
The Lipid Invasion Model: Growing Evidence for This New Explanation of Alzheimer’s Disease
Abstract
The Lipid Invasion Model (LIM) is a new hypothesis for Alzheimer’s disease (AD) which argues that AD is a result of external lipid invasion to the brain, following damage to the blood-brain barrier (BBB). The LIM provides a comprehensive explanation of the observed neuropathologies associated with the disease, including the lipid irregularities first described by Alois Alzheimer himself, and accounts for the wide range of risk factors now identified with AD, all of which are also associated with damage to the BBB. This article summarizes the main arguments of the LIM, and new evidence and arguments in support of it. The LIM incorporates and extends the amyloid hypothesis, the current main explanation of the disease, but argues that the greatest cause of late-onset AD is not amyloid-β (Aβ) but bad cholesterol and free fatty acids, let into the brain by a damaged BBB. It suggests that the focus on Aβ is the reason why we have made so little progress in treating the disease in the last 30 years. As well as offering new perspectives for further research into the diagnosis, prevention, and treatment of AD, based on protecting and repairing the BBB, the LIM provides potential new insights into other neurodegenerative diseases such as Parkinson’s disease and amyotrophic lateral sclerosis/motor neuron disease.
Alzheimer’s disease (AD) is one of the greatest challenges facing humanity today. It is the most common cause of dementia, accounting for around two-thirds of cases [1], with a global prevalence of around 24,000,000 people [2], mostly in Western countries, a figure that is growing as life expectancy continues to rise around the world [3].
There are currently no treatments that will prevent people getting the disease [4, 5], and current treatments can only delay disease progression by several months at best [6]. 99% of drug trials have failed [7], and many large pharmaceutical companies have abandoned research into AD therapies [8].
Why have we made so little progress in treating AD? An increasing number of researchers are arguing that it is because the current predominant explanation, the amyloid hypothesis, does not fully account for the disease [6, 9–11].
This paper argues that there is increasing evidence that the Lipid Invasion Model is a viable alternative hypothesis for AD.
THE AMYLOID HYPOTHESIS
The amyloid hypothesis says that AD is caused by excessive levels of a protein fragment (or peptide) amyloid-β in the brain [12]. These excessive levels of Aβ cause the amyloid plaques and tau tangles, synaptic damage, inflammatory response, and brain shrinkage that characterize AD.
Certainly, in around 5% of cases of AD, genetic mutations cause increased production of Aβ, leading to the early-onset inherited form of the disease, FAD [11–13]. However, in 95% of cases, so-called late-onset AD (LOAD), the cause of excess Aβ levels is not so clear [11].
Unlike in FAD, in many LOAD cases the brain does not show increased production of Aβ [14, 15]. This has led some researchers to propose that the cause of any excessive Aβ levels, plaques, and other Aβ aggregates may be the result of abnormally low removal of Aβ from the brain, rather than of Aβ overproduction [16–21]. At the same time, other researchers argue that there would seem to be more than an adequate number of alternative mechanisms for eradicating excess cerebral Aβ from the brain [22–25].
As well as difficulties explaining the cause of excess Aβ in LOAD, there are also doubts about how critical Aβ is to LOAD progression.
Aβ is the key component of plaques, and according to the amyloid hypothesis, plaques are a key indicator of AD. However, some recent research has shown that in many cases of LOAD, the brains have low levels of plaques [26], and that often plaques are located mainly in parts of the brain that are not associated with the memory and other cognitive problems seen in AD [15, 27]. Other research has shown that substantial numbers of plaques can be found in healthy brains, without displaying any signs of LOAD [15, 28]. So, plaques do not lead to AD in all cases.
In summary, in LOAD, which accounts for 95% of AD cases, it is not at all clear that Aβ is the key to disease development or progression. Such doubts have been reinforced by the re-evaluation in 2022 of some of the original evidence for the amyloid hypothesis [29].
The failure of the amyloid hypothesis to fully account for LOAD, many researchers argue, is the reason why, 30 years after the hypothesis first emerged, there are no truly effective treatments, and why the four most commonly used drugs for treating AD have no link to the amyloid hypothesis [30–32], three of them being derived from a previous theory, the cholinergic hypothesis [33].
A number of alternative hypotheses have since emerged, which attempt to provide a better explanation for AD. One of these is the Aβ oligomer hypothesis, which argues that smaller, more soluble Aβ aggregates, rather than amyloid plaques, are the main source of AD neuropathology. However, this only overcomes some of the shortcomings of the original amyloid hypothesis [34]. Another is the tau hypothesis [34, 35], which argues for a central role for tau protein rather than Aβ in the pathogenesis of AD. Others have proposed that it is a form of lysosomal storage disorder [36] or a novel form of diabetes [37], or that AD is caused by autophagic dysregulation [38], neuroimmunomodulation [39], or by excess exposure to aluminum [40]. However, in contrast to the LIM, none of these hypotheses provides a comprehensive account of the disease pathology and risk factors.
A NEW HYPOTHESIS: THE LIPID INVASION MODEL
The rest of this article summarizes the LIM, and how it better accounts for all aspects of AD pathology and all risk factors associated with it.
The LIM argues that AD is driven by external lipids entering the brain, as a result of damage to the blood-brain barrier. The BBB is a thick protective layer (Fig. 1) around the millions of tiny blood capillaries throughout the human brain, which prevents many substances from getting into the brain tissue (or out of it) [16, 41–45]. There are occasions when it temporarily becomes more permeable to some previously-excluded substances, including solutes, cytokines and lymphocytes, due to systemic inflammation, but it restores to normal within hours or days of inflammation terminating [46, 47].
Fig. 1
The Blood-brain barrier. The image on the left shows that brain capillaries have a protective layer and other structural arrangements that form the blood-brain barrier, whereas the image on the right shows that peripheral capillaries lack this. Used with permission of Future Science Ltd., from Mittapalli RK, Manda VK, Adkins CE, Geldenhuys WJ, Lockman PR (2010) Exploiting nutrient transporters at the blood– brain barrier to improve brain distribution of small molecules. Ther Deliv 1, 775– 784 [48]; permission conveyed through Copyright Clearance Center, Inc.
![The Blood-brain barrier. The image on the left shows that brain capillaries have a protective layer and other structural arrangements that form the blood-brain barrier, whereas the image on the right shows that peripheral capillaries lack this. Used with permission of Future Science Ltd., from Mittapalli RK, Manda VK, Adkins CE, Geldenhuys WJ, Lockman PR (2010) Exploiting nutrient transporters at the blood– brain barrier to improve brain distribution of small molecules. Ther Deliv 1, 775– 784 [48]; permission conveyed through Copyright Clearance Center, Inc.](https://content.iospress.com:443/media/jad/2023/94-2/jad-94-2-jad221175/jad-94-jad221175-g001.jpg)
Lipids are fatty substances found throughout the body [49], and there is a different system for transporting them in the brain to elsewhere in the body. Lipids in the brain are exclusively transported by lipoproteins [50, 51], whereas lipids in the rest of the body can be transported either inside of lipoproteins or independently of them. An important role of the BBB is that it separates these two different lipid transport systems [52–55].
LIPOPROTEINS
Lipoproteins could be described as lipid transport containers. As can be seen in Fig. 2, lipoproteins come in different sizes, reflecting the volume of lipids they contain [51].
Fig. 2
Lipoprotein classes in the bloodstream. The classification of the major types of lipoproteins is based on their densities. The density range for each class is shown, in addition to the lipid (red) and protein (blue) content. The diagram is not to scale. Image source: [56]; licensed under CC BY 3.0, with modifications by Jonathan Rudge.
![Lipoprotein classes in the bloodstream. The classification of the major types of lipoproteins is based on their densities. The density range for each class is shown, in addition to the lipid (red) and protein (blue) content. The diagram is not to scale. Image source: [56]; licensed under CC BY 3.0, with modifications by Jonathan Rudge.](https://content.iospress.com:443/media/jad/2023/94-2/jad-94-2-jad221175/jad-94-jad221175-g002.jpg)
Lipoproteins play a key role in heart disease [57]. The smallest lipoprotein, high-density lipoprotein (HDL), is commonly referred to as ‘good cholesterol’, whereas the larger ones to the right of the red line in this figure, are commonly referred to as ‘bad cholesterol’. The cholesterol is neither good nor bad in itself; it is the excess quantity of cholesterol they contain that leads low-density lipoprotein, intermediate-density lipoprotein, and very-low-density lipoprotein to being termed ‘bad’ [58–60]. (Chylomicrons are not considered ‘bad cholesterol’ because they do not contain much cholesterol, despite their large size [61].)
Extensive studies have revealed that the brain only generates HDL-sized lipoproteins (or ‘good cholesterol’) [50, 62]. These studies include measurements of lipoprotein distributions within the brain, including in astrocytes and other glial cells, and in cerebrospinal fluid, found mostly within the fluid-filled brain ventricles, which receive brain waste) [63].
By contrast, the rest of the body also generates the much larger lipoproteins (or ‘bad cholesterol’) [51]. Normally, these are prevented from entering the brain tissue by the BBB [53, 64].
The LIM argues that when the BBB gets damaged, it allows the larger lipoproteins (the ‘bad cholesterol’) into the brain, with the result that brain cells get overloaded with the excess cholesterol within them.
This is critical because it is generally accepted that excess cholesterol uptake by neurons is associated with Aβ formation, even if the exact mechanism involved is still a matter of debate [65–74]. Cholesterol in the brain is created by astrocytes and provided to neighboring neurons, via small HDL-like lipoproteins. If these lipoproteins are artificially loaded with excess cholesterol, neuronal Aβ levels rise [65]. A 2021 study [65] showed that cholesterol levels in neurons are normally kept very low, and that this inhibits Aβ accumulation (unless Aβ is being abnormally overproduced because of genetic mutations, as seen in FAD).
Collectively, the evidence strongly suggests that, if the tight astrocyte control of neuronal cholesterol is bypassed by entry of external lipoproteins (especially larger Apolipoprotein B-containing lipoproteins, i.e.,” bad cholesterol") through damaged portions of the BBB, it will result in overproduction and accumulation of Aβ.
The Aβ created by the excess cholesterol will typically lead to the creation of plaques. The excess cholesterol will also lead to tau hyperphosphorylation and tangle formation, either as a direct result of excess cholesterol or as an indirect result of excess Aβ [65, 75–79]. In addition, other evidence suggests that cholesterol of astrocyte origin may contribute to activation of microglia, probably tau-induced [79].
