques stimulate a chronic inflammatory reaction [84]. In other words, the activation and increased proliferation of microglia in AD [85] are thought to result from glial reactions to events related to the ongoing deposition of Aβ [86, 87]. In this regard, the high density of microglia found around Aβ plaques is consistent with their role in Aβ clearance pathways and their activation by Aβ itself [88]. Moreover, the finding that microglia possess a range of pattern recognition receptors, including Toll-like receptors, receptors for advanced glycation end products, and scavenger receptors, many of which can recognize different Aβ species through various interactions of differing affinities, support their role in Aβ clearance [89–91]. Once activated, microglia and astrocytes produce several proinflammatory signaling molecules, including cytokines, growth factors, complement molecules, cell adhesion molecules, and chemokines [84, 92, 93]. In agreement with these findings, increased levels of inflammatory cytokines and chemokines [78] as well as upregulated chemokine receptors [94], have been found in the AD brain.
Microglia and adult neurogenesis
At this point, it is relevant to the current theory to disclose that microglia have been found to have a role in adult neurogenesis.
Microglia modulates the production of new neurons
In particular, microglia can modulate the production of new neurons in the adult brain [88]. NSCs have been found to depend on signals from their niche to regulate their self-renewal, proliferation, and differentiation [88]. In the absence of microglia, NSCs progressively lose the capacity to undergo the differentiation process required for neurogenesis [95]. The role of microglia in neurogenesis can be seen as instructive, with microglial-secreted factors, such as IGF-1 and trypsinogen, having the capacity to regulate adult NSC proliferation and differentiation, promoting neurogenesis [96, 97]. Interestingly, both acute and chronic microglial activation can modulate neurogenesis [88]. In particular, “in vitro”, microglia acutely activated with lipopolysaccharide (LPS) strongly expressing IL-1, IL-6, and TNF-α have been found to reduce neural progenitor cell survival [98]. The study of some neurological disorders confirmed that overactive microglia might inhibit adult hippocampal neurogenesis [99–101]. For instance, microglia-mediated neuroinflammation can disrupt neurogenic niches and undermine the integrity of neuronal population replenishment programs [102]. Conversely, “in vitro”, microglia chronically activated by LPS with a secretory profile dominated by IL-10 and prostaglandin E2 (PGE2) are highly permissive to the neurogenic cascade [98]. The finding that microglia chronically activated with a definite secretory profile can support adult neurogenesis is consistent with the current theory in which Aβ plaques stimulate a chronic inflammatory reaction by microglia. In addition, LPS, IL-10 and PGE2 are implicated in AD [103–111].
Microglia has phagocytic activity
Of note, microglia serve a further function in adult neurogenesis. In young adult rats, approximately 9,000 new cells are generated in the SGZ each day, but nearly half do not survive [112, 113], similar to what occurs in the V-SVZ [113]. In particular, the majority of newborn neural progenitors undergo apoptosis 1–4 days after they are generated in the SGZ, and microglia phagocytose apoptotic debris from these cells to help maintain the equilibrium of the neurogenic niche [114]. Interestingly, this microglial phagocytic activity is apparently unchanged by aging or acute neuroinflammation, suggesting that it is a mechanism that promotes a homeostatic neurogenic niche in both healthy and disease states [114].
Microglia directs the migration of neuroblasts
Finally, microglia seem to have the capacity to direct the migration of neuroblasts [115]. Interestingly, some authors found in a transgenic mouse strain that depletion of microglia in the V-SVZ was linked to a marked reduction in neuroblasts reaching the OB with a concomitant accumulation of immature cells in the V-SVZ and RMS [116]. These findings suggest that microglia residing in the V-SVZ/RMS regions are critical for neuroblast survival and migration to the OB, possibly as a consequence of their release of the cytokines IL-4, IL-6, and IL-10 [116]. It is also important to note that microglia-mediated phagocytosis of neuroblasts is a rare phenomenon along the V-SVZ/RMS migratory pathway, and accordingly, markers of activated microglia, such as TREM2 and CD68, were undetectable in these regions [116]. In contrast, within the OB layers, where interneurons are continuously replaced by V-SVZ-generated precursors, microglia exhibit overt and robust phagocytosis. Therefore, microglia in the neurogenic areas of the V-SVZ/RMS are unique and specialized to support neural precursor proliferation and migration across significant distances to their final destination [117–119]. Further data supporting the role of microglia in sustaining and driving the migration of neuroblasts come from research on brain injury. In this regard, an invariant feature of damage to the CNS is the migration of microglial cells to the site of injury and their subsequent activation [120, 121]. Interestingly, several studies have shown that precursor cells preferentially migrate to sites of inflammation in animal models of multiple sclerosis and that these new cells preferentially differentiate into oligodendrocytes [122–124]. In contrast, in experimental models of more acute damage with neuronal loss, precursor cells, both extrinsically provided and endogenous precursor cells, migrated to the damaged area and differentiated into neurons [125–130]. More recently, some authors reported that precursor cells migrate from the V-SVZ and RMS to the injured cortex after traumatic brain injury (TBI) in mice and that prokineticin 2 (PROK2), a chemokine important for OB neurogenesis, is expressed exclusively by cortical microglia in the cortex as early as 24 h after injury [131]. In addition, the same authors demonstrated “in vitro” that cells expressing PROK2 directionally attract V-SVZ cells [131].
The role of astrocytes
The role of astrocytes, in addition to that of microglia, is in line with current theory, considering that astrocytes are early involved in AD [132–134] and have strong pro-neurogenic activity [135, 136].
Astrocytes in AD
Ramon y Cajal noticed reactive hypertrophic astrocytes surrounding senile plaques and blood vessels with amyloid deposits in post-mortem AD patients already in 1913 [137]. This observation has been replicated several times in AD patients’ brains [138–141] and in AD mouse models [142–144]. Within the CNS, astrocytes play a key role in the protection and repair of neuronal damage [145, 146]. Astrocytes respond to inflammatory substances and undergo a process known as reactive astrogliosis [147, 148] in various pathological conditions, including acute injury and progressive disorders such as tumors and AD [147]. Reactive astrocytes release molecules such as cytokines, chemokines, growth factors and gliotransmitters [149]. Notably, astrocytes release factors that promote axon growth, which are essential for synaptic formation and maturation in response to an injury [148, 150]. Moreover, astrocytes increase neuronal viability and mitochondrial biogenesis, protecting neural cells from oxidative stress and inflammation induced by amyloid peptides [151]. At the same time, astrocytes may exert neuroprotection at different stages of AD. Indeed, both astrogliosis and microgliosis, in response to amyloid, increase glial secretion of transforming growth factor-β (TGF-β), which protects neurons from amyloid toxicity and increases amyloid clearance by microglia [152, 153]. Furthermore, astrocytes surrounding amyloid plaques show phagocytic activity and are able to phagocytize neuritic dystrophies both in mouse models and in AD patients’ brains [154]. Indeed, astrocytes are part of the brain’s glymphatic system, a clearance system for proteins and soluble solutes [133]. The astrocyte water channel aquaporin-4, expressed at the ends of astrocytes, facilitates this process and is important for Aβ clearance [155, 156] and probably also for tau clearance [133].
Astrocytes and adult neurogenesis
Astrocyte is the main cell type in the hippocampal niche of neurogenesis by number [135]. In the molecular layer of the dentate gyrus and in the SVZ [157], astrocytes are in close contact with NSCs and contribute to the regulation of almost all stages of adult neurogenesis, from the proliferation of NSCs to the functional integration of new neurons [135]. In particular, molecules secreted by astrocytes increase the proliferation of adult NSCs. For example, in vitro studies found that the adenosine 50-triphosphate (ATP) through P2Y1-PLC-phosphatidylinositol 3-kinase (PI3K) signaling [158], the N-methyl-D-aspartate receptor co-agonist (NMDAR) D-serine [159–161] and the fibroblast growth factor-2 (FGF2) act as factors in the proliferative induction of adult NSCs [162, 163]. In addition, several miRNAs expressed in astrocyte exosomes are known to regulate adult neurogenesis [164]. In addition to affecting proliferation, molecules secreted by astrocytes can also modulate other stages of adult neurogenesis, such as the migration and differentiation of progenitors into neurons, or the maturation, synaptic integration, and survival of newborn neurons [135]. For example, the first in vitro study examining the role of astrocytes in adult neurogenesis in the hippocampus showed that astrocyte-conditioned cell culture medium increases the differentiation of NSCs into neurons [165]. Neuronal differentiation of adult NSCs is also promoted in vitro through juxtacrine signaling by astrocyte secretion of ephrin-B2 and activation of EphB4 receptors on the stem cell [166]. In addition, astrocyte-derived soluble factor thrombospondin-1 (TSP1) is known for its antiangiogenic activity and promotion of synaptogenesis during brain development [167]; it also increases adult NSC proliferation and neuronal differentiation in vitro [167]. Consistently, adult TSP1-deficient mice exhibit reduced proliferation of adult NSCs [168]. Another secretory factor, neurogenesin-1, increases the neuronal fate of newly formed hippocampal cells [169], while IL-1b and IL-6 promote neuronal differentiation of adult NSCs/progenitors in vitro [170]. Finally, D-serine released from astrocytes has been shown to control dendritic maturation and functional integration of newborn hippocampal neurons [171].
Detrimental effects of glial cells in AD
The current theory focuses mainly on the neuroprotective functions of microglia and astrocytes in AD and their proneurogenic actions. However, it is worth noting that both types of glial cells have been found to contribute to the damaging effects in AD, mainly through the promotion of innate immunity and pro-inflammation and influencing the permeability of the blood-brain barrier [172, 173]. Specifically, both microglia and astrocytes interact with Aβ, and Aβ in turn activates microglia and astrocytes through TLRs to release neuroinflammatory mediators that promote neurodegeneration [174, 175]. Pro-inflammatory cytokines decrease the phagocytic activity of microglia and probably transform microglia into pro-inflammatory phenotypes [173]. In addition, pro-inflammatory microglia increase tau phosphorylation and aggravate tau pathology [176]. At the same time, reactive astrocytes have been found to release excessive amounts of GABA and glutamate, resulting in memory impairment and synaptic loss in an animal model of AD [177, 178]. Moreover, these cells contribute to the microcirculation dysregulation and blood-brain barrier disruption, which facilitates Aβ accumulation and disease progression [179, 180]. Finally, reactive astrocytes might even pave the way for the formation of early amyloid plaques [181]. Considering that AD has a long preclinical phase, this dual role of glial cells [182] is not incompatible with the current theory that easily explains the very early stages of the disease. Moreover, some aspects of neuroinflammation, even under chronic conditions, continue to promote microglial phagocytosis and Aβ containment, resulting in a neuroprotective function [183].
THE SCALING OF MOLECULAR PATHOLOGY TO THE MACROSCOPIC BRAIN
Braak staging model
According to Braak’s neuropathological staging [184–187], pathological tau aggregates in AD develop first in nerve cells of brainstem nuclei (subcortical stages a–c) that have projections ending in the cerebral cortex [188–190]. It appears that from the locus coeruleus (LC) of the pontine tegmentum [191–196], the lesions progress to a distinct portion of the cerebral cortex, the transentorhinal cortex (TEC) [197]. In cortical projection neurons, the resultant and originally nonargyrophilic pretangle protein, during cortical stages 1a and 1b, becomes transformed into argyrophilic neurofibrillary lesions that characterize subsequent NFT stages I–VI [189]. The neurofibrillary pathology advances from the TEC (NFT stage I) into the OB [198], the entorhinal cortex (EC), and the hippocampal formation (NFT stage II). During NFT stage III, tau pathology progresses from the TEC to the laterally adjoining basal temporal neocortex, and during NFT stage IV, it extends more widely to the temporal, insular, and frontal neocortices. In NFT stage V, cases display severe involvement of most neocortical association areas, leaving only the primary fields mildly involved or intact. In the end stage, NFT stage VI, even these areas become involved. The production of abnormal tau continues from the outset until the final stage of the pathological process [188, 189, 199]. In summary, in AD, the pathology progresses anterogradely from distinct predilection sites in the lower brainstem to distant but connected regions of the cerebral cortex, and it does so sequentially with little interindividual variation, albeit at different rates [189]. Considering the mechanism implicated in tau spreading, a great deal of data suggest that transcellular propagation of tau aggregates, or seeds, could underlie disease progression [200–207].
The prion-like seeding and spreading hypothesis of tau
According to the prion-like hypothesis, pathological tau can distribute from one cell to another, thus propagating pathology from affected brain areas to interconnected healthy areas, involving mechanisms similar to those of prion diseases [208]. This hypothesis could explain the hierarchical pathway of neurodegeneration described in Braak’s scheme [184]. The prion-like hypothesis involves two main stages, namely the seeding, that is the ability of abnormal tau to convert normal tau into a pathological form, and the propagation, that is the spread of pathological tau to connected neurons [209].
Abnormal tau has seeding capacity
Several studies support a seeding capacity of tau similar to that of prions [209]. In this regard, some authors showed that injection of tau aggregates extracted from mice overexpressing mutated tau (P301S) into mice overexpressing wild-type human tau is sufficient to induce tau pathology [201]. In particular, when a tau immunodepleted extract was injected, no pathology was detected, demonstrating that tau is the responsible factor of aggregation, as later confirmed by other research groups [210–212]. Most in vitro studies showed that incubated aggregates/seeds are internalized by endocytosis and promote aggregation of overexpressed tau in cell lines [203, 204, 210, 213–221]. Evidence that tau aggregates have prion-like seeding behavior come mostly from experimental models [222]. However, there is also evidence for seeding activity in tau aggregates derived from patients with tauopathy. Indeed, sarkosyl-insoluble PHFs extracted from AD brain tissue induce seeding in cultured cells and wild-type mice [223, 224]. In addition, in brain homogenates and cerebrospinal fluid (CSF) from AD cases, tau seeds have been found to induce non-aggregated tau aggregation in FRET-based biosensor assays, particularly in regions known to be devoid of phospho-tau deposits [220, 225]. Moreover, recent work showed that human CSF from AD patients can induce tau seeding in experimental models [226].
Seeded tau aggregation is templated
Other studies demonstrated that the seeded tau aggregation is templated [222]. Some authors, for example, observed that native P301S tau seeds derived from transgenic mice brains confer their highest seeding competence to less competent recombinant P301S tau seeds when co-incubated with them in vitro [227]. Under the light microscope, tau aggregates induced in cells or in vivo have the same morphological appearance as the parent tau seed, suggesting a templated conversion mechanism [222]. This has been demonstrated in many studies from Diamond’s laboratory, in which the formation of morphologically distinct tau seeds resembles the parent tau seed both in cell culture [228] and, more recently, in vivo [225]. That a templated conversion mechanism may be relevant to tauopathies in humans has been demonstrated by studies from the laboratories of Goedert and Tolnay, in which injection of brain homogenates of different tauopathies into the brains of mice expressing unaggregated human tau resulted in the formation of only the inclusions of the corresponding tauopathy [229].
Neuroanatomical spread of tau aggregates
Trans-synaptic propagation of pathological tau has been demonstrated using a number of different approaches in transgenic mice [222]. Some authors showed not only the induction of tau aggregation in rodent brains following intracerebral injection of brain homogenates containing tau seeds, but also the time-dependent appearance of tau pathology in anatomically connected brain regions [201]. Others reported the appearance of pathological tau in areas connected to sites injected with tau seeds or tau-expressing viral vectors [230–233]. Some authors used a model in which human tau expression was restricted to the entorhinal cortex alone, showing that the tau pathology was evident in anatomically connected regions that did not express the human tau transgene [202, 205]. Further studies in tau transgenic mice indicated that tau seeds predict disease spread by appearing in brain regions before the occurrence of any other pathological changes [215]. Interestingly, this finding explains the histopathological observation made by some authors more than 20 years ago [234]. These authors reported the absence of pathological tau in a frontal cortical region that was anatomically disconnected from the limbic region following neurosurgery, decades before the patient developed AD. Conversely, the authors found an extensive tau pathology in the immediately adjacent brain regions and limbic and isocortical areas [234].
Propagation involves several steps
The propagation of pathological tau to connected neurons consists of at least four steps [235]. First, tau must be secreted or released from donor neurons; second, it must undergo aggregation before or after being released; third, tau must be taken up by recipient neurons; and fourth, tau aggregation must be induced in recipient cells [236].
Currently, there is evidence that misfolded tau is indeed secreted [209, 212, 237]. However, the nature of secreted tau is debated in the literature [222, 238]. Tau is secreted mainly in free form [239–242], but it is also found within nanotubes [243, 244] or associated with extracellular vesicles (EVs) [245], such as exosomes [242, 246, 247] and ectosomes [239]. While nanotubes may be difficult to visualize in the human brain, phospho-tau-containing EVs have been found not only in the brains of transgenic mice [248, 249], but also in peripheral fluids (CSFs) [242, 250] and blood [251–253] of AD patients [209].
Considering the uptake step of tau by an adjacent recipient cell, in vitro studies showed that extracellular aggregates of tau can be internalized by naïve cells by promoting fibrillation of intracellular tau [203, 204, 254]. Tau pathology can be transferred between co-cultured cells [203, 204, 254] and also through synaptic contacts between neurons that facilitate the propagation of pathology [255]. Intracranial or peripheral administration of pathological tau [222, 256] and in vitro experiments have shown that tau is mainly internalized by active endocytic processes [203, 257]. In particular, three types of endocytosis have been described: bulk-endocytosis, actin-dependent macropinocytosis mediated by HSPGs on the cell surface, and clathrin-mediated endocytosis [208].
Once internalized, tau can escape endosomal vesicles by inducing their rupture [258, 259] and accumulate in the cytoplasm where it becomes a potential template for tau misfolding [208]. Indeed, pathogenic misfolded tau proteins act as “seeds” that recruit soluble endogenous tau into larger aberrant conformations [260] that slowly propagate into interconnected brain regions, as demonstrated in various animal models [235]. Although the biochemical mechanisms that drive the conversion of normal tau to the pathological form remain unclear, several models of tau seeding have been proposed [261, 262]. Finally, transcellular transfer of tau aggregates between serially cultured cells in microfluidic chambers was demonstrated [263]. In addition, diffusion of tau from neuron to neuron through trans-synaptic connections via exosomes has been reported to seed aggregates [242, 249]. However, other mechanisms that do not require secretion but a direct connection between cytoplasm might be involved [222]. Indeed, a recent work showed that nanotubes promote the interneuronal transfer of tau fibrils into neurons [243, 244].
Limitations of the seeding and spreading hypothesis
Although several pieces of evidence seem to support the seeding and spreading hypothesis of tau, many points still remain to be clarified [68, 264, 265]. Firstly, some authors pointed out that the methods used and data collected in some studies supporting the hypothesis are not all without some limitations [68, 265]. Secondly, the biochemical mechanism that drives the conversion of normal tau to the pathological form is still not clear [208]. Thirdly, the exact nature of the tau seeds responsible for the propagation of tau pathology remains controversial [222]. Furthermore, the specific pathways and mechanisms underlying the spread of pathological tau, including the mechanism of releasing from donor neurons and subsequent uptake by recipient neurons in AD, remain unclear [209, 235, 266]. In addition, the molecular forms of extracellular tau are not fully understood, and the physiological or pathological functions of this extracellular tau remain unknown [266]. Further investigations are then needed to clarify the relationship between the propagation of tau aggregates and tau-induced toxicity and degeneration [222]. Furthermore, it cannot be ruled out that genetic variants identified as risk factors for tauopathies play a role in the propagation of tau pathology, but many more studies are needed to document this [222]. Finally, the contribution of selective vulnerability of neuronal populations as an alternative explanation of the spread of tau pathology needs to be clarified [222].
Microglia and astrocytes could be involved in the spread of pathological tau
It is worth noting that some studies highlighted the involvement of microglia in the spread of pathological tau [208]. Indeed, it has been reported that increased microglial activation accelerates the propagation of tau in the brain [267]. Furthermore, microglia were found to promote tau propagation [246, 268], as supported by the marked reduction in tau propagation through microglia depletion in two independent models of tauopathy [246]. The mechanism involved in the promotion of tau propagation by microglia has not been fully elucidated. However, tau was found in the EVs in the CSF of individuals with AD [239], and microglia were found to internalize tau seeds and degrade them [182]. When microglia fail to degrade these tau seeds, deleterious consequences occur, including the secretion of tau-containing exosomes that can spread to neurons [182].
Interestingly, tau was also found in astrocytes of individuals with AD [269]. Although tau was found in glial cells [270], astrocytes do not express this protein under physiological conditions [271], and the origin of tau in astrocytes in AD is still unclear [272]. One unproven possibility is that AD progression induces the translation of tau from the mRNA present in astrocytes [273]. Alternatively, astrocytes could also capture extracellular tau [228, 274, 275]. In this regard, astrocytes have specific heparin sulfate proteoglycans (HSPGs) and receptors, such as low-density lipoprotein receptor-related protein 1 (LDR1), that can mediate the uptake of tau aggregates [133]. Aggregates can be internalized and processed by various mechanisms, including lysosomal degradation. Disruption of aquaporin-4 in perivascular astrocyte ends may contribute to the disruption of tau clearance and accumulation of tau aggregates in the CNS [133]. However, the microglial and especially astrocytic mechanisms that may contribute to pathological tau seeding are not yet fully understood [133]. As for microglia, it is sufficient to mention that they can also reduce the seeding activity of tau [268, 276–278], supporting the idea that microglia are indeed able to limit or promote the spread of tau [182]. Considering astrocytes, one hypothesis is that tau pathology spreads from one astrocyte to another, possibly through astrocyte gap junction networks and tunneling nanotubes across brain regions [266, 279]. Astrocyte engulfment of tau-containing synapses may be another pathway by which astrocytes contribute to the spread of tau in AD [133].
The possible involvement of glial cells in tau spreading is consistent with the current theory. In this case, microglia and astrocytes activated by Aβ deposition would not only promote tau pathogenesis through their proneurogenic effect, but also contribute to the spread of tau aggregates in the brain.
Open questions left in the Braak model
Despite the indubitable value of the Braak staging model, some open questions remain.
First, the view that nonthalamic nuclei would be the first site of tau pathology has been questioned [280]. Indeed, these nuclei are equipped with a type of termination (i.e., nonjunctional varicosities) [191, 196, 281, 282], supporting a diffusive mode of transmission [191, 196, 281, 283–285] that is not suitable for neuron-to-neuron transmission of abnormal tau as provided by the model. Indeed, recent findings have suggested that TEC/EC are actually the first site that develops early tau pathology [286]. In particular, tau seeding activity that precedes detectable NFTs was found in the LC only after it was already prominent in the TEC/EC, i.e., at later NFT stages (IV–VI), suggesting the idea that tau seeds spread from the TEC/EC to the LC and then to more distant cortical regions [286].
Second, to date, a clear explanation of why tau pathology begins in TEC/EC seems lacking. Indeed, it is not easy to contextualize this finding in the frame of classical amyloid theory, considering that amyloid plaques first appear in the association cortices of the temporal lobe, at some distance from the TEC/EC where damaged neurons containing NFTs are first found [187, 287]. Additionally, further studies examining the early degeneration of the lateral EC have reported that levels of amyloid peptides in this region are not higher than those in other, less affected regions [288, 289].
Third, despite the evidence that early tau pathology emerges in entorhinal layer II cells at Braak stage II [186] and that these cells project to the DG by the perforant path [290–294], the DG is not affected by tau pathology at early stages. Indeed, granule cells of the DG remain uninvolved in Braak stage III, and some tau pathology emerges only at stage IV [186]. At the same time, hippocampal CA1 cells receiving projections from entorhinal layer III cells [292, 293] are impacted far earlier, starting at Braak stage II [186].
Finally, a clear and accepted explanation of the peculiar regional distribution of tau lesions and subsequent neurodegeneration in EOAD, especially in the syndromic variants of AD, is lacking in the Braak model.
A new model of tau spreading in the medial temporal lobe
The current theory provides a new model of tau spreading in the medial temporal lobe (MTL) (Fig. 1). According to the new model, tau pathology begins in NSCs within the niches of adult neurogenesis. In particular, in LOAD, the initial tau pathology, likely in the form of soluble aggregates of misfolded and hyperphosphorylated but nonfibrillar tau protein [280], originates in the SGZ in the hippocampal DG. From this site, seeds of tau pathology spread retrogradely to the EC through the perforant path. From here on out, anterograde transmission flanks retrograde transmission. Therefore, by virtue of reciprocal connections between the EC and TEC [294–297], the TEC receives tau seeds both anterogradely and retrogradely. In this regard, it is interesting to note that the EC projections to the TEC are input to layer II [296, 297], where NFTs are first found [185, 298]. Then, from EC/TEC, the pattern of distribution of tau pathology follows the Braak model. However, a further difference may occur. Indeed, according to the current model, it cannot be excluded that some further foci of degeneration could start locally in some regions of the cortex, provoked by neuroblasts carrying the seeds of tau pathology arising from the V-SVZ niche. In this case, in LOAD, the primary regional distribution pattern of tau pathology in the MTL would be complicated by concurrent foci of pathology that emerge locally in neocortical regions. In the same context, it is noteworthy that the OB is the arrival point of migrating neuroblasts from the V-SVZ along the RMS, and at the same time, many findings support early involvement of this region in AD [198, 299–301]. However, olfactory structures, including the OB, anterior olfactory nucleus, and piriform cortex, send projections to the superficial layers of EC [302, 303]. Therefore, retrograde transmission of tau pathology from the EC to the OB could be a more parsimonious explanation. The current model seems plausible and coherent with some findings in the literature. In this regard, it is worth noting that retrograde transmission has been found to be possible. Indeed, projection neurons generate long axons to transmit information from one site to another, and for this purpose, their axons have mechanisms for both anterograde and retrograde transport of various cargos [222, 280]. Moreover, the idea of retrograde transmission of tau pathology in the MTL in AD has already been suggested [304]. In addition, the pattern of connections among the DG, EC and TEC is compatible with the new model of both retrograde and anterograde transmission. In particular, the DG receives projections from entorhinal layer II cells, where tau pathology is found early in EC [186, 305]. Moreover, projections from the EC to the perirhinal cortex that includes the TEC terminate most heavily in and around layer II, where tau pathology is first found in the TEC [184, 298].
