
Interpretable ontology extension in chemistry
Abstract
Reference ontologies provide a shared vocabulary and knowledge resource for their domain. Manual construction and annotation enables them to maintain high quality, allowing them to be widely accepted across their community. However, the manual ontology development process does not scale for large domains. We present a new methodology for automatic ontology extension for domains in which the ontology classes have associated graph-structured annotations, and apply it to the ChEBI ontology, a prominent reference ontology for life sciences chemistry. We train Transformer-based deep learning models on the leaf node structures from the ChEBI ontology and the classes to which they belong. The models are then able to automatically classify previously unseen chemical structures, resulting in automated ontology extension. The proposed models achieved an overall F1 scores of 0.80 and above, improvements of at least 6 percentage points over our previous results on the same dataset. In addition, the models are interpretable: we illustrate that visualizing the model’s attention weights can help to explain the results by providing insight into how the model made its decisions. We also analyse the performance for molecules that have not been part of the ontology and evaluate the logical correctness of the resulting extension.
1.Introduction
Ontologies11 represent knowledge in a way that is both accessible to humans and is machine interpretable. Reference ontologies provide a shared vocabulary for a community, and are successfully being used in a range of different domains. Examples include the ontologies belonging to the Open Biomedical Ontologies (OBO) collection in the life sciences [48], the Financial Industry Business Ontology for the financial domain [1], and the Open Energy Ontology in the energy systems domain [8]. While these ontologies differ in many respects, they share one important feature: they are manually created by experts using a process in which each term is manually added to the ontology together with a textual definition, relevant axioms, and ideally some additional documentation. Often, this process involves extensive discussions about individual terms. Hence, developing such ontologies is a time-intensive and expensive process. This leads to a scalability challenge for ontologies that cover a large domain.
For example, the ChEBI (Chemical Entities of Biological Interest) ontology [22] is the largest and most widely used ontology for the domain of biologically relevant chemistry in the public domain. It currently (as of July 2022) contains 60,094 fully curated classes, which makes it large in comparison to other reference ontologies. ChEBI is for the most part manually maintained by a team of expert curators. This is an essential prerequisite for its success, because it allows it to capture the terminology and classification logic in use by chemistry experts. However, the number of chemicals covered by ChEBI is dwarfed by the 110 million chemicals in the PubChem database [51], which itself is not comprehensive. The manually curated portion of ChEBI only grows at a rate of approximately 100 entries per month, thus will only ever be able to cover a small fraction of the chemicals that are in its domain. Therefore, the ChEBI team is in a kind of dilemma. The manual curation process is a prerequisite to ChEBI’s success and, thus, needs to be maintained. However, the same process does not scale and, thus, limits the potential use of ChEBI. The ChEBI team tries to navigate this dilemma by extending the manually curated core part of the ontology automatically using the ClassyFire tool [13]. This approach has close to tripled ChEBI’s coverage to 160,000 classes (as of July 2022), by loading additional pre-curated content automatically. However, there are limitations to this approach. Firstly, ClassyFire uses a different underlying classification approach to ChEBI (e.g. conjugate bases and acids are not distinguished), thus, mapping from ClassyFire to ChEBI loses classification precision. More importantly, ClassyFire is rule-based and while the extension of the ontology is automated by the tool, the creation and curation of the rule set on which the tool is based is not. This limits the scalability of this approach.
Somewhat inspired by ChEBI’s workflow, we propose addressing the dilemma of choosing between manual curation and scalability by using a new kind of approach to ontology extension, which respects the design decisions of the developers of an ontology, but is not only automatically applicable, but also automatically maintainable. Our starting point is an existing, manually curated reference ontology. We suggest the use of machine learning methods to learn some of the criteria that the ontology developers adopted in the development of the ontology, and then use the learned model to extend the ontology to entities that have not yet been covered by the manual ontology development process. We will illustrate this approach in this paper for the chemistry use case by training an artificial neural network (with a Transformer-based architecture) to automate the extension of ChEBI with new classes of chemical entities. The approach has several benefits: since it builds on top of the existing ontology, the extension will preserve the manually created consensus. Moreover, the model is trained solely on the content of the ontology itself and does not rely on any external sources. Finally, as we will see, the chosen architecture allows stakeholders to interpret the predictions made by the neural network, and, thus to validate the trained model to some degree by manual inspection.
In the next two sections, we discuss related work and the overall methodology that we are using to train models for classifying new classes of chemical entity as subclasses of existing classes in ChEBI. This is followed by a presentation of the results and their evaluation, where we can show that in our latest iteration we have outperformed previous results while significantly reducing time for training. We also apply the model to a large collection of never-before-seen molecules as a case study for the real-life application of our method, and discuss our findings. Further, we address how visualising the attention within a model enables an interpretation of the results. Finally, we discuss how our system measures up against the goals for ontology extension that were formulated by [39].
We here extend the results of our previous workshop publication [36] in several ways: first, we use a new transformer model that is faster than the one used in [36] while maintaining the same level of accuracy. Secondly, we provide a more in-depth discussion of interpretability, based on the attention weights of the model. Thirdly, we evaluate the real-world performance of the model on a large dataset of chemicals from a different source to that used to develop the training, test and validation datasets.