In other words, exposure of nerve cells to the higher levels of cholesterol found within larger lipoproteins that have entered the brain through a damaged BBB can explain the presence of amyloid plaques and tau tangles in LOAD, and may also explain the excess stimulation of microglia and neuroinflammation in LOAD [80].
FREE FATTY ACIDS
However, the LIM argues that cholesterol is not the only external lipid driving AD— there is another type of lipid, free fatty acids (FFAs), a form of fatty acids (FAs), that may be a more important cause of the memory and other cognitive effects seen in the disease.
FAs can serve a number of functions in the body, including as a source of energy, as ligands that activate certain cell receptors, and as components of larger lipids (e.g., phospholipids) [81, 82]. FAs are also transported differently inside and outside the brain.
Inside the brain tissue, FAs are transported deep within lipoproteins, and similar lipid-transport particles, mostly esterified (meaning, in this case, cross-linked via a glycerol molecule) as triglycerides and diglycerides (Fig. 3a) [51]. By contrast, outside the brain tissue (primarily in the bloodstream), many FAs are transported individually, outside of lipoproteins in non-esterified form, as FFAs. These are often bound to the transporter protein serum albumin, as shown in Fig. 3b [83–85].
Fig. 3
Fatty acid transport (a) inside the brain – inside HDL-sized lipoproteins, and (b) in the bloodstream outside of the brain. Image source: (a) [86], licensed under CC BY-NC-ND 4.0.
![Fatty acid transport (a) inside the brain – inside HDL-sized lipoproteins, and (b) in the bloodstream outside of the brain. Image source: (a) [86], licensed under CC BY-NC-ND 4.0.](https://content.iospress.com:443/media/jad/2023/94-2/jad-94-2-jad221175/jad-94-jad221175-g003.jpg)
This difference is critical. The LIM argues that, when the BBB gets damaged, it lets in individual FFAs, which proceed to over-activate certain receptors in the brain. Most importantly these include the Toll-like receptor 4 (TLR4), involved in inflammation [87], and the extrasynaptic GABAA receptor, a primary receptor for γ-aminobutyric acid, the major inhibitory neurotransmitter in the brain and wider central nervous system [88]. Respectively, these cause much of the neuroinflammation and anterograde amnesia (AA) seen in AD.
The effect is, in fact, similar to what happens when the brain is exposed to chronic alcohol. This is because ethanol over-activates many of the same brain receptors as FFAs, which is why it can be very difficult to distinguish AD from dementia caused by long-term alcoholism [89]. (The molecule ethanol is known to decrease neuronal excitability when administered acutely, whilst causing hyperexcitability in chronic intermittent form [90–93].) And, as in the case of ethanol-induced dementia, AD driven primarily by FFAs, rather than by cholesterol-rich external lipoproteins, will be characterized by low levels of amyloid plaques and tau tangles.
By extension, the LIM also argues that FFAs getting into the brain may be what disrupts our body clock in AD, as alcohol does [94–97], and that entry of external FFAs into the brain will drive a local ketogenic shift in brain energy production away from the normal glucose/lactate bioenergetic dominance [98, 99], as well as causing mitochondrial toxicity and oxidative stress within neurons. These are all characteristics of AD [100–104].
That is not to say that brain exposure to peripheral FFAs will be harmful in all respects. For instance, it is unclear if an FFA-driven ketogenic shift in bioenergetics would be wholly detrimental to the brain, as there is some evidence that ketone bodies may have neuroprotective properties in patients with mild to moderate AD [105–107]. And, bioenergetics aside, there is some evidence that unsaturated fatty acids, including long-chain omega-3 FFAs, may have mild neuroprotective properties in mild cognitive impairment cases and in healthy older populations [108].
However, on balance, the evidence suggests that direct large-scale exposure of brain cells to FFAs (especially saturated FFAs) will be harmful to brain cells and may help explain many AD-associated symptoms. For instance, direct exposure of the mitochondrial electron transport chain to FFAs is associated with various forms of disruption, as explained in page 139 of [109], with neuronal mitochondria likely to be at particular risk.
Exposure to high levels of certain saturated FFAs has also been shown to activate microglia, the primary immune cell of the brain, whose overactivation is known to account for much AD-associated neuroinflammation [80]. This is explained in page 139 of [109].
Finally, FFAs have been shown to induce general anesthesia in a range of animals [114–117], albeit weakly. This is explained in page 141 of [109], and suggests that exposure of the brain to FFAs will lead to AA [88], providing an alternative explanation for its early occurrence in AD. Collectively, it can be seen how external FFAs, if able to pass continually through a disrupted BBB, will cause growing damage to the brain, adding to the damage caused by excess cholesterol from larger external lipoproteins.
In conclusion, the LIM says that AD is primarily driven by the invasion of external lipids into the brain, following damage to the BBB. Entry of ‘bad cholesterol’ causes increased Aβ production, which, in turn, results in the formation of plaques and (perhaps) tangles, triggering subsequent neuroinflammation. Additionally, the increased Aβ may contribute some further BBB damage (as explained below). Entry of FFAs results also in neuroinflammation, as well as anesthesia-related inhibition of neurogenesis, anterograde amnesia and body clock disruption, neuronal mitochondrial toxicity, and changes in bioenergetics. This explains the progressive loss of neurons and other brain cells, and overall brain shrinkage.
As such, the LIM incorporates and greatly extends the amyloid hypothesis, proposing ‘bad cholesterol’ as the primary cause of the Aβ, plaques and tau tangles found in cases of LOAD, and identifying FFAs as an additional, and perhaps even more important, driver of the damage seen in the disease. Both these external lipids enter the brain tissue due to damage to the BBB. In assigning a lesser role to amyloid plaques in disease progression, the LIM explains why some cases of LOAD do not have many plaques, and why plaques do not lead to AD in all cases.
EVIDENCE FOR THE LIM
There are three main lines of evidence that support the Lipid Invasion Model: first, the presence of lipid anomalies in AD brains; second, evidence of BBB damage in AD brains; and third the correlation between the risk factors for AD and BBB damage.
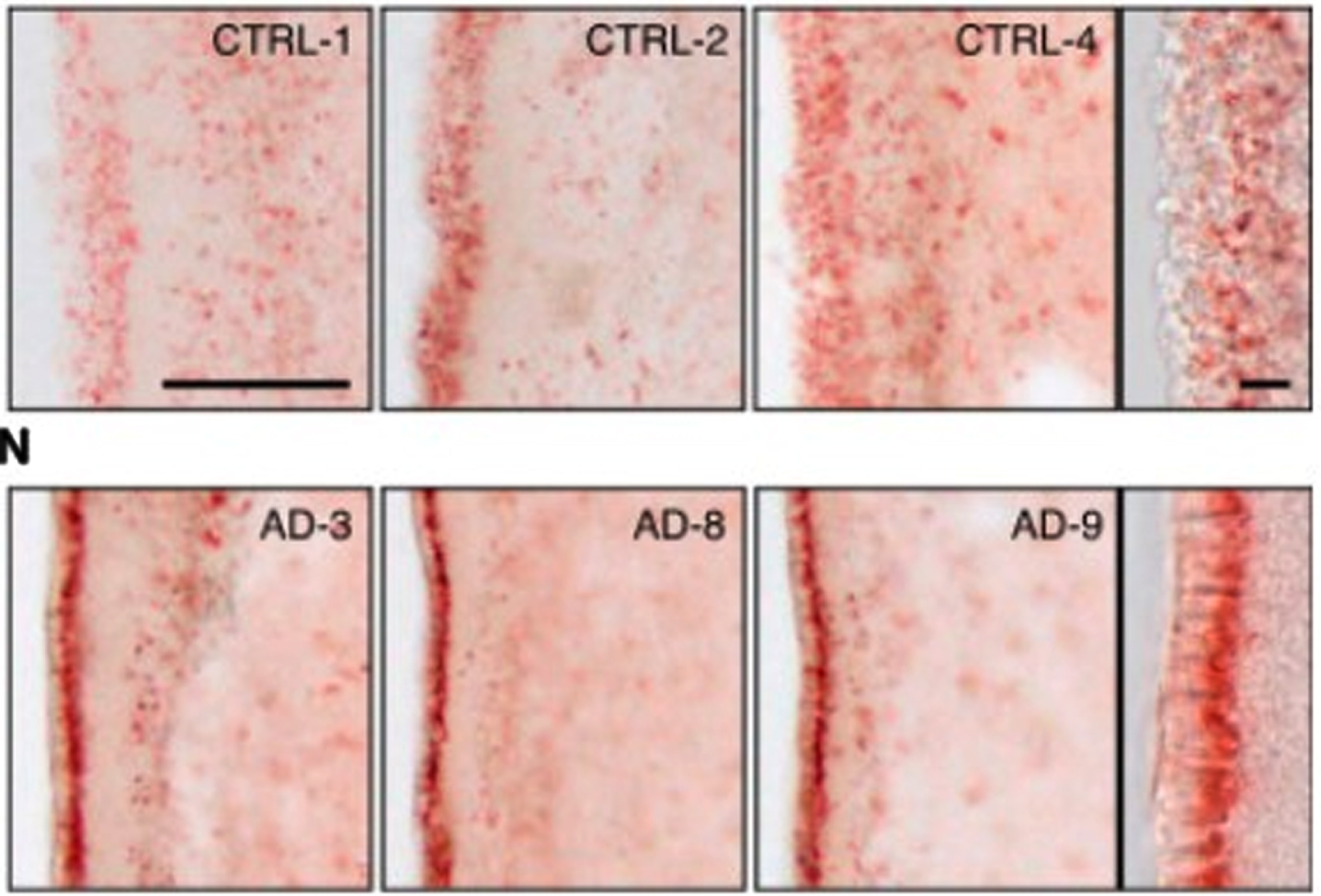
First, there is abundant evidence of the presence of lipid anomalies in the brains of AD patients [77, 118–120]. This goes back to the earliest descriptions by Dr. Alois Alzheimer himself, which contain almost as many references to lipid anomalies as to plaques and tangles [119, 121]. Other early accounts reported similar anomalies [119]. An illustration of what he and his contemporaries were referring to can be seen in the two images in Fig. 4. The brain cells in the top row are normal; the brain cells with AD in the bottom row contain excessive numbers of lipid deposits, which show up in red.
