
Primary ciliary dyskinesia (PCD): A genetic disorder of motile cilia
Abstract
Primary ciliary dyskinesia (PCD) is a genetic disorder of motile cilia. Clinical features include chronic oto-sino-pulmonary disease, laterality defects, and male fertility reflecting impaired function of respiratory cilia in the upper and lower respiratory tracts, nodal cilia in the embryonic node and sperm tails, respectively. Recent studies have identified over 40 PCD-associated genes that encode proteins involved in ciliary biogenesis, assembly, structure, or function. Mutations in these genes account for approximately 70% of PCD cases; therefore, further gene discovery is expected. The diagnosis of PCD is challenging because no single test has the required diagnostic accuracy. Recent efforts have focused on standardizing and validating a panel of tests (including assessment for key clinical features, nasal nitric oxide measurement, ciliary ultrastructure analysis, and PCD genetic testing) to be used at PCD Centers to accurately diagnose PCD. Multi-center research programs focused on PCD in North America and Europe have been crucial for PCD gene discovery, advancing our understanding of the natural history of PCD and launching multi-center clinical trials.
1Introduction
Primary ciliary dyskinesia is a heterogeneous, usually recessive, genetic disorder of motile cilia, and is characterized by a diverse array of clinical features, including chronic/recurrent upper and lower respiratory tract infections, laterality defects (including situs inversus totalis and heterotaxy), and male infertility [78]. Early names for this disorder included “Kartagener Syndrome” (triad of bronchiectasis, chronic sinusitis and situs inversus totalis) [73] and “Immotile Cilia Syndrome” (after identification of immotile respiratory cilia and immotile sperm in individuals with Kartagener Syndrome) [1, 41]. Electron microscopy studies demonstrated absence of dynein arms on ciliary cross-sections from patients with “Immotile Cilia Syndrome” [1, 41]. Similar dynein arm defects occurred in patients with bronchiectasis and sinusitis, but without situs inversus totalis [41]. Subsequent studies showed that respiratory cilia from some patients with dynein arm defects were not immotile, but were dyskinetic; hence, the names “dyskinetic cilia syndrome” [129] and “ciliary dyskinesia” [130] were introduced and then modified in 1981 to “primary ciliary dyskinesia” to reflect the congenital nature of this disorder of motile cilia [141]. Advances over the past 25 years have greatly expanded our understanding of the genetics, pathophysiology, clinical features and clinical progression of primary ciliary dyskinesia.
Primary ciliary dyskinesia (PCD) is a separate entity from the “non-motile ciliopathies” or “primary ciliopathies” that are inclusive terms used over the last 10–15 years for an array of genetic disorders of the non-motile or sensory primary cilium [33, 147]. The term non-motile ciliopathies is preferred to avoid confusion between primary ciliopathies and primary ciliary dyskinesia. While the general structure of motile and non-motile cilia are similar, each type has distinct structures and functions.
2Cilia: Structure and function
2.1Types of cilia
Cilia are highly conserved organelles that can be divided into two major types: motile cilia and non-motile (sensory) cilia. Specialized motile cilia are apparent during early embryonic development within the ventral node and hence known as “nodal cilia”.
2.1.1Motile cilia
Motile cilia are distinct structures at the apical surface of cells that beat rhythmically and are essential for muco-ciliary clearance and transport of fluid, and play a role in sexual reproduction. Motile cilia are abundant in the upper respiratory tract (including nasal passage, paranasal sinuses and Eustachian tube), lower respiratory tract (tracheobronchial tree), as well as the ependyma of the brain and the fallopian tubes [127]. Sperm tails are structurally similar to motile cilia [68].
Motile cilia typically occur as multiple protrusions oriented in an organized array. The axonemal structure of motile cilia is very similar to flagella in single-cell organisms such as the green algae, Chlamydomonas reinhardtii, that has two flagella that are used for locomotion and reproduction [69]. These similarities have provided insights into the structure, function and genetics of human motile cilia [57].
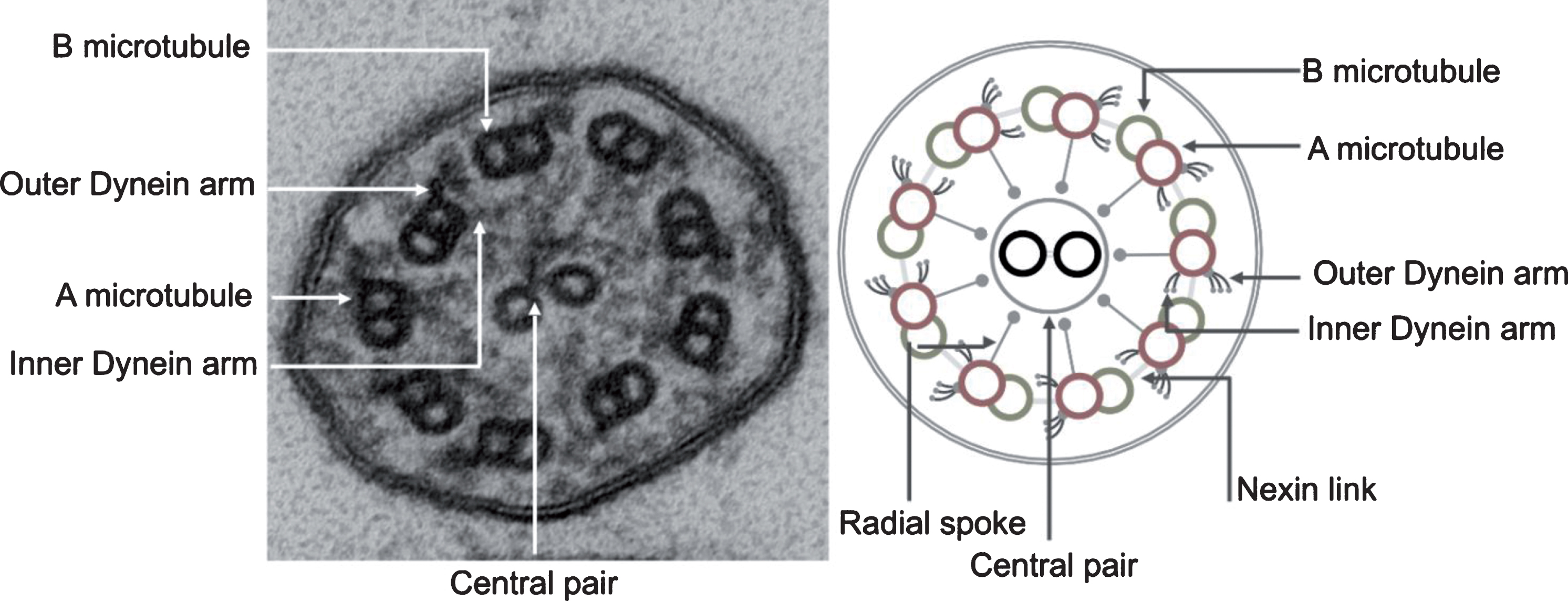
Motile cilia are complex structures composed of hundreds of proteins. A cylindrical scaffold of nine microtubule doublets encircling a central pair of microtubules creates the characteristic 9 + 2 arrangement typically seen on transmission electron microscopy [144] (Fig. 1). The basal body is a specialized structure at the base of the cilium that anchors the cilium in the apical cell membrane thereby directing the direction of ciliary beat and playing a pivotal role in assembly and transport of proteins into the cilium [122]. Emanating from each microtubule doublet are the outer and inner dynein arms that occur at distinct intervals. These dynein arms are complex structures that contain ATPase and function as “motors” for the cilia. Dynein arms emerge from the A microtubule of the outer doublets and slide onto the B-microtubule of the adjacent doublet, generating a force that translates to a bending motion of the cilium. The outer dynein arm is a large protein complex of heavy, intermediate and light dynein chains grouped into three stalks, with a tilt axis perpendicular to the microtubule. Such an arrangement allows the outer dynein arms to provide the power stroke needed for bending the cilium [69]. Inner dynein arms are composed of two stalks, and are important for controlling the rhythmic motion of cilia as part of a complex often referred to as the Nexin-dynein regulatory complex (N-DRC) [17, 57]. Cilia also have nexin links that connect adjacent outer microtubular doublets and limit the degree of sliding between microtubules, as well as radial spokes that control dynein arm activity and relay signals from the central apparatus to the dynein arms [47]. The central structure of a cilium is separated from the cell cytoplasm by a functional barrier, which regulates flux of proteins into the cilium [5, 122]. Movement of proteins along the length of the cilium is done through a specialized mechanism of intra-flagellar transport (IFT), that was first described in lower order organisms [27, 126].
Fig.1
Transmission electron photomicrograph (left) and schematic diagram (right) of a normal motor cilium in cross-section showing the ultrastructural features of the ciliary axoneme.
An airway epithelial cell has approximately 200 motile cilia that are oriented in the same direction and move in unison at a constant frequency that ranges between 8–20 Hz. Motile cilia beating is influenced by changes in the ciliary microenvironment, such that the cilia beat frequency can change in response to triphosphate nucleotides, the depth and viscosity of the apical surface fluid, changes in redox conditions, and exposure to pathogens and airborne pollutants [72, 140].
2.1.2Nodal cilia
A subtype of motile cilia, known as nodal cilia, transiently appears during early embryonic development. Nodal cilia are motile solitary cilia, with a “9 + 0” arrangement that emerges in the ventral node of the gastrula. Because these cilia lack a central pair, they have a rotatory motion that generates left-ward movement of fluid across the surface of the embryonic node. This motion is sensed by immotile sensory cilia around the rim of the node, and is thought to be responsible for determining body laterality [43, 106, 151]. In PCD associated with immotile nodal cilia, left-right orientation becomes random, resulting in laterality defects such as situs inversus totalis, situs ambiguus, and heterotaxy syndromes [8, 58, 76, 86].
2.1.3Non-motile (sensory) cilia
The non-motile cilium typically occurs as a solitary cilium per cell and serves as an “antenna” to sense the surrounding environment and transmit signals [147]. Unlike motile cilia, non-motile cilia lack dynein arms and thus are immotile. They also lack a central pair, thus forming a “9 + 0” configuration. Non-motile cilia protrude from the surface of most mammalian cells during interphase and are essential for normal development and tissue differentiation, harboring many signaling receptors such as sonic hedgehog (SHH), epidermal growth factor receptor (EGFR), and platelet-derived growth factor receptor (PDGFR) [23, 26, 51]. Non-motile cilia have specialized functions in sensory organs, forming the retinal photoreceptors and cochlear stereocilia [44, 51]. Not surprisingly, mutations affecting the function of non-motile cilia, result in multiorgan involvement, leading to syndromes and conditions that are known as non-motile ciliopathies [6, 47, 63]. These include conditions such as the autosomal recessive polycystic kidney disease and Bardet-Biedl Syndrome.
2.1.4Overlap functions
Although motile and primary sensory cilia are often organized as having separate functions, it is important to recognize that sensory receptors have also been identified on motile cilia, including bitter taste receptors [132], and the Polycystin-1 (PKD1) and polycystin-2 (PKD2) flow sensors which are associated with increased risk of bronchiectasis [70].
3Genetics of primary ciliary dyskinesia
Motile cilia are composed of hundreds of proteins. Genes encoding proteins involved in ciliary biogenesis, assembly, structure, or function are potential candidate genes for PCD. Gene discovery studies have identified 40 PCD-associated genes (Table 1). Mutations in known PCD genes account for approximately 70% of all PCD cases [81]. It is expected that more PCD genes will be identified, as more patients with clinical phenotypes suggestive of PCD are evaluated.
Table 1
Genes associated with primary ciliary dyskinesia
| Gene name | Alias | Locus | Protein location | Ultrastructure Phenotype |
| DNAH5 | Chromosome 5 | Outer dynein arm | ODA absent or truncated | |
| DNAI1 | Chromosome 9 | Outer dynein arm | ODA absent or truncated | |
| DNAI2 | Chromosome 17 | Outer dynein arm | ODA absent or truncated | |
| DNAL1 | Chromosome 14 | Outer dynein arm | ODA absent or truncated | |
| NME8 | TXNDC3 | Chromosome 7 | Outer dynein arm | ODA absent or truncated |
| DNAH8 | Chromosome 6 | Outer dynein arm | Not defined | |
| DNAH11 | Chromosome 7 | Outer dynein arm | Normal structure | |
| TTC25 | Chromosome 17 | Outer dynein arm docking | ODA absent or truncated | |
| CCDC114 | Chromosome 19 | Outer dynein arm docking | ODA absent or truncated | |
| ARMC4 | Chromosome 10 | Outer dynein arm docking | ODA absent or truncated | |
| CCDC151 | Chromosome 19 | Outer dynein arm docking | ODA absent or truncated | |
| CCDC103 | Chromosome 17 | Dynein arm attachment | ODA absent | |
| DNAH1 | Chromosome 3 | Inner dynein arm | Not defined | |
| LRRC6 | Chromosome 8 | Cytoplasmic | ODA + IDA absent or truncated | |
| DNAAF1 | LRRC50; ODA7 | Chromosome 16 | Cytoplasmic | ODA + IDA absent or truncated |
| DNAAF2 | C14orf104; KTU | Chromosome 14 | Cytoplasmic | ODA + IDA absent or truncated |
| DNAAF3 | C19orf51; PF22 | Chromosome 19 | Cytoplasmic | ODA + IDA absent or truncated |
| DNAAF4 | DYX1C1 | Chromosome 15 | Cytoplasmic | ODA + IDA absent or truncated |
| DNAAF5 | HEATR2 | Chromosome 7 | Cytoplasmic | ODA + IDA absent or truncated |
| SPAG1 | Chromosome 8 | Cytoplasmic | ODA + IDA absent or truncated | |
| ZMYND10 | Chromosome 3 | Cytoplasmic | ODA + IDA absent or truncated | |
| CFAP298 | C21orf59; kurly | Chromosome 21 | Cytoplasmic | ODA + IDA absent or truncated |
| PIH1D3 | Chromosome X | Cytoplasmic | ODA + IDA absent or truncated | |
| CFAP300 | C11orf70 | Chromosome 11 | Cytoplasmic | ODA + IDA absent or truncated |
| HYDIN | Chromosome 16 | Central pair | Normal structure | |
| STK36 | Chromosome 2 | Central pair | Normal/Central pair defect | |
| RSPH4A | Chromosome 6 | Radial spoke | Normal/Central pair defect | |
| RSPH9 | Chromosome 6 | Radial spoke | Normal/Central pair defect | |
| RSPH1 | Chromosome 21 | Radial spoke | Normal/Central pair defect | |
| RSPH3 | Chromosome 6 | Radial spoke | Normal/Central pair defect | |
| DNAJB13 | Chromosome 11 | Radial spoke | Normal/Central pair defect | |
| CCDC65 | Chromosome 12 | Nexin link | Normal structure | |
| DRC1 | CCDC164 | Chromosome 2 | Nexin link | Normal structure |
| GAS8 | Chromosome 16 | Nexin link | Normal structure | |
| CCDC39 | Chromosome 3 | N-DRC | IDA absent and microtubular disorganization | |
| CCDC40 | Chromosome 17 | N-DRC | IDA absent and microtubular disorganization | |
| CCNO | Chromosome 5 | Apical cytoplasm for centriole replication | Reduced cilia | |
| MCIDAS | Chromosome 5 | Nucleus as transcription co-regulator | Reduced cilia number | |
| RPGR | Chromosome X | Transition zone | Normal structure | |
| OFD1 | Chromosome X | Centriole | Normal structure |
ODA: outer-dynein arm; IDA: inner-dynein arm; N-DRC: nexin-dynein regulatory complex.
