Adenoviral vectors for modulation of poly(ADP-ribose) polymerase-1 (PARP1) – dependent DNA repair as a predictive tool for chemotherapy
Abstract
Loss of the DNA homologous recombination repair pathway (DHR) sensitizes cells to the effects of clastogenic chemotherapy agents when the alternative base excision DNA repair pathway (BER) is blocked by inhibitors of poly(ADP-ribose) polymerases (PARP). The mutation of BRCA is an example of defective DHR, where several PARP inhibitors are now in advanced stages of clinical trials due to their ability to form synthetic lethality with chemotherapy drugs in breast and ovarian cancer. Because the inability of cells to perform DHR is the determining factor for successful PARP inhibitor administration in a chemotherapy protocol, testing of patient cells for sensitization by PARP inhibitors would be advantageous for the prediction of therapy outcome. This proof-of-principle study set out to design recombinant adenoviruses as a simple tool for the functional block of PARP1-mediated DNA strand break repair important for the execution of BER that could potentially be used to test cultured cells from patients for the sensitizing effects of PARP inhibitors in a chemotherapy setting. Recombinant adenoviruses for the expression of the PARP1 DNA binding domain (DBD) as a dominant-negative PARP1 mutant, for full-length PARP1 expression and other control viruses were generated and used to infect normal human fibroblasts. Transgene expression and the modulation of poly(ADP-ribose) (PAR) were studied using immunofluorescence and immunoblotting techniques. Overexpression of PARP1 increased, but expression of the PARP1 DBD blocked PAR formation in response to methylnitronitrosoguanidine (MNNG) treatment of virally transduced cells. Ectopic DBD expression caused the formation of caspase 3 cleavage products of PARP1 protein, which are a marker of apoptotic cell death. Cells infected with control virus without PARP DBD did not induce caspase 3-mediated cleavage of PARP1. In conclusion, the set of adenoviral vectors presented in this study can be used to modulate PARP activity in normal human cells and may therefore be used as a tool for testing cellular sensitization by blocking DNA BER by administration of PARP inhibitors and to gain better understanding of potential PARP inhibitor resistance.
1Introduction
The management and repair of DNA damage represents an important aspect of cellular survival and the maintenance of genomic stability throughout the lifetime of the cell. Cellular DNA is constantly subjected to endogenous and environmental damage that would lead to either the demise of the cell or to mutation if left unrepaired. Overlapping and alternative DNA damage response (DDR) pathways have evolved to (a) sense DNA damage, (b) signal to halt the cell cycle and (c) activate the appropriate cellular response such as recruitment of the appropriate DNA repair enzymes needed for the alternative DNA repair processes that exist in the cell (recently reviewed in [1, 2]).
DNA damage signaling induces distinct biochemical decision steps that determine cellular fate in response to DNA damage. Depending on the severity of the damage and the intensity of the damage signal, cells can either experience temporary cell cycle arrest, undergo apoptosis, differentiate to exit the cell cycle, or they can become prematurely senescent and die. DNA single- or double- strand breaks (SB) that occur either as a direct result of the exogenous impact, such as by ionizing radiation, or as intermediate products of several DNA repair pathways, almost always play a key role in these decisions because they represent the most severe form of DNA damage.
DNA damage with the formation of single or double SB leads to the rapid induction of poly(ADP-ribosyl)ation. This post-translational protein modification is mediated by members of the poly(ADP-ribose) polymerase (PARP) enzyme family [3]. Poly(ADP-ribose) polymerase-1 (PARP1) is the most prevalent nuclear PARP. Exposure of cells to ionizing radiation, alkylating agents or oxidants lead either directly or as intermediates of cellular DNA repair efforts to the formation of DNA strand breaks, which activate PARP1 [4, 5]. The activated PARP1 modifies itself and other nuclear proteins with long, branched chains of poly(ADP-ribose) (PAR), consuming NAD+ as a substrate and releasing nicotinamide as a byproduct. The ADP-ribose polymers subsequently are quickly degraded to monomeric ADP-ribose by the activity of poly(ADP-ribose) glycohydrolase (PARG) [6, 7].
PARP1 plays important roles in the repair of DNA strand breaks which it binds to via its DNA binding domain (DBD, Fig. 1). Binding of the DBD to a DNA strand break activates the catalytic domain of PARP1 to form PAR. The biopolymer is covalently bound to glutamate and arginine residues of target proteins such as H1 and, to a lesser extent, H2A and H2B nucleosomal subunits at the damaged site as a large, highly electronegative posttranslational modification (PTM). PAR reduces or fully neutralizes the ability of these proteins and other chromatin components to bind to DNA, leading to profound local chromatin decondensation by transient removal of H1 and core histones from the DNA (reviewed in [5, 8]). PARP1 auto-modification with PAR also removes the enzyme from the DNA strand break and inhibits its enzymatic activity. The local chromatin decondensation mediated by PARP1 is an essential step in DNA repair, where repair factors need access to the damaged site [9]. In addition, PAR formation by PARP or PARP2 is involved in a myriad of other biological processes including DNA replication, transcription, and DNA unknotting mediated by topoisomerases [10–14], cell differentiation during spermiogenesis and a multitude of epigenetic events such as the regulation of RNA metabolism, the activation of ATM/ATR-dependent H2AX phosphorylation in the presence of DNA double strand breaks, and the regulation of histone demethylases such as KDM5B and KDM4D [15–21]. PARP1 and possibly PARP2 play clinically important roles in base excision repair (BER). Alkylating agents, such as they are used in cancer chemotherapy or the compound MNNG (methylnitronitrosoguanidine) used in this study, add an alkyl group to DNA bases which triggers a DNA damage response. In the BER pathway, alkylated G and T are recognized by specific glycosylases that remove altered bases to create an apurinic or apyrimidinic (AP) site but leave the sugar-phosphate backbone of the DNA intact. A specific AP endonuclease (APE1) then cleaves the DNA at the AP site to create a single-strand break (nick) which activates PARP1 [16]. Inhibition of PARP blocks BER, so that the alternative pathway, homologous recombination repair, is left as the only alternative to repair this type of DNA damage. If cells are unable to perform homologous recombination repair, such as in breast cancer cells with defective BRCA protein, they are strongly sensitized to DNA damaging agents when BER is blocked, which has been the rationale for designing highly potent and bioavailable PARP inhibitors as anticancer drugs where PARP inhibition causes synthetic lethality in combination with ionizing radiation or clastogenic antitumor drugs [22–24].
Despite the fact that highly potent PARP inhibitors exist that are currently in clinical trials for targeting DNA repair in cancer cells (reviewed in [22]), there are currently no inhibitors that are truly selective for PARP1 versus PARP2 or any of the 13 other PARP enzymes [3, 25]. This circumstance has been a problem in investigating the roles of the two PARP enzymes because PARP1 and PARP2 have partly redundant and partly specific functions in cells [26, 27]. In contrast, overexpression of a dominant negative PARP1 DNA binding domain (DBD) fragment reduces poly(ADP-ribosyl)ation, impairs efficient DNA repair after DNA damage and renders cells more susceptible to cell death [28–33] but should not affect any other PARP enzymes. The dominant-negative effect is quite pronounced because the PARP DBD has both zinc finger domains that tightly bind DNA strand breaks but no catalytic domain for auto-modification of the enzyme with PAR. By binding to the DNA SB, endogenous wild-type PARP1 can no longer bind to DNA breaks to be activated and alternative DNA repair pathways must be activated in the cell. We therefore chose to overexpress the DNA binding domain of PARP1 to specifically interfere with DNA strand break repair in normal human cells with the goal creating a test system where PARP1 activity is inhibited.
