
Role of fluid shear stress in regulating VWF structure, function and related blood disorders
Abstract
Von Willebrand factor (VWF) is the largest glycoprotein in blood. It plays a crucial role in primary hemostasis via its binding interaction with platelet and endothelial cell surface receptors, other blood proteins and extra-cellular matrix components. This protein is found as a series of repeat units that are disulfide bonded to form multimeric structures. Once in blood, the protein multimer distribution is dynamically regulated by fluid shear stress which has two opposing effects: it promotes the aggregation or self-association of multiple VWF units, and it simultaneously reduces multimer size by facilitating the force-dependent cleavage of the protein by various proteases, most notably ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type repeats, motif 1 type 13). In addition to these effects, fluid shear also controls the solution and substrate-immobilized structure of VWF, the nature of contact between blood platelets and substrates, and the biomechanics of the GpIbα–VWF bond. These features together regulate different physiological and pathological processes including normal hemostasis, arterial and venous thrombosis, von Willebrand disease, thrombotic thrombocytopenic purpura and acquired von Willebrand syndrome. This article discusses current knowledge of VWF structure–function relationships with emphasis on the effects of hydrodynamic shear, including rapid methods to estimate the nature and magnitude of these forces in selected conditions. It shows that observations made by many investigators using solution and substrate-based shearing devices can be reconciled upon considering the physical size of VWF and the applied mechanical force in these different geometries.
1.Introduction
Von Willebrand Factor (VWF) is a soluble plasma glycoprotein that is stored in the Weibel–Palade bodies of vascular endothelial cells and α-granules of megakaryocytes/platelets [1]. VWF is secreted into blood both constitutively and in response to a range of thrombotic and inflammatory stimuli [2]. Such stimuli include, but are not limited to, adrenaline, adenosine diphosphate, collagen, fibrin, histamine, thrombin and the vasopressin analog desmopressin (DDAVP). Studies with transgenic mice suggest that VWF constitutively secreted from endothelial cells, but not platelets, likely controls plasma VWF levels since animals lacking endothelial-VWF display negligible plasma VWF levels while those lacking platelet-VWF were normal [3]. Additionally, from a functional standpoint, mice lacking endothelial-, but not platelet-, VWF displayed reduced hemostatic potential and lower potential to correct bleeding defects. In general, the storage compartments of vascular endothelial cells release ultra-large VWF (ULVWF) with molecular mass >10,000 kDa upon stimulation. In blood, an enzyme called ADAMTS13, proteolytically cleaves the Tyr1605–Met1606 scissile bond in the A2-domain of this protein, to generate lower molecular mass VWF multimers in the range of 500–10,000 kDa (1–20 dimer or protomer units) [4,5]. VWF is also proteolytically cleaved by other blood enzymes, notably, plasmin [6], various neutrophil released proteases [7], and possibly also ADAM10 and/or ADAM17 [8].
Fig. 1.
Mature VWF. (A) Mature VWF is a multi-domain protein. It contains the A1, A2 and A3 domains that are flanked by two D-domain assemblies. These D-domains include the VWD, cysteine-8(C8)-fold, trypsin inhibitor-like (TIL) structure and E-modules. Following this, the protein contains six VW C-domains followed by the C-terminal cysteine knot (CTCk). Mature VWF includes 10 O-linked glycans depicted by open circle and 13 N-linked glycans depicted by filled circles. Thirteen percent of the N-glycans carry ABO(H) blood group determinants. VWF has a number of binding partners that are shown below the schematic. The A2-domain is also cleaved by the enzyme ADAMTS13. (B) Approximate representation of VWF based on existing structural knowledge. (C) Though the carbohydrates of VWF are usually represented by small lollipops, their physical dimension can be fairly large, in particular the N-linked glycans. (Colors are visible in the online version of the article; http://dx.doi.org/10.3233/BIR-15061.)
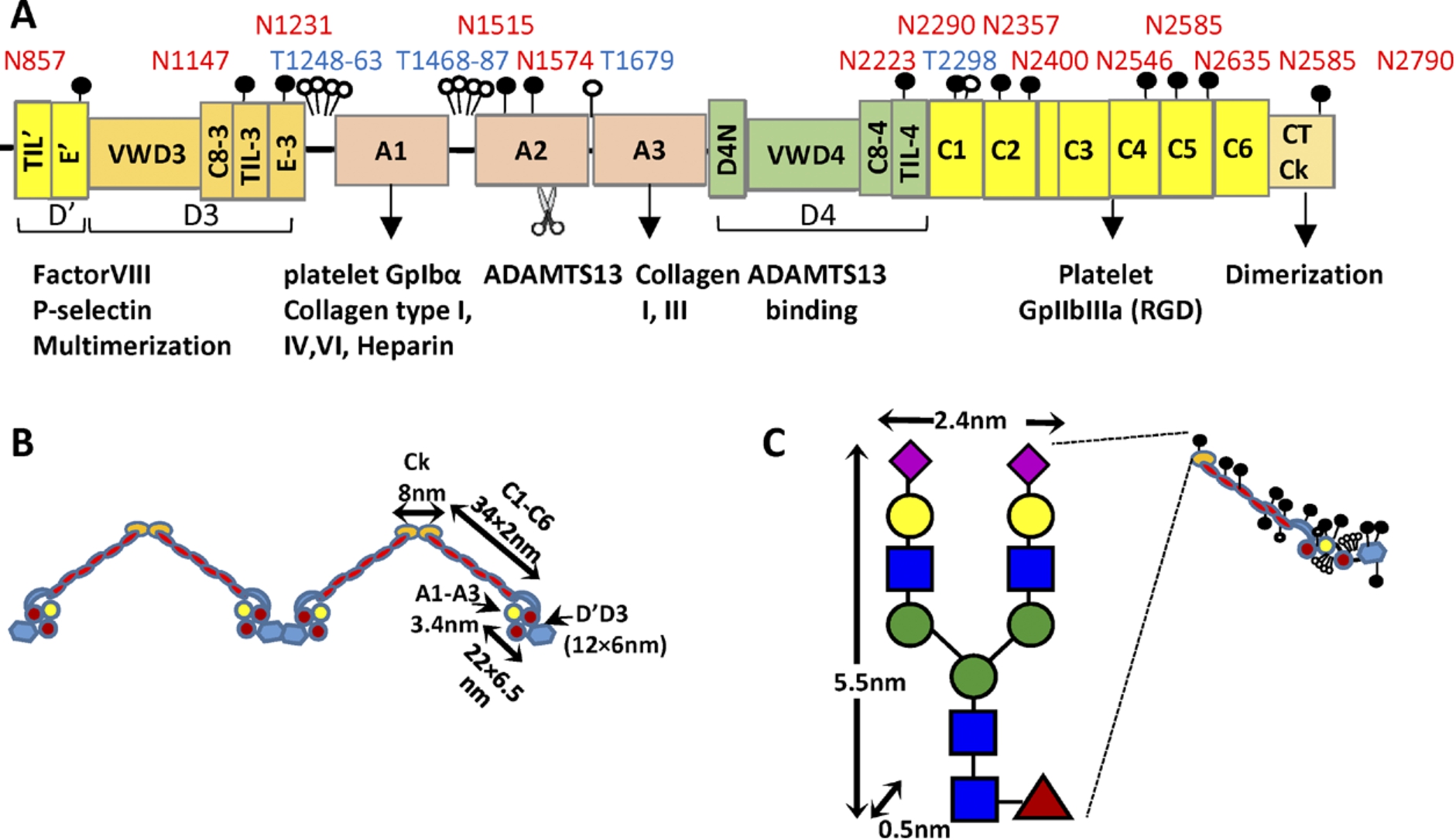
VWF is produced as a single polypeptide that subsequently releases a rather large 761 aa (amino acid) propeptide to form the 2050 aa mature protein (Fig. 1(A)). This mature protein contains multiple domains that have been elucidated based on bioinformatics sequence analysis, crystallography studies and electron microscopy investigations [9,10]. A key element of this protein includes the three VWF-A domains: VWF-A1 which principally recognizes the platelet GpIbα receptor under shear, VWF-A2 which is cleaved by ADAMTS13 to regulate protein size in circulation, and VWF-A3 which is the primary binding partner for collagen on the denuded endothelium. Each of these A-domains adopts a Rossman fold with 6 parallel β-sheets at the center surrounded by a series of α-helices [10]. A disulfide bridge between the N- and C-terminus of the A1- and A3-domains locks each of these domains in a somewhat rigid configuration. The A2-domain is however susceptible to large-scale conformation changes as the disulfide bridge in this domain is located between vicinal cysteines at the C-terminus only [11]. Surrounding the A-domains are the D′D3 and D4 assemblies, which are thought to include different structural elements including the von Willebrand D-domain (VWD), cysteine-8 (C8) -fold, trypsin inhibitor-like (TIL) structure and E-modules [9]. Also included in mature VWF are six VW C-domains and the CTCK (C-terminal cysteine knot) that aids the dimerization of VWF. At low pH (6.2), the entire assembly between CTCK and VWF-A2 is thought to zip up into a dimeric bouquet. This likely provides for molecular compaction so that the large VWF multimer can be efficiently assembled into the Weibel–Palade bodies of endothelial cells [12]. Figure 1(B) presents the overall shape of the mature VWF in a dimeric form including key dimensions used in later hydrodynamic calculations.