Mutations in PCD genes usually cause one of the “hallmark” ultrastructural axonemal defects, including absence of the outer dynein arm, absence of inner and outer dynein arms, absence of inner dynein arms associated with microtubular disorganization and/or central pair organization defects [78]. Approximately, 30% of genetically proven PCD cases have normal or normal-appearing ciliary axonemal structure which can reflect limitations of electron microscopy to detect subtle changes in structure [61, 79].
Early gene discovery relied on homozygosity mapping with sequencing of candidate genes, comparative genomics, transcriptomics, and proteomics [47, 90, 100, 115]. Recent advances in genetic approaches, such as next generation sequencing technologies including the use of whole exome sequencing, has allowed the discovery of increasing numbers of new PCD genes in subjects with symptoms suggestive of PCD [62, 80, 116] and enabled identification of PCD genes that encode non-structural proteins in cilia. As shown in Table 1, PCD-associated genes can be classified by location of protein (e.g., outer dynein arm, radial spoke, cytoplasmic) or by associated ultrastructural defect (e.g., absent outer dynein arm, absent outer and inner dynein arms, absent inner dynein arm with microtubular disorganization). For most structural protein genes, alteration in the encoded structural protein results in a distinctive ultrastructural defect (such as absent outer dynein arms with mutated DNAH5 that encodes an outer dynein arm protein). However, mutations in some genes (such as DNAH11 that encodes a different heavy chain isoform of the outer dynein arm), does not result in a detectable ultrastructural defect on transmission electron microscopy [125], yet, absence of DNAH11 is detectable by three dimensional tomography [138].
3.1Genes encoding outer dynein arm components
This group includes some of the most prevalent genes implicated in PCD. Mutations in two of these genes, DNAI1 (MIM 604366) [52, 123] and DNAH5 (MIM 603335) [110], are associated with absence of the outer dynein arms, dyskinetic and slow ciliary beat pattern and account for more than 30% of all PCD cases [90]. DNAI1 was the first identified PCD gene through a candidate gene approach based on screening mutants that cause abnormal flagellar motility in the green alga C. reinhardtii. DNAI1 mutations account for about 9% of all PCD subjects [156]. DNAH5 was first cloned using homozygosity mapping in affected endogamous families with PCD [114], and later identified by sequencing subjects with PCD, accounting for approximately 25% of PCD patients [110]. Other outer dynein arm genes, including DNAL1 (MIM 610062) [66], DNAI2 (MIM 605483) [91], and NME8 (MIM 607421) [39] are associated with outer dynein arm defect, but are much less prevalent (<3% of PCD). Mutations in DNAH11 (MIM 603339), which also encodes an outer dynein arm protein, produces an intriguing phenotype [79, 125] with normal ciliary ultrastructure, but very rapid beat frequency with subtle limitation of the bend of the cilia. Even though the ciliary beat frequency appears normal or supra-normal, mutations in DNAH11 cause a subtle abnormality in the ciliary waveform with inadequate mucociliary clearance and hence disease.
3.2Genes encoding nexin-dynein regulatory complex (N-DRC) components
Coordinated beating of motile cilia relies on the function of the N-DRC that functions as a biochemical complex maintaining the alignment between microtubule doublets during cilia beating, interacting with the radial spokes and IDA, and includes the nexin link that connects outer microtubules [16, 57]. Mutations in coiled-coil domain containing proteins, CCDC39 (MIM 613798) [98] and CCDC40 (MIM 613799) [9, 11] cause defects in both the assembly of the N-DRC and inner dynein arms, and disruption of this periodicity. The latter results in inconsistent ultrastructual abnormalities, characterized by disordered microtubules in some, but only a minority, of all cilia. Axonemal disorganization and inner dynein arm defects represent about 12% of all PCD cases, and mutations in CCDC39 and CCDC40 account for about 70% inner dynein arm defects with microtubular disorganization [2]. Interestingly, individuals with mutations in CCDC39 and CCDC40, and the axonemal disorganization and inner dynein arm defect group, have worse pulmonary function and more rapid decline compared to other PCD patients [34, 35].
3.3Genes encoding radial spoke components
This group includes RSPH9 (MIM 612648) [24], RSPH4A (MIM 612647) [24], RSPH3 (MIM 61586) [71], and RSPH1 (MIM 609314) [111]. Mutations in RSPH9 and RSPH4A, both associated with absence of some of the central pairs and motility defects [24], were identified using homozygosity mapping in consanguineous families. These proteins encode radial spoke head proteins, and screening in the alga C. reinhardtii identified mutations in orthologous genes that showed similar ultrastructural defects [154]. In PCD subjects with mutant radial spokes proteins, cilia motility is circular. Because nodal cilia do not normally have central pairs, this group of patients does not present with situs abnormalities.
3.4Genes encoding cytoplasmic proteins involved in assembly
Cilia are complex organelles, and the current paradigm is that at least part of the dynein arms are preassembled in the cytoplasm prior to transport into the ciliary axoneme [49, 126]. Recently, PCD-causing mutations in genes encoding cytoplasmic proteins that result in absence of ODA and IDA were identified, indicating that they play a role in the preassembly of dynein motor complexes. DNAAF2 (ktu; MIM 612517) was the first cytoplasmic gene to be identified in patients with PCD [115]. DNAAF2 interacts with intermediate dynein chain DNAI2 and the chaperone heat shock protein HSP70. Other PCD genes encoding cytoplasmic proteins that are associated with absence of ODA and IDA when mutated include: DNAAF1 (LRRC50, ODA7, MIM 613190) [90], LRRC6 (MIM 614930) [64, 83], DNAAF5 (HEATR2; MIM 614864) [62], DNAAF3 (PF22; MIM 614566) [100], DNAAF4 (DYX1C1; MIM 608706) [145], SPAG1 (MIM 603395) [81], CCDC103 (MIM 614677) [121], ZMYND10 (MIM 607070) [102, 155], CFAP298 (C21orf59; Kurly; MIM 615494)) [4], CFAP300 (C11orf70; MIM 618058)) [59] and PIH1D3; MIM 300933) [112, 119] (Table 1). The exact function of these proteins is not known, but evidence points to close interactions among several proteins, as well as protein complexes and heat-shock proteins suggesting that they may function as chaperones during dynein arm protein assembly [65, 92], e.g., LRRC6 for instance interacts with ZMYND10 [155]; DNAAF5 interacts with SPAG1 and DNAAF2 during the earliest stages of cilia preassembly [65]; DNAAF2 interacts with DYX1C1 and PIH1D3 at later stages of cilia preassembly, possibly acting as linker proteins [65, 119, 145]. These proteins also interact with protein chaperones HSP70 and HSP90, facilitating the organization of dynein arm assembly [25, 115, 120, 145].
3.5Genes encoding proteins involved in centriole replication
The multi-ciliated airway cell undergoes a process by which the cell centrosome multiplies to form over 200 centrioles. These centrioles form the basal bodies from which motile cilia nucleate. This process is poorly understood, but appears to be regulated by two factors, MCIDAS (MIM 614086) and CCNO (MIM 607752). While mutations in most PCD-associated genes result in structural changes in the ciliary axoneme, mutations in MCIDAS and CCNO are associated with reduced numbers of normally appearing motile cilia [15, 148]. MCIDAS is a co-regulator that regulates CCNO and FOXJ1, a master cilia regulatory protein; however, CCNO regulates centrosome replication, which is essential for the formation of multiple cilia. Patients present with a clinical phenotype compatible with PCD. TEM evaluation shows sparse cilia, thus the term “reduced generation of multiple motile cilia” disease (RGMC). RGMC should be considered part of the spectrum of PCD, and not a separate entity. The findings in these patients is consistent with the syndrome previously known as ciliary aplasia [37, 96, 128], and mutations in these factors should be considered in patients with symptoms suggestive of PCD with repeated ciliary biopsies that report inadequate numbers of motile cilia.
3.6Genes encoding centrosomal proteins and the transition zone
The cilia transition zone is an area at the base of the cilium that is active in intra-ciliary transport of signaling molecules and other components into the ciliary axoneme [105]. Many of the gene mutations associated with the transition zone have been associated with non-motile ciliopathies. Although most non-motile ciliopathy genes do not influence motile cilia function [63], one gene stands in contrast. RPGR (MIM 312610) encodes a protein in the outer segment of the retinal rod photoreceptor, which is a modified cilium. In airway epithelia, RPGR localizes to the transition zone [60] where it provides cilia stability and ciliary protein trafficking. Mutations in RPGR have also been reported in patients with symptoms combining X-linked Retinitis Pigmentosa and PCD [101]. These patients had typical PCD respiratory symptoms, associated with normal ultrastructure and, in some cases, variable cilia axonemal structural defects, including truncated or absent dynein arms or central pair defects [101].
OFD1 (MIM 300170) encodes a centrosomal protein that is also important in WNT signaling and body axis patterning. Mutations in OFD1 are associated with non-motile cilia dysfunction and X-linked oral-facial-digital type I (OFD1) syndrome [48]. Mutations in OFD1 were also reported in a few patients with OFD1 with respiratory symptoms and severely disorganized motile cilia beating [20].
4Clinical features of PCD
The clinical features of PCD reflect defective motile cilia function in the respiratory tract, sinuses, middle ear, and reproductive tract, as well as in the ventral node during embryogenesis (Table 2) [78, 137].
Table 2
Age-Related Prevalence of Clinical Features in Primary Ciliary Dyskinesia1
| PCD clinical feature | Youngest age when feature present in >50% of PCD | Youngest age when feature present in >80% of PCD |
| Neonatal respiratory distress | 12 hr of life | 24 hr of life |
| Organ laterality defects (SIT or SA) | Neonatal to school age | — |
| Recurrent otitis media with effusion | Infancy | Infancy |
| Year-round, daily cough | Infancy | Infancy |
| Year-round, daily nasal congestion | Infancy | Infancy |
| Chronic pansinusitis | Preschool | School age |
| Recurrent lower respiratory infections | Infancy | Preschool |
| Bronchiectasis | School age | Adult |
| Male infertility | — | Adult |
1Table reproduced from Shapiro, Pediatric Pulmonology, 2015 [137]. SIT, situs inversus totalis; SA, situs ambiguus.
4.1Lower respiratory tract
4.1.1Neonatal respiratory distress
Respiratory manifestations of PCD are present from birth. The majority (75–91%) of patients diagnosed with PCD have a history of unexplained neonatal respiratory distress [46, 67, 87, 107]. Despite term gestation, PCD neonates present with tachypnea and increased work of breathing, and usually require supplemental oxygen, continuous positive airway pressure ventilation (CPAP) or, in rare cases, intubation with mechanical ventilation. Frequently, these neonates will be diagnosed with transient tachypnea of the newborn (TTN) or neonatal pneumonia; however, the PCD clinical presentation differs from that of other etiologies of neonatal respiratory distress. In a case-control study comparing PCD term neonates versus term neonates with neonatal respiratory distress (disease controls), PCD presented with later onset of neonatal respiratory distress (median 12 hours of life versus 1 hour of life), required a longer duration of oxygen therapy (mean 15.2 days versus 0.8 days), and had a higher frequency of atelectasis and/or lobar collapse on chest radiographs (70% versus 0%) [103]. Mechanisms for lobar collapse may include preferential gas trapping and hyperinflation of lower lobes leading to atelectasis/compression in the upper lobes, or intraluminal airway mucous obstruction in the upper lobes [103]. Lobar collapse in the setting of unexplained neonatal respiratory distress should prompt consideration for PCD. The combination of neonatal respiratory distress and situs inversus totalis warrants PCD evaluation. Similarly, situs ambiguus with neonatal respiratory distress out of proportion to the associated cardiac lesion should prompt PCD evaluation.
4.1.2Chronic cough
Patients with PCD have persistent, year-round wet cough that typically starts before 6 months of age [87]. They have intermittent exacerbations and recurrent lower respiratory tract infections, including pneumonia and bronchitis. Nearly 80% of pre-school children with PCD have recurrent lower respiratory tract infections [78]. While cough can improve with antibiotics and aggressive airway clearance, it does not completely resolve. The most prevalent pathogens recovered in sputum from PCD children are Haemophilus influenza, Staphylococcus aureus, Streptococcus pneumonia and Moraxella catarrhalis. In children with PCD, the age-specific prevalence of Pseudomonas aeruginosa is lower than in CF, with reported prevalence of 5–7% for mucoid phenotypes in children and adolescents [107]. In adult patients with PCD, the prevalence is approximately 27% for mucoid P. aeruginosa, with increased prevalence with age, especially after 30 years [107]. In contrast to CF, cough clearance is preserved in PCD, which may decrease the risk of chronic colonization with P. aeruginosa during childhood [35]. However, after years of impaired mucociliary clearance and progressive bronchiectasis, the diseased airways of many patients with PCD develop chronic P. aeruginosa infection [152]. While rare in childhood, non-tuberculous mycobacteria has been reported in more than 10% of adults with PCD [107].
