
Differential analysis of transcriptome of psychrophilic bacteria under different culture temperatures
Abstract
BACKGROUND:
Psychrophilic bacteria can survive in a unique living environment.
OBJECTIVE:
To explore the mechanism of low temperature adaptation and the physiological function of thermophilic metabolic genes.
METHOD:
Serratia marcescens strain F13 stored in microbial laboratory was cultured at 5∘C, 10∘C and 25∘C respectively, and the obtained strains were sequenced by high-throughput transcriptome. Serratia marcescens strain CAV1761 was used as the reference strain. The data produced by transcriptome sequencing were statistically analyzed by biostatistics software such as soapnuke, soap and edger. The differentially expressed genes were found based on the gene expression, and analyzed by Gene Ontology (GO) enrichment analysis and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analysis.
RESULTS:
The results showed that there were 718 differential genes in F13-10 vs F13-5 comparison group, 1614 differential genes in F13-25 vs F13-5 comparison group and 1636 differential genes in F13-25 vs F13-10 comparison group. GO function enrichment analysis showed that the GO term mainly enriched by different genes in the three comparison groups was mostly related to the migration and transport of cellular or subcellular components, cell localization and transmembrane transporter activity, as well as cilia or flagella dependent cell movement. In the enrichment analysis of KEGG pathway, the three comparison groups all enriched the largest number of differential genes in the branch pathway of KEGG metabolism, followed by the branch pathway of environmental information processing.
CONCLUSION:
In F13-10 vs F13-5, the differential genes were mainly concentrated in 20 pathways such as ATP-binding cassette transport (ABC) transporters, thiamine metabolism and flagella assembly; In F13-25 vs F13-5, the differential genes are mainly concentrated in 20 pathways, such as (ABC) transporters, arginine and proline metabolism, two-component system and so on; In F13-25 vs F13-10, the differential genes are mainly concentrated in 20 pathways such as various types of glycan synthesis, two-component system and arginine metabolism.
1.Introduction
Psychrophilic bacteria can survive in a unique living environment [1], carry out normal physiological activities, such as maintaining material transport in the body, energy transfer and replication of genetic material, maintaining their own various biological structures and nutrient requirements in order to achieve life activities [2]. This kind of psychrophilic microorganisms is abundant and is a potential microbial resource. In the past few years, a lot of attention has been paid to the psychrophilic microorganisms that survive and reproduce in large numbers in low temperature environments [3], mainly focusing on elucidating their psychrophilic mechanism and applying them to modern industrial production.
At present, it is believed that psychrophilic bacteria mainly adapt to the low temperature environment through the following mechanisms. Low temperature resistance mechanism: straight-chain and branched-chain unsaturated fatty acids in the lipid components of the membrane can reduce the melting point of lipids, maintain good fluidity, and ensure the nutritional needs of microorganisms by secreting a large amount of extracellular lipase and protease [4]; tRNA: The content of dihydrouracil in psychrophilic bacteria tRNA is high, which helps to maintain the local conformation of tRNA; Enzyme: The molecular structure of low-temperature enzymes has good flexibility, and can quickly adjust conformation for catalytic reactions; Acid-resistant mechanism, metal ions on the surface of the membrane will exchange with hydrogen ions in an acidic environment, so as to avoid excessive hydrogen ions from causing poisonous cells; Transmembrane potential difference: acidophilic microorganisms maintain a neutral environment in the cell through the balance mechanism and the diffusion of hydrogen ions; alkali resistance mechanism: the cell wall contains a large number of acidic small molecules, which can neutralize H
Using the second-generation high-throughput sequencing technology for cDNA sequencing, the transcripts of a specific organ or tissue of a species in a certain state can be obtained comprehensively and quickly. Transcriptome research is the basis and starting point for the study of gene function and structure, and plays an important role in interpreting the functional elements of the genome and revealing the molecular composition of cells and tissues. With the development of science and technology, such research methods have become more mature, convenient and widely used.
In order to investigate the low-temperature adaptation mechanism of psychrophilic bacteria and the physiological function of psychrophilic metabolic genes, in this paper, the transcriptome sequencing of Serratia marcescens strain F13 (Serratia marcescens strain F13) cultured at different temperatures (5∘C, 10∘C, 25∘C) was carried out. Serratia marcescens strain CAV1761 (Serratia marcescens strain CAV1761) was used as a reference strain, and biostatistical software was used to statistically analyze the transcriptome sequencing data, mainly to find differentially expressed genes, and to perform GO enrichment analysis on significantly differentially expressed genes and KEGG pathway enrichment analysis.
2.Materials and methods
2.1Source of strains
The cryophilic bacterium Serratia marcescens strain F13 and the reference strain Serratia marcescens strain CAV1761 were obtained from our experiment.
2.2Resurrection and expanded culture of strains
The F13 strain was inoculated on a glucose solid culture plate and cultured upside down at 10∘C for 3–4 days. After obvious colonies grew on the plate, it was inoculated into 100 mL of glucose liquid medium and cultured at 110 rpm constant temperature shaking at low temperature (5∘C and 10∘C) and normal temperature (25∘C). The growth curve was measured using spectrophotometer turbidimetry under the condition of A600nm. Inoculate the logarithmic phase bacterial solution of the F13 strain into the sterilized Luria-Bertani (LB) culture solution, pipette it with a pipette gun at a volume ratio of 1:100 in a sterile operating table, at the three temperatures of 5∘C, 10∘C and 25∘C, respectively,shake the culture at 110 rpm in a constant temperature incubator until the best harvest period, repeat 4 times, the cultures at 5∘C were named F13_1_5, F13_2_5, F13_3_5, F13_4_5, and the cultures at 10∘C were named F13_1_10, F13_2_10, F13_3_10, F13_4_10, cultured at 25∘C were named as F13_1_25, F13_2_25, F13_3_25, F13_4_25, respectively.
