
The Case for a Novel Therapeutic Approach to Dementia: Small Molecule Hepatocyte Growth Factor (HGF/MET) Positive Modulators
Abstract
An estimated 6.5 million Americans aged 65 years or older have Alzheimer’s disease (AD), which will grow to 13.8 million Americans by 2060. Despite the growing burden of dementia, no fundamental change in drug development for AD has been seen in > 20 years. Currently approved drugs for AD produce only modest symptomatic improvements in cognition with small effect sizes. A growing mismatch exists between the urgent need to develop effective drugs for symptomatic AD and the largely failed search for disease modification. The failure rate of clinical trials in AD is high overall, and in particular for disease-modifying therapies. Research efforts in AD have focused predominantly on amyloid-β and tau pathologies, but limiting clinical research to these “classical hallmarks” of the disease does not address the most urgent patient, caregiver, or societal needs. Rather, clinical research should consider the complex pathophysiology of AD. Innovative approaches are needed that provide outside-the-box thinking, and re-imagine trial design, interventions, and outcomes as well as progress in proteomics and fluid biomarker analytics for both diagnostics and disease monitoring. A new approach offering a highly specific, yet multi-pronged intervention that exerts positive modulation on the HGF/MET neurotrophic system is currently being tested in mid-to-late-stage clinical trials in mild to moderate AD. Findings from such trials may provide data to support novel approaches for development of innovative drugs for treating AD at various disease stages, including among patients already symptomatic, and may offer benefits for other neurodegenerative diseases.
INTRODUCTION
Currently an estimated 6.5 million Americans aged 65 years or older have Alzheimer’s disease (AD), and as the population in the United States (U.S.) continues to age, it is estimated that 13.8 million Americans will have AD by 2060 [1]. Globally, it is estimated that the number of people with dementia will increase from 57 million in 2019 to 153 million by 2050 [2]. Despite the enormous personal and economic burden of AD [3, 4], and the extensive investment in drug development for AD, promising early results were recently reported with lecanemab in early AD [5], but only one product, aducanumab, has been approved in the last 19 years.
Thus, despite extensive clinical development efforts, a critical need exists for effective drugs for the treatment of AD, and it may be greatest for those with mild to moderate AD. Disease acceleration is most pronounced at this stage [6, 7] and the financial burden to payors and society is higher than in the pre-dementia stage [8]. Also, molecular approaches to clinical intervention are needed that consider the heterogeneity of causative factors in AD, along with past failed efforts while focusing on novel drugs that address biology other than amyloid-β (Aβ) and tau. This review highlights this critical need in the current landscape of clinical development, considers failed clinical trials to date, and provides insights into the need for identifying novel pharmacological approaches and study designs to enhance progress in drug development in AD.
IMPACT OF ALZHEIMER’S DISEASE
Currently, total healthcare costs for AD in the US are estimated at $300 billion, which is expected to rise to > $1 trillion by 2050 [1]. Key factors contributing to the economic burden of AD include disease severity, dependence level, rate of institutionalization, and comorbidity [4]. Furthermore, analyses of the costs of AD and dementia to society usually are underestimated because they fail to include costs for home safety modifications, adult day care, and the impact on caregiver health and productivity [9]. Considering the burgeoning prevalence of AD globally, we need to understand better both direct and indirect costs of AD to avoid an impending health care crisis [10]. Data from a claims database of outpatients at a memory clinic were used to evaluate medical costs and caregiver burden and demonstrated that direct medical costs increased as cognitive deficits increased, and importantly, caregiver burden increased with the severity of the patients’ cognitive deficits [11]. Others have reported the importance of indirect costs, which are associated with considerable societal and personal burden, including quality of life and caregiving, and are usually not factored into overall costs [12]. Costs for those with AD in the community healthcare setting were significantly lower in comparison to costs in a long-term care setting [13], and the transition into the long-term care setting increased total costs of dementia care also from a societal perspective.
Thus, any treatment that delays nursing home placement for people with AD has the potential to result in substantial economic benefits by reducing indirect costs [4, 14, 15]. Nursing home placement contributes to loss of independence and a decrease in physical and mental health with an associated increase in morbidity and mortality, and often is the most expensive option for family and caregivers. Thus, a truly effective treatment that delays progression in symptomatic AD has the potential for dramatic cost savings [4]. A number of studies have shown a delay or reduction in nursing home placement from treatment with currently approved AD therapies with cholinesterase inhibitors and memantine [16–19]. Further, a program designed to improve caregiver well-being reduced nursing home placement of persons with AD [20]. A model of the effect of reduced nursing home use with effective treatments for AD projected billions of dollars saved for reduced nursing home placement [21]. Another study of societal benefits from a treatment that slowed disease progression by 30% projected a reduction in costs of $5 trillion over 20 years in the US [22].
Over time, patients with AD often develop symptoms that are beyond the capabilities of caregivers to manage with the result that patients end up in a long-term care facility and incur an enormous economic burden [9, 11]. Thus, any delay in symptom onset or progression may result in a substantial impact on caregiver and economic burden. In particular, behavioral and psychological symptoms of dementia (BPSD) occur in 90% or more of all dementia patients and include symptoms of agitation, aggression, apathy, anxiety, depression, aberrant motor behavior, elation, irritability, disinhibition, delusions, hallucinations, and sleep or appetite changes [8, 23–25]. The BPSD spectrum is associated with poor outcomes, including increased burden among patients and caregivers, more frequent hospitalization, and increased health care costs [26]. Thus, a high medical need exists for novel drugs and non-drug therapies to improve outcomes in patients developing BPSD [8, 27]. New therapeutic approaches with the ability to address any impact of BPSD may also improve the burden of caregivers [28].
LANDSCAPE OF ALZHEIMER’S DISEASE DRUG DEVELOPMENT
Despite the growing burden of dementia worldwide, no fundamental change in the approaches to drug development for AD has occurred in > 20 years [29–32]. Current fully approved drugs for AD are neurotransmitter targeted and produce consistent but modest improvements in cognition, especially for those with advanced disease, with small effect sizes and a waning effect over time [33–36]. Some statistically significant effects on select BPSD also have been observed, e.g., with memantine [37].
A mismatch exists between the urgent need to develop effective drugs for secondary prevention and treatment of symptomatic AD and the yet largely failed track record of clinical development programs. Currently, the failure rate for clinical trials in AD is very high overall, and in particular for disease-modifying therapies [27, 29, 32, 38]. Research efforts in AD focus almost exclusively on Aβ and tau pathologies, including recent efforts with monoclonal antibodies for active or passive vaccination [30, 39–41]. Although the search for the “holy grail” of AD drug development producing tangible disease modification has been disappointing, prevention of the symptomatic stages of AD remains an important goal. For prevention trials in AD, clinically defined nearly asymptomatic subjects with pre-dementia based on experimental diagnostic criteria, rather than symptomatic patients, must be studied, which requires trials of long duration (>12 months) in large populations (>1000 patients) [27, 38, 42]. This, in turn, poses challenges for how representative these trial populations are of the majority of patients needing treatment, and the time point of diagnosis in the general population. Further, the related debate about the definition of a minimum clinically meaningful effect and relevant surrogate biomarkers is ongoing.
Despite the high failure rate in clinical trials, the majority of clinical development programs are using randomized, parallel-arm trial designs. However, alternative trial designs, such as staggered start/randomized withdrawal designs may improve the success rate of clinical trials for demonstrating efficacy by slowing progression in AD [32, 42–45]. Several challenges need to be addressed when designing clinical studies including appropriate identification of the patient population, selection of relevant clinical endpoints, and adequate duration of follow-up [38, 42, 46]. Studies in pre-dementia stage AD need to incorporate imaging and biomarkers to confirm the presence of disease prior to clinical symptoms [47] and target engagement by the investigational treatment, to select specific patient populations more likely to demonstrate clinically meaningful improvement with treatment [40, 42, 43]. Thus, disease-modifying treatments require study design features that are markedly different from symptomatic treatments [27].