4.1.3Progressive obstructive lung disease
Obstructive lung disease can develop in early life, yet the severity and progression of lung disease in PCD is highly heterogeneous. Early cross-sectional and longitudinal studies show that airflow obstruction worsens with increasing age [42, 56, 95]. One cross-sectional study in pediatric and adult subjects with PCD showed that the mean FEV1 decline (–0.8% predicted per year) was slower than that reported for CF at that time (–3.6% predicted per year) [107]. In a recent longitudinal study of children with PCD, mean annual change in percent predicted FEV1 was –0.57% per year, with children with inner dynein arm/central pair/microtubular disorganization mutations having the greatest rate of decline [35]. Similarly, in a retrospective, international cohort study of 991 children and adults with PCD, lung function was reduced in all age groups when compared to reference values, particularly for those PCD patients with microtubular defects [54].
4.1.4Bronchiectasis
Patients with PCD develop progressive bronchiectasis that is essentially universal by adulthood [18, 107]. Chest radiographic studies have demonstrated evidence of lung disease during infancy and early childhood [78]. Chest CT findings may include subsegmental atelectasis, mucous plugging, air-trapping, ground-glass opacities, peribronchial thickening, and bronchiectasis. Bronchiectasis is the permanent dilation of bronchi resulting from a vicious cycle of recurrent infection, impaired mucociliary clearance, and destruction of the bronchial wall and elastic connective tissue. Progressive airway obstruction and bronchiectasis can lead to end stage lung disease [107]. In a recent retrospective study of 151 adults with PCD with a median age of 35 years, 7-year incidence of all-cause mortality was nearly 5%, and respiratory mortality was 3.3% [131].
4.2Upper respiratory tract
4.2.1Nasal congestion and chronic sinusitis
Chronic, year-round daily nasal congestion occurs in 76–100% of children with PCD, and is frequently associated with hypoplastic frontal and sphenoid sinuses [124]. Onset typically begins within the first 6 months of life. Recurrent sinusitis is also common, and most PCD patients will have pansinusitis on computed tomography (CT) [99]. Nasal polyps can occur in up to 15% of PCD patients [22].
4.2.2Chronic otitis media
Recurrent otitis media (ROM, “glue ear”) occurs in up to 80% of PCD children [137]. In one study, 38% of PCD children required more than 30 lifetime antibiotic courses [143]. ROM complications can include conductive hearing loss, speech and language delay, and cholesteatoma formation [40]. PCD children have nearly universal conductive hearing loss due to persistent otitis media with effusion [137]. Hearing abnormalities can improve during adolescence, but may persist into adulthood [137].
4.3Laterality defects
Cilia dysfunction in embryonic nodal cells can result in laterality defects and congenital heart disease [78, 133]. Nodal cilia contain a 9 + 0 axonemal composition and lack the components of the central apparatus and radial spokes. Patients with mutations in genes that encode the central complex and radial spokes do not have situs abnormalities, since they have normal rotatory function essential for left-right asymmetry [111]. Organ laterality defects in PCD include situs inversus totalis (SIT— mirror-image arrangement), and situs ambiguus (SA— arrangement falls between mirror image and normal, Fig. 2) [133]. SIT occurs in slightly <50% of PCD patients, whereas SA occurs in at least 12% of PCD patients and is associated with a 200-fold increased probability of having structural congenital heart disease compared to the general population [19, 55, 133]. Therefore, echocardiograms are indicated to evaluate for CHD in patients with PCD and SA, while in general they are not necessary for asymptomatic patients with PCD and SIT or situs solitus. Patients with SA may have heterotaxy syndromes (i.e. intestinal malrotation, interrupted inferior vena cava, or polysplenia) and imaging studies (i.e. abdominal ultrasound and echocardiogram) should be considered.
Fig.2
Examples of laterality defects on radiology imaging in various situs groups [133]. Examples of various laterality defects on radiology imaging in PCD. Different situs arrangements found in PCD, including (A) a participant with situs solitus, or normal organ arrangement, with left cardiac apex, left-sided stomach bubble, and right-sided liver; (B) a patient with situs inversus totalis (SIT), or mirror-image organ arrangement, with right cardiac apex, right-sided stomach bubble, and left-sided liver; (C) a patient with situs ambiguus (SA), with left cardiac apex, right-sided stomach bubble, right-sided liver, and intestinal malrotation; This patient also had right-sided polysplenia visualized on a CT scan. C, cardiac apex; S, stomach; L, liver; M, intestinal malrotation. Image and legend reproduced from Shapiro et al, Chest, 2014, with permission from Elsevier [133].
![Examples of laterality defects on radiology imaging in various situs groups [133]. Examples of various laterality defects on radiology imaging in PCD. Different situs arrangements found in PCD, including (A) a participant with situs solitus, or normal organ arrangement, with left cardiac apex, left-sided stomach bubble, and right-sided liver; (B) a patient with situs inversus totalis (SIT), or mirror-image organ arrangement, with right cardiac apex, right-sided stomach bubble, and left-sided liver; (C) a patient with situs ambiguus (SA), with left cardiac apex, right-sided stomach bubble, right-sided liver, and intestinal malrotation; This patient also had right-sided polysplenia visualized on a CT scan. C, cardiac apex; S, stomach; L, liver; M, intestinal malrotation. Image and legend reproduced from Shapiro et al, Chest, 2014, with permission from Elsevier [133].](https://content.iospress.com:443/media/trd/2019/4-1-2/trd-4-1-2-trd190036/trd-4-trd190036-g002.jpg)
4.4Infertility
Males with PCD are typically infertile, but some males report normal fertility. Females may have hypofertility and increased risk of ectopic pregnancy.
4.4.1Male infertility
Sperm motility is driven by a flagellum containing motile axonemes consisting of 9 + 2 microtubules, dynein arms, and regulatory structures, yet the motor protein composition of the axoneme of the sperm tail differs from that of motile respiratory cilia [68]. Abnormal axonemal structure results in reduction or loss of motility, leading to infertility. The most frequent ultrastructural defects of the sperm flagella are outer dynein arm defects, microtubular translocation, and absent radial spokes [68]. CCDC114 mutations may have no significant effect on fertility, since there is low transcript expression of CCDC114 in testis compared to respiratory ciliated cells [116]. Genetic counseling is recommended and reproductive strategies for immotile/dysmotile sperm include in vitro fertilization and intracytoplasmic sperm injection [68].
4.4.2Female hypofertility
Females with PCD with dynein arm defects can be fertile [68]. The motor protein composition and ciliary beating of human fallopian tube cilia is identical to respiratory cilia; however, the transport of the ovum towards the uterus is not totally cilia-dependent [127]. Females with PCD may also experience increased risk of ectopic pregnancy [127].
4.5Clinical features overlapping with other disorders
PCD can co-exist with other rare diseases involving ciliary genes, including retinitis pigmentosa (an inherited cause of blindness from retinal ciliary dysfunction), and Orofaciodigital Syndrome (mental retardation, craniofacial abnormalities, macrocephaly, digital anomalies, and cystic kidneys) [150]. Overlapping ciliopathies have been reported, particularly with genetic disorders of non-motile cilia that result in cystic kidneys, cystic or cholestatic liver, skeletal malformations, developmental delay, hydrocephalus, blindness, or deafness [150]. PCD can also occur with other disease due to proximity of disease causing mutations at the same chromosomal locus, such as Cri du Chat syndrome, which results in a large deletion on chromosome 5p that includes the region of DNAH5 [136].
4.6Genotype-phenotype correlation
Genotype-phenotype relationships are emerging in PCD, particularly regarding variability in severity and progression of lung disease. Patients with a distinct ultrastructural defect (inner dynein arm defect with microtubular disorganization), that is usually linked to mutations in CCDC39 and CCDC40, typically present earlier in life, have worse pulmonary function by both functional and structural assessment (spirometry and CT scans), and poorer nutritional status compared to similar-aged patients with outer dynein arm defects [34, 35]. In contrast, patients with RSPH1 mutations typically have milder disease, higher levels of nasal nitric oxide, and better lung function [133].
5Approach to diagnosis of PCD
PCD is a rare disease and is significantly underdiagnosed. Currently in North America, there are approximately 1,000 individuals with PCD followed by PCD clinical centers; however, prevalence estimates suggest that there should be at least 16,000 individuals with PCD in this region of the world [85, 87]. Substantial advances in the definition of PCD phenotype, ciliary ultrastructure analysis, PCD genetic testing, and nasal nitric oxide (nNO) measurement have provided more accurate tools for PCD diagnosis. Interpretation of these tests requires specialized expertise. Most PCD Centers employ a panel of tests because no single test has the diagnostic accuracy needed at this time.
5.1PCD clinical phenotype
The first step in making a diagnosis of PCD is recognizing the specific clinical symptoms consistent with the disease (Table 2). Most children with PCD are diagnosed between 3 and 6 years of age, yet clinical symptoms for these children are present in the first year of life [85]. Children with PCD who do not have an organ laterality defect are diagnosed later than children who present with respiratory symptoms and situs inversus totalis [85]. These findings suggest both a need for improved understanding of the clinical phenotype of PCD and the possibility of insufficient availability and/or utilization of PCD diagnostic testing.
A detailed clinical history is the first key step to recognize individuals at high likelihood of having PCD [87]. Any patient with the Kartagener triad (bronchiectasis, chronic sinusitis and situs inversus totalis) is highly likely to have PCD. However, early diagnosis relies on early recognition of chronic sino-pulmonary disease before progression to chronic sinusitis and bronchiectasis. A challenge for pediatricians is distinguishing the chronic respiratory symptoms of PCD from the recurrent episodes of viral respiratory infections. A recent multi-center study identified four key clinical features that enhance the likelihood of PCD in children: year-round, wet-sounding cough that starts in the first 6 months; year-round nasal congestion that starts in the first 6 months of life; a history of neonatal respiratory distress requiring supplemental oxygen for more than 24 hours despite term gestation; and organ laterality defect that is typically situs inversus totalis, but may be heterotaxy or situs ambiguus [87, 137]. Other clinical features, such as chronic otitis media, are part of the PCD phenotype but have not proven to have the same discriminatory diagnostic functionality as these specific 4 features [87, 137]. Presence of any one of these key clinical features should raise suspicion of PCD (sensitivity 0.96, specificity 0.41); presence of two key clinical features warrants diagnostic testing (sensitivity 0.80, specificity 0.72); and presence of all 4 key clinical features is highly likely PCD (sensitivity 0.21, specificity 0.99) [87, 137].
5.2Ciliary biopsy
Several approaches have been used for ciliary biopsy, including scraping the respiratory epithelium from the nasal cavity (inferior surface of inferior nasal turbinate), brush biopsy from the nasal turbinate or brush biopsy of the bronchial mucosa during a bronchoscopy [113, 137, 139]. An important aspect of this procedure is obtaining an adequate sample of cilia for evaluation, accounting for changes in the epithelial surface from inflammation, including loss of cilia and other secondary changes to the cilia [113, 137].
5.2.1Ultrastructural defects in cilia
In the past, ciliary biopsy with analysis of cilia structure by electronic microscopy (EM) was the gold standard diagnostic test for PCD; however, genetic testing has shown that biallelic mutations in some of the PCD genes can be associated with normal or near-normal ciliary ultrastructure. While this finding limits the sensitivity of EM analysis, this test still has diagnostic utility [139, 146]. It is important to have enough cilia in the sample to determine if a structural abnormality is present consistently [139]. Assessment of 200 or more individual cilia in cross-section is recommended for an adequate EM evaluation [36, 78, 113]. Hallmark defects of ciliary ultrastructure that are considered diagnostic for PCD include absence (or truncation) of outer dynein arms, absence (or truncation) of both outer and inner dynein arms and absence of inner dynein arms associated with microtubular disorganization [137, 149]. Each of these hallmark defects are associated with a specific group of genes that encode either proteins that are components of the defective structures or proteins that are involved in assembly of these structures (Table 1). Central apparatus defects (missing central pair, central pair off-center, transposition of outer doublet into central position) may occur in a subset of cilia and should be considered suggestive of PCD. These central apparatus defects can occur in small subset of cilia from healthy individuals or individuals recovering from viral illnesses. Therefore, central apparatus defects are not considered diagnostic unless observed on repeated biopsies or confirmed by genetic testing that identifies biallellic mutations in a radial spoke gene (Table 1) [108]. Similarly, absence of inner dynein arms is frequently a secondary change in cilia and should not be considered diagnostic unless this defect occurs on repeated biopsies [108]. Of note, isolated inner dynein arm defects have not been associated with any of the presently defined PCD genes [108]. It is also important to remember that normal ciliary ultrastructure does not rule-out PCD, as it is estimated that up to 30% of individuals with PCD will have normal ciliary structure on EM [78].
5.2.2Altered ciliary beat pattern
Diagnosis of PCD by ciliary movement analysis requires significant expertise and careful considerations. Although once a commonly utilized diagnostic test, evaluation of ciliary movement by light microscopy should not be used as a stand-alone test to diagnose PCD [134, 137]. High-speed video microscopy (HSVM) is an evolving alternative approach to ciliary movement analyses [134, 137]. HSVM involves recording of ciliary movement at high speed and then assessing the recording at low speed for altered ciliary beat pattern. At present, limitations of HSVM include subjectivity and low inter-rater reliability for HSVM ciliary evaluations; therefore, HSVM is not recommended as a PCD confirmatory test [134]. However, when performed in specialized centers, in conjunction with EM evaluation of cilia, it can have diagnostic utility.
5.2.3Immunofluorescence using antibodies to ciliary proteins
Immunofluorescence technologies have been used in genetic discovery studies to confirm that mutations in a ciliary gene alter the encoded protein within ciliated cells. In some cases, immunofluorescence studies have identified protein alterations that are not easily detectable by EM. For example, cilia with radial spoke head defects often have near-normal appearance on EM; but, immunofluorescence studies with specific antibodies to radial spoke proteins can detect altered expression of respective proteins [50, 117]. Similarly, individuals with mutations in DNAH11 have normal ciliary ultrastructure on EM [78]; however, immunofluorescence studies demonstrate that individuals with biallelic mutations in DNAH11 have partial reduction of the outer dynein arms in only the proximal region of the cilia [38]. With further standardization and optimization of antibody panels, immunofluorescence technology has potential as a diagnostic test in the future.