Here we describe an approach using a pair of recombinant adenoviral vectors to achieve overexpression of PARP1 and PARP1 DBD in primary human fibroblasts to specifically target the repair process at the DNA strand break by modulating PARP1 activity in normal cells.
2Material and methods
2.1Construction of recombinant adenoviruses rAdV-PARP1, rAdV-DBD, rAdV-LacZ and rAdV-GFP-PARP1
Recombinant adenoviruses for the expression of human PARP1, PARP1 DNA-binding domain (DBD), green fluorescent protein (GFP) and β-galactosidase (LacZ) were constructed using the method published by He et al. 1998 [34]. The cDNA for the Parp1 and Parp1-DBD cassettes [35] (Parp1, Parp1-DBD, LacZ) were cloned into a recombination plasmid (pTrack-CMV, pShuttle-CMV [34], giving rise to plasmids pShuttle-CMV-PARP1, pShuttle-CMV-DBD, pShuttle-CMV-LacZ and pTrack-CMV-PARP1 (the latter containing a GFP transgene under the control of a CMV promoter). These plasmids were linearized using PmeI (New England Biolabs) and then co-transformed with plasmid pAdEasy-1 [34] into the recombination-competent E.coli strain BJ5183. Selection with kanamycin allowed for selection of recombinant virus plasmids pAdV-CMV-PARP1, pAdV-CMV-DBD, pAdV-CMV-LAcZ and pAd-CMV-GFP-CMV-PARP1. Colonies were expanded in 5 ml cultures and plasmids were extracted using standard alkaline lysis and analyzed for successful recombination as described [34].
For viral particle production, 911 cells [6] were seeded in 10 cm tissue culture dishes at a density of 2×106 cells per dish. Twenty-four hours after seeding, cells were transfected with 10μg of recombinant plasmid DNA cleaved with PacI (New England Biolabs), using standard calcium phosphate precipitation. Two to four days after transfection, cells started showing signs of the virus-induced cytopathic effect. Incubation was allowed for 1-2 additional days after onset of the cytopathic effect. Cells and supernatant were harvested and used for the propagation / amplification of recombinant viral particles. High titer stocks were produced using a CsCl ultracentrifugation banding purification step as described [34], and at http://www.coloncancer.org/adeasy/protocol.htm.
Purified virus suspensions were diluted in Tris-saline-glycerol buffer (10% glycerol, 100 mM Tris/HCl pH 8.5).
Titration of viral stocks was carried out on 911 cells and titers calculated using a modified TCID50 method (tissue culture infectious dose 50) by Spearman & Kärber [37]. Virus concentrations were adjusted to 1×107 plaque forming units (pfu) per ml.
Infectivity of the resulting viruses were tested on human cancer cell lines HeLa, A549 and U87MG, as well as on normal human skin fibroblasts (huSF).
2.1.1Cell culture and Infections
911 cells, (Introgen, Netherlands, now: Crucell, Netherlands) were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Gibco/BRL) supplemented with 10% fetal bovine serum (Gibco, BRL). Normal human fibroblast (designated HuFi) (Clonetics, Germany) were cultured according to the instructions of the supplier.
For infection with recombinant adenoviruses, cells were seeded at a density of 5×104 cells/well into standard 6 well plates which contained one sterile cover slip per well. After 24 h viral stock solution was added to the cells using the indicated multiplicity of infection (MOI).
2.1.2Microscopic preparations and immunofluorescence staining
One day after infection, cells were treated with 200μM MNNG (1-Methyl-3-nitro-1-nitrosog-uanidine, Sigma St. Louis) for 20 min. After treatment, cells were washed with PBS once and fixed with 4% formaldehyde in PBS for 10 min at room temperature. Fixed coverslips were incubated in 100 mM Glycine in PBS for 1 min, washed with PBS, permeabilized in 0.4% Triton X-100 in PBS for 3 min, washed and blocked in 5% bovine serum albumin in PBS for 30 min at room temperature. Immunostaining was performed using a monoclonal antibody against poly(ADP-ribose) (clone 10H, [38]) and a rabbit antiserum recognizing poly(ADP-ribose) polymerase 1 (PARP1) (Roche, Basel). A fluorescein-coupled goat anti-mouse antiserum and a rhodamine-coupled goat anti rabbit antiserum were used as secondary antibodies (Jackson ImmunoResearch Laboratories, West Grove). Coverslips were embedded in Vectashield mounting medium (Vector Laboratories, Burlingame), which contained 1μg/ml DAPI (Roche) as a DNA counterstain. Fluorescence microscopy was performed using an upright fluorescence microscope (Zeiss Axiophot).
2.2Western blot analysis
Western blot analyses of were performed using a standard SDS-PAGE method with 10% polyacrylamide gels. After electrotransfer to PVDF membranes (Merck Millipore, Billerica), PARP1 protein was detected using a polyclonal rabbit antiserum (Roche, Basel) and a secondary goat anti-rabbit antibody coupled to horseradish peroxidase (Jackson Laboratories) with an enhanced chemoluminescence (ECL) reaction.
3Results
Cell lysates from infected 911 cells yielded high virus concentrations (rAdV-PARP1 : 5×108 pfu/ml, rAdV-PARP-GFP: 4×108 pfu/ml, rAdV-DBD: 4.7×108 pfu/ml, rAdV-GFP-PARP1 : 4×108 pfu/ml) prior to cesium chloride purification. This result indicated that expression of PARP1 or PARP1 DBD did not interfere with virus propagation.
All cell types tested could be infected efficiently with varying MOI, including huSF where targeting 60% of cells in culture required an MOI of 25 pfu/cell. Infection of huSF with an MOI = 10 pfu/cell is shown in Fig. 2, demonstrating that normal human cells can be infected with high efficiency by the recombinant adenoviruses.
Depending on and varying with cell types, very high MOI (5-fold above optimum) triggered cell death due to some inherent cytotoxicity of the viral infection at excessive concentrations, particularly in HeLa cells.
We next analyzed infected cells for expression of the transgenes. Western blot analyses of normal huSF infected with the same MOI (MOI = 25 pfu/cell) of rAdV-PARP1 and rAdV-DBD adenoviruses showed several-fold overexpression of PARP1 and DBD, respectively, compared to endogenous PARP1 expression (Fig. 3). The expression of PARP1 in huSF after rAdV-GFP-PARP1 infection was only about half of the PARP1 expression in cells infected with rADV-PARP1, which can be interpreted to be due to the second CMV-driven expression cassette included in this virus, which was the GFP marker that was added to indicate infection rate using an epifluorescence microscope for live imaging.
Interestingly, the PARP1 DBD was expressed at a disproportionately higher rate than full-length PARP1 and the C2-10 antibody detected a number of shorter peptides in addition to the expected 45 kDa band. Molecular sizes of some of the additional bands are consistent with apoptotic fragment sizes after caspase 3 cleavage in the DEVD consensus motif [39], which are 43 kDa and 18 kDa for the DBD (Fig. 3, arrow, Fig. 1). Additional bands may be the result of additional protease activity or indicate aborted expression products. In addition to apoptotic fragments of full-length PARP1 protein, which are 43 kDa and 85 kDa in size, minor additional fragment sizes were observed after adenoviral expression of full-length PARP as well (Fig. 3, arrows, Fig. 1).