Another critical limitation of pre-dementia trials is the inevitable reliance on experimental diagnostic criteria, as established by the A/T/N system, which includes the classical “hallmarks” of amyloid and tau accumulation [47]. The A/T/N system characterizes individuals using biomarkers of AD pathophysiology using the Aβ pathway (A), tau-mediated pathophysiology (T), and neurodegeneration (N) [47] and is independent of clinical assessment of cognitive status [48]. The A/T/N system is supposed to provide a more precise division of the continuum of AD based on pathology but may be limiting since different biomarkers for defining A/T/N are not interchangeable. Each component of biomarkers included in the A/T/N classification system contributes differently to the staging of AD, and the optimal combinations for predicting cognition may differ by cognitive status [49]. An updated A/T/X/N system has been proposed to accommodate a broader spectrum of pathophysiology, where X represents novel candidate biomarkers for additional pathophysiological mechanisms such as neuroimmune dysregulation, synaptic dysfunction, and blood-brain barrier alterations [50].
Accumulating evidence is consistent with a complex, decades-long, cellular phase of AD that produces dysfunctional neuronal, glial, and endothelial mechanisms that contribute to irreversible brain damage [43, 51–53]. AD is described as a brain disorder that results from a complex interplay of loss of synaptic homeostasis and dysfunction in the highly interrelated endosomal/lysosomal clearance pathways in which the precursors, aggregated species, and post-translationally modified products of Aβ and tau play important roles [53]. Based on this description, the search continues for targets that substantially change the clinical course in persons with AD; more promising trial results were recently reported for lecanemab [5], an investigational humanized monoclonal antibody recognizing protofibrils and aimed to prevent deposition of Aβ. Recent advances in proteomics provide increased evidence that the pathophysiology of AD extends beyond the classical “hallmarks”, i.e., amyloid and tau accumulation, to other mechanisms resulting in neurodegeneration [39, 54–57]. For instance, persons with no cognitive impairment can demonstrate biomarker evidence of Aβ pathology and may develop no clinical manifestations of AD in their lifetime [58]. In addition, a pattern of biomarkers consistent with AD may be found in other brain diseases in which AD pathology presents as a comorbidity. Recently, the incidence of co-morbidity of AD and cerebrovascular disease and its relevance for cognitive outcome was examined in those with autopsy-confirmed AD [59]. Increased functional and cognitive decline was found in those with AD and co-morbid cerebrovascular disease, and the presence of cerebrovascular disease enhanced the effects on AD neuropathology. These repeat findings, in a substantial proportion of subjects diagnosed clinically as AD, suggest an additive and even synergistic effect of co-morbid cerebrovascular disease on the neuropathology associated with AD.
Meanwhile, it has been demonstrated that amyloid and tau accumulation poorly reflect the intricate cell-mediated pathophysiology of AD [39, 54–57, 60, 61]. Animal models have failed to adequately replicate the pathology of the condition [48, 54, 57, 62]. Such a neuron-centric, linear cascade initiated by Aβ and leading to dementia suggests a direct causality that is not compatible with clinical observations [44, 51, 54–57, 62, 63]. Focusing almost exclusively on Aβ and tau mechanisms also ignores results from postmortem studies indicating a substantial contribution from vascular processes in the pathogenesis of AD [64, 65]; more than 30% of subjects with clinically diagnosed AD are classified postmortem as mixed dementia, a fact largely ignored by clinical development, other than only their exclusion from clinical trials is sought. Instead, the long cellular phase of dysregulation of the interplay between neurons, astrocytes, microglia, inflammatory, and vascular changes suggests the need for a broader therapeutic approach [53, 67, 68]. Varied research findings support such observations, for instance elevated concentrations of TNF-α in AD brains; TNF-α was shown to mediate multiple pertinent aspects of neurodegeneration, including synaptic dysfunction and neuronal decay [69]. Despite such findings, amyloid and tau accumulation continue to be the main focus for the diagnosis and subsequent selection of patients for clinical trials [70]; and thus, amyloid and tau accumulation continue to be the most prominent targets for interventional trials in AD [29, 41, 51, 56, 57]. This traditional focus may lead to a circular logic of patient selection, therapeutic intervention, and outcomes, e.g., reduced plaque load. Instead, subject inclusion criteria for clinical trials in AD might also consider more recent insights based on proteomics, which suggest more inter-individual heterogeneity in glial subtypes than in neurons [39, 55, 71]. A combined biomarker panel could potentially better distinguish between healthy subjects, asymptomatic and symptomatic AD patient subtypes when compared to a system based on Aβ and tau alone [52, 55]. Selection of such trial populations may provide a departure from a circular logic of diagnosis and treatment based only on the amyloid hypothesis [39, 44, 55]. Thus, adoption of a broader, more holistic view of the emerging pathophysiology of AD could result in substantial changes in the research and development field and open up new avenues to success.
NOVEL APPROACHES TO CLINICAL TRIAL DESIGN FOR ALZHEIMER’S DISEASE
The lack of progress in developing effective therapies for AD may be attributed to a lack of appreciation of the complex pathophysiology of the condition, but also to clinical trial design features that are not conducive to establishing drug effectiveness [72]. Blood-based biomarkers (e.g., neurofilament light chain, glial fibrillary acidic protein) may become essential in clinical trials to identify appropriate patients, determine the status of disease, confirm target engagement, establish the optimal dose, and monitor treatment response [72]. Most novel trials use an adaptive Bayesian design to predict efficacy or failure of individual interventions; interim analyses allow early termination of a study when a predefined futility signal is detected, with an aim to accelerate the development process. Such adaptive approaches can help reduce development times but also have proven treacherous in the past.
For novel interventions to actually demonstrate clinical efficacy at later stages, a more holistic approach also to clinical trial design is warranted. This in turn necessitates a re-focus on the symptomatic stage of AD, which only represents a minority of trials conducted in the past 20 years. At the present time, and in the absence of further development of diagnostics for AD [40], using a syndromal diagnosis at the dementia stage to identify patients to enroll in clinical trials provides a means of identifying real-life populations rather than highly experimental sub-populations of patients. Clinical trials should incorporate proven trial design features of patients in the dementia stage utilizing validated endpoints to minimize the need for 12- to 24-month double-blind follow-up periods, requiring years for completion. Such designs also could include specific improvements, e.g., innovative composite endpoints like the Integrated Alzheimer’s Disease Rating Scale (iADRS) [73], the AD Composite Score (ADCOMS) [74] to improve sensitivity to change over time, and the Global Statistical Test (GST) [75], which may provide an unbiased, integrated readout of therapeutic effects; incorporating such composite endpoints may help increase credibility of the real-life utility of a novel intervention. Other examples for trial innovations are passive monitoring of motor speed using mobile phones [76] for screening purposes, or the utilization of prognostic information from subjects into randomized controlled trials to enable smaller control groups while maintaining statistical power [77].
Re-focusing on the somewhat neglected group of patients with mild to moderate AD with suitable interventions for the symptomatic stage could prove helpful, as it may provide advantages for clinical trials of investigational drugs. The mild to moderate population experiences the most accelerated progression of cognitive impairment and develops also other important, non-cognitive changes, e.g., BPSD. It is arguably the population with presently the largest unmet medical need [6, 7]. Ultimately, a development program that targets mild to moderate AD should have a greater likelihood of success because a clinical diagnosis is possible rather than reliance on indirect criteria [42] to predict cognitive decline; a drug-placebo difference is also more likely; and there is an undisputed regulatory pathway for approval, based on precedent [78].