5.3Nasal nitric oxide testing
Nitric oxide (NO) is a small diffusible gas molecule that is involved in multiple functions throughout the body. Multiple studies have demonstrated that nitric oxide within the nasal cavity (particularly within the sinuses) is high in healthy individuals and much lower (<10% of normal values) in PCD, suggesting that nasal NO (nNO) measurement may be a useful test for PCD [29, 84, 88, 135]. Nasal NO is measured by a non-invasive approach using a small nasal probe to aspirate nasal gas at a rate of approximately 5 ml/second for measurement of nitric oxide content [10, 14]. Recent guidelines recommend measuring nasal NO with a chemiluminescence analyzer [137]. Alternative nitric oxide analyzers, including hand-held electrochemical devices are under study [94, 134]. Velum closure (VC) maneuvers are used to limit “contamination” of nasal air by air exhaled by the lower airways [14]. These techniques yield reproducible “plateau” measurements in children over 5 years of age, but often are not possible in younger children, who cannot cooperate with VC maneuvers. Some centers have measured nasal NO during tidal breathing in young children who are unable to cooperate with palate closure maneuvers [10, 14]. Currently, nasal NO testing is performed on a research basis at most PCD centers secondary to lack of regulatory body approval of the device for disease specific testing.
Multi-center studies and several meta-analyses have demonstrated diagnostic utility of nasal NO testing with a cut-off value of less than 77 nl/min considered consistent with a diagnosis of PCD [29, 84, 88, 135]. Most individuals with PCD have levels much lower than this cut-off. Despite tremendous utility, nNO testing does have several important limitations. Because PCD is so rare, it is important to employ this testing in individuals with a high-suspicion of PCD based on clinical symptoms as more indiscriminate testing of individuals can result in false positives [28]. Further, while a nNO measurement less than the cut-off of 77 nl/min is consistent with a diagnosis of PCD, it is important to note that one testing measurement by itself should not be considered diagnostic of PCD [134]. Current guidelines recommend that in suspected PCD cases, the nNO measurement should be repeated at least one additional time and this repeat test should be at least one month after the initial testing [137]. Repeat testing accounts for clinical situations in which false positives can occur such as recent viral infection, acute sinusitis or occult sinus bleeding not readily assessed on clinical exam [3, 84, 88]. In addition, low nasal NO levels have been reported in cystic fibrosis [88, 107]; therefore, this disorder should be excluded if nasal NO is used as the primary diagnostic test. In an individual with a clinical phenotype consistent with PCD and with two NO measurements below the cut-off, but a normal sweat chloride test to rule out cystic fibrosis, the diagnostic accuracy of nasal NO testing is comparable to that of ciliary biopsy and genetic testing [135].
5.4Genetic testing for PCD
Genetic discovery over the past 15 years has identified over 40 PCD genes [77, 157], and several commercial panels test for the majority (>34) of these known genes [12, 134]. At present, genetic confirmation is possible for approximately 70% of individuals with PCD with present commercial panels [30, 93, 137]. Hence, an inconclusive or negative genetic test does not rule-out PCD. Of note, many of the PCD genes are very large with greater than 10 exons; therefore, it is not uncommon for PCD genetic testing to identify a single pathogenic or likely pathogenic variant in two or more different PCD genes. It is important to understand that this finding is not diagnostic of PCD. In order to improve the sensitivity of genetic testing, further research is needed to identify additional genes associated with PCD as well as to better characterize the variants of unknown significance that are frequently identified. Expanding access and cost-effectiveness of whole-exome and whole genome sequencing creates the potential for almost all individuals with PCD to have a genetic diagnosis for PCD. Furthermore, new gene discovery informs understanding of ciliary structure and function. This collective advancement not only improves PCD diagnostic capabilities, but also sets the stage for the development of targeted PCD therapies that improve specific ciliary dysfunction abnormalities.
5.5PCD diagnostic summary
At present, no test has the sensitivity and specificity to serve as a single diagnostic test for PCD. Most PCD Centers use a panel of tests. Recent Guidelines and Consensus Statements emphasize the importance of clinical phenotyping to better identify those individuals more likely to have PCD, followed by nasal NO testing and PCD genetic testing to make a confirmatory diagnosis of PCD [134, 137]. However, current diagnostic testing has limitations and must be used with caution to prevent inaccurate diagnoses.
6Approach to therapy in PCD
6.1Management of lung disease
No PCD-specific therapies are available and the evidence base with which to guide management of PCD is extremely slim. The general therapeutic approach to PCD is based on studies in cystic fibrosis and non-CF bronchiectasis. A recent state-of-the-art publication provides consensus recommendations from the PCD Foundation for monitoring and management in PCD [137]. The aims of PCD treatment are to maintain or recover lung function by early diagnosis and to minimize progression of bronchiectasis through aggressive airway clearance and antibiotic treatment of respiratory tract infections. Ideally, long-term follow-up should be at a PCD Foundation-accredited clinical center or CF Foundation-accredited CF center with established practices for monitoring lung disease with surveillance cultures, spirometry and imaging [137].
Enhanced mucus clearance is promoted with positive pressure expiratory devices, high-frequency chest compression using vest therapy, manual chest physiotherapy, autogenic drainage, postural drainage, active cycle breathing, and exercise [32]. There have been limited studies of inhaled medications in PCD. Theoretically, nebulized hypertonic saline stimulates cough and increases airway hydration, improving cough clearance. In a 3-month study, nebulized 7% hypertonic saline demonstrated improved lung function, quality of life, and reduced antibiotic use in non-CF bronchiectasis [74]. One small randomized clinical trial evaluating hypertonic saline in PCD patients demonstrated no improvement in quality of life [118]. Dornase alpha, a deoxyribonuclease that reduces mucus viscosity, is beneficial in CF. However, its use in adults with non-CF bronchiectasis was associated with increased pulmonary exacerbations and greater lung function decline in one small study [109]. Inhaled corticosteroids are not recommended routinely for patients with PCD, except for those patients with concomitant asthma or airway reactivity.
Multiple studies demonstrate that systemic antibiotics are effective for treating worsening respiratory exacerbations in CF and non-CF bronchiectasis, including PCD. Prompt and aggressive treatment of respiratory tract infections should be guided by microbiological studies using sputum culture, and/or bronchoalveolar lavage (BAL). While preliminary studies of inhaled antibiotics demonstrated no benefit with respect to lung function, one study using inhaled gentamicin demonstrated reduced pulmonary exacerbations, bacterial load, and inflammatory markers [104]. Chronic macrolide use is well-supported in the cystic fibrosis literature given its antibacterial, anti-inflammatory, and anti-quorum-sensing properties, and its association with improvement of lung function and reduction of exacerbations [31]. Similarly, macrolide use in non-CF bronchiectasis patients is associated with reduction of pulmonary exacerbations [153]. A European clinical trial is underway to assess the efficacy and safety of a 6-month course of azithromycin in PCD patients [82].
Lobectomy is not routinely recommended for patients with PCD. Careful consideration and consultation with PCD experts should be pursued, with consideration of resection reserved for those patients with severe localized bronchiectasis and recurrent febrile relapses or severe hemoptysis, despite aggressive medical management [142]. Similarly, lung transplantation is reserved for patients with PCD who have advanced lung disease.
Additional preventative measures include avoidance of smoke exposure, infection control practices, and pediatric immunizations, including pneumococcal vaccine and annual influenza vaccine [137].
6.2Management of otolargyngologic manifestations
Pediatric PCD patients should visit an otolaryngologist at least once to twice annually, whereas adult PCD patients should follow-up with an otolaryngologist as needed [137]. Otolaryngology care includes audiology assessment, consideration for pressure equalization tubes (PET) placement, and management of chronic rhinosinusitis. PCD patients have nearly universal conductive hearing loss due to persistent otitis media with effusion. Since chronic otitis media can persist into adulthood, audiology assessments, hearing aids, and communication assistance should be made available for those PCD patients with hearing loss. There is controversy over the clinical indication and benefit of pressure-equalizing tube placement. In studies assessing hearing in children with PCD, PET placement was associated with greater improvement in hearing compared to medical treatment, with normalization of hearing in 80–100% of children [53]. In contrast, the European Respiratory Society Consensus Statement recommends against placement of tubes for chronic otitis media in PCD, given the complications associated with otorrhea and likely resolution of chronic otitis media in the teenage years [7].
Initial management of sinus disease includes nasal steroids, nasal lavage, and intermittent courses of systemic antibiotics. Daily nasal saline rinses are encouraged. Nasopharyngoscopy may be utilized to assess for nasal polyps. Endoscopy sinus surgery (ESS) is often performed in PCD patients and may improve lower respiratory tract disease in some patients [97].
7PCD research and future directions
The past 15 years have brought remarkable progress to our understanding of PCD and PCD gene discovery. A major factor contributing to this progress has been the establishment of multi-center research programs in North America (Genetic Disorders of Mucociliary Clearance Consortium that is part of the Rare Diseases Clinical Research Network supported by National Institutes of Health) and in Europe (BESTCILIA program supported by European Commission) as well as multiple international collaborations involved in gene discovery. Recently, the PCD Foundation has certified and organized PCD Clinical Centers across North America and has launched a PCD Registry. These efforts laid the foundation for multi-center clinical trials that have been completed recently: a clinical trial in Europe evaluating use of chronic azithromycin in PCD and a trans-Atlantic, multi-center clinical trial evaluating agents to enhance hydration of airway secretions and, thereby, enhance cough clearance in PCD. These well-established collaborative networks are critical for advancing to the next stages of research focused on this rare disorder. Priorities for the future include further PCD gene discovery, further definition of genotype-phenotype correlations, definition of factors in early PCD that predict long-term outcomes, and definition of best outcome measures for clinical trials. This research will direct future studies to identify and test therapies to control and, possibly, correct the underlying genetic defect(s).
Addendum
Since submission of this manuscript, three additional PCD-associated genes (DNAH9, GAS2L2, LRRC56) have been reported [13, 21, 45, 89].
Acknowledgments
Supported by the National Institutes of Health: a) The Genetic Disorders of Mucociliary Clearance (U54HL096458; PI: Knowles MR) is a part of the NCATS Rare Diseases Clinical Research Network (RDCRN). RDCRN is an initiative of the Office of Rare Diseases Research (ORDR), NCATS, funded through a collaboration between NCATS and NHLBI. b) R01HL071798 (PI Knowles MR) c) National Center for Advancing Translational Sciences (NCATS), National Institutes of Health, through Grant Award Number UL1TR002489. The contents are solely the responsibility of the authors and do not necessarily represent the official views of the National Institute of Health.