Other evidence of lipid involvement in AD includes the presence of high levels of cholesterol and other lipids within amyloid plaques and in tau tangle-containing neurons in AD brains [66, 123–125]. Research in the last three years has shown that having the APOE4 variant of APOE (one of the most important AD risk factors [21]) leads to faulty lipid (especially cholesterol) handling and storage in brain cells [126–128].
Fig. 4
Evidence of lipid anomalies in the AD brain: (Top row) Only a few lipid droplets in ependymal cells lining the lateral ventricles of healthy patient brains; (Lower row) Many more lipid droplets in ependymal cells of AD patient brains. Panels at the right show representative higher-magnification images. Reprinted from Cell Stem Cell, 17(4), Hamilton LK, Dufresne M, Joppé SE, Petryszyn S, Aumont A, Calon F, Barnabé-Heider F, Furtos A, Parent M, Chaurand P, Fernandes KJL, Aberrant lipid metabolism in the forebrain niche suppresses adult neural stem cell proliferation in an animal model of Alzheimer’s disease, 397-411, Copyright (2015), with permission from Elsevier.
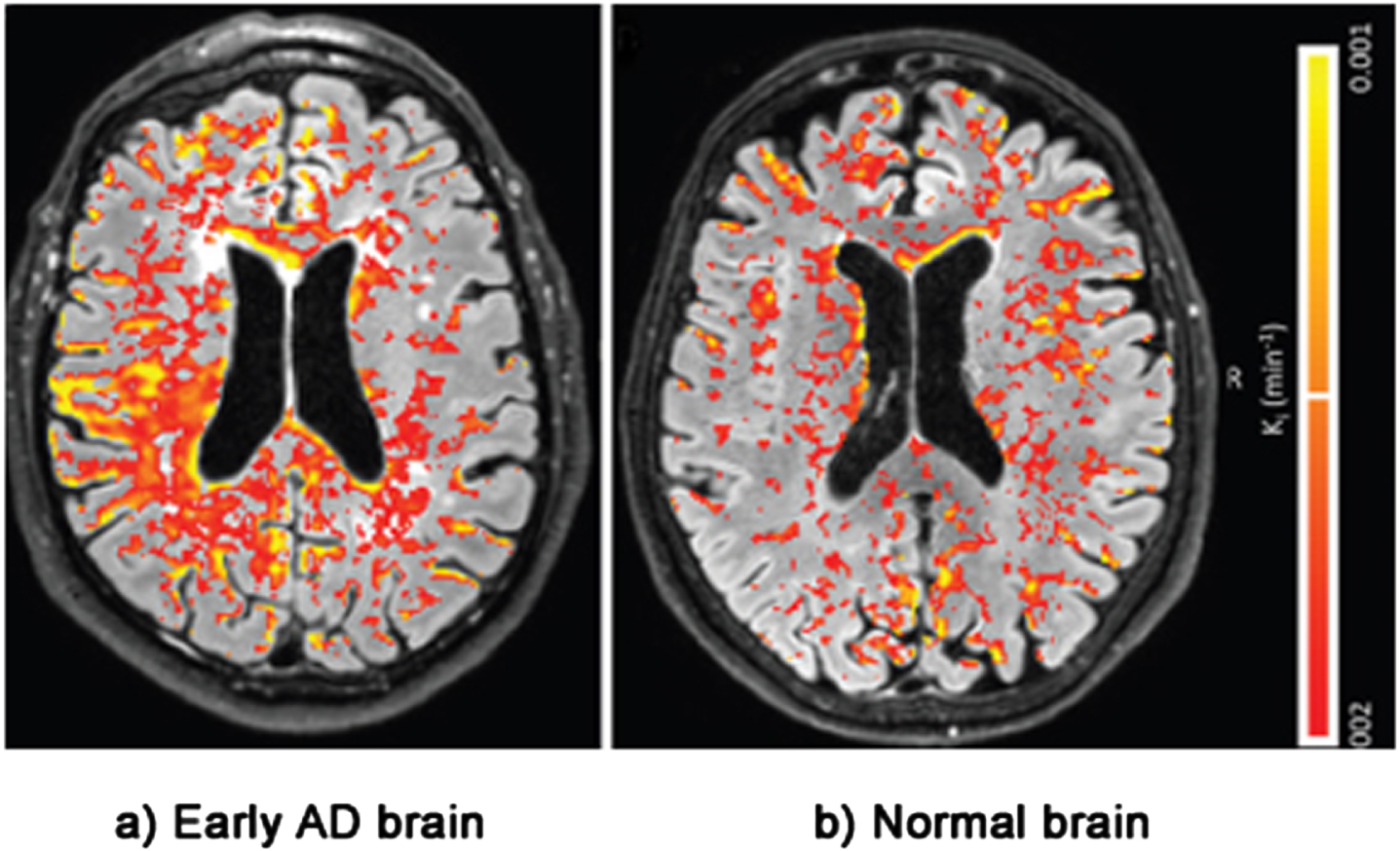
The second line of evidence for the LIM comes from physical evidence of BBB damage inside the brains of AD patients. This evidence is taken from postmortem brains, MRI and PET scans [16, 129–135]. Figure 5 shows an MRI scan of leakage through the BBB of biomarker gadobutrol in the brain of a patient with a mild form of cognitive impairment— early AD— compared with leakage in a normal brain. The BBB leakage in the early AD brain on the left is clearly substantially higher than the leakage in the normal brain on the right [136].
Fig. 5
Evidence of BBB damage in the AD brain: (a) extensive leakage of gadobutrol (an MRI contrasting agent) through a damaged BBB in brains of patients with early signs of AD; (b) less extensive leakage of the agent in brains of normal patients. Used with permission of The Radiological Society of North America, from van de Haar HJ, Burgmans S, Jansen JFA, van Osch MJP, van Buchem MA, Muller M, Hofman PAM, Verhey FRJ, Backes WH, Blood-brain barrier leakage in patients with early Alzheimer disease, Radiology (2016) 281, 527–535.
Interestingly, MRI scans of more advanced AD brains often show that the BBB has been damaged close to the location of plaques and tangles [137–139].
Other studies of AD patients [16, 130–132, 140–145] have detected the presence of proteins in the bloodstream that are normally found only in the brain tissue, and vice versa. These include the blood transport proteins Apolipoprotein B and serum albumin. Apolipoprotein B is normally found only in the larger lipoproteins (the ‘bad cholesterol’) in the external blood stream. Serum albumin is the primary transporter of FFAs, again a form of transport only found in the external bloodstream [83, 84]. Both these proteins should be stopped from entering the brain tissue by the BBB.
The third line of evidence supporting the LIM, shown in Table 1, is that all of the risk factors for AD are also risk factors for damage to the BBB. The risk factors include aging, brain trauma, high blood pressure, stress, sleep deprivation, smoking, excess alcohol, obesity, diabetes, having the APOE4 genotype, and Aβ [146–157].
Table 1
Risk factors for Alzheimer’s disease are also risk factors for blood-brain barrier (BBB) damage
| Risk Factor | Alzheimer’s disease | BBB Damage |
| Aging | ✓ | ✓ |
| Brain trauma | ✓ | ✓ |
| Hypertension | ✓ | ✓ |
| Stress | ✓ | ✓ |
| Sleep deprivation | ✓ | ✓ |
| Smoking | ✓ | ✓ |
| Excess alcohol | ✓ | ✓ |
| Obesity | ✓ | ✓ |
| Diabetes | ✓ | ✓ |
| APOE4 | ✓ | ✓ |
| Amyloid-β | ✓ | ✓ |
In LOAD (95% of AD cases), BBB damage can be caused by all the factors listed, but the primary cause is the wear and tear of aging, which is how the LIM explains why AD so disproportionately affects older people, and why the number of people affected by the disease is growing as life expectancy increases globally.
Brain trauma is also a key risk factor for BBB damage, which, the LIM argues, is why increasing numbers of contact sports players are showing signs of AD and similar dementias such as chronic traumatic encephalopathy, often at an even earlier age than FAD [158–160].
As can be seen, Aβ is also one of the risk factors for BBB damage. Aβ can damage the BBB in many ways, including: redistributing and reducing tight junction protein expression; causing the loss of pericytes, a key BBB component cell; increasing matrix metalloproteinase expression, leading to erosion of the BBB basement membrane; and promoting uncontrolled angiogenesis [41, 161–165].
In FAD AD is caused primarily by the overproduction of Aβ due to genetic mutations, as established by the amyloid hypothesis. However, the overproduction of Aβ also damages the BBB.
The fact that in FAD excess Aβ is presumably being produced from birth, and yet disease onset is typically seen only in people over 50, suggests that Aβ is only slowly damaging to the BBB, perhaps because most Aβ is locked up in plaques or other aggregates, or cleared from the brain. This may explain why attacking Aβ does not have a big impact on halting disease progression.
In summary, the LIM argues that BBB damage is the primary cause of AD overall, as shown in Table 2.
Table 2
Causes of AD according to the LIM
| Type of AD | AD driver | Immediate biological impact | Subsequent biological impact | Final biological impact | |
| FAD | LOAD | ||||
| –5% of cases | –95% of cases | ||||
| ✓ | Genes (AβPP -associated) | - Increased Aβ | - Endosomal-lysosomal disorder | - Synaptic and neuronal death | |
| ✓ | ✓ | Invasion of ‘Bad cholesterol’ - LDL, IDL, VLDL, following BBB damage | - Amyloid plaques | - Long-term memory loss | |
| - Tau tangles | - Brain shrinkage and enlarged ventricles | ||||
| - Neuroinflammation- Some modest Aβ-mediated BBB disruption | - Death | ||||
| ✓ | ✓ | Invasion of FFAs, following BBB damage | - Stimulation of brain receptors(e.g., TLR4, extrasynaptic GABAA) - Ketogenic shift in brain bioenergetics- Neuronal mitochondrial toxicity | - Neuroinflammation - Inhibition of neurogenesis - Anterograde amnesia - Body clock disruption - Oxidative stress | |
AD, Alzheimer’s disease; FAD, familial AD; LOAD, late-onset AD; LDL, low-density lipoprotein; IDL, intermediate-density lipoprotein; VLDL, very-low-density lipoprotein; BBB, blood-brain barrier.
The fact that most new drug trials have been targeted at Aβ, which, according to the LIM, is not a key driver of AD progression in 95% of cases (i.e. LOAD), could be the reason why 99% of such trials have failed, and why the most successful amyloid-related drug, lecanemab (approved for medical use in January 2023), shows only modest benefits, even when administered at the earliest stages of AD [166].