2.3Cell collection
Centrifuge the psychrophilic bacterial solution cultured at low temperature of 5∘C, 10∘C, and normal temperature of 25∘C at corresponding low temperature of 5∘C, 10∘C, normal temperature of 25∘C, and 200 rmp for 3 minutes, remove the supernatant, and repeat several times to obtain F13 three Bacteria at different temperatures were marked, quickly frozen in liquid nitrogen for 30 seconds, and then transferred to a
2.4Transcriptome analysis of F13 strain and reference strain
In the transcriptome analysis phase, the reference genome and transcriptome of the sugar-degrading bacterium Serratia marcescens strain F13, specifically the Serratia marcescens strain CAV1761, were utilized. The post-quality control sequence of each sample was then aligned with the reference genome and transcript sequence using SOAP2.2.1, resulting in a statistics table comparing Reads with the reference genome. From this alignment, the number of Reads on each transcript from every sample was determined, which in turn provided insights into transcript expression levels. Lastly, a principal component analysis (PCA) was performed on the gene expression values (FPKM) of the samples using Spss software.
(1) Psychrophilic Bacteria: Reference genome and transcriptome of the sugar-degrading bacterium Serratia marcescens strain F13: Serratia marcescens strain CAV1761. Using SOAP2.2.1, the second-generation sequence after quality control of each sample was compared with the reference genome and reference transcript sequence, and the comparison statistics table of Reads and the reference genome was obtained.
(2) Expression level analysis: using the results of alignment with the reference genome and transcriptome, the Serratia marcescens CAV1761(Serratia marcescens strain CAV1761), the number of Reads on each transcript was obtained from each sample, get transcript expression levels.
(3) Correlation analysis: The gene expression value (FPKM) of the samples was analyzed by principal component analysis (PCA) using Spss software to estimate the differences and repetitions between samples or within samples.
(4) Differential expression analysis: The R language package edgeR was used for differential analysis. The screening threshold was set at FDR (false discovery rate)
(5) GO enrichment analysis of differential genes: After the differential genes were identified, the expression differences between the samples were clarified in terms of gene functions using GO enrichment analysis.The GO enrichment method used was Hyper-geometric distribution. GO terms with an FDR
(6) KEGG enrichment analysis: KEGG (Kyoto Encyclopedia of Genes and Genomes) is the main public database on pathways. Pathway significant enrichment analysis takes KEGG Pathway as the unit, and applies hypergeometric test to find out the pathway with significant enrichment of differential genes relative to all annotated genes. The KEGG enrichment analysis method is Hyper-geometric distribution. Like the CO enrichment analysis method, pathway with FDR
2.5Statistical analysis
All statistical analyses were performed using R software (version 3.6.1). The significance of the differences between groups was determined using Student’s