References
[1] | Afzelius B.A. , A human syndrome caused by immotile cilia, Science 193: ((1976) ), 317–319. |
[2] | Antony D. , Becker-Heck A. , Zariwala M.A. , Schmidts M. , Onoufriadis A. , Forouhan M. , Wilson R. , Taylor-Cox T. , Dewar A. , Jackson C. , Goggin P. , Loges N.T. , Olbrich H. , Jaspers M. , Jorissen M. , Leigh M.W. , Wolf W.E. , Daniels M.L. , Noone P.G. , Ferkol T.W. , Sagel S.D. , Rosenfeld M. , Rutman A. , Dixit A. , O’Callaghan C. , Lucas J.S. , Hogg C. , Scambler P.J. , Emes R.D. , Uk10k , Chung E.M. , Shoemark A. , Knowles M.R. , Omran H. and Mitchison H.M. , Mutations in CCDC39 and CCDC40 are the major cause of primary ciliary dyskinesia with axonemal disorganization and absent inner dynein arms, Hum Mutat 34: ((2013) ), 462–472. |
[3] | Arnal J.F. , Flores P. , Rami J. , Murris-Espin M. , Bremont F. , Pasto I.A.M. , Serrano E. and Didier A. , Nasal nitric oxide concentration in paral sinus inflammatory diseases, Eur Respir J 13: ((1999) ), 307–312. |
[4] | Austin-Tse C. , Halbritter J. , Zariwala M.A. , Gilberti R.M. , Gee H.Y. , Hellman N. , Pathak N. , Liu Y. , Panizzi J.R. , Patel-King R.S. , Tritschler D. , Bower R. , O’Toole E. , Porath J.D. , Hurd T.W. , Chaki M. , Diaz K.A. , Kohl S. , Lovric S. , Hwang D.Y. , Braun D.A. , Schueler M. , Airik R. , Otto E.A. , Leigh M.W. , Noone P.G. , Carson J.L. , Davis S.D. , Pittman J.E. , Ferkol T.W. , Atkinson J.J. , Olivier K.N. , Sagel S.D. , Dell S.D. , Rosenfeld M. , Milla C.E. , Loges N.T. , Omran H. , Porter M.E. , King S.M. , Knowles M.R. , Drummond I.A. , Hildebrandt F. , Zebrafish ciliopathy screen plus human mutational analysis identifies C21orf59 and CCDC65 defects as causing primary ciliary dyskinesia, Am J Hum Genet 93: ((2013) ), 672–686. |
[5] | Awata J. , Takada S. , Standley C. , Lechtreck K.F. , Bellve K.D. , Pazour G.J. , Fogarty K.E. and Witman G.B. , NPHP4 controls ciliary trafficking of membrane proteins and large soluble proteins at the transition zone, J Cell Sci 127: ((2014) ), 4714–4727. |
[6] | Baker K. and Beales P.L. , Making sense of cilia in disease: The human ciliopathies, Am J Med Genet C Semin Med Genet 151C: ((2009) ), 281–295. |
[7] | Barbato A. , Frischer T. , Kuehni C.E. , Snijders D. , Azevedo I. , Baktai G. , Bartoloni L. , Eber E. , Escribano A. , Haarman E. , Hesselmar B. , Hogg C. , Jorissen M. , Lucas J. , Nielsen K.G. , O’Callaghan C. , Omran H. , Pohunek P. , Strippoli M.P. and Bush A. , Primary ciliary dyskinesia: A consensus statement on diagnostic and treatment approaches in children, Eur Respir J 34: ((2009) ), 1264–1276. |
[8] | Basu B. and Brueckner M. , Cilia multifunctional organelles at the center of vertebrate left-right asymmetry, Curr Top Dev Biol 85: ((2008) ), 151–174. |
[9] | Becker-Heck A. , Zohn I.E. , Okabe N. , Pollock A. , Lenhart K.B. , Sullivan-Brown J. , McSheene J. , Loges N.T. , Olbrich H. , Haeffner K. , Fliegauf M. , Horvath J. , Reinhardt R. , Nielsen K.G. , Marthin J.K. , Baktai G. , Anderson K.V. , Geisler R. , Niswander L. , Omran H. and Burdine R.D. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation, Nat Genet 43: ((2011) ), 79–84. |
[10] | Beydon N. , Chambellan A. , Alberti C. , de Blic J. , Clement A. , Escudier E. and Le Bourgeois M. , Technical and practical issues for tidal breathing measurements of nasal nitric oxide in children, Pediatr Pulmonol 50: ((2015) ), 1374–1382. |
[11] | Blanchon S. , Legendre M. , Copin B. , Duquesnoy P. , Montantin G. , Kott E. , Dastot F. , Jeanson L. , Cachanado M. , Rousseau A. , Papon J.F. , Beydon N. , Brouard J. , Crestani B. , Deschildre A. , Desir J. , Dollfus H. , Leheup B. , Tamalet A. , Thumerelle C. , Vojtek A.M. , Escalier D. , Coste A. , de Blic J. , Clement A. , Escudier E. and Amselem S. , Delineation of CCDC39/CCDC40 mutation spectrum and associated phenotypes in primary ciliary dyskinesia, J Med Genet 49: ((2012) ), 410–416. |
[12] | Boaretto F. , Snijders D. , Salvoro C. , Spalletta A. , Mostacciuolo M.L. , Collura M. , Cazzato S. , Girosi D. , Silvestri M. , Rossi G.A. , Barbato A. and Vazza G. , Diagnosis of primary ciliary dyskinesia by a targeted next-generation sequencing panel: Molecular and clinical findings in Italian patients, J Mol Diagn 18: ((2016) ), 912–922. |
[13] | Bonnefoy S. , Watson C.M. , Kernohan K.D. , Lemos M. , Hutchinson S. , Poulter J.A. , Crinnion L.A. , Berry I. , Simmonds J. , Vasudevan P. , O’Callaghan C. , Hirst R.A. , Rutman A. , Huang L. , Hartley T. , Grynspan D. , Moya E. , Li C. , Carr I.M. , Bonthron D.T. , Leroux M. , Boycott K.M. , Bastin P. and Sheridan E.G. , Biallelic Mutations in LRRC56, Encoding a Protein Associated with Intraflagellar Transport, Cause Mucociliary Clearance and Laterality Defects, Am J Hum Genet 103: ((2018) ), 727–739. |
[14] | Boon M. , Meyts I. , Proesmans M. , Vermeulen F.L. , Jorissen M. and De Boeck, K. , Diagnostic accuracy of nitric oxide measurements to detect primary ciliary dyskinesia, Eur J Clin Invest 44: ((2014) ), 477–485. |
[15] | Boon M. , Wallmeier J. , Ma L. , Loges N.T. , Jaspers M. , Olbrich H. , Dougherty G.W. , Raidt J. , Werner C. , Amirav I. , Hevroni A. , Abitbul R. , Avital A. , Soferman R. , Wessels M. , O’Callaghan C. , Chung E.M. , Rutman A. , Hirst R.A. , Moya E. , Mitchison H.M. , Van Daele S. , De Boeck K. , Jorissen M. , Kintner C. , Cuppens H. and Omran H. , MCIDAS mutations result in a mucociliary clearance disorder with reduced generation of multiple motile cilia, Nat Commun 5: ((2014) ), 4418. |
[16] | Bower R. , Tritschler D. , Vanderwaal K. , Perrone C.A. , Mueller J. , Fox L. , Sale W.S. and Porter M.E. , The N-DRC forms a conserved biochemical complex that maintains outer doublet alignment and limits microtubule sliding in motile axonemes, Mol Biol Cell 24: ((2013) ), 1134–1152. |
[17] | Brokaw C.J. and Kamiya R. , Bending patterns of Chlamydomonas flagella: IV. Mutants with defects in inner and outer dynein arms indicate differences in dynein arm function, Cell Motil Cytoskeleton 8: ((1987) ), 68–75. |
[18] | Brown D.E. , Pittman J.E. , Leigh M.W. , Fordham L. and Davis S.D. , Early lung disease in young children with primary ciliary dyskinesia, Pediatr Pulmonol 43: ((2008) ), 514–516. |
[19] | Brueckner M. , Heterotaxia, congenital heart disease and primary ciliary dyskinesia, Circulation 115: ((2007) ), 2793–2795. |
[20] | Budny B. , Chen W. , Omran H. , Fliegauf M. , Tzschach A. , Wisniewska M. , Jensen L.R. , Raynaud M. , Shoichet S.A. , Badura M. , Lenzner S. , Latos-Bielenska A. and Ropers H.H. , A novel X-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome, Hum Genet 120: ((2006) ), 171–178. |
[21] | Bustamante-Marin X.M. , Yin W.N. , Sears P.R. , Werner M.E. , Brotslaw E.J. , Mitchell B.J. , Jania C.M. , Zeman K.L. , Rogers T.D. , Herring L.E. , Refabert L. , Thomas L. , Amselem S. , Escudier E. , Legendre M. , Grubb B.R. , Knowles M.R. , Zariwala M.A. and Ostrowski L.E. Lack of GAS2L2 Causes PCD by Impairing Cilia Orientation and Mucociliary Clearance, Am J Hum Genet 104: ((2019) ), 229–245. |
[22] | Campbell R. , Managing upper respiratory tract complications of primary ciliary dyskinesia in children, Curr Opin Allergy Clin Immunol 12: ((2012) ), 32–38. |
[23] | Caspary T. , Larkins C.E. and Anderson K.V. , The graded response to Sonic Hedgehog depends on cilia architecture, Dev Cell 12: ((2007) ), 767–778. |
[24] | Castleman V.H. , Romio L. , Chodhari R. , Hirst R.A. , de Castro S.C. , Parker K.A. , Ybot-Gonzalez P. , Emes R.D. , Wilson S.W. , Wallis C. , Johnson C.A. , Herrera R.J. , Rutman A. , Dixon M. , Shoemark A. , Bush A. , Hogg C. , Gardiner R.M. , Reish, O. , Greene N.D. , O’Callaghan C. , Purton S. , Chung E.M. and Mitchison H.M. , Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities, Am J Hum Genet 84: ((2009) ), 197–209. |
[25] | Chen Y. , Zhao M. , Wang S. , Chen J. , Wang Y. , Cao Q. , Zhou W. , Liu J. , Xu Z. , Tong G. and Li J. , A novel role for DYX1C1, a chaperone protein for both Hsp70 and Hsp90, in breast cancer, J Cancer Res Clin Oncol 135: ((2009) ), 1265–1276. |
[26] | Christensen S.T. , Pedersen S.F. , Satir P. , Veland I.R. and Schneider L. , The primary cilium coordinates signaling pathways in cell cycle control and migration during development and tissue repair, Curr Top Dev Biol 85: ((2008) ), 261–301. |
[27] | Cole D.G. , Diener D.R. , Himelblau A.L. , Beech P.L. , Fuster J.C. and Rosenbaum J.L. , Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons, J Cell Biol 141: ((1998) ), 993–1008. |
[28] | Collins S.A. , Behan L. , Harris A. , Gove K. and Lucas J.S. The dangers of widespread nitric oxide screening for primary ciliary dyskinesia, Thorax 71: ((2016) ), 560–561. |
[29] | Collins S.A. , Gove K. , Walker W. and Lucas J.S. , Nasal nitric oxide screening for primary ciliary dyskinesia: Systematic review and meta-analysis, Eur Respir J 44: ((2014) ), 1589–1599. |
[30] | Dalrymple R.A. and Kenia P. , European Respiratory Society guidelines for the diagnosis of primary ciliary dyskinesia: A guideline review, Arch Dis Child Educ Pract Ed ((2018) ). |
[31] | Damseh N. , Quercia N. , Rumman N. , Dell S.D. and Kim R.H. , Primary ciliary dyskinesia: Mechanisms and management, Appl Clin Genet 10: ((2017) ), 67–74. |
[32] | Daniels M.L. and Noone P.G. , Genetics, diagnosis and future treatment strategies for primary ciliary dyskinesia, Expert Opin Orphan Drugs 3: ((2015) ), 31–44. |
[33] | Davenport J.R. and Yoder B.K. , An incredible decade for the primary cilium: A look at a once-forgotten organelle,F, Am J Physiol Renal Physiol 289: ((2005) ), 1159–1169. |
[34] | Davis S.D. , Ferkol T.W. , Rosenfeld M. , Lee H.S. , Dell S.D. , Sagel S.D. , Milla C. , Zariwala M.A. , Pittman J.E. , Shapiro A.J. , Carson J.L. , Krischer J.P. , Hazucha M.J. , Cooper M.L. , Knowles M.R. and Leigh M.W. , Clinical features of childhood primary ciliary dyskinesia by genotype and ultrastructural phenotype, Am J Respir Crit Care Med 191: ((2015) ), 316–324. |
[35] | Davis S.D. , Rosenfeld M. , Lee H.S. , Ferkol T.W. , Sagel S.D. , Dell S.D. , Milla C. , Pittman J.E. , Shapiro A.J. , Sullivan K.M. , Nykamp K.R. , Krischer J.P. , Zariwala M.A. , Knowles M.R. , Leigh M.W. and and Genetic Disorders of Mucociliary Clearance Consortium, Primary ciliary dyskinesia: Longitudinal study of lung disease by ultrastructure defect and genotype, Am J Respir Crit Care Med 199: ((2018) ), 190–198. |
[36] | de Iongh R.U. and Rutland J. , Ciliary defects in healthy subjects, bronchiectasis and primary ciliary dyskinesia, Am J Respir Crit Care Med 151: ((1995) ), 1559–1567. |
[37] | de Santi M.M. , Gardi C. , Barlocco G. , Canciani M. , Mastella G. and Lungarella G. , Cilia-lacking respiratory cells in ciliary aplasia, Biol Cell 64: ((1988) ), 67–70. |
[38] | Dougherty G.W. , Loges N.T. , Klinkenbusch J.A. , Olbrich H. , Pennekamp P. , Menchen T. , Raidt J. , Wallmeier J. , Werner C. , Westermann C. , Ruckert C. , Mirra V. , Hjeij R. , Memari Y. , Durbin R. , Kolb-Kokocinski A. , Praveen K. , Kashef M.A. , Kashef S. , Eghtedari F. , Haffner K. , Valmari P. , Baktai G. , Aviram M. , Bentur L. , Amirav I. , Davis E.E. , Katsanis N. , Brueckner M. , Shaposhnykov A. , Pigino G. , Dworniczak B. and Omran H. , DNAH11 localization in the proximal region of respiratory cilia defines distinct outer dynein arm complexes, Am J Respir Cell Mol Biol 55: ((2016) ), 213–224. |
[39] | Duriez B. , Duquesnoy P. , Escudier E. , Bridoux A.M. , Escalier D. , Rayet I. , Marcos E. , Vojtek A.M. , Bercher J.F. and Amselem S. , A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia, Proc Natl Acad Sci U S A 104: ((2007) ), 3336–3341. |
[40] | el-Sayed Y. , al-Sarhani A. and al-Essa A.R. , Otological manifestations of primary ciliary dyskinesia, Clin Otolaryngol Allied Sci 22: ((1997) ), 266–270. |