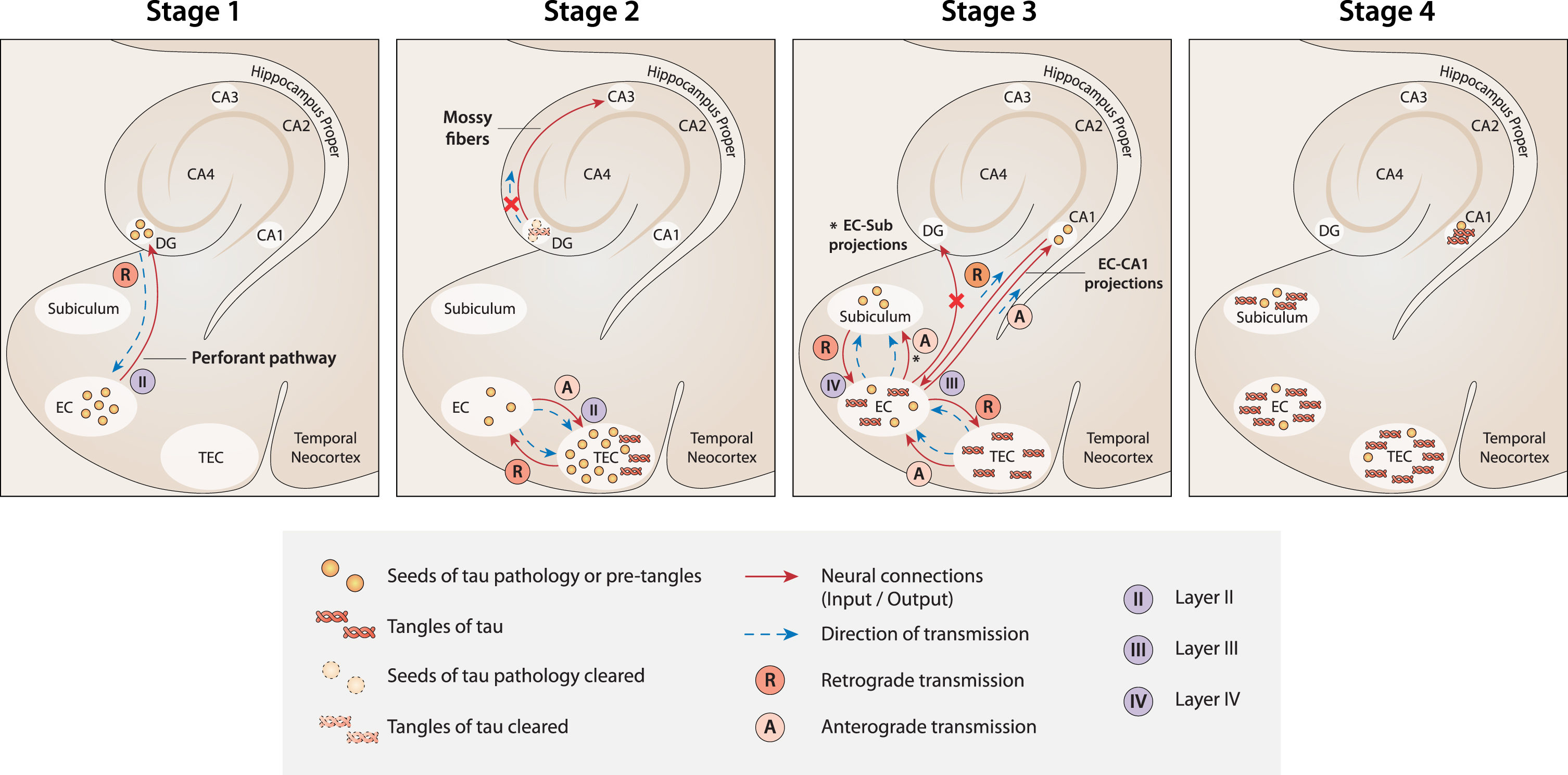
Fig. 1
A new model of the spread of tau pathology in the MTL according to current theory. Stage 1: The first seeds of tau pathology develop in NSCs in the SGZ niche of the DG. Then, they spread from the DG to the EC by retrograde transmission along the connections of the perforant pathway. Stage 2: Seeds of tau pathology spread from the EC to the TEC by both anterograde and retrograde transmission along the multiple connections between the two regions. Because of the massive load of tau pathology accumulated in the TEC, tangles develop here first. At the same time, tangle formation is suppressed (or delayed) in the DG because of the strong clearance activity that usually occurs in the neurogenesis niches. As a result, the transmission of tau pathology from the DG to CA3 is nullified (red cross). Stage 3: Seeds of tau pathology spread from the EC to the CA1 and subiculum along the EC-CA1 and EC-subiculum projections, respectively. In contrast, the seeds of tau pathology would not spread anterogradely to the DG (red cross), because the neuronal connections between the EC and DG are already deteriorated at this stage due to the transmission of tau pathology in the reverse (retrograde) direction in the previous stage (Stage 1). Stage 4. When tau tangles emerge in the CA1 and subiculum (starting from Braak stage II), the DG and CA3 are not yet affected. Figure 1 was produced by Antonio Garcia, scientific illustrator from Bio-Graphics.
Possible solution to the questions left in Braak staging
Interestingly, the current model seems to offer possible explanations for the open questions left in Braak staging.
First, it is coherent with the recent view considering the LC and other nonthalamic nuclei as not the primary sites for tau spreading in the MTL. At the same time, the core hypothesis of tau pathogenesis as linked to adult neurogenesis and migration offers a speculative explanation for the emergence of tau pathology in these sites. In fact, the LC is highly connected to the hypothalamus, and constitutive neurogenesis in the adult hypothalamus of mammals, including rodents, rats, mice, voles [306–314], and sheep [315], has been documented [316].
Second, according to the current model, tau pathology emerges first in the TEC because this region receives the massive seeds of tau pathology from the EC, both anterogradely and retrogradely, through the multiple reciprocal connections between the two regions. Conversely, the EC receives the seeds of tau pathology from the DG only by retrograde transmission (Fig. 1, Stage 1). Therefore, in the current model, NFTs emerge first in TEC because the initial load of tau pathology would be greater than in the EC (Fig. 1, Stage 2).
Third, the absence of tau pathology in the DG at early Braak stages is due to the strong activity of clearance usually occurring in the neurogenic niches [74, 75]. According to this view, although seeds of tau pathology originate in NSCs in the SGZ niche and spread retrogradely toward the EC by a perforant path, the formation and accumulation of NFTs is suppressed or at least delayed in NSCs in the DG (Fig. 1, Stage 2). For the same reason, CA3 [118], which receives excitatory outputs from the DG, is not impacted by tau pathology at early stages. Instead, tau pathology emerges first in CA3 at Braak stages III-IV [186]. In the same context, it is relevant to note that adult-generated neurons in the SGZ receive local connections from multiple types of GABAergic interneurons [317], whose inputs to the niche are fundamental for maintaining a healthy level of neurogenesis under normal conditions [318, 319]. Interestingly, these same GABAergic interneurons have been shown to be particularly vulnerable to AD pathologies, such as NFTs of phosphorylated tau protein [317, 320–323]. Therefore, GABAergic interneurons could be plausible candidates to convey transmission of the first tau seeds originating in SGZ NSCs. Strictly related to the previous point, the current model seems to offer a plausible explanation for why tau pathology spreads early from the EC overall to the hippocampal CA1 region, while it seems not to target the DG, despite that the EC and the DG are highly connected by the perforant path. Damage to the perforant path between the lateral EC and DG occurs unusually early in AD [324]. The long axons of projection neurons are in fact not well equipped to degrade or eliminate pathological proteinaceous aggregates [189, 325]. Based on the current model, the seeds of tau pathology, after which the EC, and more so the TEC, have been impacted, would spread anterogradely from the EC to multiple regions, as in the classical Braak model. However, at this point, the current view predicts that the DG would be primarily disconnected from the EC because the entorhinal perforant projections toward the DG would already be deteriorated due to the precedent retrograde transmission of tau pathology along the same projections in the opposite direction, from the DG to the EC, during the first stage of disease. Consequently, the connections between the EC and CA1 (and subiculum) are unique undamaged fibers in the perforant path available for tau seed transmission at this stage (Fig. 1, Stage 3).
Finally, the current model seems to offer a plausible explanation for the peculiar distribution of tau pathology in EOAD. Individuals with EOAD may present with striking neurobehavioral phenotypes, reflecting damage to the language systems [326], visual systems [327], or frontal-executive systems [328]. In general, EOAD is more likely to present with atypical clinical phenotypes than LOAD patients [329]. In one study, approximately 25% of EOAD patients presented with a nonamnestic phenotype in whom visual or apraxic and language phenotypes predominated [330]. In another study, almost 60–70% of EOAD patients exhibited atypical patterns of brain atrophy [329]. Interestingly, the focus and system-specific neurobehavioral features in EOAD variants do not reflect regional accentuation of Aβ, but they do show strong correlations with the pattern of glucose hypometabolism and atrophy [331–334]. More generally, despite their differences, in autosomal dominant AD (ADAD), EOAD and LOAD, the distribution of Aβ deposition throughout the brain is similar (with the exception of Aβ deposition in the striatum in ADAD), affecting large confluent areas of the association cortex and overlapping with a set of brain regions active at rest [335–339]. Therefore, phenotypic heterogeneity in AD is not easy to explain considering both the frame of the classical amyloid theory and the primary pattern of regional distribution of tau pathology starting from the MTL according to Braak staging. In the current model, EOAD and LOAD exhibit different regional distributions of tau pathology and subsequent degeneration because the V-SVZ niche is primarily active in EOAD, while the SGZ niche is primarily active in LOAD. Accordingly, EOAD especially impacts regions on the dorsal cortex, whereas LOAD impacts the MTL [340] (Fig. 2). Moreover, activated microglia surrounding Aβ plaques release chemokines that attract and drive migrating neuroblasts toward the regions of Aβ deposition, similar to what happens in brain injury [125–131]. Consequently, especially in EOAD, migrating neuroblasts deviate from the RMS to the OB and take different paths toward various regions of the cortex, carrying the seeds of tau pathology to those locations. In summary, the redirection of migrating neuroblasts to multiple possible destinations in the cortex is the basis of heterogeneity in the regional distribution of tau pathology and subsequent degeneration in atypical EOAD syndromes (Fig. 2). In addition, as already reported, long-distance migration throughout the inhibitory environment of the adult brain could contribute to augmenting tau phosphorylation.
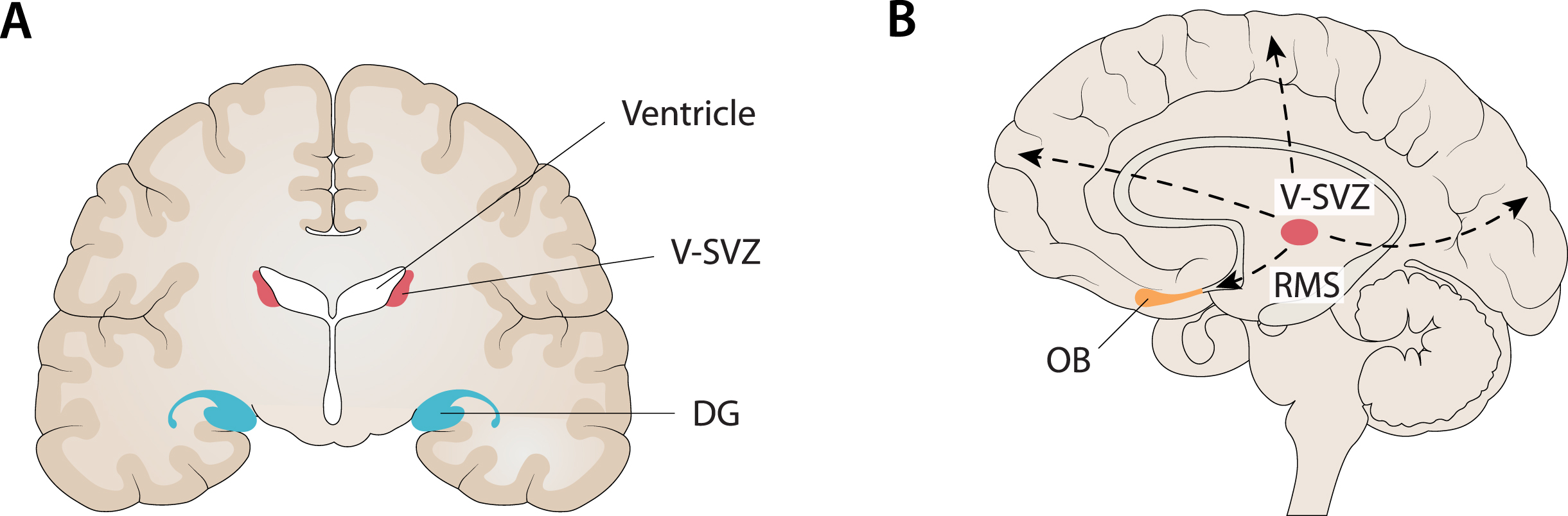
Fig. 2
Compatibility between the localization of the main niches of adult neurogenesis and the core regions targeted in AD. A) One of the main niches of adult neurogenesis is the sub-granular zone (SGZ) of the dentate gyrus (DG) in the hippocampus. At the same time, the hippocampus is the first major region targeted in AD, especially when late-onset AD (LOAD) is considered. B) Another main niche in adult neurogenesis is the ventricular subventricular zone (V-SVZ) along the lateral ventricles. From this niche, through several long migrations to the cortex (dashed lines), it is possible to reach every cortical region (e.g., frontal, fronto-parietal, occipital) that is targeted by AD, especially when considering early-onset AD (EOAD) and syndromic variants of AD. In addition, it is noteworthy that the olfactory bulb (OB) is the end point of neuroblasts migrating from the V-SVZ along the rostral migratory stream (RMS) and, at the same time, many findings support an early involvement of this region in AD. Figure 2 was produced by Antonio Garcia, scientific illustrator from Bio-Graphics.
Interestingly, recent in vivo tau-PET imaging studies in AD have revealed substantial heterogeneity in tau deposition patterns with significant deviations from Braak’s scheme [334, 341, 342]. These findings are in line with the four subtypes previously identified from neuropathology and neuroimaging studies based on the distribution of NFTs and patterns of brain atrophy, respectively: hippocampal-sparing AD, limbic-predominant AD, typical AD, and minimal atrophy AD [343–354]. In addition, some studies confirmed atypical patterns of tau deposition with elevated tau-PET signal in the occipital and parietal cortex [355], left temporo-parietal areas (logopenic) [356] and, similarly, perirolandic areas (corticobasal syndrome due to AD) [357, 358] reflecting the clinical variants most frequently associated with EOAD [359, 360]. Interestingly, some of these tau-PET imaging studies in AD found that pathological tau accumulates in the associative cortex, completely sparing the hippocampus [342, 350, 359, 361, 362]. This finding strongly supports the idea of distinct foci of early tau deposition and multiple pathways of tau diffusion in AD, including cases without any involvement of the hippocampus and/or entorhinal cortex [342, 348].
This scenario does not fit well with Braak’s staging system and seems more consistent with the current theory that predicts different niches of adult neurogenesis and multiple pathways of migration to the cortex.
Cortical arealization in development and AD
Considering that migration paths throughout the cortex, including the RMS, are mostly quiescent in the human adult cortex, the current model would be plausible only provided that a strong pro-neurogenic action would foster neurogenesis and support highly demanding migrations. In this respect, the accumulation of Aβ deposits and consequent microglial activation are key factors. However, the distribution of Aβ throughout the brain is diffuse and similar in LOAD, EOAD, and FAD, as well as in AD variants. Therefore, it is not plausible that microglia surrounding Aβ plaques signal a precise direction to migrating neuroblasts, similar to focal brain insults, such as stroke. Furthermore, the current model cannot explain why only some directions of migration are undertaken—those corresponding to the paths toward the regions impacted in well-known AD variants, e.g., posterior, frontal, and left perisylvian—and not others. At this point, I speculate that not only should a further source of signals drive migration throughout the cortex in EOAD but also that this source should contain information about brain topography, likely at the macroscopic level of hemispheres, lobes, and gyres, to efficiently work. Surprisingly, I found that the program under cortical arealization in development perfectly fits this idea. In particular, there is a complex mechanism regulating the progressive patterning and correct localization of brain areas during development [363], which necessarily uses some spatial information related to brain topography to work. This mechanism would be mostly, even if not exclusively, under the genetic control of factors with discrete expression in the cortical field (protomap models). Moreover, some findings have suggested that the main spatial information used is related to simple brain axes. In particular, animal studies have demonstrated that there is an anterior-posterior (A-P) gradient of gene expression of morphogens or transcription factors, such that specific genetic factors enlarge rostral (motor) areas at the expense of caudal (sensory) areas, and vice versa [363]. In addition to this A-P gradient, there is evidence for graded expression patterns along with other distributions, including the medial-lateral (M-L) and dorsal-ventral (D-V) axes.
The failure of certain processes (e.g., cell proliferation, migration, and abnormal organization) during cortex development has been associated with several cortical malformations [364]. What is interesting is that most malformations do not involve the entire cortex uniformly but have regions of maximum severity. For example, some malformations (schizencephaly, megalencephaly) may alternately involve one or both hemispheres. Another type of malformation (e.g., lissencephaly) may have two forms, one with maximum severity in the frontal lobes and the other with maximum severity in the occipital lobes [364]. Another more diverse malformation (i.e., polymicrogyria) shows a highly heterogeneous topographic distribution (e.g., frontal, frontoparietal, perisylvian, parasagittal parieto-occipital, parietal, generalized), with a predilection for the perisylvian cortex [365]. As might be expected considering that the malformations are due to the failure of certain processes during cortical development, by observing the distribution over the cortex of some of these developmental malformations, we can easily recognize the structure of the A-P, D-V, and M-L axes underlying the cortical arealization process (Fig. 3). Interestingly, the regions targeted by degeneration during early stages in AD, considering LOAD, EOAD, FAD and all the syndromic variants together, seem to be arranged at opposite locations along the same A-P, D-V, and M-L brain axes [366] (Fig. 3). In other words, AD (and more specifically EOAD) and the program of cortical arealization in development seem to use the same alphabet of spatial information on brain topography (Fig. 3).
Fig. 3
Cortical arealization in development and AD appear to share the same alphabet of spatial information about brain topography. The coarse distribution over the cortex of brain malformations due to the failure of the arealization program during development and the key regions targeted by AD, considering all phenotypes, seem to follow the same few topographical instructions related to anterior-posterior, medial-lateral, dorsal-ventral brain axes and a simple left-right hemisphere specification. Figure 3 was produced by Antonio Garcia, scientific illustrator from Bio-Graphics. This is a modified version of Fig. 1 in Abbate (2018) [366].
![Cortical arealization in development and AD appear to share the same alphabet of spatial information about brain topography. The coarse distribution over the cortex of brain malformations due to the failure of the arealization program during development and the key regions targeted by AD, considering all phenotypes, seem to follow the same few topographical instructions related to anterior-posterior, medial-lateral, dorsal-ventral brain axes and a simple left-right hemisphere specification. Figure 3 was produced by Antonio Garcia, scientific illustrator from Bio-Graphics. This is a modified version of Fig. 1 in Abbate (2018) [366].](https://content.iospress.com:443/media/jad/2023/93-4/jad-93-4-jad221279/jad-93-jad221279-g003.jpg)
In summary, I speculate that in EOAD, when neuroblasts leave the V-SVZ niche and start migrating, the signals from microglia activated by Aβ deposition provoke path redirection from the RMS and, at the same time, sustain long-distance migration. However, reactivation of the genetic program of arealization during development would contribute to signaling the direction for migrating neuroblasts to follow.
DISCUSSION
Adult neurogenesis in brain injury and AD
The study of adult neurogenesis and migration in brain injury, keeping in mind the peculiarity of Aβ deposition compared to other types of injury, seems to support the notions of increased neurogenesis, promotion, and redirection of neuroblast migration, as well as reactivation of quiescent paths, recognized in the current theory. Indeed, in rodents, various pathological changes and injuries, e.g., ischemia or TBI, stimulate neurogenesis in the V-SVZ [367–369] and in the DG [370–374]. In addition, in the injured adult brain, neuroblasts generated in the V-SVZ migrate toward the site of injury [125, 126, 375–377], driven by various guidance cues, such as chemoattractants secreted by injury-activated astrocytes, microglia, and vascular endothelial cells in the injured area [73, 378]. Accordingly, multiple studies have shown significant intensification of neuroblast migration [125, 126, 367, 379] under these conditions. Therefore, the migratory paths from the V-SVZ, which are largely quiescent in the adult brain [380], could be reactivated in response to injury [367].
Over the years epileptic seizures, as well as stroke and TBI, were demonstrated to provoke functional alterations in the hippocampal neurogenic cascade that were characterized under the umbrella term “aberrant neurogenesis” [381, 382]. In particular, aberrant neurogenesis encompasses multiple (dys)functional outcomes, including excessive activation of NSCs [383, 384], alterations in NSC fate [383, 385] with a shift from neurogenesis to astrogenesis, downregulation of the proliferative capacity of NSCs, neural progenitor cells or neuroblasts [385–387], abnormal development and length of the dendritic tree of newborn neurons [388, 389], and ectopic migration of newborn neurons [390]. Interestingly, some authors, both in postmortem AD patients and in a transgenic (3xTg) AD mouse model, found that hyperphosphorylated tau, especially when expressed in GABAergic interneurons in the DG, was related to multiple alterations in SGZ NSCs strictly resembling the cardinal features of aberrant neurogenesis [317]. This finding suggests that tau-mediated aberrant neurogenesis also occurs in AD.
The link between AD and adult neurogenesis
A link between AD and adult neurogenesis has been recognized for some time. Indeed, AD and adult neurogenesis are not only linked by common sites where early pathology occurs and newly born neurons integrate in the preexisting circuitry (e.g., MTL, OB) but also share a number of common molecules in both processes [391–395]. In particular, molecular players in AD, including apolipoprotein E (ApoE), APP, and presenilin 1 (PS1), as well as their metabolites, play a role in adult neurogenesis [392, 394]. Further critical signals in AD have been found to regulate neurogenesis, such tau [38, 317, 396], Notch1 [384, 397], cyclic AMP response element binding protein (CREB) [398–406], and Wnt/β-catenin [407–410]. Furthermore, some authors have shown that blocking adult hippocampal neurogenesis in an AD mouse model exacerbated neuronal loss and cognitive impairment, while inducing adult hippocampal neurogenesis together with brain-derived neurotrophic factor improved cognition in AD mice [411]. Another study observed markers of increased neurogenesis in the DG of rare “resilient” individuals who remained cognitively intact, despite the presence of neuropathological features associated with AD, compared to AD and mild cognitive impairment patients [412]. Therefore, the prevailing view in the field is that impaired neurogenesis is a key contributing factor to AD pathology-driven neuronal dysfunction [394, 413–415]. Actually, the study of adult neurogenesis in postmortem AD patients and AD animal models has yielded conflicting results, frequently reporting a decrease [392, 416–420], but sometimes also an increase [392, 420, 421], in adult neurogenesis. The current theory predicts a complex relationship between the rate of neurogenesis and AD, depending on disease stage. In the first stage, there is a long-lasting tonic phase of reactive neurogenesis promoted by activated microglia triggered by Aβ deposition. Accordingly, the rate of neurogenesis is augmented, and tau pathogenesis is amplified. Then, the accumulation of tau aggregates in the niches start to have detrimental effects, likely ultimately reducing the rate of neurogenesis. This represents a phase of aberrant neurogenesis in AD that is most likely tau-mediated. A further phase could start when degeneration first occurs in the brain. Indeed, similar to what happens in stroke or TBI, cell death in the injured area stimulates the release of signals that could provoke a second cycle of aberrant neurogenesis in AD. The contrasting results found in the study of neurogenesis in AD seem to reflect this suspected complex relationship.
Adult neurogenesis, AD, and primary age-related tauopathy
The relative independence between the two processes driving amyloid and tau pathology in the new paradigm allows us to consider tau pathogenesis without Aβ deposition. Consequently, the finding of tau pathology uncoupled from amyloid pathology, as found, for example, in primary age-related tauopathy (PART) [422], seems compatible with the current paradigm. Moreover, the current model seems to fit well in explaining tau pathogenesis in PART. Indeed, the topography of tau lesions in PART is consistent with a possible origin in the NSCs within the SGZ niche. In particular, NFT changes in PART are usually restricted to the MTL and adjacent regions [132, 422, 423]. Later age of onset [422, 424–427], as well as limited spreading of tau pathology outside the MTL in PART compared to AD, would be due to the lack of a promoting effect on both neurogenesis and tau spreading by microglia in the absence of Aβ deposition. Therefore, the core hypothesis of tau pathogenesis as linked to adult neurogenesis and migration seems capable of combining AD and PART in a unique scenario. Some significant similarities identified between the two diseases support this idea. For example, NFTs in both disorders are identical, sharing both 3 repeat and 4 repeat tau isoforms and a 22–25 nm paired helical filamentous ultrastructure [422, 424, 428]. Moreover, phosphorylated tau lesions have the same topographic distribution in both PART and early AD [184, 424, 429]. In more detail, it has been reported that neurons in layer II of the TEC, a crucial region of early involvement in AD [184, 298], are also affected by neurofibrillary degeneration in PART [184, 298].
Adult neurogenesis, AD, and chronic traumatic encephalopathy
Unexpectedly, some findings from the study of a different disease, chronic traumatic encephalopathy (CTE) [430], seem to support the core hypothesis of tau pathogenesis as linked to adult neurogenesis and migration. CTE is a progressive tauopathy with distinctive clinical and pathological features that occurs after repetitive mild TBIs [430]. Microscopically, CTE is characterized primarily by NFTs and astrocytic tangles, with a relative absence of Aβ peptide deposits [431–447]. The evidence suggests that CTE begins focally, usually perivascularly, especially around small cerebral vessels, and at the depths of the sulci in the cerebral cortex [430, 446, 448–450]. Interestingly, some data have shown that the perivascular regions and the depths of the cerebral sulci are the most stressed regions, when the brain is subjected to rapid acceleration, deceleration, or rotational forces, such as occurs in mild TBI [451]. Thus, the highest concentration of phosphorylated tau correlates to the highest areas of stress in CTE [451]. Subsequently, pathological NFTs spread from these areas to adjacent superficial cortical layers. Considering the findings of an increase in the generation of new neurons in the niches [368, 452] and redirection of neuroblast migration to the injury site after TBI [73], V-SVZ neuroblast migration is ultimately redirected to the perivascular regions and the depths of the cerebral sulci in CTE because these regions are the primary sites of injury. Therefore, the current model of tau pathogenesis based on adult neurogenesis and migration in a special case of brain injury fits well for explaining the peculiar sites of pathological tau deposition in CTE. Indeed, the distinct feature of prominent periventricular NFTs in CTE is in agreement with the location of the V-SVZ niche. Therefore, this model also seems to combine neurodegeneration (AD) with traumatic degenerative dementia (CTE) in a unique scenario. The fact that the isoform profile and phosphorylation state of CTE are very similar to those in AD [285, 308, 309] agrees with this idea. In particular, neuronal tau pathology in CTE shows immunoreactivity to both 3R and 4R tau, as in AD [430, 453, 454]. In addition, tau in both AD and CTE is phosphorylated at the same amino acids, including tau phosphorylated at threonine 231, and all six isoforms are present, leading to the observation that NFTs associated with AD are indistinguishable from those that occur in TBI [453].