IMPLICATIONS OF THE LIM FOR AD RESEARCH
The Lipid Invasion Model has been developed from piecing together the results of hundreds of research reports on AD published over the last 40 years. There are elements that need to be tested with empirical evidence. For example, further studies are needed to confirm reports of the presence of Apolipoprotein B and excess cholesterol in close proximity to amyloid plaques and to sites of BBB disruption, and to establish the underlying mechanism linking them [65, 123, 124, 143, 145, 167]. Also, to rule out the possibility that proximity of BBB damage to elevated Aβ and plaque levels may be the result of reduced Aβ drainage, rather than of excess Aβ production. The University of Reading in the UK is researching this.
However, if the model is correct, it means that if we want to make progress in treating AD, we should reduce our research focus on Aβ and pay more attention to the BBB.
The first step is enabling identification of BBB damage using scanning and leakage markers, such as gadobutrol. This can be a good predictor of having AD, when memory problems are only just beginning to emerge, as was shown in Fig. 5.
Thereafter, research is needed to find measures to protect the BBB, and treatments to repair any damage that has already occurred. This will not be easy, given the complexity of the BBB and our limited understanding of it.
Increasing evidence suggests that encouraging lifestyle changes can help prevent BBB damage, since many of the risk factors for BBB damage and AD are also lifestyle factors (as shown in Table 1) [149, 168–171].
This is reinforced by the findings from one of the largest longitudinal studies ever within the field of dementia (29,072 adults studied over 10 years), published in January 2023 [172]. This shows that adoption of certain lifestyle factors (a healthy diet, taking regular physical exercise, not drinking alcohol and not smoking, as well as active cognitive activity and social contact) can significantly slow cognitive decline with age, both in the presence and absence of APOE4.
Other recent studies have suggested that certain foods and dietary supplements can protect the BBB, and even repair it [173–184]. And there has been growing interest in how a healthy, more varied diet can promote a healthy gut microbiome, which has been shown to protect and maintain the BBB [180, 185–187]. This may help to explain why the longitudinal study into cognitive decline mentioned above [172] identified diet as the most important of the lifestyle factors under study for slowing memory decline with age.
POSSIBLE WIDER IMPLICATIONS OF THE LIM
The current version of the LIM is focused on AD. However, the model may have wider implications. The LIM suggests that external lipid invasion through a damaged BBB may well explain many cases of Parkinson’s disease and ALS/motor neuron disease. As in AD, affected brains in both diseases show lipid anomalies [188–191], and, as is reported so often in the press, participants in sports in which head trauma is a frequent event, such as boxing, football and rugby, have a higher risk of getting both these neurodegenerative diseases [158, 192–194], as well as AD and similar dementias such as chronic traumatic encephalopathy [158, 159].
CONCLUSION
The LIM provides a new and comprehensive explanation of the neuropathologies and risk factors associated with AD, and offers new insights for future research into the disease, focusing on protecting and repairing the BBB rather than attacking Aβ.
ACKNOWLEDGMENTS
The author would like to thank the following who provided helpful advice and support during the writing of this article: Cherry Mill, MA; Dr. Francesco Tamagnini; Professor Peter Jeavons; Dr. Louise Johnson; Andrew & Christina Hughes-Nind; Mrs. Eveline Rudge
I would also like to thank the reviewers for their very helpful critiques and suggestions in the preparation of this paper.
FUNDING
The author has no funding to report.
CONFLICT OF INTEREST
The author has no conflict of interest to report.
REFERENCES
[1] | Garre-Olmo J ((2018) ) Epidemiology of Alzheimer’s disease and other dementias. Rev Neurol 66: , 377–386. |
[2] | Javaid SF , Giebel C , Khan MA and Hashim MJ ((2021) ) Epidemiology of Alzheimer’s disease and other dementias: Rising global burden and forecasted trends [version 1; peer review: 1 approved with reservations]. F1000Research 10: , 425. https://doi.org/10.12688/f1000research.50786.1. |
[3] | Zhang X-X , Tian Y , Wang Z-T , Ma Y-H , Tan L , Yu J-T ((2021) ) The epidemiology of Alzheimer’s disease modifiable risk factors and prevention. J Prev Alzheimers Dis 8: , 313–321. |
[4] | Abbott A ((2022) ) Could drugs prevent Alzheimer’s? These trials aim to find out. Nature 603: , 216–219. |
[5] | nhs.uk, Can dementia be prevented, Last updated December 21, 2017, Accessed on December 21, 2017. |
[6] | Castellani RJ , Perry G ((2012) ) Pathogenesis and disease-modifying therapy in Alzheimer’s disease: The flat line of progress. Arch Med Res 43: , 694–698. |
[7] | Cummings J , Feldman HH , Scheltens P ((2019) ) The “rights” of precision drug development for Alzheimer’s disease. Alzheimers Res Ther 11: , 76. |
[8] | Quartz, Why the pharmaceutical industry is giving up the search for an Alzheimer’s cure, Last updated May 20, 2018, Accessed on May 20, 2018. |
[9] | Makin S ((2018) ) The amyloid hypothesis on trial. Nature 559: , S4–S7. |
[10] | Herrup K ((2015) ) The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 18: , 794–799. |
[11] | Ricciarelli R , Fedele E ((2017) ) The amyloid cascade hypothesis in Alzheimer’s disease: It’s time to change our mind. Curr Neuropharmacol 15: , 926–935. |
[12] | Hardy J ((2009) ) The amyloid hypothesis for Alzheimer’s disease: A critical reappraisal. J Neurochem 110: , 1129–1134. |
[13] | Wu L , Rosa-Neto P , Hsiung G-YR , Sadovnick AD , Masellis M , Black SE , Jia J , Gauthier S ((2012) ) Early-onset familial Alzheimer’s disease (EOFAD). Can J Neurol Sci 39: , 436–445. |
[14] | Bowman GL , Quinn JF ((2008) ) Alzheimer’s disease and the blood-brain barrier: Past, present and future. Aging Health 4: , 47–55. |
[15] | Caselli RJ , Knopman DS , Bu G ((2020) ) An agnostic reevaluation of the amyloid cascade hypothesis of Alzheimer’s disease pathogenesis: The role of APP homeostasis. Alzheimers Dement 16: , 1582–1590. |
[16] | Sweeney MD , Sagare AP , Zlokovic BV ((2018) ) Blood– brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol 14: , 133–150. |
[17] | Lam FC , Liu R , Lu P , Shapiro AB , Renoir J-M , Sharom FJ , Reiner PB ((2001) ) β-Amyloid efflux mediated by p-glycoprotein. J Neurochem 76: , 1121–1128. |
[18] | Ma Q , Zhao Z , Sagare AP , Wu Y , Wang M , Owens NC , Verghese PB , Herz J , Holtzman DM , Zlokovic BV ((2018) ) Blood-brain barrier-associated pericytes internalize and clear aggregated amyloid-β42 by LRP1-dependent apolipoprotein E isoform-specific mechanism. Mol Neurodegener 13: , 57. |
[19] | Wei W , Bodles-Brakhop AM , Barger SW ((2016) ) A role for P-glycoproteinin clearance of alzheimer amyloid β-peptide from the brain. Curr Alzheimer Res 13: , 615–620. |
[20] | Zhao Z , Sagare AP , Ma Q , Halliday MR , Kong P , Kisler K , Winkler EA , Ramanathan A , Kanekiyo T , Bu G , Owens NC , Rege SV , Si G , Ahuja A , Zhu D , Miller CA , Schneider JA , Maeda M , Maeda T , Sugawara T , Ichida JK , Zlokovic BV ((2015) ) Central role for PICALM in amyloid– β blood– brain barrier transcytosis and clearance. Nat Neurosci 18: , 978–987. |
[21] | Liu C-C , Kanekiyo T , Xu H , Bu G ((2013) ) Apolipoprotein E and Alzheimer disease: Risk, mechanisms, and therapy. Nat Rev Neurol 9: , 106–118. |
[22] | Takechi R , Galloway S , Pallebage-Gamarallage MMS , Lam V , Mamo JCL ((2010) ) Dietary fats, cerebrovasculature integrity and Alzheimer’s disease risk. Prog Lipid Res 49: , 159–170. |
[23] | Strazielle N , Ghersi-Egea JF ((2000) ) Choroid plexus in the central nervous system: Biology and physiopathology. J Neuropathol Exp Neurol 59: , 561–574. |
[24] | Crossgrove JS , Li GJ , Zheng W ((2005) ) The choroid plexus removes β-amyloid from brain cerebrospinal fluid. Exp Biol Med (Maywood) 230: , 771–776. |
[25] | Iwata N , Tsubuki S , Takaki Y , Watanabe K , Sekiguchi M , Hosoki E , Kawashima-Morishima M , Lee H-J , Hama E , Sekine-Aizawa Y , Saido TC ((2000) ) Identification of the major Aβ1– 42-degrading catabolic pathway in brain parenchyma: Suppression leads to biochemical and pathological deposition. Nat Med 6: , 143–150. |
[26] | Monsell SE , Kukull WA , Roher AE , Maarouf CL , Serrano G , Beach TG , Caselli RJ , Montine TJ , Reiman EM ((2015) ) APOE4 carriers and non-carriers with the clinical diagnosis of Alzheimer’s dementia and minimal amyloid plaques. JAMA Neurol 72: , 1124–1131. |
