
The autodigestion hypothesis: Proteolytic receptor cleavage in rheological and cardiovascular cell dysfunction1
Abstract
Transformation of circulating leukocytes from a dormant into an activated state with changing rheological properties leads to a major shift of their behavior in the microcirculation. Low levels of pseudopod formation or expression of adhesion molecules facilitate relatively free passage through microvessels while activated leukocytes with pseudopods and enhanced levels of adhesion membrane proteins become trapped in microvessels, attach to the endothelium and migrate into the tissue. The transformation of leukocytes into an activated state is seen in many diseases. While mechanisms for activation due to infections, tissue trauma, as well as non-physiological biochemical or biophysical exposures are well recognized, the mechanisms for activation in many diseases have not been conclusively liked to these traditional mechanisms and remain unknown. We summarize our recent evidence suggesting a major and surprising role of digestive enzymes in the small intestine as root causes for leukocyte activation and microvascular disturbances. During normal digestion of food digestive enzymes are compartmentalized in the lumen of the intestine by the mucosal epithelial barrier. When permeability of this barrier increases, these powerful degrading enzymes leak into the wall of the intestine and into the systemic circulation. Leakage of digestive enzymes occurs for example in physiological shock and multi-organ failure. Entry of digestive enzymes into the wall of the small intestine leads to degradation of the intestinal tissue in an autodigestion process. The digestive enzymes and tissue/food fragments generate not only activate leukocytes but also cause numerous cell dysfunctions. For example, proteolytic destruction of membrane receptors, plasma proteins and other biomolecules occurs. We conclude that escape of digestive enzymes from the intestinal track serves as a major source of cell dysfunction, morbidity and even mortality, including abnormal leukocyte activation seen in rheological studies.

1.Introduction
With deep gratitude I thank the International Society for Biorheology for selection of my name as the Poiseuille Award winner. As student of the microcirculation it is a special privilege to be associated with the name of Jean Léonard Marie Poiseuille, whose famous law embraces some of the most fundamental aspects of hemodynamics in small blood vessels. It is a special honor to join the list of esteemed Poiseuille Award winners.
The study of blood rheology has served as point of departure to understand important aspects about living tissues in molecular detail. As student I had the great fortune to join Benjamin Zweifach and Yuan-Cheng Fung at the University of California San Diego and later as postdoctoral fellow continue this work with Shu Chien and Richard Skalak at Columbia University, all pioneers in blood rheology. I studied leukocyte rheology, at a time when blood rheology was largely focused on red cells and platelets. The interest in leukocyte rheology was only starting. The biophysical and rheological properties of leukocytes were largely unknown and there was an opportunity to develop a deeper understanding of their behavior in-vivo.
In the following I will give a brief summary of selected observations about these cells in the microcirculation. The study of the rheological properties of leukocytes in-vitro and in-vivo opened an important gateway to analyze disease states at a new level of detail. Indeed, the study of leukocytes led eventually after a long and winding exploratory tour to a new idea about the origin of human disease and death, which we designated as “Autodigestion”. It would not have been possible without an understanding of leukocytes in-vivo.
2.Microvascular perfusion and leukocytes activation
Leukocytes have the ability to obstruct the microcirculation and generate states of immune-incompetence. In spite of their relatively small numbers in the blood circulation, leukocytes can play a major role in perfusion of a capillary network. Their morphology [1,2] and stiff cytoplasmic viscoelastic properties is dramatically different compared to that of the highly deformable red cells [3–5]. In addition leukocytes have the ability to spontaneously change their cytoplasmic properties transforming from a viscoelastic to an elastic cytoplasm by localized actin polymerization and cross linking, a process that leads often to projection of stiff pseudopods. Leukocytes control membrane adhesion by expression and activation of specific membrane receptors which in turn orchestrate a sequence of events that serves to attach the cell to the endothelium and facilitate transendothelial migration, a process that has been subject to numerous investigations [6–12].
Circulating leukocytes control microvascular perfusion in capillaries, and they do so by influencing the motion of the red cells [13,14]. Leukocytes (e.g. granulocytes or monocytes) in single file capillaries are less deformed compared to the highly flexible red cells and therefore move slower [15]. The velocity difference between the two cell types leads to formation of “leukocyte–erythrocyte trains” [16] visible to a person in their own retina [17]. The leukocytes disturbed the slower moving red cells, an effect that causes an increase of the apparent viscosity controlled by the red cells in capillaries with an increase in whole organ hydrodynamic resistance [13,14].
A severe microvascular disturbance by leukocyte occurs when they are trapped in capillaries. Leukocytes that obstruct capillaries generate the capillary no-reflow phenomenon [18]. Capillary obstruction can occur by multiple mechanisms, when the microvascular perfusion pressure is reduced [19–21], when capillary endothelium projects pseudopods [22] or undergoes apoptosis [23], and/or when leukocytes become activated. The leukocyte activation is detected by the presence of cells with pseudopods, by an altered expression of membrane adhesion molecules, by spontaneous degranulation, and even by biochemical indicators such as enhanced release of oxygen free radicals, all of which without indications for cell death (apoptosis or necrosis). Activation with pseudopod formation retards leukocytes in the microcirculation, may cause their entrapment and further raises the hemodynamic resistance [14,24]. Activated leukocytes can be observed in many different diseases as well as in the presence of risk factors for disease [25–29]. This evidence that the state of activation controls basic hemodynamics brought to light the importance of leukocyte activation in disease.
3.Transformation of the leukocyte rheological properties in disease
Leukocyte, in the free circulation or when attached to the endothelium, become activated as an early step in the inflammatory cascade and eventual tissue repair. While leukocytes require several cellular mechanisms to migrate or to phagocytize, in the free circulation most of these mechanisms remain in a relatively dormant state. Specifically, it is rare to find in a normal microcirculation of a healthy individual leukocytes with pseudopods (Fig. 1). In fact the fluid shear stress in the circulation serves to stimulate retraction of pseudopods [30] by a mechanism that involves membrane receptors [31] and interaction with red cells [32]. Fluid shear stress at levels encountered in the circulation also reduce membrane adhesion [33]. The majority of leukocytes collected by venous phlebotomy of non-symptomatic control individuals shows low levels of leukocyte activation.
Fig. 1.