HGF/MET: PRECLINICAL FINDINGS RELEVANT TO ALZHEIMER’S DISEASE
The complex multifaceted processes that contribute to neurodegeneration in AD warrant novel treatment approaches that seek to improve the health and function of neurons and supporting glia. Promoting the activity of neurotrophic factors, including the hepatocyte growth factor (HGF), is an example for a therapeutic approach that is neuroprotective and stimulates regenerative mechanisms [79]. HGF is a ubiquitous signaling protein that activates the receptor tyrosine kinase MET [80, 81]. As a potent neurotrophic growth factor, HGF is involved in numerous processes including embryonic and organ development, regeneration, and inflammation [82], and is upstream of other important trophic factors in the central nervous system, e.g., brain-derived neurotrophic factor (BDNF). HGF enhances neuronal survival and regeneration including hippocampal, midbrain dopaminergic, cerebral cortical, motor, sensory, and cerebellar granular neurons [79, 80]. HGF signaling exhibits pro-neural and pro-cognitive effects, which offer the potential for treating the neurodegenerative cascade observed in AD while promoting neuronal survival [79, 83–86]. HGF signaling is also active in glial cells [87] where it modulates the expression of glial-specific glutamate transporters in astrocytes, which may reduce glutamate cytotoxicity [88].
The effects of increasing HGF signaling have been evaluated in vitro and in vivo. In vitro, HGF enhanced synaptic long-term potentiation in the CA1 region of the hippocampus [89], and overexpression of HGF in vivo improved memory and learning after cerebral infarction in rats [90]. Augmentation of the HGF/MET system slowed disease progression and restored function in rodent models of AD [91] and other neurodegenerative disorders in non-human primate models [92–96]. Elevated concentrations of HGF in the CSF of AD subjects have been reported and may be a result of blood-brain barrier dysfunction or a response to neurological damage due to AD pathology [97–99]. Conversely, MET expression is markedly decreased in AD brains, particularly in hippocampal pyramidal neurons, suggesting that this reduced MET activity may negatively affect hippocampal neuron survival and function in AD [100]. In AD, the underlying brain pathology is characterized by loss of synaptic connections [101–104]. HGF, as a key neurotrophic factor, enhances synaptogenesis and neuroplasticity, which may have widespread functional relevance in the symptomatology of AD [79, 80].
Multiple studies were conducted to identify brain-penetrant small molecules that activate the HGF/MET system, and to evaluate the neuroactivity of a highly potent target compound, fosgonimeton (previously ATH-1017), in vivo, and to optimize its pharmacological characteristics [105]. These preclinical studies demonstrated the activity of fosgo-AM, the active metabolite of the prodrug, fosgonimeton. Upon peripheral administration, fosgo-AM activates the brain HGF/MET system and restores cognitive function in experimental animals at clinically relevant exposures. These results supported further preclinical study with fosgo-AM in other neurotrophic models in vitro, and models of neurological disorders in vivo. After treatment with fosgo-AM, primary hippocampal neurons demonstrated enhanced synaptogenesis and neurite outgrowth. In an LPS-induced model of cognitive impairment in mice, cognitive deficits
Fig. 1
Positive Modulation of HGF/MET by Fosgonimeton. Hypothesized fast-onset effect of fosgonimeton mediated via the NMDA receptor. This mechanism of action represents a departure from drugs acting on the neurotransmitter level only, as the specific HGF/MET interaction could lead to multimodal downstream effects on processes known to be affected in various neurodegenerative conditions.
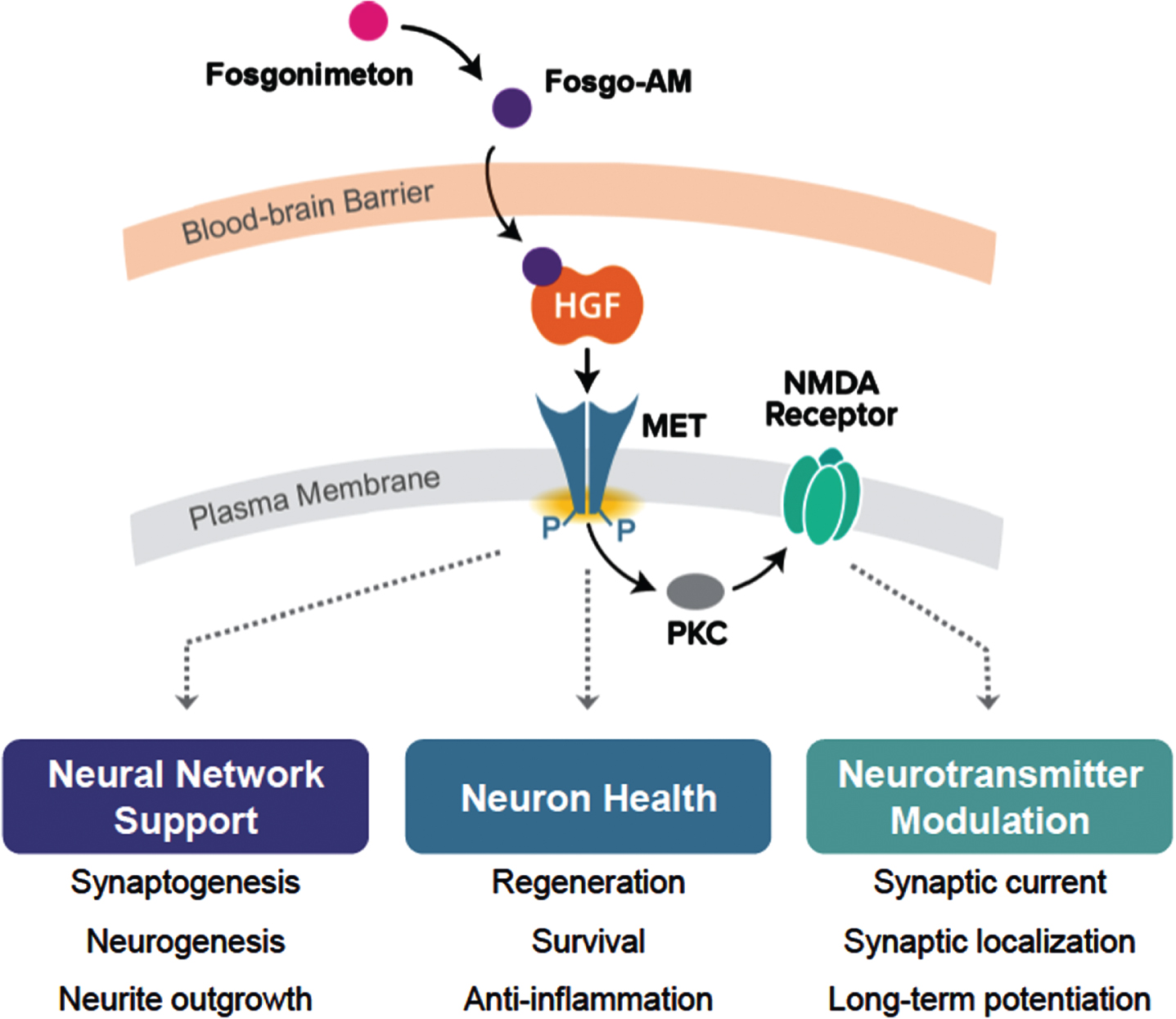
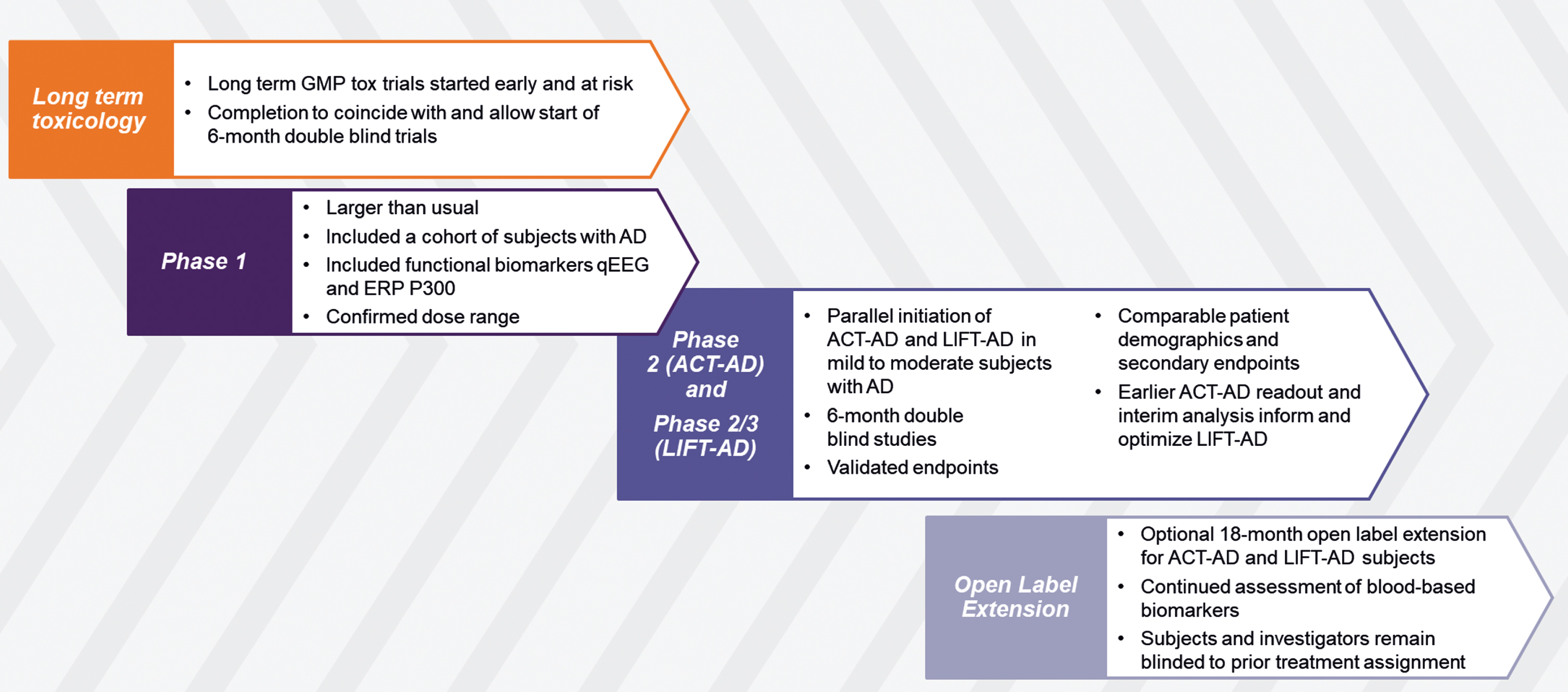
Fig. 2
Clinical Development of Fosgonimeton for Alzheimer’s Disease.
In summary, and in light of the above considerations on druggable targets in AD, the results of these preclinical studies support the potential of positively modulating the HGF/MET neurotrophic system in AD, and of fosgonimeton, as a novel compound and intervention with the potential to address several known aspects contributing to neurodegeneration.
CURRENT CLINICAL DEVELOPMENT OF COMPOUNDS PROMOTING HGF/MET FOR ALZHEIMER’S DISEASE
Fosgonimeton, a highly specific, small-molecule positive modulator of the HGF/MET neurotrophic system, is currently in late-stage clinical development for AD. To accelerate the development process (Fig. 2), the sponsor, Athira Pharma, Inc. (Athira) has taken an atypical approach by initiating long term toxicology studies at risk in parallel to Phase I, with results coinciding with the read out from human pharmacology. This Phase Ia/b study had a standard placebo-controlled single ascending dose (SAD)/multiple ascending dose (MAD) design. The study also protocolled a separate cohort of subjects with AD to receive a fixed dose of 40 mg once daily by s.c. injection for 9 days. Quantitative EEG and event-related potential (ERP) P300 latency were measured over time to support human blood-brain barrier penetration of fosgonimeton, and increase confidence in potential pro-cognitive effects, respectively [107].
