
Effects of in vitro short- and long-term treatment with telomerase inhibitor in U-251 glioma cells
Abstract
BACKGROUND:
The inhibition of the enzyme telomerase (TERT) has been widely investigated as a new pharmacological approach for cancer treatment, but its real potential and the biochemical consequences are not totally understood.
OBJECTIVE:
Here, we investigated the effects of the telomerase inhibitor MST-312 on a human glioma cell line after both short- and long-term (290 days) treatments.
METHODS:
Effects on cell growth, viability, cell cycle, morphology, cell death and genes expression were assessed.
RESULTS:
We found that short-term treatment promoted cell cycle arrest followed by apoptosis. Importantly, cells with telomerase knock-down revealed that the toxic effects of MST-312 are partially TERT dependent. In contrast, although the long-term treatment decreased cell proliferation at first, it also caused adaptations potentially related to treatment resistance and tumor aggressiveness after long time of exposition.
CONCLUSIONS:
Despite the short-term effects of telomerase inhibition not being due to telomere erosion, they are at least partially related to the enzyme inhibition, which may represent an important strategy to pave the way for tumor growth control, especially through modulation of the non-canonical functions of telomerase. On the other hand, long-term exposure to the inhibitor had the potential to induce cell adaptations with possible negative clinical implications.
1Introduction
A pharmacological approach that has been widely investigated for cancer treatment is the target strategy of telomerase reverse transcriptase (TERT) inhibition. The expression of this enzyme is an important hallmark in most human cancers [1, 2]. This leads to the maintenance of telomeric DNA, allowing the bypass of senescence and the immortalization of cancer cell [3]. Importantly, several studies have demonstrated that inhibiting telomerase seems to be effective in controlling some types of cancer cell lines, including osteosarcoma [4], prostrate [5] and pancreas [6]. Moreover, clinical trials were developed to evaluate the effects of telomerase inhibition in different cancer types, including hematological, lung and breast cancer [7].
Most studies with telomerase inhibitors focus on their effects on telomeres shortening and consequent induction of senescence or apoptosis [8–11]. However, these effects are expected to occur after a long-term exposure to telomerase inhibitors, which still has its biochemical consequences largely unknown. In fact, despite important advances in the field, our understanding on the long-term effects of telomerase inhibition as a therapeutic strategy to treat cancer is still very limited.
Overall, in this work we investigated the effects of the telomerase inhibitor MST-312 [12], a synthetic compound derived from epigallocatechin gallate, on the human glioma cell line U-251. This cell line was chosen to be the model due to its convenient characteristics (hTERT expression, high proliferative pattern and susceptibility to transfection), but also because gliomas, and especially glioblastomas, remain a clinical challenge that demands the development of new drugs [13, 14]. We analyzed the consequences of short-term and long-term telomerase inhibition treatment on cancer cells growth, morphology, cell cycle, viability, and its association with histone methyltransferases (HMTs) gene expression, aiming to increase knowledge about the mechanism of action of MST-312 and to investigate the potential of short-term inhibition of telomerase as therapeutic strategy.
2Methods
2.1Cell culture and short-term treatment with MST-312
Telomerase positive human glioma U-251 cells were cultured at 37 °C with Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 44 mM NaHCO3, 10 %fetal bovine serum, 100 IU/mL penicillin and 100μg/mL streptomycin in a humidified atmosphere of 5%CO2 and 95%air. The culture medium was changed every 2 days.
MST-312 (Sigma Aldrich) was diluted in dimethyl sulfoxide (DMSO) to a 50 mM concentration and stored at –20°C. The short-term treatment was performed when cells (3.1×104 /cm2) were plated in 96-wells plate. After 24 h, U-251 cells were treated with increasing concentrations of MST-312 (0.25, 0.5, 1, 2, 5, 10, 15, 25, 35 and 50μM) for 24 h, 48 h and 72 h. DMSO (0.1%) was present in each treated group and in the vehicle control group.
2.2Long-term treatment with MST-312
We used the sub-toxic concentration of 2μM of MST-312 for the long-term treatment in U-251 cells. Cells were grown in 10 cm plates in the continuous presence of the drug or in 0.01%DMSO for 290 days. Trypan blue dye exclusion assay was used to monitor cell viability. When plates reached 70%confluence, cells were detached using 0.05%trypsin, washed with 20%EDTA in PBS and centrifuged at 168 g for 6 minutes. The pellet was suspended, trypan blue was added (0.05%), and cells were counted using a hemocytometer. Blue stained cells were considered unviable. The number of living cells was registered for population doubling calculation and a total of 5,000 viable cells were seeded in a new plate to continue the long-term treatment.
2.3Cell viability analysis
As mentioned previously, to evaluate MST-312 short-term treatment, the U-251 cells were cultured with increasing concentrations of the drug during 24 h, 48 h and 72 h. On the other hand, to evaluate drug resistance using higher doses of MST-312, cells continuously treated with 2μM of MST-312 were collected from the primary experiment in different time points and were re-challenged with 5, 25 or 50μM of MST-312 for 48 h. Both dose response curve of MST-312 and evaluation of resistance were measured by cell viability assay in 96-well plates (N = 8), using the classic MTT method. The cells were incubated with MTT (1 mg/mL) for 2 hours and then lysed (20%SDS and 50%dimethylformamide). After overnight complete formazan crystals solubilization, the absorbance was measured at 540 nm. Cell viability was expressed as percentage of the control (0.1%DMSO), considered as 100%. Cytotoxic effect was confirmed by trypan blue exclusion assay. In this case, after treatments, the culture medium was collected, and cells were removed from plates using 0.05%trypsin, 20%EDTA in PBS for 5 min. Cells were harvested and centrifuged at 150 g for 6 min. Then, the pellet was suspended and mixed with fresh culture medium, and 0.05%trypan blue was added. Five minutes after, the cells were counted in a hemocytometer.
2.4Cell morphology assessment
Cell morphology was registered by phase-contrast microscopy (NIKON Eclipse TS100) and recorded in digital micrographs. The polarity index (PIn) was adopted as quantitative parameter to analyze intergroup differences. For this purpose, the PIn was defined as the ratio between the longest axis and its longest perpendicular (measured in μm) of a single cell. At least twenty cells were measured randomly in each group.
2.5Determination of doubling time (DT)
Cell proliferation profile was accessed by estimating DT (time, in hours, to duplicate the number of cells in culture). Cells were cultured in 96-well plates at sub-confluence conditions (1,000 cells/well) and maintained at standard conditions for 1 week. The medium was changed after the fourth day and cell viability was analyzed by MTT assay, in eight wells at several points of timeline (at approximately 12 hours intervals). After that, data were plotted in a graph and fitted to a nonlinear regression with exponential equation to estimate DT, using GraphPad Prim 5.0 software.
2.6Cell cycle and cell death analyses
The mechanism of cell death induced by short-term treatment was accessed using FITC-Annexin V and propidium iodide (PI) staining approach (annexin-V FITC-Kit, BioSource, Camarillo, USA), that identifies both apoptotic (annexin-V +) and necrotic cells (PI + and annexin-V -). Moreover, analysis of DNA content was performed to assess the cell cycle phase distribution. After the treatments, cells were harvested by trypsinization, fixed with cold 70%ethanol, and stained with PI. A fluorescence-activated flow cytometer FACScalibur (Becton-Dickinson, FranklinLakes, NJ, USA) and the Cell Quest™3.1 software was used for both analyses, considering at least 10,000 events acquired in FL-1 and FL-2 channels.