[41] | Eliasson R. , Mossberg B. , Camner P. and Afzelius B.A. , The immotile-cilia syndrome. A congenital ciliary abnormality as an etiologic factor in chronic airway infections and male sterility, N Engl J Med 297: ((1977) ), 1–6. |
[42] | Ellerman A. and Bisgaard H. , Longitudinal study of lung function in a cohort of primary ciliary dyskinesia, Eur Respir J 10: ((1997) ), 2376–2379. |
[43] | Essner J.J. , Vogan K.J. , Wagner M.K. , Tabin C.J. , Yost H.J. and Brueckner M. , Conserved function for embryonic nodal cilia, Nature 418: ((2002) ), 37–38. |
[44] | Falk N. , Losl M. , Schroder N. and Giessl A. , Specialized cilia in mammalian sensory systems, Cells 4: ((2015) ), 500–519. |
[45] | Fassad M.R. , Shoemark A. , Legendre M. , Hirst R.A. , Koll F. , Borgne P. , Louis B. , Daudvohra F. , Patel M.P. , Thomas L. , Dixon M. , Burgoyne T. , Hayes J. , Nicholson A.G. , Cullup T. , Jenkins L. , Carr S.B. , Aurora P. , Lemullois M. , Aubusson-Fleury A. , Papon J.F. , O’Callaghan C. , Amselem, S. , Hogg C. , Escudier E. , Tassin A.M. and Mitchison H.M. , Mutations in Outer Dynein Arm Heavy Chain DNAH9 Cause Motile Cilia Defects and Situs Inversus, Am J Hum Genet 103: ((2018) ), 984–994. |
[46] | Ferkol T. and Leigh M. , Primary ciliary dyskinesia and newborn respiratory distress, Semin Perinatol 30: ((2006) ), 335–340. |
[47] | Ferkol T.W. and Leigh M.W. , Ciliopathies: The central role of cilia in a spectrum of pediatric disorders, J Pediatr 160: ((2012) ), 366–371. |
[48] | Ferrante M.I. , Zullo A. , Barra A. , Bimonte S. , Messaddeq N. , Studer M. , Dolle P. and Franco B. , Oral-facial-digital type I protein is required for primary cilia formation and left-right axis specification, Nat Genet 38: ((2006) ), 112–117. |
[49] | Fowkes M.E. and Mitchell D.R. , The role of preassembled cytoplasmic complexes in assembly of flagellar dynein subunits, Mol Biol Cell 9: ((1998) ), 2337–2347. |
[50] | Frommer A. , Hjeij R. , Loges N.T. , Edelbusch C. , Jahnke C. , Raidt J. , Werner C. , Wallmeier J. , Grosse-Onnebrink J. , Olbrich H. , Cindric S. , Jaspers M. , Boon M. , Memari Y. , Durbin R. , Kolb-Kokocinski A. , Sauer S. , Marthin J.K. , Nielsen K.G. , Amirav I. , Elias N. , Kerem E. , Shoseyov D. , Haeffner K. and Omran H. , Immunofluorescence analysis and diagnosis of primary ciliary dyskinesia with radial spoke defects, Am J Respir Cell Mol Biol 53: ((2015) ), 563–573. |
[51] | Gerdes J.M. , Davis E.E. and Katsanis N. , The vertebrate primary cilium in development, homeostasis and disease, Cell 137: ((2009) ), 32–45. |
[52] | Guichard C. , Harricane M.C. , Lafitte J.J. , Godard P. , Zaegel M. , Tack V. , Lalau G. and Bouvagnet P. , Axonemal dynein intermediate-chain gene (DNAI1) mutations resultinversus and primary ciliary dyskinesia (Kartagener syndrome), Am J Hum Genet 68: ((2001) ), 1030–1035. |
[53] | Hadfield P.J. , Rowe-Jones J.M. , Bush A. and Mackay I.S. , Treatment of otitis media with effusion in children with primary ciliary dyskinesia, Clin Otolaryngol Allied Sci 22: ((1997) ), 302–306. |
[54] | Halbeisen F.S. , Goutaki M. , Spycher B.D. , Amirav I. , Behan L. , Boon M. , Hogg C. , Casaulta C. , Crowley S. , Haarman E.G. , Karadag B. , Koerner-Rettberg C. , Loebinger M.R. , Mazurek H. , Morgan L. , Nielsen K.G. , Omran H. , Santamaria F. , Schwerk N. , Thouvenin G. , Yiallouros P. , Lucas J.S. , Latzin P. and Kuehni C.E. , Lung function in patients with primary ciliary dyskinesia: An iPCD Cohort study, Eur Respir J 52: ((2018) ). |
[55] | Harrison M.J. , Shapiro A.J. and Kennedy M.P. , Congenital heart disease and primary ciliary dyskinesia, Paediatr Respir Rev 18: ((2016) ), 25–32. |
[56] | Hellinckx J. , Demedts M. and De Boeck K. , Primary ciliary dyskinesia: Evolution of pulmonary function, Eur J Pediatr 157: ((1998) ), 422–426. |
[57] | Heuser T. , Raytchev M. , Krell J. , Porter M.E. and Nicastro D. , The dynein regulatory complex is the nexin link and a major regulatory node in cilia and flagella, J Cell Biol 187: ((2009) ), 921–933. |
[58] | Hirokawa N. , Tanaka Y. , Okada Y. and Takeda S. , Nodal flow and the generation of left-right asymmetry, Cell 125: ((2006) ), 33–45. |
[59] | Hoben I.M. , Hjeij R. , Olbrich H. , Dougherty G.W. , Nothe-Menchen T. , Aprea I. , Frank D. , Pennekamp P. , Dworniczak B. , Wallmeier J. , Raidt J. , Nielsen K.G. , Philipsen M.C. , Santamaria F. , Venditto L. , Amirav I. , Mussaffi H. , Prenzel F. , Wu K. , Bakey Z. , Schmidts M. , Loges N.T. and Omran H. , Mutations in C11orf70 cause primary ciliary dyskinesia with randomization of left/right body ssymmetry due to defects of outer and inner dynein arms, Am J Hum Genet 102: ((2018) ), 973–984. |
[60] | Hong D.H. , Pawlyk B. , Sokolov M. , Strissel K.J. , Yang J. , Tulloch B. , Wright A.F. , Arshavsky V.Y. and Li T. , RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia, Invest Ophthalmol Vis Sci 44: ((2003) ), 2413–2421. |
[61] | Horani A. , Brody S.L. , Ferkol T. , Shoseyov D. , Wasserman M.G. , Ta-shma A. , Wilson K.S. , Bayly P.V. , Amirav I. , Cohen-Cymberknoh M. , Dutcher S.K. , Elpeleg O. and Kerem E. , CCDC65 mutation causes primary ciliary dyskinesia with normal ultrastructure and hyperkinetic cilia, PLoS One 8: ((2013) ), e72299. |
[62] | Horani A. , Druley T.E. , Zariwala M.A. , Patel A.C. , Levinson B.T. , Van Arendonk L.G. , Thornton K.C. , Giacalone J.C. , Albee A.J. , Wilson K.S. , Turner E.H. , Nickerson D.A. , Shendure J. , Bayly P.V. , Leigh M.W. , Knowles M.R. , Brody S.L. , Dutcher S.K. and Ferkol T.W. , Whole-exome capture and sequencing identifies HEATR2 mutation as a cause of primary ciliary dyskinesia, Am J Hum Genet 91: ((2012) ), 685–693. |
[63] | Horani A. and Ferkol T.W. , Primary ciliary dyskinesia and associated sensory ciliopathies, Expert Rev Respir Med 10: ((2016) ), 569–576. |
[64] | Horani A. , Ferkol T.W. , Shoseyov D. , Wasserman M.G. , Oren Y.S. , Kerem B. , Amirav I. , Cohen-Cymberknoh M. , Dutcher S.K. , Brody S.L. , Elpeleg O. and Kerem E. , LRRC6 mutation causes primary ciliary dyskinesia with dynein arm defects, PLoS One 8: ((2013) ), e59436. |
[65] | Horani A. , Ustione A. , Huang T. , Firth A.L. , Pan J. , Gunsten S.P. , Haspel J.A. , Piston D.W. and Brody S.L. , Establishment of the early cilia preassembly protein complex during motile ciliogenesis, Proc Natl Acad Sci U S A 115: ((2018) ), E1221–E1228. |
[66] | Horvath J. , Fliegauf M. , Olbrich H. , Kispert A. , King S.M. , Mitchison H. , Zariwala M.A. , Knowles M.R. , Sudbrak R. , Fekete G. , Neesen J. , Reinhardt R. and Omran H. , Identification and analysis of emal dynein light chain axon1 in primary ciliary dyskinesia patients, Am J Respir Cell Mol Biol 33: ((2005) ), 41–47. |
[67] | Hossain T. , Kappelman M.D. , Perez-Atayde A.R. , Young G.J. , Huttner K.M. and Christou H. , Primary ciliary dyskinesia as a cause of neonatal respiratory distress: Implications for the neonatologist, J Perinatol 23: ((2003) ), 684–687. |
[68] | Inaba K. and Mizuno K. , Sperm dysfunction and ciliopathy, Reprod Med Biol 15: ((2016) ), 77–94. |
[69] | Ishikawa T. , Sakakibara H. and Oiwa K. , The architecture of outer dynein arms, J Mol Biol 368: ((2007) ), 1249–1258. |
[70] | Jain R. , Javidan-Nejad C. , Alexander-Brett J. , Horani A. , Cabellon M.C. , Walter M.J. and Brody S.L. , Sensory functions of motile cilia and implication for bronchiectasis, Front Biosci (Schol Ed) 4: ((2012) ), 1088–1098. |
[71] | Jeanson L. , Copin B. , Papon J.F. , Dastot-Le Moal F. , Duquesnoy P. , Montantin G. , Cadranel J. , Corvol H. , Coste A. , Desir J. , Souayah A. , Kott E. , Collot N. , Tissier S. , Louis B. , Tamalet A. , de Blic J. , Clement A. , Escudier E. , Amselem S. and Legendre M. , RSPH3 mutations cause primary ciliary dyskinesia with central-complex defects and a near absence of radial spokes, Am J Hum Genet 97: ((2015) ), 153–162. |
[72] | Johnson N.T. , Villalon M. , Royce F.H. , Hard R. and Verdugo P. , Autoregulation of beat frequency in respiratory ciliated cells. Demonstration by viscous loading, Am Rev Respir Dis 144: ((1991) ), 1091–1094. |
[73] | Kartagener M. , Zur pathogenese der bronkiectasien. Bronkiectasien bei situs viscerum inversus, Beitr Klin Tuberk 82: ((1933) ), 489–501. |
[74] | Kellett F. and Robert N.M. , Nebulised 7% hypertonic saline improves lung function and quality of life in bronchiectasis, Respir Med 105: ((2011) ), 1831–1835. |
[75] | Kennedy M.P. , Noone P.G. , Leigh M.W. , Zariwala M.A. , Minnix S.L. , Knowles M.R. and Molina P.L. , High-resolution CT of patients with primary ciliary dyskinesia, AJR Am J Roentgenol 188: ((2007) ), 1232–1238. |
[76] | Kennedy M.P. , Omran H. , Leigh M.W. , Dell S. , Morgan L. , Molina P.L. , Robinson B.V. , Minnix S.L. , Olbrich H. , Severin T. , Ahrens P. , Lange L. , Morillas H.N. , Noone P.G. , Zariwala M.A. and Knowles M.R. , Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia, Circulation 115: ((2007) ), 2814–2821. |
[77] | Kim R.H. , D A.H. , Cutz E. , Knowles M.R. , Nelligan K.A. , Nykamp K. , Zariwala M.A. and Dell S.D. , The role of molecular genetic analysis in the diagnosis of primary ciliary dyskinesia, Ann Am Thorac Soc 11: ((2014) ), 351–359. |
[78] | Knowles M.R. , Daniels L.A. , Davis S.D. , Zariwala M.A. and Leigh M.W. , Primary ciliary dyskinesia. Recent advances in diagnostics, genetics and characterization of clinical disease, Am J Respir Crit Care Med 188: ((2013) ), 913–922. |
[79] | Knowles M.R. , Leigh M.W. , Carson J.L. , Davis S.D. , Dell S.D. , Ferkol T.W. , Olivier K.N. , Sagel S.D. , Rosenfeld M. , Burns K.A. , Minnix S.L. , Armstrong M.C. , Lori A. , Hazucha M.J. , Loges N.T. , Olbrich H. , Becker-Heck A. , Schmidts M. , Werner C. , Omran H. and Zariwala M.A. , Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure, Thorax 67: ((2011) ), 433–441. |
[80] | Knowles M.R. , Leigh M.W. , Ostrowski L.E. , Huang L. , Carson J.L. , Hazucha M.J. , Yin W. , Berg J.S. , Davis S.D. , Dell S.D. , Ferkol T.W. , Rosenfeld M. , Sagel S.D. , Milla C.E. , Olivier K.N. , Turner E.H. , Lewis A.P. , Bamshad M.J. , Nickerson D.A. , Shendure J. , Zariwala M.A. and Genetic Disorders of Mucociliary Clearance Consortium, Exome sequencing identifies mutations in CCDC114 as a cause of primary ciliary dyskinesia, Am J Hum Genet 92: ((2013) ), 99–106. |
[81] | Knowles M.R. , Ostrowski L.E. , loges N.T. , Hurd T.W. , Leigh M. , Huang B.Q. , Wolf W.E. , Carson J.L. , Hazucha M.J. , Yin W. , Davis S.D. , Dell S.D. , Ferkol T. , Sagel S.D. , Olivier K.N. , Jahnke C. , Olbrich H. , Werner C. , Raidt J. , Wallmeier J. , Pennekamp P. , Dougherty G.W. , Hjeij R. , Gee H.Y. , Otto E.A. , Halbritter J. , Chaki M. , Diaz K.A. , Braun D.A. , Porath J.D. , Schueler M. , Baktai G. , Griese M. , Turner E.H. , Lewis A.P. , Bamshad M.J. , Nickerson D.A. , Hildebrandt F. , Shendure J. , Omran H. and Zariwala M. , Mutations in SPAG1 cause primary ciliary dyskinesia associated with defective outer and inner dynein arms, Am J Hum Genet 93: ((2013) ), 711–720. |
[82] | Kobbernagel H.E. , Buchvald F.F. , Haarman E.G. , Casaulta C. , Collins S.A. , Hogg C. , Kuehni C.E. , Lucas J.S. , Omran H. , Quittner A.L. , Werner C. and Nielsen K.G. , Study protocol, rationale and recruitment in a European multi-centre randomized controlled trial to determine the efficacy and safety of azithromycin maintenance therapy for 6 months in primary ciliary dyskinesia, BMC Pulm Med 16: ((2016) ), 104. |
[83] | Kott E. , Duquesnoy P. , Copin B. , Legendre M. , Dastot-Le Moal F. , Montantin G. , Jeanson L. , Tamalet A. , Papon J.F. , Siffroi J.P. , Rives N. , Mitchell V. , de Blic J. , Coste A. , Clement A. , Escalier D. , Toure A. , Escudier E. and Amselem S. , Loss-of-function mutations in LRRC6, a gene essential for proper axonemal assembly of inner and outer dynein arms, cause primary ciliary dyskinesia, Am J Hum Genet 91: ((2012) ), 958–964. |