Upon completion of the Phase Ia/b study, Phase II (ACT-AD; NCT04491006), and Phase II/III (LIFT-AD; NCT04488419) studies of fosgonimeton were launched; both are 26-week double-blind, placebo-controlled trials in subjects with mild to moderate AD, with a largely mirrored design but different objectives. While ACT-AD (N = 77, completed) was powered to replicate and extend the Phase Ib observation on ERP P300 latency in AD subjects [107], LIFT-AD (N revised upon interim analysis, recruiting) is powered as a potentially confirmatory trial. A unique feature is that both trials were started in parallel rather than sequentially, to allow results from ACT-AD analysis to ascertain the presence of positive biological signals (on cognition, function, and biomarkers) and be utilized for optimization of conduct and analysis of LIFT-AD [108, 109]. The primary endpoint for ACT-AD was ERP P300 latency, a functional measure of working memory processing speed that has recently been reviewed as a novel non-invasive, objective neuroplasticity biomarker [110]; secondary endpoints measuring cognition, function, and behavior were also included in the study. The primary endpoint for LIFT-AD is the composite Global Statistical Test [75], which is a mathematical algorithm that combines the scores from cognition (AD Assessment Scale-Cognitive Subscale [ADAS-Cog11]), and either global impression of change (AD Cooperative Study-Clinical Global Impression of Change [ADCS-CGIC]), or function (AD Cooperative Study-Activities of Daily Living [ADCS-ADL23]). Topline results from ACT-AD were announced in June and August 2022 [108, 109], and a manuscript is in preparation. Data for LIFT-AD are currently expected in 2023. An open label extension following the completion of either ACT-AD or LIFT-AD is currently ongoing, allowing eligible and interested participants to receive up to an additional 18 months of open label treatment.
Two other plasmid-mediated products that interact with HGF, from independent companies, are also in clinical development. HGF plasmid (AMG0001, AnGes, Inc. and Mitsubishi Tanabe) is DNA-based and encodes the human HGF gene. It is administered intramuscularly into the lower limb and is targeted to improve peripheral vascularization. AMG001 is approved in Japan and in late-stage development in the US for the long-term treatment of chronic arterial occlusive disease and arteriosclerosis obliterans, resulting in lower limb ulcers.
VM202 (Helixmith Co., Ltd.) is non-viral plasmid DNA product designed to express recombinant HGF protein peripherally in nerve and Schwann cells to promote nerve system regeneration and induce the formation of microvascular blood vessels. VM202 is being evaluated for the treatment of diabetic peripheral neuropathy, diabetic foot ulcer, amyotrophic lateral sclerosis, claudication, Charcot Marie Tooth disease, and coronary artery disease (Helixmith Co., Ltd.). At least 10 clinical studies have been completed with VM202. Both plasmid-based drugs increase vascular perfusion, which confirms the pharmacological effects of HGF/MET modulation. In common with AMG0001, VM202 is a large molecule that does not penetrate the CNS and therefore is not applicable for the treatment of AD or other CNS neurodegenerative conditions.
SUMMARY AND CONCLUSIONS
In the AD arena, limiting diagnosis and therapy to investigate the “classical hallmarks” of the disease may not sufficiently address the most urgent patient, caregiver, or societal needs for new, safe, and effective, and accessible therapies which include the symptomatic stages; this continued focus on the pre-dementia stages may lead to a circular logic in selecting experimental patient populations, therapeutic interventions, and outcomes. It is further proposed that clinical research should also focus on recent, novel insights in the pathophysiology of AD, in particular, based on proteomics, which may provide a broader characterization of the complex disease processes in AD, and potentially allow for a better delineation of study populations in the future [30, 39, 53, 111, 112].
A re-focused and innovative approach to developing effective drugs for AD is needed that incorporates novel elements in the study design including patient selection, type of interventions, and also key outcomes. For example, a highly specific, yet multi-pronged intervention that exerts positive modulation of HGF/MET is currently being tested in clinical trials. Findings from these trials could provide support for differentiated approaches for clinical development of innovative drugs for treating AD and may offer a way forward to achieving effective treatment for this devastating disease that has evaded research for too long.