2.Related work
In this paper, we present a methodology for ontology extension, which can be considered as a kind of ontology learning. Ontology learning has been an active area of research for more than two decades [3,4,6,32,39] and a number of automated ontology generators have been developed. A recent publication [39] defined a list of six desirable goals for ontology learning methods: they should support expressive languages, require a small amount of time and training data, require limited or no human intervention, support unsupervised learning, handle inconsistencies and noise, and their results should be interpretable. We will reflect upon these goals in the conclusion section. Note that in this paper, we use the terms explainability and interpretability interchangeably. Sometimes explainability is considered to be a stronger form of interpretability [18].
Different approaches to ontology learning and ontology extension have been considered in the literature. We will discuss them in the next three subsections.
2.1.Text-based approaches
Text-based ontology extension systems are largely based on the lexical features of ontology classes (e.g. names and synonyms), using sophisticated tools to match those to an available corpus of text. Artificial intelligence is used to analyse corpora of relevant literature in order to extract important terms and their relations. For example, in [2] a learning system is proposed that is able to identify entity mentions in a corpus of text (e.g. PubMed publications), recognise those that could be added to a given ontology, and suggest parents for them. Other approaches use the ontology as a seed to identify terms that are important for the target domain [2,5,29,30,40,45,54].22 These are then used to guide the same approaches that are applied in ontology learning ‘from scratch’. The resulting extensions may potentially introduce biases and noise from the literature into the ontology. This even holds for approaches like [40] that use definitions of terms from Wikipedia and web articles — these definitions are not necessarily well-aligned with the ontology. These approaches reflect rather than resolve the inherent ambiguities and differences in language use that exist within different communities of domain experts, or even within single communities. The resolution of these ambiguities is an essential part of the ontology development process that involves extensive in-depth communication with and between domain experts [8]. As a result, many approaches involve several manual steps, in which experts evaluate concepts and related phrases to manually sort out inconsistencies [27]. The involvement of human experts has the advantage of providing quality control, but is labour intensive and, thus, costly. To sum up, text-based approaches to ontology extension either risk reducing the quality of manually curated ontologies, or they are still time and labour intensive.
Instead of relying on text corpora, therefore, our goal is to rely only on the content of the ontology that is being extended, in particular on the structured annotations it contains. The research question becomes: Given the design decisions by the ontology developers, how would they extend their ontology to cover a novel entity? That is, based on the internal structure of an ontology and its structural annotations, how can we predict, for a given new class, subsumption and other axioms that connect the new class to the ontology? Techniques answering these questions should be based on complex features of the existing ontology classifications and associated data. Our goal is to use a manually created ontology as the core of an automatically generated extended ontology that is consistent with the manually maintained ontology and improves in its quality when the manually created ontology grows. This leads to structure-based classification of novel entities.
2.2.Structure-based classification
Chemical ontologies provide stable identifiers and a shared vocabulary for biochemical ‘small molecules’ and their functions. The ChEBI reference ontology, mentioned above, has been widely adopted and can be considered the “gold standard” chemical ontology in the public domain. It is used for informatics applications such as bioinformatics and systems biology analyses of metabolism, biological data integration, natural language processing, and as a chemistry component for semantic web applications (e.g. [17,23,24,38]).
Chemical entities are classified based on features of their chemical structures. The chemical graph formalism encodes the chemical structures of small molecules as graphs of atoms connected via bonds, which may form rings or cycles. Chemical classes can be defined based on overall patterns or sub-graphs within these structures, whose sub-graphs may themselves contain structural sub-units. Although chemical classification is typically based on structural features, full computable structural definitions for classes –which would support the use case of ontology extension through automated reasoning– are seldom formally captured in chemical ontologies, as the underlying logical formalisms are not able to encompass the full range of relevant structural features [21,26,33]. Thus, chemical ontologies typically only reflect a partial axiomatization for the chemical domain. Various extensions to the OWL language have been explored in order to allow a fuller set of structural features to be exposed to the computable classification hierarchy, including description graphs (our work [19]), logic programs [34], and nonmonotonic existential rules [33]. Nevertheless, these formalisms have not seen widespread adoption, in part as they are not supported by the wide range of tools that are available for OWL, and in part because performance remains a challenge. Thus, they have not been able to scale to the use case of extending real-world ontologies such as ChEBI. Moreover, chemists themselves often only have implicit knowledge (e.g. about which functional groups or structural patterns are most relevant for a classification) that is hard to make explicit.
On the other hand, a rich algorithmic toolkit for structural definitions of chemical classes has arisen in the cheminformatics domain, based around generalisations of the chemical graph formalism for structures. Consequently, several attempts have been made to combine weakly axiomatised chemical ontologies with fast cheminformatics (algorithmic) chemical graph operations in hybrid systems that are able to expose some of the chemical knowledge in ontology axioms, but ultimately perform performance-intensive operations such as graph substructure or superstructure matching algorithmically, that is, outside of the ontology. One of the first such attempts was “CO” [15], an ontology of 260 classes based on combinations of chemical functional groups generated with the cheminformatics “checkmol” software. Later, we have developed this into the more complete approach reported in [9] which used a custom general molecular fragmentation algorithm to enumerate all the parts of each molecular entity and assert those as axioms in a resulting OWL ontology. However, this strategy quickly creates a combinatorial explosion of content, which becomes inefficient as the size of the knowledge base grows.
Fig. 1.
The class of naphthionic acids as represented in ChEBI. The bottom of the picture shows the structural annotation with SMILES. These annotations will be used by our system to predict subsumption relations.