3.Results and analysis
3.1Sequencing data quality assessment
Raw reads were filtered to obtain Clean reads with a length of 150 bp. The Q20 is greater than 96%, and the Q30 is greater than 88%, meeting the quality requirements for subsequent sequencing analysis. The content distribution of bases A, T, G, and C was generally at 25%, indicating good sequencing quality. The sequencing quality distribution showed that the actual coverage of Reads generated by the transcription library construction experiment and sequencing had high coverage depth and uniformity.
The closer to the 5’ end or 3’ end of the transcript, the lower the average sequencing depth. According to statistics on the number of sequences that have mismatches and the number of mismatches is less than or equal to 5 compared to the genome, 5∘C accounts for 41%, 10∘C accounts for 37%, and 25∘C accounts for 34%. The comparison of Reads and reference transcripts shows that the number of sequenced sequences that can be mapped to the genome accounts for 58% at 5∘C, 47% and 53% at 10∘C and 25∘C, respectively; According to statistics on the number of sequences that have mismatches and the number of mismatches is less than or equal to 5 compared to the genome, 5∘C accounts for 28%, 10∘C accounts for 29%, and 25∘C accounts for 23%.
3.2PCA analysis
In this study, we firstly do PCA (Principal Component Analysis) analysis was performed on the transcript differences of samples at 5∘C, 10∘C, and 25∘C to examine the overall patterns of gene expression and to identify the major sources of variation in the data. PCA analysis was performed on the transcript differences of samples at 5∘C, 10∘C, and 25∘C, and the analysis results are shown in Fig. 1. It can be seen from the PCA diagram that the samples in the three groups of low temperature 5∘C, 10∘C, and normal temperature 25∘C are gathered together. It can be seen that the parallelism of the samples in the group is good; The samples in the groups were scattered and far away. It can be seen that the expression patterns of F13 were significantly different at different temperatures (5∘C, 10∘C, 25∘C).
Figure 1.
PCA plot of all samples.
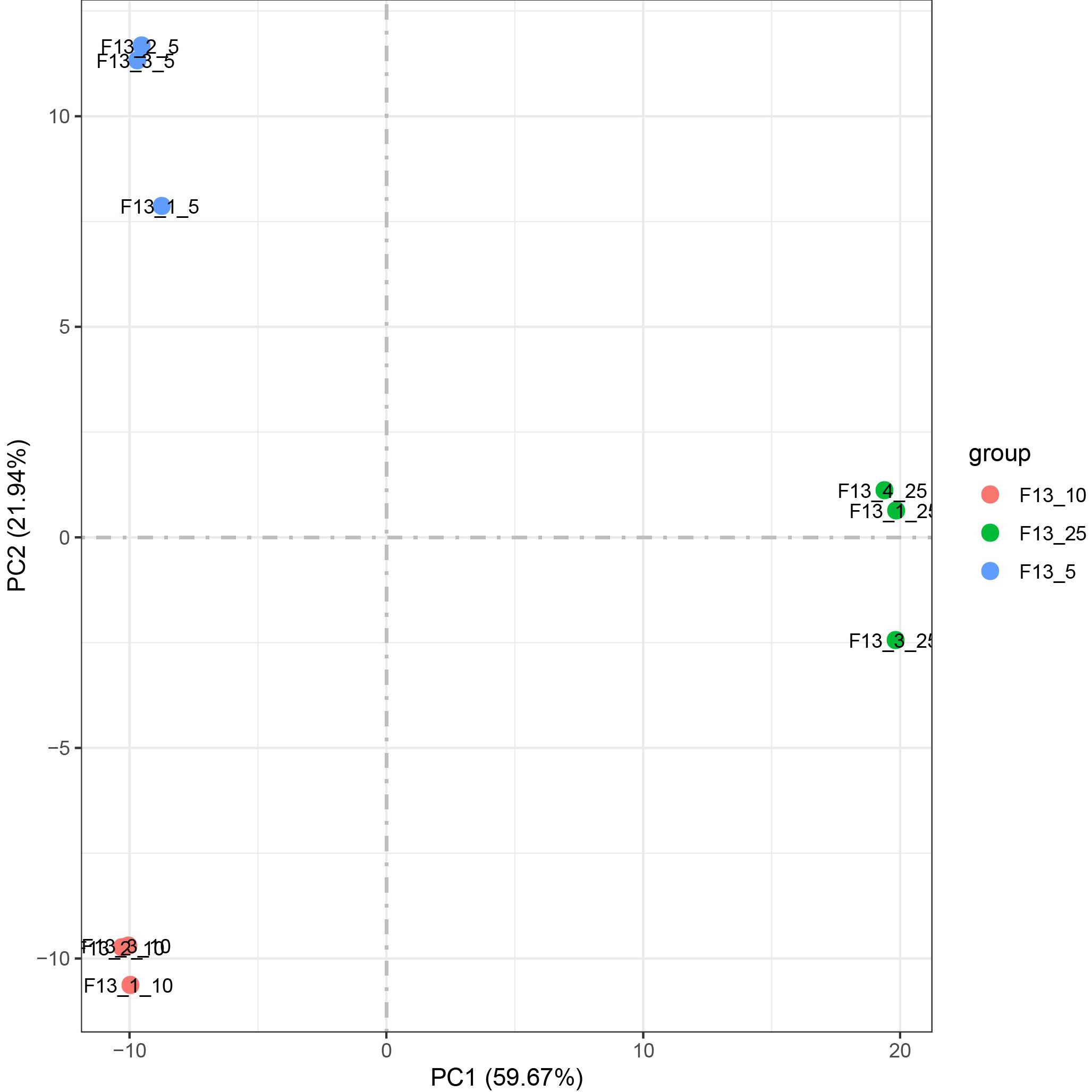
Figure 2.
Sample correlation heat map. The branches represent clustering branches, and the color from blue to red indicates that the correlation gradually increases.
3.3Differential expression analysis
3.3.1Differential gene screening
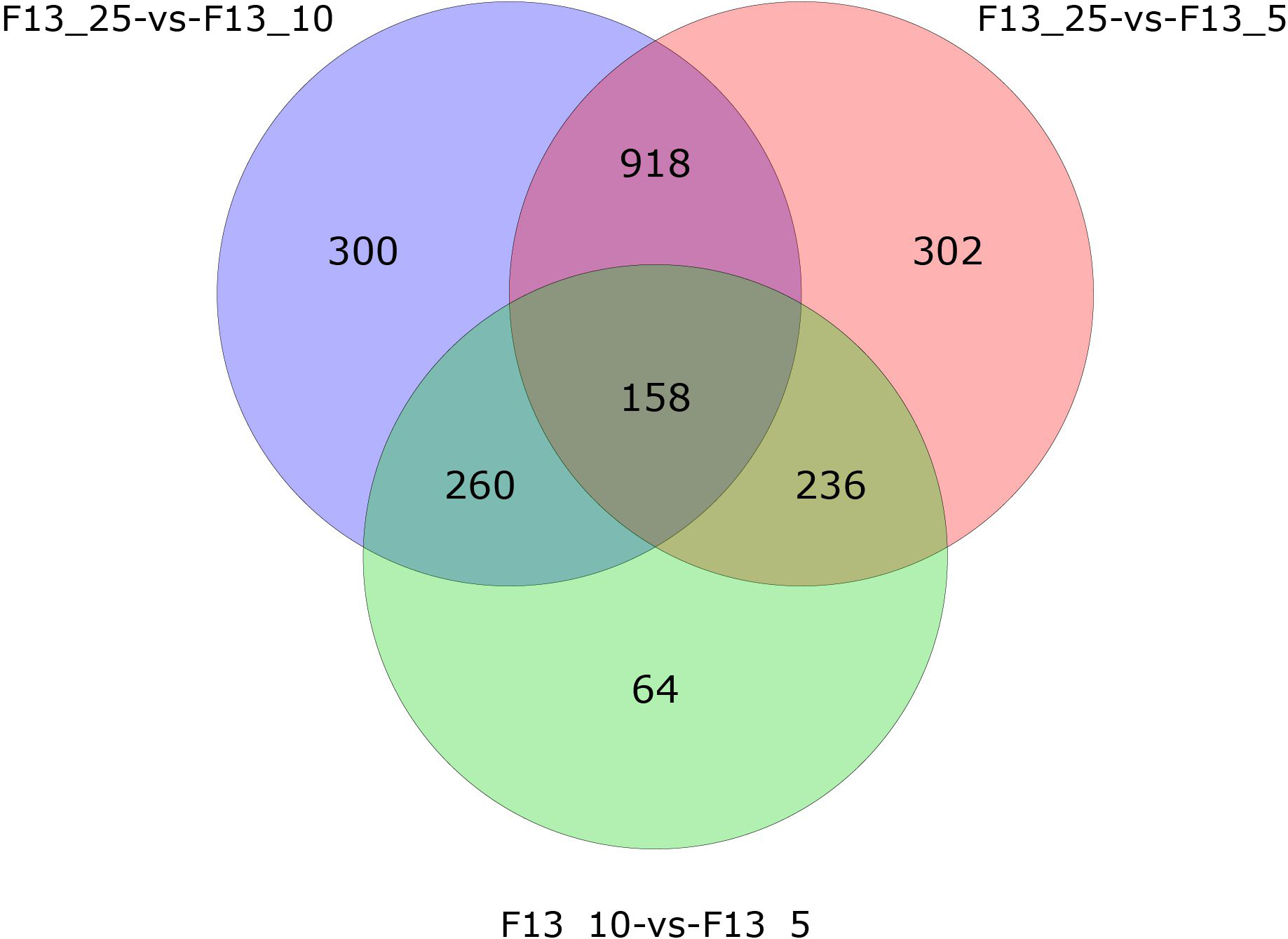
Transcriptome sequencing was performed on the F13 and CAV1761 strains, and the sequencing data were analyzed to identify differentially expressed genes. Under the culture conditions of 25∘C and 10∘C, there were 777 up-regulated genes and 859 down-regulated genes. Under the culture conditions of 25∘C and 10∘C, there were 797 up-regulated genes and 817 down-regulated genes. Compared with 10∘C and 5∘C culture conditions, there were 404 up-regulated genes and 314 down-regulated genes. A Venn diagram showed the number of differentially expressed genes between groups. The number of differentially expressed genes shared between the three comparison groups was 158.