Scanning electron micrograph of fresh, untreated, circulating leukocytes (predominantly neutrophils and monocytes) in venous blood derived from the free circulation. Cells in the top row have low levels of pseudoformation and represent a non-activated (passive) state, and in bottom row exhibit an activated state with extensive pseudopods. Leukocytes of non-symptomatic healthy controls are rare in non-symptomatic control individuals but occur more frequently in circulating leukocytes of individuals with disease symptoms. Activation of circulating leukocytes may be accompanied by expression of membrane adhesion molecules, endothelial adhesion, oxygen free radical formation, degranulation, depletion glycocalyx, reduced response to fluid shear stress and other forms of cell activity. Length of bars is 1 μm.
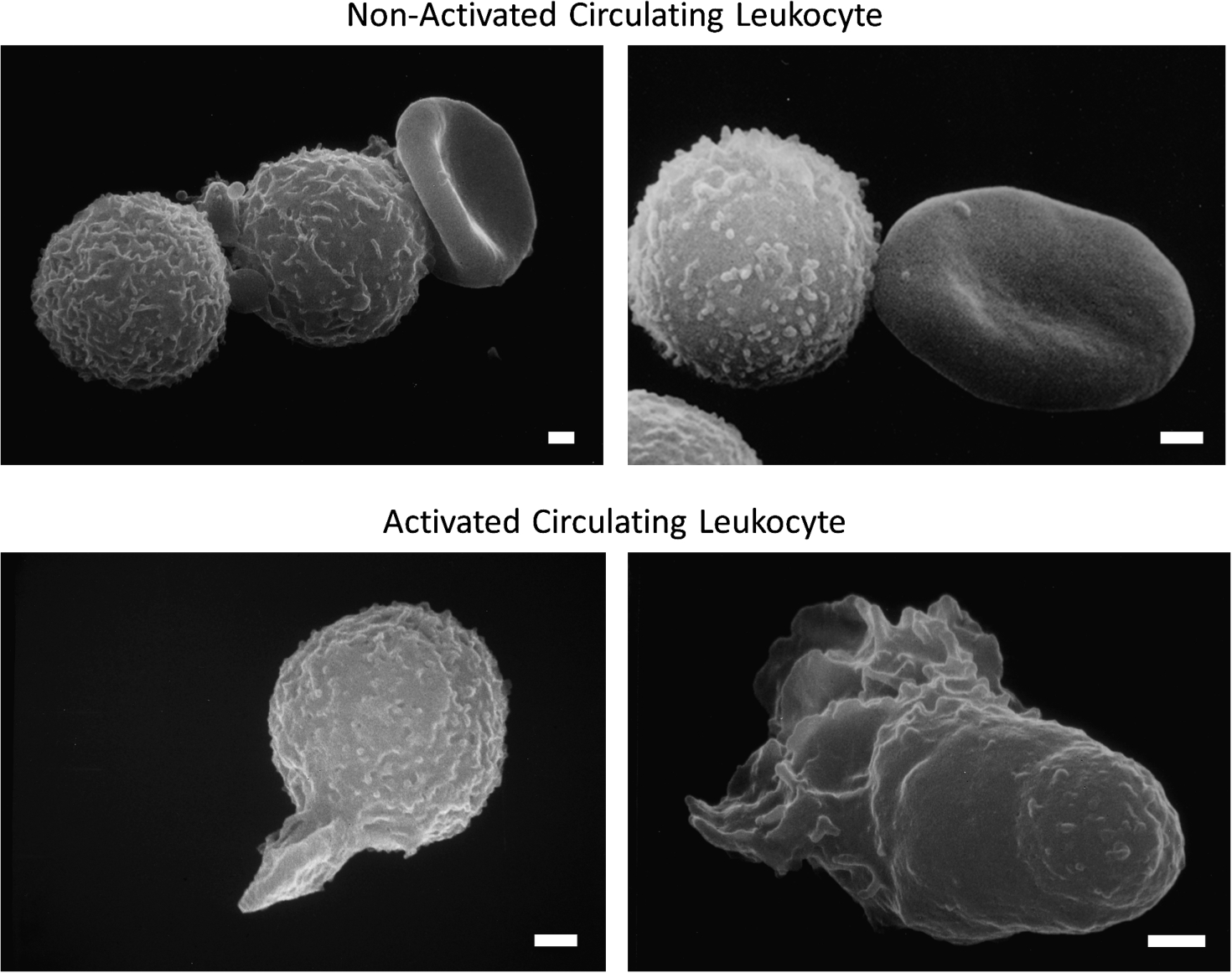
In contrast, in diseases or in the presence of risks for diseases the number of circulating leukocytes forming pseudopods, or expressing oxygen free radicals, is notably increased [26,34–36]. The enhanced level of leukocyte activation is detected in freshly drawn blood samples, even though activated leukocytes may not stay in the active circulation due to capillary entrapment. Evidence for leukocyte activation can also be detected by use of naive leukocytes from a healthy donor exposed in-vitro to plasma from patients. In experimental hemorrhagic shock, during a transient (1 to 2 hours) reduction of the central arterial blood pressure it is possible to predict with high probability the mortality 24 hours after shock by the level of leukocyte activation before shock [37]. These observations suggest that the study of leukocyte activation can serve as a tool to explore mechanisms leading to acute organ failure in shock, and thereby provide insight into a severe pathogenic event.
We took advantage of this opportunity to search for mechanisms for the activation derived from the plasma during hemorrhagic shock. The activation is easily detectable and reproducible when plasma collected during shock is exposed to naive donor leukocytes.
4.Mechanisms leading to leukocyte activation
Healthy animals have a low levels of cell activation. In contrast less than an hour after central blood pressure reduction elevated levels of cell activation are detected. This can be see on leukocytes from unseparated venous blood, but is also observed when plasma of animals collected during the shock period is separated and applied to naive donor control leukocytes [38] indicating that a factor in plasma is involved in the activation. An understanding of the mechanism(s) that causes this activation seemed to us essential to identify the source for microvascular defects in hemorrhagic shock.