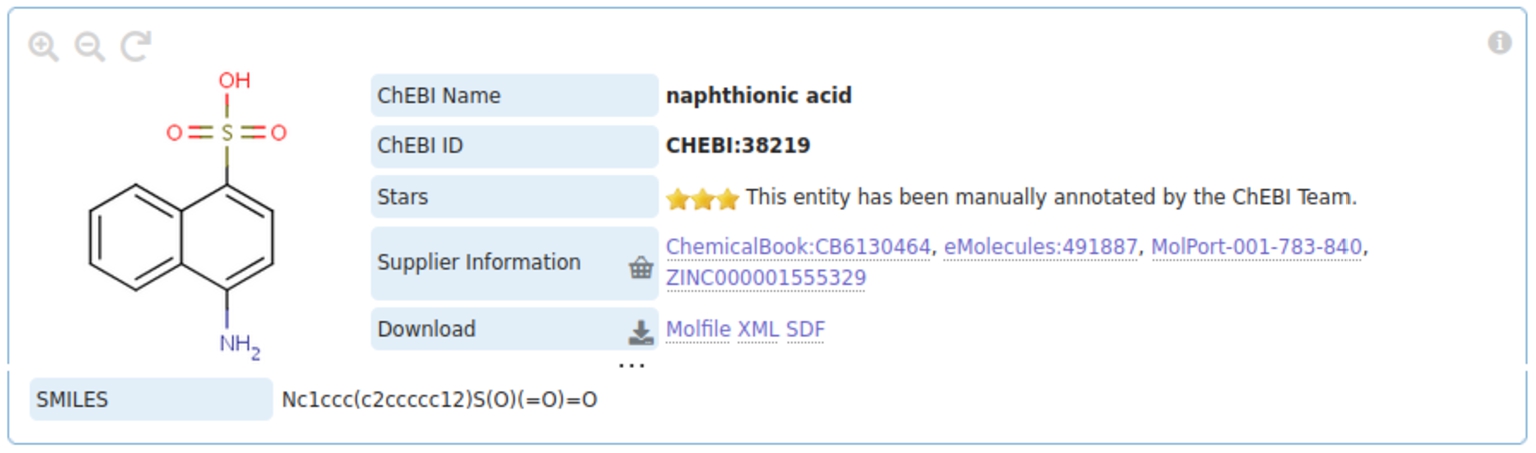
The SMILES language is a chemical line notation widely used for compact chemical structure representation in cheminformatics, in which characters representing atoms are laid out in a sequence in which adjacency typically represents a chemical bond [52]. Thus, the SMILES string ‘CCC’ represents three Carbon atoms bonded together, such as in the small hydrocarbon molecule propane. This representation of chemicals has been used as a foundation for numerous chemistry-based ML approaches and other chemistry based systems. The ChEBI ontology uses SMILES strings as structural annotations for classes of molecules as shown in Fig. 1.
The SMILES language has an extension, SMARTS, which allows ambiguities and patterns in, and logical combinations of, chemical structural elements, to be represented in a way that allows for the definition of classes of chemicals. This language has been used to define chemical classes in chemical ontologies such as OntoChem’s SODIAC [7] and more recently the ClassyFire application [13]. At the time of writing, ClassyFire is the state of the art tool for structure-based chemical ontology classification, in terms of size (9,000 definition rules, and an associated ontology of 4,825 classes) and adoption, and is used in the automated extension of the ChEBI ontology. However, ClassyFire does not harness logic-based automated reasoning: even though it uses an ontology to structure the classes that it supports, the SMARTS chemical class definitions are not integrated with the definitions of ontology classes, nor are the rules for selecting the most appropriate parent when several structural matches are obtained integrated into the ontology. Thus, the tool operates as an algorithmic “black box”, in that the rules it uses are not semantically accessible, the associated chemical ontology still has to be maintained manually, and updating the integrated knowledge system can only be accomplished by updating the custom software suite. Learning approaches promise to overcome these problems.
2.3.Machine learning and deep learning approaches
Deep learning has been used in chemistry in various ways, e.g. for protein structure prediction, drug design, property prediction and even synthesis planning [12,35]. The DeepChem library [42] is an open-source machine learning framework that has gained widespread adoption in the life science community as it offers a variety of tools and algorithms to facilitate chemical property analysis. MoleculeNet [53], which serves as a benchmark and curated dataset collection for molecular machine learning, is also released as a part of the DeepChem library.
Deep learning has also been used for classification of molecules in chemical ontologies (which in turn can be used for ontology extension). In [25], a back-propagating artificial neural network is applied to classify natural products, that is, secondary metabolites largely of plant origin. Named NPClassifier, it is trained on a dataset of around 73,000 natural products sourced from public databases including Pubchem, ChEBI, and the Universal Natural Products Database. The hierarchy into which these molecules were organised consisted of three hierarchical levels: 7 Pathways, 70 Superclasses, and 653 Classes. Rather than training a single model for the full prediction task, they used three single-task models – one model for each of the classification hierarchical levels. They report promising performance in a direct comparison to ClassyFire for a selection of classes. However, the restriction to only natural products (a subset of organic molecules) and to only three hierarchical levels addresses an artificially simpler task than the general problem of classification in chemical ontologies, where classes can be arranged in a hierarchy of arbitrary depth and reflect a wider chemical diversity.
In [14], machine learning was used to predict class membership directly from mass spectrometry features in an untargeted metabolomics study. This is an important use case, as in untargeted metabolomics there are often many features which relate to ‘unknown’ molecular entities and thus are not mapped to defined molecular entities about which metabolic information is known, however, they may nevertheless share detectable chemical classes. In this effort, the chemical fingerprint was used as an intermediary structural representation for learning purposes: support vector machines were used to predict chemical fingerprints from mass spectrometry features, and a deep neural network was then used to predict class membership from the resulting fingerprints. However, their system predicts class membership only for a subset of the classes belonging to the chemical ontology underlying ClassyFire, and moreover does not attempt to extend the ontology itself.