Figure 2 shows a Venn diagram showing the number of differentially expressed genes between groups, the number of differentially expressed genes in unique transcripts and the number of differentially expressed genes in common transcripts between the three comparison groups of F13_25 vs F13_10, F13_25 vs F13_5 and F13_10 vs F13_5 can be visualized. The number of differentially expressed genes shared between F13_25 vs F13_10, F13_25 vs F13_5 and F13_10 vs F13_5 is 158, the number of unique differentially expressed genes in F13_25 vs F13_10 is 300, the number of unique differentially expressed genes in F13_25 vs F13_5 is 302, and the unique number of differentially expressed genes in F13_10 vs F13_5 is 64.
3.3.2Heat map analysis
From the differential gene expression clustering heat map, it can be seen that there are a large number of genes with significant differences in gene expression between F13_25 and F13_10, F13_25 and F13_5, and F13_10 and F13_5, and genes with similar expression patterns are clustered on the left into a subcluster.
The expression pattern clustering analysis of the three groups of samples at culture temperatures of 5∘C, 10∘C, and 25∘C was performed, and Fig. 3 was obtained. Blue represents low-expression genes, and red represents high-expression genes.
3.4GO and KEGG enrichment analysis of differential genes
In this study, GO and KEGG pathway enrichment analysis were performed to identify the biological functions and pathways associated with the differentially expressed genes in Serratia marcescens strain F13 under low temperature conditions. As shown in the GO classification histogram of differential genes in Fig. 4A, the GO database belongs to the Gene Ontology database, including gene molecular functions, cellular components, and biological processes. Serratia marcescens strain CAV1761 was used as the reference transcriptome.
The differential expressed genes are mainly focused on cellular process, metabolic process and location GO terms belong to biological processes, and focused on cellular anatomical entities GO term belongs to cellular components, and focused on catalytic activity, binding and transporter activity GO terms belong to gene molecular functions in the three comparison groups.
In organisms, different genes coordinate with each other to perform their biological functions, and the most important biochemical metabolic pathways and signal transduction pathways in which differentially expressed genes participate can be determined through significant enrichment of pathways. Taking Serratia marcescens strain CAV1761 as the reference transcriptome, and using the differential genes of F13 for KEGG enrichment analysis, the KEGG classification histogram of differentially expressed genes in Figure 4B was drawn. The differential genes of the three comparison groups of F13-10 vs F13-5,
Figure 3.
Venn diagram of the number distribution of differentially expressed genes between groups. With
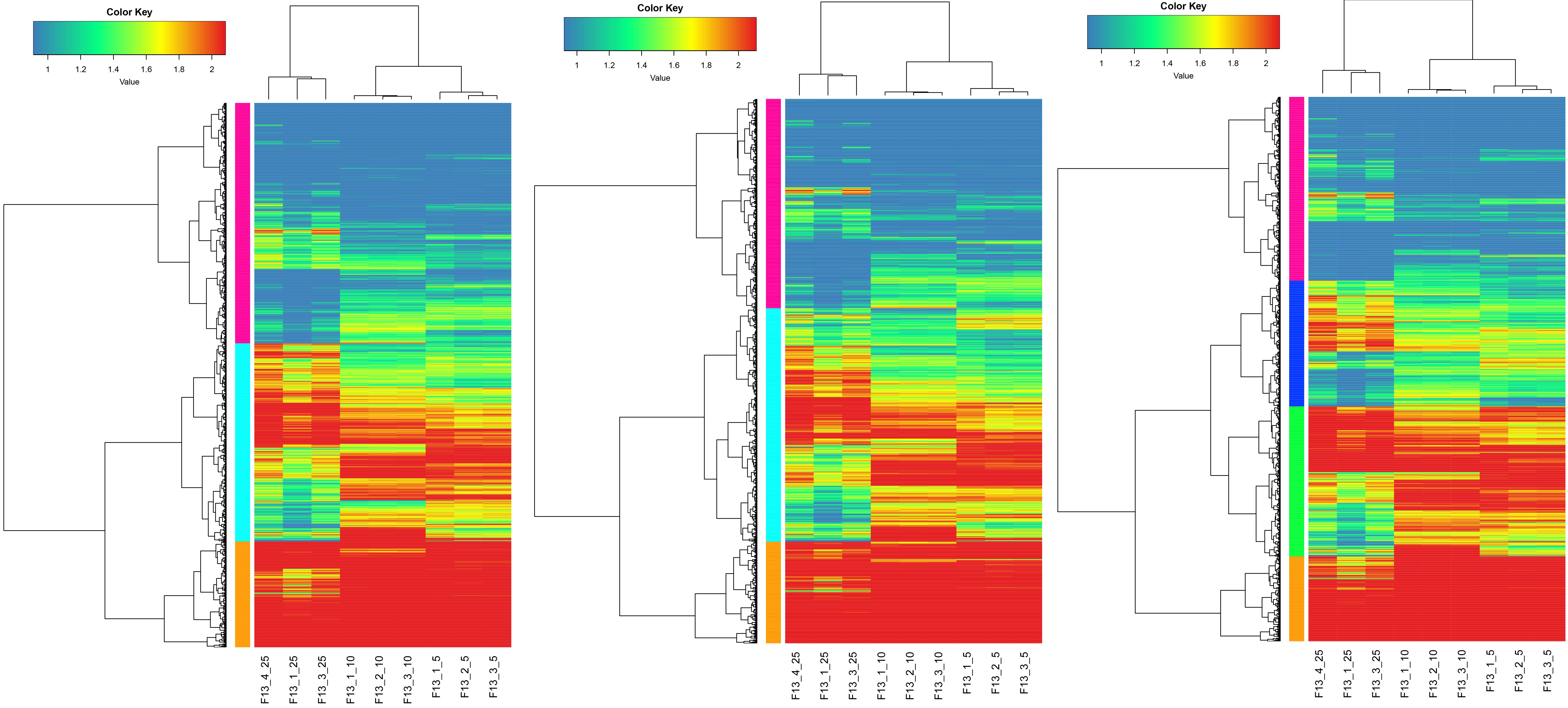
Figure 4.