[27] | Jansen WJ , Ossenkoppele R , Knol DL , Tijms BM , Scheltens P , Verhey FRJ , Visser PJ ((2015) ) Prevalence of cerebral amyloid pathology in persons without dementia. JAMA 313: , 1924–1938. |
[28] | Arboleda-Velasquez JF , Lopera F , O’Hare M , Delgado-Tirado S , Marino C , Chmielewska N , Saez-Torres KL , Amarnani D , Schultz AP , Sperling RA , Leyton-Cifuentes D , Chen K , Baena A , Aguillon D , Rios-RomenetsS , Giraldo M , Guzmán-Vélez E , Norton DJ , Pardilla-Delgado E , Artola A , Sanchez JS , Acosta-Uribe J , Lalli M , Kosik KS , Huentelman MJ , Zetterberg H , Blennow K , Reiman RA , Luo J , Chen Y , Thiyyagura P , Su Y , Jun GR , Naymik M , Gai X , Bootwalla M , Ji J , Shen L , Miller JB , Kim LA , Tariot PN , Johnson KA , Reiman EM , Quiroz YT ((2019) ) Resistance to autosomal dominant Alzheimer’s disease in an APOE3Christchurch homozygote: A case report. Nat Med 25: , 1680–1683. |
[29] | Pillar C ((2022) ) Potential fabrication in research images threatens key theory of Alzheimer’s disease. Science. https://www.science.org/content/article/potential-fabrication-research-images-threatens-key-theory-alzheimers-disease. |
[30] | National Health Service, Alzheimer’s disease - Treatment, https://www.nhs.uk/conditions/alzheimers-disease/treatment/. |
[31] | National Institute on Aging, How Is Alzheimer’s Disease Treated? https://www.nia.nih.gov/health/how-alzheimers-disease-treated. |
[32] | Bores GM , Huger FP , Petko W , Mutlib AE , Camacho F , Rush DK , Selk DE , Wolf V , Kosley RW , Davis L , Vargas HM ((1996) ) Pharmacological evaluation of novel Alzheimer’s disease therapeutics: Acetylcholinesterase inhibitors related to galanthamine. J Pharmacol Exp Ther 277: , 728–738. |
[33] | Contestabile A ((2011) ) The history of the cholinergic hypothesis. Behav Brain Res 221: , 334–340. |
[34] | Kametani F , Hasegawa M ((2018) ) Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front Neurosci 12: , 25. |
[35] | Maccioni RB , Farías G , Morales I , Navarrete L ((2010) ) The revitalized tau hypothesis on Alzheimer’s disease. Arch Med Res 41: , 226–231. |
[36] | Lambeth TR , Riggs DL , Talbert LE , Tang J , Coburn E , Kang AS , Noll J , Augello C , Ford BD , Julian RR ((2019) ) Spontaneous isomerization of long-lived proteins provides a molecular mechanism for the lysosomal failure observed in Alzheimer’s disease. ACS Cent Sci 5: , 1387–1395. |
[37] | de la Monte SM , Wands JR ((2008) ) Alzheimer’s disease is type 3 diabetes-evidence reviewed. J Diabetes Sci Technol 2: , 1101–1113. |
[38] | Lee J-H , Yang D-S , Goulbourne CN , Im E , Stavrides P , Pensalfini A , Chan H , Bouchet-Marquis C , Bleiwas C , Berg MJ , Huo C , Peddy J , Pawlik M , Levy E , Rao M , Staufenbiel M , Nixon RA ((2022) ) Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat Neurosci 25: , 688–701. |
[39] | Morales I , Farías G , Maccioni RB ((2010) ) Neuroimmunomodulation in the pathogenesis of Alzheimer’s disease. Neuroimmunomodulation 17: , 202–204. |
[40] | Tomljenovic L ((2011) ) Aluminum and Alzheimer’s disease: After a century of controversy, is there a plausible link? J Alzheimers Dis 23: , 567–598. |
[41] | Winkler EA , Sagare AP , Zlokovic BV ((2014) ) The pericyte: A forgotten cell type with important implications for Alzheimer’s disease? Brain Pathol 24: , 371–386. |
[42] | Zhao Z , Nelson AR , Betsholtz C , Zlokovic BV ((2015) ) Establishment and dysfunction of the blood-brain barrier. Cell 163: , 1064–1078. |
[43] | Fakhrejahani E , Toi M ((2012) ) Tumor angiogenesis: Pericytes and maturation are not to be ignored. J Oncol 2012: , 261750. |
[44] | Cabezas R , Ávila M , Gonzalez J , El-Bachá RS , Báez E , García-Segura LM , Jurado Coronel JC , Capani F , Cardona-Gomez GP , Barreto GE ((2014) ) Astrocytic modulation of blood brain barrier:Perspectives on Parkinson’s disease. Front Cell Neurosci 8: , 211. |
[45] | Kisler K , Nelson AR , Montagne A , Zlokovic BV ((2017) ) Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer’s disease. Nat Rev Neurosci 18: , 419–434. |
[46] | Galea I ((2021) ) The blood– brain barrier in systemic infection and inflammation. Cell Mol Immunol 18: , 2489–2501. |
[47] | Varatharaj A , Galea I ((2017) ) The blood-brain barrier in systemic inflammation. Brain Behav Immun 60: , 1–12. |
[48] | Mittapalli RK , Manda VK , Adkins CE , Geldenhuys WJ , Lockman PR ((2010) ) Exploiting nutrient transporters at the blood– brain barrier to improve brain distribution of small molecules. Ther Deliv 1: , 775–784. |
[49] | LIPID MAPS® Lipidomics Gateway. https://www.lipidmaps.org/data/structure/. |
[50] | Ladu MJ , Reardon C , Eldik LV , Fagan AM , Bu G , Holtzman D , Getz GS ((2000) ) Lipoproteins in the central nervous system. Ann N Y Acad Sci 903: , 167–175. |
[51] | Feingold KR , Grunfeld C ((2000) ) Introduction to lipids and lipoproteins. In Endotext, FeingoldKR, AnawaltB, BoyceA, ChrousosG, DunganK, GrossmanA, HershmanJM, KaltsasG, KochC, KoppP, KorbonitsM, McLachlanR, MorleyJE, NewM, PerreaultL, PurnellJ, RebarR, SingerF, TrenceDL, VinikA, WilsonDP, eds. MDText.com, Inc., South Dartmouth (MA). |
[52] | Jeske DJ , Dietschy JM ((1980) ) Regulation of rates of cholesterol synthesis in vivo in the liver and carcass of the rat measured using [3H]water. J Lipid Res 21: , 364–376. |
[53] | Dietschy JM , Turley SD ((2004) ) Cholesterol metabolism in the central nervous system during early development and in the mature animal. J Lipid Res 45: , 1375. |
[54] | Hamilton J , Brunaldi K ((2007) ) A model for fatty acid transport into the brain. J Mol Neurosci 33: , 12–17. |
[55] | Zhang J , Liu Q ((2015) ) Cholesterol metabolism and homeostasis in the brain. Protein Cell 6: , 254–264. |
[56] | Jairam V , Uchida K , Narayanaswami V ((2012) ) Pathophysiology of Lipoprotein Oxidation, IntechOpen. |
[57] | Linton MF , Yancey PG , Davies SS , Jerome WG , Linton EF , Song WL , Doran AC , Vickers KC ((2019) ) The Role of Lipids and Lipoproteins in Atherosclerosis, MDText.com, Inc. |
[58] | Yoshida H , Ito K , Manita D , Sato R , Hiraishi C , Matsui S , Hirowatari Y ((2021) ) Clinical significance of intermediate-density lipoprotein cholesterol determination as a predictor for coronary heart disease risk in middle-aged men. Front Cardiovasc Med 8: , 756057. |
[59] | Nordestgaard BG , Lewis B ((1991) ) Intermediate density lipoprotein levels are strong predictors of the extent of aortic atherosclerosis in the St. Thomas’s Hospital rabbit strain. Atherosclerosis 87: , 39–46. |
[60] | Shoji T , Nishizawa Y , Kawagishi T , Kawasaki K , Taniwaki H , Tabata T , Inoue T , Morii H ((1998) ) Intermediate-density lipoprotein as an independent risk factor for aortic atherosclerosis in hemodialysis patients. J Am Soc Nephrol 9: , 1277–1284. |
[61] | Huang TC , Kuksis A ((1967) ) A comparative study of the lipids of chylomicron membrane and fat core and of the lymph serum of dogs. Lipids 2: , 443–452. |
[62] | Vitali C , Wellington CL , Calabresi L ((2014) ) HDL and cholesterol handling in the brain. Cardiovasc Res 103: , 405–413. |
[63] | Yu C , Youmans KL , LaDu MJ ((2010) ) Lipoprotein remodelling in the periphery: A model for the brain? Biochim Biophys Acta 1801: , 819–823. |
[64] | Björkhem I , Meaney S ((2004) ) Brain cholesterol: Long secret life behind a barrier. Arteriosclerosis Thrombosis Vasc Biol 24: , 806–815. |
[65] | Wang H , Kulas JA , Wang C , Holtzman DM , Ferris HA , Hansen SB ((2021) ) Regulation of beta-amyloid production in neurons by astrocyte-derived cholesterol Proc Natl Acad Sci U S A 118: , e2102191118. |
[66] | Xiong H , Callaghan D , Jones A , Walker DG , Lue L-F , Beach TG , Sue LI , Woulfe J , Xu H , Stanimirovic DB , Zhang W ((2008) ) Cholesterol retention in Alzheimer’s brain is responsible for high β- and γ-secretase activities and Aβ production. Neurobiol Dis 29: , 422–437. |
[67] | Wolozin B ((2004) ) Cholesterol and the biology of Alzheimer’s disease. Neuron 41: , 7–10. |
[68] | Bodovitz S , Klein WL ((1996) ) Cholesterol modulates -secretase cleavage of amyloid precursor protein. J Biol Chem 271: , 4436–4440. |
[69] | Kojro E , Gimpl G , Lammich S , Marz W , Fahrenholz F ((2001) ) Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha-secretase ADAM 10. Proc Natl Acad Sci U S A 98: , 5815–5820. |
[70] | Ehehalt R , Keller P , Haass C , Thiele C , Simons K ((2003) ) Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J Cell Biol 160: , 113–123. |
[71] | Vetrivel KS , Thinakaran G ((2010) ) Membrane rafts in Alzheimer’s disease beta-amyloid production. Biochim Biophys Acta 1801: , 860–867. |
[72] | Arriagada C , Astorga C , Atwater I , Rojas E , Mears D , Caviedes R , Caviedes P ((2007) ) Endosomal abnormalities related to amyloid precursor protein in cholesterol treated cerebral cortex neuronal cells derived from trisomy 16 mice, an animal model of Down syndrome. Neurosci Lett 423: , 172–177. |