In order to determine whether the observed PARP cleavage, which has been widely used as a molecular marker of apoptosis in cells, was caused by cytotoxicity of the adenoviral infection per se or by effect of the overexpressed DBD, cells were infected with increasing MOI of the viruses (Fig. 4). The beta galactosidase-expressing rAdV-LacZ was used as a control virus for viral toxicity in these experiments. At MOI = 100 pfu/cell, infection with the rAdV-LacZ virus showed toxicity as indicated by the overall reduced protein concentration despite correction for cell numbers loaded per lane, suggesting that cells underwent cytopathic effect by the infection and lytic cell death. PARP cleavage was not enhanced at any MOI using this virus (Fig. 4, right panel, which shows extended exposure of the ECL reaction to the X-ray film to demonstrate absence of increased PARP cleavage. In contrast, expression of the PARP1 DBD increased PARP cleavage even at low MOI = 1 (Fig. 4, right panel). At MOI = 10, significant PARP cleavage product of 87 kDa was detected (Fig. 4, right panel). These results indicate that DBD overexpression was associated with extensive PARP cleavage in huSF at higher MOI, which was not primarily due to adenoviral toxicity but to expression of PARP1 DBD. Overexpression of PARP1 and DBD expression was also confirmed at the level of individual cells using immunofluorescence staining (Fig. 5).
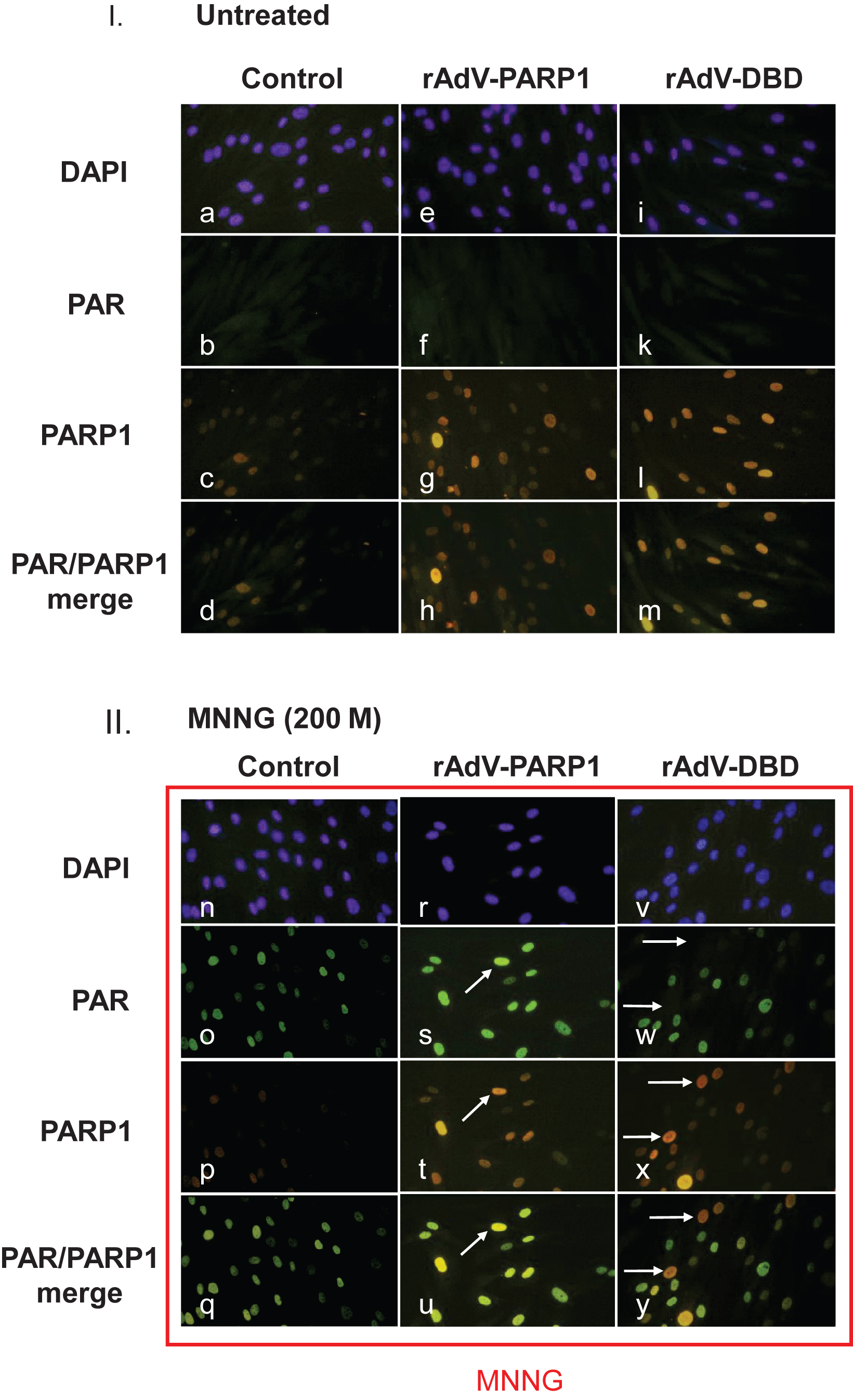
Next, we investigated whether PAR formation is altered in normal cells overexpressing PARP1 or the PARP1 DBD. Cultured normal human skin fibroblasts were transduced with rAdV-PARP1 or rAdV-DBD 24 hours prior to treatment with the alkylating agent MNNG. This compound adds alkyl groups to the O6 of guanine (G) and O4 of thymine (T) bases in DNA, which triggers DNA base excision repair (BER) and PARP activation. PAR synthesis in response to MNNG treatment was detected in mock-infected (using Tris-saline-glycerol buffer), normal control cells but not in these normal cells mock-treated with phosphate-buffered saline (PBS) buffer which was used as a solvent for MNNG (Fig. 5). PAR synthesis was strongly increased in rAdV-PARP1 infected, MNNG-treated fibroblasts but effectively suppressed below detection level in rAdV-DBD infected cells (Fig. 5, u, y). These data show that ectopic PARP1 overexpression and DBD expression using adenoviral vectors can be used to elevate or suppress cellular PAR levels in response to genotoxic stress.
4Discussion
The rAdV-DBD vectors described in this report were able to block PARP1-dependent activity at DNA strand breaks in the presence of the alkylating agent MNNG. Viruses causing overexpression of PARP1 resulted in elevated PAR formation in transduced cells under the same conditions. We propose that together with the infection control virus rAdV-LacZ this pair of viruses may be a unique tool to investigate PARP1-dependent DNA repair.
We set out to develop a system to investigate consequences of impaired versus improved DNA strand break repair in primary human cells. In the present study we addressed how this could be achieved by targeting the poly(ADP-ribose) pathway in cells that are difficult to transfect using adenoviral vectors.