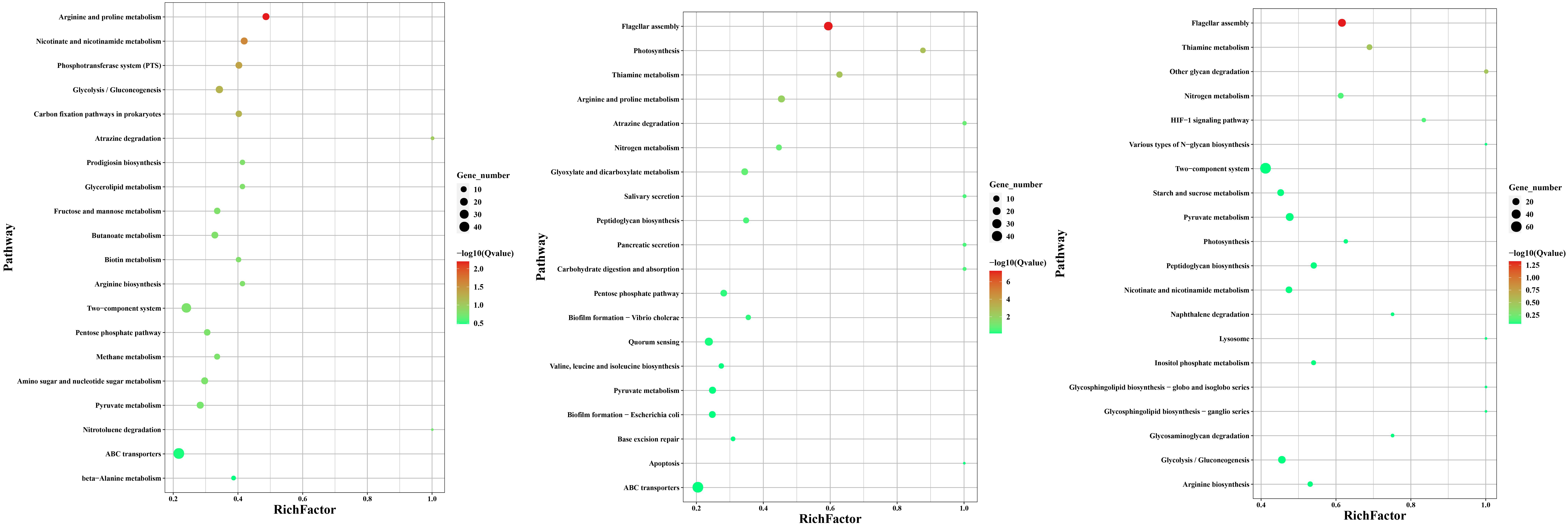
GO enrichment scatter plot of differentially expressed genes. A: The vertical axis represents the name of the GO term, and the name is sorted according to the Qvalue from small to large. The horizontal axis represents the enrichment factor. The larger the enrichment factor, the greater the degree of enrichment; the size of the point represents the number of differentially expressed genes in this GO term how much to count; The color of the point corresponds to different Qvalue values, ranging from 0 to 1, the closer to zero, the closer to the lighter color, the more significant the enrichment, and select the 20 GO terms with the most significant enrichment (if the enrichment If there are less than 20 terms, all will be displayed), and the GO enrichment scatter diagram of differentially expressed genes in Fig. 4 is drawn.
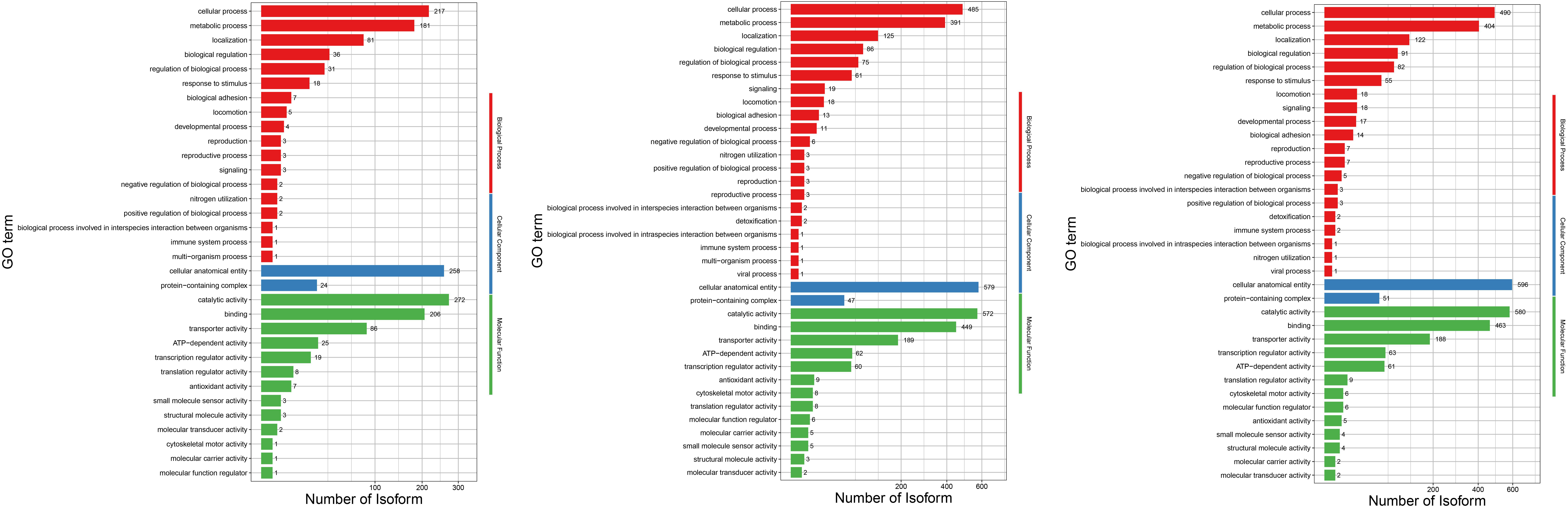
Figure 4.