2.7Evaluation of gene expression profile
Total RNA extraction was performed in three time points of the long-term treatment using TRIzol Reagent (Thermo Fisher Scientific, Waltham, MA, USA), and mRNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. Gene expression profile of HMTs SUV39H1, SUV420H1 and SUV420H2, and TERT was accessed by qPCR. Reactions were performed on a StepOnePlus ™ Real-Time PCR System using TaqMan Universal PCR MasterMix and TaqMan Gene Expression Assays. The probes (Thermo Fisher Scientific, Waltham, MA, USA) were Hs00957892_m1 (SUV39H1), Hs00992344_m1 (SUV420H1), Hs00261961_m1 (SUV420H2), Hs00972656_m1 (hTERT) and Hs99999903_m1(β-actin). Amplification conditions were as follow: 2 minutes at 50°C and 10 minutes at 95°C on holding stage, and then 40 cycles (95°C for 15 s and 60°C for 1 min). To determine the relative quantification (RQ) of gene expression, the data were analyzed using the comparative quantification Ct method (ΔΔCt). The amount of each mRNA, normalized to the endogenous reference (β-actin) and relative to a calibrator was converted into relative quantification by the formula: 2–ΔΔCt.
2.8Measurement of average telomeric DNA length
First, for genomic DNA extraction, cells were trypsinized and washed with PBS. Then, lysis solution (2%SDS) was added, followed by buffer containing 20 mmol/l EDTA and 50 mmol/l Tris. DNA was precipitated with 5 mol/l sodium chloride and ethanol, suspended in water, quantified using a nanospectrophotometer and stocked under –80°C. The relative quantification of the telomere content in DNA samples was performed by real-time PCR. Each 25μl qPCR reaction contains Taq DNA polymerase (0.75 U) in 1×Taq buffer, 0.2 mmol/l each dATP, dCTP, dGTP, dTTP, 1.5 mmol/l MgCl 1.5 mmol/l MgCl2, EvaGreen (Biotium, Fremont, CA, USA) 1×, 10 mmol/l dithiothreitol, 0.5 ml DMSO, 5 ml DNA template, and primer sets. Two reactions were performed: telomere and the single copy gene (SCG) 36B4. The final primer concentrations were as follows: telomere: forward 0.3 mmol/l and reverse 0.4 mmol/l; 36B4: forward 0.3 mmol/l and reverse 0.5 mmol/l. The primer sequences were the same as those described by Lau et al. [15] The PCR conditions were as follows: telomere: 95°C for 15 min, 40 cycles of 95° C for 15 s, 54°C for 30 s and 72°C for 2 min, 36B4: 95°C for 15 min, 35 cycles of 95°C 15 s, 58°C for 30 s, and 72°C for 1 min. For data analysis, the telomere product was normalized with the SCG product.
2.9Production of transient TERT knock down cell line
U-251 cells were transfected with TERT shRNA plasmid (sc-36641-SH, Santa Cruz Biotechnology®), using the Lipofectamine 3000 protocol, submitted to selection with puromycin for five days and then grown until reach 70%confluence. A scramble shRNA plasmid was used as control. Decreased gene expression level was confirmed by qPCR, as previously described. Complementarily, single cells were isolated in 96 well plates by calculated dilution, and viable colonies were selected to develop clonal confluent cultures, in which TERT stable suppression was investigated.
2.10Statistical analysis
Data were tested for normal distribution, by the D’Agostino and Pearson normality test and analyses of skewness and kurtosis, when applicable. Data were expressed as average±SD (qPCR), mean±SEM or median and ranges according to the distribution. The statistical analysis was performed using GraphPad Prism version 5.00 for Windows, GraphPad Software, San Diego California USA. The statistical approach adopted for each analysis is described in the figure legends. Non-parametric tests were used for data with non-normal distribution. Experiments were performed at least in triplicate. Probability values of p < 0.05 were accepted as indication of statistically significant difference. IC50 (inhibitory concentration) and DT were calculated by non-linear regression of the data.
3Results
3.1MST-312 long–term treatment decreases cell proliferation, leading to adaptations, significant morphological alterations, and resistance in U-251 cells
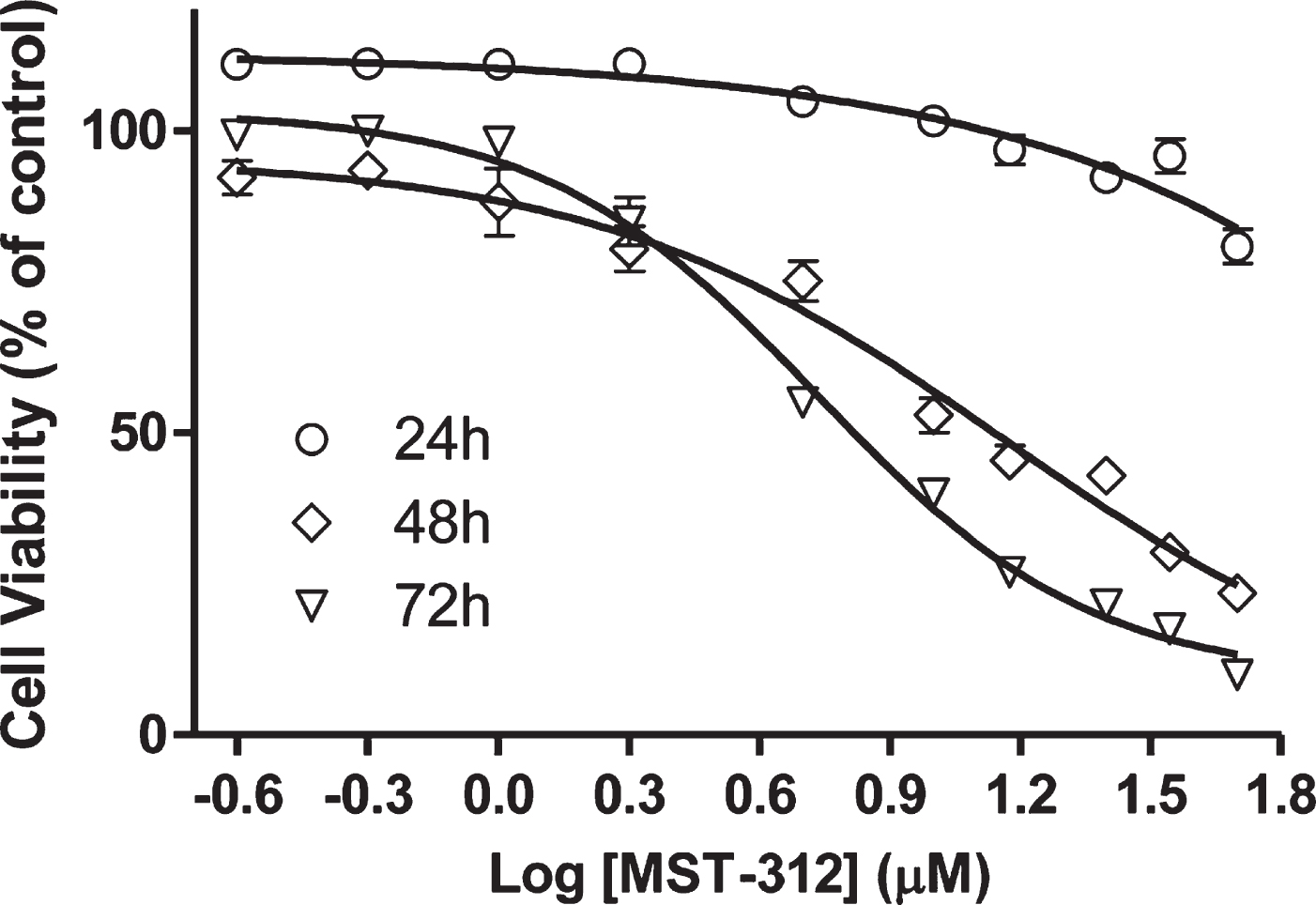
We initially determined the toxic profile of MST-312 towards U-251 cells by plotting dose-response curves. The data shows that the drug induced a significant decrease on cell viability in a concentration-dependent way, even under short-term exposure of 24 hours (Fig. 1). A sub-toxic low concentration (2μM) was used to perform the long-term exposure experiment. This concentration is known to be sufficient to promote telomerase inhibition in cell cultures [12].