[84] | Kouis P. , Papatheodorou S.I. and Yiallouros P.K. , Diagnostic accuracy of nasal nitric oxide for establishing diagnosis of primary ciliary dyskinesia: A meta-analysis, BMC Pulm Med 15: ((2015) ), 153. |
[85] | Kuehni C.E. , Frischer T. , Strippoli M.P. , Maurer E. , Bush A. , Nielsen K.G. , Escribano A. , Lucas J.S. , Yiallouros P. , Omran H. , Eber E. , O’Callaghan C. , Snijders D. , Barbato A. and Task E.R.S. , Force on Primary Ciliary Dyskinesia in Children, Factors influencing age at diagnosis of primary ciliary dyskinesia in European children, Eur Respir J 36: ((2010) ), 1248–1258. |
[86] | Larkins C.E. , Long A.B. and Caspary T. , Defective Nodal and Cerl2 expression in the Arl13b(hnn) mutant node underlie its heterotaxia, Dev Biol 367: ((2012) ), 15–24. |
[87] | Leigh M.W. , Ferkol T.W. , Davis S.D. , Lee H.S. , Rosenfeld M. , Dell S.D. , Sagel S.D. , Milla C. , Olivier K.N. , Sullivan K.M. , Zariwala M.A. , Pittman J.E. , Shapiro A.J. , Carson J.L. , Krischer J. , Hazucha M.J. and Knowles M.R. , Clinical features and associated likelihood of primary ciliary dyskinesia in children and adolescents, Ann Am Thorac Soc 13: ((2016) ), 1305–1313. |
[88] | Leigh M.W. , Hazucha M.J. , Chawla K.K. , Baker B.R. , Shapiro A.J. , Brown D.E. , Lavange L.M. , Horton B.J. , Qaqish B. , Carson J.L. , Davis S.D. , Dell S.D. , Ferkol T.W. , Atkinson J.J. , Olivier K.N. , Sagel S.D. , Rosenfeld M. , Milla C. , Lee H.S. , Krischer J. , Zariwala M.A. and Knowles M.R. , Standardizing nasal nitric oxide measurement as a test for primary ciliary dyskinesia, Ann Am Thorac Soc 10: ((2013) ), 574–581. |
[89] | Loges N.T. , Antony D. , Maver A. , Deardorff M.A. , Gulec E.Y. , Gezdirici A. , Nothe-Menchen T. , Hoben I.M. , Jelten L. , Frank D. , Werner C. , Tebbe J. , Wu K. , Goldmuntz E. , Cuturilo G. , Krock B. , Ritter A. , Hjeij R. , Bakey Z. , Pennekamp P. , Dworniczak B. , Brunner H. , Peterlin B. , Tanidir C. , Olbrich H. , Omran H. and Schmidts M. , Recessive DNAH9 Loss-of-Function Mutations Cause Laterality Defects and Subtle Respiratory Ciliary-Beating Defects, Am J Hum Genet 103: ((2018) ), 995–1008. |
[90] | Loges N.T. , Olbrich H. , Becker-Heck A. , Haffner K. , Heer A. , Reinhard C. , Schmidts M. , Kispert A. , Zariwala M.A. , Leigh M.W. , Knowles M.R. , Zentgraf H. , Seithe H. , Nurnberg G. , Nurnberg P. , Reinhardt R. and Omran H. , Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects, Am J Hum Genet 85: ((2009) ), 883–889. |
[91] | Loges N.T. , Olbrich H. , Fenske L. , Mussaffi H. , Horvath J. , Fliegauf M. , Kuhl H. , Baktai G. , Peterffy E. , Chodhari R. , Chung E.M. , Rutman A. , O’Callaghan C. , Blau H. , Tiszlavicz L. , Voelkel K. , Witt M. , Zietkiewicz E. , Neesen J. , Reinhardt R. , Mitchison H.M. and Omran H. , DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm, Am J Hum Genet 83: ((2008) ), 547–558. |
[92] | Mali G.R. , Yeyati P.L. , Mizuno S. , Dodd D.O. , Tennant P.A. , Keighren M.A. , Zur Lage P. , Shoemark A. , Garcia-Munoz A. , Shimada A. , Takeda H. , Edlich F. , Takahashi S. , von Kreigsheim A. , Jarman A.P. and Mill P. , ZMYND10 functions in a chaperone relay during axonemal dynein assembly, Elife 7: ((2018) ). |
[93] | Marshall C.R. , Scherer S.W. , Zariwala M.A. , Lau L. , Paton T.A. , Stockley T. , Jobling R.K. , Ray P.N. , Knowles M.R. , Forge Canada Consortium, Hall D.A. , Dell S.D. and Kim R.H. , Whole-exome sequencing and targeted copy number analysis in primary ciliary dyskinesia, G3 (Bethesda) 5: ((2015) ), 1775–1781. |
[94] | Marthin J.K. and Nielsen K.G. , Hand-held tidal breathing nasal nitric oxide measurement— A promising targeted case-finding tool for the diagnosis of primary ciliary dyskinesia, PLoS One 8: ((2013) ), e57262. |
[95] | Marthin J.K. , Petersen N. , Skovgaard L.T. and Nielsen K.G. , Lung function in patients with primary ciliary dyskinesia: A cross-sectional and 3-decade longitudinal study, Am J Respir Crit Care Med 181: ((2010) ), 1262–1268. |
[96] | Matwijiw I. , Thliveris J.A. and Faiman C. , Aplasia of nasal cilia with situs inversus, azoospermia and normal sperm flagella: A unique variant of the immotile cilia syndrome, J Urol 137: ((1987) ), 522–524. |
[97] | Mener D.J. , Lin S.Y. , Ishman S.L. and Boss E.F. , Treatment and outcomes of chronic rhinosinusitis in children with primary ciliary dyskinesia: Where is the evidence? A qualitative systematic review, Int Forum Allergy Rhinol 3: ((2013) ), 986–991. |
[98] | Merveille A.C. , Davis E.E. , Becker-Heck A. , Legendre M. , Amirav I. , Bataille G. , Belmont J. , Beydon N. , Billen F. , Clement A. , Clercx C. , Coste A. , Crosbie R. , de Blic J. , Deleuze S. , Duquesnoy P. , Escalier D. , Escudier E. , Fliegauf M. , Horvath J. , Hill K. , Jorissen M. , Just J. , Kispert A. , Lathrop M. , Loges N.T. , Marthin J.K. , Momozawa Y. , Montantin G. , Nielsen K.G. , Olbrich H. , Papon J.F. , Rayet I. , Roger G. , Schmidts M. , Tenreiro H. , Towbin J.A. , Zelenika D. , Zentgraf H. , Georges M. , Lequarre A.S. , Katsanis N. , Omran H. and Amselem S. , CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs, Nat Genet 43: ((2011) ), 72–78. |
[99] | Mirra V. , Werner C. and Santamaria F. , Primary ciliary dyskinesia: An update on clinical aspects, genetics, diagnosis and future treatment strategies, Front Pediatr 5: ((2017) ), 135. |
[100] | Mitchison H.M. , Schmidts M. , Loges N.T. , Freshour J. , Dritsoula A. , Hirst R.A. , O’Callaghan C. , Blau H. , Al Dabbagh M. , Olbrich H. , Beales P.L. , Yagi T. , Mussaffi H. , Chung E.M. , Omran H. and Mitchell D.R. , Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia, Nat Genet 44: ((2012) ), 381–389, S381–382. |
[101] | Moore A. , Escudier E. , Roger G. , Tamalet A. , Pelosse B. , Marlin S. , Clement A. , Geremek M. , Delaisi B. , Bridoux A.M. , Coste A. , Witt M. , Duriez B. , Amselem S. , RPGR is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa, J Med Genet 43: ((2006) ), 326–333. |
[102] | Moore D.J. , Onoufriadis A. , Shoemark A. , Simpson M.A. , Zur Lage P.I. , de Castro S.C. , Bartoloni L. , Gallone G. , Petridi S. , Woollard W.J. , Antony D. , Schmidts M. , Didonna T. , Makrythanasis P. , Bevillard J. , Mongan N.P. , Djakow J. , Pals G. , Lucas J.S. , Marthin J.K. , Nielsen K.G. , Santoni F. , Guipponi M. , Hogg C. , Antonarakis S.E. , Emes R.D. , Chung E.M. , Greene N.D. , Blouin J.L. , Jarman A.P. and Mitchison H.M. , Mutations in ZMYND10, a gene essential for proper axonemal assembly of inner and outer dynein arms in humans and flies, cause primary ciliary dyskinesia, Am J Hum Genet 93: ((2013) ), 346–356. |
[103] | Mullowney T. , Manson D. , Kim R. , Stephens D. , Shah V. and Dell S. , Primary ciliary dyskinesia and neonatal respiratory distress, Pediatrics 134: ((2014) ), 1160–1166. |
[104] | Murray M.P. , Govan J.R. , Doherty C.J. , Simpson A.J. , Wilkinson T.S. , Chalmers J.D. , Greening A.P. , Haslett C. and Hill A.T. , A randomized controlled trial of nebulized gentamicin in non-cystic fibrosis bronchiectasis, Am J Respir Crit Care Med 183: ((2011) ), 491–499. |
[105] | Nachury M.V. , The molecular machines that traffic signaling receptors into and out of cilia, Curr Opin Cell Biol 51: ((2018) ), 124–131. |
[106] | Nonaka S. , Tanaka Y. , Okada Y. , Takeda S. , Harada A. , Kanai Y. , Kido M. and Hirokawa N. , Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein, Cell 95: ((1998) ), 829–837. |
[107] | Noone P.G. , Leigh M.W. , Sannuti A. , Minnix S.L. , Carson J.L. , Hazucha M. , Zariwala M.A. and Knowles M.R. , Primary ciliary dyskinesia: Diagnostic and phenotypic features, Am J Respir Crit Care Med 169: ((2004) ), 459–467. |
[108] | O’Callaghan C. , Rutman A. , Williams G.M. and Hirst R.A. , Inner dynein arm defects causing primary ciliary dyskinesia: Repeat testing required, Eur Respir J 38: ((2011) ), 603–607. |
[109] | O’Donnell A.E. , Barker A.F. , Ilowite J.S. and Fick R.B. , Treatment of idiopathic bronchiectasis with aerosolized recombinant human DNase I. rhDNase Study Group, Chest 113: ((1998) ), 1329–1334. |
[110] | Olbrich H. , Haffner K. , Kispert A. , Volkel A. , Volz A. , Sasmaz G. , Reinhardt R. , Hennig S. , Lehrach H. , Konietzko N. , Zariwala M. , Noone P.G. , Knowles M. , Mitchison H.M. , Meeks M. , Chung E.M. , Hildebrandt F. , Sudbrak R. and Omran H. , Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry, Nat Genet 30: ((2002) ), 143–144. |
[111] | Olbrich H. , Schmidts M. , Werner C. , Onoufriadis A. , Loges N.T. , Raidt J. , Banki N.F. , Shoemark A. , Burgoyne T. , Al Turki S. , Hurles M.E. , Kohler G. , Schroeder J. , Nurnberg G. , Nurnberg P. , Chung E.M. , Reinhardt R. , Marthin J.K. , Nielsen K.G. , Mitchison H.M. and Omran H. , Recessive HYDIN mutations cause primary ciliary dyskinesia without randomization of left-right body asymmetry, Am J Hum Genet 91: ((2012) ), 672–684. |
[112] | Olcese C. , Patel M.P. , Shoemark A. , Kiviluoto S. , Legendre M. , Williams H.J. , Vaughan C.K. , Hayward J. , Goldenberg A. , Emes R.D. , Munye M.M. , Dyer L. , Cahill T. , Bevillard J. , Gehrig C. , Guipponi M. , Chantot S. , Duquesnoy P. , Thomas L. , Jeanson L. , Copin B. , Tamalet A. , Thauvin-Robinet C. , Papon J.F. , Garin A. , Pin I. , Vera G. , Aurora P. , Fassad M.R. , Jenkins L. , Boustred C. , Cullup T. , Dixon M. , Onoufriadis A. , Bush A. , Chung E.M. , Antonarakis S.E. , Loebinger M.R. , Wilson R. , Armengot M. , Escudier E. , Hogg C. , Group U.K.R. , Amselem S. , Sun Z. , Bartoloni L. , Blouin J.L. , Mitchison H.M. , X-linked primary ciliary dyskinesia due to mutations in the cytoplasmic axonemal dynein assembly factor PIH1D3, Nat Commun 8: ((2017) ), 14279. |
[113] | Olin J.T. , Burns K. , Carson J.L. , Metjian H. , Atkinson J.J. , Davis S.D. , Dell S.D. , Ferkol T.W. , Milla C.E. , Olivier K.N. , Rosenfeld M. , Baker B. , Leigh M.W. , Knowles M.R. , Sagel S.D. and Genetic Disorders of Mucociliary Clearance Consortium, Diagnostic yield of nasal scrape biopsies in primary ciliary dyskinesia: A multicenter experience, Pediatr Pulmonol 46: ((2011) ), 483–488. |
[114] | Omran H. , Haffner K. , Volkel A. , Kuehr J. , Ketelsen U.P. , Ross U.H. , Konietzko N. , Wienker T. , Brandis M. and Hildebrandt F. , Homozygosity mapping of a gene locus for primary ciliary dyskinesia on chromosome 5p and identification of the heavy dynein chain DNAH5 as a candidate gene, Am J Respir Cell Mol Biol 23: ((2000) ), 696–702. |
[115] | Omran H. , Kobayashi D. , Olbrich H. , Tsukahara T. , Loges N.T. , Hagiwara H. , Zhang Q. , Leblond G. , O’Toole E. , Hara C. , Mizuno H. , Kawano H. , Fliegauf M. , Yagi T. , Koshida S. , Miyawaki A. , Zentgraf H. , Seithe H. , Reinhardt R. , Watanabe Y. , Kamiya R. , Mitchell D.R. and Takeda H. , Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins, Nature 456: ((2008) ), 611–616. |
[116] | Onoufriadis A. , Paff T. , Antony D. , Shoemark A. , Micha D. , Kuyt B. , Schmidts M. , Petridi S. , Dankert-Roelse J.E. , Haarman E.G. , Daniels J.M. , Emes R.D. , Wilson R. , Hogg C. , Scambler P.J. , Chung E.M. , Uk10K , Pals G. and Mitchison H.M. , Splice-site mutations in the axonemal outer dynein arm docking complex gene CCDC114 cause primary ciliary dyskinesia, Am J Hum Genet 92: ((2013) ), 88–98. |
[117] | Onoufriadis A. , Shoemark A. , Schmidts M. , Patel M. , Jimenez G. , Liu H. , Thomas B. , Dixon M. , Hirst R.A. , Rutman A. , Burgoyne T. , Williams C. , Scully J. , Bolard F. , Lafitte J.J. , Beales P.L. , Hogg C. , Yang P. , Chung E.M. , Emes R.D. , O’Callaghan C. , Uk10K , Bouvagnet P. and Mitchison H.M. , Targeted NGS gene panel identifies mutations in RSPH1 causing primary ciliary dyskinesia and a common mechanism for ciliary central pair agenesis due to radial spoke defects, Hum Mol Genet 23: ((2014) ), 3362–3374. |