ACKNOWLEDGMENTS
The authors would like to acknowledge the editorial assistance of Richard Perry, PharmD and Josh Pan, PhD, in the preparation of this manuscript, which was supported by Athira Pharma, Inc., Bothell, WA.
The ACT-AD trial is supported by a grant from the National Institute on Aging of the National Institutes of Health under Award Number R01AG06268. The information presented in this manuscript is solely the responsibility of Athira and does not necessarily represent the official views of the National Institutes of Health.
FUNDING
The authors have no funding to report for the development of this manuscript.
CONFLICT OF INTEREST
HJM and KJC are employees of and hold stock in Athira Pharma, Inc.
DATA AVAILABILITY
No primary data were presented in this review; all cited data referenced from peer-reviewed publications.
REFERENCES
[1] | Alzheimer’s Association ((2022) ) Alzheimer’s disease facts and figures. Alzheimers Dement 18: , 700–789. |
[2] | GBD 2019 Dementia Forecasting Collaborators ((2022) ) Estimation of the global prevalence of dementia in 2019 and forecasted prevalence in 2050: An analysis for the Global Burden of Disease Study 2019. Lancet Public Health 7: , e105–e125. |
[3] | Scheltens P , De Strooper B , Kivipelto M , Holstege H , Chetelat G , Teunissen CE , Cummings J , van der Flier WM ((2021) ) Alzheimer’s disease. Lancet 397: , 1577–1590. |
[4] | Tahami Monfared AA , Byrnes MJ , White LA , Zhang Q ((2022) ) The humanistic and economic burden of Alzheimer’s disease. Neurol Ther 11: , 553–569. |
[5] | van Dyck CH , Swanson CJ , Aisen P , Bateman RJ , Chen C , Gee M , Kanekiyo M , Li D , Reyderman L , Cohen S , Froelich L , Katayama S , Sabbagh M , Vellas B , Watson D , Dhadda S , Irizarry M , Kramer LD , Iwatsubo T ((2022) ) Lecanemab in early Alzheimer’s disease. N Engl J Med 388: , 9–21. |
[6] | Caroli A , Frisoni GB ; Alzheimer’s Disease Neuroimaging Initiative ((2010) ) The dynamics of Alzheimer’s disease biomarkers in the Alzheimer’s Disease Neuroimaging Initiative cohort. Neurobiol Aging 31: , 1263–1274. |
[7] | Ower AK , Hadjichrysanthou C , Gras L , Goudsmit J , Anderson RM , de Wolf F ; Alzheimer’s Disease Neuroimaging Initiative ((2018) ) Temporal association patterns and dynamics of amyloid-β and tau in Alzheimer’s disease. Eur J Epidemiol 33: , 657–666. |
[8] | Cerejeira J , Lagarto L , Mukaetova-Ladinska EB ((2012) ) Behavioral and psychological symptoms of dementia. Front Neurol 3: , 73. |
[9] | Deb A , Thornton JD , Sambamoorthi U , Innes K ((2017) ) Direct and indirect cost of managing Alzheimer’s disease and related dementias in the United States. Expert Rev Pharmacoecon Outcomes Res 17: , 189–202. |
[10] | El-Hayek YH , Wiley RE , Khoury CP , Daya RP , Ballard C , Evans AR , Karran M , Molinuevo JL , Norton M , Atri A ((2019) ) Tip of the iceberg: Assessing the global socioeconomic costs of Alzheimer’s disease and related dementias and strategic implications for stakeholders. J Alzheimers Dis 70: , 323–341. |
[11] | Dauphinot V , Potashman M , Levitchi-Benea M , Su R , Rubino I , Krolak-Salmon P ((2022) ) Economic and caregiver impact of Alzheimer’s disease across the disease spectrum: A cohort study. Alzheimers Res Ther 14: , 34. |
[12] | Wong W ((2020) ) Economic burden of Alzheimer disease and managed care considerations. Am J Manag Care 26(8 Suppl): , S177–S183. |
[13] | Wübker A , Zwakhalen SM , Challis D , Suhonen R , Karlsson S , Zabalegui A , Soto M , Saks K , Sauerland D ((2015) ) Costs of care for people with dementia just before and after nursing home placement: Primary data from eight European countries. Eur J Health Econ 16: , 689–707. |
[14] | Spiegl K , Luttenberger K , Graessel E , Becker L , Scheel J , Pendergrass A ((2021) ) Predictors of institutionalization in users of day care facilities with mild cognitive impairment to moderate dementia. BMC Health Serv Res 21: , 1009. |
[15] | Weimer DL , Sager MA ((2009) ) Early identification and treatment of Alzheimer’s disease: Social and fiscal outcomes. Alzheimers Dement 5: , 215–226. |
[16] | Feldman HH , Pirttila T , Dartigues JF , Everitt B , Van Baelen B , Schwalen S , Kavanagh S ((2009) ) Treatment with galantamine and time to nursing home placement in Alzheimer’s disease patients with and without cerebrovascular disease. Int J Geriatr Psychiatry 24: , 479–488. |
[17] | Wattmo C , Londos E , Minthon L ((2018) ) Short-term response to cholinesterase inhibitors in Alzheimer’s disease delays time to nursing home placement. Curr Alzheimer Res 15: , 905–916. |
[18] | Wimo A , Winblad B , Stoeffler A , Wirth Y , Moebius HJ ((2003) ) Resource utilisation and cost analysis of memantine in patients with moderate to severe Alzheimer’s disease. Pharmacoeconomics 21: , 327–340. |
[19] | Zhu CW , Sano M ((2006) ) Economic considerations in the management of Alzheimer’s disease. Clin Interv Aging 1: , 143–154. |
[20] | Mittelman MS , Haley WE , Clay OJ , Roth DL ((2006) ) Improving caregiver well-being delays nursing home placement of patients with Alzheimer disease. Neurology 67: , 1592–1599. |
[21] | Lam J , Jun H , Cho SK , Hanson M , Mattke S ((2021) ) Projection of budgetary savings to US state Medicaid programs from reduced nursing home use due to an Alzheimer’s disease treatment. Alzheimers Dement 13: , e12159. |
[22] | Prados MJ , Liu Y , Jun H , Lam J , Mattke S ((2022) ) Projecting the long-term societal value of a disease-modifying treatment for Alzheimer’s disease in the United States. Alzheimers Dement 18: , 142–151. |
[23] | Cummings J , Lai TJ , Hemrungrojn S , Mohandas E , Yun Kim S , Nair G , Dash A ((2016) ) Role of donepezil in the management of neuropsychiatric symptoms in Alzheimer’s disease and dementia with Lewy bodies. CNS Neurosci Ther 22: , 159–166. |
[24] | Kwon CY , Lee B ((2021) ) Prevalence of behavioral and psychological symptoms of dementia in community-dwelling dementia patients: A systematic review. Psychiatry 12: , 741059. |
[25] | Mukherjee A , Biswas A , Roy A , Biswas S , Gangopadhyay G , Das SK ((2017) ) Behavioural and psychological symptoms of dementia: Correlates and impact on caregiver distress. Dement Geriatr Cogn Dis Extra 7: , 354–365. |
[26] | Yunusa I , Alsumali A , Garba AE , Regestein QR , Eguale T ((2019) ) Assessment of reported comparative effectiveness and safety of atypical antipsychotics in the treatment of behavioral and psychological symptoms of dementia: A network meta-analysis. JAMA Netw Open 2: , e190828. |