[73] | Rushworth JV , Hooper NM ((2010) ) Lipid rafts: Linking Alzheimer’s amyloid-β production, aggregation, and toxicity at neuronal membranes. Int J Alzheimers Dis 2011: , e603052. |
[74] | Habchi J , Chia S , Galvagnion C , Michaels TCT , Bellaiche MMJ , Ruggeri FS , Sanguanini M , Idini I , Kumita JR , Sparr E , Linse S , Dobson CM , Knowles TPJ , Vendruscolo M ((2018) ) Cholesterol catalyses Aβ42 aggregation through a heterogeneous nucleation pathway in the presence of lipid membranes. Nat Chem 10: , 673–683. |
[75] | Hurtado DE , Molina-Porcel L , Iba M , Aboagye AK , Paul SM , Trojanowski JQ , Lee VM-Y ((2010) ) Aβ accelerates the spatiotemporal progression of tau pathology and augments tau amyloidosis in an Alzheimer mouse model. Am J Pathol 177: , 1977–1988. |
[76] | Bittar A , Bhatt N , Kayed R ((2020) ) Advances and considerations in AD tau-targeted immunotherapy. Neurobiol Dis 134: , 104707. |
[77] | Burns M , Duff K ((2002) ) Cholesterol in Alzheimer’s disease and tauopathy. Ann N Y Acad Sci 977: , 367–375. |
[78] | Puglielli L , Tanzi RE , Kovacs DM ((2003) ) Alzheimer’s disease: The cholesterol connection. Nat Neurosci 6: , 345–351. |
[79] | Wang C , Xiong M , Gratuze M , Bao X , Shi Y , Andhey PS , Manis M , Schroeder C , Yin Z , Madore C , Butovsky O , Artyomov M , Ulrich JD , Holtzman DM ((2021) ) Selective removal of astrocytic APOE4 strongly protects against tau-mediated neurodegeneration and decreases synaptic phagocytosis by microglia. Neuron 109: , 1657–1674e7. |
[80] | Hensley K ((2010) ) Neuroinflammation in Alzheimer’s disease: Mechanisms, pathologic consequences, and potential for therapeutic manipulation. J Alzheimers Dis 21: , 1–14. |
[81] | Calder PC ((2015) ) Functional roles of fatty acids and their effects on human health. JPEN J Parenter Enteral Nutr 39: , 18S–32S. |
[82] | Burdge GC , Calder PC ((2015) ) Introduction to fatty acids and lipids. World Rev Nutr Diet 112: , 1–16. |
[83] | van der Vusse GJ ((2009) ) Albumin as fatty acid transporter. Drug Metab Pharmacokinetics 24: , 300–307. |
[84] | Vance DE , Vance JE ((2008) ) Biochemistry of lipids, lipoproteins, and membranes, Elsevier. |
[85] | Spector AA ((1975) ) Fatty acid binding to plasma albumin. J Lipid Res 16: , 165–179. |
[86] | Nazir S , Jankowski V , Bender G , Zewinger S , Rye K-A , van der Vorst E ((2020) ) Interaction between high-density lipoproteins and inflammation: Function matters more than concentration!. Adv Drug Deliv Rev 159: , 94–119. |
[87] | Alfonso-Loeches S , Pascual-Lucas M , Blanco AM , Sanchez-Vera I , Guerri C ((2010) ) Pivotal role of TLR4 receptors in alcohol-induced neuroinflammation and brain damage. J Neurosci 30: , 8285–8295. |
[88] | Bonin RP , Orser BA ((2008) ) GABAA receptor subtypes underlying general anesthesia. Pharmacol Biochem Behav 90: , 105–112. |
[89] | Alcohol-Related Dementia and Early-Onset Alzheimer’s Disease. https://pro.psycom.net/assessment-diagnosis-adherence/alcohol-use-disorder-related-dementia-alzheimers. |
[90] | Nimitvilai S , Lopez MF , Mulholland PJ , Woodward JJ ((2016) ) Chronic intermittent ethanol exposure enhances the excitability and synaptic plasticity of lateral orbitofrontal cortex neurons and induces a tolerance to the acute inhibitory actions of ethanol. Neuropsychopharmacology 41: , 1112–1127. |
[91] | Pleil KE , Lowery-Gionta EG , Crowley NA , Li C , Marcinkiewcz CA , Rose JH , McCall NM , Maldonado-Devincci AM , Morrow AL , Jones SR , Kash TL ((2015) ) Effects of chronic ethanol exposure on neuronal function in the prefrontal cortex and extended amygdala. Neuropharmacology 99: , 735–749. |
[92] | Cannady R , Nimitvilai-Roberts S , Jennings SD , Woodward JJ , Mulholland PJ ((2020) ) Distinct region- and time-dependent functional cortical adaptations in C57BL/6J mice after short and prolonged alcohol drinking. eNeuro 7: , ENEURO.0077-20.2020. |
[93] | Kang M-H , Spigelman I , Olsen RW ((1998) ) Alteration in the sensitivity of GABAA receptors to allosteric modulatory drugs in rat hippocampus after chronic intermittent ethanol treatment. Alcohol Clin Exp Res 22: , 2165–2173. |
[94] | McElroy B , Zakaria A , Glass JD , Prosser RA ((2009) ) Ethanol modulates mammalian circadian clock phase resetting through extrasynaptic gaba receptor activation. Neuroscience 164: , 842–848. |
[95] | Ruby CL , Prosser RA , DePaul MA , Roberts RJ , Glass JD ((2009) ) Acute ethanol impairs photic and nonphotic circadian phase resetting in the Syrian hamster. Am J Physiol Regul Integr Comp Physiol 296: , R411–R418. |
[96] | Brager AJ , Ruby CL , Prosser RA , Glass JD ((2011) ) Acute ethanol disrupts photic and serotonergic circadian clock phase-resetting in the mouse. Alcohol Clin Exp Res 35: , 1467–1474. |
[97] | Prosser RA , Glass JD ((2015) ) Assessing ethanol’s actions in the suprachiasmatic circadian clock using in vivo and in vitro approaches. Alcohol 49: , 321–339. |
[98] | Le Foll C , Levin BE ((2016) ) Fatty acid-induced astrocyte ketone production and the control of food intake. Am J Physiol Regul Integr Comp Physiol 310: , R1186–R1192. |
[99] | Patil S , Melrose J , Chan C ((2007) ) Involvement of astroglial ceramidein palmitic acid-induced Alzheimer-like changes in primary neurons. Eur J Neurosci 26: , 2131–2141. |
[100] | Yao J , Rettberg JR , Klosinski LP , Cadenas E , Brinton RD ((2011) ) Shift in brain metabolism in late onset Alzheimer’s disease: Implications for biomarkers and therapeutic interventions. Mol Aspects Med 32: , 247–257. |
[101] | Ding F , Yao J , Rettberg JR , Chen S , Brinton RD ((2013) ) Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: Implication for bioenergetic intervention. PLoS One 8: , e79977. |
[102] | Sheehan JP , Swerdlow RH , Miller SW , Davis RE , Parks JK , Parker WD , Tuttle JB ((1997) ) Calcium homeostasis and reactive oxygen species production in cells transformed by mitochondria from individuals with sporadic Alzheimer’s disease. J Neurosci 17: , 4612–4622. |
[103] | Bojarski L , Herms J , Kuznicki J ((2008) ) Calcium dysregulation in Alzheimer’s disease. Neurochem Int 52: , 621–633. |
[104] | Wang W , Zhao F , Ma X , Perry G , Zhu X ((2020) ) Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol Neurodegener 15: , 30. |
[105] | Henderson ST , Vogel JL , Barr LJ , Garvin F , Jones JJ , Costantini LC ((2009) ) Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr Metab 6: , 31. |
[106] | Roopashree PG , Shetty SS , Suchetha Kumari N ((2021) ) Effect of medium chain fatty acid in human health and disease. J Funct Foods 87: , 104724. |
[107] | Krikorian R , Shidler MD , Dangelo K , Couch SC , Benoit SC , Clegg DJ ((2012) ) Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol Aging 33: , 425.e19–425.e27. |
[108] | Wood AHR , Chappell HF , Zulyniak MA ((2022) ) Dietary and supplemental long-chain omega-3 fatty acids as moderators of cognitive impairment and Alzheimer’s disease. Eur J Nutr 61: , 589–604. |
[109] | Rudge JD ((2022) ) A new hypothesis for Alzheimer’s disease: The lipid invasion model. J Alzheimers Dis Rep 6: , 129–161. |
[110] | Patil S , Chan C ((2005) ) Palmitic and stearic fatty acids induce Alzheimer-like hyperphosphorylation of tau in primary rat cortical neurons. Neurosci Lett 384: , 288–293. |
[111] | Patil S , Sheng L , Masserang A , Chan C ((2006) ) Palmitic acid-treated astrocytes induce BACE1 upregulation and accumulation of C-terminal fragment of APP in primary cortical neurons. Neurosci Lett 406: , 55–59. |
[112] | Patil S , Balu D , Melrose J , Chan C ((2008) ) Brain region-specificity of palmitic acid-induced abnormalities associated with Alzheimer’s disease. BMC Res Notes 1: , 20. |
[113] | Patil S ((2007) ) Involvement of Saturated Fatty Acids in Causing Pathophysiological and Metabolic Changes Associated with Alzheimer’s Disease, ProQuest. |
[114] | Samson FE , Dahl N , Dahl DR ((1956) ) A study on the narcotic action of the short chain fatty acids, J Clin Invest 35: , 1291–1298. |
[115] | White RP , Samson FE ((1956) ) Effects of fatty acid anions on the electroencephalogram of unanesthetized rabbits. Am J Physiol 186: , 271–274. |
[116] | McCandless DW ((1985) ) Octanoic acid-induced coma and reticular formation energy metabolism. Brain Res 335: , 131–137. |
[117] | Matsuzaki M , Takagi H ((1967) ) Para-sleep induction by sodium butyrate in acute brain stem preparations (Cats). Brain Res 4: , 223–242. |
[118] | Burns MP , Noble WJ , Olm V , Gaynor K , Casey E , LaFrancois J , Wang L , Duff K ((2003) ) Co-localization of cholesterol, apolipoprotein E and fibrillar Aβ in amyloid plaques. Mol Brain Res 110: , 119–125. |
[119] | Foley P ((2010) ) Lipids in Alzheimer’s disease: A century-old story. Biochim Biophys Acta 1801: , 750–753. |
[120] | Di Paolo G , Kim T-W ((2011) ) Linking lipids to Alzheimer’s disease: Cholesterol and beyond. Nat Rev Neurosci 12: , 284–296. |