Adult neurogenesis, AD, and the antimicrobial protection hypothesis
The current theory does not focus on the process that drives the initial deposition of Aβ. Consequently, it is compatible with a recent etiologic model of AD, namely the Antimicrobial Protection hypothesis, which views Aβ deposition in a new light compared to the classic amyloid hypothesis [455]. In this model, Aβ deposition is an innate immune response that normally protects against genuine, or misperceived, microbial infection in the brain. Aβ first traps and neutralizes invading pathogens in Aβ. Fibrillation of Aβ stimulates neuroinflammatory pathways that help fight infection and clear Aβ/pathogen deposits. In AD, chronic activation of this pathway leads to sustained inflammation and neurodegeneration. This new model is supported by several lines of evidence.
Indeed, many studies documented the presence of abnormal levels of pathogens in the AD brain, including viral, bacterial, and fungal infections [456], particularly herpes simplex virus type 1 (HSV1) [457], Chlamydia pneumoniae, and several types of spirochetes [458–461].
Most importantly, Aβ showed consistent antimicrobial activity. Soscia et al. (2010) [462] were the first to demonstrate antibacterial and antifungal activity of Aβ peptide against numerous pathogens. These authors found that Aβ can act as an antimicrobial peptide and that Aβ deposition can be rapidly induced in mice, in Caenorhabditis elegans models, and in AD-based neural cell models as an innate immune defense mechanism against microbial pathogens [462, 463]. Interestingly, synthetic Aβ can reduce the growth of common pathogens up to 200-fold in vitro [462]. Some authors reported that Aβ peptide strongly inhibits the infectivity of influenza A virus in cell culture [464], while others [465, 466] reported similar results for herpes simplex virus type 1 (HSV-1). Further studies showed that Aβ peptides can protect the host against brain infections with Salmonella enterica serovar Typhimurium, HSV-1, and HHV-6 [463, 467].
Moreover, several evidence have mainly linked HSV-1 infections to the pathogenesis of AD [468–470]. In fact, HSV-1 DNA has been detected more frequently in the brains of AD patients than in healthy controls and has been found to be co-localized with Aβ [471]. In addition, some studies verified the presence of IgM anti-HSV-1 antibodies in most people with AD [472]. Moreover, high titers of anti-HSV-1 antibodies have been found to be positively correlated with the development of AD-like cognitive dysfunction [473], with symptoms of mild cognitive impairment [474], and with bilateral temporal and orbitofrontal cortical gray matter volume [475]. In addition, some studies suggested that in people carrying the APOE ɛ4 allele and, therefore, predisposed to develop AD, HSV-1 infection significantly increases the risk of AD [476, 477]. Furthermore, HSV-1 was shown to produce calcium-dependent GSK-3β activation, which results in hyperphosphorylation of tau and AβPP proteins as well as Aβ accumulation [478, 479]. Also, HSV-1 reactivation was associated with neuroinflammation and the appearance of several markers of neurodegeneration [478–480]. In addition, the brain regions mainly affected during acute encephalitis produced by active replication of HSV-1 in neuronal cells of the brain (herpes simplex encephalitis) [481], both in humans and in experimental rodent models, are the same regions impaired in AD (limbic system, frontal and temporal cortex) [482–488]. Consistently, a high percentage of brains of elderly people contain latent HSV-1 DNA especially in CNS regions critically involved in AD [476, 489]. More recently, some authors found that a mouse model of HSV-1 infection and recurrent reactivation showed a picture resembling the phenotype of sporadic AD [490]. Indeed, after infection and multiple rounds of reactivation of the virus promoting its spread within the brain, infected mice showed accumulation of Aβ and hyperphosphorylated tau proteins in several brain areas, including the hippocampus, and these molecular changes were accompanied by memory deficits [490].
Interestingly, the current adult neurogenesis theory of AD is not only compatible with the antimicrobial protection hypothesis, but also shows some relevant points of convergence with it. It is noteworthy that HSV-1 latency has been observed mainly within the lateral ventricles in the SVZ, hippocampus, and brainstem before being detected in the neurons of the trigeminal ganglion [491, 492], and also in the olfactory bulbs, frontal cortex, and cerebellum in some studies [492]. More generally, the olfactory nerve, which leads to the lateral entorhinal cortex, is a portal of entry of HSV-1 [493] and other viruses [494], as well as Chlamydia pneumoniae, into the brain [495]. In addition, brainstem areas that harbor latent HSV directly irrigate these brain regions [190], and from the brainstem, neurons project to the thalamus and eventually reach the sensory cortex. Thus, it is interesting to note that the sites of HSV-1 latency (hippocampus and SVZ) overlap with the major niches of adult neurogenesis, and the pathways of HSV-1 infection seem consistent with the network of connected regions considered relevant in current theory in both LOAD (hippocampus - EC - OB - brainstem) and EOAD (SVZ - OB). Based on the finding that the preferred sites of HSV-1 latency are in the niches of adult neurogenesis (hippocampus and SVZ), ependymal cells and neural progenitor cells turn out to be highly susceptible to HSV-1 infection [491, 496–498]. Indeed, HSV-1 readily replicates in these cells during acute encephalitis [496, 498], and viral lytic-associated proteins were detected in these cells during latency [498]. The presence of HSV-1 in lateral ventricle ependymal cells and neural progenitor cells during latent infection alters the proliferation of NPCs as a consequence of fibroblast growth factor 2 deficiency [499], whereas HSV-1 replication during acute encephalitis results in their loss and altered differentiation [496, 498]. Interestingly, a recent study showed that HSV-1 affects adult hippocampal neurogenesis in vitro and in vivo by reducing the proliferation of NSCs and their neuronal differentiation in the SGZ of the hippocampal DG, through intracellular accumulation of Aβ, without inducing cell death [500, 501]. Indeed, anti-Aβ antibodies or experimental mouse models lacking APP (and thus unable to form Aβ) reverse the impairment of neurogenesis induced by HSV-1 infection [500]. Furthermore, impairment of adult hippocampal neurogenesis occurs when cognitive dysfunction induced by HSV-1 infection is not yet present, suggesting a role of adult hippocampal neurogenesis in the pathogenesis of AD [501].
Main unresolved questions in the field and possible solutions
The scenario proposed in the current theory seems to offer possible explanations for some unresolved questions in dementia and AD research (Table 1).
Table 1
Unresolved questions in the field and possible explanations according to current theory
| Open question in the field | Possible solution according to current theory | |
| Aβ-tau interaction | While amyloid and tau pathology are clearly critical in the pathogenesis of AD, a major unresolved question is how the two interact? | Amyloid and tau have a twofold interaction. At a very early preclinical stage, Aβ deposition has an indirect microglia-mediated effect of promoting adult neurogenesis and supporting migration, with the effect of amplifying hyperphosphorylation of tau in niches or during long migrations. Later, when the two are in close interaction, Aβ has the documented downstream effect on tau pathogenesis. |
| Difference between EOAD and LOAD | Why do tau seem to accumulate preferentially in the memory system in aging and LOAD, but predominates in a number of other neural systems in EOAD, although both disorders show a generalized pattern of Aβ distribution? | Tau pathology develops in the niches of adult neurogenesis in all types of AD, but in EOAD the V-SVZ is particularly active. NSCs carrying the seeds of tau pathology migrate from this niche to different regions of the cortex driven by complex signals after microglia activated by Aβ deposition redirect migration from the canonical RMS. In LOAD, in accordance with an age-related decline in neurogenesis, the SGZ of the hippocampal DG remains mainly, or often exclusively, active. NSCs briefly migrate to the granular layer in the DG, where the seeds of tau pathology begin to develop and spread within the MTL. |
| Phenotypic heterogeneity in (EO)AD | Why do different (EO)AD syndromes exhibit different distributions of tau despite similar distributions of Aβ? | Microglia activated by Aβ deposition promote neurogenesis so that more newborn neurons are present mainly in the V-SVZ niche in the EOAD. When these neuroblasts leave the V-SVZ niche and begin to migrate, microglia signals cause a redirection of the pathway from the canonical RMS and support long-distance migration. Meanwhile, the genetic program of arealization during development is reactivated and helps signal the direction for migrating neuroblasts to follow. Specifically, a mechanism based on gradients of gene expression of morphogens or transcription factors specifies certain localizations along the simple brain axes A-P, D-V and L-M. Thus, some neuroblasts carrying the seeds of tau pathology migrate to these locations in the cortex and, when they arrive at their final destination in the occipital, parietal, frontal, or left perisylvian cortex, a focus of degeneration begins to develop. |
| Scaling of molecular pathology to the macroscopic brain | Why does AD start in TEC? | According to current theory, adult NSCs in the SGZ niche are the first cells that carry the initial seeds of tau pathology. However, NFT formation is suppressed and delayed in the DG because of the strong clearance activity in the niche. The seeds of tau pathology, probably after some GABAergic interneurons carry transmission from SGZ NSCs, spread retrogradely from the DG to the EC along the perforant pathway connections. Then, the seeds of tau pathology spread from EC to TEC through both retrograde and anterograde transmission due to the multiple reciprocal connections between the two regions. Thus, the TEC receives a massive load of tau pathology from the EC, and this fact would explain why NFTs appear first here. |
| Not specificity of tau pathology in AD | Why is tau pathology in CTE, PART and AD so similar? | According to the new theory, tau pathology would develop in the niches of adult neurogenesis in all three diseases. However, in CTE, migration is redirected to sites of injury that lie at the depth of sulci and at the perivascular level. In PART, the lack of a promoter effect on both neurogenesis and tau spreading by microglia, in the absence of Aβ deposition, causes limited spread of tau pathology outside MTL compared with AD. |
| Selective vulnerability (region) | Why some definite brain regions but not others are impacted by degeneration? | The selection of a particular brain region as a target would be determined by the localization of the SGZ niche in the MTL or the direction of migrations to the cortex made by neuroblasts carrying the seeds of tau pathology from the V-SVZ. Thus, the selection of a cortical region depends on the location of the niches or the combined outcome of the complex signals guiding the migrating neuroblasts rather than on a regional vulnerability. Therefore, the concept of regional selective vulnerability appears superfluous in the new scenario. |
| Selective vulnerability (cell) | Why proteins that usually show widespread expression should accumulate in one set of cells but not in apparently similar neighboring cells? | Tau pathology develops in NSCs when neurons are immature and undifferentiated and their fate, as well as their final localization, have not yet been fully decided. Therefore, in this scenario, the concept of selective cellular vulnerability appears superfluous |
| Preponderance in humans | Why AD develops mainly in humans although all vertebrates produce APP, β-secretase, Aβ, and tau protein, and neurogenesis and neuronal migration in the adult brain are well conserved from fish to primates? | The cause is the incredible development of the neocortex in humans compared with other vertebrates. Both processes implicated in the pathogenesis of AD according to current theory, i.e., metabolism driving amyloid deposition and adult neurogenesis/migration driving tau pathogenesis, are particularly stressed in the extended and interconnected cortex of the human brain. The longevity revolution, another distinct aspect of humans, is likely to contribute to the extremes of this scenario. |
EOAD, early onset Alzheimer’s disease; LOAD, late onset Alzheimer’s disease; V-SVZ, ventricular subventricular zone; NSCs, neural stem cells; RMS, rostral migratory stream; SGZ, subgranular zone; DG, dentate gyrus; MTL, medial temporal lobe; A-P, anterior-posterior; D-V, dorsal-ventral; M-L, medial-lateral; NFTs, neurofibrillary tangles; EC, entorhinal cortex; TEC, transentorhinal cortex; CTE, chronic traumatic encephalopathy; PART, primary age-related tauopathy.
Aβ-tau interaction
While amyloid and tau pathology are clearly critical in the pathogenesis of AD, a major unresolved question at this time is how the two interact [2, 335]. According to the current theory, amyloid and tau have a twofold interaction. At a very early preclinical stage, Aβ deposition has an indirect microglia-mediated effect of fostering adult neurogenesis and supporting migration, amplifying tau hyperphosphorylation in the niches or during long migrations. Later, when the two are in close interaction, Aβ has a documented downstream effect on tau pathogenesis. The idea that Aβ deposition acts as a driving force for tau pathogenesis by fostering neurogenesis and supporting migration is consistent with several findings showing that Aβ deposition facilitates both the pathogenicity [502–506] and spread [335, 507, 508] of tau in the brain, even when Aβ and tau are not topographically closely related [509, 510].
Phenotypic heterogeneity in AD
Comparisons between EOAD and LOAD are particularly puzzling because, although both disorders show a generalized pattern of Aβ distribution, tau seems to accumulate preferentially in the memory system in aging and LOAD but predominates in a number of other neural systems in EOAD [335, 508]. Even within EOAD, different syndromes exhibit different distributions of tau, despite similar distributions of Aβ [335, 508]. The hypothesis of tau pathogenesis as linked to adult neurogenesis seems to combine EOAD and LOAD, as well as different EOAD syndromes, in a unique scenario. Tau pathology develops in the niches of adult neurogenesis in all types of AD, but in EOAD, it is especially active in the V-SVZ. Some neuroblasts carrying the seeds of tau pathology migrate from this niche toward different regions of the cortex guided by complex signals, after which microglia activated by Aβ deposition redirect migration from the RMS toward the site of injury, similar to what occurs in stroke or TBI. When these neuroblasts arrive at target regions in the occipital, parietal, frontal, or left perisylvian cortex, degeneration begins to develop. Therefore, in this model, a common mechanism through distinct migratory paths would explain the phenotypic heterogeneity in EOAD. In LOAD, in agreement with an age-related decline in neurogenesis [511–514], it remains active, especially, or often exclusively, the SGZ of the hippocampal DG. Neuroblasts migrate a brief distance to the granular layer in the DG, where the seeds of tau pathology start to develop and spread inside the MTL.
Not specificity of tau pathology in AD
The model of tau pathogenesis proposed in the current theory seems not to be specific for AD. For example, it was able to combine three different diseases, i.e., AD, CTE, and PART, into a unique scenario. Equally, it could be conveniently applied to other proteinopathies in different neurodegenerative diseases. In this regard, considering, for example, Parkinson’s disease dementia and limbic-predominant age-related TDP-43 encephalopathy, it is intriguing to report that there is some evidence of the presence of neurogenic niches in mammals in the substantia nigra and amygdala [316].
Selective vulnerability
The clinical manifestation of a particular neurodegenerative disease reflects the region of the brain and the specific population of cells within it that are affected [515]. Major neurodegenerative diseases differ from each other not only in the type of pathological protein that accumulates but also in the regions impacted and the types of neurons that are vulnerable. Why proteins that usually show widespread expression should accumulate in one set of cells but not in apparently similar neighboring cells and why some definite brain regions but not others are impacted are fundamental questions remaining in the field [515]. These questions have been primarily conceptualized in the notion of selective vulnerability (cellular and regional) [515–517]. The current conceptualization seems to offer a simple and unexpectedly rapid solution to the question of selective vulnerability in dementia. Indeed, the selection of a definite brain region as a target would be determined either by localization of the neurogenic niches or by direction of the migration paths toward the cortex taken by neuroblasts carrying the seeds of tau pathology. Therefore, the selection would be established in advance or actively driven rather than emerging passively by virtue of regional vulnerability. In other words, according to the current theory, there would not be any regional vulnerability to determine. We can draw the same conclusion considering selective cell vulnerability. Indeed, according to the main hypothesis, tau pathology develops in NSCs when neurons are immature, and their fate, as well as final localization, have not yet been totally decided. In this context, it is evident that the concept of selective cell vulnerability appears unnecessary.
Preponderance in humans
The current view suggests a possible explanation for another relevant question in the field. In particular, although all vertebrates produce AβPP, β-secretase, Aβ, and tau protein [518–520] and neurogenesis and neuronal migration in the adult brain are well conserved from fish to primates [73, 521–523], AD develops mainly in humans. The reason is most likely found in the primary distinctive aspect of the human brain, that is, the incredible development of the neocortex. In this regard, it is worth noting that both the processes implicated in AD pathogenesis in the current theory, i.e., metabolism driving amyloid deposition and adult neurogenesis/migration driving tau pathogenesis, are particularly stressed in an extended and interconnected cortex. In particular, advanced cognitive performance is highly demanding on the metabolism. At the same time, adult neurogenesis strains try to supply an adequate pool of new neurons to guarantee high plasticity and repair from injury. Finally, neuronal migration is long-distance and demanding. The longevity revolution [524], a further distinct aspect of humans, likely contributes to extremes in this scenario. Therefore, amyloid deposition and NFT formation are inevitable consequences in extremely old and advanced human brains. Unfortunately, amyloid and tau pathology interact, with the first fosters and amplifying the second, resulting in emergent AD.
Limitations
The current theory is highly speculative. Many parts of the scenario depicted are based on suggestions and hypotheses that require more concrete evidence. Moreover, I did not find any data in the literature supporting some of the hypotheses proposed. For example, one hypothesis is that long migration of neuroblasts from the V-SVZ to different regions of the cortex are implicated in the development of EOAD. However, evidence for organized, long-distance migration of newly generated neurons in the adult human brain is lacking [525–528]. Additionally, useful methods and techniques are needed to verify some hypotheses proposed in the theory that are not yet available. In particular, the study of adult neurogenesis in humans faces many challenges. For example, it is currently not possible to confirm the existence of adult neurogenesis in the living human brain [529]. Additionally, some authors noticed that adult hippocampal neurogenesis markers degrade rapidly in fixed postmortem tissues and could thus be undetectable if the tissues are not stored and processed quickly [530]. Moreover, considering the study of neurogenesis in AD, other authors observed that data from postmortem human tissues are intrinsically controversial and difficult to interpret because as a rule, postmortem material reflects the late stages of the disease [420]. In addition, artifacts and misinterpretations can arise due to the stage of the disease and treatments provided [420]. Furthermore, these discrepancies also depend on the methods used for labeling proliferating cells [420].
ACKNOWLEDGMENTS
The theory presented in this article was awarded one of four gold prizes in the Oskar Fischer Prize competition organized by the University of Texas, College of Sciences, at San Antonio in June 2022. The prize was awarded thanks to a generous donation from Dr. James Truchard. I would like to express my immense gratitude to Dr. James Truchard and all the UTSA staff involved in organizing the competition. I would also like to express my sincere congratulations to the other winners of the Oskar Fischer Prize competition. Finally, I would like to thank Antonio Garcia, Bio-Graphics illustrator for the valuable work in making the Figures.
FUNDING
The author has no funding to report.
CONFLICT OF INTEREST
Carlo Abbate is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.
REFERENCES
[1] | Masters CL , Selkoe DJ ((2012) ) Biochemistry of amyloid β-protein and amyloid deposits in Alzheimer disease. Cold Spring Harb Perspect Med 2: , a006262. |
[2] | Lane CA , Hardy J , Schott JM ((2018) ) Alzheimer’s disease. Eur J Neurol 25: , 59–70. |
[3] | Hardy JA , Higgins GA ((1992) ) Alzheimer’s disease: The amyloid cascade hypothesis. Science 256: , 184–185. |
[4] | Hardy J , Selkoe DJ ((2002) ) The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 297: , 353–356. |
[5] | Gotz J , Chen F , van Dorpe J , Nitsch RM ((2001) ) Formation of neurofibrillary tangles in P301 l tau transgenic mice induced by Abeta 42 fibrils. Science 293: , 1491–1495. |
[6] | Bolmont T , Clavaguera F , Meyer-Luehmann M , Herzig MC , Radde R , Staufenbiel M , Lewis J , Hutton M , Tolnay M , Jucker M ((2007) ) Induction of tau pathology by intracerebral infusion of amyloid-beta containing brain extract and by amyloid-beta deposition in APP×Tau transgenic mice. Am J Pathol 171: , 2012–2020. |
[7] | Vergara C , Houben S , Suain V , Yilmaz Z , De Decker R , Dries VV , Boom A , Mansour S , Leroy K , Ando K , Brion J-P ((2019) ) Amyloid-β pathology enhances pathological fibrillary tau seeding induced by Alzheimer PHF in vivo. Acta Neuropathol 137: , 397–412. |
[8] | Bennett RE , DeVos SL , Dujardin S , Corjuc B , Gor R , Gonzalez J , Roe AD , Frosch MP , Pitstick R , Carlson GA , Hyman BT ((2017) ) Enhanced tau aggregation in the presence of amyloid β. Am J Pathol 187: , 1601–1612. |