The viruses we constructed are intended to serve many purposes. First, they should be a suitable research tool to investigate biological functions of impaired versus enhanced PAR formation in response to genotoxic agents in normal cells. Investigating poly(ADP-ribose) (PAR) metabolism in primary cells has been difficult due to a lack of appropriate genetic tools by which PAR levels can be modulated in cells that are difficult or impossible to transfect. Second, we propose to use them to screen patient cells for sensitization by PARP inhibition, without the expected development of resistance against PARP inhibitor drugs [40]. Recent progress in the understanding of PARP functions in DNA repair, as outlined above have put PARP inhibition at the forefront of anticancer drug development (reviewed in [41]). To determine which patients would be suitable depends on the ability of the ovarian or breast cancer cells to perform homologous recombination as an alternate pathway to BER. The viral vectors presented here may be used as part of a homologous recombination deficiency test to determine whether a cancer patient would be a good candidate for treatment with PARP inhibitors. Previous and ongoing investigations to develop a laboratory test that detects homologous recombination deficiency use deep sequencing-based genomic analysis of the loss of heterozygosity, as they are caused for example by the mutation of BRCA1/2 genes [42, 43]. In contrast, blocking PARP activation using DBD expression followed by low dose exposure to radiation or radiomimetic chemicals inducing DNA strand breaks would identify cells that are hypersensitive compared to appropriate controls as a quick first round screening approach.
The disruption of different Parp genes, including Parp1 and Parp2 has been performed in mice [44–46]. Each of the individual knock-out mice has a relatively mild phenotype but disruption of both genes is lethal at an early embryonic stage [26], suggesting common overlapping as well as individual functions of the two enzymes. In addition, gene knock-down strategies have also been used to target PARP1 expression, but in general these studies have shown that loss of the individual PARP enzymes and their pharmacological block have very different consequences and phenotypes [32, 33, 47].
Although PARP1 overexpression, as well as its functional downregulation has been performed in a great variety of animal cancer cells and in mouse models, research on human cells has been limited mainly to cancer cell lines. Non-immortalized, normal cell populations are mostly difficult to transfect using conventional methods and their limited proliferation capacity in culture hinders extended selection procedures in order to generate stably transfected cell clones, which is another problem effectively addressed by this study.
While there are many endogenous and environmental substances that inhibit PARP1 [48], there is currently no pharmacological solution to increase PARP activity in cells. Poly(ADP-ribosyl)ation has been shown to be of major importance in the general maintenance of chromosomal integrity and genomic stability, particularly under conditions of low or moderate genotoxic stress. In contrast, extremely high levels of PARP1 activation, often designated as “overactivation”, lead to depletion of cellular NAD+ and ATP pools and necrotic cell death [1], reviewed in [2].
The overexpression of PARP1 has been used previously to upregulate steady state levels of PAR [35, 49]. Increased PARP1 expression paradoxically resulted in an increased sensitivity of cells towards ionizing irradiation, probably due to excessive NAD depletion due to an exaggerated PAR formation, but also simultaneously to a reduction of sister chromatid exchange (SCE) frequencies after low DNA damage by alkylating agents, suggesting an improved processing of DNA strand breaks in cells [49] or an involvement in homologous recombination repair, or both. PARP1/2 and PAR-mediated chromatin decondensation is very transient due the enzyme PAR glycohydrolase (PARG), which quickly digests PAR shortly after its formation. The interplay of PARG and PARP1/2 in response to DNA damage involving strand breaks leads to a highly dynamic metabolism of NAD+, PAR and monomeric ADP-ribose where the half-life of PAR is in the range of seconds to minutes [50]. The resulting dramatic NAD depletion and PAR interaction with specific mitochondrial can lead to a quasi-necrotic, and not entirely understood, death of cells with excessive DNA strand breakage due to mitochondrial release of apoptosis inducing factor (AIF) and cytochrome C [3, 51–54]. This type of cell death is more typical in certain primary cells that proliferate only slowly or not at all, like cardiomyocytes, or that do not typically undergo apoptosis, such as fibroblasts. However, human fibroblast transduced with rAd-DBD showed an increase in apoptotic cleavage of PARP1 and displayed an increase in cell death. Fibroblasts typically undergo senescence instead of apoptosis, suggesting that caspase3-dependent PARP cleavage may also be a marker of other types of cell death in normal cells. DBD expression was found to be toxic where very high MOI was used, corresponding to toxicity of DBD itself, not of virus, as judged by a control LacZ virus.