[118] | Paff T. , Daniels J.M. , Weersink E.J. , Lutter R. , Vonk Noordegraaf A. and Haarman E.G. , A randomised controlled trial on the effect of inhaled hypertonic saline on quality of life in primary ciliary dyskinesia, Eur Respir J 49: ((2017) ). |
[119] | Paff T. , Loges N.T. , Aprea I. , Wu K. , Bakey Z. , Haarman E.G. , Daniels J.M.A. , Sistermans E.A. , Bogunovic N. , Dougherty G.W. , Hoben I.M. , Grosse-Onnebrink J. , Matter A. , Olbrich H. , Werner C. , Pals G. , Schmidts M. , Omran H. , Micha D. , Mutations in PIH1D3 cause X-linked primary ciliary dyskinesia with outer and inner dynein arm defects, Am J Hum Genet 100: ((2017) ), 160–168. |
[120] | Pal M. , Morgan M. , Phelps S.E. , Roe S.M. , Parry-Morris S. , Downs J.A. , Polier S. , Pearl L.H. and Prodromou C. , Structural basis for phosphorylation-dependent recruitment of Tel2 to Hsp90 by Pih1, Structure 22: ((2014) ), 805–818. |
[121] | Panizzi J.R. , Becker-Heck A. , Castleman V.H. , Al-Mutairi D.A. , Liu Y. , Loges N.T. , Pathak N. , Austin-Tse C. , Sheridan E. , Schmidts M. , Olbrich H. , Werner C. , Haffner K. , Hellman N. , Chodhari R. , Gupta A. , Kramer-Zucker A. , Olale F. , Burdine R.D. , Schier A.F. , O’Callaghan C. , Chung E.M. , Reinhardt R. , Mitchison H.M. , King S.M. , Omran H. and Drummond I.A. , CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms, Nat Genet 44: ((2012) ), 714–719. |
[122] | Pazour G.J. and Bloodgood R.A. , Targeting proteins to the ciliary membrane, Curr Top Dev Biol 85: ((2008) ), 115–149. |
[123] | Pennarun G. , Escudier E. , Chapelin C. , Bridoux A.M. , Cacheux V. , Roger G. , Clement A. , Goossens M. , Amselem S. and Duriez B. , Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia, Am J Hum Genet 65: ((1999) ), 1508–1519. |
[124] | Pifferi M. , Bush A. , Caramella D. , Di Cicco M. , Zangani M. , Chinellato I. , Macchia P. and Boner A.L. , Agenesis of paral sinuses and nasal nitric oxide in primary ciliary dyskinesia, Eur Respir J 37: ((2011) ), 566–571. |
[125] | Pifferi M. , Michelucci A. , Conidi M.E. , Cangiotti A.M. , Simi P. , Macchia P. and Boner A.L. , New DNAH11 mutations in primary ciliary dyskinesia with normal axonemal ultrastructure, Eur Respir J 35: ((2010) ), 1413–1416. |
[126] | Piperno G. and Mead K. , Transport of a novel complex in the cytoplasmic matrix of Chlamydomonas flagella, Proc Natl Acad Sci U S A 94: ((1997) ), 4457–4462. |
[127] | Raidt J. , Werner C. , Menchen T. , Dougherty G.W. , Olbrich H. , Loges N.T. , Schmitz R. , Pennekamp P. and Omran H. , Ciliary function and motor protein composition of human fallopian tubes, Hum Reprod 30: ((2015) ), 2871–2880. |
[128] | Richard S. , Nezelof C. , Pfister A. , de Blic J. , Scheinmann P. and Paupe J. , Congenital ciliary aplasia in two siblings. A primitive disregulation of ciliogenesis? Pathol Res Pract 185: ((1989) ), 181–183. |
[129] | Rossman C.M. , Forrest J.B. , Lee R.M. and Newhouse M.T. , The dyskinetic cilia syndrome. Ciliary motility in immotile cilia syndrome, Chest 78: ((1980) ), 580–582. |
[130] | Rutland J. and Cole P. , Ciliary dyskinesia, Lancet 316: ((1980) ), 859. |
[131] | Shah A. , Shoemark A. , MacNeill S.J. , Bhaludin B. , Rogers A. , Bilton D. , Hansell D.M. , Wilson R. and Loebinger M.R. , A longitudinal study characterising a large adult primary ciliary dyskinesia population, Eur Respir J 48: ((2016) ), 441–450. |
[132] | Shah A.S. , Ben-Shahar Y. , Moninger T.O. , Kline J.N. and Welsh M.J. , Motile cilia of human airway epithelia are chemosensory, Science 325: ((2009) ), 1131–1134. |
[133] | Shapiro A.J. , Davis S.D. , Ferkol T. , Dell S.D. , Rosenfeld M. , Olivier K.N. , Sagel S.D. , Milla C. , Zariwala M.A. , Wolf W. , Carson J.L. , Hazucha M.J. , Burns K. , Robinson B. , Knowles M.R. and Leigh M.W. , and Genetic Disorders of Mucociliary Clearance Consortium, Laterality defects other than situs inversus totalis in primary ciliary dyskinesia: Insights into situs ambiguus and heterotaxy, Chest 146: ((2014) ), 1176–1186. |
[134] | Shapiro A.J. , Davis S.D. , Polineni D. , Manion M. , Rosenfeld M. , Dell S.D. , Chilvers M.A. , Ferkol T.W. , Zariwala M.A. , Sagel S.D. , Josephson M. , Morgan L. , Yilmaz O. , Olivier K.N. , Milla C. , Pittman J.E. , Daniels M.L.A. , Jones M.H. , Janahi I.A. , Ware S.M. , Daniel S.J. , Cooper M.L. , Nogee L.M. , Anton B. , Eastvold T. , Ehrne L. , Guadagno E. , Knowles M.R. , Leigh M.W. , Lavergne V. and American Thoracic Society Assembly on Pediatrics, Diagnosis of primary diliary dyskinesia. An official American Thoracic Society Clinical Practice Guideline, Am J Respir Crit Care Med 197: ((2018) ), e24–e39. |
[135] | Shapiro A.J. , Josephson M. , Rosenfeld M. , Yilmaz O. , Davis S.D. , Polineni D. , Guadagno E. , Leigh M.W. , Lavergne V. , Accuracy of nasal nitric oxide measurement as a diagnostic test for primary ciliary dyskinesia. A systematic review and meta-analysis, Ann Am Thorac Soc 14: ((2017) ), 1184–1196. |
[136] | Shapiro A.J. , Weck K.E. , Chao K.C. , Rosenfeld M. , Nygren A.O. , Knowles M.R. , Leigh M.W. and Zariwala M.A. , Cri du chat syndrome and primary ciliary dyskinesia: A common genetic cause on chromosome 5p, J Pediatr 165: ((2014) ), 858–861. |
[137] | Shapiro A.J. , Zariwala M.A. , Ferkol T. , Davis S.D. , Sagel S.D. , Dell S.D. , Rosenfeld M. , Olivier K.N. , Milla C. , Daniel S.J. , Kimple A.J. , Manion M. , Knowles M.R. , Leigh M.W. and Genetic Disorders of Mucociliary Clearance Consortium, Diagnosis, monitoring and treatment of primary ciliary dyskinesia: PCD Foundation consensus recommendations based on state of the art review, Pediatr Pulmonol 51: ((2016) ), 115–132. |
[138] | Shoemark A. , Burgoyne T. , Kwan R. , Dixon M. , Patel M.P. , Rogers A.V. , Onoufriadis A. , Scully J. , Daudvohra F. , Cullup T. , Loebinger M.R. , Wilson R. , Chung E.M.K. , Bush A. , Mitchison H.M. and Hogg C. , Primary ciliary dyskinesia with normal ultrastructure: Three-dimensional tomography detects absence of DNAH11, Eur Respir J 51: ((2018) ). |
[139] | Shoemark A. , Dixon M. , Corrin B. and Dewar A. , Twenty-year review of quantitative transmission electron microscopy for the diagnosis of primary ciliary dyskinesia, J Clin Pathol 65: ((2012) ), 267–271. |
[140] | Simet S.M. , Sisson J.H. , Pavlik J.A. , Devasure J.M. , Boyer C. , Liu X. , Kawasaki S. , Sharp J.G. , Rennard S.I. and Wyatt T.A. , Long-term cigarette smoke exposure in a mouse model of ciliated epithelial cell function, Am J Respir Cell Mol Biol 43: ((2010) ), 635–640. |
[141] | Sleigh M.A. , Primary ciliary dyskinesia, Lancet 2: ((1981) ), 476. |
[142] | Smit H.J. , Schreurs A.J. , Van den Bosch J.M. and Westermann C.J. , Is resection of bronchiectasis beneficial in patients with primary ciliary dyskinesia? Chest 109: ((1996) ), 1541–1544. |
[143] | Sommer J.U. , Schafer K. , Omran H. , Olbrich H. , Wallmeier J. , Blum A. , Hormann K. and Stuck B.A. , ENT manifestations in patients with primary ciliary dyskinesia: Prevalence and significance of otorhinolaryngologic co-morbidities, Eur Arch Otorhinolaryngol 268: ((2011) ), 383–388. |
[144] | Sorokin S.P. , Reconstructions of centriole formation and ciliogenesis in mammalian lungs, J Cell Sci 3: ((1968) ), 207–230. |
[145] | Tarkar A. , Loges N.T. , Slagle C.E. , Francis R. , Dougherty G.W. , Tamayo J.V. , Shook B. , Cantino M. , Schwartz D. , Jahnke C. , Olbrich H. , Werner C. , Raidt J. , Pennekamp P. , Abouhamed M. , Hjeij R. , Kohler G. , Griese M. , Li Y. , Lemke K. , Klena N. , Liu X. , Gabriel G. , Tobita K. , Jaspers M. , Morgan L.C. , Shapiro A.J. , Letteboer S.J. , Mans D.A. , Carson J.L. , Leigh M.W. , Wolf W.E. , Chen S. , Lucas J.S. , Onoufriadis A. , Plagnol V. , Schmidts M. , Boldt K. , Uk10K, Roepman R. , Zariwala M.A. , Lo C.W. , Mitchison H.M. , Knowles M.R. , Burdine R.D. , Loturco J.J. and Omran H. , DYX1C1 is required for axonemal dynein assembly and ciliary motility, Nat Genet 45: ((2013) ), 995–1003. |
[146] | Theegarten D. and Ebsen M. , Ultrastructural pathology of primary ciliary dyskinesia: Report about 125 cases in Germany, Diagn Pathol 6: ((2011) ), 115. |
[147] | Tobin J.L. and Beales P.L. , The nonmotile ciliopathies, Genet Med 11: ((2009) ), 386–402. |
[148] | Wallmeier J. , Al-Mutairi D.A. , Chen C.T. , Loges N.T. , Pennekamp P. , Menchen T. , Ma L. , Shamseldin H.E. , Olbrich H. , Dougherty G.W. , Werner C. , Alsabah B.H. , Kohler G. , Jaspers M. , Boon M. , Griese M. , Schmitt-Grohe S. , Zimmermann T. , Koerner-Rettberg C. , Horak E. , Kintner C. , Alkuraya F.S. and Omran H. , Mutations in CCNO result in congenital mucociliary clearance disorder with reduced generation of multiple motile cilia, Nat Genet 46: ((2014) ), 646–651. |
[149] | Wallmeier J. , Shiratori H. , Dougherty G.W. , Edelbusch C. , Hjeij R. , Loges N.T. , Menchen T. , Olbrich H. , Pennekamp P. , Raidt J. , Werner C. , Minegishi K. , Shinohara K. , Asai Y. , Takaoka K. , Lee C. , Griese M. , Memari Y. , Durbin R. , Kolb-Kokocinski A. , Sauer S. , Wallingford J.B. , Hamada H. and Omran H. , TTC25 deficiency results in defects of the outer dynein arm docking machinery and primary ciliary dyskinesia with left-right body asymmetry randomization, Am J Hum Genet 99: ((2016) ), 460–469. |
[150] | Ware S.M. , Aygun M.G. and Hildebrandt F. , Spectrum of clinical diseases caused by disorders of primary cilia, Proc Am Thorac Soc 8: ((2011) ), 444–450. |
[151] | Watanabe D. , Saijoh Y. , Nonaka S. , Sasaki G. , Ikawa Y. , Yokoyama T. and Hamada H. , The left-right determinant Inversin is a component of node monocilia and other 9+0 cilia, Development 130: ((2003) ), 1725–1734. |
[152] | Wijers C.D. , Chmiel J.F. and Gaston B.M. , Bacterial infections in patients with primary ciliary dyskinesia: Comparison with cystic fibrosis, Chron Respir Dis 14: ((2017) ), 392–406. |
[153] | Wong C. , Jayaram L. , Karalus N. , Eaton T. , Tong C. , Hockey H. , Milne D. , Fergusson W. , Tuffery C. , Sexton P. , Storey L. and Ashton T. , Azithromycin for prevention of exacerbations in non-cystic fibrosis bronchiectasis (EMBRACE): A randomised, double-blind, placebo-controlled trial, Lancet 380: ((2012) ), 660–667. |
[154] | Yang P. , Diener D.R. , Yang C. , Kohno T. , Pazour G.J. , Dienes J.M. , Agrin N.S. , King S.M. , Sale W.S. , Kamiya R. , Rosenbaum J.L. and Witman G.B. , Radial spoke proteins of Chlamydomonas flagella, J Cell Sci 119: ((2006) ), 1165–1174. |
[155] | Zariwala M.A. , Gee H.Y. , Kurkowiak M. , Al-Mutairi D.A. , Leigh M.W. , Hurd T.W. , Hjeij R. , Dell S.D. , Chaki M. , Dougherty G.W. , Adan M. , Spear P.C. , Esteve-Rudd J. , Loges N.T. , Rosenfeld M. , Diaz K.A. , Olbrich H. , Wolf W.E. , Sheridan E. , Batten T.F. , Halbritter J. , Porath J.D. , Kohl S. , Lovric S. , Hwang D.Y. , Pittman J.E. , Burns K.A. , Ferkol T.W. , Sagel S.D. , Olivier K.N. , Morgan L.C. , Werner C. , Raidt J. , Pennekamp P. , Sun Z. , Zhou W. , Airik R. , Natarajan S. , Allen S.J. , Amirav I. , Wieczorek D. , Landwehr K. , Nielsen K. , Schwerk N. , Sertic J. , Kohler G. , Washburn J. , Levy S. , Fan S. , Koerner-Rettberg C. , Amselem S. , Williams D.S. , Mitchell B.J. , Drummond I.A. , Otto E.A. , Omran H. , Knowles M.R. and Hildebrandt F. , ZMYND10 is mutated in primary ciliary dyskinesia and interacts with LRRC6, Am J Hum Genet 93: ((2013) ), 336–345. |
[156] | Zariwala M.A. , Leigh M.W. , Ceppa F. , Kennedy M.P. , Noone P.G. , Carson J.L. , Hazucha M.J. , Lori A. , Horvath J. , Olbrich H. , Loges N.T. , Bridoux A.M. , Pennarun G. , Duriez B. , Escudier E. , Mitchison H.M. , Chodhari R. , Chung E.M. , Morgan L.C. , de Iongh R.U. , Rutland J. , Pradal U. , Omran H. , Amselem S. and Knowles M.R. , Mutations of DNAI1 in primary ciliary dyskinesia: Evidence of founder effect in a common mutation, Am J Respir Crit Care Med 174: ((2006) ), 858–866. |
[157] | Zariwala M.A. , Omran H. and Ferkol T.W. , The emerging genetics of primary ciliary dyskinesia, Proc Am Thorac Soc 8: ((2011) ), 430–433. |