Our recent work explored the applicability of machine learning and deep learning to chemical ontology extension based on chemical structures [20]. In order to allow machine learning from the existing ontology, which is inherently unbalanced, we introduced an ontology sub-sampling approach which selected a specified number of members in a balanced way for a subset of ontology classes. We then used the associated chemical structures as inputs to train a multi-label classifier able to predict ontology class membership based on chemical structural features. In [20], we compared several different families of “classical” classifiers including logistic regression, random forests, and support vector machines, as well as a deep neural network in the form of a long short-term memory network (LSTM). We also compared chemical fingerprint-based inputs and inputs based on embedding the SMILES representation of chemical structure. In this study, we found that the LSTM was the best-performing approach, clearly outperforming also ClassyFire. Moreover, no single approach gave the best results under all conditions. The performance of the LSTM was satisfactory as a whole, but several specific limitations were identified. In particular, the model failed to provide any prediction for a certain subset of input molecules. Furthermore, LSTMs embed their inputs into internal representations that are not interpretable.
The current contribution harnesses two Transformer-based architectures and describes how the attention weights of the resulting model can provide insights into how the model made its decisions. Furthermore, by using transfer learning, a broader applicability of this data- and compute-hungry method becomes computationally more feasible. One of our Transformer models is based on the ChemBERTa [10] architecture. This chemistry-focused Transformer-based architecture has been successfully employed for toxicity prediction. In the context of this work, it has been trained for the first time on the structural annotations of an existing chemical ontology. The learning method biases the classifier towards the ontology’s internal structure, yielding a model that is in line with the domain experts’ conceptualisation as represented in the existing ontology. The resulting model is then used to integrate previously unseen classes into the ontology.
3.Methodology
Our goal is to train a system that automatically extends the ChEBI ontology with new classes of chemical entities (such as molecules) based on the design decisions that are implicitly reflected in the structure of ChEBI and the structural annotations associated with its classes. Thus, for our work we take the ‘upper level’ of the ontology, which contains generic distinctions (such as that between atom, molecular entity, and group, or that between metal atom and nonmetal atom), as given. Moreover, ChEBI is relatively weakly axiomatised, consisting largely of a taxonomy supplemented by existentially restricted relationships. All chemical entities in ChEBI are represented as classes and there are no individuals in the ontology. Thus, the ontology extension task consists of adding classes and subsumption relationships.
In the following section, we will introduce the two models that we are going to use in this paper (ELECTRA and RoBERTa), the three datasets (
Our focus is the extension of the ChEBI ontology with classes of chemical entities that may be characterised by a SMILES string, i.e., they are associated with a specific chemical structure. Chemical structures can typically only be fully specified for well-defined classes of chemical entity, thus, although these classes are not necessarily leaf nodes in the ontological hierarchy, they nevertheless tend to be in the ‘lower’ (more specific) part of the hierarchy.
The learning task for chemical ontology extension may be characterised as follows: Given a class of chemical entities (characterised by a SMILES string), what are its optimal superclasses in ChEBI?
While our goal is – from an ontological point of view – to extend the ChEBI ontology with new classes (i.e., adding new subsumptions), from a machine learning perspective we turned this problem into a multi-label classification task, for which we prepare an appropriate learning dataset from the ontology.
Hierarchical chemical classifications should group chemical compounds in a scientifically valid and meaningful way [7,21]. Each chemical entity has many structural features which contribute to its potential structure-based classification and structures that determine different classes may occur in a single molecule. Thus, ChEBI contains classes that overlap (i.e. share members or subclasses). The ChEBI ontology provides two separate classification hierarchies for the chemical entities: one based on their structures and another based on their functions or uses. In the current work, we focus on the structure-based sub-ontology. Entities in the structure-based sub-ontology are often associated with specifications of their molecular structures, particularly – but not exclusively – the leaf nodes within the classification hierarchy. In ChEBI, a chemical entity with a defined structure can be the classification parent for another structurally defined entity, since all entities are classes according to the ChEBI ontology, and there can be different levels of specificity even amongst structurally defined classes. To formulate a supervised machine learning problem, however, we need to create a distinction between those entities with chemical structures that form the input for learning, and the chemical classes that they belong to that form the learning target. This distinction is created by sampling structurally defined entities only from the ontology leaf nodes, and using the higher-up classes in the hierarchy as targets for the prediction.
As mentioned above, the SMILES notation is analogous to a language to describe atoms and their bonds within a chemical structure. Intuitively, this leads to a correspondence between the processing of chemical structures in this type of representation, and natural language processing [46]. Therefore, architectures that have been successfully applied to language-based problems can also be employed for this multi-label prediction task. One of these successful architectures is RoBERTa [31], whose architecture offers a learning paradigm that enables pre-training the model on unlabeled data and then fine-tuning it for the ultimately desired task. Fine-tuning can be done by adding one additional layer to the pre-trained model, without requiring major modifications to the model’s architecture. RoBERTa is trained on two unsupervised tasks: Masked Language Modeling (MLM), in which some tokens are randomly removed from the input sequences and the model will learn to predict masked tokens, and a binary classification task that predicts whether or not the second sentence in the input sequence follows the first sentence in the original text. The ELECTRA model [11] is an extension of the RoBERTa model, which offers several improvements to the pre-training strategy. The most notable of these changes is that tokens are not just masked, but replaced by other tokens. The model has to predict which tokens have been replaced.