Taken together, utilization of this novel Adenovirus 5-PARP expression system should promote research on the effects of an altered poly(ADP-ribosyl)ation capacity in a wide variety of primary cell types, including those of human origin, and may be useful for screening of DNA repair capacity of normal cells, for example within a personalized medicine program.
References
[1] | Roos WP , Thomas AD , Kaina B . DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer (2016) ;16: (1):20–33. |
[2] | Matt S , Hofmann TG . The DNA damage-induced cell death response: A roadmap to kill cancer cells. Cell Mol Life Sci CMLS (2016) . |
[3] | Hassa PO , Hottiger MO . The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front Biosci J Virtual Libr (2008) ;13: :3046–82. |
[4] | Burkle A . Poly(APD-ribosyl)ation, a DNA damage-driven protein modification and regulator of genomic instability. Cancer Lett (2001) ;163: (1):1–5. |
[5] | Burkle A , Virag L . Poly(ADP-ribose): PARadigms and PARadoxes. Mol Aspects Med (2013) . |
[6] | Meyer RG , Meyer-Ficca ML , Jacobson EL , Jacobson MK . Human poly(ADP-ribose) glycohydrolase (PARG) gene and the common promoter sequence it shares with inner mitochondrial membrane translocase 23 (TIM23). Gene (2003) ;314: :181–90. |
[7] | Meyer RG , Meyer-Ficca ML , Whatcott CJ , Jacobson EL , Jacobson MK . Two small enzyme isoforms mediate mammalian mitochondrial poly(ADP-ribose) glycohydrolase (PARG) activity. Exp Cell Res (2007) ;313: (13):2920–36. |
[8] | Meyer-Ficca ML , Meyer RG , Jacobson EL , Jacobson MK . Poly(ADP-ribose) polymerases: Managing genome stability. Int J Biochem Cell Biol (2005) ;37: (5):920–6. |
[9] | Rouleau M . Poly(ADP-ribosyl)ated chromatin domains: Access granted. J Cell Sci (2004) ;117: (6):815–25. |
[10] | Meyer-Ficca ML , Lonchar JD , Ihara M , Meistrich ML , Austin CA , Meyer RG . Poly(ADP-ribose) polymerases PARP1 and PARP2 modulate topoisomerase II beta (TOP2B) function during chromatin condensation in mouse spermiogenesis. Biol Reprod (2011) ;84: (5):900–9. |
[11] | Bauer PI , Chen HJ , Kenesi E , Kenessey I , Buki KG , Kirsten E , et al. Molecular interactions between poly(ADP-ribose) polymerase (PARP I) and topoisomerase I (Topo I): Identification of topology of binding. FEBS Lett (2001) ;506: (3):239–42. |
[12] | Tramontano F , Di Meglio S , Quesada P . Co-localization of poly(ADPR)polymerase 1 (PARP-1) poly(ADPR)polymerase 2 (PARP-2) and related proteins in rat testis nuclear matrix defined by chemical cross-linking. J Cell Biochem (2005) ;94: (1):58–66. |
[13] | Lis JT , Kraus WL . Promoter cleavage: A topoIIbeta and PARP-1 collaboration. Cell (2006) ;125: (7):1225–7. |
[14] | Malanga M , Althaus FR . Poly(ADP-ribose) reactivates stalled DNA topoisomerase I and Induces DNA strand break resealing. J Biol Chem (2004) ;279: (7):5244–8. |
[15] | Hottiger MO . Nuclear ADP-ribosylation and its role in chromatin plasticity, cell differentiation, and epigenetics. Annu Rev Biochem (2015) ;84: :227–63. |
[16] | Bai P . Biology of poly(ADP-ribose) polymerases: The factotums of cell maintenance. Mol Cell (2015) ;58: (6):947–58. |
[17] | Bock FJ , Todorova TT , Chang P . RNA regulation by poly(ADP-ribose) polymerases. Mol Cell (2015) ;58: (6):959–69. |
[18] | Haince JF , Kozlov S , Dawson VL , Dawson TM , Hendzel MJ , Lavin MF , et al. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J Biol Chem (2007) ;282: (22):16441–53. |
[19] | Guleria A , Chandna S . ATM kinase: Much more than a DNA damage responsive protein. DNA Repair (2016) ;39: :1–20. |
[20] | Goldberg M . KDM4D crosstalks with PARP1 and RNA at DNA DSBs. Cell Cycle (2015) ;14: (10):1495–5. |
[21] | Krishnakumar R , Kraus WL . PARP-1 regulates chromatin structure and transcription through a KDM5B-dependent pathway. Mol Cell (2010) ;39: (5):736–49. |
[22] | Curtin N . PARP inhibitors for anticancer therapy. Biochem Soc Trans (2014) ;42: (1):82–8. |
[23] | Bryant HE , Schultz N , Thomas HD , Parker KM , Flower D , Lopez E , et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature (2005) ;434: (7035):913–7. |
[24] | Farmer H , McCabe N , Lord CJ , Tutt AN , Johnson DA , Richardson TB , et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature (2005) ;434: (7035):917–21. |
[25] | Hottiger MO , Hassa PO , Luscher B , Schuler H , Koch-Nolte F . Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem Sci (2010) ;35: (4):208–19. |
[26] | Menissier de Murcia J , Ricoul M , Tartier L , Niedergang C , Huber A , Dantzer F , et al. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J (2003) ;22: (9):2255–63. |
[27] | Yelamos J , Schreiber V , Dantzer F . Toward specific functions of poly(ADP-ribose) polymerase-2. Trends Mol Med (2008) ;14: (4):169–78. |
[28] | Hans MA , Muller M , Meyer-Ficca M , Burkle A , Kupper JH . Overexpression of dominant negative PARP interferes with tumor formation of HeLa cells in nude mice: Evidence for increased tumor cell apoptosis in vivo . Oncogene (1999) ;18: (50):7010–5. |
[29] | Rudat V , Bachmann N , Küpper JH , Weber KJ . Overexpression of the DNA-binding domain of poly(ADP-ribose) polymerase inhibits rejoining of ionizing radiation-induced DNA double-strand breaks. Int J Radiat Biol (2001) ;77: (3):303–7. |
[30] | Rudat V , Küpper JH , Weber KJ . Trans-dominant inhibition of poly(ADP-ribosyl)ation leads to decreased recovery from ionizing radiation-induced cell killing. Int J Radiat Biol (1998) ;73: (3):325–30. |
[31] | Kupper JH , van Gool L , Burkle A . Molecular genetic systems to study the role of poly(ADP-ribosyl)ation in the cellular response to DNA damage. Biochimie (1995) ;77: (6):450–5. |