Fig. 1
Effects of MST-312 on cell viability. Analysis of cell viability by MTT assay after treatment with MST-312 at increasing concentrations showing a dose-dependent and time-dependent drug toxicity. The IC50 was estimated by non-linear regression. The IC50 and R2 values were 13.88μM / 0.90 and 6.56μM / 0.98 for the 48 h and 72 h curves, respectively. It was not possible to calculate the IC50 for the 24 h curve due to the low toxicity in these conditions.
As expected for telomerase inhibitors, the MST-312 showed no significant effect on culture growth during the initial 3 months of treatment (Fig. 2A). However, in the following months it induced a sustained decrease on the proliferative rate (Fig. 2A), defining the responding phase of the experiment. This phase was accompanied by development of drug resistance toward higher concentrations of MST-312 and remarkable morphological changes, leading to cells with fusiform shape (Fig. 2B,C) The responding phase lasted approximately one hundred days resulting in a significant decrease in proliferation. After this period, the cell culture recovered its original proliferation rate, measured by slope, and typical morphology. In fact, the substantial morphological change in cultured cells, that happened when the drug most impacted the proliferation, became evident by polarity index calculation used as the morphological quantitative parameter (Fig. 3A). There was no detectable alteration in TERT mRNA levels (Fig. 3B) on MST-312 long-term treated cultured cells compared to DMSO control.
Fig. 2
Effects of long-term treatment with MST-312 on U251 cells. (A) Cell growth in cultures long-term treated with DMSO [0.01%DMSO (ltt)] or MST-312 [2μM MST-312 (ltt)] was followed up for 290 days in order to calculate the population doubling time (a growth rate indicator). According to this parameter, the long-term treatment period can be divided into three phases: 1: both groups exhibited the same growth pattern; 2: after 100 days of treatment, the MST-312 treated cells proliferated at a significantly lower rate; 3: finally the MST-312 treated cells recovered the original growth rate (graph slope). (B) This long-term exposition also led to the acquisition of resistance to high concentration of MST-312 showed by MTT assay, but it was restricted to the responding (2) phase. Data are expressed as mean with SEM and analyzed by ANOVA followed by Dunnett’s post-test. (C) The 2μM MST-312 (ltt) treated cells showed remarkable morphological changes under phase-contrast microscopy, during the responding (2) phase.
![Effects of long-term treatment with MST-312 on U251 cells. (A) Cell growth in cultures long-term treated with DMSO [0.01%DMSO (ltt)] or MST-312 [2μM MST-312 (ltt)] was followed up for 290 days in order to calculate the population doubling time (a growth rate indicator). According to this parameter, the long-term treatment period can be divided into three phases: 1: both groups exhibited the same growth pattern; 2: after 100 days of treatment, the MST-312 treated cells proliferated at a significantly lower rate; 3: finally the MST-312 treated cells recovered the original growth rate (graph slope). (B) This long-term exposition also led to the acquisition of resistance to high concentration of MST-312 showed by MTT assay, but it was restricted to the responding (2) phase. Data are expressed as mean with SEM and analyzed by ANOVA followed by Dunnett’s post-test. (C) The 2μM MST-312 (ltt) treated cells showed remarkable morphological changes under phase-contrast microscopy, during the responding (2) phase.](https://content.iospress.com:443/media/tub/2021/43-1/tub-43-1-tub211515/tub-43-tub211515-g002.jpg)
Fig. 3
Effects of long-term exposure to MST-312 on cell morphology, telomerase expression and resistance to cytotoxic effects of the compound. (A) Dramatic changes on cell morphology, accessed by polarity index quantification, during the responding phase of long-term treatment. After 200 days of treatment, no difference on this morphological parameter between control and treated group was found (*** two-tailed p value < 0.001; Mann-Whitney test). (B) The treatment did not change levels of TERT mRNA. (C) Phase contrast micrographs show the resistance of long term MST-312 treated cells to cytotoxic effects of the drug at high concentration, which remains as characteristic of the culture at the end of experiment, showed by trypan blue exclusion assay (D).
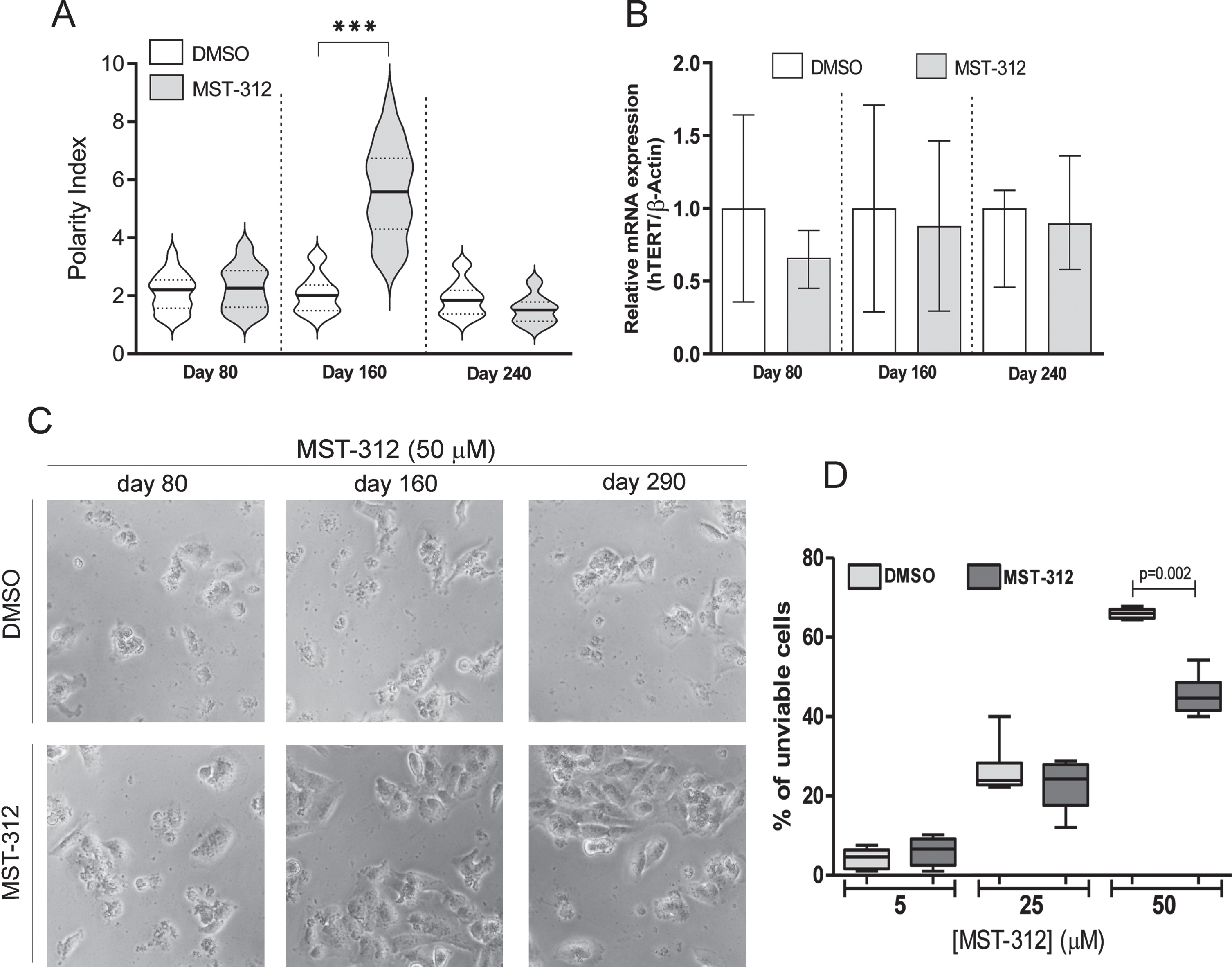
Interestingly, the resistance to deleterious actions of high concentration of MST-312 remained as characteristic of the culture even at the very end of the long-term experiment (Fig. 3C,D).