Along the protein backbone, VWF is extensively glycosylated with 13 N-linked and 10 O-linked glycans identified to date [13–15]. Among these, the most dominant N-glycans contain
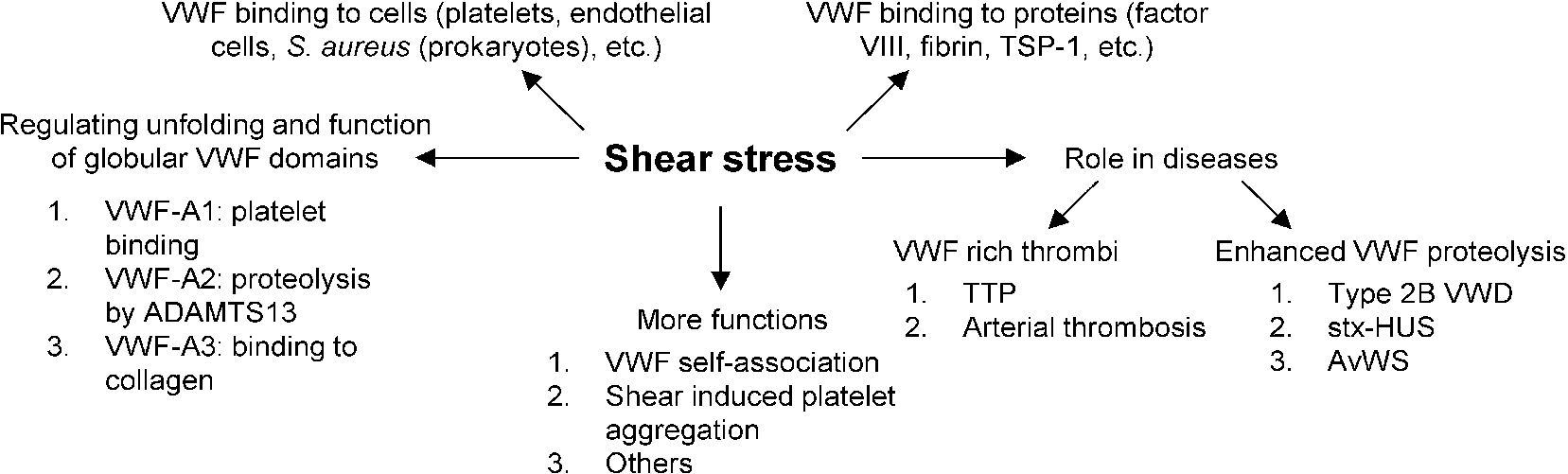
The primary role of VWF is to maintain healthy hemostasis in the vasculature where high shear stress conditions are encountered [1]. It does so by acting as a mediator of platelet–subendothelium interaction, platelet activation and cell aggregation [25]. VWF is also a carrier of clotting Factor VIII and helps prolong its half-life in circulation by protecting it from proteolytic degradation, ultimately delivering it to sites of vascular damage. Since the biological function of VWF is tightly regulated by the applied hydrodynamic stress, the current review examines the relationship between VWF and shear stress, with a focus on VWF-related thrombotic and bleeding disorders. Figure 2 summarizes the different roles that shear stress plays in VWF physiology.
Fig. 2.
Role of shear stress in VWF related biology. Shear stress exerts force on multimeric VWF and causes structural changes in globular A1, A2 and A3 domains, allowing them to carry out their respective functions. Shear stress also regulates the binding of VWF to various plasma proteins and surface receptors on platelets, endothelial cells and even prokaryotic cells in circulation. In a number of different blood disorders, high shear stress either causes enhanced VWF proteolysis (bleeding disorders) or the formation of VWF-rich thrombi (thrombotic disorders). Finally, shear stress is important for VWF and platelet aggregation in physiology. Abbreviations: Shiga-toxin-hemolytic uremic syndrome (stx-HUS), thrombotic thrombocytopenic purpura (TTP), acquired von Willebrand syndrome (AvWS).
2.Hemodynamics in circulation
Table 1
Typical range of wall shear rates and stresses*
| Blood vessel | Mean wall shear rate (/s) | Mean wall shear stress (dyn/cm2)** |
| Large arteries | 300–800 | 13.5–36 |
| Arterioles | 450–1,600 | 20.2–72 |
| Veins | 15–200 | 0.7–9 |
| Stenotic vessels | 800–10,000 | 36–450 |
* Values presented are mean values for indicated vessels. Higher flows can be locally observed and these can also be enhanced during human pathologies
** Shear stress (τ) = viscosity (μ) · shear rate (γ) with blood viscosity = 0.045 poise.
Blood flows through vessels due to a pressure gradient [26]. This results in the application of tangential forces in the direction of flow that have a ‘shearing’ effect. Tensile and circumferential stresses are also applied on the vascular walls which cause vessel distention. At low shear rates below 50–100/s, blood has a non-Newtonian, shear-thinning character with apparent viscosity decreasing upon increasing shear rate. Here, the applied shear stress (τ) versus shear rate (γ) relationship can be described using the Casson equation:
Nevertheless since the shear stress is highest in the arterioles where the effects of VWF are most prominent, under most conditions, it is appropriate to consider blood as a Newtonian fluid when studying VWF biophysics. Additionally, in capillaries and small vessels, the physical dimension of single VWF multimers is far less than the vessel diameter and thus one would expect VWF to respond to the local shear environment rather than vessel dimensions. Experiments where media viscosity have been varied also demonstrate that VWF function is regulated by fluid shear stress which controls the applied mechanical force rather than shear rate which controls protein–protein contact [29,30]. Thus, ideally, different results of VWF structure–function relationship would be better compared upon considering the magnitude of applied shear stress, and even better by considering the nature of the mechanical force applied on the protein or molecular bond in different shearing assays/conditions.
3.Common shearing devices used to study platelet-VWF function
The cone and plate viscometer and the parallel-plate flow chamber represent the two most common fluid shearing devices used to study the response of VWF and platelets to hydrodynamic forces. In this regard, the viscometer is used to shear small volumes of cells/proteins in confined volumes. Since all cells in this device are subjected to identical shear stresses, multi-parametric flow cytometry analysis of withdrawn samples is possible [31–33]. Such a device is ideal when studying VWF structure–function relationships in solution [29,31,34], for example in the context of analyzing the effects of mechanical circulatory support devices on VWF self-association and acquired von Willebrand syndrome (AVWS). The parallel plate flow chamber system, on the other hand, is an open-system where platelet adhesion to immobilized VWF and platelet thrombus formation on substrates bearing collagen can be studied. This flow geometry mimics the geometry of cellular interaction with the vessel wall.