[27] | Cummings J ((2021) ) New approaches to symptomatic treatments for Alzheimer’s disease. Neurodegener 16: , 2. |
[28] | Kim B , Noh GO , Kim K ((2021) ) Behavioural and psychological symptoms of dementia in patients with Alzheimer’s disease and family caregiver burden: A path analysis. Geriatr 21: , 160. |
[29] | Ayton S , Bush AI ((2021) ) Beta-amyloid: The known unknowns. Ageing Res Rev 65: , 101212. |
[30] | Frisoni GB , Altomare D , Thal DR , Ribaldi F , van der Kant R , Ossenkoppele R , Blennow K , Cummings J , van Duijn C , Nilsson PM , Dietrich PY , Scheltens P , Dubois B ((2022) ) The probabilistic model of Alzheimer disease: The amyloid hypothesis revised. Nat Rev Neurosci 23: , 53–66. |
[31] | Reiss AB , Montufar N , DeLeon J , Pinkhasov A , Gomolin IH , Glass AD , Arain HA , Stecker MM ((2021) ) Alzheimer disease clinical trials targeting amyloid: Lessons learned from success in mice and failure in humans. Neurologist 26: , 52–61. |
[32] | Sabbagh MN , Hendrix S , Harrison JE ((2019) ) FDA position statement “Early Alzheimer’s disease: Developing drugs for treatment, Guidance for Industry”. Alzheimers Dement 5: , 13–19. |
[33] | Birks J (2006) Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst Rev, CD005593. |
[34] | Budd Haeberlein S , Salloway S , Aisen P , Chalkias S , Chen T , Cohen S , Dent G , Hansson O , Harrison K , von Hehn C , Iwatsubo T , Mallinckrodt C , Mummery CJ , Muralidharan KK , Nestorov I , Nisenbaum L , Rajagovindan R , Skordos L , Tian Y , van Dyck CH , Vellas B , Wu S , Zhu Y , Sandrock A Evaluation of aducanumab efficacy in early Alzheimer’s disease. Paper presented at 15th International Conference on Alzheimer’s & Parkinson’s Diseases virtual conference, March 9-14, 2021. |
[35] | McShane R , Westby MJ , Roberts E , Minakaran N , Schneider L , Farrimond LE , Maayan N , Ware J , Debarros J ((2019) ) Memantine for dementia. Cochrane Database Syst Rev 3: , CD003154. |
[36] | Rockwood K ((2004) ) Size of the treatment effect on cognition of cholinesterase inhibition in Alzheimer’s disease. J Neurol Neurosurg Psychiatry 75: , 677–685. |
[37] | Gauthier S , Wirth Y , Moebius HJ ((2005) ) Effects of memantine on behavioural symptoms in Alzheimer’s disease patients: An analysis of the Neuropsychiatric Inventory (NPI) data of two randomised, controlled studies. Int J Geriatr Psychiatry 20: , 459–464. |
[38] | Cummings J , Feldman HH , Scheltens P ((2019) ) The “rights” of precision drug development for Alzheimer’s disease. Alzheimers Res Therapy 11: , 76. |
[39] | Rayaprolu S , Higginbotham L , Bagchi P , Watson CM , Zhang T , Levey AI , Rangaraju S , Seyfried NT ((2021) ) Systems-based proteomics to resolve the biology of Alzheimer’s disease beyond amyloid and tau. Neuropsychopharmacology 46: , 98–115. |
[40] | van Bokhoven P , de Wilde A , Vermunt L , Leferink PS , Heetveld S , Cummings J , Scheltens P , Vijverberg EGB ((2021) ) The Alzheimer’s disease drug development landscape. Alzheimers Res Ther 13: , 186. |
[41] | Vignon A , Salvador-Prince L , Lehmann S , Perrier V , Torrent J ((2021) ) Deconstructing Alzheimer’s disease: How to bridge the gap between experimental models and the human pathology? Int J Mol Sci 22: , 8769. |
[42] | de Aquino CH ((2021) ) Methodological issues in randomized clinical trials for prodromal Alzheimer’s and Parkinson’s disease. Front Neurol 12: , 694329. |
[43] | Aisen PS , Bateman RJ , Carrillo M , Doody R , Johnson K , Sims JR , Sperling R , Vellas B ((2021) ) Platform trials to expedite drug development in Alzheimer’s disease: A report from the EU/US CTAD Task Force. J Prev Alzheimers Dis 8: , 306–312. |
[44] | Cummings J , Ritter A , Zhong K ((2018) ) Clinical trials for disease-modifying therapies in Alzheimer’s disease: A primer, lessons learned, and a blueprint for the future. Alzheimers Dis 64: , S3–S22. |
[45] | Leber P ((1996) ) Observations and suggestions on antidementia drug development. Alzheimer Dis Assoc Disord 10: , 31–35. |
[46] | Cummings J , Aisen PS , DuBois B , Frölich L , Jack CR Jr , Jones RW , Morris JC , Raskin J , Dowsett SA , Scheltens P ((2016) ) Drug development in Alzheimer’s disease: The path to 2025. Alzheimers Res Ther 8: , 39. |
[47] | Jack CR Jr , Bennett DA , Blennow K , Carrillo MC , Feldman HH , Frisoni GB , Hampel H , Jagust WJ , Johnson KA , Knopman DS , Petersen RC , Scheltens P , Sperling RA , Dubois B ((2016) ) A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology 87: , 539–547. |
[48] | Grøntvedt GR , Lauridsen C , Berge G , White LR , Salvesen Ø , Bråthen G , Sando SB ((2020) ) The amyloid, tau, and neurodegeneration (A/T/N) classification applied to a clinical research cohort with long-term follow-up. J Alzheimers Dis 74: , 829–37. |
[49] | Lin RR , Xue YY , Li XY , Chen YH , Tao QQ , Wu ZY ((2021) ) Optimal combinations of AT(N) biomarkers to determine longitudinal cognition in the Alzheimer’s disease. Front Aging Neurosci 13: , 718959. |
[50] | Hampel H , Cummings J , Blennow K , Gao P , Jack CR Jr , Vergallo A ((2021) ) Developing the ATX(N) classification for use across the Alzheimer disease continuum. Nat Rev Neurol 17: , 580–589. |
[51] | Decourt B , D’Souza GX , Shi J , Ritter A , Suazo J , Sabbagh MN ((2022) ) The cause of Alzheimer’s disease: The theory of multipathology convergence to chronic neuronal stress. Aging Dis 13: , 37–60. |
[52] | Johnson ECB , Dammer EB , Duong DM , Ping L , Zhou M , Yin L , Higginbotham LA , Guajardo A , White B , Troncoso JC , Thambisetty M , Montine TJ , Lee EB , Trojanowski JQ , Beach TG , Reiman EM , Haroutunian V , Wang M , Schadt E , Zhang B , Dickson DW , Ertekin-Taner N , Golde TE , Petyuk VA , De Jager PL , Bennett DA , Wingo TS , Rangaraju S , Hajjar I , Shulman JM , Lah JJ , Levey AI , Seyfried NT ((2020) ) Large-scale proteomic analysis of Alzheimer’s disease brain and cerebrospinal fluid reveals early changes in energy metabolism associated with microglia and astrocyte activation. Med 26: , 769–780. |
[53] | Knopman DS , Amieva H , Petersen RC , Chételat G , Holtzman DM , Hyman BT , Nixon RA , Jones DT ((2021) ) Alzheimer disease. Nat Rev Dis Primers 7: , 33. |
[54] | Boder EJ , Banerjee IA ((2021) ) Alzheimer’s disease: Current perspectives and advances in physiological modeling. Bioengineering 8: , 211. |
[55] | Higginbotham L , Ping L , Dammer EB , Duong DM , Zhou M , Gearing M , Hurst C , Glass JD , Factor SA , Johnson ECB , Hajjar I , Lah JJ , Levey AI , Seyfried NT ((2020) ) Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Sci Adv 6: , eaaz9360. |