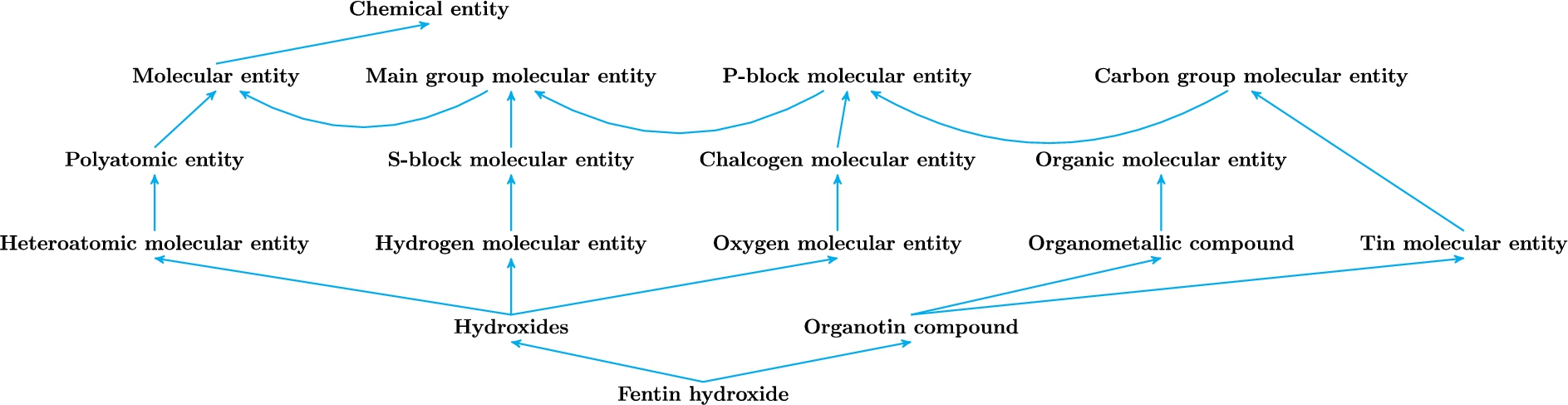
Since chemical structures in ChEBI typically belong to several ontology classes, the problem of automated chemical entity categorization can be viewed as a multi-label prediction task. Figure 2 shows the fentin hydroxide molecule and its parents in the ChEBI ontology: organotin compound and hydroxides.
Fig. 2.
Fentin hydroxide and its hierarchical classes. Blue lines indicate the sub-class relationships.
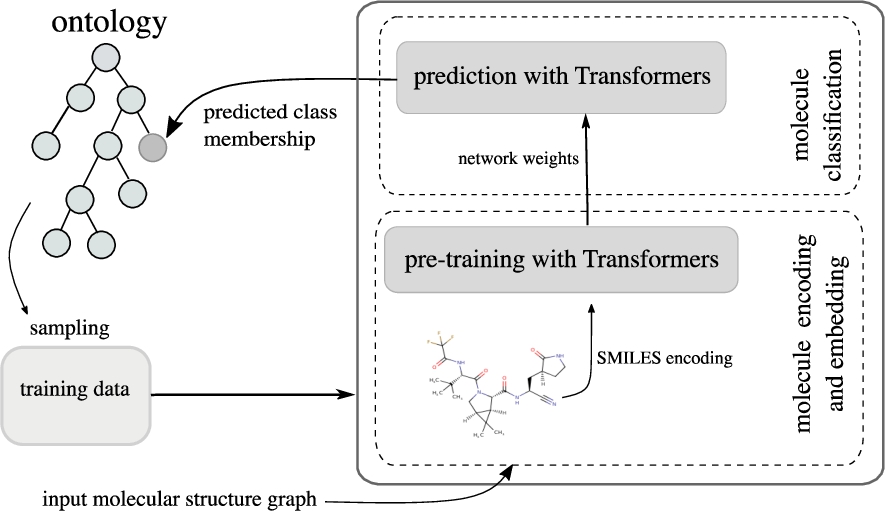
Our approach33 trains a RoBERTa and an ELECTRA model on SMILES strings, and then predicts multiple chemical class memberships. The overall architecture is shown in Fig. 3, where “Transformer” can be either “RoBERTa” or “ELECTRA”.
Fig. 3.
Architecture of our ontology extension with Transformer-based models.
3.1.Datasets
Our training setup uses four different datasets. These datasets are sourced from the ChEBI ontology or from the PubChem database as depicted in Figure 4. Whilst PubChem has a much larger variety of different chemicals, molecules are not necessarily associated with their chemical classes as they are in ChEBI. Therefore, molecules that have been extracted from PubChem cannot be used for the fine-tuning task, as this would require chemical classes as labels. It is however possible to harness the diversity of the PubChem database during the unlabeled pre-training task and additionally it provides a good source of additional chemical structures for manual evaluation.
Fig. 4.
The data generation process.