[9] | Héraud C , Goufak D , Ando K , Leroy K , Suain V , Yilmaz Z , DeDecker R , Authelet M , Laporte V , Octave J-N , Brion J-P ((2014) ) Increased misfolding and truncation of tau in APP/PS1/tau transgenicmice compared to mutant tau mice. Neurobiol Dis 62: , 100–112. |
[10] | Hurtado DE , Molina-Porcel L , Iba M , Aboagye AK , Paul SM , Trojanowski JQ , Lee VM-Y ((2010) ) Abeta accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am J Pathol 177: , 1977–1988. |
[11] | Lewis J , Dickson DW , Lin WL , Chisholm L , Corral A , Jones G , Yen S-H , Sahara N , Skipper L , Yager D , Eckman C , Hardy J , Hutton M , Mcgowan E ((2001) ) Enhanced neurofibrillary degeneration in transgenic miceexpressing mutant tau and APP. Science 293: , 1487–1491. |
[12] | Perez M , Ribe E , Rubio A , Lim F , Moran MA , Ramos PG , Ferrer I , Islab MTG , Avila J ((2005) ) Characterization of a double (amyloid precursor protein-tau) transgenic: Tau phosphorylation and aggregation. Neuroscience 130: , 339–347. |
[13] | Pooler AM , Polydoro M , Maury EA , Nicholls SB , Reddy SM , Wegmann S , William C , Saqran L , Cagsal-Getkin O , Pitstick R , Beier DR , Carlson GA , Spires-Jones TL , Hyman BT ((2015) ) Amyloid accelerates tau propagation and toxicity in a model of early Alzheimer’s disease. Acta Neuropathol Commun 3: , 14. |
[14] | Seino Y , Kawarabayashi T , Wakasaya Y , Watanabe M , Takamura A , Yamamoto-Watanabe Y , Tomoko Kurata T , Koji Abe K , Ikeda M , Westaway D , Murakami T , St. George Hyslop P , Matsubara E , Shoji M ((2010) ) Amyloid beta accelerates phosphorylation of tau and neurofibrillary tangle formation in an amyloid precursor protein and tau double-transgenic mouse model. J Neurosci Res 88: , 3547–3554. |
[15] | Stancu IC , Ris L , Vasconcelos B , Marinangeli C , Goeminne L , Laporte V , Laetitia E. , Haylani LE , Julien Couturier J , Olivier Schakman O , Gailly P , Pierrot N , Kienlen-Campard P , Octave J-N , Dewachter I ((2014) ) Tauopathy contributes to synaptic and cognitive deficits in a murine model for Alzheimer’s disease. FASEB J 28: , 2620–2631. |
[16] | Terwel D , Muyllaert D , Dewachter I , Borghgraef P , Croes S , Devijver H , Van Leuven F ((2008) ) Amyloid activates GSK-3beta to aggravate neuronal tauopathy in bigenic mice. Am J Pathol 172: , 786–798. |
[17] | Takashima A , Sato M , Mercken M , Tanaka S , Kondo S , Honda T , Sato K , Murayama M , Noguchi K , Nakazato Y , Takahashi H ((1996) ) Localization of Alzheimer-associated presenilin 1 in transfected COS-7 cells. Biochem Biophys Res Commun 227: , 423–426. |
[18] | Takashima A , Noguchi K , Michel G , Mercken M , Hoshi M , Ishiguro K , Imahori K ((1996) ) Exposure of rat hippocampal neurons to amyloid beta peptide (25-35) induces the inactivation of phosphatidyl inositol-3 kinase and the activation of tau protein kinase I/glycogen synthase kinase-3beta. Neurosci Lett 203: , 33–36. |
[19] | Llorens-Martin M , Jurado J , Hernandez F , Avila J ((2014) ) GSK-3beta, a pivotal kinase in Alzheimer disease. Front Mol Neurosci 7: , 46. |
[20] | Leroy K , Yilmaz Z , Brion JP ((2007) ) Increased level of active GSK-3beta in Alzheimer’s disease and accumulation in argyrophilic grains and in neurones at different stages of neurofibrillary degeneration. Neuropathol Appl Neurobiol 33: , 43–55. |
[21] | Avila J , Hernandez F ((2007) ) GSK-3 inhibitors for Alzheimer’sdisease. Expert Rev Neurother 7: , 1527–1533. |
[22] | Engel T , Goni-Oliver P , Lucas JJ , Avila J , Hernandez F ((2006) ) Chronic lithium administration to FTDP-17 tau and GSK-3beta overexpressing mice prevents tau hyperphosphorylation and neurofibrillary tangle formation, but pre-formed neurofibrillary tangles do not revert. J Neurochem 99: , 1445–1455. |
[23] | Hernandez F , de Barreda EG , Fuster-Matanzo A , Goni-Oliver P , Lucas JJ , Avila J ((2009) ) The role of GSK3 in Alzheimer disease. Brain Res Bull 80: , 248–250. |
[24] | Hernandez F , Gomez de Barreda E , Fuster-Matanzo A , Lucas JJ , Avila J ((2010) ) GSK3: A possible link between beta amyloid peptide and tau protein. Exp Neurol 223: , 322–325. |
[25] | Jin M , Shepardson N , Yang T , Chen G , Walsh D , Selkoe DJ ((2011) ) Soluble amyloid-protein dimers isolated from Alzheimer cortex directly induce tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci U S A 108: , 5819–5824. |
[26] | Cohen AD , Price JC , Weissfeld LA , James J , Rosario BL , Bi W , Nebes RD , Saxton JA , Snitz BE , Aizenstein HA , Wolk DA , DeKosky ST , Mathis CA , Klunk WE ((2009) ) Basal cerebral metabolism may modulate the cognitive effects of Aβ in mild cognitive impairment: An example of brain reserve. J Neurosci 29: , 14770–14778. |
[27] | Johnson SC , Christian BT , Okonkwo OC , Oh JM , Harding S , Xu G , Hillmer AT , Wooten DW , Murali D , Barnhart TE , Hall LT , Racine AM , Klunk WE , Mathis CA , Bendlin BB , Gallagher CL , Carlsson CM , Rowley HA , Hermann BP , Dowling NM , Asthana S , Sager MA ((2014) ) Amyloid burden and neural function in people at risk for Alzheimer’s disease. Neurobiol Aging 35: , 576–584. |
[28] | Oh H , Madison C , Baker S , Rabinovici G , Jagust W ((2016) ) Dynamic relationships between age, amyloid-β deposition, and glucose metabolism link to the regional vulnerability to Alzheimer’s disease. Brain 139: , 2275–2289. |
[29] | Scopa C , Marrocco F , Latina V , Ruggeri F , Corvaglia V , La Regina F , Ammassari-Teule M , Middei S , Amadoro G , Meli G , Scardigli R , Cattaneo A ((2020) ) Impaired adult neurogenesis is an early event in Alzheimer’s disease neurodegeneration, mediated by intracellular Aβ oligomers. Cell Death Differ 27: , 934–948. |
[30] | Lu M , Kosik KS ((2001) ) Competition for microtubule-binding with dual expression of tau missense and splice isoforms. Mol Biol Cell 12: , 171–184. |
[31] | Avila J , Lucas JJ , Perez M , Hernandez F ((2004) ) Role of tau protein in both physiological and pathological conditions. Physiol Rev 84: , 361–384. |
[32] | Bullmann T , de Silva R , Holzer M , Mori H , Arendt T ((2007) ) Expression of embryonic tau protein isoforms persist during adult neurogenesis in the hippocampus. Hippocampus 17: , 98–102. |
[33] | Brion JP , Smith C , Couck AM , Gallo JM , Anderton BH ((1993) ) Developmental changes in tau phosphorylation: Fetal tau is transiently phosphorylated in a manner similar to paired helical filament-tau characteristic of Alzheimer’s disease. J Neurochem 61: , 2071–2080. |
[34] | Brion JP , Octave JN , Couck AM ((1994) ) Distribution of the phosphorylated microtubule-associated protein tau in developing cortical neurons. Neuroscience 63: , 895–909. |
[35] | Yu Y , Run X , Liang Z , Li Y , Liu F , Liu Y , Iqbal K , Grundke-Iqbal I , Gong CX ((2009) ) Developmental regulation of tau phosphorylation, tau kinases, and tau phosphatases. J Neurochem 108: , 1480–1494. |
[36] | Lovestone S , Hartley CL , Pearce J , Anderton BH ((1996) ) Phosphorylation of tau by glycogen synthase kinase-3 beta in intact mammalian cells: The effects on the organization and stability of microtubules. Neuroscience 73: , 1145–1157. |
[37] | Kenessey A , Yen SH ((1993) ) The extent of phosphorylation of fetal tau is comparable to that of PHF-tau from Alzheimer paired helical filaments. Brain Res 629: , 40–46. |
[38] | Fuster-Matanzo A , Llorens-Martin M , Jurado-Arjona J , Avila J , Hernandez F ((2012) ) Tau protein and adult hippocampal neurogenesis. Front Neurosci 6: , 104. |
[39] | Fuster-Matanzo A , De Barreda EG , Dawson HN , Vitek MP , Avila J , Hernandez F ((2009) ) Function of tau protein in adult newborn neurons. FEBS Lett 583: , 3063–3068. |
[40] | Llorens-Martin M , Teixeira CM , Fuster-Matanzo A , Jurado-Arjona J , Borrell V , Soriano E , Avila J , Hernandez F ((2012) ) Tau isoform with three microtubule binding domains is a marker of new axons generated from the subgranular zone in the hippocampal dentate gyrus: Implications for Alzheimer’s disease. J Alzheimers Dis 29: , 921–930. |
[41] | Teixeira CM , Pallas-Bazarra N , Bolos M , Terreros-Roncal J , Avila J , Llorens-Martín M ((2018) ) Untold new beginnings: Adulthippocampal neurogenesis and Alzheimer’s disease. J Alzheimers Dis 64: , S497–S505. |
[42] | Hong XP , Peng CX , Wei W , Tian Q , Liu YH , Cao FY , Wang Q , Wang JZ ((2011) ) Relationship of adult neurogenesis with tau phosphorylationand GSK-3beta activity in subventricular zone. Neurochem Res 36: , 288–296. |
[43] | Grundke-Iqbal I , Iqbal K , Quinlan M , Tung YC , Zaidi MS , Wisniewski HM ((1986) ) Microtubule-associated protein tau. A component of Alzheimer paired helical filaments. J Biol Chem 261: , 6084–6089. |
[44] | Grundke-Iqbal I , Iqbal K , Tung YC , Quinlan M , Wisniewski HM , Binder LI ((1986) ) Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A 83: , 4913–4917. |
[45] | Ihara Y , Nukina N , Miura R , Ogawara M ((1986) ) Phosphorylated tauprotein is integrated into paired helical filaments in Alzheimer’sdisease. J Biochem (Tokyo) 99: , 1807–1810. |
[46] | Montejo de Garcini E , Serrano L , Avila J ((1986) ) Self-assembly of microtubule associated protein tau into filaments resembling those found in Alzheimer disease. Biochem Biophys Res Commun 141: , 790–796. |
[47] | Lee VM , Balin BJ , Otvos L Jr, Trojanowski JQ ((1991) ) A68: A major subunit of paired helical filaments and derivatized forms of normal tau. Science 251: , 675–678. |
[48] | Goedert M , Jakes R , Spillantini MG , Hasegawa M , Smith MJ , Crowther RA ((1996) ) Assembly of microtubule associated protein tau into Alzheimer-like filaments induced by sulphated glycosaminoglycans. Nature 383: , 550–553. |
[49] | Goedert M ((1993) ) Tau protein and the neurofibrillary pathology of Alzheimer’s disease. Trends Neurosci 16: , 460–465. |
[50] | Morishima-Kawashima M , Hasegawa M , Takio K , Suzuki M , Yoshida H , Watanabe A , Titani K , Ihara Y ((1995) ) Hyperphosphorylation of tau in PHF. Neurobiol Aging 16: , 365–371. |
[51] | Ishiguro K , Omori A , Takamatsu M , Sato K , Arioka M , Uchida T , Imahori K ((1992) ) Phosphorylation sites on tau by tau protein kinase I, a bovine derived kinase generating an epitope of paired helical filaments. Neurosci Lett 148: , 202–206. |
[52] | Ishiguro K , Takamatsu M , Tomizawa K , Omori A , Takahashi M , Arioka M , Uchida T , Imahori K ((1992) ) Tau protein kinase I converts normal tau protein into A68-like component of paired helical filaments. J Biol Chem 267: , 10897–10901. |
[53] | Wang JZ , Wu Q , Smith A , Grundke-Iqbal I , Iqbal K ((1998) ) Tau is phosphorylated by GSK-3 at several sites found in Alzheimer disease and its biological activity markedly inhibited only after it is prephosphorylated by A-kinase. FEBS Lett 436: , 28–34. |
[54] | Liu SJ , Zhang AH , Li HL , Wang Q , Deng HM , Netzer WJ , Xu H , Wang JZ ((2003) ) Overactivation of glycogen synthase kinase-3 by inhibition of phosphoinositol-3 kinase and protein kinase C leads to hyperphosphorylation of tau and impairment of spatial memory. J Neurochem 87: , 1333–1344. |
[55] | Plattner F , Angelo M , Giese KP ((2006) ) The roles of cyclin-dependent kinase 5 and glycogen synthase kinase 3 in tau hyperphosphorylation. J Biol Chem 281: , 25457–25465. |
[56] | Takahashi M , Tomizawa K , Kato R , Sato K , Uchida T , Fujita SC , Imahori K ((1994) ) Localization and developmental changes of tau protein kinase I/glycogen synthase kinase-3 beta in rat brain. J Neurochem 63: , 245–255. |
[57] | Leroy K , Brion JP ((1999) ) Developmental expression and localization of glycogen synthase kinase-3beta in rat brain. J Chem Neuroanat 16: , 279–293. |
[58] | Hong XP , Peng CX , Wei W , Tian Q , Liu YH , Yao XQ , Zhang Y , Cao F-Y , Wang Q , Wang J-Z ((2010) ) Essential role of tau phosphorylation in adult hippocampal neurogenesis. Hippocampus 20: , 1339–1349. |
[59] | Gong CX , Liu F , Grundke-Iqbal I , Iqbal K ((2006) ) Dysregulation of protein phosphorylation/dephosphorylation in Alzheimer’s disease: A therapeutic target. J Biomed Biotechnol 31825: , 31825. |
[60] | Alonso Del A C , Li B , Grundke-Iqbal I , Iqbal K ((2008) ) Mechanism of tau-induced neurodegeneration in Alzheimer disease and related tauopathies. Curr Alzheimer Res 5: , 375–384. |
[61] | Avila J ((2006) ) Tau phosphorylation and aggregation in Alzheimer’s disease pathology. FEBS Lett 580: , 2922–2927. |
[62] | Gong CX , Iqbal K ((2008) ) Hyperphosphorylation of microtubule associated protein tau: A promising therapeutic target for Alzheimer disease. Curr Med Chem 15: , 2321–2328. |
[63] | Iqbal K , Grundke-Iqbal I , Zaidi T , Merz PA , Wen GY , Shaikh SS , Wisniewski HM , Alafuzoff I , Winblad B ((1986) ) Defective brain microtubule assembly in Alzheimer’s disease. Lancet 2: , 421–426. |
[64] | Yoshida H , Ihara Y ((1993) ) Tau in paired helical filaments is functionally distinct from fetal tau: Assembly incompetence of paired helical filament-tau. J Neurochem 61: , 1183–1186. |
[65] | Alonso AD , Zaidi T , Grundke-Iqbal I , Iqbal K ((1994) ) Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A 91: , 5562–5566. |
[66] | Alonso AD , Zaidi T , Novak M , Grundke-Iqbal I , Iqbal K ((2001) ) Hyperphosphorylation induces self-assembly of tau into tangles ofpaired helical filaments/straight filaments. Proc Natl Acad SciU S A 98: , 6923–6928. |
[67] | Alonso AD , Li B , Grundke-Iqbal I , Iqbal K ((2006) ) Polymerization of hyperphosphorylated tau into filaments eliminates its inhibitory activity. Proc Natl Acad Sci U S A 23: , 8864–8869. |
[68] | Castellani RJ , Perry G ((2019) ) Tau biology, tauopathy, traumatic brain injury, and diagnostic challenges. J Alzheimers Dis 67: , 447–467. |
[69] | Morris M , Knudsen GM , Maeda S , Trinidad JC , Ioanoviciu A , Burlingame AL , Mucke L ((2015) ) Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat Neurosci 18: , 1183–1189. |
[70] | Liu F , Li B , Tung EJ , Grundke-Iqbal I , Iqbal K , Gong CX ((2007) ) Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur J Neurosci 26: , 3429–3436. |
[71] | Wang JZ , Grundke-Iqbal I , Iqbal K ((2007) ) Kinases and phosphatasesand tau sites involved in Alzheimer neurofibrillary degeneration. Eur J Neurosci 25: , 59–68. |
[72] | Ghashghaei HT , Lai C , Anton ES ((2007) ) Neuronal migration in the adult brain: Are we there yet? Nat Rev Neurosci 8: , 141–151. |
[73] | Kaneko N , Sawada M , Sawamoto K ((2017) ) Mechanisms of neuronal migration in the adult brain. J neurochem 141: , 835–847. |
[74] | Casares-Crespo L , Calatayud-Baselga I , García-Corzo L , Mira H ((2018) ) On the role of basal autophagy in adult neural stem cells andneurogenesis. Front Cell Neurosci 12: , 339. |
[75] | Hong CJ , Park H , Yu SW ((2016) ) Autophagy for the quality control of adult hippocampal neural stem cells. Brain Res 1649: , 166–172. |
[76] | Moore Z , Taylor JM , Crack PJ ((2019) ) The involvement of microglia in Alzheimer’s disease: A new dog in the fight. Br J Pharmacol 176: , 3533–3543. |
[77] | Femminella GD , Dani M , Wood M , Fan Z , Calsolaro V , Atkinson R , Edginton T , Hinz R , Brooks DJ , Edison P ((2019) ) Microglial activation in early Alzheimer trajectory is associated with higher gray matter volume. Neurology 92: , 1331–1343. |
[78] | Heneka MT , Kummer MP , Latz E ((2014) ) Innate immune activation in neurodegenerative disease. Nat Rev Immunol 14: , 463–477. |
[79] | Calsolaro V , Edison P ((2016) ) Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimers Dement 12: , 719–732. |
[80] | Femminella GD , Ninan S , Atkinson R , Fan Z , Brooks DJ , Edison P ((2016) ) Does microglial activation influence hippocampal volume and neuronal function in Alzheimer’s disease and Parkinson’s disease dementia? J Alzheimers Dis 51: , 1275–1289. |
[81] | Hamelin L , Lagarde J , Dorothee G , Leroy C , Labit M , Comley RA , Cruz de Souza L , Corne H , Dauphinot L , Bertoux M , Dubois B , Gervais P , Colliot O , Potier MC , Bottlaender M , Sarazin M , the Clinical IMABio3 team ((2016) ) Early and protective microglial activation in Alzheimer’s disease: A prospective study using 18 F-DPA-714 PET imaging. Brain 139: , 1252–1264. |
[82] | Selkoe DJ ((1994) ) Amyloid beta-protein precursor: New clues to the genesis of Alzheimer’s disease. Curr Opin Neurobiol 4: , 708–716. |
[83] | Mrak RE , Griffin WS ((2005) ) Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging 26: , 349–354. |
[84] | Akiyama H , Barger S , Barnum S , Bradt B , Bauer J , Cole GM , Cooper NR , Eikelenboom P , Emmerling M , Fiebich BL , Finch CE , Frautschy S , Griffin WST , Hampel H , Hull M , Landreth G , Lue L-F , Mrak R , Mackenzie IR , McGeer PL , O’Banion MK , Pachter J , Pasinetti G , Plata-Salaman C , Rogers J , Rydel R , Shen Y , Streit W , Strohmeyer R , Tooyoma I , Van Muiswinkel FL , Veerhuis R , Walker D , Webster S , Wegrzyniak B , Wenk G , Wyss-Coray T ((2000) ) Inflammation and Alzheimer’s disease. Neurobiol Aging 21: , 383–421. |
[85] | Gomez-Nicola D , Fransen NL , Suzzi S , Perry VH ((2013) ) Regulation of microglial proliferation during chronic neurodegeneration. J Neurosci 33: , 2481–2493. |
[86] | Wyss-Coray T ((2006) ) Inflammation in Alzheimer disease: Driving force, bystander or beneficial response? Nat Med 12: , 1005–1015. |
[87] | Heneka MT , O’Banion MK ((2007) ) Inflammatory processes in Alzheimer’s disease. J Neuroimmunol 184: , 69–91. |
[88] | Gray SC , Kinghorn KJ , Woodling NS ((2020) ) Shifting equilibriums in Alzheimer’s disease: The complex roles of microglia in neuroinflammation, neuronal survival and neurogenesis. Neural Regener Res 15: , 1208. |
[89] | El Khoury J , Hickman SE , Thomas CA , Cao L , Silverstein SC , Loike JD ((1996) ) Scavenger receptor-mediated adhesion of microglia to β-amyloid fibrils. Nature 382: , 716–719. |
[90] | Landreth GE , Reed-Geaghan EG ((2009) ) Toll-like receptors in Alzheimer’s disease. Curr Top Microbiol Immunol 336: , 137–153. |
[91] | Venegas C , Heneka MT ((2017) ) Danger-associated molecular patterns in Alzheimer’s disease. J Leukoc Biol 101: , 87–98. |
[92] | Gebicke-Haerter PJ ((2001) ) Microglia in neurodegeneration: Molecular aspects. Microsc Res Tech 54: , 47–58. |
[93] | Streit WJ ((2002) ) Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 40: , 133–139. |
[94] | Mines M , Ding Y , Fan GH ((2007) ) The many roles of chemokine receptors in neurodegenerative disorders: Emerging new therapeutical strategies. Curr Med Chem 14: , 2456–2470. |
[95] | Walton NM , Sutter BM , Laywell ED , Levkoff LH , Kearns SM , Marshall GP 2nd, Scheffler B , Steindler DA ((2006) ) Microglia instruct subventricular zone neurogenesis. Glia 54: , 815–825. |
[96] | Nikolakopoulou AM , Dutta R , Chen Z , Miller RH , Trapp BD ((2013) ) Activated microglia enhance neurogenesis via trypsinogen secretion. Proc Natl Acad Sci U S A 110: , 8714–8719. |
[97] | Mir S , Cai W , Carlson SW , Saatman KE , Andres DA ((2017) ) IGF-1 mediated neurogenesis involves a novel RIT1/Akt/Sox2 cascade. Sci Rep 7: , 3283. |
[98] | Cacci E , Ajmone-Cat MA , Anelli T , Biagioni S , Minghetti L ((2008) ) In vitro neuronal and glial differentiation from embryonic or adult neural precursor cells are differently affected by chronic or acute activation of microglia. Glia 425: , 412–425. |
[99] | Ekdahl CT , Claasen JH , Bonde S , Kokaia Z , Lindvall O ((2003) ) Inflammation is detrimental for neurogenesis in adult brain. Proc Natl Acad Sci U S A 100: , 13632–13637. |
[100] | Pluchino S , Muzio L , Imitola J , Deleidi M , Alfaro-Cervello C , Salani G , Porcheri C , Brambilla E , Cavasinni F , Bergamaschi A , Garcia-Verdugo JM , Comi G , Khoury SJ , Martino G ((2008) ) Persistent inflammation alters the function of the endogenous brain stem cell compartment. Brain 131: , 2564–2578. |
[101] | Tepavcević V , Lazarini F , Alfaro-Cervello C , Kerninon C , Yoshikawa K , Garcia-Verdugo JM , Lledo PM , Nait-Oumesmar B , Baron-VanEvercooren A ((2011) ) Inflammation-induced subventricular zonedysfunction leads to olfactory deficits in a targeted mouse model ofmultiple sclerosis. J Clin Invest 121: , 4722–4734. |
[102] | Fan LW , Pang Y ((2017) ) Dysregulation of neurogenesis by neuroinflammation: Key differences in neurodevelopmental and neurological disorders. Neural Regen Res 12: , 366–371. |
[103] | Bhattacharjee S , Lukiw WJ ((2013) ) Alzheimer’s disease and the microbiome. Front Cell Neurosci 7: , 153. |
[104] | Bagyinszky E , Giau VV , Shim K , Suk K , An SSA , Kim S ((2017) ) Role of inflammatory molecules in the Alzheimer’s disease progression and diagnosis. J Neurol Sci 376: , 242–254. |
[105] | Zhao Y , Cong L , Jaber V , Lukiw WJ ((2017) ) Microbiome-derived lipopolysaccharide enriched in the perinuclear region of Alzheimer’s disease brain. Front Immunol 8: , 1064. |
[106] | Zhao Y , Jaber V , Lukiw WJ ((2017) ) Secretory products of the human GI tract and their potential impact on Alzheimer’s disease (AD): Detection of lipopolysaccharide (LPS) in AD Hippocampus. Front Cell Infect Microbiol 7: , 318. |
[107] | Lio D , Licastro F , Scola L , Chiappelli M , Grimaldi LM , Crivello A , Colonna-Romano G , Candore G , Franceschi C , Caruso C ((2003) ) Interleukin-10 promoter polymorphism in sporadic Alzheimer’s disease. Genes Immun 4: , 234–238. |
[108] | Arosio B , Trabattoni D , Galimberti L , Bucciarelli P , Fasano F , Calabresi C , Cazzullo CL , Vergani C , Annoni G , Clerici M ((2004) ) Interleukin-10 and interleukin-6 gene polymorphisms as risk factors for Alzheimer’s disease. Neurobiol Aging 25: , 1009–1015. |
[109] | Speciale L , Calabrese E , Saresella M , Tinelli C , Mariani C , Sanvito L , Longhi R , Ferrante P ((2007) ) Lymphocyte subset patterns and cytokine production in Alzheimer’s disease patients. Neurobiol Aging 28: , 1163–1169. |
[110] | Montine TJ , Sidell KR , Crews BC , Markesbery WR , Marnett LJ , Roberts LJ , Morrow JD ((1999) ) Elevated cerebrospinal fluid prostaglandin E2 levels in patients with probable Alzheimer’s disease. Neurology 53: , 1495–1498. |
[111] | Combrinck M , Williams J , De Berardinis MA , Warden D , Puopolo M , Smith AD , Minghetti L ((2006) ) Levels of CSF prostaglandin E2, cognitive decline, and survival in Alzheimer’s disease. J Neurol Neurosurg Psychiatry 77: , 85–88. |
[112] | Cameron HA , McKay RD ((2001) ) Adult neurogenesis produces a large pool of new granule cells in the dentate gyrus. J Comp Neurol 435: , 406–417. |
[113] | Abrous DN , Koehl M , Le Moal M ((2005) ) Adult neurogenesis: From precursors to network and physiology. Physiol Rev 85: , 523–569. |
[114] | Sierra A , Encinas JM , Deudero JJ , Chancey JH , Enikolopov G , Overstreet-Wadiche LS , Tsirka SE , Maletic-Savatic M ((2010) ) Microglia shape adult hippocampal neurogenesis through apoptosis-coupled phagocytosis. Cell Stem Cell 7: , 483–495. |
[115] | Aarum J , Sandberg K , Haeberlein SLB , Persson MA ((2003) ) Migration and differentiation of neural precursor cells can be directed by microglia. Proc Natl Acad Sci U S A 100: , 15983–15988. |
[116] | Xavier ALR , Kress BT , Goldman SA , de Menezes JRL , Nedergaard M ((2015) ) A distinct population of microglia supports adult neurogenesis in the subventricular zone. J Neurosci 35: , 11848–11861. |
[117] | Menezes JR , Smith CM , Nelson KC , Luskin MB ((1995) ) The division of neuronal progenitor cells during migration in the neonatal mammalian forebrain. Mol Cell Neurosci 6: , 496–508. |
[118] | Zhao C , Teng EM , Summers RG Jr, Ming GL , Gage FH ((2006) ) Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J Neurosci 26: , 3–11. |