[56] | Mullard A ((2021) ) Failure of first anti-tau antibody in Alzheimer disease highlights risks of history repeating. Nat Rev Drug Discov 20: , 3–5. |
[57] | Oxford AE , Stewart ES , Rohn TT ((2020) ) Clinical trials in Alzheimer’s disease: A hurdle in the path of remedy. Int J Alzheimers Dis 2020: , 5380346. |
[58] | Dubois B , Villain N , Frisoni GB , Rabinovici GD , Sabbagh M , Cappa S , Bejanin A , Bombois S , Epelbaum S , Teichmann M , Habert MO , Nordberg A , Blennow K , Galasko D , Stern Y , Rowe CC , Salloway S , Schneider LS , Cummings JL , Feldman HH ((2021) ) Clinical diagnosis of Alzheimer’s disease: Recommendations of the International Working Group. Lancet Neurol 20: , 484–496. |
[59] | Frank B , Ally M , Tripodis Y , Puzo C , Labriolo C , Hurley L , Martin B , Palmisano J , Chan L , Steinberg E , Turk K , Budson A , OÂ’Connor M , Au R , Qiu WQ , Goldstein L , Kukull W , Kowall N , Killiany R , Stern R , Stein T , McKee A , Mez J , Alosco M ((2022) ) Trajectories of cognitive decline in brain donors with autopsy-confirmed Alzheimer disease and cerebrovascular disease. Neurology 98: , e2454–e2464. |
[60] | Kametani F , Hasegawa M ((2018) ) Reconsideration of amyloid hypothesis and tau hypothesis in Alzheimer’s disease. Front Neurosci 12: , 25. |
[61] | Sabbagh M , Miller J , Jones S , Ritter A , Shi J , DeCourt B , Wint D ((2021) ) Does informant-based reporting of cognitive decline correlate with age-adjusted hippocampal volume in mild cognitive impairment and Alzheimer’s disease? J Alzheimers Dis Rep 5: , 207–211. |
[62] | Cummings J ((2018) ) Lessons learned from Alzheimer disease: Clinical trials with negative outcomes. Clin Transl Sci 11: , 147–152. |
[63] | Herrup K ((2022) ) Fallacies in neuroscience: The Alzheimer’s Edition. eNeuro 9: , ENEURO.0530-21.2021. |
[64] | Iacono D , Resnick SM , O’Brien R , Zonderman AB , An Y , Pletnikova O , Rudow G , Crain B , Troncoso JC ((2014) ) Mild cognitive impairment and asymptomatic Alzheimer disease subjects: Equivalent β-amyloid and tau loads with divergent cognitive outcomes. J Neuropathol Exp Neurol 73: , 295–304. |
[65] | Perez-Nievas BG , Stein TD , Tai HC , Dols-Icardo O , Scotton TC , Barroeta-Espar I , Fernandez-Carballo L , de Munain EL , Perez J , Marquie M , Serrano-Pozo A , Frosch MP , Lowe V , Parisi JE , Petersen RC , Ikonomovic MD , López OL , Klunk W , Hyman BT , Gómez-Isla T ((2013) ) Dissecting phenotypic traits linked to human resilience to Alzheimer’s pathology. Brain 136: , 2510–2526. |
[66] | Chen SJ , Tsai HH , Tsai LK , Tang SC , Lee BC , Liu HM , Yen RF , Jeng JS ((2019) ) Advances in cerebral amyloid angiopathy imaging. Ther Adv Neurol Disord 12: , 1756286419844113. |
[67] | Klohs J ((2019) ) An integrated view on vascular dysfunction in Alzheimer’s disease. Dis 19: , 109–127. |
[68] | Zhao M , Jiang XF , Zhang HQ , Sun JH , Pei H , Ma LN , Cao Y , Li H ((2021) ) Interactions between glial cells and the blood-brain barrier and their role in Alzheimer’s disease. Ageing Res Rev 72: , 101483. |
[69] | Chang R , Yee KL , Sumbria RK ((2017) ) Tumor necrosis factor α inhibition for Alzheimer’s disease. J Cent Nerv Syst Dis 9: , 1179573517709278. |
[70] | Jacobs N , Theunissen B ((2022) ) It’s groundhog day! What can the history of science say about the crisis in Alzheimer’s disease research? J Alzheimers Dis 90: , 1401–1415. |
[71] | Duara R , Barker W ((2022) ) Heterogeneity in Alzheimer’s disease diagnosis and progression rates: Implications for therapeutic trials. Neurotherapeutics 19: , 8–25. |
[72] | Yiannopoulou KG , Anastasiou AI , Zachariou V , Pelidou SH ((2019) ) Reasons for failed trials of disease-modifying treatments for Alzheimer disease and their contribution in recent research. Biomedicines 7: , 97. |
[73] | Wessels AM , Andersen SW , Dowsett SA , Siemers ER ((2018) ) The Integrated Alzheimer’s Disease Rating Scale (iADRS) Findings from the EXPEDITION3 Trial. J Prev Alzheimers Dis 5: , 134–136. |
[74] | Wang J , Logovinsky V , Hendrix SB , Stanworth SH , Perdomo C , Xu L , Dhadda S , Do I , Rabe M , Luthman J , Cummings J , Satlin A ((2016) ) ADCOMS: A composite clinical outcome for prodromal Alzheimer’s disease trials. J Neurol Neurosurg Psychiatry 87: , 993–999. |
[75] | O’Brien PC ((1984) ) Procedures for comparing samples with multiple endpoints. Biometrics 40: , 1079–1087. |
[76] | NQMedical, AP/PD2021. https://www.prweb.com/releases/nq_medical_a_digital_biomarker_discovery_platform_unveils_latest_alzheimers_disease_clinical_trial_results_at_ad_pd_2021_international_conference/prweb17785720.htm |
[77] | Unlearn, AP/PD2021. https://www.unlearn.ai/post/ad-pd-2021-unlearn-will-present-novel-ai-driven-approaches-to-enabling-smaller-more-efficient-alzheimers-clinical-trials |
[78] | Food and Drug Administration Guidance for Industry (2018) Early Alzheimer’s Disease: Developing Drugs for Treatment. |
[79] | Funakoshi H , Nakamura T ((2011) ) Hepatocyte growth factor (HGF): Neutrophic functions and therapeutic implications for neuronal injury/disease. Curr Signal Tranduct Ther 6: , 156–167. |
[80] | Desole C , Gallo S , Vitacolonna A , Montarolo F , Bertolotto A , Vivien D , Comoglio P , Crepaldi T ((2021) ) HGF and MET: From brain development to neurological disorders. Front Cell Dev Biol 9: , 683609. |
[81] | Nakamura T , Mizuno S ((2010) ) The discovery of hepatocyte growth factor (HGF) and its significance for cell biology, life sciences and clinical medicine. Proc Jpn Acad Ser B Phys Biol Sci 86: , 588–610. |
[82] | Nakamura T , Sakai K , Nakamura T , Matsumoto K ((2011) ) Hepatocyte growth factor twenty years on: Much more than a growth factor. J Gastroenterol Hepatol 26 Suppl 1: , 188–202. |
[83] | Akita H , Takagi N , Ishihara N , Takagi K , Murotomi K , Funakoshi H , Matsumoto K , Nakamura T , Takeo S ((2008) ) Hepatocyte growth factor improves synaptic localization of the NMDA receptor and intracellular signaling after excito- toxic injury in cultured hippocampal neurons. Exp Neurol 210: , 83–94. |
[84] | Maina F , Klein R ((1999) ) Hepatocyte growth factor, a versatile signal for developing neurons. Nat Neurosci 2: , 213–217. |
[85] | Nicoleau C , Benzakour O , Agasse F , Thiriet N , Petit J , Prestoz L , Roger M , Jaber M , Coronas V ((2009) ) Endogenous hepatocyte growth factor is a niche signal for subventricular zone neural stem cell amplification and self-renewal. Stem Cells 27: , 408–419. |
[86] | Tyndall SJ , Walikonis RS ((2006) ) The receptor tyrosine kinase Met and its ligand hepatocyte growth factor are clustered at excitatory synapses and can enhance clustering of synaptic proteins. Cell Cycle 5: , 1560–1568. |