3.2U-251 cells presented longer telomeres and increased HMTs genes expression during MST-312 responding phase
As expected, the telomeres of cells treated with the telomerase inhibitor were shortened in the first months of treatment. However, at the end of phase 2 the average telomeres length in the treated culture was approximately five times longer than control. Furthermore, after recovering original proliferative rate, the telomere lengths were similar in MST-312 and DMSO treated cultures (Fig. 4A). Also, the expression of SUV39H1, SUV420H1 and SUV420H2 HMTs (which are associated with genetic stability and telomere recombination) was significantly increased in the responding phase of the long-term exposure to MST-312 (Fig. 4B). It was not observed any significant HMTs gene expression alteration neither with 80 days of culture nor with 240 days, as described with other parameters previously.
Fig. 4
Effects of long-term exposure to MST-312 on average telomere length and expression of specific methyltransferases. After a retraction of approximately 70%in the first 100 days, the longest telomeres were observed in the end of responding (2) phase. In the end of the experiment, the average telomeres lengths were the same in control and treated groups (A). Coincidently, the expression of SUV39H1, SUV420H1 and SUV420H2 histone methyltransferases were increased in the responding phase, but not in early or late phases of the long-term exposure to MST-312 (B). There were no changes in the expression levels on DMOS control groups (data not shown).
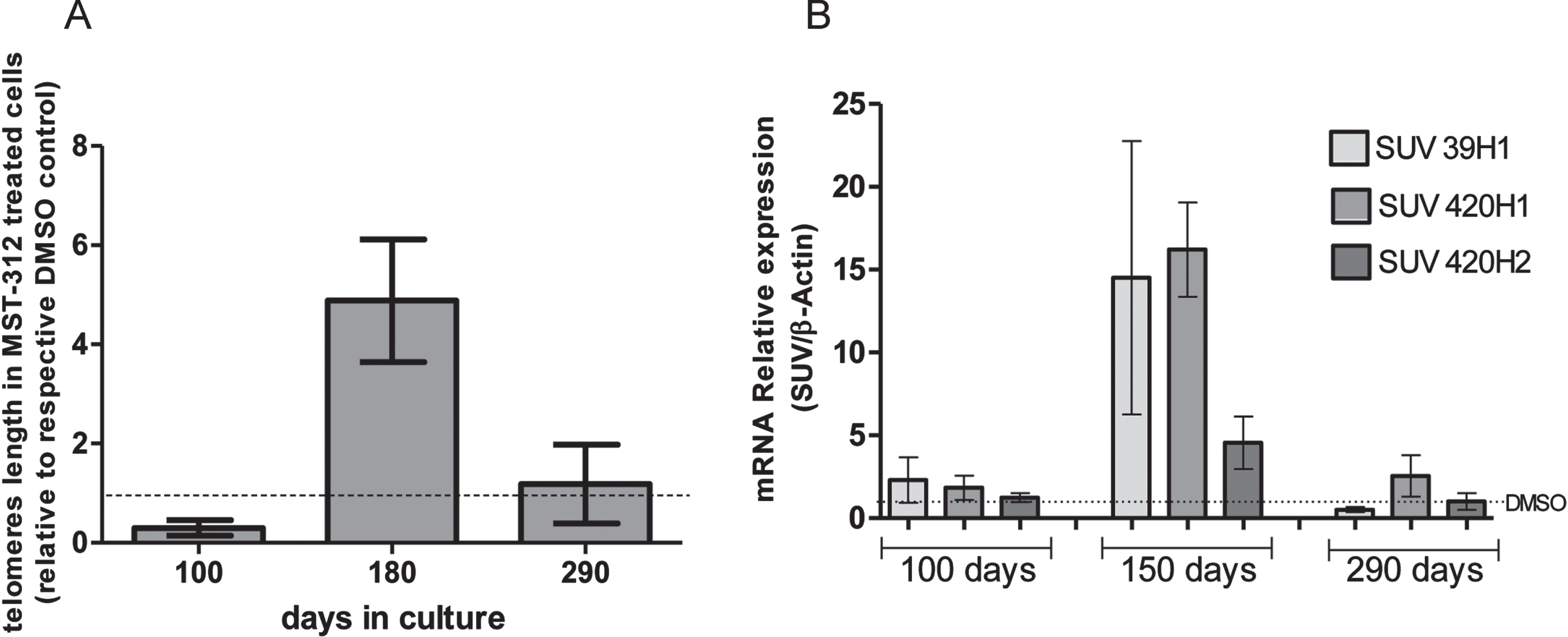
Fig. 5
Toxic effect of MST-312 at low concentration on U-251 cells. (A) MTT assay showing no toxicity of 2μM of MST-312 after 24 h treatment, but with decrease on cell viability after 48 h treatment (* two-tailed p value < 0.05; Mann-Whitney test). (B) Assessment of apoptotic (Propidium Iodide negative annexin V positive) cells after 24 h and 48 h exposition to the drug (** two-tailed p value < 0.01; unpaired t-test), indicating apoptosis induction. (C) Gate with representative events of cell cycle analysis. (D) Representative graph showing cell cycle arrest in G0/G1 phase after 24 h treatment with 2μM of MST-312. (E) Quantification of the effect (N = 3, ** two-tailed p value < 0.01; ***p < 0.001; compared to respective controls - unpaired t-test).