Continued. B: The vertical axis is the name of the KEGG metabolic pathway, and the horizontal axis is the number of transcripts annotated to this pathway. The KEGG pathways involved in transcripts are divided into five branches: cellular processes, environmental information processing, genetic information processing, metabolism, and organic systems.
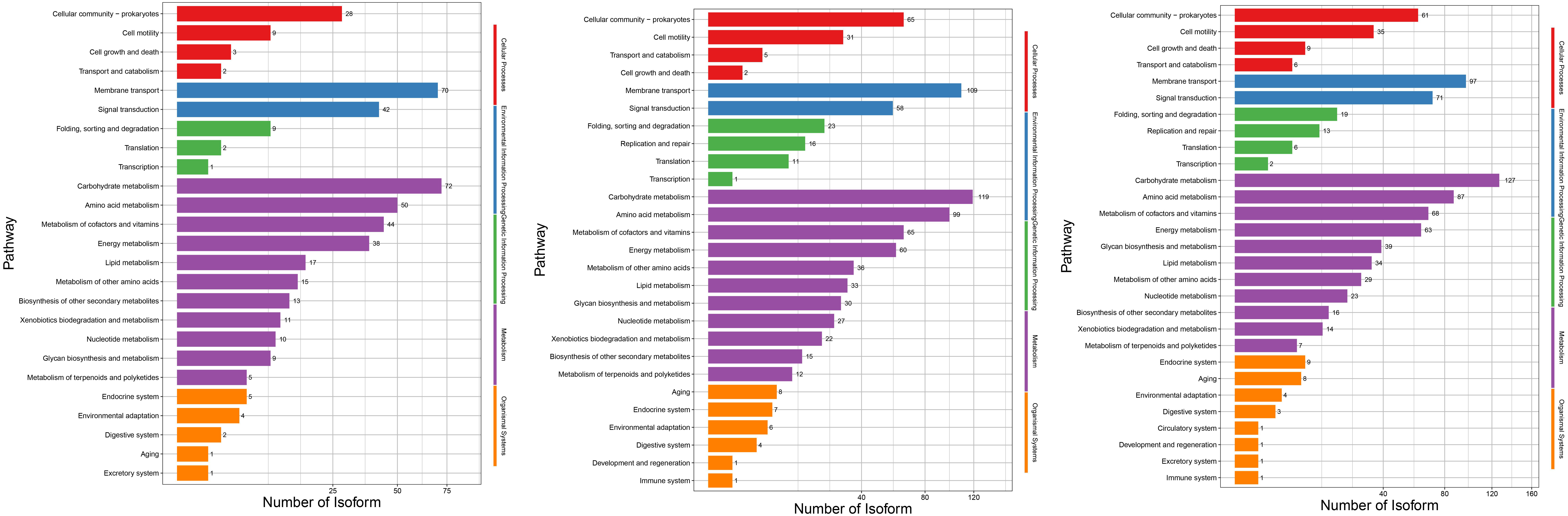
Figure 5.
KEGG enrichment scatter diagram of differentially expressed genes. Note: The vertical axis represents the name of each pathway, the order of Qvalue is from small to large, and the horizontal axis represents the enrichment factor. The larger the enrichment factor, the greater the degree of enrichment; The size of the points in the figure indicates the number of differentially expressed genes in this GO term; points with different colors have different Qvalue values, and the value range is between 0 and 1. The closer to 0, the more significant the enrichment.
F13-25 vs F13-5, and F13-25 vs F13-10 were annotated into 25, 27, and 29 KEGG secondary pathways, respectively. In the three comparison groups, the number of differential genes enriched in the KEGG metabolic (Metabolism) branch pathway was the largest, followed by the Environmental Information Processing branch pathway. Among the branched pathways of Metabolism, the secondary pathways with the largest number of differential genes are carbohydrate metabolism and amino acid metabolism; In the environmental information processing branch pathway, the secondary pathway with the largest number of differential genes is membrane transport and signal transduction.
As shown in the KEGG enrichment scatter diagram of differentially expressed genes in Fig. 5, 20 pathways with significant differential gene enrichment were selected. The enrichment degree of KEGG is represented by enrichment factor, Qvalue and the number of genes enriched in this metabolic pathway. In F13-10 vs F13-5, the differential genes were mainly concentrated in 20 pathways such as ABC transporters, Thiamine metabolism and flagellar assembly; In F13-25 vs F13-5, the differential genes were mainly concentrated in 20 pathways, such as ABC tranporters, arginine and proline metabolism, and two-component system; In F13-25 vs F13-10, the differential genes were mainly concentrated in 20 pathways including various types of glycan synthesis, two-component system, and arginine metabolism.
4.Discussion
Low temperatures affect cell integrity, solute dissolution rates, membrane fluidity, enzyme activity, and interactions between macromolecules [6, 7]. In order to survive, psychrophilic bacteria must have corresponding adaptive strategies to deal with the adverse effects of low temperature environment. In this study, we sequenced the transcriptome of Serratia marcescens strain F13, a Serratia marcescens strain of psychrotrophic bacteria, cultured at different temperatures of 5∘C, 10∘C and 25∘ C.
During the cold shock process [8], there were 1636 differentially expressed genes in the F13-25 vs F13-10 comparison group, and the culture temperature was lowered from 25∘C to 10∘C, the expression of 777 genes was up-regulated, and the expression of 859 genes was down-regulated quantity. The results of this study provide valuable insights into the molecular mechanisms underlying the cold adaptation of Serratia marcescens strain F13. The transcriptome analysis revealed a total of 1,042 differentially expressed genes under low temperature conditions, including 558 up-regulated genes and 484 down-regulated genes. This is consistent with the research results of Chen et al., Teoh et al., and Frank et al. [9, 10, 11], F13 also changes the expression of a large number of genes to make cells enter the low-temperature growth period in a short period of time.