3.1.Cone and plate viscometer
The viscometer is a shearing device containing a rotating cone with angle α and radius R, placed over a stationary plate. Fluid flow is initiated by imparting an angular velocity of Ω (rad/s) to the cone in the azimuthal or ϕ direction. At low angular velocities, the flow is one dimensional with only a rotational component, i.e. only the fluid velocity in the ϕ-direction (w) is non-zero [35,36]. The corresponding flow in the vertical/θ-direction (v) and radial/r-direction (u) are zero. Thus the velocity gradient tensor G at a radial distance of r and angle β is given by:
This is called primary flow. For small cone angles, the single non-zero component can be written as
The cone–plate viscometer is the preferred geometry in many biorheology studies due to the uniformity and simplicity of flow, as opposed to parallel-plate or couette type devices where the applied shear rate varies with position in the sample [37]. Nevertheless, when the angular velocity is high and/or cone angle is large, non-uniform flow occurs due to the higher centrifugal forces which push the fluid radially out near the cone surface. The requirement of continuity then causes a radial inward motion near the plate and this sets up fluid circulation or secondary flow [35,36]. The consequence of secondary flow is that it causes the applied shear rate/stress to vary with position within the viscometer. The average shear stress in this device is however close to the primary flow and time-varying shear stresses have negligible effects on platelet-VWF function likely because these effects are volume-averaged [29].
3.2.Parallel-plate flow cell
Parallel-plate flow chambers are nowadays commonly made using optically transparent polydimethylsiloxane, with a rectangular cross-section using standard photolithography methods. Here, for a microfluidic flow cell with length L (along x-axis), height h (along z axis) and width w (y-axis), such that
When studying the effects of fluid shear in typical flow chambers, in addition to the applied force, the applied shear rate/stress also affects the flux of cells in the microscope’s region of interest depending on cell trajectory, the geometry of the flow inlet and the distance from the inlet [39].
4.Nature of mechanical forces applied on VWF and platelet receptor GpIbα
Cells like platelets and biomolecules like VWF are constantly exposed to hydrodynamic forces in circulation. These forces are important regulators of normal human physiology and pathophysiology. The nature of these fluid forces vary depending on the flow geometry since VWF can interact with platelets both in solution and after it is immobilized on substrates.
In solution (i.e. conditions resembling the cone–plate viscometer geometry). VWF and platelets, free in blood, experience periodic variations in normal forces that attempt to either pull them apart (i.e. tensile forces) or push them together (i.e. compressive forces). These biological entities are also subjected to shear forces that act in a tangential direction [35,40–42]. Such force oscillations rapidly change directions in blood flow with cycles of tensile/compressive/shear forces reoccurring every ∼10–20 ms when the local shear rate is 1000/s, with force-cycle times varying inversely with G. In addition, proteins like free VWF in solution are also subjected to Brownian motion that can affect the nature of force loading [41]. Regardless of the above complexity, it would be helpful to obtain estimates of peak forces applied on various biological entities subjected to flow since this can provide bounds on the magnitude of mechanical stresses applied under a variety of experimental and physiological conditions. Such estimations require simplifying assumptions like the idealization that VWF and platelets behave either as simple spheres or spheres connected by string(s).
Fig. 3.
Similarity of geometry in biological particles of interest: (A)–(D) Biological particles of interest can be approximated as rigid dumbbells composed of two (un)equal spheres linked by a thin tether in many cases. (E)–(F) In other cases, these entities can be modeled as beads or disks attached to a wall by a string.
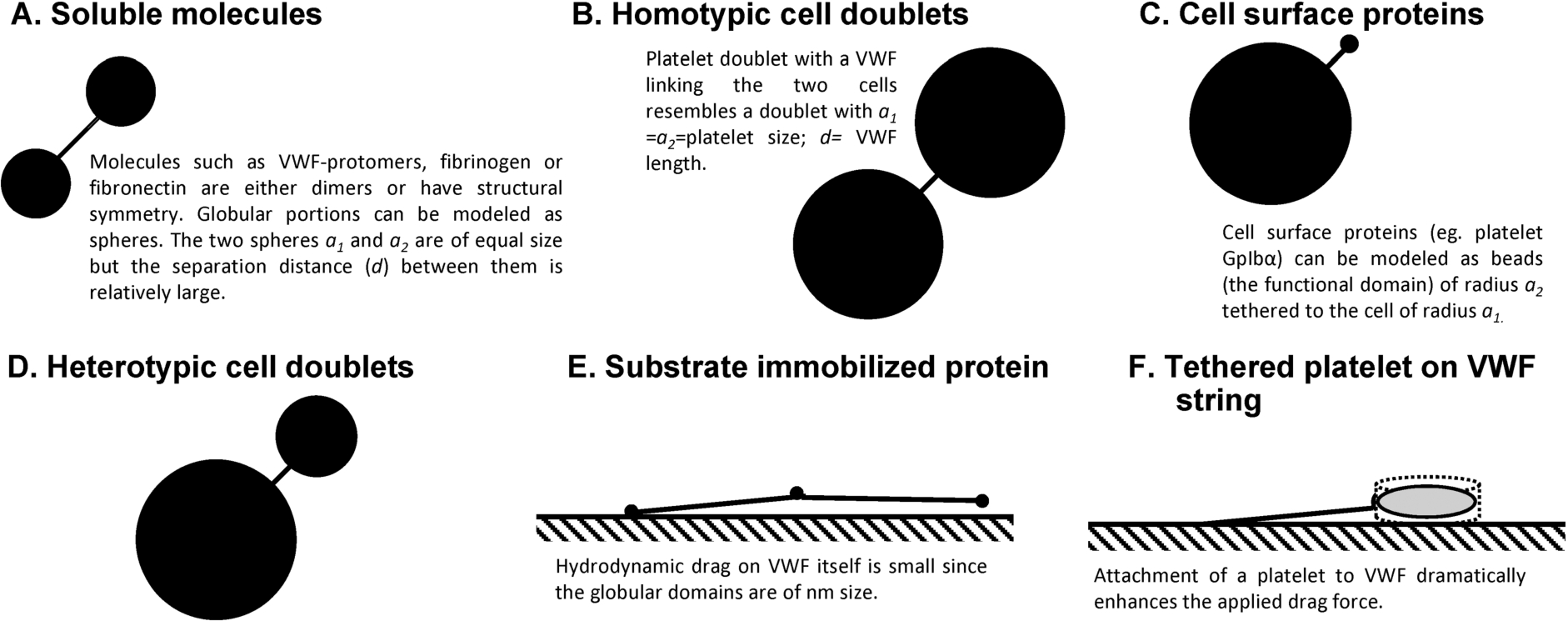
When VWF is subjected to stretching under flow, the maximum force applied is tensile in nature. Shear forces are of smaller magnitude. These tensile forces have a tendency to unfold the protein [41]. If VWF is modeled to behave as a doublet of spheres of radii
Table 2
Typical magnitude of force
| Parameter | VWF protomer | VWF multimer | Platelet doublet | GpIbα on platelet (no VWF) | GpIbα on platelet (with VWF) |
| Dumbbell radius, | 0.013 | – | 1.5 | 1.5 | 1.5 |
| Dumbbell radius, | 0.013 | – | 1.5 | 0.0035 | 0.1 |
| Separation distance, d (µm) | 0.094 | – | 4.03 | 0.0315 | 0.0315 |
| Radius ratio, | 1 | – | 1 | 0.0023 | 0.067 |
| Dimensionless separation distance, | 7.23 | – | 2.68 | 0.02 | 0.02 |
| Force coefficient ( | 60 | – | 20 | 0.005 | 0.35 |
| Peak force, | 0.01 | – | 45 | 0.011 | 0.79 |
| Peak force (F)*(pN) | 0.1–0.2 | 20–40** | 450 | 0.1–0.2 | ∼8 |
* Assumes
** Estimates peak force on fully extended 40-mer VWF with 20 protomer units.
Unlike VWF, the peak force applied on platelet GpIbα would be a ‘shear force’ since the size of the two spheres linked by the tether are highly unequal (i.e.