[119] | Platel JC , Dave KA , Bordey A ((2008) ) Control of neuroblast production and migration by converging GABA and glutamate signals in the postnatal forebrain. J Physiol 586: , 3739–3743. |
[120] | Aloisi F ((2001) ) Immune function of microglia. Glia 36: , 165–179. |
[121] | Streit WJ ((2000) ) Microglial response to brain injury: A brief synopsis. Toxicol Pathol 28: , 28–30. |
[122] | Pluchino S , Quattrini A , Brambilla E , Gritti A , Salani G , Dina G , Galli R , Del Carro U , Amadio S , Bergami A , Furlan R , Comi G , Vescovi AL , Martino G ((2003) ) Injection of adult neurospheres induces recovery in a chronic model of multiple sclerosis. Nature 422: , 688–694. |
[123] | Ben-Hur T , Einstein O , Mizrachi-Kol R , Ben-Menachem O , Reinhartz E , Karussis D , Abramsky O ((2003) ) Transplanted multipotential neuralprecursor cells migrate into the inflamed white matter in responseto experimental autoimmune encephalomyelitis. Glia 41: , 73–80. |
[124] | Picard-Riera N , Decker L , Delarasse C , Goude K , Nait-Oumesmar B , Liblau R , Pham-Dinh D , Baron-Van Evercooren A ((2002) ) Experimental autoimmune encephalomyelitis mobilizes neural progenitors from the subventricular zone to undergo oligodendrogenesis in adult mice. Proc Natl Acad Sci U S A 99: , 13211–13216. |
[125] | Arvidsson A , Collin T , Kirik D , Kokaia Z , Lindvall O ((2002) ) Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med 8: , 963–970. |
[126] | Nakatomi H , Kuriu T , Okabe S , Yamamoto SI , Hatano O , Kawahara N , Tamura A , Kirino T , Nakafuku M ((2002) ) Regeneration of hippocampal pyramidal neurons after ischemic brain injury by recruitment of endogenous neural progenitors. Cell 110: , 429–441. |
[127] | Young MJ , Ray J , Whiteley SJ , Klassen H , Gage FH ((2000) ) Neuronal differentiation and morphological integration of hippocampal progenitor cells transplanted to the retina of immature and mature dystrophic rats. Mol Cell Neurosci 16: , 197–205. |
[128] | Aboody KS , Brown A , Rainov NG , Bower KA , Liu S , Yang W , Small JE , Herrlinger U , Ourednik V , Black PMcL , Breakefield XO , Snyder EY ((2000) ) Neural stem cells display extensive tropism for pathology in adult brain: Evidence from intracranial gliomas. Proc Natl AcadSci U S A 97: , 12846–12851. |
[129] | Snyder EY , Yoon C , Flax JD , Macklis JD ((1997) ) Multipotent neural precursors can differentiate toward replacement of neurons undergoing targeted apoptotic degeneration in adult mouse neocortex. Proc Natl Acad Sci U S A 94: , 11663–11668. |
[130] | Svendsen CN , Caldwell MA , Shen J , ter Borg MG , Rosser AE , Tyers P , Karmiol S , Dunnett SB ((1997) ) Long-term survival of human central nervous system progenitor cells transplanted into a rat model of Parkinson’s disease. Exp Neurol 148: , 135–146. |
[131] | Mundim MV , Zamproni LN , Pinto AAS , Galindo LT , Xavier AM , Glezer I , Porcionatto M ((2019) ) A new function for Prokineticin 2: Recruitment of SVZ-derived neuroblasts to the injured cortex in a mouse model of traumatic brain injury. Mol Cell Neurosci 94: , 1–10. |
[132] | Monterey MD , Wei H , Wu X , Wu JQ ((2021) ) The many faces of astrocytes in Alzheimer’s disease. Front Neurol 12: , 619626. |
[133] | Reid MJ , Beltran-Lobo P , Johnson L , Perez-Nievas BG , Noble W ((2020) ) Astrocytes in tauopathies. Front Neurol 11: , 572850. |
[134] | Preman P , Alfonso-Triguero M , Alberdi E , Verkhratsky A , Arranz AM ((2021) ) Astrocytes in Alzheimer’s disease: Pathological significance and molecular pathways. Cell 10: , 540. |
[135] | Cassé F , Richetin K , Toni N ((2018) ) Astrocytes’ contribution toadult neurogenesis in physiology and Alzheimer’s disease. Front Cell Neurosci 12: , 432. |
[136] | Sung PS , Lin PY , Liu CH , Su HC , Tsai KJ ((2020) ) Neuroinflammation and neurogenesis in Alzheimer’s disease and potential therapeutic approaches. Int J Mol Sci 21: , 701. |
[137] | Garcia-Marin V , Garcia-Lopez P , Freire M ((2007) ) Cajal’s contributions to the study of Alzheimer’s disease. J Alzheimers Dis 12: , 161–174. |
[138] | Bouvier DS , Jones EV , Quesseveur G , Davoli MA , Ferreira TA , Quirion R , Mechawar N , Murai KK ((2016) ) High resolution dissection of reactive glial nets in Alzheimer’s disease. Sci Rep 6: , 24544. |
[139] | Osborn LM , Kamphuis W , Wadman WJ , Hol EM ((2016) ) Astrogliosis: An integral player in the pathogenesis of Alzheimer’s disease. Prog Neurobiol 144: , 121–141. |
[140] | Serrano-Pozo A , Muzikansky A , Gómez-Isla T , Growdon JH , Betensky RA , Frosch MP , Hyman BT ((2013) ) Differential relationships of reactive astrocytes and microglia to fibrillar amyloid deposits in Alzheimer disease. J Neuropathol Exp Neurol 72: , 462–471. |
[141] | Beach T , McGeer E ((1988) ) Lamina-specific arrangement of astrocytic gliosis and senile plaques in Alzheimer’s disease visual cortex. Brain Res 463: , 357–361. |
[142] | Verkhratsky A , Zorec R , Rodríguez JJ , Parpura V ((2016) ) Astroglia dynamics in ageing and Alzheimer’s disease. Curr OpinPharm 26: , 74–79. |
[143] | Rodríguez JJ , Olabarria M , Chvatal A , Verkhratsky A , Rodr JJ ((2008) ) Astroglia in dementia and Alzheimer’s disease. Cell Death Differ 16: , 378–385. |
[144] | Verkhratsky A , Zorec R , Parpura V ((2017) ) Stratification of astrocytes in healthy and diseased brain. Brain Pathol 27: , 629–644. |
[145] | Frost GR , Li YM ((2017) ) The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol 7: , 170228. |
[146] | Burda JE , Sofroniew MV ((2014) ) Reactive gliosis and the multicellular response to CNS damage and disease. Neuron 81: , 229–248. |
[147] | Escartin C , Galea E , Lakatos A , O’Callaghan JP , Petzold GC , Serrano-Pozo A , Steinhäuser C , Volterra A , Carmignoto G , Agarwal A , Allen NJ , Araque A , Barbeito L , Barzilai A , Bergles DE , Bonvento G , Butt AM , Chen W-T , Cohen-Salmon M , Cunningham C , Deneen B , DeStrooper B , Díaz-Castro B , Farina C , Freeman M , Gallo V , Goldman JE , Goldman SA , Götz M , Gutiérrez A , Haydon PG , Heiland DH , Hol EM , Holt MG , Iino M , Kastanenka KV , Kettenmann H , Khakh BS , Koizumi S , Lee CJ , Liddelow SA , MacVicar BA , Magistretti P , Messing A , Mishra A , Molofsky AV , Murai KK , Norris CM , Okada S , Oliet SHR , Oliveira JF , Panatier A , Parpura V , Pekna M , Pekny M , Pellerin L , Perea G , Pérez-Nievas BG , Pfrieger FW , Poskanzer KE , Quintana FJ , Ransohoff RM , Riquelme-Perez M , Robel S , Rose CR , Rothstein JD , Rouach N , Rowitch DH , Semyanov A , Sirko S , Sontheimer H , Swanson RA , Vitorica J , Wanner I-B , Wood LB , Wu J , Zheng B , Zimmer ER , Zorec R , Sofroniew MV , Verkhratsky A ((2021) ) Reactiveastrocyte nomenclature, definitions, and future directions. NatNeurosci 24: , 312–325. |
[148] | Pekny M , Pekna M , Messing A , Steinhauser C , Lee JM , Parpura V , Hol EM , Sofroniew MV , Verkhratsky A ((2016) ) Astrocytes: A central element in neurological diseases. Acta Neuropathol 131: , 323–345. |
[149] | Li K , Li J , Zheng J , Qin S ((2019) ) Reactive astrocytes in neurodegenerative diseases. Aging Dis 10: , 664–675. |
[150] | Chen Y , Swanson RA ((2003) ) Astrocytes and brain injury. J Cereb Blood Flow Metab 23: , 137–149. |
[151] | Verkhratsky A , Olabarria M , Noristani HN , Yeh CY , Rodriguez JJ ((2010) ) Astrocytes in Alzheimer’s disease. Neurotherapeutics 7: , 399–412. |
[152] | Diniz LP , Tortelli V , Matias I , Morgado J , Araujo APB , Melo HM , Da Silva GSS , Alves-Leon SV , De Souza JM , Ferreira ST , De Felice FG , Gomes FCA ((2017) ) Astrocyte transforming growth factor beta 1 protects synapses against Abeta oligomers in Alzheimer’s disease model. J Neurosci 37: , 6797–6809. |
[153] | Lian H , Zheng H ((2016) ) Signaling pathways regulating neuron-glia interaction and their implications in Alzheimer’s disease. J Neurochem 136: , 475–491. |
[154] | Gomez-Arboledas A , Davila JC , Sanchez-Mejias E , Navarro V , Nuñez-Diaz C , Sanchez-Varo R , Sanchez-Mico MV , Trujillo-Estrada L , Fernandez-Valenzuela JJ , Vizuete M , Comella JX , Galea E , Vitorica J , Gutierrez A ((2018) ) Phagocytic clearance of presynaptic dystrophies by reactive astrocytes in Alzheimer’s disease. Glia 66: , 637–653. |
[155] | Iliff JJ , Wang M , Liao Y , Plogg BA , Peng W , Gundersen GA , Benveniste H , Vates GE , Deane R , Goldman SA , Nagelhus EA , Nedergaard M ((2012) ) A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid-β. Sci Transl Med 4: , 147ra111. |
[156] | Benarroch EE ((2007) ) Aquaporin-4, homeostasis, and neurologic disease. Neurology 69: , 2266–2268. |
[157] | Lim DA , Alvarez-Buylla A ((1999) ) Interaction between astrocytes and adult subventricular zone precursors stimulates neurogenesis. Proc Natl Acad Sci U S A 96: , 7526–7531. |
[158] | Cao X , Li L-P , Qin X-H , Li S-J , Zhang M , Wang Q , Hu H-H , Fang Y-Y , Gao Y-B , Li X-W , Sun L-R , Xiong W-C , Gao T-M , Zhu X-H ((2013) ) Astrocytic adenosine 5’-triphosphate release regulates the proliferation of neural stem cells in the adult hippocampus. Stem Cells 31: , 1633–1643. |
[159] | Mothet J-P , Pollegioni L , Ouanounou G , Martineau M , Fossier P , Baux G ((2005) ) Glutamate receptor activation triggers a calcium-dependent and SNARE protein-dependent release of the gliotransmitter D-serine. Proc Natl Acad Sci U S A 102: , 5606–5611. |
[160] | Papouin T , Henneberger C , Rusakov DA , Oliet SHR ((2017) ) Astroglial versus neuronal D-serine: Fact checking. Trends Neurosci 40: , 517–520. |
[161] | Sultan S , Gebara E , Toni N ((2013) ) Doxycycline increases neurogenesis and reduces microglia in the adult hippocampus. Front Neurosci 7: , 131. |
[162] | Palmer TD , Ray J , Gage FH ((1995) ) FGF-2-responsive neuronal progenitors reside in proliferative and quiescent regions of the adult rodent brain. Mol Cell Neurosci 6: , 474–486. |
[163] | Kirby ED , Muroy SE , Sun WG , Covarrubias D , Leong MJ , Barchas LA , Kaufer D ((2013) ) Acute stress enhances adult rat hippocampalneurogenesis and activation of newborn neurons via secretedastrocytic FGF2. eLife 2: , e00362. |
[164] | Jovıcíc A , Gitler AD ((2017) ) Distinctrepertoires of microRNAs present in mouse astrocytes compared toastrocyte-secreted exosomes. PLoS One 12: , e0171418. |
[165] | Song H , Stevens CF , Gage FH ((2002) ) Astroglia induce neurogenesis from adult neural stem cells. Nature 417: , 39–44. |
[166] | Ashton RS , Conway A , Pangarkar C , Bergen J , Lim K-I , Shah P , Bissell M , Schaffer DV ((2012) ) Astrocytes regulate adult hippocampal neurogenesis through ephrin-B signaling. Nat Neurosci 15: , 1399–1406. |
[167] | Christopherson KS , Ullian EM , Stokes CCA , Mullowney CE , Hell JW , Agah A , Lawler J , Mosher DF , Bornstein P , Barres BA ((2005) ) Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 120: , 421–433. |
[168] | Lu Z , Kipnis J ((2010) ) Thrombospondin 1-a key astrocyte-derived neurogenic factor. FASEB J 24: , 1925–1934. |
[169] | Ueki T , Tanaka M , Yamashita K , Mikawa S , Qiu Z , Maragakis NJ , Hevner RF , Miura N , Sugimura H , Sato K ((2003) ) A novel secretory factor, Neurogenesin-1, provides neurogenic environmental cues for neural stem cells in the adult hippocampus. J Neurosci 23: , 11732–11740. |
[170] | Barkho BZ , Song H , Aimone JB , Smrt RD , Kuwabara T , Nakashima K , Gage FH , Zhao X ((2006) ) Identification of astrocyte-expressed factors that modulate neural stem/progenitor cell differentiation. Stem Cells Dev 15: , 407–421. |
[171] | Sultan S , Li L , Moss J , Petrelli F , Cassé F , Gebara E , Lopatar J , Pfrieger FW , Bezzi P , Bischofberger J , Toni N ((2015) ) Synapticintegration of adult-born hippocampal neurons is locally controlledby astrocytes. Neuron 88: , 957–972. |
[172] | Leng F , Edison P ((2021) ) Neuroinflammation and microglial activationin Alzheimer disease: Where do we go from here? Nat Rev Neurol 17: , 157–172. |
[173] | Kwon HS , Koh SH ((2020) ) Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl Neurodegener 9: , 1–12. |
[174] | Kempuraj D , Thangavel R , Natteru PA , Selvakumar GP , Saeed D , Zahoor H , Zaheer S , Iyer SS , Zaheer A ((2016) ) Neuroinflammation induces neurodegeneration. J Neurol Neurosurg Spine 20161: , 1003. |
[175] | Glass CK , Saijo K , Winner B , Marchetto MC , Gage FH ((2010) ) Mechanisms underlying inflammation in neurodegeneration. Cell 140: , 918–934. |
[176] | Lee DC , Rizer J , Selenica ML , Reid P , Kraft C , Johnson A , Blair L , Gordon MN , Dickey CA , Morgan D ((2010) ) LPS- induced inflammation exacerbates phospho-tau pathology in rTg4510 mice. J Neuroinflammation 7: , 56. |
[177] | Jo S , Yarishkin O , Hwang YJ , Chun YE , Park M , Woo DH , Bae JY , Kim T , Lee J , Chun H , Park HJ , Lee DY , Hong J , Kim HY , Oh S-J , Park SJ , Lee H , Yoon B-E , Kim YS , Jeong Y , Shim I , Bae YC , Cho J , Kowall NW , Ryu H , Hwang E , Kim D , Lee CJ ((2014) ) GABA from reactive astrocytes impairs memory in mouse models of Alzheimer’s disease. Nat Med 20: , 886–896. |
[178] | Chang J , Liu F , Lee M , Wu B , Ting K , Zara JN , Soo C , Al Hezaimi K , Zou W , Chen X , Mooney DJ , Wang C-Y ((2013) ) NF-κB inhibits osteogenic differentiation of mesenchymal stem cells by promoting β-catenin degradation. Proc Natl Acad Sci U S A 110: , 9469–9474. |
[179] | Winkler EA , Nishida Y , Sagare AP , Rege SV , Bell RD , Perlmutter D , Sengillo JD , Hillman S , Kong P , Nelson AR , Sullivan JS , Zhao Z , Meiselman HJ , Wenby RB , Soto J , Abel ED , Makshanoff J , Zuniga E , De Vivo DC , Zlokovic BV ((2015) ) GLUT1 reductions exacerbate Alzheimer’s disease vasculo-neuronal dysfunction and degeneration. Nat Neurosci 18: , 521–530. |
[180] | Kisler K , Nelson AR , Montagne A , Zlokovic BV ((2017) ) Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat Rev Neurosci 18: , 419–434. |
[181] | Heneka MT , Sastre M , Dumitrescu-Ozimek L , Dewachter I , Walter J , Klockgether T , Van Leuven F ((2005) ) Focal glial activation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J Neuroinflammation 2: , 22. |
[182] | Odfalk KF , Bieniek KF , Hopp SC ((2022) ) Microglia: Friend and foe in tauopathy. Prog Neurobiol 216: , 102306. |
[183] | Da Gray SC , Kinghorn KJ , Woodling NS ((2020) ) Shifting equilibriums in Alzheimer’s disease: The complex roles of microglia in neuroinflammation, neuronal survival and neurogenesis. Neural Regen Res 15: , 120. |
[184] | Braak H , Braak E ((1991) ) Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82: , 239–259. |
[185] | Braak H , Braak E ((1997) ) Frequency of stages of Alzheimer-related lesions in different age categories. Neurobiol Aging 18: , 351–357. |
[186] | Braak H , Alafuzoff I , Arzberger T , Kretzschmar H , Del Tredici K ((2006) ) Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112: , 389–404. |
[187] | Braak H , Thal DR , Ghebremedhin E , Del Tredici K ((2011) ) Stages of the pathologic process in Alzheimer disease: Age categories from 1 to 100 years. J Neuropathol Exp Neurol 70: , 960–969. |
[188] | Braak H , Del Tredici K ((2011) ) The pathological process underlying Alzheimer’s disease in individuals under thirty. Acta Neuropathol 121: , 171–181. |
[189] | Braak H , Del Tredici K ((2015) ) Neuroanatomy and pathology of sporadic Alzheimer’s disease. Adv Anat Embryol Cell Biol 215: , 1–162. |
[190] | Braak H , Del Tredici K ((2015) ) The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease. Brain 138: , 2814–2833. |
[191] | Marien MR , Colpaert FC , Rosenquist AC ((2004) ) Noradrenergic mechanisms in neurodegenerative diseases: A theory. Brain Res Rev 45: , 38–78. |
[192] | Aston-Jones G , Cohen JD ((2005) ) Adaptive gain and the role of the locus coeruleus-norepinephrine system in optimal performance. J Comp Neurol 493: , 99–110. |
[193] | Samuels ER , Szabadi E ((2009) ) Functional neuroanatomy of the noradrenergic locus coeruleus: Its roles in the regulation of arousal and autonomic function. Part I: Principles of functional organization. Curr Neuropharmacol 6: , 235–253. |
[194] | Sara SJ ((2009) ) The locus coeruleus and noradrenergic modulation of cognition. Nature Rev Neurosci 10: , 211–223. |
[195] | Counts SE , Mufson EJ ((2012) ) Locus coeruleus. In The human nervous system, 3rd ed, Mai JK, Paxinos G, eds. Academic, New York, pp. 425–438. |
[196] | O’Donnell J , Zeppenfeld D , McConnell E , Pena S , Nedergaard M ((2012) ) Norepinephrine: A neuromodulator that boosts the function of multiple cell types to optimize CNS performance. Neurochem Res 37: , 2496–2512. |
[197] | Braak H , Braak E ((1992) ) The human entorhinal cortex: Normal morphology and lamina-specific pathology in various diseases. Neurosci Res 15: , 6–31. |
[198] | Attems J , Jellinger KA ((2006) ) Olfactory tau pathology in Alzheimer’s disease and mild cognitive impairment. Clin Neuropathol 25: , 265–271. |
[199] | Braak H , Del Tredici K ((2014) ) Are cases with tau pathology in theabsence of Ab deposits part of the AD-related pathological process? Acta Neuropathol 128: , 767–772. |
[200] | Furman JL , Vaquer-Alicea J , White CL , Cairns NJ , Nelson PT , Diamond MI ((2017) ) Widespread tau seeding activity at early Braak stages. Acta Neuropathol 133: , 91–100. |
[201] | Clavaguera F , Bolmont T , Crowther RA , Abramowski D , Frank S , Probst A , Fraser G , Stalder AK , Beibel M , Staufenbiel M , Jucker M , Goedert M , Tolnay M ((2009) ) Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol 11: , 909–913. |
[202] | de Calignon A , Polydoro M , Suárez-Calvet M , William C , AdamowiczDavid H , Kathy KJ , Pitstick R , Sahara N , Ashe KH , Carlson GA , Spires-Jones TL , Hyman BT ((2012) ) Propagation of tau pathology in amodel of early Alzheimer’s disease. Neuron 73: , 685–697. |
[203] | Frost B , Jacks RL , Diamond MI ((2009) ) Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem 284: , 12845–12852. |
[204] | Guo JL , Lee VMY ((2011) ) Seeding of normal tau by pathological tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem 286: , 15317–15331. |
[205] | Liu L , Drouet V , Wu JW , Witter MP , Small SA , Clelland C , Duff K ((2012) ) Trans-synaptic spread of tau pathology in vivo. PLoS One 7: , e31302. |
[206] | Prusiner SB ((1984) ) Some speculations about prions, amyloid, andAlzheimer’s disease. N Engl J Med 310: , 661–663. |
[207] | Weaver CL , Espinoza M , Kress Y , Davies P ((2000) ) Conformational change as one of the earliest alterations of tau in Alzheimer’s disease. Neurobiol Aging 21: , 719–727. |
[208] | d ‘Errico P , Meyer-Luehmann M ((2020) ) Mechanisms of pathogenic tau and Aβ protein spreading in Alzheimer’s disease. Front Aging Neurosci 12: , 265. |
[209] | Colin M , Dujardin S , Schraen-Maschke S , Meno-Tetang G , Duyckaerts C , Courade JP , Buée L ((2020) ) From the prion-like propagationhypothesis to therapeutic strategies of anti-tau immunotherapy. Acta Neuropathol 139: , 3–25. |
[210] | Nobuhara CK , DeVos SL , Commins C , Wegmann S , Moore BD , Roe AD , Costantino I , Frosch MP , Pitstick R , Carlson GA , Hock C , Nitsch RM , Montrasio F , Grimm J , Cheung AE , Dunah AW , Wittmann M , Bussiere T , Weinreb PH , Hyman BT ((2017) ) Tau antibody targeting pathological species blocks neuronal uptake and interneuron propagation of tau in vitro. Am J Pathol 187: , 1399–1412. |
[211] | Vandermeeren M , Borgers M , Van Kolen K , Theunis C , Vasconcelos B , Bottelbergs A , Wintmolders C , Daneels G , Willems R , Dockx K , Delbroek L , Marreiro A , Ver Donck L , Sousa C , Nanjunda R , Lacy E , Van De Casteele T , Van Dam D , De Deyn PP , Kemp JA , Thomas MJ , Mercken MH ((2018) ) Anti-tau monoclonal antibodies derived from soluble and filamentous tau show diverse functional properties in vitro and in vivo. J Alzheimers Dis 65: , 265–281. |
[212] | Yanamandra K , Kfoury N , Jiang H , Mahan TE , Ma S , Maloney SE , Wozniak DF , Diamond MI , Holtzman DM ((2013) ) Anti-tau antibodies that block tau aggregate seeding in vitro markedly decrease pathology and improve cognition in vivo. Neuron 80: , 402–414. |
[213] | Evans LD , Wassmer T , Fraser G , Smith J , Perkinton M , Billinton A , Livesey FJ ((2018) ) Extracellular monomeric and aggregated tau efficiently enter human neurons through overlapping but distinct pathways. Cell Rep 22: , 3612–3624. |
[214] | Holmes BB , Devos SL , Kfoury N , Li M , Jacks R , Yanamandra K , Ouidja MO , Brodsky FM , Marasa J , Bagchi DP , Kotzbauer PT , Miller TM , Papy-Garcia D , Diamond MI ((2013) ) Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A 110: , E3138–3147. |
[215] | Holmes BB , Furman JL , Mahan TE , Yamasaki TR , Mirbaha H , Eades WC , Belaygorod L , Cairns NJ , Holtzman DM , Diamond MI ((2014) ) Proteopathic tau seeding predicts tauopathy in vivo. Proc Natl Acad Sci U S A 111: , E4376–4385. |
[216] | Nonaka T , Watanabe ST , Iwatsubo T , Hasegawa M ((2010) ) Seeded aggregation and toxicity of alpha-synuclein and tau: Cellular models of neurodegenerative diseases. J Biol Chem 285: , 34885–34898. |
[217] | Reilly P , Winston CN , Baron KR , Trejo M , Rockenstein EM , Akers JC , Kfoury N , Diamond M , Masliah E , Rissman RA , Yuan SH ((2017) ) Novel human neuronal tau model exhibiting neurofibrillary tangles and transcellular propagation. Neurobiol Dis 106: , 222–234. |
[218] | Santa-Maria I , Varghese M , Ksiezak-Reding H , Dzhun A , Wang J , Pasinetti GM ((2012) ) Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of tau protein in aggresomes. J Biol Chem 287: , 20522–20533. |
[219] | Takahashi M , Miyata H , Kametani F , Nonaka T , Akiyama H , Hisanaga S , Hasegawa M ((2015) ) Extracellular association of APP and tau fibrils induces intracellular aggregate formation of tau. Acta Neuropathol 129: , 895–907. |
[220] | Takeda S , Commins C , DeVos SL , Nobuhara CK , Wegmann S , Roe AD , Costantino I , Fan Z , Nicholls SB , Sherman AE , Trisini Lipsanopoulos AT , Scherzer CR , Carlson GA , Pitstick R , Peskind ER , Raskind MA , Li G , Montine TJ , Frosch MP , Hyman BT ((2016) ) Seed-competent high-molecular-weight tau species accumulates in the cerebrospinal fluid of Alzheimer’s disease mouse model and human patients. Ann Neurol 80: , 355–367. |
[221] | Takeda S , Wegmann S , Cho H , DeVos SL , Commins C , Roe AD , Nicholls SB , Carlson GA , Pitstick R , Nobuhara CK , Costantino I , Frosch MP , Müller DJ , Irimia D , Hyman BT ((2015) ) Neuronal uptake and propagation of a rare phosphorylated high-molecular-weight tau derived from Alzheimer’s disease brain. Nat Commun 6: , 849. |
[222] | Mudher A , Colin M , Dujardin S , Medina M , Dewachter I , Naini A , Mandelkow E-M , Mandelkow E , Buée L , Goedert M , Brion JP ((2017) ) What is the evidence that tau pathology spreads through prion-likepropagation? Acta Neuropathol Commun 5: , 99. |
[223] | Audouard E , Houben S , Masaracchia C , Yilmaz Z , Suain V , Authelet M , De Decker R , Buée L , Boom A , Leroy K , Ando K , Brion J-P ((2016) ) High-molecular-weight paired helical filaments from Alzheimer braininduce seeding of wild-type mouse tau into an argyrophillic 4R taupathology in vivo. Am J Pathol 186: , 2709–2722. |
[224] | Guo JL , Narasimhan S , Changolkar L , He Z , Stieber A , Zhang B , Gathagan RJ , Iba M , McBride JD , Trojanowski JQ , Lee VMY ((2016) ) Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med 213: , 2635–2654. |
[225] | Kaufman SK , Thomas TL , Del Tredici K , Braak H , Diamond MI ((2017) ) Characterization of tau prion seeding activity and strains from formaldehyde-fixed tissue. Acta Neuropathol Commun 5: , 41. |
[226] | Skachokova Z , Martinisi A , Flach M , Sprenger F , Naegelin Y , Steiner-Monard V ((2019) ) Cerebrospinal fluid from Alzheimer’s disease patients promotes tau aggregation in transgenic mice. Acta Neuropathol Commun 7: , 72. |