The GO and KEGG pathway enrichment analysis identified several biological processes and pathways associated with the differentially expressed genes, including metabolic processes, biosynthesis of secondary metabolites, and microbial metabolism in diverse environments. Dolhi et al. [12] found that Chlamydomonas raudensis UWO241 compensated for the loss of enzyme activity by increasing the gene expression abundance and protein concentration of Rubisco, compensated for the decrease in enzyme catalytic activity at low temperature, and maintained cell growth. Liu et al. [13] studied the comparative genomics of 21 strains of psychrophilic psychrobacter Cryobacterium and found that the number of motility and chemotaxis genes in the genome of psychrophilic bacteria was significantly more than that of mesophilic low-temperature bacteria. The KEGG scatter plot shows that most of the differentially expressed genes in the F13-25 vs F13-10 comparison group are enriched in multiple glycan metabolism pathways and flagellar assembly pathways (Flagellar assembly Pathway). Therefore, it is speculated that when the culture temperature dropped from normal temperature 25∘C to low temperature 10∘C, F13 obtained energy by enhancing cell movement and adjusting the synthesis and decomposition of various glycans to adapt to the low temperature environment. When the culture temperature continued to drop from 10∘C to 5∘C, most of the differential genes in the F13-10 vs F13-5 comparison group were enriched in the ABC transporter (ABC tranporters) pathway, multiple signal transduction pathways and multiple biofilm formation pathways. The ABC transporter is a large number and diverse transport ATPase on the bacterial plasma membrane, which catalyzes the turnover of lipids into the phospholipid bilayer, and is of certain importance to the occurrence and function maintenance of the membrane.
The PCA analysis showed that the expression patterns of F13 were significantly different at different temperatures (5∘C, 10∘C, 25∘C), indicating that the cold adaptation of F13 involves complex regulatory mechanisms. The analysis of cold adaptation-related genes identified several genes involved in the biosynthesis of compatible solutes, cold shock proteins, and membrane transporters, which were significantly up-regulated under low temperature conditions. The analysis of transcription factors identified several differentially expressed TFs that may play important roles in regulating the cold adaptation-related genes.
From the above results, it can be speculated that: under the low-temperature culture environment, F13 cells increase the expression level of cilia or flagella-dependent cell motility-related genes to increase activity and obtain more nutrients; by enhancing transport-related enzymes and proteins, and the expression level of transmembrane transporter is used to improve the active transport efficiency of nutrient absorption for the metabolism of bacteria under low temperature conditions [14, 15], the decrease in protein and enzyme activity at low temperatures is compensated by increasing the relative abundance of key proteins and enzymes. Lauro et al., Santos et al., and De Maayer et al. [16, 17, 18] found that low temperature can cause the up-regulation of cell membrane synthesis-related genes (such as fatty acids, phospholipid fatty acids, peptidoglycan, and glycosyltransferases) and membrane transport proteins to deal with the adverse factors of lower cell membrane lysis rate caused by low temperature. The KEGG classification histogram results in this study showed that the three comparison groups of F13-10 vs F13-5, F13-25 vs F13-5 and F13-25 vs F13-10 enriched the number of differentially expressed genes in the branch pathway of environmental information processing Membrane transport pathway and signal transduction pathway were the most abundant, and the number of differential genes enriched in the two pathways, indicating that F13 needs to open a large number of transmembrane transport channels to adapt to the low temperature stress environment at 5∘C. For different degrees of low temperature stress, F13 adopted different strategies to adapt to the low temperature environment. The two-component system signaling system in bacteria is involved in many environmental stress responses, including temperature, pH, osmotic pressure, and oxidative stress [19]. In recent years, two-component systems involved in low temperature adaptation have been reported in multiple microbial cells. For example, the DesK/DesR two-component system in Bacillus subtilis, the CasKR two-component system in Bacillus cereus, the CB02306/CBO2307 two-component system in botulinum, and the CheA/CheY two-component system in Yersinia pseudotuberculosis system, the two-component system plays an important role in restoring membrane fluidity at low temperatures [20, 21, 22, 23]. Therefore, it can be speculated that under low temperature culture conditions, F13 can solve the problems of reduced membrane transport rate and metabolic rate caused by low temperature through strategies such as enhancing cell movement, strengthening signal transduction, increasing membrane area, and maintaining membrane fluidity.
5.Conclusion
The results of this study provide valuable insights into the molecular mechanisms underlying the cold adaptation of Serratia marcescens strain F13 and identify potential targets for improving the cold tolerance of bacteria. The findings of this study may have important implications for the development of new strategies for enhancing the cold tolerance of microorganisms, which could have significant applications in various fields, including biotechnology, food preservation, and environmental remediation. However, further experimental validation is needed to confirm the roles of the identified genes and pathways in cold adaptation, and to explore their potential applications in biotechnology and other fields.
Availability of data and materials
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
Funding
This work was supported by a grant from the Hulunbuir Science and Technology Plan Project of Inner Mongolia (Grant no. SF2022005).
Author contributions
Study concept: Chun-Guang Xu, Li-Xia Yang, Rui Zhu.
Study design: Chun-Guang Xu, Li-Xia Yang, Rui Zhu, Jie Zhang, Yan Li, Chun-Fang Chao.
Data acquisition: Li-Xia Yang, Rui Zhu, Jie Zhang, Yan Li.
Quality control of data and algorithms: Li-Xia Yang, Rui Zhu, Jie Zhang, Yan Li.
Statistical analysis: Rui Zhu, Jie Zhang, Yan Li, Chun-Fang Chao.
Manuscript preparation and editing: Chun-Guang Xu, Li-Xia Yang.
Obtain financing: Chun-Guang Xu.
All authors critically revised the manuscript for intellectual content and approved the final version to be published.
Conflict of interest
None of the authors have any personal, financial, commercial, or academic conflicts of interest to report.