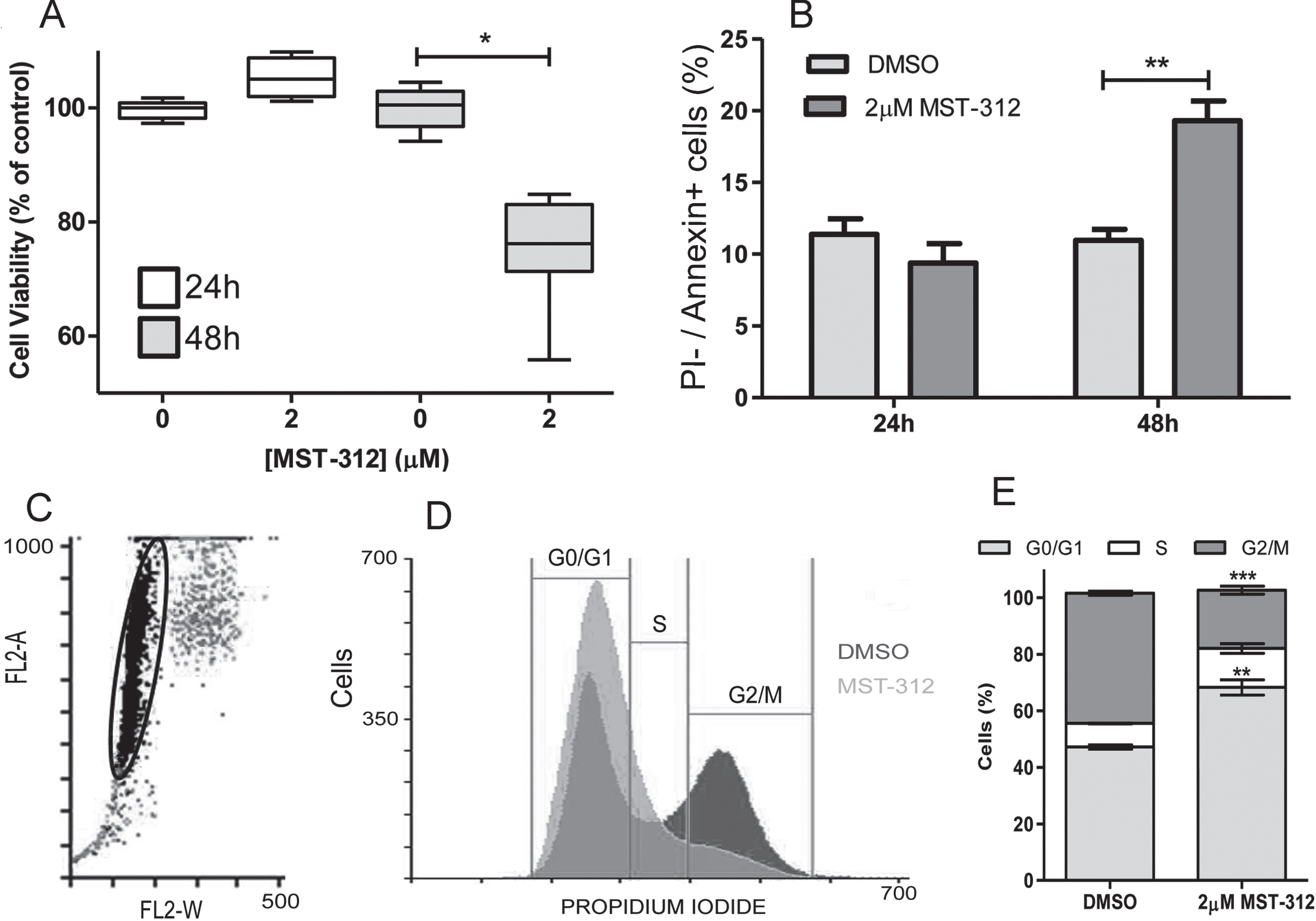
3.3Short-term treatment with MST-312 promotes apoptosis that is preceded by cell cycle arrest in U-251 cell line
Given that long-term treatment did not prove to be the best telomerase based antitumor strategy, and considering the dose-dependent toxic effect observed under the short-term treatment, we decided to investigate the mechanism of cell death induced by MST-312 in this scenario. Results from Annexin V/ Propidium iodide staining ratified the data from viability assay (Fig. 5A) suggesting that 2μM of the telomerase inhibitor used was able to significantly induce apoptosis, but only after 48 h treatment (Fig. 5B). Interestingly, 2μM of MST-312 induced cell cycle arrest in G0/G1 phase after 24 h of treatment (when no cytotoxic effect was observed), affecting 68%of treated cells compared to 47%of control cells (Fig. 5C, D, E), leading us to hypothesize that the cell death may be secondary to the impacts of telomerase inhibition on cell cycle.
3.4TERT knock down promotes resistance of U-251 cells to MST-312
To evaluate the relevance of telomerase inhibition to cytotoxic action of the drug, we generated TERT knock down U-251 cells by transient inhibition of TERT mRNA expression (Fig. 6A). These cells with reduced telomerase expression were significantly more resistant (50.7±4.2%of viable cells) to the highest concentration of MST-312 (50μM) compared to control cells (27.9±3.5%of viable cells) (Fig. 6B and C), despite the similar IC50 values (Table 1).
Fig. 6
Toxic effect of MST-312 on TERT knock-down U-251 cells. (A) Characterization of knock-down cells by qPCR showing low TERT expression in TERT -shRNA plasmid transfected cells compared to control-shRNA plasmid transfected cells. (B) 72 h dose-response curve (MTT assay, the control is not shown due to the use of log of concentrations - basal viability values in both control and KD groups were comparable) showing that TERT knock-down cells are more resistant to MST-312 effects, which was also clearly demonstrated by phase-contrast microscopy analysis (C). (D) In addition, clonal cultures with high TERT expression exhibited greater susceptibility to high concentration of the drug (50μM) (72 h treatment; * two-tailed p value < 0.05; Mann-Whitney test).