[227] | Falcon B , Cavallini A , Angers R , Glover S , Murray TK , Barnham L , Jackson S , O’Neill MJ , Isaacs AM , Hutton ML , Szekeres PG , Goedert M , Bose S ((2015) ) Conformation determines the seeding potencies of native and recombinant tau aggregates. J Biol Chem 290: , 1049–1065. |
[228] | Sanders DW , Kaufman SK , DeVos SL , Sharma AM , Mirbaha H , Li A , Barker SJ , Foley AC , Thorpe JR , Serpell LC , Miller TM , Grinberg LT , Seeley WW , Diamond MI ((2014) ) Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron 82: , 1271–1288. |
[229] | Clavaguera F , Akatsu H , Fraser G , Crowther RA , Frank S , Hench J , Probst A , Winkler DT , Reichwald J , Staufenbiel M , Ghetti B , Goedert M , Tolnay M ((2013) ) Brain homogenates from human tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A 110: , 9535–9540. |
[230] | Ahmed Z , Cooper J , Murray TK , Garn K , McNaughton E , Clarke H , Parhizkar S , Ward MA , Cavallini A , Jackson S , Bose S , Clavaguera F , Tolnay M , Lavenir I , Goedert M , Hutton ML , O’Neill MJ ((2014) ) A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: The pattern of spread is determined by connectivity, not proximity. Acta Neuropathol 127: , 667–683. |
[231] | Dujardin S , Lécolle K , Caillierez R , Bégard S , Zommer N , Lachaud C , Carrier S , Dufour N , Aurégan G , Winderickx J , Hantraye P , Déglon N , Colin M , Buée L ((2014) ) Neuron-to-neuron wild-type tau protein transfer through atrans-synaptic mechanism: Relevance to sporadic tauopathies. Acta Neuropathol Commun 2: , 14. |
[232] | Iba M , McBride JD , Guo JL , Zhang B , Trojanowski JQ , Lee VM-Y ((2015) ) Tau pathology spread in PS19 tau transgenic mice following locus coeruleus (LC) injections of synthetic tau fibrils is determined by the LC’s afferent and efferent connections. Acta Neuropathol 130: , 349–362. |
[233] | Peeraer E , Bottelbergs A , Van Kolen K , Stancu I-C , Vasconcelos B , Mahieu M , Duytschaever H , Ver Donck L , Torremans A , Sluydts E , Van Acker N , Kemp JA , Mercken M , Brunden KR , Trojanowski JQ , Dewachter I , Lee VMY , Moechars D ((2015) ) Intracerebral injection of preformed synthetic tau fibrils initiates widespread tauopathy and neuronal loss in the brains of tau transgenic mice. Neurobiol Dis 73: , 83–95. |
[234] | Duyckaerts C , Uchihara T , Seilhean D , He Y , Hauw J-J ((1997) ) Dissociation of Alzheimer type pathology in a disconnected piece of cortex. Acta Neuropathol 93: , 501–507. |
[235] | Zhang H , Cao Y , Ma L , Wei Y , Li H ((2021) ) Possible mechanisms of tau spread and toxicity in Alzheimer’s disease. Front Cell Dev Biol 9: , 707268. |
[236] | Kanmert D , Cantlon A , Muratore C , Jin M , O’malley T , Lee G , Young-Pearse TL , Selkoe DJ , Walsh DM ((2015) ) C-terminally truncated forms of tau, but not full-length tau or its C-terminal fragments, are released from neurons independently of cell death. J Neurosci 35: , 10851–10865. |
[237] | Yanamandra K , Patel TK , Jiang H , Schindler S , Ulrich JD , Boxer AL , Miller BL , Kerwin DR , Gallardo G , Stewart F , Finn MB , Cairns NJ , Verghese PB , Fogelman I , West T , Braunstein J , Robinson G , Keyser J , Roh J , Knapik SS , Hu Y , Holtzman DM ((2017) ) Anti-tau antibody administration increases plasma tau in transgenic mice and patients with tauopathy. Sci Transl Med 9: , eaal2029. |
[238] | Perez M , Medina M , Hernandez F , Avila J ((2018) ) Secretion offull-length tau or tau fragments in cell culture models. Propagationof tau in vivo and in vitro. Biomol Concepts 9: , 1–11. |
[239] | Dujardin S , Begard S , Caillierez R , Lachaud C , Delattre L , Carrier S , Loyens A , Galas M-C , Bousset L , Melki R , Aurégan G , Hantraye P , Brouillet E , Buée L , Colin M ((2014) ) Ectosomes: A newmechanism for non-exosomal secretion of tau protein. PLoS One 9: , e100760. |
[240] | Katsinelos T , Zeitler M , Dimou E , Karakatsani A , Muller HM , Nachman E , Steringer JP , Ruiz de Almodovar C , Nickel W , Jahn TR ((2018) ) Unconventional secretion mediates the trans-cellular spreading of tau. Cell Rep 23: , 2039–2055. |
[241] | Merezhko M , Brunello CA , Yan X , Vihinen H , Jokitalo E , Uronen R-L , Huttunen HJ ((2018) ) Secretion of tau via an unconventional non-vesicular mechanism. Cell Rep 25: , 2027–2035. |
[242] | Wang Y , Balaji V , Kaniyappan S , Kruger L , Irsen S , Tepper K , Chandupatla RR , Maetzler W , Schneider A , Mandelkow E , Mandelkow E-M ((2017) ) The release and trans-synaptic transmission of Tau via exosomes. Mol Neurodegener 12: , 5. |
[243] | Abounit S , Wu JW , Duff K , Victoria GS , Zurzolo C ((2016) ) Tunneling nanotubes: A possible highway in the spreading of tau and other prion-like proteins in neurodegenerative diseases. Prion 10: , 344–351. |
[244] | Tardivel M , Begard S , Bousset L , Dujardin S , Coens A , Melki R , Buée L , Colin M ((2016) ) Tunneling nanotube (TNT)-mediatedneuron-to neuron transfer of pathological Tau protein assemblies. Acta Neuropathol Commun 4: , 117. |
[245] | Perez M , Avila J , Hernandez F ((2019) ) Propagation of tau via extracellular vesicles. Front Neurosci 13: , 698. |
[246] | Asai H , Ikezu S , Tsunoda S , Medalla M , Luebke J , Haydar T , Wolozin B , Butovsky O , Kügler S , Ikezu T ((2015) ) Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat Neurosci 18: , 1584–1593. |
[247] | Simón D , García-García E , Royo F , Falcón-Pérez JM , Avila J ((2012) ) Proteostasis of tau. Tau overexpression resultsin its secretion via membrane vesicles. FEBS Lett 586: , 47–54. |
[248] | Baker S , Polanco JC , Götz J ((2016) ) Extracellular vesicles containing P301L mutant tau accelerate pathological tau phosphorylation and oligomer formation but do not seed mature neurofibrillary tangles in ALZ17 mice. J Alzheimers Dis 54: , 1207–1217. |
[249] | Polanco JC , Scicluna BJ , Hill AF , Götz J ((2016) ) Extracellular vesicles isolated from the brains of rTg4510 mice seed tau protein aggregation in a threshold-dependent manner. J Biol Chem 291: , 12445–12466. |
[250] | Saman S , Kim W , Raya M , Visnick Y , Miro S , Jackson B , McKee AC , Alvarez VE , Lee NCY , Hall GF ((2012) ) Exosome-associated tau is secreted in tauopathy models and is selectively phosphorylated in cerebrospinal fluid in early Alzheimer disease. J Biol Chem 287: , 3842–3849. |
[251] | Fiandaca MS , Kapogiannis D , Mapstone M , Boxer A , Eitan E , Schwartz JB , Abner EL , Petersen RC , Federoff HJ , Miller BL , Goetzl EJ ((2015) ) Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement 11: , 600–607. |
[252] | Guix FX , Corbett GT , Cha DJ , Mustapic M , Liu W , Mengel D , Chen Z , Aikawa E , Young-Pearse T , Kapogiannis D , Selkoe DJ , Walsh DM ((2018) ) Detection of aggregation-competent tau in neuron-derived extracellular vesicles. Int J Mol Sci 19: , 663. |
[253] | Winston CN , Goetzl EJ , Akers JC , Carter BS , Rockenstein EM , Galasko D , Masliah E , Rissman RA ((2016) ) Prediction of conversion from mild cognitive impairment to dementia with neuronally derived blood exosome protein profile. Alzheimers Dement (Amst) 3: , 63–72. |
[254] | Guo JL , and Lee VM-Y ((2013) ) Neurofibrillary tangle-like tau pathology induced by synthetic tau fibrils in primary neurons over-expressing mutant tau. FEBS Lett 587: , 717–723. |
[255] | Calafate S , Buist A , Miskiewicz K , Vijayan V , Daneels G , de Strooper B , de Wit J , Verstreken P , Moechars D ((2015) ) Synaptic contacts enhance cell-to-cell tau pathology propagation. Cell Rep 11: , 1176–1183. |
[256] | Clavaguera F , Hench J , Lavenir I , Schweighauser G , Frank S , Goedert M , Tolnay M ((2014) ) Peripheral administration of tau aggregates triggers intracerebral tauopathy in transgenic mice. Acta Neuropathol 127: , 299–301. |
[257] | Wu JW , Herman M , Liu L , Simoes S , Acker CM , Figueroa H , Steinberg JI , Margittai M , Kayed R , Zurzolo C , Di Paolo G , Duff KE ((2013) ) Small misfolded tau species are internalized via bulk endocytosis and anterogradely and retrogradely transported in neurons. J Biol Chem 288: , 1856–1870. |
[258] | Calafate S , Flavin W , Verstreken P , Moechars D ((2016) ) Loss of Bin1 promotes the propagation of tau pathology. Cell Rep 17: , 931–940. |
[259] | Flavin WP , Bousset L , Green ZC , Chu Y , Skarpathiotis S , Chaney MJ , Kordower JH , Melki R , Campbell EM ((2017) ) Endocytic vesicle rupture is a conserved mechanism of cellular invasion by amyloid proteins. Acta Neuropathol 134: , 629–653. |
[260] | Jucker M , Walker L ((2013) ) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501: , 45–51. |
[261] | Congdon EE , Kim S , Bonchak J , Songrug T , Matzavinos A , Kuret J ((2008) ) Nucleation-dependent tau filament formation: The importance of dimerization and an estimation of elementary rate constants. J Biol Chem 283: , 13806–13816. |
[262] | Mirbaha H , Chen D , Morazova OA , Ruff KM , Sharma AM , Liu X , Goodarzi M , Pappu RV , Colby DW , Mirzaei H , Joachimiak LA , Diamond MI ((2018) ) Inert and seed-competent tau monomers suggest structural origins of aggregation. eLife 7: , e36584. |
[263] | Wu JW , Hussaini SA , Bastille IM , Rodriguez GA , Mrejeru A , Rilett K , Sanders DW , Cook C , Fu H , Boonen RACM , Herman M , Nahmani E , Emrani S , Figueroa YH , Diamond MI , Clelland CL , Wray S , Duff KE ((2016) ) Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci 19: , 1085–1092. |
[264] | Walsh DM , Selkoe DJ ((2016) ) A critical appraisal of the pathogenic protein spread hypothesis of neurodegeneration. Nat Rev Neurosci 17: , 251–260. |
[265] | Lewis J , Dickson DW ((2016) ) Propagation of tau pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol 131: , 27–48. |
[266] | Wei Y , Liu M , Wang D ((2022) ) The propagation mechanisms of extracellular tau in Alzheimer’s disease. J Neurol 269: , 1164–1181. |
[267] | Maphis N , Xu G , Kokiko-Cochran ON , Jiang S , Cardona A , Ransohoff RM , Lamb BT , Bhaskar K ((2015) ) Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 138: , 1738–1755. |
[268] | Hopp SC , Lin Y , Oakley D , Roe AD , DeVos SL , Hanlon D , Hyman BT ((2018) ) The role of microglia in processing and spreading of bioactive tau seeds in Alzheimer’s disease. J Neuroinflammation 15: , 269. |
[269] | Kovacs GG , Ferrer I , Grinberg LT , Alafuzoff I , Attems J , Budka H , Cairns NJ , Crary JF , Duyckaerts C , Ghetti B , Halliday GM , Ironside JW , Love S , Mackenzie IR , Munoz DG , Murray ME , Nelson PT , Takahashi H , Trojanowski JQ , Ansorge O , Arzberger T , Baborie A , Beach TG , Bieniek KF , Bigio EH , Bodi I , Dugger BN , Feany M , Gelpi E , Gentleman SM , Giaccone G , Hatanpaa KJ , Heale R , Hof PR , Hofer M , Hortobágyi T , Jellinger K , Jicha GA , Ince P , Kofler J , Kövari E , Kril JJ , Mann DM , Matej R , McKee AC , McLean C , Milenkovic I , Montine TJ , Murayama S , Lee EB , Rahimi J , Rodriguez RD , Rozemüller A , Schneider JA , Schultz C , Seeley W , Seilhean D , Smith C , Tagliavini F , Takao M , Thal DR , Toledo JB , Tolnay M , Troncoso JC , Vinters HV , Weis S , Wharton SB , White III CL , Wisniewski T , Woulfe JM , Yamada M , Dickson DW ((2015) ) Aging-relatedtau astrogliopathy (ARTAG): Harmonized evaluation strategy. Acta Neuropathol 2: , 87–102. |
[270] | LoPresti P , Szuchet S , Papasozomenos SC , Zinkowski RP , Binder LI ((1995) ) Functional implications for the microtubule-associated protein tau: Localization in oligodendrocytes. Proc Natl Acad Sci U S A 92: , 10369–10373. |
[271] | Müller R , Heinrich M , Heck S , Blohm D , Richter-Landsberg C ((1997) ) Expression of microtubule-asssciated proteins MAP2 and tau in cultured rat brain oligodendrocytes. Cell Tissue Res 288: , 239–249. |
[272] | Richetin K , Steullet P , Pachoud M , Perbet R , Parietti E , Maheswaran M , Eddarkaoui S , Bégard S , Pythoud C , Rey M , Caillierez R , Do KQ , Halliez S , Bezzi P , Buée L , Leuba G , Colin M , Toni N , Déglon N ((2020) ) Tau accumulation in astrocytes of the dentategyrus induces neuronal dysfunction and memory deficits inAlzheimer’s disease. Nat Neurosci 23: , 1567–1579. |
[273] | Boisvert MM , Erikson GA , Shokhirev MN , Allen NJ ((2018) ) The aging astrocyte transcriptome from multiple regions of the mouse brain. Cell Rep 22: , 269–285. |
[274] | Yamada K , Cirrito JR , Stewart FR , Jiang H , Finn MB , Holmes BB , Binder LI , Mandelkow E-M , Diamond MI , Lee VM-Y , Holtzman DM ((2011) ) In vivo microdialysis reveals age-dependent decrease of brain interstitial fluid tau levels in P301S human tau transgenic mice. J Neurosci 31: , 13110–13117. |
[275] | Perea JR , López E , Díez-Ballesteros JC , Ávila J , Hernández F , Bolós M ((2019) ) Extracellular monomeric tau isinternalized by astrocytes. Front Neurosci 13: , 442. |
[276] | Andersson CR , Falsig J , Stavenhagen JB , Christensen S , Kartberg F , Rosenqvist N , Finsen B , Pedersen JT ((2019) ) Antibody-mediated clearance of tau in primary mouse microglial cultures requires Fcκ-receptor binding and functional lysosomes. Sci Rep 9: , 4658. |
[277] | Luo W , Liu W , Hu X , Hanna M , Caravaca A , Paul SM ((2015) ) Microglial internalization and degradation of pathological tau is enhanced by an anti-tau monoclonal antibody. Sci Rep 5: , 11161. |
[278] | Majerova P , Zilkova M , Kazmerova Z , Kovac A , Paholikova K , Kovacech B , Zilka N , Novak M ((2014) ) Microglia display modest phagocytic capacity for extracellular tau oligomers. J Neuroinflamm 11: , 161. |
[279] | Narasimhan S , Guo JL , Changolkar L , Stieber A , McBride JD , Silva LV , He Z , Zhang B , Gathagan RJ , Trojanowski JQ , Lee VMY ((2017) ) Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J Neurosci 37: , 11406–11423. |
[280] | Braak H , Del Tredici K ((2016) ) Potential pathways of abnormal tau and α-synuclein dissemination in sporadic Alzheimer’s and Parkinson’s diseases. Cold Spring Harb Perspect Biol 8: , a023630. |
[281] | Agnati LF , Bjekle B , Fuxe K ((1995) ) Volume versus wiring transmission in the brain: A new theoretical frame of neuropsychopharmacology.. Med Res Rev 15: , 33–45. |
[282] | Nieuwenhuys R (1999) Structure and organisation of fibre systems. In The Central Nervous System of Vertebrates, Vol. 1, Nieuwenhuys R, Ten Donkelaar JH, Nicholson C, eds. Springer, Berlin, pp. 113-157. |
[283] | Kalaria RN , Stockmeier CA , Harik SI ((1989) ) Brain microvessels are innervated by locus ceruleus noradrenergic neurons. Neurosci Lett 97: , 203–208. |
[284] | Fuxe K , Borroto-Escuela DO , Romero-Fernandez W , Diaz-Cabiale Z , Rivera A , Ferraro L , Tanganelli S , Tarakanov AO , Garriga P , Narvaez JA , Ciruela F , Guescini M , Agnati LF ((2012) ) Extrasynaptic neurotransmission in the modulation of brain function. Focus on striatal neuronal-glial networks. Front Physiol 3: , 136. |
[285] | Morrison JH , Foote SL , O’Connor D , Bloom FE ((1982) ) Laminar, tangential and regional organization of the nordrenergic innervation of monkey cortex: Dopamin-bhydroxylase immunohistochemistry. Brain Res Bull 9: , 309–319. |
[286] | Kaufman SK , Del Tredici K , Thomas TL , Braak H , Diamond MI ((2018) ) Tau seeding activity begins in the transentorhinal/entorhinal regions and anticipates phospho-tau pathology in Alzheimer’s disease and PART. Acta Neuropathol 136: , 57–67. |
[287] | Morris GP , Clark IA , Vissel B ((2018) ) Questions concerning the role of amyloid in the definition, aetiology and diagnosis of Alzheimer’s disease. Acta Neuropathol 136: , 663–689. |
[288] | Khan UA , Liu L , Provenzano FA , Diego E , Berman DE , Caterina P , Profaci CP , Sloan R , Mayeux R , Duff KE , Small SA ((2014) ) Molecular drivers and cortical spread of lateral entorhinal cortex dysfunction in preclinical Alzheimer’s disease. Nat Neurosci 17: , 304–311. |
[289] | Stranahan AM , Mattson MP , Alexis M ((2010) ) Selective vulnerability of neurons in layer II of the entorhinal cortex during aging and Alzheimer’s disease. Neural Plast 2010: , 108190. |
[290] | van Groen T , Miettinen P , Kadish I ((2003) ) The entorhinal cortex of the mouse: Organization of the projection to the hippocampal formation. Hippocampus 13: , 133–149. |
[291] | Witter MP , Van Hoesen GW , Amaral DG ((1989) ) Topograpical organization of the entorhinal projection to the dentate gyrus of the monkey. J Neurosci 9: , 216–228. |
[292] | Witter MP , Naber PA , Van Haeften T , Machielsen WCM , Rombouts SARB , Barkhof F , Scheltens P , Lopes Da Silva FH ((2000) ) Cortico-hippocampal communication by way of parallel parahippocampal-subicular pathways. Hippocampus 10: , 398–410. |
[293] | Vago DR , Kesner RP ((2008) ) Disruption of the direct perforant path input to the CA1 subregion of the dorsal hippocampus interferes with spatial working memory and novelty detection. Behav Brain Res 189: , 273–283. |
[294] | Da Insausti R , Amaral DG ((2008) ) Entorhinal cortex of the monkey: IV. Topographical and laminar organization of cortical afferents. J Comp Neurol 509: , 608–641. |
[295] | Suzuki WA , Amaral DG ((1994) ) Topographic organization of the reciprocal connections between the monkey entorhinal cortex and the perirhinal and parahippocampal cortices. J Neurosci 14: , 1856–1877. |
[296] | Suzuki WA , Amaral DG ((1994) ) Perirhinal and parahippocampal cortices of the macaque monkey: Cortical afferents. J Comp Neurol 350: , 497–533. |
[297] | Suzuki WA , Naya Y ((2014) ) The Perirhinal cortex. Annu Rev Neurosci 37: , 39–53. |
[298] | Simić G , Leko MB , Wray S , Harrington CR , Delalle I , Jovanov-Milosevic N , Bazadona D , Buée L , de Silva R , DiGiovanni G , Wischik CM , Hof PR ((2017) ) Monoaminergic neuropathologyin Alzheimer’s disease. Prog Neurobiol 151: , 101–138. |
[299] | Christen-Zaech S , Kraftsik R , Pillevuit O , Kiraly M , Martins R , Khalili K , Miklossy J ((2003) ) Early olfactory involvement in Alzheimer’s disease. Can J Neurol Sci 30: , 20–25. |
[300] | Attems J , Lintner F , Jellinger K ((2005) ) Olfactory involvement in aging and Alzheimer’s disease: An autopsy study. J Alzheimers Dis 7: , 149–157. |
[301] | Murphy C ((2019) ) Olfactory and other sensory impairments in Alzheimer disease. Nat Rev Neurol 15: , 11–24. |
[302] | Haberly LB , Price JL ((1978) ) Association and commissural fiber systems of the olfactory cortex of the rat. J Comp Neurol 181: , 781–807. |
[303] | Kosel KC , Van Hoesen GW , West JR ((1981) ) Olfactory bulb projections to the parahippocampal area of the rat. J Comp Neurol 198: , 467–482. |
[304] | Young JK ((2020) ) Neurogenesis makes a crucial contribution to the neuropathology of Alzheimer’s disease. J Alzheimers Dis Rep 4: , 365–371. |
[305] | Braak H , Braak E ((1985) ) On areas of transition between entorhinal allocortex and temporal isocortex in the human brain. Normal morphology and lamina-specific pathology in Alzheimer’s disease. Acta Neuropathol 68: , 325–332. |
[306] | Pencea V , Bingaman KD , Wiegand SJ , Luskin MB ((2001) ) Infusion of brain-derived neurotrophic factor into the lateral ventricle of the adult rat leads to new neurons in the parenchyma of the striatum, septum, thalamus, and hypothalamus. J Neurosci 21: , 6706–6717. |
[307] | Kokoeva MV , Yin H , Flier JS ((2005) ) Neurogenesis in the hypothalamus of adult mice: Potential role in energy balance. Science 310: , 679–683. |
[308] | Xu Y , Tamamaki N , Noda T , Kimura K , Itokazu Y , Matsumoto N , Dezawa M , Ide C ((2005) ) Neurogenesis in the ependymal layer of the adult rat 3rd ventricle. Exp Neurol 192: , 251–264. |
[309] | Matsuzaki K , Katakura M , Hara T , Li G , Hashimoto M , Shido O ((2009) ) Proliferation of neuronal progenitor cells and neuronal differentiation in the hypothalamus are enhanced in heat-acclimated rats. Pflugers Arch 458: , 661–673. |
[310] | Perez-Martin M , Cifuentes M , Grondona JM , Lopez-Avalos MD , Gomez-Pinedo U , Garcia-Verdugo JM , Fernandez-Llebrez P ((2010) ) IGF-I stimulates neurogenesis in the hypothalamus of adult rats. Eur J Neurosci 31: , 1533–1548. |
[311] | Pierce AA , Xu AW ((2010) ) De novo neurogenesis in adult hypothalamus as a compensatory mechanism to regulate energy balance. J Neurosci 30: , 723–730. |
[312] | Lee DA , Blackshaw S ((2012) ) Functional implications of hypothalamic neurogenesis in the adult mammalian brain. Int J Dev Neurosci 30: , 615–621. |
[313] | Li J , Tang Y , Cai D ((2012) ) IKKb/NF-kB disrupts adult hypothalamic neural stem cells to mediate a neurodegenerative mechanism of dietary obesity and pre-diabetes. Nat Cell Biol 14: , 999–1012. |
[314] | Werner L , Muller-Fielitz H , Ritzal M , Werner T , Rossner M , Schwaninger M ((2012) ) Involvement of doublecortin-expressing cells in the arcuate nucleus in body weight regulation. Endocrinology 153: , 2655–2664. |
[315] | Migaud M , Batailler M , Pillon D , Franceschini I , Malpaux B ((2011) ) Seasonal changes in cell proliferation in the adult sheep brain and pars tuberalis. J Biol Rhythms 26: , 486–496. |
[316] | Feliciano DM , Bordey A , Bonfanti L ((2015) ) Noncanonical sites of adult neurogenesis in the mammalian brain. Cold Spring Harb Perspect Biol 7: , a018846. |
[317] | Zheng J , Li H-L , Tian N , Liu F , Wang L , Yin Y , Yue L , Ma L , Wan Y , Wang J-Z ((2020) ) Interneuron accumulation of phosphorylated tau impairs adult hippocampal neurogenesis by suppressing GABAergic transmission. Cell Stem Cell 26: , 331–345. |
[318] | Dong J , Pan Y-B , Wu X-R , He L-N , Liu X-D , Feng D-F , Xu T-L , Sun S , Xu N-J ((2019) ) A neuronal molecular switch through cell-cell contact that regulates quiescent neural stem cells. Sci Adv 5: , eaav4416. |
[319] | Song J , Zhong C , Bonaguidi MA , Sun GJ , Hsu D , Gu Y , Meletis K , Huang ZJ , Ge S , Enikolopov G , Deisseroth K , Luscher B , Christian KM , Ming G-L , Song H ((2012) ) Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature 489: , 150–154. |
[320] | Andrews-Zwilling Y , Bien-Ly N , Xu Q , Li G , Bernardo A , Yoon SY , Zwilling D , Yan TX , Chen L , Huang Y ((2010) ) Apolipoprotein E4 causes age- and tau-dependent impairment of GABAergic interneurons, leading to learning and memory deficits in mice. J Neurosci 30: , 13707–13717. |
[321] | Li G , Bien-Ly N , Andrews-Zwilling Y , Xu Q , Bernardo A , Ring K , Halabisky B , Deng C , Mahley RW , Huang Y ((2009) ) GABAergic interneuron dysfunction impairs hippocampal neurogenesis in adult apolipoprotein E4 knockin mice. Cell Stem Cell 5: , 634–645. |
[322] | Najm R , Jones EA , Huang Y ((2019) ) Apolipoprotein E4, inhibitory network dysfunction, and Alzheimer’s disease. Mol Neurodegener 14: , 24. |
[323] | Wang C , Najm R , Xu Q , Jeong DE , Walker D , Balestra ME , Yoon SY , Yuan H , Li G , Miller ZA , Miller BL , Malloy MJ , Huang Y ((2018) ) Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nat Med 24: , 647–657. |
[324] | Morrone CD , Bazzigaluppi P , Beckett TL , Hill ME , Koletar MM , Stefanovic B , McLaurin J ((2020) ) Regional differences in Alzheimer’s disease pathology confound behavioural rescue after Amyloid-attenuation. Brain 143: , 359–373. |
[325] | Braak H , Del Tredici K ((2009) ) Neuroanatomy and pathology of sporadic Parkinson’s disease. Adv Anat Embryol Cell Biol 201: , 1–119. |
[326] | Gorno-Tempini ML , Hillis AE , Weintraub S , Kertesz A , Mendez M , Cappa SF , Ogar JM , Rohrer JD , Black S , Boeve BF , Manes F , Dronkers NF , Vandenberghe R , Rascovsky K , Patterson K , Miller BL , Knopman DS , Hodges JR , Mesulam MM , Grossman M ((2011) ) Classification of primary progressive aphasia and its variants. Neurology 76: , 1006–1014. |
[327] | Mendez MF , Ghajarania M , Perryman KM ((2002) ) Posterior cortical atrophy: Clinical characteristics and differences compared to Alzheimer’s disease. Dement Geriatr Cogn Disord 14: , 33–40. |