Immobilized on substrate (i.e. conditions resembling the flow chamber geometry). When immobilized on substrates like the surface of endothelial cells or exposed sub-cellular matrix proteins, the drag force on VWF can be substantial and this can lead to the formation of elongated strings [45] and fiber meshes [46,47]. The drag force applied on a particle subjected to hydrodynamic force is
Resting platelets can be modeled as rigid disks with dimensions, radius
5.VWF conformation change in response to fluid shear stress
A number of studies have determined that VWF can unfold upon the application of fluid shear both in solution and when immobilized on substrates. The studies performed in solution have utilized fluorescence microscopy [50], small-angle neutron scattering (SANS) [51] and fluorescence spectroscopy [52]. The substrate based assays utilized atomic force microscopy (AFM) [53] and noted the formation of long, oriented bundles or meshes of VWF-strings on endothelial cell [45] and collagen substrates [46].
In solution, studies employing single molecule fluorescence microscopy have suggested the sharp, well-defined, large scale extension of VWF with end-to-end lengths up to 10 µm at shear rates >5000/s (shear stress ∼40–50 dyn/cm2) [50]. The structural transitions reported by others in water-glycerol buffers subjected to Poiseuille flow are smaller with a maximum extension up to 2 µm being measured at 104 dyn/cm2 [54]. Only a small fraction (10–15%) of VWF in solution was found to exhibit elongation in this study. Other studies that employ SANS also report only relatively small changes in VWF conformation under hydrodynamic shear at an applied shear stress of 30 dyn/cm2 [51]. Larger structural transitions in VWF were noted above 60 dyn/cm2, when the binding of the fluorescent probe bis-ANS to nonpolar cavities exposed upon shearing VWF was used to quantify structural transitions [52]. In this study, VWF was observed to gradually hide the exposed hydrophobic pockets over the time course of minutes to hours. A similar shear stress of 60 dyn/cm2 was also observed to be necessary for efficient VWF self-association in solution as measured using dynamic and static light scattering [29].
On substrates, using AFM, Siedlecki et al. demonstrated the unfolding of VWF bound to a hydrophobic surface (OTS-SAM, octadecyltrichlorosilane) above a critical shear stress of 35 dyn/cm2 [53]. This result is different from observations by others who show that VWF under shear or in the presence of fixed platelets does not undergo conformation changes upon immobilization on collagen type-I [55]. Thus, immobilized-VWF response to fluid shear may be highly-substrate dependent, and this is evident upon comparing VWF conformation on hydrophobic OTS versus hydrophilic mica substrates [56]. Here, VWF only rarely displays extended structures on OTS-glass with more extended conformations being noted on mica. In addition to these biophysical studies, the formation of VWF-platelet strings under shear flow on endothelial cells has been demonstrated by Dong et al. [45]. Further, the formation of extended bundles of VWF on collagen under shear is reported [46,47]. Whereas VWF string formation on endothelial cells occurs at physiological shear stresses <5 dyn/cm2, VWF bundles on collagen require higher stresses of ∼35–100 dyn/cm2.
Overall shear stresses above 60 dyn/cm2 was typically necessary to initiate VWF structural changes both when the protein was bound to substrates and when it was sheared in solution, except for the studies on endothelial cells. Based on hydrodynamic calculations, the force for the pure protein under these conditions is very small in the order of 0.1–0.5 pN. This suggests two possibilities: (i) Very small forces themselves are sufficient to support VWF structural changes. (ii) The very small forces are necessary nucleating events that trigger larger structural changes due to the self-association of VWF both in solution [29] and on substrates [46,47].
6.VWF self-association regulated by hydrodynamic shear
The homotypic interaction between VWF in solution and VWF immobilized on substrates, a process called VWF self-association, was shown to contribute to thrombus formation in a parallel plate flow chamber based model of vascular injury [57]. Studies performed in the cone–plate viscometer demonstrated a similar VWF self-association process using purified human proteins, and presented evidence that this is a fluid shear dependent phenomenon that is optimally triggered in solution above 50–60 dyn/cm2 [29]. VWF self-association also takes place on platelet GpIbα under similar shears, since VWF variants lacking an A1-domain can bind platelet GpIbα only when shear-mixed with wild-type multimeric human protein [31]. This functional self-association contributes to shear induced platelet activation. Similar observations have been reported by others who demonstrate the formation of bundles or networks of VWF both on collagen substrates [46] and endothelial cells possibly via binding
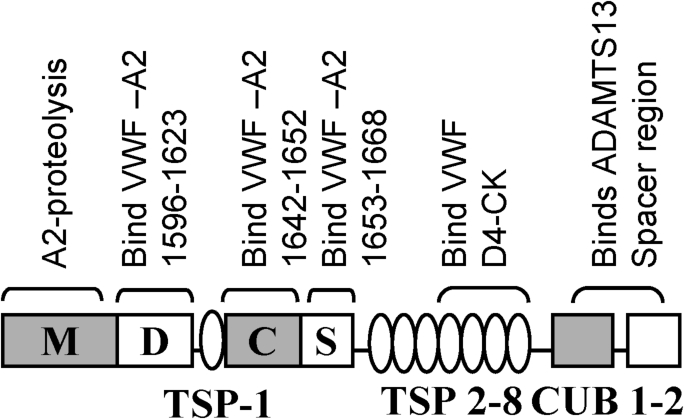
The mechanism for VWF self-association is not well established since specific inhibitors remain unavailable. The strongest, current mechanistic model to explain this phenomenon is based on the concept that the lateral association of VWF under shear is driven by disulfide exchange [59,60]. According to this, unpaired cysteines are present in native VWF but absent when the protein is subjected to shear when it self-associates [60,61]. Thus, blocking free-thiols on VWF impairs the binding of VWF to platelets [60]. Similarly, the addition of N-ethylmaleimide (NEM) inhibits VWF string formation on endothelial cells [61]. By performing mass spectrometry analysis, these authors suggest that the exchangeable Cys-residues in VWF include either some or all 9 Cys in VWF-D3 and C-domains: Cys889, Cys898, Cys2448, Cys2451, Cys2490, Cys2491, Cys2453, Cys2528 and Cys2533. Extending these observations, Ganderton et al. [59] showed that the VWF-C2 domain forms di- and tri-mers upon expression in HEK cells. Mutation of Cys2431 or Cys2451 to alanine in this recombinant fragment resulted in the production of monomers. This led the investigators to conclude that disulfide exchange involves the formation of Cys2431–Cys2431 and Cys2431–Cys2451 bonds between adjacent VWF multimers. Additional mechanisms for the alteration of VWF size based on cysteine modifications have also proposed based on the potential roles for thrombospondin-1 [62], ADAMTS13 [63] and N-acetylcysteine/NAC [64]. Interestingly, Bao et al. [63] show that VWF string formation on endothelial substrates and platelet adhesion can be inhibited by truncated, proteolytically inactive forms of ADAMTS13 that only contain the C-terminal domains stretching from TSP5 to CUB2 [63] (Fig. 4). This inhibition function is released upon addition of NEM, thus implying a free-thio dependent cell adhesion mechanism.
Fig. 4.
Domain level organization of ADAMTS-13 showing potential binding interactions with VWF. ADAMTS-13, like VWF, contains multiple subunits including a divalent-ion dependent metalloprotease domain (M) followed by a disintegrin-like (D), thrombospodin-1 repeat (TSP-1), cysteine-rich (C), spacer (S), seven more TSPs and two CUB domains. The protease has several VWF-binding exosites that bind both VWF-A2 and the D4-CK segment of VWF as indicated.