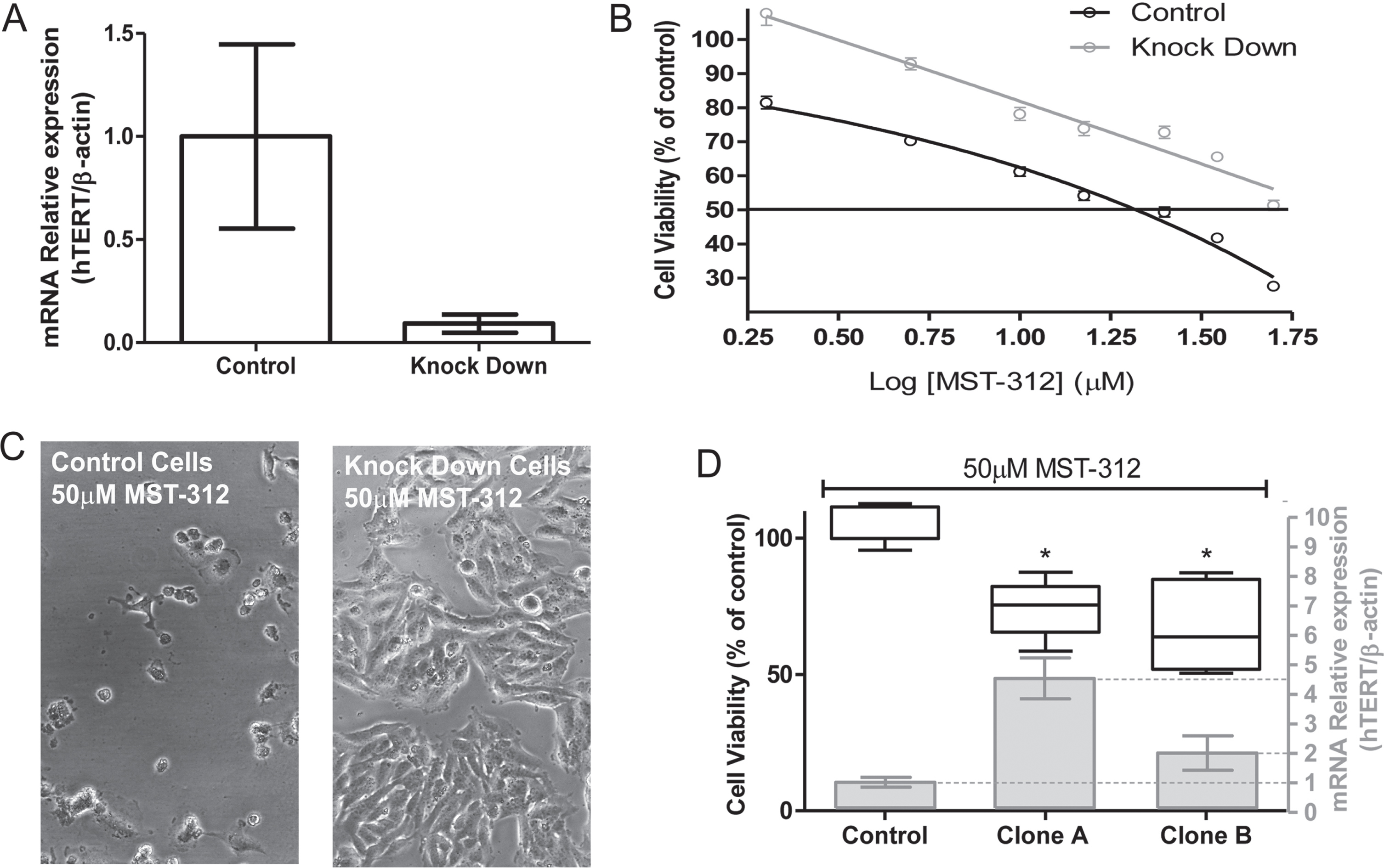
Table 1
Characteristics of U-251cell cultures with different patterns of TERT expression
| Sample | IC50 (72 h) | TERT (relative expression) | Doubling Time (hours) |
| U-251 parental | 6.56 | 1 | 47.65 |
| U-251 control-shRNA | 14.21 | 0.99 | 53.86 |
| U-251 TERT knock down | NC* | 0.09 | 70.25 |
| U-251 clone A | 15.64 | 4.86 | 71.24 |
| U-251 clone B | 10.26 | 2.1 | 50.45 |
TERT knock down in U-251 cells. U-251 TERT knock down cells, showing reduced TERT expression, were resistant to toxic activity of MST-312 compared to control transfected cells, even when compared to cultures with similar proliferative pattern. *not calculable.
We attempted to generate stable TERT knock-out clonal cells. However, the process of clonal selection was not satisfactory. The colonies that spontaneously rescued TERT expression were the only ones that thrived. Two of these clonal cultures (clones A and B) showed TERT expression higher than the control (scramble shRNA) and the parental cell line (Table 1). Corroborating with the data from TERT knock down cells, both clones with increased TERT expression were more sensitive to the toxic effects of MST-312 at the highest concentration (Fig. 6D). Taking into account the mechanism previously described, the toxic effect of MST-312 may be partially cell cycle dependent, so that a low proliferation rate could explain the weaker response of knock down cells to the drug. However, the calculation of Doubling Time for all cells involved in the experiments clarified that there is no correlation between proliferative profile and response to MST-312 (Table 1). The knock down and clone A cells, for instance, have approximately the same DT, but different susceptibility to telomerase inhibitor.
4Discussion
The study of telomerase as pharmacological target is strategic due to the cancer-related nature of this protein. Since its first description as telomerase inhibitor, MST-312 has had its activity tested in several experimental models [16–18]. Our data reinforce the potential of these inhibitors for the development of new anticancer drug, since interference in telomerase actions led to cytotoxicity. In this model, the compound showed a time and dose-dependent toxic effect against U-251 glioma cells at short-term exposure, suggesting a telomere-shortening independent mechanism. The lowest toxic concentration among those tested was chosen for a very long-term exposure, which can be divided into three different phases, early and late phases with no apparent response, and an intermediate period of time that we called responding phase, in which morphology changes and other biochemical markers alterations could be observed. This pattern is suggestive of adaptation by selective pressure leading to resistant cells [19].
As previously demonstrated for breast cancer cells [19], after a reduction of average telomeres length provoked by telomerase inhibition on U-251 cells, if time enough is given to the culture, a reversion of this condition can be observed, with increase of telomeres length, probably by selection of long telomeres clones and finally recue of original phenotype. This finding reinforce the idea that traditional dogma (of treating cancer by induction of telomeric crisis) can represent a recipe for therapeutic failure and a selective pressure for adaptations with possible negative clinical meaning.
Keeping longest telomeres requires biochemical adaptations. The telomeres have heterochromatic features and the main histone marks that characterize, heterochromatin formation are the trimethylation of histone H3 at lysine 9 (H3K9me3) and of histone H4 at lysine 20 (H4K20me3). The histone methyltransferases (HMT) known to catalyze these modifications are the SUV39H1/H2 and SUV420H1/H2. In fact, HMT have shown important roles in the maintenance of cell viability under genomic stress [20–22]. The HMT SUV420H and SUV39H HMTs were shown to be involved in telomere recombination [23]. SUV39H1, for instance, is associated with aberrant telomere elongation [24], besides acquisition of altered and complex karyotypes in chronic lymphocytic leukemia [21]. We addressed the expression of the HMT SUV39H1, SUV420H1 and SUV420H2 in long-term treated cells. Interestingly, all of them were overexpressed in long telomeres culture, but went back to its original levels in the end of the experiment, when cells recovered the initial telomeres lengths and proliferation rate.
However, it is important to take into account the role of HMT in epigenetic control of genes expression and cell signaling pathways relevant for tumor growth [25]. Inactivating mutation of SUV420H1 in tumor subpopulations, for instance, can abrogate DNA repair and it was associated with increased invasion and migration in glioma [26]. Then, the described changes in HMT expression levels can directly impact in a number of biochemical processes, leading to cells with a new set of properties, including susceptibility to apoptosis. In fact, a globally plastic and open chromatin found in stem cell-like subpopulation of human glioblastoma cells was linked to the reduced chromatin-binding affinity of SUV39H1 with consequent reduction of its repressive histone H3 lysine 9 trimethylation (H3K9me3) mark, which prevented apoptosis [27], and which up-regulation OF SUV39H1 may compensate it.
Given the relevance of telomerase for the maintenance of proliferative rate in most tumor cells, it is easy to understand that inhibition of its activity is a strong biochemical pressure that can lead to acquisition of resistance through more than one mechanism in cancer cells, such as the development of Alternative Lengthening of Telomeres (ALT), dysregulated transcriptional networks related to mitochondrial biology [28], and increasing of TERT expression in response to long-term treatment [29], which was not the case in our model. Other mechanism could include telomeric heterochromatin regulation, with protection and maintenance of telomere length by epigenetic regulators such as the SUV HMTs [23, 24], which would explain the observed upregulation of the SUV39H1, SUV420H1 and SUV420H2 enzymes in an attempt to scape cell death caused by telomere erosion, leading to the observed low rate of cell proliferation during the responding phase and to the acquisition of resistance to high concentration of MST-312. Corroborating with these findings, although studies are necessary for the HMTs, there are evidence that cancer cells can become tolerant to drugs through dynamic epigenetic chromatin remodeling [30, 31]. All of this data together show that the long term needed for strategies aiming telomere erosion represents a limitation for the development of clinically useful therapies. However, recent evidence has shown multiple roles of telomerase that are not related to telomere lengthening, the so called non-canonical functions [32, 33], implicating this enzyme, for example, with cell cycle control [34]. Therefore, telomerase remains an interesting potential target for new anticancer drugs development [35–37].