Our group and others searched over several years for specific biochemical inflammatory mediators(s) that may be present in the plasma and responsible for activation of leukocytes. Our initial hypothesis followed traditional thinking that there is a mediator in the plasma (e.g. endotoxins, complement components, reactive oxygen species, lipid mediators) that may be responsible for the activation [39]. Some of these mediators may not only serve to activate but may also be cytotoxic by themselves or as a mixture of different mediators [40,41]. We and others found mediators in the plasma (e.g. complement components, fibronectin fragments). But an important control experiment by removal of the mediators from plasma (e.g. by immunoaffinity columns) or blockade of their signaling pathways (e.g. by receptor blockade) did not eliminate the ability of the shock plasma to activate leukocytes, undermining the hypothesis that single biochemical mediator(s) were causing the activation. We were not able to abolish activation of naive cell exposed to plasma collected during hemorrhagic shock. The specific mechanism(s) by which plasma may produce cell activation remained uncertain. After years of attempts to identify the inflammatory mediators with classical biochemical approaches [39] we recognized the need for a new approach to this fundamental problem in the pathogenesis of hemorrhagic shock.
5.Cell activation in hemorrhagic shock
Even relatively short periods of hemorrhagic shock (∼40 minutes; [38]) can lead to elevated levels of leukocyte activation in the circulation. This evidence suggests that little de-novo gene expression and protein biosynthesis is required to generate the mediator in shock plasma. Since hemorrhagic shock is induced merely by central blood pressure reduction and no biochemical agents are administered to produce organ failure (as compared with endotoxic shock, in which endotoxin needs to be introduced into the circulation), cell activation is caused by a mechanism that involves biomolecules already present in a normal animal before shock.
What could be the source? We obtained an important lead by closer examination of different organs.
6.Pancreatic digestive enzymes
In hemorrhagic shock with central blood pressure reduction all organs are temporarily hypo-perfused with reduced oxygen delivery. Thus any organ, tissue or cell may become a source of mediators that causes the leukocyte activation. But the particular organ(s) that may be involved were unknown. This opportunity lead to a systematic search among organs and served to identify the pancreas as a major source for mediator of leukocyte activation in shock [42,43]. In addition to the pancreas, the intestine also served as a major source, but only if it contains digestive enzymes. If digestive enzymes are rinsed out of the lumen of the small intestine, the wall of the intestine by itself produces low leukocyte activation, comparable to other organs. This evidence led us to the hypothesis that digestive enzymes synthesized in the pancreas and discharged into the small intestine, as part of daily digestion, may in fact be a source for mediators of the leukocyte activation.
We demonstrated that pancreatic digestive enzymes, including trypsin, are able to produce powerful activating mediators not only for leukocytes but also for other cell types in the microcirculation and surrounding tissues [44]. These mediators may be hydrophilic (e.g. unbound free fatty acids) or hydrophobic (e.g. proteolytically generated peptides) and produce a pathophysiologic response sufficient to cause lethal shock [45–47].
The evidence points directly at a central role for the digestive enzymes in multi-organ failure. They are largely synthesized in the pancreas and discharged into the duodenum at the proximal segment of the small intestine as part of daily digestion where they are fully activated, relatively non-specific and concentrated. What had started as a narrow search for mediators of leukocyte activation led to a mechanism with a much broader spectrum of activities, in line with the many pathophysiological events in shock.
7.Escape of digestive enzymes across the mucosal barrier
While normally contained in the lumen of the small intestine, in shock the pancreatic digestive enzymes escape across the mucosal barrier into the wall of the intestine and from there into the systemic circulation [48,49]. The mucosal barrier consists mainly of the intestinal epithelium with interdispersed goblet cells coated with a continuous mucin layer. The mucin layer forms a barrier for the digestive enzymes, like trypsin, chymotrypsin or elastase with molecular weights in the range between 20K and 30K Dalton. Escape of digestive enzymes out of the lumen of the intestine due to an enhanced mucosal barrier permeability [48] leads to their passage into the interstitial tissue (lamina propria) of the intestinal villi. Zymography shows digestive enzyme activity inside the villi and even in the muscularis and outer serosa layers.
In the intestinal wall digestive enzymes gain access to the venous system of the intestine, the intestinal lymphatics, and to the peritoneum. Thus all possible anatomical pathways serve digestive enzymes to escape from the lumen of the intestine into the systemic circulation [49]. Digestive enzymes utilize mostly convective transport during escape from the lumen of the intestine into the central circulation, transport by diffusion is relatively inefficient considering their molecular weight.
8.Autodigestion
During leakage from the intestinal lumen active digestive enzymes cause major morphological damage to the mucosal barrier [48,50,51]. The tissue degradation starts at the tip of villi (Fig. 2), a tissue region with frequent apoptosis, and may progress until the entire villi structures are degraded [50,52] including blood vessels and lymphatics in their interior. The intestinal barrier may become degraded, exposing autologous tissue without protection from the undiluted and high concentrations of the digestive enzymes. There can be a catastrophic digestion of the intestine.
Fig. 2.
Example of small intestine histological section before (top image) and after hemorrhagic shock (with 35 mmHg mean arterial pressure for 2 hours) (bottom). Note the extensive damage to the tip of the intestinal villi (arrows), a feature that is due to entry of digestive enzymes into the wall of the intestine and causes extensive autodigestion of intestinal tissue, actual loss of epithelium and connective tissue in the villi and swelling of the muscularis layer (small arrow). The autodigestion of tissue is minimized if digestive enzymes in the lumen of the small intestine are inhibited. The autodigestion of intestinal tissue is present in many forms of shock in addition to hemorrhagic shock [51]. Length of bars is 100 μm.
![Example of small intestine histological section before (top image) and after hemorrhagic shock (with 35 mmHg mean arterial pressure for 2 hours) (bottom). Note the extensive damage to the tip of the intestinal villi (arrows), a feature that is due to entry of digestive enzymes into the wall of the intestine and causes extensive autodigestion of intestinal tissue, actual loss of epithelium and connective tissue in the villi and swelling of the muscularis layer (small arrow). The autodigestion of tissue is minimized if digestive enzymes in the lumen of the small intestine are inhibited. The autodigestion of intestinal tissue is present in many forms of shock in addition to hemorrhagic shock [51]. Length of bars is 100 μm.](https://content.iospress.com:443/media/bir/2016/53-5-6/bir-53-5-6-bir17131/bir-53-bir17131-g002.jpg)
9.Proteolytic receptor cleavage and cell dysfunction
Escape of digestive proteases, like trypsin, out of the small intestine leads to activation of other enzymes, e.g. matrix-metallo proteinases (MMPs). In the presence of an ischemic intestine the proMMP-9 of neutrophils is activated into active MMP-9 [53] in many organs, e.g. liver, lung, heart and brain [54].