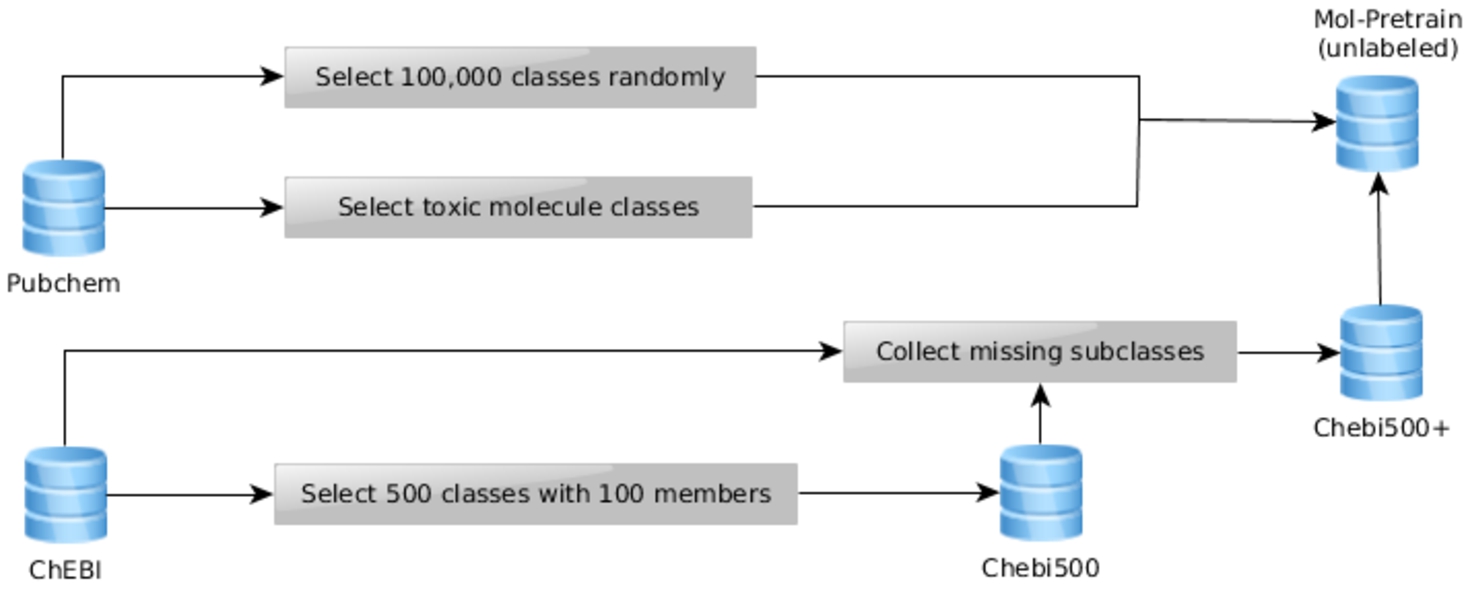
To test our use case of ontology extension with a dataset of chemicals not yet present in ChEBI that is based on a plausible real-world use-case, we selected all chemicals from PubChem which have a hazard class annotation (
We also randomly sampled 100,000 molecules from the broader PubChem database and merged the result with the previously discussed “hazardous” dataset and all molecules from the ChEBI ontology. The resulting Mol-Pretrain dataset has been used to pre-train the final ELECTRA model.
Finally, the fine-tuning is based on two different datasets, both of which have been sampled from the ChEBI ontology. To use the existing ontology classification as input to the learning task, the ontology first has to be transformed into an appropriate form. The ontology classification is inherently unbalanced, as different classes have different numbers of members and are partially overlapping. In previous research, we explored different more traditional approaches [20]. These techniques were often more susceptible to imbalances in the dataset. It was therefore necessary to define a sampling strategy to select leaf node entities and classes to minimize the impact of imbalances and overlaps on the training. As described in [20], we first order the classes by size ascending, then select 100 distinct members for each class without selecting the same member for any two classes even if in practice the classes do share members. In order to be able to compare our results to our earlier findings, we have used the same dataset44 and sampling strategy as was used in [20]. Using only the hierarchical sub-class relations in the ChEBI ontology, this dataset was created by randomly sampling leaf node molecular entities from higher-level classes that they are subclasses of, using an algorithm that aimed to minimize (as far as possible) class overlap while ensuring at least 100 individuals per class. The algorithm is described in Section 3 of [20]. The resulting
Fig. 5.