7.VWF proteolysis in solution, on platelets and on the endothelium
Fluid shear augments the proteolysis of the cryptic Tyr1605–Met1606 bond located within the VWF A2-domain using the metalloprotease ADAMTS13. This protease is partially active in blood plasma and it is chiefly secreted from hepatic stellate cells residing in the liver [5,65,66]. It is also secreted by endothelial cells [67–69] and platelets [70,71]. The importance of this cleavage process is highlighted by observations that VWF is currently the only known substrate for ADAMTS13. The absence of this enzyme activity results in enhanced VWF multimer size in circulation and a relatively rare, but life-threatening, bleeding disorder called thrombotic thrombocytopenic purpura (TTP). Whereas, VWF coexists with active ADAMTS13 in plasma it remains uncleaved unless the Tyr–Met scissile bond is exposed by mechanical forces or biomolecular binding to platelet GPIbα, collagen or endothelial cells.
Several unique structural features within the VWF-A2 domain and on the larger multimeric VWF are considered to regulate its proteolysis by ADAMTS13 [72]. Most significantly, unlike the other globular VWF A-domains (A1 and A3) which contain a disulfide-bond bridging two cysteines located at the N- and C-termini, VWF-A2 contains vicinal cysteines at Cys1669 and Cys1670. In this regard, while the absence of the disulfide bond across the domain allows the unfolding of the VWF-A2 domain, the vicinal cysteine acts as a force regulatable barrier for this process. In support of this, VWF-A2 domain constructs containing mutations in Cys1669 and/or Cys1670 display enhanced ADAMTS13 mediated proteolysis, compared to molecules with the intact vicinal cysteines [73]. Additional features that are thought to regulate the VWF-A2 folding–unfolding transition, based on crystal structure data [72], include: (i) The lack of well-formed α4-helix. Due to this, the extensive α4-associated hydrogen bond network that is observed in the VWF A1- and A3-domains is missing in VWF-A2. This may promote domain unfolding and proteolysis. (ii) The poor packing of the
The observation that the proteolysis of the isolated VWF-A2 domain proceeds more efficiently at physiological pH compared to the multimeric VWF protein, suggests that other structural features beyond VWF-A2 may regulate its cleavage kinetics [80]. In this regard, VWF and ADAMTS13 make extensive molecular interactions at a variety of exosite regions located both within and outside the A2-domain [81] (Fig. 4). First, VWF domains D4–CK interact with TSP5–TSP8 domains of ADAMTS13, while its CUB domain partially masks the proteolytic cleavage by binding to the ADAMTS13 spacer segment [82,83]. The binding of the VWF-D4 domain to ADAMTS13 releases this allosteric inhibition. Then, when the VWF-A2 domain unravels on the application of shear, additional exosite regions on VWF-A2 recognize the spacer and disintegrin-like domains of ADAMTS13. Finally, proteolysis occurs following the correct positioning of the catalytic residue Glu225 of ADAMTS13 over the Y1605–M1606 cleavage site [81]. In addition to protein based interactions, the carbohydrates of VWF also regulate proteolysis. For example, the removal of N-glycans [20] or mutation of O-linked glycosylations at the A1–A2 region [21] renders VWF more prone to ADAMTS13-mediated cleavage. Finally, the structural features of VWF and ADAMTS13, a variety of other blood proteins also control VWF proteolysis rates including Thrombospondin-1/TSP-1 which reduces proteolysis by acting as a competitive inhibitor of VWF binding, and proteases such as thrombin and plasmin which inactivate ADAMTS13 [84–86].
VWF proteolysis by ADAMTS13 upon shear application is reported to occur in solution [87], on the platelet surface [88] and on VWF bound to the endothelium [45]. The force required for such cleavage lies in the range of 7–14 pN, based on single molecule spectroscopy studies that assayed single VWF-A2 domain unfolding [11,79]. This force is larger in the case of the A1–A3 tri-domain complex [89]. Based on the hydrodynamic force estimation concepts described earlier [41], we estimate that the forces applied on VWF in solution would rarely exceed 10 pN. Consistent with this, VWF structure changes while occurring in solution may not be so pervasive as to account for all the loss of high molecular weight VWF multimers [51,54]. While forces of the same magnitude as in solution are applied on VWF bound to platelets, the applied force easily exceeds 50 pN when VWF is strung between two platelets at a local shear stress of 10 dyn/cm2 [41]. At this shear stress, however, the direction and nature of force changes rapidly as individual force oscillation cycles last <10 milliseconds [41]. Extended VWF strings on the endothelium can also experiences forces greater than 10 pN when bound with platelets and thus ADAMTS13 dependent cleavage on the vascular endothelium is reported both in vitro [45] and in vivo. [90]. Overall, VWF string cleavage could occur on VWF bound to platelets and the vascular endothelium though the relative importance of these mechanisms and the features accounting for the exquisite protein multimer distribution in blood remains to be determined.
8.VWF–GpIbα binding regulated by fluid shear
The binding of the VWF A1-domain to platelet GpIbα controls platelet translocation/rolling interactions during primary hemostasis. This allows for cell–substrate contact, platelet activation via collagen receptors, and stable cell adhesion (reviewed by [91]). Adherent and activated platelets with exposed phosphatidylserine then provide the substrate for the coagulation cascade. This molecular interaction is unique in that the number of platelets recruited by immobilized VWF increases with the magnitude of applied fluid shear stress from 0.5 to 5 dyn/cm2 before decreasing at higher shear stresses [25]. Mean platelet translocation velocity is also invariant at higher shears between ∼15–60 dyn/cm2. A number of structural features at the N-terminus of the VWF-A1 domain may control such binding to platelet GpIbα. Most notably, the D′D3-domain sterically regulates VWF-A1 binding to GpIbα [92,93]. Further, the binding of the VWF propeptide (VWFpp) to this D′D3-domain further reduces A1-GpIbα binding interactions [94]. The A2-domain of VWF is also thought to affect GpIbα-A1 binding [95], though its relative contribution with respect to the N-terminal flanking structural features like D′D3 remains unknown.
The A1–GpIbα bond strength is in the range of ∼10–20 pN, with applied mechanical force affecting both the on- and off-rate of this molecular interaction [96]. Thus, the force required for unbinding A1–GpIbα binding is approximately the same as that required for A2-domain unfolding. Using different measurement methods, the force dependent binding-unbinding data for A1–GpIbα has been interpreted to describe both a slip-bond [96] and also as a catch-bond [30]. In this regard, a slip bond is a conventional bond where the lifetime of a molecular bond, or the length of time that the bond will withstand mechanical force loading prior to rupture, decreases continuously upon increasing force. On the other hand, a catch bond results in an increase in bond lifetime with force. Besides this molecular biophysics, additional factors like VWF avidity due to multimer formation, domain–domain interactions and platelet deformation also likely influence the nature of platelet adhesion at sites of arterial thrombosis.
9.Conclusion
In summary, this review presents a perspective on how hydrodynamic shear forces regulate VWF function during hemostasis and thrombosis. In doing so, it demonstrates how theoretical estimates regarding the nature and magnitude of applied hydrodynamic forces can explain and unify a variety of biological observations under different experimental scenarios. As new biological knowledge emerges, this framework may be enhanced to allow the design of new therapies that are fluid shear responsive as opposed to current approaches that only focus on perturbing specific biochemical pathways.
Acknowledgement
Supported by NIH grant HL77258.