[121] | Stelzmann RA , Schnitzlein HN , Murtagh FR ((1995) ) An English translation of Alzheimer’s 1907 paper, “Über eine eigenartige Erkankung der Hirnrinde.”, Clin Anat 8: , 429–431. |
[122] | Hamilton LK , Dufresne M , Joppé SE , Petryszyn S , Aumont A , Calon F , Barnabé-Heider F , Furtos A , Parent M , Chaurand P , Fernandes KJL ((2015) ) Aberrant lipid metabolism in the forebrain niche suppresses adult neural stem cell proliferation in an animal modelof Alzheimer’s disease. Cell Stem Cell 17: , 397–411. |
[123] | Kiskis J , Fink H , Nyberg L , Thyr J , Li J-Y , Enejder A ((2015) ) Plaque-associated lipids in Alzheimer’s diseased brain tissue visualized by nonlinear microscopy. Sci Rep 5: , 13489. |
[124] | Mori T , Paris D , Town T , Rojiani AM , Sparks DL , Delledonne A , Crawford F , Abdullah LI , Humphrey JA , Dickson DW , Mullan MJ ((2001) ) Cholesterol accumulates in senile plaques of Alzheimer disease patients and in transgenic APPsw mice. J Neuropathol Exp Neurol 60: , 778–785. |
[125] | Distl R , Meske V , Ohm TG ((2001) ) Tangle-bearing neurons contain more free cholesterol than adjacent tangle-free neurons. Acta Neuropathol 101: , 547–554. |
[126] | Blanchard JW , Akay LA , Davila-Velderrain J , von Maydell D , Mathys H , Davidson SM , Effenberger A , Chen C-Y , Maner-Smith K , Hajjar I , Ortlund EA , Bula M , Agbas E , Ng A , Jiang X , Kahn M , Blanco-Duque C , Lavoie N , Liu L , Reyes R , Lin Y-T , Ko T , R’Bibo L , Ralvenius WT , Bennett DA , Cam HP , Kellis M , Tsai L-H ((2022) ) APOE4 impairs myelination via cholesterol dysregulation in oligodendrocytes. Nature 611: , 769–779. |
[127] | Sienski G , Narayan P , Bonner JM , Kory N , Boland S , Arczewska AA , Ralvenius WT , Akay L , Lockshin E , He L , Milo B , Graziosi A , Baru V , Lewis CA , Kellis M , Sabatini DM , Tsai L-H , Lindquist S ((2021) ) APOE4 disrupts intracellular lipid homeostasis in human iPSC-derived glia. Sci Transl Med 13: , eaaz4564. |
[128] | Tcw J , Qian L , Pipalia NH , Chao MJ , Liang SA , Shi Y , Jain BR , Bertelsen SE , Kapoor M , Marcora E , Sikora E , Andrews EJ , Martini AC , Karch CM , Head E , Holtzman DM , Zhang B , Wang M , Maxfield FR , Poon WW , Goate AM ((2022) ) Cholesterol and matrisome pathways dysregulated in astrocytes and microglia. Cell 185: , 2213–2233.e25. |
[129] | Montagne A , Zhao Z , Zlokovic BV ((2017) ) Alzheimer’s disease: A matter of blood– brain barrier dysfunction? J Exp Med 214: , 3151–3169. |
[130] | Zipser BD , Johanson CE , Gonzalez L , Berzin TM , Tavares R , Hulette CM , Vitek MP , Hovanesian V , Stopa EG ((2007) ) Microvascular injury and blood-brain barrier leakage in Alzheimer’s disease. Neurobiol Aging 28: , 977–986. |
[131] | Sengillo JD , Winkler EA , Walker CT , Sullivan JS , Johnson M , Zlokovic BV ((2013) ) Deficiency in mural vascular cells coincides with blood-brain barrier disruption in Alzheimer’s disease: Pericytes in Alzheimer’s disease. Brain Pathol 23: , 303–310. |
[132] | Ryu JK , McLarnon JG ((2009) ) A leaky blood-brain barrier, fibrinogen infiltration and microglial reactivity in inflamed Alzheimer’s disease brain. J Cell Mol Med 13: , 2911–2925. |
[133] | Nation DA , Sweeney MD , Montagne A , Sagare AP , D’Orazio LM , Pachicano M , Sepehrband F , Nelson AR , Buennagel DP , Harrington MG , Benzinger TLS , Fagan AM , Ringman JM , Schneider LS , Morris JC , Chui HC , Law M , Toga AW , Zlokovic BV ((2019) ) Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med 25: , 270–276. |
[134] | Miners JS , Kehoe PG , Love S , Zetterberg H , Blennow K ((2019) ) CSF evidence of pericyte damage in Alzheimer’s disease is associated with markers of blood-brain barrier dysfunction and disease pathology. Alzheimers Res Ther 11: , 81–86. |
[135] | Blennow K , Wallin A , Fredman P , Karlsson I , Gottfries CG , Svennerholm L ((1990) ) Blood-brain barrier disturbance in patients with Alzheimer’s disease is related to vascular factors. Acta Neurol Scand 81: , 323–326. |
[136] | van de Haar HJ , Burgmans S , Jansen JFA , van Osch MJP , van Buchem MA , Muller M , Hofman PAM , Verhey FRJ , Backes WH ((2016) ) Blood-brain barrier leakage in patients with early Alzheimer disease. Radiology 281: , 527–535. |
[137] | Perlmutter LS , Helena CC ((1990) ) Microangiopathy, the vascular basement membrane and Alzheimer’s disease: A review. Brain Res Bull 24: , 677–686. |
[138] | Nehra G , Bauer B , Hartz AMS ((2022) ) Blood-brain barrier leakage in Alzheimer’s disease: From discovery to clinical relevance. Pharmacol Ther 234: , 108119. |
[139] | Scholz W ((1938) ) Studien zur pathologie der hirngefäße II. Z Gesamte Neurol Psychiatr 162: , 694–715. |
[140] | Cullen KM , Kócsi Z , Stone J ((2005) ) Pericapillary haem-rich deposits: Evidence for microhaemorrhages in aging human cerebral cortex. J Cereb Blood Flow Metab 25: , 1656–1667. |
[141] | Fiala M , Liu QN , Sayre J , Pop V , Brahmandam V , Graves MC , Vinters HV ((2002) ) Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood-brain barrier. Eur J Clin Invest 32: , 360–371. |
[142] | Hultman K , Strickland S , Norris EH ((2013) ) The APOE ɛ4/ɛ4 genotype potentiates vascular fibrin(ogen) deposition in amyloid-laden vessels in the brains of Alzheimer’s disease patients. J Cereb Blood Flow Metab 33: , 1251–1258. |
[143] | Takechi R , Galloway S , Pallebage-Gamarallage MMS , Wellington CL , Johnsen RD , Dhaliwal SS , Mamo JCL ((2010) ) Differential effects of dietary fatty acids on the cerebral distribution of plasma-derived apo B lipoproteins with amyloid-β. Br J Nutr 103: , 652–662. |
[144] | Skillbäck T , Delsing L , Synnergren J , Mattsson N , Janelidze S , Nägga K , Kilander L , Hicks R , Wimo A , Winblad B , Hansson O , Blennow K , Eriksdotter M , Zetterberg H ((2017) ) CSF/serum albumin ratio in dementias: A cross-sectional study on 1861 patients. Neurobiol Aging 59: , 1–9. |
[145] | Namba Y , Tsuchiya H , Ikeda K ((1992) ) Apolipoprotein B immunoreactivity in senile plaque and vascular amyloids and neurofibrillary tangles in the brains of patients with Alzheimer’s disease. Neurosci Lett 134: , 264–266. |
[146] | Kivipelto M , Ngandu T , Fratiglioni L , Viitanen M , Kåreholt I , Winblad B , Helkala E-L , Tuomilehto J , Soininen H , Nissinen A ((2005) ) Obesity and vascular risk factors at midlife and the risk of dementia and Alzheimer disease. Arch Neurol 62: , 1556–1560. |
[147] | Rhea EM , Salameh TS , Logsdon AF , Hanson AJ , Erickson MA , Banks WA ((2017) ) Blood-brain barriers in obesity. AAPS J 19: , 921–930. |
[148] | Salloway S , Gur T , Berzin T , Tavares R , Zipser B , Correia S , Hovanesian V , Fallon J , Kuo-Leblanc V , Glass D , Hulette C , Rosenberg C , Vitek M , Stopa E ((2002) ) Effect of APOE genotype on microvascular basement membrane in Alzheimer’s disease. J Neurol Sci 204: , 183–187. |
[149] | Mazzone P , Tierney W , Hossain M , Puvenna V , Janigro D , Cucullo L ((2010) ) Pathophysiological impact of cigarette smoke exposure on the cerebrovascular system with a focus on the blood-brain barrier: Expanding the awareness of smoking toxicity in an underappreciated area. Int J Environ Res Public Health 7: , 4111–4126. |
[150] | Prasad S , Sajja RK , Naik P , Cucullo L ((2014) ) Diabetes mellitus and blood-brain barrier dysfunction: An overview. J Pharmacovigil 2: , 125. |
[151] | Alluri H , Wiggins-Dohlvik K , Davis ML , Huang JH , Tharakan B ((2015) ) Blood-brain barrier dysfunction following traumatic brain injury. Metab Brain Dis 30: , 1093–1104. |
[152] | Girouard H ((2016) ) Hypertension and the Brain as an End-Organ Target, Springer. |
[153] | Montagne A , Nation DA , Sagare AP , Barisano G , Sweeney MD , Chakhoyan A , Pachicano M , Joe E , Nelson AR , D’Orazio LM , Buennagel DP , Harrington MG , Benzinger TLS , Fagan AM , Ringman JM , Schneider LS , Morris JC , Reiman EM , Caselli RJ , Chui HC , TCW J , Chen Y , Pa J , Conti PS , Law M , Toga AW , Zlokovic BV ((2020) ) APOE4 leads to blood-brain barrier dysfunction predicting cognitive decline. Nature 581: , 71–76. |
[154] | Hurtado-Alvarado G , Domínguez-Salazar E , Pavon L , Velázquez-Moctezuma J , Gómez-González B ((2016) ) Blood-brain barrier disruption induced by chronic sleep loss:Low-grade inflammation may be the link. J Immunol Res 2016: , e4576012. |
[155] | Dudek KA , Dion-Albert L , Lebel M , LeClair K , Labrecque S , Tuck E , Perez CF , Golden SA , Tamminga C , Turecki G , Mechawar N , Russo SJ , Menard C ((2020) ) Molecular adaptations of the blood– brain barrier promote stress resilience vs. depression. Proc Natl Acad Sci U S A 117: , 3326–3336. |
[156] | Welcome MO , Mastorakis NE ((2020) ) Stress-induced blood brain barrier disruption: Molecular mechanisms and signaling pathways. Pharmacol Res 157: , 104769. |
[157] | Gosselet F , Saint-Pol J , Candela P , Fenart L ((2013) ) Amyloid-β peptides, Alzheimer’s disease and the blood-brain barrier. Curr Alzheimer Res 10: , 1015–1033. |