The short-term experiments demonstrated that MST-312 was able to interfere with the cell cycle leading to apoptosis in our in vitro model. In fact, telomerase has been implicated in cell cycle regulation [38], and many other telomerase inhibitors have been shown to act on cell cycle, including the specific inhibitor Imetelstat [39–41]. Moreover, the ability to induce G2/M arrest was previously described for MST-312 [17], contrasting with our findings (G0/G1 arrest) and suggesting that this mechanism of action may be tumor or even cell line specific. It is true that most of the classical anticancer drugs act, directly or indirectly, on cell cycle, but very few of them in a cancer-specific target like telomerase, evidencing the importance of our findings. In summary, it seems that, in the short term, the main mechanism for drug toxicity is its interference in the cell division and that at long term, the inhibition of telomerase triggers a compensatory mechanism that activates epigenetic regulators of heterochromatin for telomere maintenance and, ultimately, resistance against MST-312. This interference in cell cycle could also explain the chemosensitizer activity already demonstrated for MST-312 [42]. Finally, the lack of specificity of MST-312 can represent a bias when its anti-telomerase properties are under investigation. However, although the toxic effects of MST-312 were not totally reversed with the reduction of TERT expression, it clearly induced a significant decrease in the response to the inhibitor.
Therefore, our results showed that long-term exposure to the MST-312 resulted in the induction of cell adaptations with possible negative clinical implications, adding arguments against inducing telomere erosion as antitumor mechanism of drugs (beyond the long periods of treatment required). On the other hand, telomerase inhibition by small molecules may represent an important strategy for the development of new pharmacological therapies due to modulation of the non-canonical functions of the enzyme, and considering its high potential to specifically target tumor cells.
Acknowledgments
We would like to thank Professor Luis Alexandre Muehlmann and Professor Rodrigo Haddad for their valorous contribution.
This work was supported by Federal District Research Support Foundation (FAPDF) and the National Council for Scientific and Technological Development (CNPq).
Author contributions
CONCEPTION: DMO, THAM and DAR
DATA CURATION: THAM and AFRG
ANALYSIS OF DATA: AESC, DMO and GPFL
PREPARATION OF THE MANUSCRIPT: FSA, FPS, DMO, THAM and DAR
REVISION FOR IMPORTANT INTELLECTUAL CONTENT: FSA, FPS, DMO, THAM and DAR
Conflict of interests
The authors declare that there is no conflict of interest.
Data availability
All data generated or analyzed during this study are included in this published article.
References
[1] | Ruden M , Puri N . Novel anticancer therapeutics targeting telomerase. Cancer Treat. (2013) ;39: (5):444–56. doi: 10.1016/j.ctrv.2012.06.007 |
[2] | Alnafakh Rafah AA , Adishesh M , Button L , et al. Telomerase and Telomeres in Endometrial Cancer. Frontiers in Oncology. (2019) ;9: (17):344. doi: 10.3389/fonc.2019.00344 |
[3] | Maciejowski J , De Lange T . Telomeres in cancer: tumour suppression and genome instability. Nature Reviews Molecular Cell Biology. (2017) ;18: (3):175–86. doi: 10.1038/nrm.2016.171 |
[4] | Hu Y , Bobb D , He J , et al. The HSP90 inhibitor alvespimycin enhances the potency of telomerase inhibition by imetelstat in human osteosarcoma. Cancer Biology & Therapy. (2015) ;16: (6):949–57. doi: 10.1080/15384047.2015.1040964 |
[5] | Marian CO , Wright WE , Shay JW . The effects of telomerase inhibition on prostate tumor-initiating cells. International Journal of Cancer. 2010. Doi:10.1002/ijc.25043 |
[6] | Burchett KM , Yan Y , Ouellette MM . Telomerase Inhibitor Imetelstat (GRN163L) Limits the Lifespan of Human Pancreatic Cancer Cells. PLoS ON. (2014) ;9: (1):e85155. doi: 10.1371/journal.pone.0085155 |
[7] | Jafri MA , Ansari SA , Alqahtani MH , et al. Roles of telomeres and telomerase in cancer, and advances in telomerase targeted therapies. Genome Medicine. 2016. doi:10.1186/s13073-016-0324-x |
[8] | Ivancich M , Schrank Z , Wojdyla L , et al. Treating cancer by targeting telomeres and telomerase. Antioxidants. (2017) ;6: (1):15. doi: 10.3390/antiox6010015 |
[9] | Udroiu I , Marinaccio J , Sgura A . Epigallocatechin-3-gallate induces telomere shortening and clastogenic damage in glioblastoma cells. Environmental and Molecular Mutagenesis. (2019) ;60: (8):683–92. doi: 10.1002/em.22295 |
[10] | Ding X , Cheng J , Pang Q , et al. BIBRa Selective Telomerase Inhibitor, Enhances Radiosensitivity of Non-Small Cell Lung Cancer Through Increasing Telomere Dysfunction and ATM/CHK1 Inhibition. International Journal of Radiation. Oncology* Biology* Physics. (2019) ;105: (4):861–74. doi: 10.1016/j.ijrob2019.08.009 |
[11] | Chen ZF , Qin QP , Qin J , et al. Stabilization of G-quadruplex DNA, inhibition of telomerase activity, and tumor cell apoptosis by organoplatinum (II) complexes with oxoisoaporphine. Journal of Medicinal Chemistry. (2015) ;58: (5):2159–79. doi: 10.1021/jm5012484 |
[12] | Seimiya H , Oh-hara T , Suzuki T , et al. Telomere shortening and growth inhibition of human cancer cells by novel synthetic telomerase inhibitors MST-312, MST-295, and MST-1991, Mol Cancer Ther. (2002) ;1: (9):657–65. |
[13] | Bush NAO , Chang SM , Berger S . Current and future strategies for treatment of glioma. Neurosurgical Review. (2016) ;40: (1):1–14. doi: 10.1007/s10143-016-0709-8 |
[14] | Sharma M , Juthani R , Vogelbaum M . Updated response assessment criteria for high-grade glioma: beyond the MacDonald criteria. Chin Clin Oncol. (2017) ;6: (4): 37 1–13. doi: 10.21037/cco.2017.06.26 |
[15] | Lau LM , Dagg RA , Henson JD , et al. Detection of alternative lengthening of telomeres by telomere quantitative PCR. Nucleic Acids Res. (2013) ;41: :e34. doi: 10.1093/nar/gks781 |