References
1 | [[1]] Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. (1998) ;67: :395–424. |
2 | [[2]] Nightingale T, Cutler D. The secretion of von Willebrand factor from endothelial cells; an increasingly complicated story. J Thromb Haemost. (2013) ;11: (Suppl. 1):192–201. Epub 2013/07/17. |
3 | [[3]] Kanaji S, Fahs SA, Shi Q, Haberichter SL, Montgomery RR. Contribution of platelet vs. endothelial VWF to platelet adhesion and hemostasis. J Thromb Haemost. (2012) ;10: (8):1646–1652. Epub 2012/05/31. |
4 | [[4]] Plaimauer B, Zimmermann K, Volkel D, Antoine G, Kerschbaumer R, Jenab P, et al. Cloning, expression, and functional characterization of the von Willebrand factor-cleaving protease (ADAMTS13). Blood. (2002) ;100: (10):3626–3632. |
5 | [[5]] Soejima K, Mimura N, Hirashima M, Maeda H, Hamamoto T, Nakagaki T, et al. A novel human metalloprotease synthesized in the liver and secreted into the blood: Possibly, the von Willebrand factor-cleaving protease? J Biochem. (2001) ;130: (4):475–480. |
6 | [[6]] Hamilton KK, Fretto LJ, Grierson DS, McKee PA. Effects of plasmin on von Willebrand factor multimers. Degradation in vitro and stimulation of release in vivo. J Clin Invest. (1985) ;76: (1):261–270. Epub 1985/07/01. |
7 | [[7]] Raife TJ, Cao W, Atkinson BS, Bedell B, Montgomery RR, Lentz SR, et al. Leukocyte proteases cleave von Willebrand factor at or near the ADAMTS13 cleavage site. Blood. (2009) ;114: (8):1666–1674. Epub 2009/06/23. |
8 | [[8]] Lancellotti S, Dragani A, Ranalli P, Petrucci G, Basso M, Tartaglione R, et al. Qualitative and quantitative modifications of von Willebrand factor in patients with essential thrombocythemia and controlled platelet count. J Thromb Haemost. (2015) ;13: (7):1226–1237. |
9 | [[9]] Zhou YF, Eng ET, Zhu J, Lu C, Walz T, Springer TA. Sequence and structure relationships within von Willebrand factor. Blood. (2012) ;120: (2):449–458. |
10 | [[10]] Huizinga EG, Martijn van der Plas R, Kroon J, Sixma JJ, Gros P. Crystal structure of the A3 domain of human von Willebrand factor: Implications for collagen binding. Structure. (1997) ;5: (9):1147–1156. Epub 1997/10/23. |
11 | [[11]] Zhang X, Halvorsen K, Zhang CZ, Wong WP, Springer TA. Mechanoenzymatic cleavage of the ultralarge vascular protein von Willebrand factor. Science. (2009) ;324: (5932):1330–1334. |
12 | [[12]] Zhou YF, Eng ET, Nishida N, Lu C, Walz T, Springer TA. A pH-regulated dimeric Bouquet in the structure of von Willebrand factor. EMBO J. (2011) ;30: (19):4098–4111. |
13 | [[13]] Titani K, Kumar S, Takio K, Ericsson LH, Wade RD, Ashida K, et al. Amino acid sequence of human von Willebrand factor. Biochemistry. (1986) ;25: (11):3171–3184. |
14 | [[14]] Canis K, McKinnon TA, Nowak A, Haslam SM, Panico M, Morris HR, et al. Mapping the N-glycome of human von Willebrand factor. Biochem J. (2012) ;447: (2):217–228. |
15 | [[15]] Canis K, McKinnon TA, Nowak A, Panico M, Morris HR, Laffan M, et al. The plasma von Willebrand factor O-glycome comprises a surprising variety of structures including ABH antigens and disialosyl motifs. J Thromb Haemost. (2010) ;8: (1):137–145. |
16 | [[16]] Pegon JN, Kurdi M, Casari C, Odouard S, Denis CV, Christophe OD, et al. Factor VIII and von Willebrand factor are ligands for the carbohydrate-receptor Siglec-5. Haematologica. (2012) ;97: (12):1855–1863. |
17 | [[17]] Saint-Lu N, Oortwijn BD, Pegon JN, Odouard S, Christophe OD, de Groot PG, et al. Identification of galectin-1 and galectin-3 as novel partners for von Willebrand factor. Arterioscler Thromb Vasc Biol. (2012) ;32: (4):894–901. |
18 | [[18]] Padilla A, Moake JL, Bernardo A, Ball C, Wang Y, Arya M, et al. P-selectin anchors newly released ultralarge von Willebrand factor multimers to the endothelial cell surface. Blood. (2004) ;103: (6):2150–2156. |
19 | [[19]] Rydz N, Swystun LL, Notley C, Paterson AD, Riches JJ, Sponagle K, et al. The C-type lectin receptor CLEC4M binds, internalizes, and clears von Willebrand factor and contributes to the variation in plasma von Willebrand factor levels. Blood. (2013) ;121: (26):5228–5237. |
20 | [[20]] McKinnon TA, Chion AC, Millington AJ, Lane DA, Laffan MA. N-linked glycosylation of VWF modulates its interaction with ADAMTS13. Blood. (2008) ;111: (6):3042–3049. |
21 | [[21]] Nowak AA, McKinnon TA, Hughes JM, Chion AC, Laffan MA. The O-linked glycans of human von Willebrand factor modulate its interaction with ADAMTS13. J Thromb Haemost. (2014) ;12: (1):54–61. |
22 | [[22]] Schulte am Esch J 2nd, Robson SC, Knoefel WT, Eisenberger CF, Peiper M, Rogiers X. Impact of O-linked glycosylation of the VWF-A1-domain flanking regions on platelet interaction. Br J Haematol. (2005) ;128: (1):82–90. |
23 | [[23]] Nowak AA, Canis K, Riddell A, Laffan MA, McKinnon TA. O-linked glycosylation of von Willebrand factor modulates the interaction with platelet receptor glycoprotein Ib under static and shear stress conditions. Blood. (2012) ;120: (1):214–222. |
24 | [[24]] Groeneveld DJ, van Bekkum T, Cheung KL, Dirven RJ, Castaman G, Reitsma PH, et al. No evidence for a direct effect of von Willebrand factor’s ABH blood group antigens on von Willebrand factor clearance. J Thromb Haemost. (2015) ;13: (4):592–600. |
25 | [[25]] Savage B, Saldivar E, Ruggeri ZM. Initiation of platelet adhesion by arrest onto fibrinogen or translocation on von Willebrand factor. Cell. (1996) ;84: (2):289–297. |
26 | [[26]] Kroll MH, Hellums JD, McIntire LV, Schafer AI, Moake JL. Platelets and shear stress. Blood. (1996) ;88: (5):1525–1541. Epub 1996/09/01. |
27 | [[27]] Goldsmith HL, Turitto VT. Rheological aspects of thrombosis and haemostasis: Basic principles and applications. ICTH-Report – Subcommittee on Rheology of the International Committee on Thrombosis and Haemostasis. Thromb Haemost. (1986) ;55: (3):415–435. Epub 1986/06/30. |
28 | [[28]] Lipowsky HH, Kovalcheck S, Zweifach BW. The distribution of blood rheological parameters in the microvasculature of cat mesentery. Circ Res. (1978) ;43: (5):738–749. |
29 | [[29]] Shankaran H, Alexandridis P, Neelamegham S. Aspects of hydrodynamic shear regulating shear-induced platelet activation and self-association of von Willebrand factor in suspension. Blood. (2003) ;101: (7):2637–2645. |
30 | [[30]] Yago T, Lou J, Wu T, Yang J, Miner JJ, Coburn L, et al. Platelet glycoprotein Ibalpha forms catch bonds with human WT vWF but not with type 2B von Willebrand disease vWF. J Clin Invest. (2008) ;118: (9):3195–3207. Epub 2008/08/30. |
31 | [[31]] Dayananda KM, Singh I, Mondal N, Neelamegham S. Von Willebrand factor self-association on platelet GpIbalpha under hydrodynamic shear: Effect on shear-induced platelet activation. Blood. (2010) ;116: (19):3990–3998. |