[328] | Ossenkoppele R , Pijnenburg YAL , Perry DC , Cohn-Sheehy BI , Scheltens NME , Vogel JW , Kramer JH , van der Vlies AE , La Joie R , Rosen HJ , van der Flier WM , Grinberg LT , Rozemuller AJ , Huang EJ , van Berckel BNM , Miller BL , Barkhof F , Jagust WJ , Scheltens P , Seeley WW , Rabinovici GD ((2015) ) The behavioural/dysexecutive variant of Alzheimer’s disease: Clinical, neuroimaging and pathological features. Brain 138: , 2732–2749. |
[329] | Risacher SL , Anderson WH , Charil A , Castelluccio PF , Shcherbinin S , Saykin AJ , Schwarz AJ , For the Alzheimer’s Disease Neuroimaging Initiative ((2017) ) Alzheimer disease brain atrophy subtypes are associated with cognition and rate of decline. Neurology 89: , 2176–2186. |
[330] | Koedam ELGE , Lauffer V , Van Der Vlies AE , Van Der Flier WM , Scheltens P , Pijnenburg YAL ((2010) ) Early-versus late-onset Alzheimer’s disease: More than age alone. J Alzheimers Dis 19: , 1401–1408. |
[331] | Rabinovici GD , Jagust WJ , Furst AF , Ogar JM , Racine CA , Mormino EC , O’Neil JP , Lal RA , Dronkers NF , Miller BL , Gorno-Tempini ML ((2008) ) Aβ amyloid and glucose metabolism in three variants of primary progressive aphasia. Ann Neurol 64: , 388–401. |
[332] | de Souza LC , Corlier F , Habert M-O , Uspenskaya O , Maroy R , Lamari F , Chupin M , Lehéricy S , Colliot O , Hahn-Barma V , Samri D , Dubois B , Bottlaender M , Sarazin M ((2011) ) Similar amyloid- β burdenin posterior cortical atrophy and Alzheimer’s disease. Brain 134: , 2036–2043. |
[333] | Lehmann M , Ghosh PM , Madison C , Laforce R Jr , Corbetta-Rastelli C , Weiner MW , Greicius MD , Seeley WW , Gorno-Tempini ML , Rosen HJ , Miller BL , Jagust WJ , Rabinovici GD ((2013) ) Diverging patterns ofamyloid deposition and hypometabolism in clinical variants ofprobable Alzheimer’s disease. Brain 136: , 844–858. |
[334] | Ossenkoppele R , Schonhaut DR , Schöll M , Lockhart SN , Ayakta N , Baker SL , O’Neil JP , Janabi M , Lazaris A , Cantwell A , Vogel J , Santos M , Miller ZA , Bettcher BM , Vossel KA , Kramer JH , Gorno-Tempini ML , Miller BL , Jagust WJ , Rabinovici GD ((2016) ) Tau PET patterns mirror clinical and neuroanatomical variability in Alzheimer’s disease. Brain 139: , 1551–1567. |
[335] | Jagust W ((2018) ) Imaging the evolution and pathophysiology of Alzheimer disease. Nat Rev Neurosci 19: , 687–700. |
[336] | Buckner RL , Snyder AZ , Shannon BJ , LaRossa G , Sachs R , Fotenos AF , Sheline YI , Klunk WE , Mathis CA , Morris JC , Mintun MA ((2005) ) Molecular, structural, and functional characterization of Alzheimer’s disease: Evidence for a relationship between default activity, amyloid, and memory. J Neurosci 25: , 7709–7717. |
[337] | Buckner RL , Sepulcre J , Talukdar T , Krienen FM , Liu H , Hedden T , Andrews-Hanna JR , Sperling RA , Johnson KA ((2009) ) Cortical hubs revealed by intrinsic functional connectivity: Mapping, assessment of stability, and relation to Alzheimer’s disease. J Neurosci 29: , 1860–1873. |
[338] | Daianu M , Jahanshad N , Nir TM , Jack CR Jr., Weiner MW , Bernstein MA , Thompson PM , the Alzheimer’s Disease Neuroimaging Initiative ((2015) ) Rich club analysis in the Alzheimer’s disease connectome reveals a relatively undisturbed structural core network. Hum Brain Mapp 36: , 3087–3103. |
[339] | Elman JA , Madison CM , Baker SL , Vogel JW , Marks SM , Crowley S , O’Neil JP , Jagust WJ ((2016) ) Effects of beta-amyloid on resting state functional connectivity within and between networks reflect known patterns of regional vulnerability. Cereb Cortex 26: , 695–707. |
[340] | van der Flier WM , Pijnenburg YA , Fox NC , Scheltens P ((2011) ) Early-onset versus late-onset Alzheimer’s disease: The case of the missing APOE ɛ4 allele. Lancet Neurol 10: , 280–288. |
[341] | Ossenkoppele R , Lyoo CH , Sudre CH , van Westen D , Cho H , Ryu YH , Choi JY , Smith R , Strandberg O , Palmqvist S , Westman E , Tsai R , Kramer J , Boxer AL , Gorno-Tempini ML , La Joie R , Miller BL , Rabinovici GD , Hansson O ((2020) ) Distinct tau PET patterns in atrophy-defined subtypes of Alzheimer’s disease. Alzheimers Dement 16: , 335–344. |
[342] | Franzmeier N , Dewenter A , Frontzkowski L , Dichgans M , Rubinski A , Neitzel J , Smith R , Strandberg O , Ossenkoppele R , Buerger K , Duering M , Hansson O , Ewers M ((2020) ) Patient-centered connectivity-based prediction of tau pathology spread in Alzheimer’s disease. Sci adv 6: , eabd1327. |
[343] | Ferreira D , Nordberg A , Westman E ((2020) ) Biological subtypes of Alzheimer’s disease: A systematic review and meta-analysis. Neurology 94: , 436–448. |
[344] | Habes M , Grothe MJ , Tunc B , Mcmillan C , Wolk DA , Davatzikos C ((2020) ) Disentangling heterogeneity in Alzheimer’s disease and related dementias using data-driven methods. Biol Psychiatry 88: , 70–82. |
[345] | Hanna Al-Shaikh FS , Duara R , Crook JE , Lesser ER , Schaeverbeke J , Hinkle KM , Ross OA , Ertekin-Taner N , Pedraza O , Dickson DW , Graff-Radford NR , Murray ME ((2020) ) Selective vulnerability of thenucleus basalis of meynert among neuropathologic subtypes ofAlzheimer disease. JAMA Neurol 77: , 225–233. |
[346] | Jellinger KA ((2021) ) Pathobiological subtypes of alzheimer disease. Dement Geriatr Cogn Disord 49: , 321–333. |
[347] | Murray ME , Graf-Radford NR , Ross OA , Petersen RC , Duara R , Dickson DW ((2011) ) Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: A retrospective study. Lancet Neurol 10: , 785–796. |
[348] | Ferreira D , Mohanty R , Murray ME , Nordberg A , Kantarci K , Westman E ((2022) ) The hippocampal sparing subtype of Alzheimer’s disease assessed in neuropathology and in vivo tau positron emission tomography: A systematic review. Acta Neuropathol Commun 10: , 166. |
[349] | Charil A , Shcherbinin S , Southekal S , Devous MD , Mintun M , Murray ME , Miller BB , Schwarz AJ ((2019) ) Tau subtypes of Alzheimer’s disease determined in vivo using fortaucipir PET imaging. J Alzheimers Dis 71: , 1037–1048. |
[350] | Mohanty R , Mårtensson G , Poulakis K , Muehlboeck J-S , Rodriguez-Vieitez E , Chiotis K , Grothe MJ , Nordberg A , Ferreira D ((2020) ) Comparison of subtyping methods for neuroimaging studies in Alzheimer’s disease: A call for harmonization. Brain Commun 2: , 192. |
[351] | La Joie R , Visani AV , Lesman-Segev OH , Baker SL , Edwards L , Iaccarino L , Soleimani-Meigooni DN , Mellinger T , Janabi M , Miller ZA , Perry DC , Pham J , Strom A , Gorno-Tempini ML , Rosen HJ , Miller BL , Jagust WJ , Rabinovici GD ((2021) ) Association of APOE4 and clinical variability in Alzheimer disease with the pattern of tau- and amyloid-PET. Neurology 96: , e650–e661.c. |
[352] | Xia C , Makaretz SJ , Caso C , McGinnis S , Gomperts SN , Sepulcre J , Gomez-Isla T , Hyman BT , Schultz A , Vasdev N , Johnson KA , Dickerson BC ((2017) ) Association of in vivo AV-1451 tau PET imaging results with cortical atrophy and symptoms in typical and atypical Alzheimer disease. JAMA Neurol 74: , 427–436. |
[353] | Sintini I , Schwarz CG , Martin PR , Graff-Radford J , Machulda MM , Senjem ML , Reid RI , Spychalla AJ , Drubach DA , Lowe VJ , Jack CR Jr , Josephs KA , Whitwell JL ((2019) ) Regional multimodal relationships between tau, hypometabolism, atrophy, and fractional anisotropy in atypical Alzheimer’s disease. Hum Brain Mapp 40: , 1618–1631. |
[354] | Nasrallah IM , Chen YS , Hsieh M-K , Phillips JS , Ternes K , Stockbower GE , Sheline Y , McMillan CT , Grossman M , Wolk DA ((2018) ) 18F-flortaucipir PET/MRI correlations in nonamnestic and amnestic variants of Alzheimer disease. J Nucl Med 59: , 299–306. |
[355] | Day GS , Gordon BA , Jackson K , Christensen JJ , Rosana Ponisio M , Su Y , Ances BM , Benzinger TLS , Morris JC ((2017) ) Tau-PET binding distinguishes patients with early-stage posterior cortical atrophy from amnestic Alzheimer disease dementia. Alzheimer Dis Assoc Disord 31: , 87–93. |
[356] | Josephs KA , Martin PR , Botha H , Schwarz CG , Duffy JR , Clark HM , Machulda MM , Graff-Radford J , Weigand SD , Senjem ML , Utianski RL , Drubach DA , Boeve BF , Jones DT , Knopman DS , Petersen RC , Jack CR Jr , Lowe VJ , Whitwell JL ((2018) ) [18F] AV-1451 tau-PET and primary progressive aphasia. Ann Neurol 83: , 599–611. |
[357] | Smith R , Schöll M , Widner H , van Westen D , Svenningsson P , Hägerström D , Ohlsson T , Jögi J , Nilsson C , Hansson O ((2017) ) In vivo retention of 18F-AV-1451 in corticobasal syndrome. Neurology 89: , 845–853. |
[358] | Ali F , Whitwell J , Martin P , Senjem M , Knopman D , Jack C , Lowe VJ , Petersen RC , Boeve BF , Josephs KA ((2018) ) [18F] AV-1451 uptake in corticobasal syndrome: The influence of beta-amyloid and clinical presentation. J Neurol 265: , 1079–1088. |
[359] | Vogel JW , Young AL , Oxtoby NP , Smith R , Ossenkoppele R , Strandberg OT , La Joie R , Aksman LM , Grothe MJ , Iturria-Medina Y , the Alzheimer’s Disease Neuroimaging Initiative, Pontecorvo MJ , Devous MD , Rabinovici GD , Alexander DC , Lyoo CH , Evans AC , Hansson O ((2021) ) Four distinct trajectories of tau deposition identified in Alzheimer’s disease. Nat Med 27: , 871–881. |
[360] | Sirkis DW , Bonham LW , Johnson TP , La Joie R , Yokoyama JS ((2022) ) Dissecting the clinical heterogeneity of early-onset Alzheimer’sdisease. Mol Psychiatry 27: , 2674–2688. |
[361] | Whitwell JL , Graf-Radford J , Tosakulwong N , Weigand SD , Machulda M , Senjem ML , Schwarz CG , Spychalla AJ , Jones DT , Drubach DA , Knopman DS , Boeve BF , Ertekin-Taner N , Petersen RC , Lowe VJ , Jack CR Jr , Josephs KA ((2018) ) [18F]AV-1451 clustering of entorhinal and cortical uptake in Alzheimer’s disease. Ann Neurol 83: , 248–257. |
[362] | Schwarz AJ , Yu P , Miller BB , Shcherbinin S , Dickson J , Navitsky M , Joshi AD , Devous MD , Mintun MS ((2016) ) Regional profles of the candidate tau PET ligand 18F-AV-1451 recapitulate key features of Braak histopathological stages. Brain 139: , 1539–1550. |
[363] | Alfano C , Studer M ((2013) ) Neocortical arealization: Evolution, mechanisms, and open 200 questions. Dev Neurobiol 73: , 411–447. |
[364] | Kanekar S , Gent M ((2011) ) Malformations of cortical development. Semin Ultrasound CT MR 32: , 211–227. |
[365] | Leventer RJ , Jansen A , Pilz DT , Stoodley N , Marini C , Dubeau F , Malone J , Mitchell LA , Mandelstam S , Scheffer IE , Berkovic SF , Andermann F , Andermann E , Guerrini R , Dobyns WB ((2010) ) Clinical and imaging heterogeneity of polymicrogyria: A study of 328 patients. Brain 133: , 1415–1427. |
[366] | Abbate C ((2018) ) Topographic markers drive proteinopathies to selection of target brain areas at onset in neurodegenerative dementias. Front Aging Neurosci 10: , 308. |
[367] | Khodanovich M , Nemirovich-Danchenko NM ((2019) ) New neurons in the post-ischemic and injured brain: Migrating or resident? Front Neurosci 13: , 588. |
[368] | Sun D ((2016) ) Endogenous neurogenic cell response in the mature mammalian brain following traumatic injury. Exp Neurol 275: , 405–410. |
[369] | Marques BL , Carvalho GA , Freitas EMM , Chiareli RA , Barbosa TG , DiAraújo AGP , Nogueira YL , Ribeiro RI , Parreira RC , Vieira MS , Resende RR , Gomez RS , Oliveira-Lima OC , Pinto MCX ((2019) ) The role ofneurogenesis in neurorepair after ischemic stroke. Semin Cell Dev Biol 95: , 98–110. |
[370] | Schmidt W , Reymann KG ((2002) ) Proliferating cells differentiate into neurons in the hippocampal CA1 region of gerbils after global cerebral ischemia. Neurosci Lett 334: , 153–156. |
[371] | Bendel O , Bueters T , von Euler M , Ove Ogren S , Sandin J , von Euler G ((2005) ) Reappearance of hippocampal CA1 neurons after ischemia is associated with recovery of learning and memory. J Cereb Blood Flow Metab 25: , 1586–1595. |
[372] | Wojcik L , Sawicka A , Rivera S , Zalewska T ((2009) ) Neurogenesis in gerbil hippocampus following brain ischemia: Focus on the involvement of metalloproteinases. Acta Neurobiol Exp 69: , 52–61. |
[373] | Khodanovich MY , Kisel AA , Chernysheva GA , Smolyakova VI , Savchenko RR , Plotnikov MB ((2016) ) Effect of fluoxetine on neurogenesis in hippocampal dentate gyrus after global transient cerebral ischemia in rats. Bull Exp Biol Med 161: , 351–354. |
[374] | Khodanovich MY , Kisel AA , Kudabaeva MS , Chernysheva GA , Smolyakova VI , Glazacheva VY , Wasserlauf I , Pishchelko A , Plotnikov M , Yarnykh V ((2018) ) Abnormal migration of immature neurons in the global cerebral ischemia model. Am Neurol Assoc Ann Meet 84: , 154–155. |
[375] | Parent JM , Vexler ZS , Gong C , Derugin N , Ferriero DM ((2002) ) Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann Neurol 52: , 802–813. |
[376] | Jin K , Sun Y , Xie L , Peel A , Mao XO , Batteur S , Greenberg DA ((2003) ) Directed migration of neuronal precursors into the ischemic cerebral cortex and striatum. Mol Cell Neurosci 24: , 171–189. |
[377] | Yamashita T , Ninomiya M , Hernández Acosta P , García-Verdugo JM , Sunabori T , Sakaguchi M , Adachi K , Kojima T , Hirota Y , Kawase T , Araki N , Abe K , Okano H , Sawamoto K ((2006) ) Subventricularzone-derived neuroblasts migrate and differentiate into matureneurons in the post-stroke adult striatum. J Neurosci 26: , 6627–6636. |
[378] | Hayashi Y , Jinnou H , Sawamoto K , Hitoshi S ((2018) ) Adult neurogenesis and its role in brain injury and psychiatric diseases. J Neurochem 147: , 584–594. |
[379] | Thored P , Arvidsson A , Cacci E , Ahlenius H , Kallur T , Darsalia V , Ekdahl CT , Kokaia Z , Lindvall O ((2006) ) Persistent production of neurons from adult brain stem cells during recovery after stroke. Stem Cells 24: , 739–747. |
[380] | Fuentealba LC , Rompani SB , Parraguez JI , Obernier K , Romero R , Cepko CL , Alvarez-Buylla A ((2015) ) Embryonic origin of postnatal neural stem cells. Cell 161: , 1644–1655. |
[381] | Bielefeld P , Dura I , Danielewicz J , Lucassen PJ , Baekelandt V , Abrous DN , Encinas JM , Fitzsimons CP ((2019) ) Insult-induced aberrant hippocampal neurogenesis: Functional consequences and possible therapeutic strategies. Behav Brain Res 372: , 112032. |
[382] | Jessberger S , Nakashima K , Clemenson GD , Mejia E , Mathews E , Ure K , Ogawa S , Sinton CM , Gage FH , Hsieh J ((2007) ) Epigenetic modulation of seizure induced neurogenesis and cognitive decline. J Neurosci 27: , 5967–5975. |
[383] | Sierra A , Martín-Suárez S , Valcárcel-Martín R , Pascual-Brazo J , Aelvoet S-A , Abiega O , Deudero JJ , Brewster AL , Bernales I , Anderson AE , Baekelandt V , Maletić-Savatić M , Encinas JM ((2015) ) Neuronal hyperactivity accelerates depletion ofneural stem cells and impairs hippocampal neurogenesis. Cell Stem Cell 16: , 488–503. |
[384] | Lugert S , Basak O , Knuckles P , Haussler U , Fabel K , Götz M , Haas CA , Kempermann G , Taylor V , Giachino C ((2010) ) Quiescent and active hippocampal neural stem cells with distinct morphologies respond selectively to physiological and pathological stimuli and aging. Cell Stem Cell 6: , 445–456. |
[385] | Bielefeld P , Schouten M , Meijer GM , Breuk MJ , Geijtenbeek K , Karayel S , Tiaglik A , Vuuregge AH , Willems RAL , Witkamp D , Lucassen PJ , Encinas JM , Fitzsimons CP ((2019) ) Co-administration of anti microRNA-124 and -137 oligonucleotides prevents hippocampal neural stem cell loss upon non-convulsive seizures. Front Mol Neurosci 12: , 31. |
[386] | Parent JM , Yu TW , Leibowitz RT , Geschwind DH , Sloviter RS , Lowenstein DH ((1997) ) Dentate granule cell neurogenesis is increased by seizures and contributes to aberrant network reorganization in the adult rat hippocampus. J Neurosci 17: , 3727–3738. |
[387] | Jessberger S , Römer B , Babu H , Kempermann G ((2005) ) Seizures induce proliferation and dispersion of doublecortin-positive hippocampal progenitor cells. Exp Neurol 196: , 342–351. |
[388] | Overstreet-Wadiche LS , Bromberg DA , Bensen AL , Westbrook GL ((2006) ) Seizures accelerate functional integration of adult-generated granule cells. J Neurosci 26: , 4095–4103. |
[389] | LaSarge CL , Pun RYK , Muntifering MB , Danzer SC ((2016) ) Disrupted hippocampal network physiology following PTEN deletion from newborn dentate granule cells. Neurobiol Dis 96: , 105–114. |
[390] | McCloskey DP , Hintz TM , Pierce JP , Scharfman HE ((2006) ) Stereological methods reveal the robust size and stability of ectopic hilar granule cells after pilocarpine induced status epilepticus in the adult rat. Eur J Neurosci 24: , 2203–2210. |
[391] | Kaneko N , Sawamoto K ((2009) ) Adult neurogenesis and its alteration under pathological conditions. Neurosci Res 63: , 155–164. |
[392] | Lazarov O , Marr RA ((2010) ) Neurogenesis and Alzheimer’s disease: At the crossroads. Exp Neurol 223: , 267–281. |
[393] | Lazarov O , Marr RA ((2013) ) Of mice and men: Neurogenesis, cognition and Alzheimer’s disease. Front Aging Neurosci 5: , 43. |
[394] | Mu Y , Gage FH ((2011) ) Adult hippocampal neurogenesis and its role in Alzheimer’s disease. Mol Neurodegener 6: , 85. |
[395] | Winner B , Kohl Z , Gage FH ((2011) ) Neurodegenerative disease and adult neurogenesis. Eur J Neurosci 33: , 1139–1151. |
[396] | Bolos M , Pallas-Bazarra N , Terreros-Roncal J , Perea JR , Jurado-Arjona J , Avila J , Llorens-Martın M ((2017) ) Soluble tau hasdevastating effects on the structural plasticity of hippocampalgranule neurons. Transl Psychiatry 7: , 1267. |
[397] | Pierfelice T , Alberi L , Gaiano N ((2011) ) Notch in the vertebrate nervous system: An old dog with new tricks. Neuron 69: , 840–855. |
[398] | Kandel ER ((2012) ) The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain 5: , 14. |
[399] | Ge S , Goh EL , Sailor KA , Kitabatake Y , Ming GL , Song H ((2006) ) GABA regulates synaptic integration of newly generated neurons in the adult brain. Nature 439: , 589–593. |
[400] | Jagasia R , Steib K , Englberger E , Herold S , Faus-Kessler T , Saxe M , Gage FH , Song H , Lie DC ((2009) ) GABA-cAMP response element-binding protein signaling regulates maturation and survival of newly generated neurons in the adult hippocampus. J Neurosci 29: , 7966–7977. |
[401] | Herold S , Jagasia R , Merz K , Wassmer K , Lie DC ((2011) ) CREB signalling regulates early survival, neuronal gene expression and morphological development in adult subventricular zone neurogenesis. Mol Cell Neurosci 46: , 79–88. |
[402] | Merz K , Herold S , Lie DC ((2011) ) CREB in adult neurogenesis masterand partner in the development of adult-born neurons? Eur JNeurosci 33: , 1078–1086. |
[403] | Vitolo OV , Sant’Angelo A , Costanzo V , Battaglia F , Arancio O , Shelanski M ((2002) ) Amyloid beta peptide inhibition of the PKA/CREB pathway and long-term potentiation: Reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci U S A 99: , 13217–13221. |
[404] | Ma QL , Harris-White ME , Ubeda OJ , Simmons M , Beech W , Lim GP , Teter B , Frautschy SA , Cole GM ((2007) ) Evidence of Abeta- and transgene-dependent defects in ERK-CREB signaling in Alzheimer’s models. J Neurochem 103: , 1594–1607. |
[405] | Caccamo A , Maldonado MA , Bokov AF , Majumder S , Oddo S ((2010) ) CBP gene transfer increases BDNF levels and ameliorates learning and memory deficits in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci U S A 107: , 22687–22692. |
[406] | Bartolotti N , Segura L , Lazarov O ((2015) ) Diminished CRE-induced plasticity is linked to memory deficits in familial Alzheimer’s disease mice. J Alzheimers Dis 50: , 477–489. |
[407] | Chenn A , Walsh CA ((2003) ) Increased neuronal production, enlarged forebrains and cytoarchitectural distortions in beta-catenin overexpressing transgenic mice. Cereb Cortex 13: , 599–606. |
[408] | Sato N , Meijer L , Skaltsounis L , Greengard P , Brivanlou AH ((2004) ) Maintenance of pluripotency in human and mouse embryonic stem cells through activation of Wnt signaling by a pharmacological GSK-3-specific inhibitor. Nat Med 10: , 55–63. |
[409] | Lie DC , Colamarino SA , Song HJ , Désiré L , Mira H , Consiglio A , Lein ES , Jessberger S , Lansford H , Dearie AR , Gage FH ((2005) ) Wntsignalling regulates adult hippocampal neurogenesis. Nature 437: , 1370–1375. |
[410] | Shimizu T , Kagawa T , Inoue T , Nonaka A , Takada S , Aburatani H , Taga T ((2008) ) Stabilized beta-catenin functions through TCF/LEF proteins and the Notch/RBP-Jkappa complex to promote proliferation and suppress differentiation of neural precursor cells. Mol Cell Biol 28: , 7427–7441. |
[411] | Choi SH , Bylykbashi E , Chatila ZK , Lee SW , Pulli B , Clemenson GD , Kim E , Rompala A , Oram MK , Asselin C , Aronson J , Zhang C , Miller SJ , Lesinski A , Chen JW , Kim DY , Van Praag H , Spiegelman BM , Gage FH , Tanzi RE ((2018) ) Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 361: , eaan8821. |
[412] | Briley D , Ghirardi V , Woltjer R , Renck A , Zolochevska O , Taglialatela G , Micci MA ((2016) ) Preserved neurogenesis in non-demented individuals with AD neuropathology. Sci Rep 6: , 27812. |
[413] | Horgusluoglu E , Nudelman K , Nho K , Saykin AJ ((2017) ) Adult neurogenesis and neurodegenerative diseases: A systems biology perspective. Am J Med Genet B Neuropsychiatr Genet 174: , 93–112. |
[414] | Hamilton LK , Joppe SE , L MC , Fernandes KJ ((2013) ) Aging and neurogenesis in the adult forebrain: What we have learned and where we should go from here. Eur J Neurosci 37: , 1978–1986. |
[415] | Kuhn HG , Cooper-Kuhn CM , Boekhoorn K , Lucassen PJ ((2007) ) Changes in neurogenesis in dementia and Alzheimer mouse models: Are they functionally relevant? Eur Arch Psychiatry Clin Neurosci 257: , 281–289. |
[416] | Boekhoorn K , Joels M , Lucassen PJ ((2006) ) Increased proliferation reflects glial and vascular-associated changes, but not neurogenesis in the presenile Alzheimer hippocampus. Neurobiol Dis 24: , 1–14. |
[417] | Ziabreva I , Perry E , Perry R , Minger SL. , Ekonomou A , Przyborski S , Ballard C ((2006) ) Altered neurogenesis in Alzheimer’s disease. J Psychosom Res 61: , 311–316. |
[418] | Wang R , Dineley KT , Sweatt JD , Zheng H ((2004) ) Presenilin 1 familial Alzheimer’s disease mutation leads to defective associative learning and impaired adult neurogenesis. Neuroscience 126: , 305–312. |
[419] | Zhang C , McNeil E , Dressler L , Siman R ((2006) ) Long-lasting impairment in hippocampal neurogenesis associated with amyloid deposition in a knock-in mouse model of familial Alzheimer’s disease. Exp Neurol 204: , 77–87. |
[420] | Rodríguez JJ , Verkhratsky A ((2011) ) Neurogenesis in Alzheimer’sdisease. J Anat 219: , 78–89. |
[421] | Jin K , Galvan V , Xie L , Mao XO , Gorostiza OF , Bredesen DE , Greenberg DA ((2004) ) Enhanced neurogenesis in Alzheimer’s disease transgenic (PDGF-APPSw,Ind) mice. Proc Natl Acad Sci U S A 101: , 13363–13367. |
[422] | Crary JF , Trojanowski JQ , Schneider JA , Abisambra JF , Abner EL , Alafuzoff I , Arnold SE , Attems J , Beach TG , Bigio EH , Cairns NJ , Dickson DW , Gearing M , Grinberg LT , Hof PR , Hyman BT , Jellinger K , Jicha GA , Kovacs GG , Knopman DS , Kofler J , Kukull WA , Mackenzie IR , Masliah E , McKee A , Montine TJ , Murray ME , Neltner JH , Santa-Maria I , Seeley WW , Serrano-Pozo A , Shelanski ML , Stein T , Takao M , Thal DR , Toledo JB , Troncoso JC , Vonsattel JP , White CL 3rd, Wisniewski T , Woltjer RL , Yamada M , Nelson PT ((2014) ) Primary age-related tauopathy (PART): A common pathology associated with human aging. Acta Neuropathol 128: , 755–766. |