[87] | Yamada T , Tsubouchi H , Daikuhara Y , Prat M , Comoglio PM , McGeer PL , McGeer EG ((1004) ) Immunohistochemistry with antibodies to hepatocyte growth factor and its receptor protein (c-MET) in human brain tissues. Brain Res 637: , 308–312. |
[88] | Sun W , Funakoshi H , Nakamura T ((2002) ) Localization and functional role of hepatocyte growth factor (HGF) and its receptor c-met in the rat developing cerebral cortex. Brain Res Mol Brain Res 103: , 36–48. |
[89] | Akimoto M , Baba A , Ikeda-Matsuo Y , Yamada MK , Itamura R , Nishiyama N , Ikegaya Y , Matsuki N ((2004) ) Hepatocyte growth factor as an enhancer of NMDA currents and synaptic plasticity in the hippocampus. Neuroscience 128: , 155–162. |
[90] | Shimamura M , Sato N , Waguri S , Uchiyama Y , Hayashi T , Iida H , Nakamura T , Ogihara T , Kaneda Y , Morishita R ((2006) ) Gene transfer of hepatocyte growth factor gene improves learning and memory in the chronic stage of cerebral infarction. Hypertension 47: , 742–751. |
[91] | Takeuchi D , Sato N , Shimamura M , Kurinami H , Takeda S , Shinohara M , Suzuki S , Kojima M , Ogihara T , Morishita R ((2008) ) Alleviation of Aβ-induced cognitive impairment by ultrasound-mediated gene transfer of HGF in a mouse model. Gene Therapy 15: , 561–571. |
[92] | Cohen JA ((2013) ) Mesenchymal stem cell transplantation in multiple sclerosis. J Neurol Sci 333: , 43–49. |
[93] | Doeppner TR , Kaltwasser B , ElAli A , Zechariah A , Hermann D , Bähr M ((2011) ) Acute hepatocyte growth factor treatment induces long-term neuroprotection and stroke recovery via mechanisms involving neural precursor cell proliferation and differentiation. J Cereb Blood Flow Metab 31: , 1251–1262. |
[94] | Kitamura K , Fujiyoshi K , Yamane J , Toyota F , Hikishima K , Nomura T , Funakoshi H , Nakamura T , Aoki M , Toyama Y , Okano H , Nakamura M ((2011) ) Human hepatocyte growth factor promotes functional recovery in primates after spinal cord injury. PLoS One 6: , e27706. |
[95] | Koike H , Ishida A , Shimamura M , Mizuno S , Nakamura T , Ogihara T , Kaneda Y , Morishita R ((2006) ) Prevention of onset of Parkinson’s disease by gene transfer of human hepatocyte growth factor in rodent model: A model of gene therapy for Parkinson’s disease. Gene Therapy 13: , 1639–1644. |
[96] | Sun W , Funakoshi H , Nakamura T ((2002) ) Overexpression of HGF retards disease progression and prolongs life span in a transgenic mouse model of ALS. J Neurosci 22: , 6537–6548. |
[97] | Fenton H , Finch PW , Rubin JS , Rosenberg JM , Taylor WG , Kuo-Leblanc V , Rodriguez-Wolf M , Baird A , Schipper HM , Stopa EG ((1998) ) Hepatocyte growth factor (HGF/SF) in Alzheimer’s disease. Brain Res 779: , 262–270. |
[98] | Tsuboi Y , Kakimoto K , Nakajima M , Akatsu H , Yamamoto T , Ogawa K , Ohnishi T , Daikuhara Y , Yamada T ((2003) ) Increased hepatocyte growth factor level in cerebrospinal fluid in Alzheimer’s disease. Acta Neurol Scand 107: , 81–86. |
[99] | Zhao LJ , Wang ZT , Ma YH , Zhang W , Dong Q , Yu JT , Tan L ; Alzheimer’s Disease Neuroimaging Initiative ((2021) ) Associations of the cerebrospinal fluid hepatocyte growth factor with Alzheimer’s disease pathology and cognitive function. BMC Neurol 21: , 387. |
[100] | Hamasaki H , Honda H , Suzuki SO , Hokama M , Kiyohara Y , Nakabeppu Y , Iwaki T ((2014) ) Down-regulation of MET in hippocampal neurons of Alzheimer’s disease brains. Neuropathology 34: , 84–90. |
[101] | Bell KFS , Ducatenzeiler A , Ribeiro-da-Silva A , Duff K , Bennett DA , Claudio Cuello A ((2006) ) The amyloid pathology progresses in a neurotransmitter-specific manner. Neurobiol Aging 27: , 1644–1657. |
[102] | Jackson J , Jambrina E , Li J , Marston H , Menzies F , Phillips K , Gilmour G ((2019) ) Targeting the synapse in Alzheimer’s disease. Front Neurosci 13: , 735. |
[103] | Mufson EJ , Binder L , Counts SE , DeKosky ST , de Toledo-Morrell L , Ginsberg SD , Ikonomovic MD , Perez SE , Scheff SW ((2012) ) Mild cognitive impairment: Pathology and mechanisms. Acta Neuropathol 123: , 13–30. |
[104] | Selkoe DJ ((2002) ) Alzheimer’s disease is a synaptic failure. Science 298: , 789–791. |
[105] | Johnston JL , Reda SM , Setti SE , Taylor RW , Berthiaume A-A , Walker WE , Wu W , Moebius HJ , Church KJ (2022) Fosgonimeton, a novel positive hepatocyte modulator of theHGF/METsystem, promotes neurotrophic and precognitive effects in models of dementia. Neurotherapeutics, doi.org/10.1007/s13311-022-01325-5. |
[106] | Reda S , Johnston J , Taylor RW , Church K ((2022) ) Fosgonimeton, a novel, small molecule positive modulator of the HGF/MET system is neuroprotective in primary neuron culture. Alzheimers Dement 18: (Suppl 10), e065874. |
[107] | Hua X , Church K , Walker W , Hostis P , Viardot G , Danjou P , Hendrix S , Moebius HJ ((2022) ) Safety, tolerability, pharmacokinetics, and pharmacodynamics of the positive modulator of HGF/MET, fosgonimeton, in healthy volunteers and subjects with Alzheimer’s disease: Randomized, placebo-controlled, double-blind, phase I clinical trial. J Alzheimers Dis 86: , 1399–1413. |
[108] | Moebius HJ , Church K , Ooi K-B (2022) Study design and participant characteristics of a Phase 2 trial of fosgonimeton, a novel treatment for mild to moderate Alzheimer’s disease. Presented at the AD/PD 2022 International Conference, Barcelona, Spain. |
[109] | Moebius HJ , Bernick CB , Winner P , Maalouf J , Ooi K-B , Dickson SP , Hendrix SB , Church K , Olichney JM ((2022) ) ACT-AD: Fosgonimeton in mild-to-moderate Alzheimer’s disease –First results of a randomized, placebo-controlled, 26-week Phase 2 proof-of-concept trial. Alzheimers Dement 18: (Suppl 10), e061572. |
[110] | Olichney J , Xia J , Church KJ , Moebius HJ ((2022) ) Predictive power of cognitive biomarkers in neurodegenerative disease drug development: Utility of the P300 event-related potential. Neural Plast 2022: , 2104880. |
[111] | Hampel H , Nisticò R , Seyfried NT , Levey AI , Modeste E , Lemercier P , Baldacci F , Toschi N , Garaci F , Perry G , Emanuele E , Valenzuela PL , Lucia A , Urbani A , Sancesario GM , Mapstone M , Corbo M , Vergallo A , Lista S ; Alzheimer Precision Medicine Initiative (APMI) ((2021) ) Omics sciences for systems biology in Alzheimer’s disease: State-of-the-art of the evidence. Ageing Res Rev 69: , 101346. |
[112] | Cummings J , Lanctôt K (2022) Phase II Drug Development for Alzheimer’s Disease: A Panel Discussion. Presented at the Alzheimer’s Association International Conference. |