32 | [[32]] Neelamegham S, Taylor AD, Burns AR, Smith CW, Simon SI. Hydrodynamic shear shows distinct roles for LFA-1 and Mac-1 in neutrophil adhesion to intercellular adhesion molecule-1. Blood. (1998) ;92: (5):1626–1638. Epub 1998/08/26. |
33 | [[33]] Neelamegham S, Taylor AD, Hellums JD, Dembo M, Smith CW, Simon SI. Modeling the reversible kinetics of neutrophil aggregation under hydrodynamic shear. Biophys J. (1997) ;72: (4):1527–1540. Epub 1997/04/01. |
34 | [[34]] Peterson DM, Stathopoulos NA, Giorgio TD, Hellums JD, Moake JL. Shear-induced platelet aggregation requires von Willebrand factor and platelet membrane glycoproteins Ib and IIb-IIIa. Blood. (1987) ;69: (2):625–628. Epub 1987/02/01. |
35 | [[35]] Shankaran H, Neelamegham S. Effect of secondary flow on biological experiments in the cone–plate viscometer: Methods for estimating collision frequency, wall shear stress and inter-particle interactions in non-linear flow. Biorheology. (2001) ;38: (4):275–304. Epub 2001/10/24. |
36 | [[36]] Shankaran H, Neelamegham S. Nonlinear flow affects hydrodynamic forces and neutrophil adhesion rates in cone–plate viscometers. Biophys J. (2001) ;80: (6):2631–2648. Epub 2001/05/24. |
37 | [[37]] Bird RD, Stewart WE, Lightfoot EN. Transport phenomena. 2nd ed. Wiley; (2006) . 905 p. |
38 | [[38]] Bruus H. In: Theoretical microfluidics. Oxford master series in condensed matter physics. Oxford University; (2007) . p. 45–51. |
39 | [[39]] Zhang Y, Neelamegham S. Estimating the efficiency of cell capture and arrest in flow chambers: Study of neutrophil binding via E-selectin and ICAM-1. Biophys J. (2002) ;83: (4):1934–1952. Epub 2002/09/27. |
40 | [[40]] Arp P, Mason S. The kinetics of flowing dispersions: VIII. Doublets of rigid spheres (theoretical). Journal of colloid and interface science. (1977) ;61: (1):21–43. |
41 | [[41]] Shankaran H, Neelamegham S. Hydrodynamic forces applied on intercellular bonds, soluble molecules, and cell-surface receptors. Biophys J. (2004) ;86: (1 Pt 1):576–588. |
42 | [[42]] Tha SP, Goldsmith HL. Interaction forces between red cells agglutinated by antibody. I. Theoretical Biophys J. (1986) ;50: (6):1109–1116. Epub 1986/12/01. |
43 | [[43]] Fowler WE, Fretto LJ, Hamilton KK, Erickson HP, McKee PA. Substructure of human von Willebrand factor. J Clin Invest. (1985) ;76: (4):1491–1500. Epub 1985/10/01. |
44 | [[44]] Singh I, Shankaran H, Beauharnois ME, Xiao Z, Alexandridis P, Neelamegham S. Solution structure of human von Willebrand factor studied using small angle neutron scattering. J Biol Chem. (2006) ;281: (50):38266–38275. Epub 2006/10/21. |
45 | [[45]] Dong JF, Moake JL, Nolasco L, Bernardo A, Arceneaux W, Shrimpton CN, et al. ADAMTS13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood. (2002) ;100: (12):4033–4039. Epub 2002/10/24. |
46 | [[46]] Barg A, Ossig R, Goerge T, Schneider MF, Schillers H, Oberleithner H, et al. Soluble plasma-derived von Willebrand factor assembles to a haemostatically active filamentous network. Thromb Haemost. (2007) ;97: (4):514–526. Epub 2007/03/30. |
47 | [[47]] Colace TV, Diamond SL. Direct observation of von Willebrand factor elongation and fiber formation on collagen during acute whole blood exposure to pathological flow. Arterioscler Thromb Vasc Biol. (2013) ;33: (1):105–113. |
48 | [[48]] Clift R, Grace JR, Weber ME. Bubbles, drops and particles. New York, USA: Academic Press; (1978) . |
49 | [[49]] Goldman AJ, Cox RG, Brenner H. Slow viscous motion of a sphere parallel to a plane wall – II. Couette flow. Chemical Engineering Science. (1967) ;22: :653–660. |
50 | [[50]] Schneider SW, Nuschele S, Wixforth A, Gorzelanny C, Alexander-Katz A, Netz RR, et al. Shear-induced unfolding triggers adhesion of von Willebrand factor fibers. Proc Natl Acad Sci USA. (2007) ;104: (19):7899–7903. |
51 | [[51]] Singh I, Themistou E, Porcar L, Neelamegham S. Fluid shear induces conformation change in human blood protein von Willebrand factor in solution. Biophys J. (2009) ;96: (6):2313–2320. |
52 | [[52]] Themistou E, Singh I, Shang C, Balu-Iyer SV, Alexandridis P, Neelamegham S. Application of fluorescence spectroscopy to quantify shear-induced protein conformation change. Biophys J. (2009) ;97: (9):2567–2576. |
53 | [[53]] Siedlecki CA, Lestini BJ, Kottke-Marchant KK, Eppell SJ, Wilson DL, Marchant RE. Shear-dependent changes in the three-dimensional structure of human von Willebrand factor. Blood. (1996) ;88: (8):2939–2950. |
54 | [[54]] Vergauwe RM, Uji-i H, De Ceunynck K, Vermant J, Vanhoorelbeke K, Hofkens J. Shear-stress-induced conformational changes of von Willebrand factor in a water-glycerol mixture observed with single molecule microscopy. J Phys Chem B. (2014) ;118: (21):5660–5669. |
55 | [[55]] Novak L, Deckmyn H, Damjanovich S, Harsfalvi J. Shear-dependent morphology of von Willebrand factor bound to immobilized collagen. Blood. (2002) ;99: (6):2070–2076. Epub 2002/03/06. |
56 | [[56]] Raghavachari M, Tsai H, Kottke-Marchant K, Marchant RE. Surface dependent structures of von Willebrand factor observed by AFM under aqueous conditions. Colloids Surf B Biointerfaces. (2000) ;19: (4):315–324. Epub 2000/11/07. |
57 | [[57]] Savage B, Sixma JJ, Ruggeri ZM. Functional self-association of von Willebrand factor during platelet adhesion under flow. Proc Natl Acad Sci USA. (2002) ;99: (1):425–430. |
58 | [[58]] Huang J, Roth R, Heuser JE, Sadler JE. Integrin alpha(v)beta(3) on human endothelial cells binds von Willebrand factor strings under fluid shear stress. Blood. (2009) ;113: (7):1589–1597. Epub 2008/10/18. |
59 | [[59]] Ganderton T, Wong JW, Schroeder C, Hogg PJ. Lateral self-association of VWF involves the Cys2431–Cys2453 disulfide/dithiol in the C2 domain. Blood. (2011) ;118: (19):5312–5318. |
60 | [[60]] Choi H, Aboulfatova K, Pownall HJ, Cook R, Dong JF. Shear-induced disulfide bond formation regulates adhesion activity of von Willebrand factor. J Biol Chem. (2007) ;282: (49):35604–35611. |
61 | [[61]] Li Y, Choi H, Zhou Z, Nolasco L, Pownall HJ, Voorberg J, et al. Covalent regulation of ULVWF string formation and elongation on endothelial cells under flow conditions. J Thromb Haemost. (2008) ;6: (7):1135–1143. |
62 | [[62]] Pimanda JE, Ganderton T, Maekawa A, Yap CL, Lawler J, Kershaw G, et al. Role of thrombospondin-1 in control of von Willebrand factor multimer size in mice. J Biol Chem. (2004) ;279: (20):21439–21448. |
63 | [[63]] Bao J, Xiao J, Mao Y, Zheng XL. Carboxyl terminus of ADAMTS13 directly inhibits platelet aggregation and ultra large von Willebrand factor string formation under flow in a free-thiol-dependent manner. Arterioscler Thromb Vasc Biol. (2014) ;34: (2):397–407. |