Another consequence of the escape of pancreatic digestive enzymes out of the lumen of the intestine is that they cleave membrane receptors. Serine proteases, like trypsin or chymotrypsin, are initially extracellular and active to cleave any protein; this includes the ability to clip the extracellular domain of membrane receptors. We have shown in-vitro that extracellular digestive enzymes impair the ability of endothelial cells to align in the direction of fluid shear and they cleave the glycocalyx and membrane receptors (e.g. VEGR-2 or the insulin receptor) [55].
Entry of digestive proteases into the systemic circulation leads to cleavage of the extracellular domain of the insulin receptor and consequently loss of the ability to bind insulin and signal glucose transport. The consequence is hyperglycemia with attenuated response to an insulin bolus, i.e. acute insulin resistance. Blockade of the digestive proteases in the lumen of the small intestine prevents elevation of the digestive enzyme activity in the systemic circulation, and abolishes the insulin receptor cleavage and with it indicators for insulin resistance [56]. Insulin resistance in a chronic model is also inhibited by prolonged protease inhibition [57].
The ability of the digestive enzymes to directly or indirectly (via activation of MMPs) compromise cell dysfunctions by proteolytic destruction of membrane receptors appears to be a universal phenomenon. It may underlie many pathogenic mechanisms in multi-organ dysfunction and failure, including rheological dysfunctions. For example, the fluid shear stress response of leukocytes with retraction of pseudopods is compromised by MMPs due to cleavage of the FMLP-receptor (FPR) [58]. The glycocalyx on red cells [59] and on the endothelium can be cleaved by MMPs [60–62]. We detected cleavage of the inter-epithelial adhesion receptor E-cadherin in the intestine, occludin, VE-cadherin, and VEGFR2 in the lung after hemorrhagic shock [63]. The toll-like receptor in the intestine is cleaved during ischemia (TLR4) [48]. There seem to be a large number of membrane receptors whose extracellular domains may be clipped by MMPs, digestive proteases and others.
10.Blockade of digestive enzymes in the lumen of the small intestine
Enteral blockade of the digestive enzymes in the lumen of the intestine during shock has many beneficial effects in addition to prevention of rheological dysfunction or the acute insulin resistance. It serves to preserve organ morphology, attenuate the cell damage in the small intestine and its villi structures [51,64,65]. It reduces hemorrhagic lesion formation in the microvasculature with escape of whole blood into the surrounding tissue (e.g. in the lung, heart muscle), attenuates damage to the cardiac muscle and serves to maintain systemic blood pressure in different forms of shock. Enteral blockade of digestive enzymes decreases long-term mortality rates and enhances the recovery rate of body weight and intestinal function in relatively severe forms of hemorrhagic and septic shock [51]. The broad protection against organ failure and mortality is provided by different types of molecules used to block digestive proteases.
Further insight into the critical role of digestive enzymes for the outcome of septic shock is illustrated by enteral digestive protease inhibition in the case of gram-negative endotoxin shock. These experiments support the idea that endotoxin per se is not solely responsible for organ failure and instead utilizes another pathway to facilitate organ failure. Intravenous endotoxin administration causes elevation of the permeability in the endothelium and epithelium. The elevated permeability of the mucosal barrier allows escape of the digestive enzymes out of the lumen of the small intestine into the central circulation. The subsequent organ failure and death is then mediated by digestive proteases, in line with the evidence that their enteral blockade significantly attenuates organ failure in endotoxic shock [65]. This interesting observation suggests the hypothesis that infectious agents (and not just endotoxins) induce a breakdown of the mucosal barrier which then allows the leakage of the digestive enzymes out of the lumen of the small intestine. Thus the pathogenic effects of infectious agents may require an involvement of digestive enzymes. This idea needs to be tested in the future and may open new treatment opportunities for infections.
Formal experiments to test enteral blockade of digestive enzymes as a clinical intervention remain to be carried out. In 2010 enteral blockade of the pancreatic digestive proteases was successfully applied the first time in a septic patient. As noted in this report [66] enteral infusion of a trypsin inhibitor (gabexate Mesilate) resulted in a rapid reversal of the indicators of a systemic inflammatory response and the sequelae of septic shock within 72 hours.
11.What mechanisms may open the mucosal barrier?
These results support the hypothesis that escape of digestive enzymes across the mucosal barrier in the intestine is a problem that different forms of shock may have in common. Any breach of the mucosal barrier allows these enzymes to escape with potentially deadly consequences [67].
The question then arises, what mechanism may open the mucosal barrier? The answer to this important question likely depends on specific circumstances and the type of inflammatory mediators present. The barrier can be opened by reduced perfusion and reduction of ATP levels – be it by local vascular occlusions in the intestine or by central blood pressure reduction, such as splanchnic artery occlusion or hemorrhagic shock, respectively. The barrier can also be opened without ischemia in the presence of inflammatory mediators, such as endotoxins and many others, that target inflammatory pathways in the epithelium or epithelial cell apoptosis. Furthermore, the mucosal barrier may also be opened by cytotoxic mediators that are generated during digestion of food (e.g. unbound free fatty acids) and that are small enough to breach the mucosal barrier to cause death in epithelial cells. Free fatty acids are derived from breakdown of triglycerides by pancreatic digestive lipase. If free fatty acids remain unbound (as compared to micelle formation) they are cytotoxic since they dissolve in lipid bilayers and destroy them [68]. It is interesting to note in this regard that removal of food and partially digested food from the lumen of the intestine reduces injury to the intestinal wall in hemorrhagic shock [63].
During normal digestion, the transport of digestive enzymes across the mucosal barrier is to a large degree blocked by the mucin layer and the epithelium [48]. In a normal intestine dextran at molecular weight of 20 kDa, comparable to the molecular weight of pancreatic trypsin, only minimally passes across the mucin layer [50]. This observation is also supported by direct protease measurements in the intestinal wall of control animals suggesting that the digestive enzymes per se are normally kept to a large degree by the mucin barrier in the lumen of the intestine [48]. Mucin, as a highly hydrated molecule, seems to be optimized to prevent its cleavage by digestive enzymes in the lumen of the intestine.