Distribution of members per class, ordered from the classes with the lowest to the highest number of members. Top: member counts in the dataset
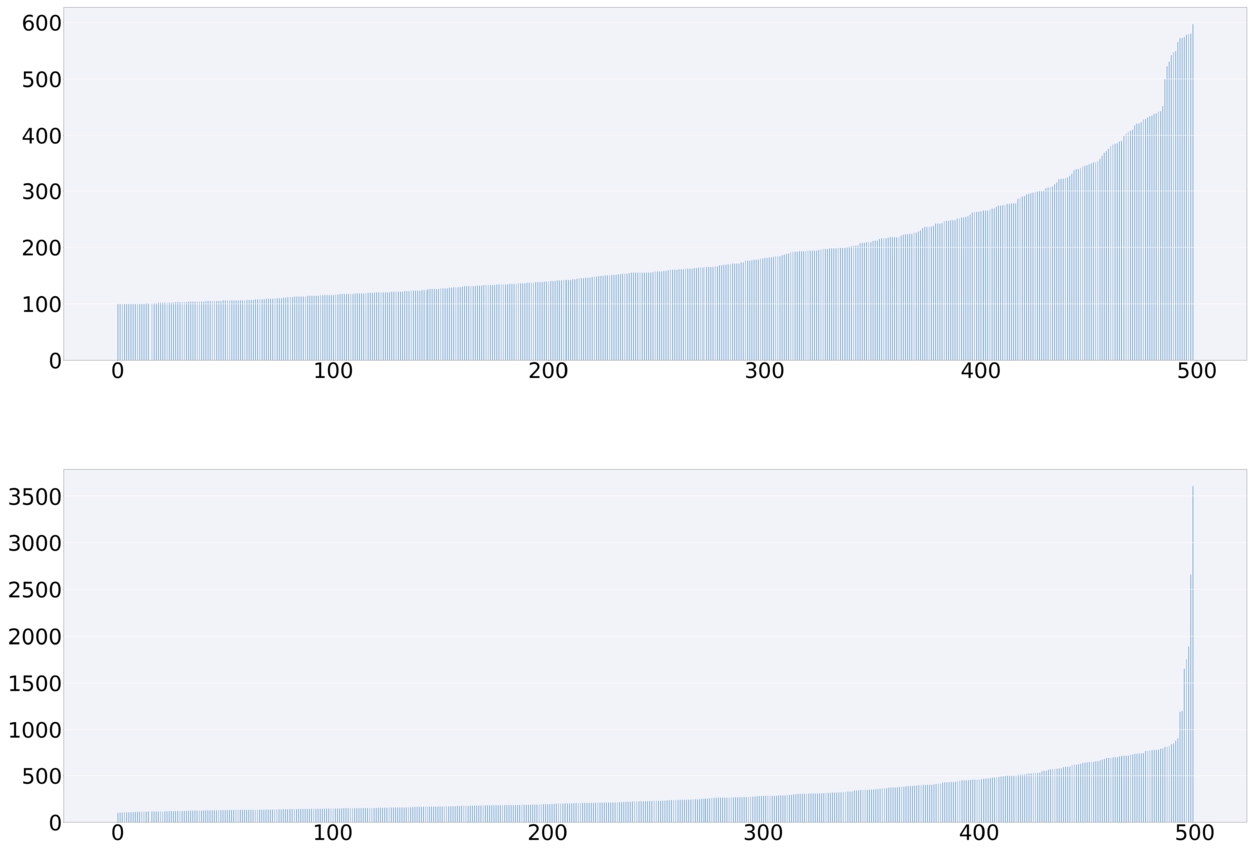
In our past work, we explored methods that were sensitive to such imbalances and required less data [20]. This work focuses on deep learning models that have a higher number of trainable parameters and, therefore, require a larger training set. To address this, we created a second dataset without sub-sampling only 100 members for the selected classes – including the same classes as in [20], but for each class including all possible molecules that belong to this class in ChEBI. The resulting
3.2.Input encodings
Tokenization is a pre-processing step to create a vocabulary (a set of unique tokens) from textual data. These tokens can be generated on different levels of the textual inputs. In this work, we will use two different tokenisation approaches (BPE and CHEM), which we will introduce in the following section. CHEM is a variant of one of the most common tokenisation methods: Character-level tokenisation, which splits texts at the character level and aims to embed these individually.
However, individual characters do not carry any chemically meaningful information. Therefore, the CHEM tokenizer parses the SMILES string according to its concrete syntax and uses the resulting syntactic categories as tokens. This means that tokens consist of single atoms including their respective charges and isotopes (e.g. “[Br]”, “[B]”, “[Fe2+]”), bonds (e.g. “-”, “=”) and ring and branch references. Figure 6 shows an example of this tokenisation.
However, as chemical classes may consist of many atoms, this low-level encoding will lead to large input sizes, which may increase the computational cost of training deep neural networks. Embeddings of whole words lead to smaller input sizes, but cannot handle words that have not been seen during training. Sub-word tokenization strategies, such as Byte Pair Encoding (BPE), aim to find the middle ground between these approaches. They break a text sequence into sub-words that can then be recombined to generate embeddings for previously unseen words. The BPE algorithm begins by counting the number of times each character pair appears in the dataset. After each iteration, the most frequently occurring pairings are merged and added to the vocabulary. For the next iteration, the two characters in each pairing are combined and considered as a single unit. The performed merge operations are stored and used when the tokenization algorithm confronts an unknown token. So, by considering the possible merges for the character pairs in an unknown token, this unknown token can be broken into smaller known tokens.
While the BPE technique performed well on our classification task, its tokenization strategy of grouping certain character pairs does not allow deeper insights into the influence that some singular atoms had on the model’s predictions. Thus, given a token OCB it would not be possible to ascertain whether the model based its prediction on Oxygen, Carbon, Barium or a combination of those. Further, since the BPE tokenizer is based on subword frequency, it does not respect the intended semantics of the chemical abbreviations. E.g., BPE might treat in OC(CCl)CCl the occurrences of [CC] as a token, in spite of the fact that in CCl the first C represents a carbon and Cl represents a chlorine atom, and the l on its own has no meaning. Another downside are the kinds of tokens that have been derived by the BPE tokenizer. The token vocabulary frequently contained SMILES specific reference characters that capture branches, cycles or molecule charges resulting in chemically infeasible tokens. Figure 6 exemplifies the tokenization of a SMILES string. It can be seen that the BPE tokenizer merges a charge indicator (“-” and a adjacent double bound into a single token (“-])=”). The chemical tokenizer, on the other hand, groups them with the corresponding atom.
Fig. 6.
Comparison of BPE tokenizer (top) and chemical tokenizer (bottom).