64 | [[64]] Chen J, Reheman A, Gushiken FC, Nolasco L, Fu X, Moake JL, et al. N-acetylcysteine reduces the size and activity of von Willebrand factor in human plasma and mice. J Clin Invest. (2011) ;121: (2):593–603. |
65 | [[65]] Uemura M, Tatsumi K, Matsumoto M, Fujimoto M, Matsuyama T, Ishikawa M, et al. Localization of ADAMTS13 to the stellate cells of human liver. Blood. (2005) ;106: (3):922–924. |
66 | [[66]] Zhou W, Inada M, Lee TP, Benten D, Lyubsky S, Bouhassira EE, et al. ADAMTS13 is expressed in hepatic stellate cells. Lab Invest. (2005) ;85: (6):780–788. |
67 | [[67]] Turner N, Nolasco L, Tao Z, Dong JF, Moake J. Human endothelial cells synthesize and release ADAMTS13. J Thromb Haemost. (2006) ;4: (6):1396–1404. |
68 | [[68]] Tati R, Kristoffersson AC, Stahl AL, Morgelin M, Motto D, Satchell S, et al. Phenotypic expression of ADAMTS13 in glomerular endothelial cells. PLoS One. (2011) ;6: (6):e21587. |
69 | [[69]] Shang D, Zheng XW, Niiya M, Zheng XL. Apical sorting of ADAMTS13 in vascular endothelial cells and Madin–Darby canine kidney cells depends on the CUB domains and their association with lipid rafts. Blood. (2006) ;108: (7):2207–2215. |
70 | [[70]] Suzuki M, Murata M, Matsubara Y, Uchida T, Ishihara H, Shibano T, et al. Detection of von Willebrand factor-cleaving protease (ADAMTS13) in human platelets. Biochem Biophys Res Commun. (2004) ;313: (1):212–216. |
71 | [[71]] Liu L, Choi H, Bernardo A, Bergeron AL, Nolasco L, Ruan C, et al. Platelet-derived VWF-cleaving metalloprotease ADAMTS13. J Thromb Haemost. (2005) ;3: (11):2536–2544. |
72 | [[72]] Zhang Q, Zhou YF, Zhang CZ, Zhang X, Lu C, Springer TA. Structural specializations of A2, a force-sensing domain in the ultralarge vascular protein von Willebrand factor. Proc Natl Acad Sci USA. (2009) ;106: (23):9226–9231. |
73 | [[73]] Lynch CJ, Lane DA, Luken BM. Control of VWF A2 domain stability and ADAMTS13 access to the scissile bond of full-length VWF. Blood. (2014) ;123: (16):2585–2592. |
74 | [[74]] Stewart DE, Sarkar A, Wampler JE. Occurrence and role of cis peptide bonds in protein structures. J Mol Biol. (1990) ;214: (1):253–260. |
75 | [[75]] Ramachandran GN, Mitra AK. An explanation for the rare occurrence of cis peptide units in proteins and polypeptides. J Mol Biol. (1976) ;107: (1):85–92. |
76 | [[76]] Brandts JF, Halvorson HR, Brennan M. Consideration of the possibility that the slow step in protein denaturation reactions is due to cis-trans isomerism of proline residues. Biochemistry. (1975) ;14: (22):4953–4963. |
77 | [[77]] Valiaev A, Lim DW, Oas TG, Chilkoti A, Zauscher S. Force-induced prolyl cis-trans isomerization in elastin-like polypeptides. J Am Chem Soc. (2007) ;129: (20):6491–6497. |
78 | [[78]] Zhou M, Dong X, Baldauf C, Chen H, Zhou Y, Springer TA, et al. A novel calcium-binding site of von Willebrand factor A2 domain regulates its cleavage by ADAMTS13. Blood. (2011) ;117: (17):4623–4631. |
79 | [[79]] Jakobi AJ, Mashaghi A, Tans SJ, Huizinga EG. Calcium modulates force sensing by the von Willebrand factor A2 domain. Nat Commun. (2011) ;2: :385. |
80 | [[80]] Dayananda KM, Gogia S, Neelamegham S. Escherichia coli-derived von Willebrand factor-A2 domain fluorescence/Forster resonance energy transfer proteins that quantify ADAMTS13 activity. Anal Biochem. (2011) ;410: (2):206–213. |
81 | [[81]] Crawley JT, de Groot R, Xiang Y, Luken BM, Lane DA. Unraveling the scissile bond: How ADAMTS13 recognizes and cleaves von Willebrand factor. Blood. (2011) ;118: (12):3212–3221. |
82 | [[82]] Muia J, Zhu J, Gupta G, Haberichter SL, Friedman KD, Feys HB, et al. Allosteric activation of ADAMTS13 by von Willebrand factor. Proc Natl Acad Sci USA. (2014) ;111: (52):18584–18589. Epub 2014/12/17. |
83 | [[83]] South K, Luken BM, Crawley JT, Phillips R, Thomas M, Collins RF, et al. Conformational activation of ADAMTS13. Proc Natl Acad Sci USA. (2014) ;111: (52):18578–18583. Epub 2014/12/17. |
84 | [[84]] Bonnefoy A, Daenens K, Feys HB, De Vos R, Vandervoort P, Vermylen J, et al. Thrombospondin-1 controls vascular platelet recruitment and thrombus adherence in mice by protecting (sub)endothelial VWF from cleavage by ADAMTS13. Blood. (2006) ;107: (3):955–964. |
85 | [[85]] Crawley JT, Lam JK, Rance JB, Mollica LR, O’Donnell JS, Lane DA. Proteolytic inactivation of ADAMTS13 by thrombin and plasmin. Blood. (2005) ;105: (3):1085–1093. |
86 | [[86]] Wang A, Liu F, Dong N, Ma Z, Zhang J, Su J, et al. Thrombospondin-1 and ADAMTS13 competitively bind to VWF A2 and A3 domains in vitro. Thromb Res. (2010) ;126: (4):e260–e265. |
87 | [[87]] Tsai HM. Physiologic cleavage of von Willebrand factor by a plasma protease is dependent on its conformation and requires calcium ion. Blood. (1996) ;87: (10):4235–4244. |
88 | [[88]] Shim K, Anderson PJ, Tuley EA, Wiswall E, Sadler JE. Platelet-VWF complexes are preferred substrates of ADAMTS13 under fluid shear stress. Blood. (2008) ;111: (2):651–657. |
89 | [[89]] Wu T, Lin J, Cruz MA, Dong JF, Zhu C. Force-induced cleavage of single VWFA1A2A3 tridomains by ADAMTS13. Blood. (2010) ;115: (2):370–378. Epub 2009/11/10. |
90 | [[90]] Chauhan AK, Goerge T, Schneider SW, Wagner DD. Formation of platelet strings and microthrombi in the presence of ADAMTS13 inhibitor does not require P-selectin or beta3 integrin. J Thromb Haemost. (2007) ;5: (3):583–589. Epub 2006/12/15. |
91 | [[91]] Jackson SP. Arterial thrombosis – Insidious, unpredictable and deadly. Nat Med. (2011) ;17: (11):1423–1436. Epub 2011/11/09. |
92 | [[92]] Ulrichts H, Udvardy M, Lenting PJ, Pareyn I, Vandeputte N, Vanhoorelbeke K, et al. Shielding of the A1 domain by the D′D3 domains of von Willebrand factor modulates its interaction with platelet glycoprotein Ib-IX-V. J Biol Chem. (2006) ;281: (8):4699–4707. |
93 | [[93]] Madabhushi SR, Zhang C, Kelkar A, Dayananda KM, Platelet NS. GpIba binding to von Willebrand factor under fluid shear: Contributions of the D′D3-domain, A1-domain flanking peptide and O-linked glycans. J Am Heart Assoc. (2014) ;3: (5):e001420. Epub 2014/10/25. |
94 | [[94]] Madabhushi SR, Shang C, Dayananda KM, Rittenhouse-Olson K, Murphy M, Ryan TE, et al. Von Willebrand factor (VWF) propeptide binding to VWF D′D3 domain attenuates platelet activation and adhesion. Blood. (2012) ;119: (20):4769–4778. |
95 | [[95]] Martin C, Morales LD, Cruz MA. Purified A2 domain of von Willebrand factor binds to the active conformation of von Willebrand factor and blocks the interaction with platelet glycoprotein Ibalpha. J Thromb Haemost. (2007) ;5: (7):1363–1370. |
96 | [[96]] Kim J, Zhang CZ, Zhang X, Springer TA. A mechanically stabilized receptor-ligand flex-bond important in the vasculature. Nature. (2010) ;466: (7309):992–995. |