This evidence indicates that a separate mechanism in the mucin/epithelial layers is involved with elevation of the mucosal barrier permeability for molecules the size of digestive enzymes. The mucin layer and epithelium of the mucosal barrier require metabolic and signaling processes to maintain a state of low permeability to digestive enzymes; failure of any one of them serves to open pathways for transport across the mucin and the epithelial cells into the underlying connective tissue in the intestinal villi (e.g. the lamina propria). They include metabolic support for ATP production, intact cell adhesion mechanisms (e.g. adherence and tight junctions by E-cadherins, occludin, claudin, JAM, desmosomes), synthesis of mucin and integrin attachment of epithelial cells to the basal lamina and the lamina propria.
Failure of the mucin layer provides trypsin and the other digestive enzymes access to intercellular junctions and cleavage of the extracellular domain of junction proteins and thereby starts the process that leads to breakdown of the mucosal barrier. Digestive enzymes destroy the epithelial lining and its tight junction proteins [69]. Such autodigestion is a catastrophic event since the mucosal barrier is then breached so that all wall structures of the intestine are exposed without a barrier to the digestive enzymes in the lumen [52].
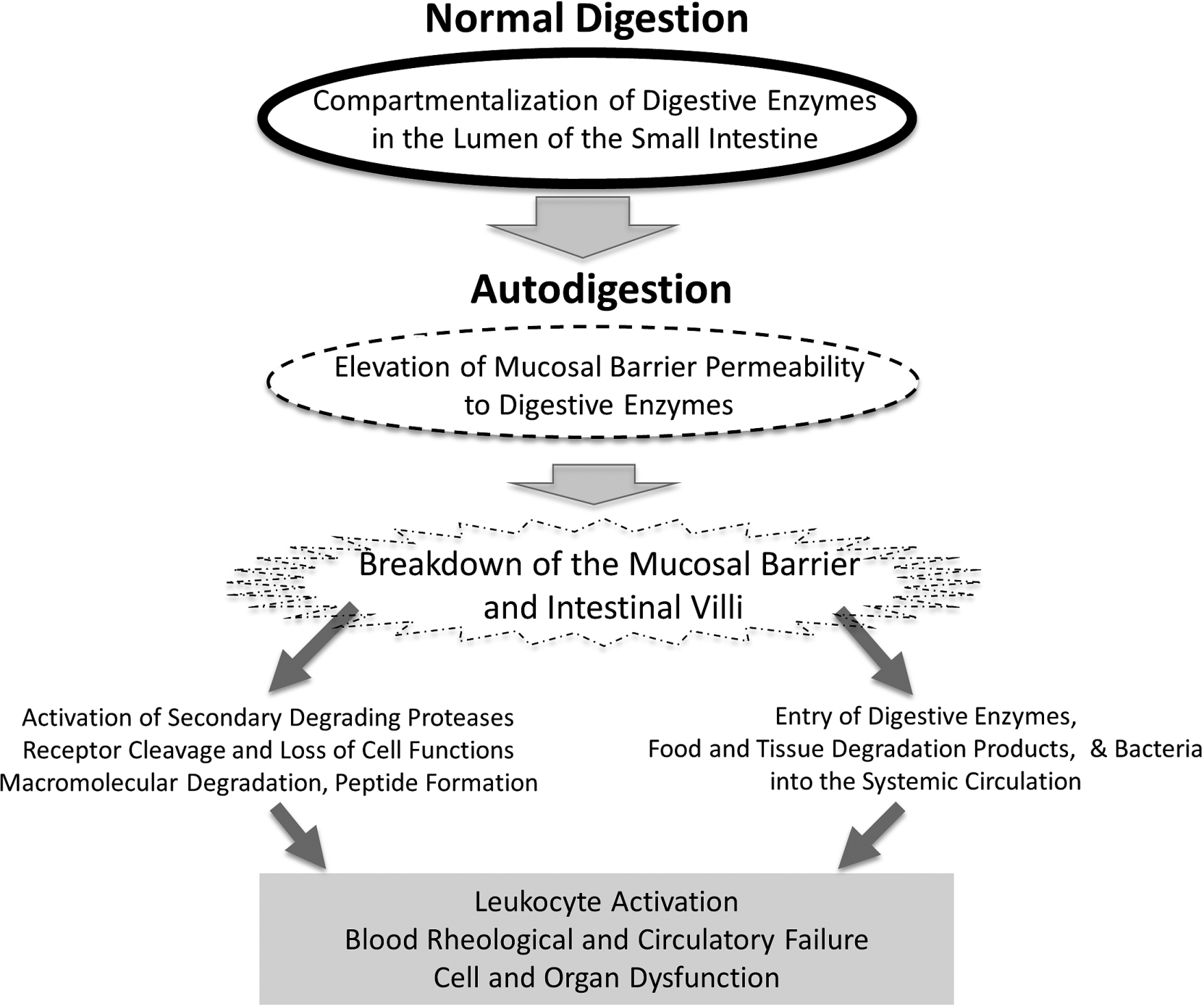
Fig. 3.
Schematic diagram of key events during the transition from normal digestion into autodigestion and its extensive consequences in the central circulation, including leukocyte activation, blood rheological changes to the point of full organ failure.
12.Conclusion
What started decades ago as a question about the mechanisms for leukocyte activation in plasma has led to a surprising new focus: digestive enzymes. Synthesized in the pancreas and carried in the small intestine as part of normal daily digestion, these powerful enzymes may escape across the mucosal barrier and play a central role in cell and organ dysfunction. In the systemic circulation they may degrade membrane components, which in turn triggers diverse cell dysfunctions. For example, cleavage of the ectodomain of membrane receptors on leukocytes impairs their response to fluid shear stress or adhesion to the endothelium, and cleavage of the insulin receptor leads to insulin resistance. Cleavage of the glycocalyx influences red cell aggregation and other biorheological properties of blood. In physiological forms of shock we now have evidence to indicate that besides cell dysfunctions digestive enzymes can cause organ failure and death (Fig. 3). It is interesting to think about the possibility that digestive enzymes not only digest food but may also autodigest one’s own tissues. If this idea is confirmed, we may have gained fundamental new insight into disease and death.