References
[1] | Cavicchioli R. On the concept of a psychrophile. ISME J. (2016) ; 10: (4): 793-795. doi: 10.1038/ismej.2015.160. |
[2] | Garcia-Lopez E, Alcazar P, Cid C. Identification of biomolecules involved in the adaptation to the environment of cold-loving microorganisms and metabolic pathways for their production. Biomolecules. (2021) ; 11: (8): 1155. Published 2021 Aug 4. doi: 10.3390/biom11081155. |
[3] | Aksakal R, Mertens C, Soete M, Badi N, Du Prez F. Applications of discrete synthetic macromolecules in life and materials science: Recent and future trends. Adv Sci (Weinh). (2021) ; 8: (6): 2004038. Published 2021 Jan 25. doi: 10.1002/advs.202004038. |
[4] | Roldán DM, Carrizo D, Sánchez-García L, Menes RJ. Diversity and effect of increasing temperature on the activity of methanotrophs in sediments of fildes peninsula freshwater lakes, king george island, antarctica. Front Microbiol. (2022) ; 13: : 822552. Published 2022 Mar 17. doi: 10.3389/fmicb.2022.822552. |
[5] | Liu X, Sun W, Jian S, Chao J. Passive protective ability of the outer membrane protein PF1380 of Pseudomonas fluorescens against the major pathogenic bacteria of freshwater aquaculture in fish. Aquaculture Reports. (2022) ; 22: : 100985. doi: 10.1016/j.aqrep.2021.100985. |
[6] | Singh V. Microbial genes responsible for cold adaptation. Survival Strategies in Cold-adapted Microorganisms. (2022) ; 153-171. doi: 10.1007/978-981-16-2625-8_7. |
[7] | Piette F, D’Amico S, Mazzucchelli G, Danchin A, Leprince P, Feller G. Life in the cold: A proteomic study of cold-repressed proteins in the antarctic bacterium pseudoalteromonas haloplanktis TAC125. Appl Environ Microbiol. (2011) ; 77: (11): 3881-3883. doi: 10.1128/AEM.02757-10. |
[8] | Tomlinson BR, Denham GA, Torres NJ, et al. Assessing the Role of Cold-Shock Protein C: A Novel Regulator of Acinetobacter baumannii Biofilm Formation and Virulence. Infect Immun. (2022) ; 90: (10): e0037622. doi: 10.1128/iai.00376-22. |
[9] | Chen Z, Yu H, Li L, Hu S, Dong X. The genome and transcriptome of a newly described psychrophilic archaeon, Methanolobus psychrophilus R15, reveal its cold adaptive characteristics. Environ Microbiol Rep. (2012) ; 4: (6): 633-641. doi: 10.1111/j.1758-2229.2012.00389.x. |
[10] | Teoh CP, Lavin P, Yusof NA, Gonzalez M. Transcriptomics analysis provides insights into the heat adaptation strategies of an Antarctic bacterium, Cryobacterium sp. SO1. Polar Biology. (2023) ; 46: (3): 185-197. doi: 10.1007/s00300-023-03115-x. |
[11] | Frank S, Schmidt F, Klockgether J, et al. Functional genomics of the initial phase of cold adaptation of Pseudomonas putida KT2440. FEMS Microbiol Lett. (2011) ; 318: (1): 47-54. doi: 10.1111/j.1574-6968.2011.02237.x. |
[12] | Dolhi JM, Maxwell DP, Morgan-Kiss RM. The Antarctic Chlamydomonas raudensis: An emerging model for cold adaptation of photosynthesis. Extremophiles. (2013) ; 17: (5): 711-722. doi: 10.1007/s00792-013-0571-3. |
[13] | Liu Y, Shen L, Zeng Y, Xing T, Xu B, Wang N. Genomic insights of cryobacterium isolated from ice core reveal genome dynamics for adaptation in glacier. Front Microbiol. (2020) ; 11: : 1530. Published 2020 Jul 14. doi: 10.3389/fmicb.2020.01530. |
[14] | Siddiqui KS, Williams TJ, Wilkins D, et al. Psychrophiles. Annual Review of Earth and Planetary Sciences. (2013) ; 41: : 87-115. |
[15] | Agrawal R, Goyal VD, Kumar A, et al. Two-domain aminopeptidase of M1 family: Structural features for substrate binding and gating in absence of C-terminal domain. J Struct Biol. (2019) ; 208: (1): 51-60. doi: 10.1016/j.jsb.2019.07.010. |
[16] | Lauro FM, Tran K, Vezzi A, Vitulo N, Valle G, Bartlett DH. Large-scale transposon mutagenesis of Photobacterium profundum SS9 reveals new genetic loci important for growth at low temperature and high pressure. J Bacteriol. (2008) ; 190: (5): 1699-1709. doi: 10.1128/JB.01176-07. |
[17] | Santos T, Viala D, Chambon C, Esbelin J, Hébraud M. Listeria monocytogenes Biofilm Adaptation to Different Temperatures Seen Through Shotgun Proteomics. Front Nutr. (2019) ; 6: : 89. Published 2019 Jun 14. doi: 10.3389/fnut.2019.00089. |
[18] | De Maayer P, Anderson D, Cary C, Cowan DA. Some like it cold: Understanding the survival strategies of psychrophiles. EMBO Rep. (2014) ; 15: (5): 508-517. doi: 10.1002/embr.201338170. |
[19] | Monteagudo-Cascales E, Santero E, Canosa I. The Regulatory Hierarchy Following Signal Integration by the CbrAB Two-Component System: Diversity of Responses and Functions. Genes (Basel). (2022) ; 13: (2): 375. Published 2022 Feb 18. doi: 10.3390/genes13020375. |
[20] | Fernández P, Porrini L, Albanesi D, et al. Transmembrane Prolines Mediate Signal Sensing and Decoding in Bacillus subtilis DesK Histidine Kinase. mBio. (2019) ; 10: (6): e02564-19. Published 2019 Nov 26. doi: 10.1128/mBio.02564-19. |
[21] | Diomandé SE, Chamot S, Antolinos V, et al. The CasKR two-component system is required for the growth of mesophilic and psychrotolerant Bacillus cereus strains at low temperatures. Appl Environ Microbiol. (2014) ; 80: (8): 2493-2503. doi: 10.1128/AEM.00090-14. |
[22] | Derman Y, Isokallio M, Lindström M, Korkeala H. The two-component system CBO2306/CBO2307 is important for cold adaptation of Clostridium botulinum ATCC 3502. Int J Food Microbiol. (2013) ; 167: (1): 87-91. doi: 10.1016/j.ijfoodmicro.2013.06.004. |
[23] | Palonen E, Lindström M, Karttunen R, Somervuo P, Korkeala H. Expression of signal transduction system encoding genes of Yersinia pseudotuberculosis IP32953 at 28C and 3C. PLoS One. (2011) ; 6: (9): e25063. doi: 10.1371/journal.pone.0025063. |