[32] | Molinete M , Vermeulen W , Burkle A , Menissier-de Murcia J , Kupper JH , Hoeijmakers JH , et al. Overproduction of the poly(ADP-ribose) polymerase DNA-binding domain blocks alkylation-induced DNA repair synthesis in mammalian cells. EMBO J (1993) ;12: (5):2109–17. |
[33] | Kupper JH , de Murcia G , Burkle A . Inhibition of poly(ADP-ribosyl)ation by overexpressing the poly(ADP-ribose) polymerase DNA-binding domain in mammalian cells. J Biol Chem (1990) ;265: (31):18721–4. |
[34] | He TC , Zhou S , da Costa LT , Yu J , Kinzler KW , Vogelstein B . A simplified system for generating recombinant adenoviruses. Proc Natl Acad Sci U S A (1998) ;95: (5):2509–14. |
[35] | Van Gool L , Meyer R , Tobiasch E , Cziepluch C , Jauniaux JC , Mincheva A , et al. Overexpression of human poly(ADP-ribose) polymerase in transfected hamster cells leads to increased poly(ADP-ribosyl)ation and cellular sensitization to gamma irradiation. Eur J Biochem FEBS (1997) ;244: (1):15–20. |
[36] | Fallaux FJ , Kranenburg O , Cramer SJ , Houweling A , van Ormondt H , Hoeben RC , et al. Characterization of 911: A new helper cell line for the titration and propagation of early region 1-deleted adenoviral vectors. Hum Gene Ther (1996) ;7: (2):215–22. |
[37] | Hamilton MA , Russo RC , Thurston RV . Trimmed Spearman-Karber method for estimating median lethal concentrations in toxicity bioassays. Environ Sci Technol (1977) ;11: (7):714–9. |
[38] | Kawamitsu H , Hoshino H , Okada H , Miwa M , Momoi H , Sugimura T . Monoclonal antibodies to poly(adenosine diphosphate ribose) recognize different structures. Biochem John Wiley Sons (1984) ;23: (16):3771–7. |
[39] | Lazebnik YA , Kaufmann SH , Desnoyers S , Poirier GG , Earnshaw WC . Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature (1994) ;371: (6495):346–7. |
[40] | Kaye SB . Progress in the treatment of ovarian cancer-lessons from homologous recombination deficiency-the first 10 years. Ann Oncol Off J Eur Soc Med Oncol ESMOi (2016) ;27: (Suppl 1):1–3. |
[41] | De Vos M , Schreiber V , Dantzer F . The diverse roles and clinical relevance of PARPs in DNA damage repair: Current state of the art. Biochem Pharmacol (2012) ;84: (2):137–46. |
[42] | Timms KM , Abkevich V , Hughes E , Neff C , Reid J , Morris B , et al. Association of BRCA1/2 defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res BCR (2014) ;16: (6):475. |
[43] | Jenner ZB , Sood AK , Coleman RL . Evaluation of rucaparib and companion diagnostics in the PARP inhibitor landscape for recurrent ovarian cancer therapy. Future Oncol Lond Engl (2016) . |
[44] | Ame JC , Rolli V , Schreiber V , Niedergang C , Apiou F , Decker P , et al. PARP-2, A novel mammalian DNA damage-dependent poly(ADP-ribose) polymerase. J Biol Chem (1999) ;274: (25):17860–8. |
[45] | Shall S , de Murcia G . Poly(ADP-ribose) polymerase- What have we learned from the deficient mouse model? Mutat Res (2000) ;460: (1):1–15. |
[46] | Schreiber V , Ame JC , Dolle P , Schultz I , Rinaldi B , Fraulob V , et al. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem (2002) ;277: (25):23028–36. |
[47] | Sriram CS , Jangra A , Bezbaruah BK , V AK , Sykam S . Alternative mechanisms of inhibiting activity of poly (ADP-ribose) polymerase-1. Front Biosci Sch Ed (2016) ;8: :123–8. |
[48] | Banasik M , Stedeford T , Strosznajder RP . Natural inhibitors of poly(ADP-ribose) polymerase-1. Mol Neurobiol (2012) ;46: (1):55–63. |
[49] | Meyer R , Muller M , Beneke S , Kupper JH , Burkle A . Negative regulation of alkylation-induced sister-chromatid exchange by poly(ADP-ribose) polymerase-1 activity. Int J Cancer J Int Cancer (2000) ;88: (3):351–5. |
[50] | Alvarez-Gonzalez R , Althaus FR . Poly(ADP-ribose) catabolism in mammalian cells exposed to DNA-damaging agents. Mutat Res (1989) ;218: (2):67–74. |
[51] | Yu SW , Wang H , Poitras MF , Coombs C , Bowers WJ , Federoff HJ , et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science (2002) ;297: (5579):259–63. |
[52] | Alvarez-Gonzalez R , Eichenberger R , Althaus FR . Poly(ADP-ribose) biosynthesis and suicidal NAD+ depletion following carcinogen exposure of mammalian cells. Biochem Biophys Res Commun (1986) ;138: (3):1051–7. |
[53] | Koh DW , Dawson TM , Dawson VL . Mediation of cell death by poly(ADP-ribose) polymerase-1. Pharmacol Res Off J Ital Pharmacol Soc (2005) ;52: (1):5–14. |
[54] | Andrabi SA , Kim NS , Yu SW , Wang H , Koh DW , Sasaki M , et al. Poly(ADP-ribose) (PAR) polymer is a death signal. Proc Natl Acad Sci U S A (2006) ;103: (48):18308–13. |
Figures and Tables
Fig.1
Features and approximate sizes of PARP1, PARP1 DBD and their apoptotic fragments. Caspase-3 cleavage of PARP1 takes place in the DEVD G amino acid sequence in the middle of the bipartite nuclear localization signal (NLS). The recognition site of the PARP1 specific antibody (clone C2-10) is depicted by a star. The molecular size of PARP1 protein is 113 kDa and the PARP1 apoptotic fragment recognized by the C2-10 antibody is ∼85 kDa. PARP1 DBD is 44 kDa in size and the apoptotic fragment of PARP-1 DBD recognized by C2-10 is ∼18 kDa.
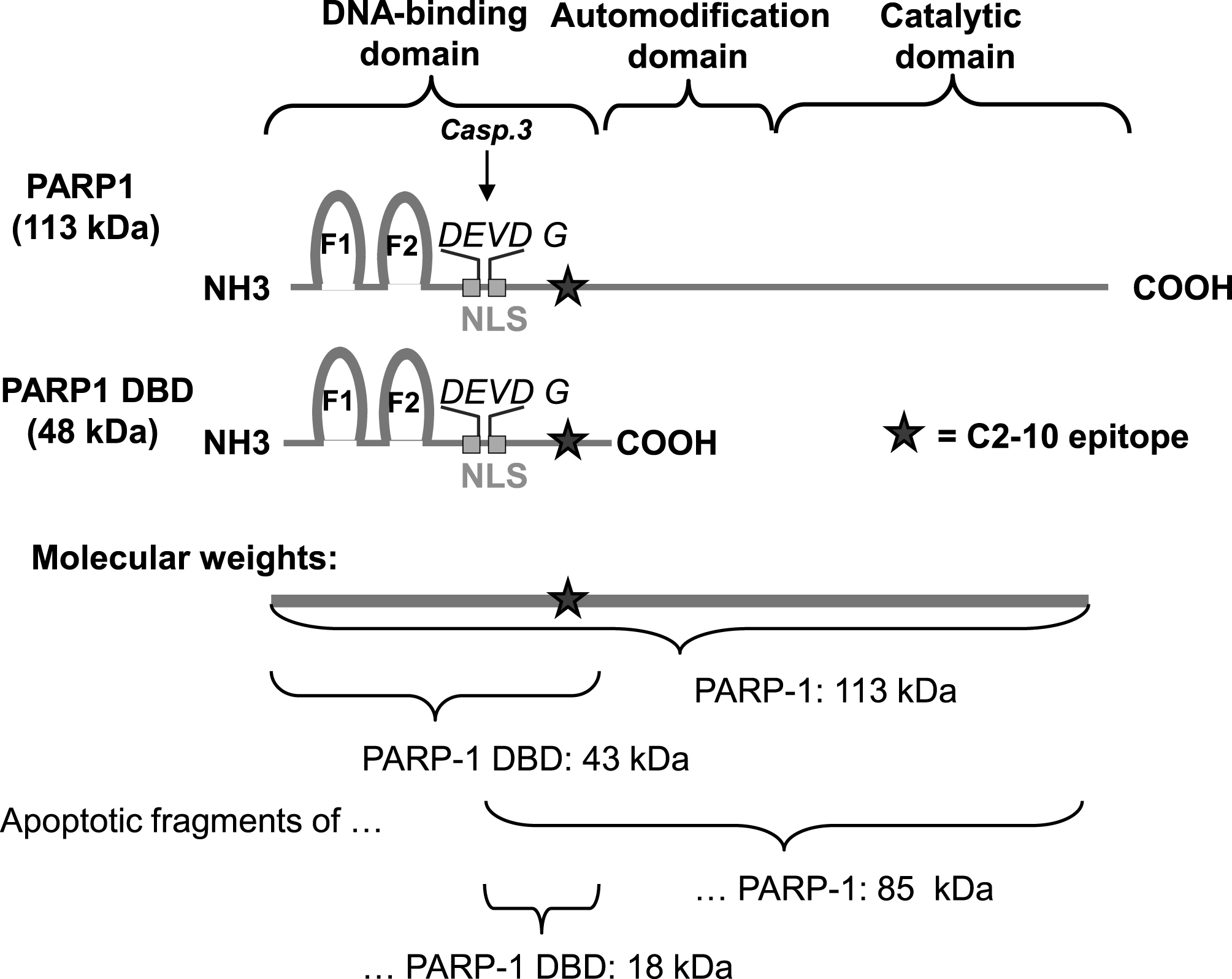
Fig.2
Normal human skin fibroblasts are efficiently transduced by the recombinant adenovirus. Primary fibroblast were transduced with rAdV-PARP-GFP (MOI = 25). GFP expression was analyzed by fluorescence microscopy 24 h after transduction. Cells successfully transduced expressed a GFP reporter (B). Phase contrast image of the same cells (A).