Acknowledgements
I thank Dr. Erik Kistler, Marisol Chang, Angelina Altshuler, Alex Penn, Rafi Mazor, Mr. Frank A. DeLano and Marco Santamaria-Aviles for many discussions regarding the autodigestion hypothesis. Supported by NIH Grant GM 85072 and by an unrestricted gift from Leading Biosciences Inc.
Conflict of interest
Dr. Schmid-Schönbein owns equity in InflammaGen, a company by Leading Biosciences Inc., which develops therapy for shock patients.
References
[1] | Schmid-Schönbein GW, Shih YY, Chien S. Morphometry of human leukocytes. Blood. (1980) ;56: (5):866–875. |
[2] | Schmid-Schönbein GW, Jan J-M, Skalak R, Chien S. Deformation of leukocytes on a hematological blood film. Biorheology. (1984) ;21: :767–781. |
[3] | Schmid-Schönbein GW, Sung K-LP, Tözeren H, Chien S, Skalak R. Passive mechanical properties of human leukocytes. Biophys J. (1981) ;36: :243–256. doi:10.1016/S0006-3495(81)84726-1. |
[4] | Dong C, Skalak R, Sung K-LP, Schmid-Schönbein GW, Chien S. Passive deformation analysis of human leukocytes. J Biomech Eng. (1988) ;110: :27–36. doi:10.1115/1.3108402. |
[5] | Warnke KC, Skalak TC. In vivo measurement of leukocyte viscosity during capillary plugging. J Biomech Eng. (1992) ;114: (4):533–538. doi:10.1115/1.2894107. |
[6] | Konstantopoulos K, Kukreti S, McIntire LV. Biomechanics of cell interactions in shear fields. Adv Drug Deliv Rev. (1998) ;33: (1–2):141–164. doi:10.1016/S0169-409X(98)00024-6. |
[7] | Schmid-Schönbein GW, Skalak R, Sung K-LP, Chien S. Human leukocytes in the active state. In: Bagge U, Born GVR, Gaehtgens P, editors. White Blood Cells, Morphology and Rheology as Related to Function. The Hague: Martinus Nijhoff; (1982) . p. 21–31. |
[8] | Simon SI, Green CE. Molecular mechanics and dynamics of leukocyte recruitment during inflammation. Annu Rev Biomed Eng. (2005) ;7: :151–185. doi:10.1146/annurev.bioeng.7.060804.100423. |
[9] | Luo BH, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. (2007) ;25: :619–647. doi:10.1146/annurev.immunol.25.022106.141618. |
[10] | Shin HY, Simon SI, Schmid-Schönbein GW. Fluid shear-induced activation and cleavage of CD18 during pseudopod retraction by human neutrophils. J Cell Physiol. (2008) Feb;214: (2):528–536. |
[11] | Lawrence MB, Springer TA. Leukocytes roll on a selectin at physiologic flow rates: Distinction from and prerequisite for adhesion through integrins. Cell. (1991) ;65: (5):859–873. doi:10.1016/0092-8674(91)90393-D. |
[12] | Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) ;7: (9):678–689. doi:10.1038/nri2156. |
[13] | Thompson TN, La Celle PL, Cokelet GR. Perturbation of red cell flow in small tubes by white blood cells. Pflügers Arch. (1989) ;413: :372–377. doi:10.1007/BF00584486. |
[14] | Helmke BP, Sugihara-Seki M, Skalak R, Schmid-Schönbein GW. A mechanism for erythrocyte-mediated elevation of apparent viscosity by leukocytes in vivo without adhesion to the endothelium. Biorheology. (1998) ;35: (6):437–448. doi:10.1016/S0006-355X(99)80021-3. |
[15] | Mayrovitz HN. The relationship between leukocytes and erythrocytes velocity in arterioles. In: Bagge U, Born GVR, Gaehtgens P, editors. White Blood Cells, Morphology and Rheology as Related to Function. The Hague: Martinus Nijhoff; (1982) . |
[16] | Schmid-Schönbein GW, Usami S, Skalak R, Chien S. The interaction of leukocytes and erythrocytes in capillary and postcapillary vessels. Microvasc Res. (1980) ;19: (1):45–70. doi:10.1016/0026-2862(80)90083-7. |
[17] | Sinclair SH, Azar-Cavanagh M, Soper KA, Tuma RF, Mayrovitz HN. Investigation of the source of the blue field entoptic phenomenon. Invest Ophthalmol Vis Sci. (1989) ;30: (4):668–673. |
[18] | Del Zoppo GJ, Schmid-Schönbein GW, Mori E, Copeland BR, Chang C-M. Polymorphonuclear leukocytes occlude capillaries following middle cerebral artery occlusion and reperfusion. Stroke. (1991) ;22: :1276–1283. doi:10.1161/01.STR.22.10.1276. |
[19] | Bagge U, Amundson B, Lauritzen C. White blood cell deformability and plugging of skeletal muscle capillaries in hemorrhagic shock. Acta Physiologica Scandinavica. (1980) ;108: (2):159–163. doi:10.1111/j.1748-1716.1980.tb06513.x. |
[20] | Engler RL, Schmid-Schönbein GW, Pavelec RS. Leukocyte capillary plugging in myocardial ischemia and reperfusion in the dog. Am J Pathol. (1983) ;111: :98–111. |
[21] | Barroso-Aranda J, Schmid-Schönbein GW, Zweifach BW, Engler RL. Granulocytes and the no-reflow phenomenon in irreversible hemorrhagic shock. Circ Res. (1988) ;63: :437–447. doi:10.1161/01.RES.63.2.437. |
[22] | Lee J, Schmid-Schönbein GW. Biomechanics of skeletal muscle capillaries: Hemodynamic resistance, endothelial distensibility, and pseudopod formation. Ann Biomed Eng. (1995) ;23: (3):226–246. doi:10.1007/BF02584425. |
[23] | Tran ED, Schmid-Schönbein GW. An in-vivo analysis of capillary stasis and endothelial apoptosis in a model of hypertension. Microcirculation. (2007) ;14: :1–12. doi:10.1080/10739680601072099. |