[423] | Bell WR , An Y , Kageyama Y , English C , Rudow GL , Pletnikova O , Thambisetty M , O’Brien R , Moghekar AR , Albert MS , Rabins PV , Resnick SM , Troncoso JG ((2019) ) Neuropathologic, genetic, and longitudinal cognitive profiles in primary age-related tauopathy (PART) and Alzheimer’s disease. Alzheimers Dement 15: , 8–16. |
[424] | Jellinger KA , Alafuzoff I , Attems J , Beach TG , Cairns NJ , Crary JF , Dickson DW , Hof PR , Hyman BT , Jack CR Jr., Jicha GA , Knopman DS , Kovacs GG , Mackenzie IR , Masliah E , Montine TJ , Nelson PT , Schmitt F , Schneider JA , Serrano-Pozo A , Thal DR , Toledo JB , Trojanowski JQ , Troncoso JC , Vonsattel JP , Wisniewski T ((2015) ) PART, a distinct tauopathy, different from classical sporadic Alzheimer disease. Acta Neuropathol 129: , 757–762. |
[425] | Tomlinson BE , Blessed G , Roth M ((1968) ) Observations on the brains of non-demented old people. J Neurol Sci 7: , 331–356. |
[426] | Jellinger KA , Bancher C ((1998) ) Senile dementia with tangles (tangle predominant form of senile dementia). Brain Pathol 8: , 367–376. |
[427] | Neltner JH , Abner EL , Jicha GA , Schmitt FA , Patel E , Poon LW , Marla G , Green RC , Davey A , Johnson MA , Jazwinski SM , Kim S , Davis D , Woodard JL , Kryscio RJ , Van Eldik LJ , Nelson PT ((2016) ) Brain pathologies in extreme old age. Neurobiol Aging 37: , 1–11. |
[428] | Santa-Maria I , Haggiagi A , Liu X , Wasserscheid J , Nelson PT , Dewar K , Clark LN , Crary JF ((2012) ) The MAPT H1 haplotype is associated with tangle-predominant dementia. Acta Nneuropathol 124: , 693–704. |
[429] | Braak H , Braak E ((1995) ) Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol Aging 16: , 271–278. |
[430] | McKee AC , Stern RA , Nowinski CJ , Stein TD , Alvarez VE , Daneshvar DH , Lee HS , Wojtowicz SM , Hall G , Baugh CM , Riley DO , Kubilus CA , Cormier KA , Jacobs MA , Martin BR , Abraham CR , Ikezu T , Reichard RR , Wolozin BL , Budson AE , Goldstein LE , Kowall NW , Cantu RC ((2013) ) The spectrum of disease in chronic traumatic encephalopathy. Brain 136: , 43–64. |
[431] | Corsellis JA , Brierley JB ((1959) ) Observations on the pathology of insidious dementia following head injury. J Ment Sci 105: , 714–720. |
[432] | Corsellis JA , Bruton CJ , Freeman-Browne D ((1973) ) The aftermath of boxing. Psychol Med 3: , 270–303. |
[433] | Hof PR , Knabe R , Bovier P , Bouras C ((1991) ) Neuropathological observations in a case of autism presenting with self-injury behaviour. Acta Neuropathol 82: , 321–326. |
[434] | Geddes JF , Vowles GH , Nicoll JA , Revesz T ((1999) ) Neuronal cytoskeletal changes are an early consequence of repetitive head injury. Acta Neuropathol 98: , 171–178. |
[435] | Omalu BI , DeKosky ST , Hamilton RL , Minster RL , Kamboh MI , Shakir AM , Wecht CH ((2006) ) Chronic traumatic encephalopathy in a national football league player: Part II. Neurosurgery 59: , 1086–1092. |
[436] | Omalu BI , DeKosky ST , Minster RL , Kamboh MI , Hamilton RL , Wecht CH ((2005) ) Chronic traumatic encephalopathy in a national football league player. Neurosurgery 57: , 128–134. |
[437] | Omalu BI , Bailes J , Hammers JL , Fitzsimmons RP ((2010) ) Chronic traumatic encephalopathy, suicides and parasuicides in professional American athletes: The role of the forensic pathologist. Am J Forensic Med Pathol 31: , 130–132. |
[438] | McKee AC , Cantu RC , Nowinski CJ , Hedley-Whyte ET , Gavett BE , Budson AE , Santini VE , Lee H-S , Kubilus CA , Stern RA ((2009) ) Chronic traumatic encephalopathy in athletes: Progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol 68: , 709–735. |
[439] | McKee AC , Gavett BE , Stern RA , Nowinski CJ , Cantu RC , Kowall NW , Perl DP , Hedley-Whyte ET , Price B , Sullivan C , Morin P , Lee H-S , Kubilus CA , Daneshvar DH , Wulff M , Budson AE ((2010) ) TDP-43 proteinopathy and motor neuron disease in chronic traumatic encephalopathy. J Neuropathol Exp Neurol 69: , 918–929. |
[440] | Gavett BE , Stern RA , Cantu RC , Nowinski CJ , McKee AC ((2010) ) Mild traumatic brain injury: A risk factor for neurodegeneration. Alzheimers Res Ther 2: , 18. |
[441] | Gavett BE , Stern RA , McKee AC ((2011) ) Chronic traumatic encephalopathy: A potential late effect of sport-related concussive and subconcussive head trauma. Clin Sports Med 30: , 179–188. |
[442] | Daneshvar DH , Baugh CM , Nowinski CJ , McKee AC , Stern RA , Cantu RC ((2011) ) Helmets and mouth guards: The role of personal equipment in preventing sport-related concussions. Clin Sports Med 30: , 145–163. |
[443] | Daneshvar DH , Nowinski CJ , McKee AC , Cantu RC ((2011) ) The epidemiology of sport-related concussion. Clin Sports Med 30: , 1–17. |
[444] | Costanza A , Weber K , Gandy S , Bouras C , Hof PR , Giannakopoulos G , Canuto A ((2011) ) Contact sport-related chronic traumatic encephalopathy in the elderly: Clinical expression and structural substrates. Neuropathol Appl Neurobiol 37: , 570–584. |
[445] | Stern RA , Riley DO , Daneshvar DH , Nowinski CJ , Cantu RC , McKee AC ((2011) ) Long-term consequences of repetitive brain trauma: Chronic traumatic encephalopathy. PM R 3: , S460–S467. |
[446] | Goldstein LE , Fisher AM , Tagge CA , Zhang XL , Velisek L , Sullivan JA , Upreti C , Kracht JM , Ericsson M , Wojnarowicz MW , Goletiani CJ , Maglakelidze GM , Casey N , Moncaster JA , Minaeva O , Moir RD , Nowinski CJ , Stern RA , Cantu RC , Geiling J , Blusztajn JK , Wolozin BL , Ikezu T , Stein TD , Budson AE , Kowall NW , Chargin D , Sharon A , Saman S , Hall GF , Moss WC , Cleveland RO , Tanzi RE , Stanton PK , Mckee AC ((2012) ) Chronic traumatic encephalopathy in blast-exposed military veterans and a blast neurotrauma mouse model. Sci Transl Med 4: , 134ra60. |
[447] | Saing T , Dick M , Nelson PT , Kim RC , Cribbs DH , Head E ((2012) ) Frontal cortex neuropathology in dementia pugilistica. J Neurotrauma 29: , 1054–1070. |
[448] | Smith DH , Johnson VE , Stewart W ((2013) ) Chronic neuropathologies ofsingle and repetitive TBI: Substrates of dementia? Nat Rev Neurol 9: , 211–221. |
[449] | Sponheim SR , McGuire KA , Kang SS , Davenport ND , Aviyente S , Bernat EM , Lim KO ((2011) ) Evidence of disrupted functional connectivity inthe brain after combatrelated blast injury. Neuroimage 54: (Suppl 1), S21–S29. |
[450] | McKee AC , Cairns NJ , Dickson DW , Folkerth RD , Keene CD , Litvan I , Perl DP , Stein TD , Vonsattel JP , Stewart W , Tripodis Y , Crary JF , Bieniek KF , Dams-O’Connor K , Alvarez VE , Gordon WA , group TC ((2016) ) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131: , 75–86. |
[451] | Vile AR , Atkinson L ((2017) ) Chronic traumatic encephalopathy: The cellular sequela to repetitive brain injury. J Clin Neurosci 41: , 24–29. |
[452] | Kernie SG , Parent JM ((2010) ) Forebrain neurogenesis after focal Ischemic and traumatic brain injury. Neurobiol Dis 37: , 267–274. |
[453] | Schmidt ML , Zhukareva V , Newell KL , Lee V , Trojanowski J ((2001) ) Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer’s disease. Acta Neuropathol 101: , 518–524. |
[454] | McKee AC , Daneshvar DH , Alvarez VE , Stein TD ((2014) ) The neuropathology of sport. Acta Neuropathol 127: , 29–51. |
[455] | Moir RD , Lathe R , Tanzi RE ((2018) ) The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement 14: , 1602–1614. |
[456] | Lathe R , Sapronova A , Kotelevtsev Y ((2014) ) Atherosclerosis and Alzheimer’s diseases with a common cause? Inflammation, oxysterols, vasculature. BMC Geriatr 14: , 36. |
[457] | Jamieson GA , Maitland NJ , Wilcock GK , Craske J , Itzhaki RF ((1991) ) Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J Med Virol 33: , 224–227. |
[458] | De Chiara G , Marcocci ME , Sgarbanti R , Civitelli L , Ripoli C , Piacentini R , Garaci E , Grassi C , Palamara AT ((2012) ) Infectious agents and neurodegeneration. Mol Neurobiol 46: , 614–638. |
[459] | Itzhaki RF ((2014) ) Herpes simplex virus type 1 and Alzheimer’s disease: Increasing evidence for a major role of the virus. Front Aging Neurosci 6: , 202. |
[460] | Balin BJ , Hudson AP ((2014) ) Etiology and pathogenesis of late-onset Alzheimer’s disease. Curr Allergy Asthma Rep 14: , 417. |
[461] | Miklossy J ((2015) ) Historic evidence to support a causal relationship between spirochetal infections and Alzheimer’s disease. Front Aging Neurosci 7: , 46. |
[462] | Soscia SJ , Kirby JE , Washicosky KJ , Tucker SM , Ingelsson M , Hyman B , Burton MA , Goldstein LE , Duong S , Tanzi RE , Moir RD ((2010) ) The Alzheimer’s disease-associated amyloid β-protein is an antimicrobial peptide. PLoS One 5: , e9505. |
[463] | Kumar DKV , Choi HS , Washicosky KJ , Eimer WA , Tucker S , Ghofrani J , Lefkowitz A , Mccoll G , Goldstein LE , Tanzi RE , Moir RD ((2016) ) Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med 8: , 340ra72. |
[464] | White MR , Kandel R , Tripathi S , Condon D , Qi L , Taubenberger J , Hartshorn KL ((2014) ) Alzheimer’s associated β-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS One 9: , e101364. |
[465] | Bourgade K , Garneau H , Giroux G , Le Page AY , Bocti C , Dupuis G , Frost EH , Fülöp T Jr. ((2015) ) β-Amyloid peptides display protective activity against the human Alzheimer’s disease-associated herpes simplex virus-1. Biogerontology 16: , 85–98. |
[466] | Bourgade K , Le Page A , Bocti C , Witkowski JM , Dupuis G , Frost EH , Fülöp T Jr ((2016) ) Protective effect of amyloid-b peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J Alzheimers Dis 50: , 1227–1241. |
[467] | Eimer WA , Vijaya Kumar DK , Navalpur Shanmugam NK , Rodriguez AS , Mitchell T , Washicosky KJ , György B , Breakefield XO , Tanzi RE , Moir RD ((2018) ) Alzheimer’s disease-associated β-amyloid is rapidly seeded by herpes viridae to protect against brain infection. Neuron 99: , 56.e3–63.e3. |
[468] | Piacentini R , De Chiara G , Li Puma DD , Ripoli C , Marcocci ME , Garaci E , Palamara AT , Grassi C ((2014) ) HSV-1 and Alzheimer’s disease: More than a hypothesis. Front Pharmacol 5: , 97. |
[469] | Duarte LF , Farías MA , Álvarez DM , Bueno SM , Riedel CA , González PA ((2019) ) Herpes simplex virus type 1 infection of thecentral nervous system: Insights into proposed interrelationshipswith neurodegenerative disorders. Front Cell Neurosci 13: , 46. |
[470] | Itzhaki RF , Lathe R , Balin BJ , Ball MJ , Bearer EL , Braak H , Bullido MJ , Carter C , Clerici M , Cosby SL , Del Tredici K , Field H , Fulop T , Grassi C , Griffin WST , Haas J , Hudson AP , Kamer AR , Kell DB , Licastro F , Letenneur L , Lövheim H , Mancuso R , Miklossy J , Otth C , Palamara AT , Perry G , Preston C , Pretorius E , Strandberg T , Tabet N , Taylor-Robinson SD , Whittum-Hudson JA ((2016) ) Microbes and Alzheimer’s disease. J Alzheimers Dis 51: , 979. |
[471] | Wozniak MA , Mee AP , Itzhaki RF ((2009) ) Herpes simplex virus type 1 DNA is located within Alzheimer’s disease amyloid plaques. J Pathol 217: , 131–138. |
[472] | Lövheim H , Gilthorpe J , Adolfsson R , Elgh F ((2014) ) Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimers Dement 11: , 593–599. |
[473] | Letenneur L , Pérès K , Fleury H , Garrigue I , Barberger-Gateau P , Helmer C , Orgogozo J-M , Gauthier S , Dartigues J-F ((2008) ) Seropositivity to herpes simplex virus antibodies and risk ofAlzheimer’s disease: A population-based cohort study. PLoS One 3: , e3637. |
[474] | Kobayashi N , Nagata T , Shinagawa S , Oka N , Shimada K , Shimizu A , Tatebayashi Y , Yamada H , Nakayama K , Kondo K , Tatebayashi Y , Yamada H , Nakayama K , Kondo K ((2013) ) Increase in the IgG avidity index due to herpes simplex virus type 1 reactivation and its relationship with cognitive function in amnestic mild cognitive impairment and Alzheimer’s disease. Biochem Biophys Res Commun 430: , 907–911. |
[475] | Mancuso R , Baglio F , Cabinio M , Calabrese E , Hernis A , Nemni R , Clerici M ((2014) ) Titers of herpes simplex virus type 1 antibodies positively correlate with grey matter volumes in Alzheimer’s disease. J Alzheimers Dis 38: , 741–745. |
[476] | Itzhaki RF , Lin WR , Shang D , Wilcock GK , Faragher B , Jamieson GA ((1997) ) Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet 349: , 241–244. |
[477] | Itzhaki RF , Lin WR ((1998) ) Herpes simplex virus type I in brain and the type 4 allele of the apolipoprotein E gene are a combined risk factor for Alzheimer’s disease. Biochem. Soc Trans 26: , 273–277. |
[478] | Zambrano A , Solis L , Salvadores N , Cortés M , Lerchundi R , Otth C ((2008) ) Neuronal cytoskeletal dynamic modification andneurodegeneration induced by infection with herpes simplex virustype 1. J Alzheimers Dis 14: , 259–269. |
[479] | Wozniak MA , Frost AL , Itzhaki RF ((2009) ) Alzheimer’s disease specific tau phosphorylation is induced by herpes simplex virus type 1. J Alzheimers Dis 16: , 341–350. |
[480] | Martin C , Aguila B , Araya P , Vio K , Valdivia S , Zambrano A , Concha MI , Otth C ((2013) ) Inflammatory and neurodegeneration markers during asymptomatic HSV-1 reactivation. J Alzheimers Dis 39: , 849–859. |
[481] | Gnann JW , Whitley RJ ((2017) ) Herpes simplex encephalitis: An update. Curr Infect Dis Rep 19: , 13. |
[482] | Ball MJ ((1982) ) Limbic predilection in Alzheimer dementia: Is reactivated herpesvirus involved? Can J Neurol Sci 9: , 303–306. |
[483] | Damasio AR , Van Hoesen GW ((1985) ) The limbic system and the localisation of herpes simplex encephalitis. J Neurol Neurosurg Psychiatry 48: , 297–301. |
[484] | Caparros-Lefebvre D , Girard-Buttaz I , Reboul S , Lebert F , Cabaret M , Verier A , Steinling M , Pruvo JP , Petit H ((1996) ) Cognitive and psychiatric impairment in herpes simplex virus encephalitis suggest involvement of the amygdalo-frontal pathways. J Neurol 243: , 248–256. |
[485] | Beffert U , Bertrand P , Champagne D , Gauthier S , Poirier J ((1998) ) HSV-1 in brain and risk of Alzheimer’s disease. Lancet 351: , 1330–1331. |
[486] | Wu HM , Huang CC , Chen SH , Liang YC , Tsai JJ , Hsieh CL , Hsu K-S ((2003) ) Herpes simplex virus type 1 inoculation enhances hippocampal excitability and seizure susceptibility in mice. Eur J Neurosci 18: , 3294–3304. |
[487] | Taylor SW , Lee DH , Jackson AC ((2007) ) Herpes simplex encephalitis presenting with exclusively frontal lobe involvement. J Neurovirol 13: , 477–481. |
[488] | Ando Y , Kitayama H , Kawaguchi Y , Koyanagi Y ((2008) ) Primary target cells of herpes simplex virus type 1 in the hippocampus. Microbes Infect 10: , 1514–1523. |
[489] | Jamieson GA , Maitland NJ , Wilcock GK , Yates CM , Itzhaki RF ((1992) ) Herpes simplex virus type 1 DNA is present in specific regions of brain from aged people with and without senile dementia of the Alzheimer type. J Pathol 167: , 365–368. |
[490] | De Chiara G , Piacentini R , Fabiani M , Mastrodonato A , Marcocci ME , Limongi D , Napoletani G , Protto V , Coluccio P , Celestino I , Li Puma DD , Grassi C , Palamara AT ((2019) ) Recurrent herpes simplex virus-1 infection induces hallmarks of neurodegeneration and cognitive deficits in mice. PLoS Pathog 15: , e1007617. |
[491] | Menendez CM , Jinkins JK , Carr DJJ ((2016) ) Resident T cells are unable to control herpes simplex virus-1 activity in the brain ependymal region during latency. J Immunol 197: , 1262–1275. |
[492] | Yao H-W , Ling P , Tung Y-Y , Hsu S-M , Chen S-H ((2014) ) In vivoreactivation of latent herpes simplex virus 1 in mice can occur inthe brain before occurring in the trigeminal ganglion. J Virol 88: , 11264–11270. |
[493] | Mori I , Nishiyama Y , Yokochi T , Kimura Y ((2005) ) Olfactory transmission of neurotropic viruses. J Neurovirol 11: , 129–137. |
[494] | Gillet L , Frederico B , Stevenson PG ((2015) ) Host entry bygamma-herpesviruses - lessons from animal viruses? Curr OpinVirol 15: , 34–40. |
[495] | Little CS , Bowe A , Lin R , Litsky J , Fogel RM , Balin BJ , Fresa-Dillon KL ((2005) ) Age alterations in extent and severity of experimental intranasal infection with Chlamydophila pneumoniae in BALB/c mice. Infect Immun 73: , 1723–1734. |
[496] | Conrady CD , Zheng M , van Rooijen N , Drevets DA , Royer D , Alleman A , Carr DJ ((2013) ) Microglia and a functional type I IFN pathway are required to counter HSV-1-driven brain lateral ventricle enlargement and encephalitis. J Immunol 190: , 2807–2817. |
[497] | Braun E , Zimmerman T , Hur TB , Reinhartz E , Fellig Y , Panet A , Steiner I ((2006) ) Neurotropism of herpes simplex virus type 1 in brain organ cultures. J Gen Virol 87: , 2827–2837. |
[498] | Chucair-Elliott AJ , Conrady C , Zheng M , Kroll CM , Lane TE , Carr DJ ((2014) ) Microglia-induced IL-6 protects against neuronal loss following HSV-1 infection of neural progenitor cells. Glia 62: , 1418–1434. |
[499] | Rotschafer JH , Hu S , Little M , Erickson M , Low WC , Cheeran MC ((2013) ) Modulation of neural stem/progenitor cell proliferation during experimental herpes simplex encephalitis is mediated by differential FGF-2 expression in the adult brain. Neurobiol Dis 58: , 144–155. |
[500] | Li Puma DD , Piacentini R , Leone L , Gironi K , Marcocci ME , De Chiara G , Palamara AT , Grassi C ((2019) ) Herpes simplex virus type-1 infection impairs adult hippocampal neurogenesis via amyloid-β protein accumulation. Stem Cells 37: , 1467–1480. |
[501] | Li Puma DD , Piacentini R , Grassi C ((2021) ) Does impairment of adult neurogenesis contribute to pathophysiology of Alzheimer’s disease? A still open question. Front Mol Neurosci 13: , 578211. |
[502] | Quiroz YT , Sperling RA , Norton DJ , Baena A , Arboleda-Velasquez JF , Cosio D , Schultz A , Lapoint M , Guzman-Velez E , Miller JB , Kim LA , Chen K , Tariot PN , Lopera F , Reiman EM , Johnson KA ((2018) ) Association between amyloid and tau accumulation in young adults with autosomal dominant Alzheimer disease. JAMA Neurol 75: , 548–556. |
[503] | Wang L , Benzinger TL , Su Y , Christensen J , Friedrichsen K , Aldea P , McConathy J , Cairns NJ , Fagan AM , Morris JC , Ances BM ((2016) ) Evaluation of tau imaging in staging Alzheimer disease and revealing interactions between β-amyloid and tauopathy. JAMA Neurol 73: , 1070–1077. |
[504] | Bischof GN , Jessen F , Fliessbach K , Dronse J , Hammes J , Neumaier B , Onur O , Fink GR , Kukolja J , Drzezga A , van Eimeren T , for the Alzheimer’s Disease Neuroimaging Initiative ((2016) ) Impact of tau and amyloid burden on glucose metabolism in Alzheimer’s disease. Ann Clin Transl Neurol 3: , 934–939. |
[505] | Hanseeuw BJ , Betensky RA , Schultz AP , Papp KV , Mormino EC , Sepulcre J , Bark JS , Cosio DM , LaPoint M , Chhatwal JP , Rentz DM , Sperling RA , Johnson KA ((2017) ) Fluorodeoxyglucose metabolism associated with tau-amyloid interaction predicts memory decline. Ann Neurol 81: , 583–596. |
[506] | Adams JN , Lockhart SN , Li L , Jagust WJ ((2018) ) Relationships between tau and glucose metabolism reflect Alzheimer’s disease pathology in cognitively normal older adults. Cereb Cortex 29: , 1997–2009. |
[507] | Jacobs HIL , Hedden T , Schultz AP , Sepulcre J , Perea RD , Amariglio RE , Papp KV , Rentz DM , Sperling RA , Johnson KA ((2018) ) Structural tract alterations predict downstream tau accumulation in amyloid positive older individuals. Nat Neurosci 21: , 424–431. |
[508] | Villemagne VL , Doré V , Burnham SC , Masters CL , Rowe CC ((2018) ) Imaging tau and amyloid-β proteinopathies in Alzheimerdisease and other conditions. Nat Rev Neurol 14: , 225–236. |
[509] | Sepulcre J , Schultz AP , Sabuncu M , Gomez-Isla T , Chhatwal J , Becker A , Sperling R , Johnson KA ((2016) ) In vivo tau, amyloid, and gray matter profiles in the aging brain. J Neurosci 36: , 7364–7374. |
[510] | Lockhart SL , Schöll M , Baker SL , Ayakta N , Swinnerton KN , Bell RK , Mellinger TJ , Shah VD , O’Neil JP , Janabi M , Jagust WJ ((2017) ) Amyloid and tau PET demonstrate region-specific associations in normal older people. Neuroimage 150: , 191–199. |
[511] | Demars MP , Hollands C , Zhao KDT , Lazarov O ((2013) ) Soluble amyloid precursor protein-a rescues age-linked decline in neural progenitor cell proliferation. Neurobiol Aging 34: , 2431–2440. |
[512] | Aizawa K , Ageyama N , Terao K , Hisatsune T ((2011) ) Primate-specific alterations in neural stem/progenitor cells in the aged hippocampus. Neurobiol Aging 32: , 140–150. |
[513] | Leuner B , Kozorovitskiy Y , Gross CG , Gould E ((2007) ) Diminished adult neurogenesis in the marmoset brain precedes old age. Proc Natl Acad Sci U S A 104: , 17169–17173. |
[514] | Bergmann O , Spalding KL , Frisen J ((2015) ) Adult neurogenesis in humans. Cold Spring Harb Perspect Biol 7: , a018994. |
[515] | Fu H , Hardy J , Duff KE ((2018) ) Selective vulnerability in neurodegenerative diseases. Nat Neurosci 21: , 1350–1358. |
[516] | Mattsson N , Schott JM , Hardy J , Turner MR , Zetterberg H ((2016) ) Selective vulnerability in neurodegeneration: Insights from clinical variants of Alzheimer’s disease. J Neurol Neurosurg Psychiatry 87: , 1000–1004. |
[517] | Mrdjen D , Fox EJ , Bukhari SA , Montine KS , Bendall SC , Montine TJ ((2019) ) The basis of cellular and regional vulnerability in Alzheimer’s disease. Acta Neuropathol 138: , 729–749. |
[518] | Brothers HM , Gosztyla ML , Robinson SR ((2018) ) The physiological roles of amyloid-β peptide hint at new ways to treat Alzheimer’s disease. Front Aging Neurosci 10: , 118. |
[519] | Takuma H , Arawaka S , Mori H ((2003) ) Isoforms changes of tau protein during development in various species. Dev Brain Res 142: , 121–127. |
[520] | Arendt T , Stieler J , Ueberham U ((2017) ) Is sporadic Alzheimer′ s disease a developmental disorder? J Neurochem 143: , 396–408. |
[521] | Garcia-Verdugo JM , Ferron S , Flames N , Collado L , Desfilis E , Font E ((2002) ) The proliferative ventricular zone in adult vertebrates: A comparative study using reptiles, birds, and mammals. Brain Res Bull 57: , 765–775. |
[522] | Sawada M , Sawamoto K ((2013) ) Mechanisms of neurogenesis in the normal and injured adult brain. Keio J Med 62: , 13–28. |
[523] | Paredes MF , Sorrells SF , Garcia-Verdugo JM , Alvarez-Buylla A ((2016) ) Brain size and limits to adult neurogenesis. J Comp Neurol 524: , 646–664. |
[524] | Christensen K , Doblhammer G , Rau R , Vaupel JW ((2009) ) Ageing populations: The challenges ahead. Lancet 374: , 1196–208. |
[525] | Sanai N , Tramontin AD , Quiñones-Hinojosa A , Barbaro NM , Gupta N , Kunwar S , Lawton MT , McDermott MW , Parsa AT , Verdugo JM-G , Berger MS , Alvarez-Buylla A ((2004) ) Unique astrocyte ribbon in adult human brain contains neural stem cells but lacks chain migration. Nature 427: , 740–744. |
[526] | Palmer TD , Schwartz PH , Taupin P , Kaspar B , Stein SA , Gage FH ((2001) ) Cell culture. Progenitor cells from human brain after death. Nature 411: , 42–43. |
[527] | Nunes MC , Singh Roy N , Keyoung HM , Goodman RR , McKhann II G , Jiang L , Kang J , Nedergaard M , Goldman SA ((2003) ) Identification and isolation of multipotential neural progenitor cells from the subcortical white matter of the adult human brain. Nat Med 9: , 439–447. |
[528] | Eriksson PS , Perfilieva E , Björk-Eriksson T , Alborn A-M , Nordborg C , Peterson DA , Gage FH ((1998) ) Neurogenesis in the adult human hippocampus. Nat Med 4: , 1313–1317. |
[529] | Choi SH , Tanzi RE ((2019) ) Is Alzheimer’s disease a neurogenesis disorder? Cell Stem Cell 25: , 7–8. |
[530] | Kempermann G , Gage FH , Aigner L , Song H , Curtis MA , Thuret S , Kuhn HG , Jessberger S , Frankland PW , Cameron HA , Gould E , Hen R , Abrous DN , Toni N , Schinder AF , Zhao X , Lucassen PJ , Frisén J ((2018) ) Human adult neurogenesis: Evidence and remaining questions. Cell Stem Cell 23: , 25–30. |