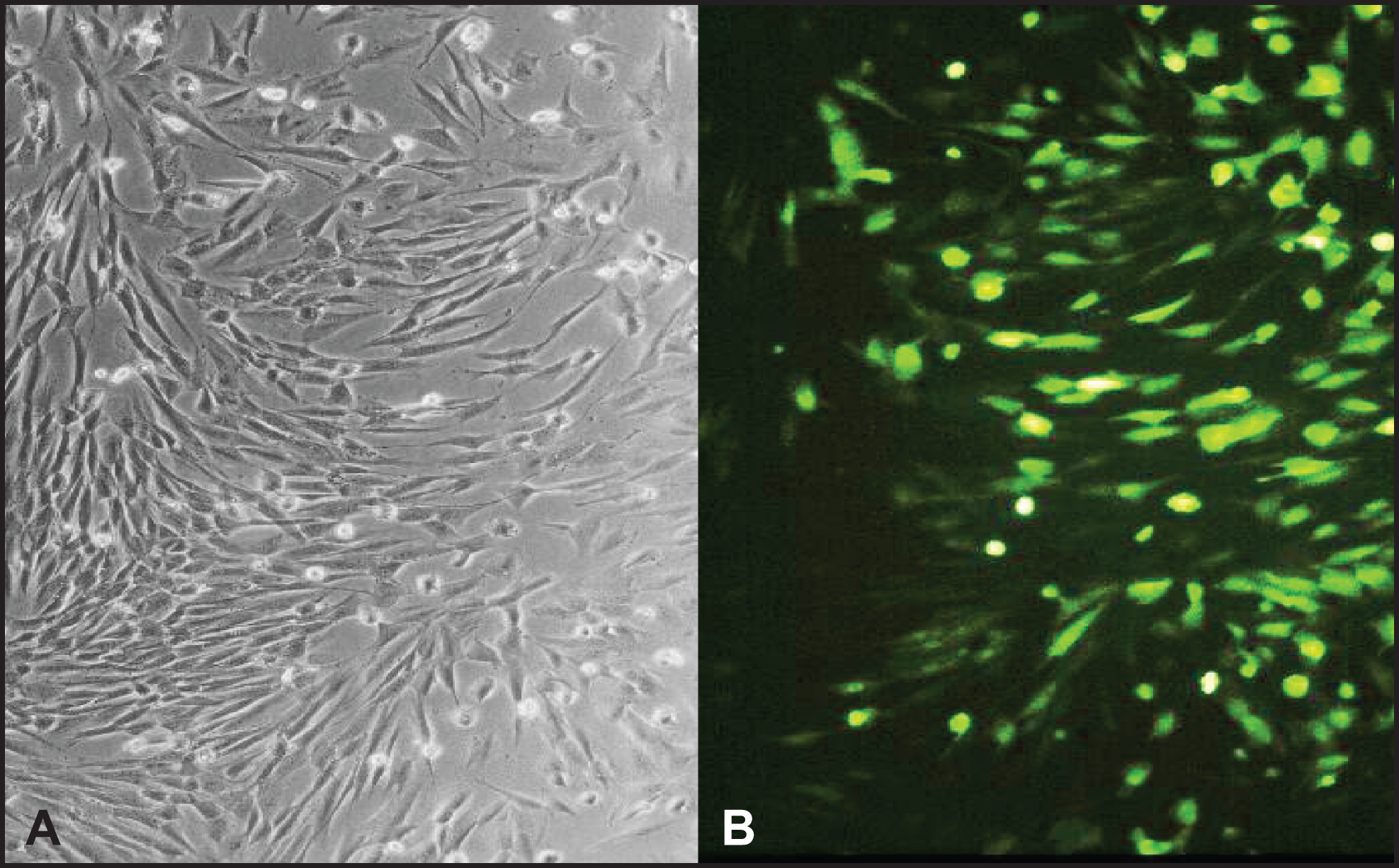
Fig.3
Transduced cells overexpress PARP1 and PARP1 DBD. Normal human fibroblast cells were transduced with rAdV-DBD, rAdV-PARP-GFP and rAdV-PARP (MOI = 25 each). Cell lysates were prepared 24 h after transduction and 1×105 cells were analyzed by western blot analysis using antibody C2-10 to detect PARP1 and DBD. Overexpression of PARP1 DBD and PARP1 of the expected sizes was observed at 45 kDa and 113 kDa, respectively. Apoptotic fragments (arrows) and proteolytic cleavage products of both PARP-1 DBD and PARP-1 were also detectable. Note the slightly lower expression levels obtained with rAd-PARP1-GFP as compared to rAd-PARP1.
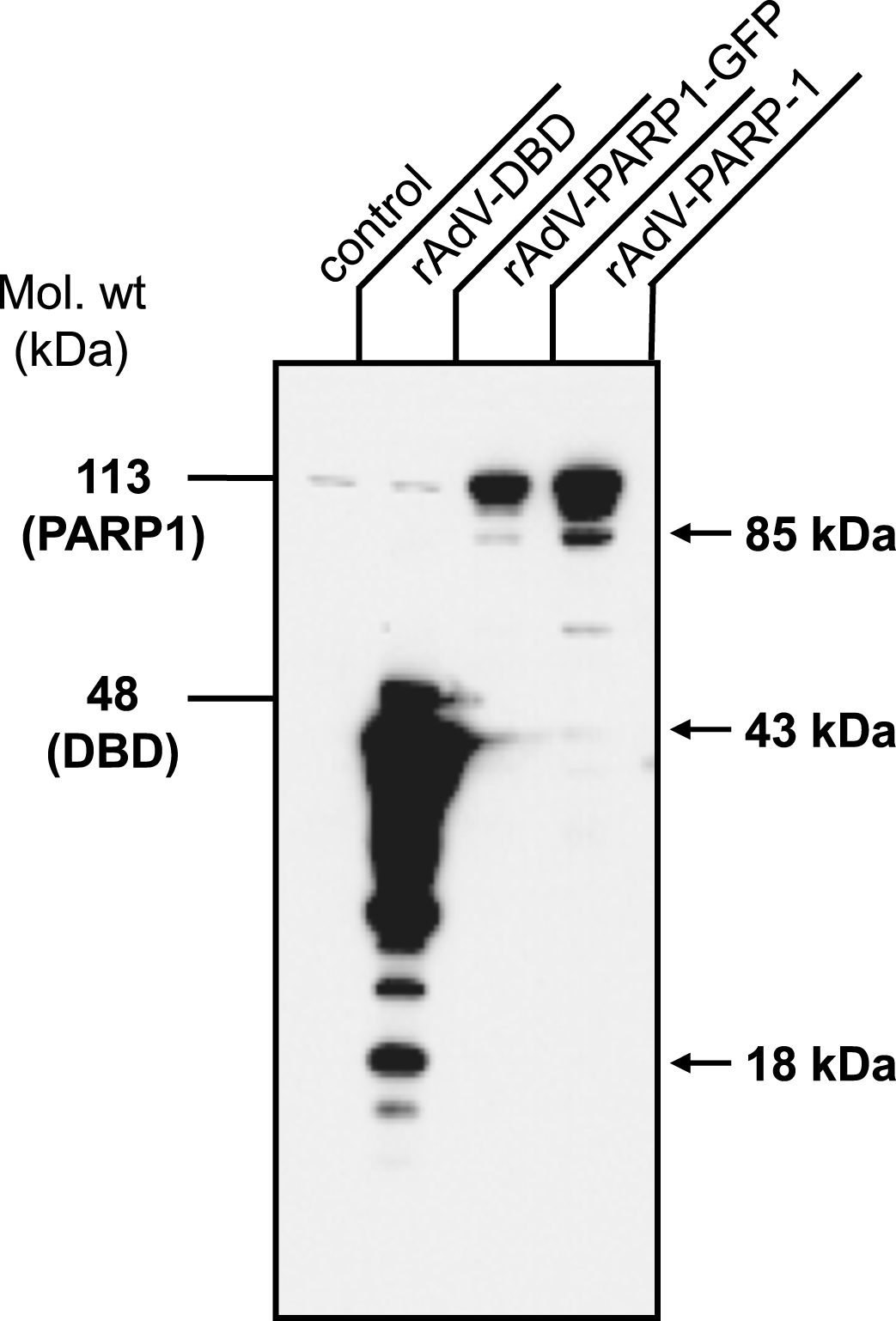
Fig.4
Adenoviral transduction of the dominant negative PARP-1 DNA binding domain induces cell death at high MOI. One million normal human skin fibroblasts were seeded and transduced with rAdV-LacZ and rAdV-DBD at MOI 1, 2, 10 and 100. One day after transduction, cells were trypsinized and cell lysates were prepared and analyzed by Western blot using the PARP1 specific antibody (clone C2-10). Cells transduced with rAdV-DBD at MOI = 100 showed significant cell death and therefore the protein content in this lysate was less than in the other samples, in which no significant cell death was observed. In cells transduced with rAdV-DBD at MOI 1, 10 and 100 Western Blot analysis confirmed very strong expression of PARP1 DBD (low exposure blot, arrowhead). With increasing MOI an increase in PARP1 apoptotic cleavage fragment (below, upper arrow) and PARP1 DBD apoptotic cleavage fragment (lower arrow) was visible in the longer exposed blot. In addition to the apoptotic fragment PARP1 DBD transduction at MOI = 10 also produces several other degradation fragments. Transduction with comparable MOI of rAdV-LacZ did not lead to an increased PARP-1 cleavage or cell death.