[24] | Helmke BP, Bremner SN, Zweifach BW, Skalak R, Schmid-Schönbein GW. Mechanisms for increased blood flow resistance due to leukocytes. Am J Physiol. (1997) ;273: (6 Pt 2):H2884-90. |
[25] | Schmid-Schönbein GW, Engler RL. Perspectives of leukocyte activation in the microcirculation. Biorheology. (1990) ;27: (6):859–869. |
[26] | Chang RR, Chien NT, Chen CH, Jan KM, Schmid-Schönbein GW, Chien S. Spontaneous activation of circulating granulocytes in patients with acute myocardial and cerebral diseases. Biorheology. (1992) ;29: (5–6):549–561. |
[27] | Pitzer JE, Del Zoppo GJ, Schmid-Schönbein GW. Neutrophil activation in smokers. Biorheology. (1996) ;33: (1):45–58. doi:10.1016/0006-355X(96)00004-2. |
[28] | Schmid-Schönbein GW, Kistler EB, Hugli TE. Mechanisms for cell activation and its consequences for biorheology and microcirculation: Multi-organ failure in shock. Biorheology. (2001) ;38: (2–3):185–201. |
[29] | Mazzoni MC, Schmid-Schönbein GW. Mechanisms and consequences of cell activation in the microcirculation. Cardiovasc Res. (1996) ;32: (4):709–719. doi:10.1016/S0008-6363(96)00146-0. |
[30] | Moazzam F, DeLano FA, Zweifach BW, Schmid-Schönbein GW. The leukocyte response to fluid stress. Proceedings of the National Academy of Sciences of the United States of America. (1997) ;94: (10):5338–5343. doi:10.1073/pnas.94.10.5338. |
[31] | Makino A, Prossnitz ER, Bünemann, Wang JM, Yao W, Schmid-Schönbein GW. G protein-coupled receptors serve as mechanosensors for fluid shear stress in neutrophils. Am J Physiol Cell Physiol. (2006) ;290: :C1633–C1639. doi:10.1152/ajpcell.00576.2005. |
[32] | Komai Y, Schmid-Schönbein GW. De-activation of neutrophils in suspension by fluid shear stress: A requirement for erythrocytes. Ann Biomed Eng. (2005) ;33: (10):1375–1386. doi:10.1007/s10439-005-6768-6. |
[33] | Fukuda S, Schmid-Schönbein GW. Regulation of CD18 expression on neutrophils in response to fluid shear stress. Proc Natl Acad Sci USA. (2003) ;100: (23):13152–13157. doi:10.1073/pnas.2336130100. |
[34] | Fukuda S, Yasu T, Kobayashi N, Ikeda N, Schmid-Schönbein GW. Contribution of fluid shear response in leukocytes to hemodynamic resistance in the spontaneously hypertensive rat. Circ Res. (2004) ;95: (1):100–108. doi:10.1161/01.RES.0000133677.77465.38. |
[35] | Pitzer JE, Del Zoppo GJ, Schmid-Schönbein GW. Neutrophil activation in smokers. Biorheology. (1996) ;33: :45–58. doi:10.1016/0006-355X(96)00004-2. |
[36] | Edwards KM, Sheu B, Hong S, Penn AH, Schmid-Schönbein GW, Mills PJ. Leukocyte membrane bleb and pseudopod formation in hypertension. J Hum Hypertens. (2010) ;24: (10):684–686. |
[37] | Barroso-Aranda J, Schmid-Schönbein GW. Transformation of neutrophils as indicator of irreversibility in hemorrhagic shock. Am J Physiol. (1989) ;257: :H846–H852. |
[38] | Barroso-Aranda J, Zweifach BW, Mathison JC, Schmid-Schönbein GW. Neutrophil activation, tumor necrosis factor, and survival after endotoxic and hemorrhagic shock. Journal of Cardiovascular Pharmacology. (1995) ;25: (Suppl 2(7)):S23–S29. doi:10.1097/00005344-199500252-00006. |
[39] | Schmid-Schönbein GW. A journey with Tony Hugli up the inflammatory cascade towards the auto-digestion hypothesis. Int Immunopharmacol. (2007) ;7: (14):1845–1851. doi:10.1016/j.intimp.2007.07.015. |
[40] | Ginsburg I, Kohen R. Cell damage in inflammatory and infectious sites might involve a coordinated “cross-talk” among oxidants, microbial haemolysins and ampiphiles, cationic proteins, phospholipases, fatty acids, proteinases and cytokines (an overview). Free Radic Res. (1995) ;22: (6):489–517. doi:10.3109/10715769509150323. |
[41] | Ginsburg I, Misgav R, Pinson A, Varani J, Ward PA, Kohen R. Synergism among oxidants, proteinases, phospholipases, microbial hemolysins, cationic proteins, and cytokines. Inflammation. (1992) ;16: (5):519–538. doi:10.1007/BF00918977. |
[42] | Kistler EB, Hugli TE, Schmid-Schönbein GW. The pancreas as a source of cardiovascular cell activating factors. Microcirculation. (2000) ;7: (3):183–192. doi:10.1111/j.1549-8719.2000.tb00119.x. |
[43] | Waldo SW, Rosario HS, Penn AH, Schmid-Schönbein GW. Pancreatic digestive enzymes are potent generators of mediators for leukocyte activation and mortality. Shock. (2003) ;20: (2):138–143. doi:10.1097/01.shk.0000073866.47824.ae. |
[44] | Kistler EB, Lefer AM, Hugli TE, Schmid-Schönbein GW. Plasma activation during splanchnic arterial occlusion shock. Shock. (2000) ;14: (1):30–34. doi:10.1097/00024382-200014010-00006. |
[45] | Kramp WJ, Waldo S, Schmid-Schönbein GW, Hoyt D, Coimbra R, Hugli TE. Characterization of two classes of pancreatic shock factors: Functional differences exhibited by hydrophilic and hydrophobic shock factors. Shock. (2003) ;20: (4):356–362. doi:10.1097/01.shk.0000082442.66379.90. |
[46] | Penn AH, Hugli TE, Schmid-Schönbein GW. Pancreatic enzymes generate cytotoxic mediators in the intestine. Shock. (2007) ;27: (3):296–304. doi:10.1097/01.shk.0000235139.20775.7f. |
[47] | Alsaigh T, Chang M, Richter M, Mazor R, Kistler EB. In vivo analysis of intestinal permeability following hemorrhagic shock. World J Crit Care Med. (2015) ;4: (4):287–295. doi:10.5492/wjccm.v4.i4.287. |