[158] | Mackay DF , Russell ER , Stewart K , MacLean JA , Pell JP , Stewart W ((2019) ) Neurodegenerative disease mortality among former professional soccer players. N Engl J Med 381: , 1801–1808. |
[159] | Stein TD , Alvarez VE , McKee AC ((2014) ) Chronic traumatic encephalopathy: A spectrum of neuropathological changes following repetitive brain trauma in athletes and military personnel. Alzheimers Res Ther 6: , 4. |
[160] | Gavett BE , Stern RA , McKee AC ((2011) ) Chronic traumatic encephalopathy: A potential late effect of sport-related concussive and subconcussive head trauma. Clin Sports Med 30: 179–xi. |
[161] | Carrano A , Hoozemans JJM , van der Vies SM , Rozemuller AJM , van Horssen J , de Vries HE ((2011) ) Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal 15: , 1167–1178. |
[162] | Hartz AMS , Bauer B , Soldner ELB , Wolf A , Boy S , Backhaus R , Mihaljevic I , Bogdahn U , Klünemann HH , Schuierer G , Schlachetzki F ((2012) ) Amyloid-β contributes to blood– brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke 43: , 514–523. |
[163] | Tai LM , Holloway KA , Male DK , Loughlin AJ , Romero IA ((2010) ) Amyloid-β-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J Cell Mol Med 14: , 1101–1112. |
[164] | Biron KE , Dickstein DL , Gopaul R , Jefferies WA ((2011) ) Amyloid triggers extensive cerebral angiogenesis causing blood brain barrier permeability and hypervascularity in Alzheimer’s disease. PLoS One 6: , e23789. |
[165] | Brkic M , Balusu S , Wonterghem EV , Gorlé N , Benilova I , Kremer A , Hove IV , Moons L , Strooper BD , Kanazir S , Libert C , Vandenbroucke RE ((2015) ) Amyloid β oligomers disrupt blood– CSF barrierintegrity by activating matrix metalloproteinases. J Neurosci 35: , 12766–12778. |
[166] | Biogen ((2022) ) Lecanemab Confirmatory Phase 3 Clarity AD Study met primary endpoint, showing highly statistically significant reduction of clinical decline in large global clinical study of 1,795 participants with Early Alzheimer’s disease, https://investors.biogen.com/news-releases/news-release-details/lecanemab-confirmatory-phase-3-clarity-ad-study-met-primary. |
[167] | Takechi R , Galloway S , Pallebage-Gamarallage M , Wellington C , Johnsen R , Mamo JC ((2009) ) Three-dimensional colocalization analysis of plasma-derived apolipoprotein B with amyloid plaques in APP/PS1 transgenic mice. Histochem Cell Biol 131: , 661–666. |
[168] | Dhana K , Evans DA , Rajan KB , Bennett DA , Morris MC ((2020) ) Healthy lifestyle and the risk of Alzheimer dementia. Neurology 95: , e374–e383. |
[169] | Małkiewicz MA , Szarmach A , Sabisz A , Cubała WJ , Szurowska E , Winklewski PJ ((2019) ) Blood-brain barrier permeability and physicalexercise. J Neuroinflammation 16: , 15. |
[170] | Starr JM , Wardlaw J , Ferguson K , MacLullich A , Deary IJ , Marshall I ((2003) ) Increased blood– brain barrier permeability in type II diabetes demonstrated by gadolinium magnetic resonance imaging. J Neurol Neurosurg Psychiatry 74: , 70–76. |
[171] | Wei J , Dai Y , Wen W , Li J , Ye LL , Xu S , Duan DD ((2021) ) Blood-brain barrier integrity is the primary target of alcohol abuse. Chem Biol Interact 337: , 109400. |
[172] | Jia J , Zhao T , Liu Z , Liang Y , Li F , Li Y , Liu W , Li F , Shi S , Zhou C , Yang H , Liao Z , Li Y , Zhao H , Zhang J , Zhang K , Kan M , Yang S , Li H , Liu Z , Ma R , Lv J , Wang Y , Yan X , Liang F , Yuan X , Zhang J , Gauthier S , Cummings J ((2023) ) Association between healthy lifestyle and memory decline in older adults: 10 year, population based, prospective cohort study. BMJ 380: , e072691. |
[173] | Solfrizzi V , Panza F , Frisardi V , Seripa D , Logroscino G , Imbimbo BP , Pilotto A ((2011) ) Diet and Alzheimer’s disease risk factors or prevention: The current evidence. Expert Rev Neurotherap 11: , 677–708. |
[174] | Medina-Vera I , Sanchez-Tapia M , Noriega-López L , Granados-Portillo O , Guevara-Cruz M , Flores-López A , Avila-Nava A , Fernández ML , Tovar AR , Torres N ((2019) ) A dietaryintervention with functional foods reduces metabolic endotoxaemiaand attenuates biochemical abnormalities by modifying faecalmicrobiota in people with type 2 diabetes. Diabetes Metab 45: , 122–131. |
[175] | Gardener S , Gu Y , Rainey-Smith SR , Keogh JB , Clifton PM , Mathieson SL , Taddei K , Mondal A , Ward VK , Scarmeas N , Barnes M , Ellis KA , Head R , Masters CL , Ames D , Macaulay SL , Rowe CC , Szoeke C , Martins RN ((2012) ) Adherence to a Mediterranean diet and Alzheimer’s disease risk in an Australian population, Transl Psychiatry 2: , e164-e164. |
[176] | Xie Y , Yan L , Zeng H , Chen W , Lu J-H , Wan J-B , Su H , Yao X ((2020) ) Fish oil protects the blood– brain barrier integrity in a mouse model of Alzheimer’s disease. Chin Med 15: , 29. |
[177] | Takechi R , Pallebage-Gamarallage MM , Lam V , Giles C , Mamo JC ((2013) ) Nutraceutical agents with anti-inflammatory properties prevent dietary saturated-fat induced disturbances in blood– brain barrier function in wild-type mice. J Neuroinflammation 10: , 842. |
[178] | Nerurkar PV , Johns LM , Buesa LM , Kipyakwai G , Volper E , Sato R , Shah P , Feher D , Williams PG , Nerurkar VR ((2011) ) Momordica charantia (bitter melon) attenuates high-fat diet-associated oxidative stress and neuroinflammation. J Neuroinflammation 8: , 64. |
[179] | Novochadlo M , Goldim MP , Bonfante S , Joaquim L , Mathias K , Metzker K , Machado RS , Lanzzarin E , Bernades G , Bagio E , Garbossa L , de Oliveira Junior AN , da Rosa N , Generoso J , Fortunato JJ , Barichello T , Petronilho F ((2021) ) Folic acid alleviates the blood brain barrier permeability and oxidative stress and prevents cognitive decline in sepsis-surviving rats. Microvasc Res 137: , 104193. |
[180] | Parker A , Fonseca S , Carding SR ((2020) ) Gut microbes and metabolites as modulators of blood-brain barrier integrity and brain health. Gut Microbes 11: , 135–157. |
[181] | Kaddoumi A , Denney TS , Deshpande G , Robinson JL , Beyers RJ , Redden DT , Praticò D , Kyriakides TC , Lu B , Kirby AN , Beck DT , Merner ND ((2022) ) Extra-virgin olive oil enhances the blood-brain barrier function in mild cognitive impairment: A randomized controlled trial. Nutrients 14: , 5102. |
[182] | Gardener H , Scarmeas N , Gu Y , Boden-Albala B , Elkind MSV , Sacco RL , DeCarli C , Wright CB ((2012) ) Mediterranean diet and white matter hyperintensity volume in the Northern Manhattan Study. Arch Neurol 69: , 251–256. |
[183] | Lin D-T , Kao N-J , Cross T-WL , Lee W-J , Lin S-H ((2022) ) Effects of ketogenic diet on cognitive functions of mice fed high-fat-high-cholesterol diet. J Nutr Biochem 104: , 108974. |
[184] | Hajiluian G , Nameni G , Shahabi P , Mesgari-Abbasi M , Sadigh-Eteghad S , Farhangi MA ((2017) ) Vitamin D administration, cognitive function, BBB permeability and neuroinflammatory factors in high-fat diet-induced obese rats. Int J Obes (Lond) 41: , 639–644. |
[185] | Kowalski K , Mulak A ((2019) ) Brain-gut-microbiota axis in Alzheimer’s disease. J Neurogastroenterol Motil 25: , 48–60. |
[186] | Braniste V , Al-Asmakh M , Kowal C , Anuar F , Abbaspour A , Tóth M , Korecka A , Bakocevic N , Ng LG , Kundu P , Gulyás B , Halldin C , Hultenby K , Nilsson H , Hebert H , Volpe BT , Diamond B , Pettersson S ((2014) ) The gut microbiota influences blood-brain barrierpermeability in mice, Sci Transl Med 6: , 263ra158. |
[187] | Michel L , Prat A ((2016) ) One more role for the gut: Microbiota and blood brain barrier. Ann Transl Med 4: , 15. |
[188] | Schmitt F , Hussain G , Dupuis L , Loeffler J-P , Henriques A ((2014) ) A plural role for lipids in motor neuron diseases: Energy, signaling and structure. Front Cell Neurosci 8: , 25. |
[189] | Pennetta G , Welte MA ((2018) ) Emerging links between lipid droplets and motor neuron diseases. Dev Cell 45: , 427–432. |
[190] | Fabelo N , Martín V , Santpere G , Marín R , Torrent L , Ferrer I , Díaz M ((2011) ) Severe alterations in lipid composition of frontal cortex lipid rafts from Parkinson’s disease and incidental Parkinson’s disease. Mol Med 17: , 1107–1118. |
[191] | Xicoy H , Wieringa B , Martens GJM ((2019) ) The role of lipids in Parkinson’s disease. Cells 8: , 27. |
[192] | Chiò A , Benzi G , Dossena M , Mutani R , Mora G ((2005) ) Severely increased risk of amyotrophic lateral sclerosis among Italian professional football players. Brain 128: , 472–476. |
[193] | Blecher R , Elliott MA , Yilmaz E , Dettori JR , Oskouian RJ , Patel A , Clarke A , Hutton M , McGuire R , Dunn R , DeVine J , Twaddle B , Chapman JR ((2019) ) Contact sports as a risk factor for amyotrophic lateral sclerosis: A systematic review. Global Spine J 9: , 104–118. |
[194] | Lolekha P , Phanthumchinda K , Bhidayasiri R ((2010) ) Prevalence and risk factors of Parkinson’s disease in retired Thai traditional boxers. Mov Disord 25: , 1895–1901. |