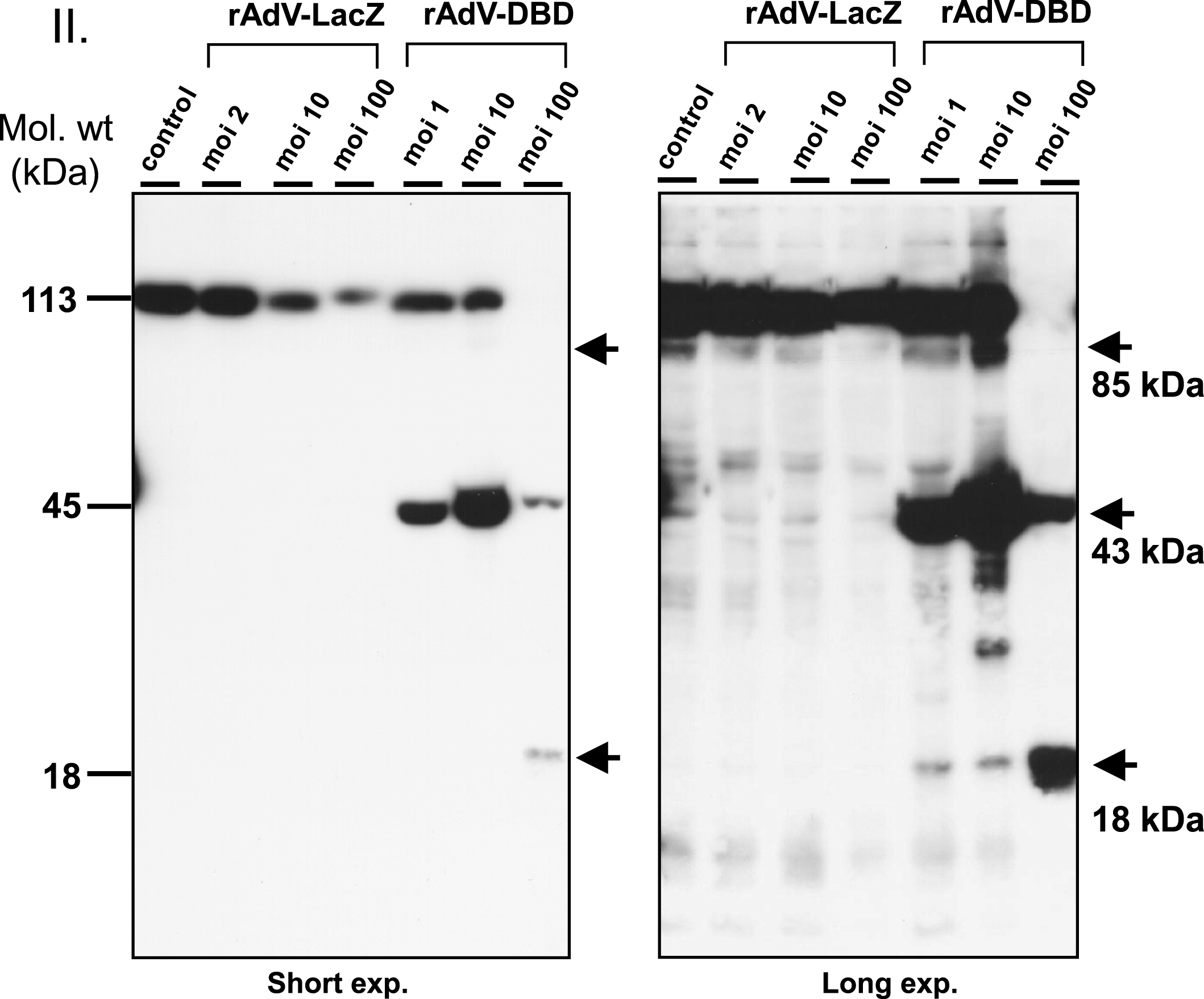
Fig.5
Poly(ADP-ribose) formation is altered in cells overexpressing PARP1 and PARP1 DBD. Normal human skin fibroblasts were transduced with rAdV-DBD and rAdV-PARP (MOI = 25 each). Twenty-four hours after transduction, cells were treated with 200μM MNNG for 20 min. Cells were washed with PBS, fixed with 4% formaldehyde and immunofluorescence staining was performed using a monoclonal antibody (10H) to detect poly(ADP-ribose) (PAR) and a polyclonal rabbit antiserum to detect PARP1. Secondary antibodies used were goat anti-mouse (fluorescein coupled (FITC, green) and goat anti-rabbit (rhodamine-coupled (TRITC, red). DNA was counterstained with DAPI. I. Fibroblast transduced with either rAdV-PARP or rAdV-DBD showed stronger nuclear PARP1 signals (g and l, respectively) than control cells (c). Without exogenously induced DNA damage, no increased PAR formation could be observed in control cells and in PARP1 and PARP1 DBD overexpressing cells (c, f, k). II. After MNNG induced DNA damage, control cells showed strong nuclear poly(ADP-ribose) signals (o). The PARP1 signal in these cells are very strong (p). Therefore the merge of PAR signals and PARP1 signals shows a green nuclear color (q). In cells overexpressing PARP1, the PAR signals (s) were stronger in cells which also showed a stronger PARP1 specific staining (compare arrows in s and t), indicating that a high expression level due to the transduction leads to an increase in PAR formation. In the merged image, these cells appear yellowish, while cells with lower PARP1 expression levels appear greenish (u). In cells which overexpressed the dominant negative DBD of PARP1, cells with strong PARP1 DBD signals (arrows in x) lacked PAR signals (arrows in w). In the merged image (y), these cells therefore appeared red (arrows), while cells without strong DBD signals appeared greenish due to the ADP-ribose polymer signals.