[48] | Chang M, Alsaigh T, Kistler EB, Schmid-Schönbein GW. Breakdown of mucin as barrier to digestive enzymes in the ischemic rat small intestine. PLoS One. (2012) ;7: (6):e40087. |
[49] | Altshuler AE, Penn AH, Yang JA, Kim GR, Schmid-Schönbein GW. Protease activity increases in plasma, peritoneal fluid, and vital organs after hemorrhagic shock in rats. PLoS One. (2012) ;7: (3):e32672. |
[50] | Chang M, Kistler EB, Schmid-Schönbein GW. Disruption of the mucosal barrier during gut ischemia allows entry of digestive enzymes into the intestinal wall. Shock. (2012) ;37: (3):297–305. doi:10.1097/SHK.0b013e318240b59b. |
[51] | DeLano FA, Hoyt DB, Schmid-Schönbein GW. Pancreatic digestive enzyme blockade in the intestine increases survival after experimental shock. Sci Transl Med. (2013) ;5: (169):169ra11. doi:10.1126/scitranslmed.3005046. |
[52] | Fitzal F, DeLano FA, Young C, Schmid-Schönbein GW. Improvement in early symptoms of shock by delayed intestinal protease inhibition. Arch Surg. (2004) ;139: (9):1008–1016. doi:10.1001/archsurg.139.9.1008. |
[53] | Rosario HS, Waldo SW, Becker SA, Schmid-Schönbein GW. Pancreatic trypsin increases matrix metalloproteinase-9 accumulation and activation during acute intestinal ischemia-reperfusion in the rat. Am J Pathol. (2004) ;164: (5):1707–1716. doi:10.1016/S0002-9440(10)63729-7. |
[54] | Altshuler AE, Penn AH, Yang JA, Kim GR, Schmid-Schönbein GW. Protease activity increases in plasma, peritoneal fluid, and vital organs after hemorrhagic shock in rats. PLoS One. (2012) ;7: (3):e32672. |
[55] | Altshuler AE, Morgan MJ, Chien S, Schmid-Schönbein GW. Proteolytic activity attenuates the response of endothelial cells to fluid shear stress. Cell Mol Bioeng. (2012) ;5: (1):82–91. doi:10.1007/s12195-011-0207-6. |
[56] | DeLano FA, Zhang H, Tran EE, Zhang C, Schmid-Schönbein GW. New hypothesis for insulin resistance in hypertension due to receptor cleavage. Expert Review of Endocrinology and Metabolism. (2010) ;5: (1):149–158. doi:10.1586/eem.09.64. |
[57] | Delano FA, Schmid-Schönbein GW. Proteinase activity and receptor cleavage. Mechanism for insulin resistance in the spontaneously hypertensive rat. Hypertension. (2008) ;52: (2):415–423. |
[58] | Chen AY, DeLano FA, Valdez SR, Ha JN, Shin HY, Schmid-Schönbein GW. Receptor cleavage reduces the fluid shear response in neutrophils of the spontaneously hypertensive rat. Am J Physiol Cell Physiol. (2010) ;299: (6):C1441–C1449. doi:10.1152/ajpcell.00157.2010. |
[59] | Pot C, Chen AY, Ha JN, Schmid-Schönbein GW. Proteolytic cleavage of the red blood cell glycocalyx in a genetic form of hypertension. J Cellular and Molecular Bioengineering. (2011) ;4: :678–692. doi:10.1007/s12195-011-0180-0. |
[60] | Mulivor AW, Lipowsky HH. Role of glycocalyx in leukocyte-endothelial cell adhesion. Am J Physiol Heart Circ Physiol. (2002) ;283: (4):H1282–H1291. doi:10.1152/ajpheart.00117.2002. |
[61] | Mulivor AW, Lipowsky HH. Inflammation- and ischemia-induced shedding of venular glycocalyx. Am J Physiol Heart Circ Physiol. (2004) ;286: (5):H1672–H1680. doi:10.1152/ajpheart.00832.2003. |
[62] | Tuma M, Canestrini S, Alwahab Z, Marshall J. Trauma and endothelial glycocalyx: The microcirculation helmet? Shock. (2016) ;46: (4):352–527. |
[63] | Altshuler AE, Richter MD, Modestino AE, Penn AH, Heller MJ, Schmid-Schönbein GW. Removal of luminal content protects the small intestine during hemorrhagic shock but is not sufficient to prevent lung injury. Physiol Rep. (2013) ;1: (5):e00109. |
[64] | Mitsuoka H, Kistler EB, Schmid-Schönbein GW. Generation of in vivo activating factors in the ischemic intestine by pancreatic enzymes. Proc Natl Acad Sci USA. (2000) ;97: (4):1772–1777. doi:10.1073/pnas.97.4.1772. |
[65] | Fitzal F, Delano FA, Young C, Rosario HS, Junger WG, Schmid-Schönbein GW. Pancreatic enzymes sustain systemic inflammation after an initial endotoxin challenge. Surgery. (2003) ;134: (3):446–456. doi:10.1067/S0039-6060(03)00168-5. |
[66] | Lee YT, Wei J, Chuang YC, Chang CY, Chen IC, Weng CF, Schmid-Schönbein GW. Successful treatment with continuous enteral protease inhibitor in a patient with severe septic shock. Transplant Proc. (2012) ;44: (3):817–819. doi:10.1016/j.transproceed.2012.03.032. |
[67] | Kistler EB, Alsaigh T, Chang M, Schmid-Schönbein GW. Impaired small-bowel barrier integrity in the presence of lumenal pancreatic digestive enzymes leads to circulatory shock. Shock. (2012) ;38: (3):262–267. doi:10.1097/SHK.0b013e31825b1717. |
[68] | Penn AH, Schmid-Schönbein GW. The intestine as source of cytotoxic mediators in shock: Free fatty acids and degradation of lipid-binding proteins. Am J Physiol Heart Circ Physiol. (2008) ;294: (4):H1779–H1792. doi:10.1152/ajpheart.00902.2007. |
[69] | Altshuler AE, Lamadrid I, Li D, Ma SR, Kurre L, Schmid-Schönbein GW, Penn AH. Transmural intestinal wall permeability in severe ischemia after enteral protease inhibition. PLoS One. (2014) ;9: (5):e96655. |