[16] | Gurung RL , Lim SN , Low GK , Hande MP . MST-312 Alters Telomere Dynamics, Gene Expression Profiles and Growth in Human Breast Cancer Cells. J Nutrigenet Nutrigenomics. (2014) ;7: (4-6):283–98. doi: 10.1159/000381346 |
[17] | Fatemi A , Safa M , Kazemi A . MST-312 induces G2/M cell cycle arrest and apoptosis in APL cells through inhibition of telomerase activity and suppression of NF-κB pathway. Tumour Biol. (2015) ;36: (11):8425–37. doi: 10.1007/s13277-015-3575-z |
[18] | Ameri Z , Ghiasi S , Farsinejad A , et al. Telomerase inhibitor MST-312 induces apoptosis of multiple myeloma cells and down-regulation of anti-apoptotic, proliferative and inflammatory genes. Life Sci. (2019) ;228: (1):66–71. doi: 10.1016/j.lfs.2019.04.060 |
[19] | Morais KDS , Arcanjo DDS , de Faria Lopes GP , et al. Long-term in vitro treatment with telomerase inhibitor MST-312 induces resistance by selecting long telomeres cells. Cell Biochem Funct. (2019) ;37: (4):273–80. doi: 10.1002/cbf.3398 |
[20] | Vajen B , Modlich U , Schienke A , et al. Histone methyltransferase Suv39h1 deficiency prevents Myc-induced chromosomal instability in murine myeloid leukemias. Genes Chromosomes Cancer. (2013) ;52: (4):423–30. doi: 10.1002/gcc.22040 |
[21] | Carvalho Alves-Silva J , do Amaral Rabello D , Oliveira Bravo M , et al. Aberrant levels of SUV39H1 and SUV39H2 methyltransferase are associated with genomic instability in chronic lymphocytic leukemia. Environ Mol Mutagen. (2017) ;58: (9):654–61. doi: 10.1002/em.22128 |
[22] | Bromberg KD , Mitchell TR , Upadhyay AK , et al. The SUV4-20 inhibitor A-196 verifies a role for epigenetics in genomic integrity. Nat Chem Biol. (2017) ;13: (3):317–24. doi: 10.1038/nchembio.2282 |
[23] | Benetti R , Gonzalo S , Jaco I , et al. Suv4-20h deficiency results in telomere elongation and derepression of telomere recombination. J Cell Biol. (2007) ;178: (6):925–36. doi: 10.1083/jcb.200703081 |
[24] | García-Cao M , O’Sullivan R , Peters AH , et al. Epigenetic regulation of telomere length in mammalian cells by the Suv39h1 and Suv39h2 histone methyltransferases. Nat Genet. (2004) ;36: (1):94–9. https://doi.org/10.1038/ng1278 |
[25] | Vougiouklakis T , Sone K , Saloura V , et al. SUV420H1 enhances the phosphorylation and transcription of ERK1 in cancer cells. Oncotarget. (2015) ;6: (41):43162–71. doi: 10.18632/oncotarget.6351 |
[26] | Vinci M , Burford A , Molinari V , et al. Functional diversity and cooperativity between subclonal populations of pediatric glioblastoma and diffuse intrinsic pontine glioma cells. Nat Med. (2018) ;24: (8):1204–15. doi: 10.1038/s41591-018-0086-7 |
[27] | Mallm JP , Windisch P , Biran A , et al. Glioblastoma initiating cells are sensitive to histone demethylase inhibition due to epigenetic deregulation. Int J Cancer. (2020) ;146: (5):1281–92. doi: 10.1002/ijc.32649 |
[28] | Hu J , Hwang SS , Liesa M , et al. Antitelomerase therapy provokes ALT and mitochondrial adaptive mechanisms in cancer. Cell. (2012) ;148: (4):651–63. doi: 10.1016/j.cell.2011.12.028 |
[29] | Morais KS , Guimarães AFR , Ramos DAR , et al. Long-term exposure to MST-312 leads to telomerase reverse transcriptase overexpression in MCF-7 breast cancer cells. Anticancer Drugs. (2017) ;28: (7):750–6. doi: 10.1097/CAD.0000000000000508 |
[30] | Yan H , Chen X , Zhang Q , et al. Drug-tolerant cancer cells show reduced tumor-initiating capacity: depletion of CD44 cells and evidence for epigenetic mechanisms. PLoS One. (2439) ;6: (9):e7. doi: 10.1371/journal.pone.0024397 |
[31] | Sharma SV , Lee DY , Li B , et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. (2010) ;141: (1):69–80. https://doi.org/10.1016/j.cell.2010.02.027 |
[32] | Thompson CAH and Wong JMY , Non-canonical Functions of Telomerase Reverse Transcriptase: Emerging Roles and Biological Relevance. Curr Top Med Chem. (2020) ;20: (6):498–507. doi: 10.2174/1568026620666200131125110 |
[33] | Ghareghomi S , Ahmadian S , Zarghami N , Kahroba H . Fundamental insights into the interaction between telomerase/TERT and intracellular signaling pathways. Biochimie. (2021) ;181: :12–24. doi: 10.1016/j.biochi.2020.11.015 |
[34] | Fleisig HB and Wong JMY , Telomerase promotes efficient cell cycle kinetics and confers growth advantage to telomerase-negative transformed human cells. Oncogene. (2012) ;31: (8):954–65. doi: 10.1038/onc.2011.292 |
[35] | Berei J , Eckburg A , Miliavski E , et al. Potential Telomere-Related Pharmacological Targets. Curr Top Med Chem. (2020) ;20: (6):458–84. doi: 10.2174/1568026620666200109114339 |
[36] | Choi SH , Cho KJ , Yun SH , et al. HKR3 regulates cell cycle through the inhibition of hTERT in hepatocellular carcinoma cell lines. J Cancer. (2020) ;11: (9):2442–52. doi: 10.7150/jca.39380 |
[37] | Ningarhari M , Caruso S , Hirsch TZ , et al. Telomere length is key to hepatocellular carcinoma diversity and telomerase addiction is an actionable therapeutic target. J Hepatol.. (2021) ;74: (5):1155–66. doi: 10.1016/j.jhe2020.11.052 |
[38] | Noureen N , Wu S , Lv Y , et al. Integrated analysis of telomerase enzymatic activity unravels an association with cancer stemness and proliferation. Nat Commun. (2021) ;12: (1):139. doi: 10.1038/s41467-020-20474-9 |
[39] | Thompson CAH , Gu A , Yang SY , et al. Transient Telomerase Inhibition with Imetelstat Impacts DNA Damage Signals and Cell-Cycle Kinetics. Mol Cancer Res. (2018) ;16: (8):1215–25. doi: 10.1158/1541-7786.MCR-17-0772 |
[40] | Relitti N , Saraswati AP , Federico S , et al. Telomerase-based Cancer Therapeutics: A Review on their Clinical Trials. Curr Top Med Chem. (2020) ;20: (6):433–57. doi: 10.2174/1568026620666200102104930 |
[41] | Samad MA , Saiman MZ , Abdul Majid N , et al. Berberine Inhibits Telomerase Activity and Induces Cell Cycle Arrest and Telomere Erosion in Colorectal Cancer Cell Line, HCT 116. Molecules. (2021) ;26: (2):376. doi: 10.3390/molecules26020376 |
[42] | Ghasemimehr N , Farsinejad A , Mirzaee Khalilabadi R , et al. The telomerase inhibitor MST-312 synergistically enhances the apoptotic effect of doxorubicin in pre-B acute lymphoblastic leukemia cells. Biomed Pharmacother. (2018) ;106: :1742–50. doi: 10.1016/j.biopha.2018.07.140 |