Table 1
(Hyper-)parameters of the models
| Parameter | RoBERTa | ELECTRA |
| Number of attention heads | 12 | 8 |
| Number of hidden layers | 6 | 6 |
| Neurons in hidden layer | 512 | 256 |
| Dropout for attention probabilities | 0.1 | 0.1 |
| Activation function in the encoder | gelu | gelu |
| Activation for the classification layer | sigmoid | sigmoid |
| Number of epochs in pre-training | 100 | 100 |
| Number of epochs in fine-tuning | 100 | 100 |
| Loss function for pre-training | BCE | BCEWithLogits |
| Loss function for fine-tuning | BCEWithLogits | BCEWithLogits |
| Optimizer | Adam | Adam |
3.3.Model training and evaluation
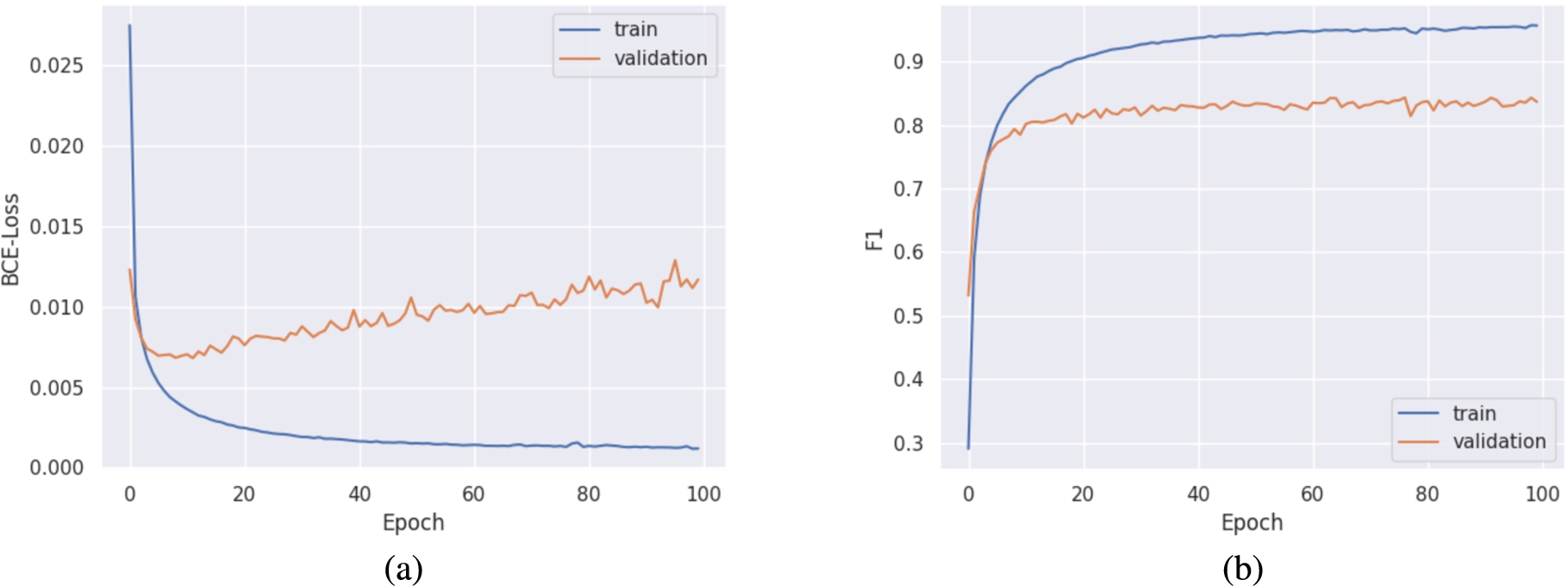
To train the model, we used two GPUs (GeForce GTX Titan X, 12 GB GDDR5). Table 1 shows the hyper-parameters for our model. The parameters for RoBERTa were based on the respective publication [10]. Electra then derived from that configuration, the number of attention heads and size of hidden layers were derived from the model’s defaults. Both models require different kinds of pre-training. RoBERTa was pre-trained on a dynamically masked set of SMILES strings and ELECTRA on SMILES with randomly substituted tokens for 100 epochs each (unsupervised). As discussed in Section 3, the pre-trained model provides a good starting point for training a model on a related task. This starting point incorporates the trained weights of the model. Furthermore, we validated the model on a separate dataset after each training epoch. The validation during training has no effect on the model’s trained weights, nevertheless, it helps in adjusting the model’s hyper-parameters. For the final multi-label classification task, we loaded the pre-trained model and trained it for 100 epochs with the class labels (supervised). The respective setup that performed best on the validation set has then been selected for further analysis. Figure 7 (a) shows the train and validation loss during the fine-tuning step. Similarly, Fig. 7 (b) shows the F1 score after fine-tuning.
Fig. 7.
Train and validation results for ELECTRA on
3.4.Case study for ontology extension
We applied the model to predict parent classes amongst the 500 ChEBI classes on which the model was trained, for the 140,913 classes in the dataset of hazardous molecules extracted from PubChem. Subsequently, we created a composite ontology consisting of (a) the subset of ChEBI corresponding to our 500 classes and the additional classes needed to complete their classification hierarchy to the root of the ontology, (b) together with the newly created classes for each input hazard molecule, connected by (c) asserted subclass relationships to those classes to which the model predicts they belong. We are then able to use logical reasoning to validate the extended ontology together with the set of disjointness axioms that is associated with ChEBI (as described in [16]). Disjointness axioms specify classes that should not overlap or share any members. They are thus useful to detect invalid predictions: in cases where multiple superclasses have been predicted for a single molecule spanning classes that are flagged as disjoint, at least one of the predictions is incorrect.
4.Results
4.1.Model training and evaluation
Fig. 8.
Part of the extended ontology. Existing subsumption relations (black) have been enriched with new subclasses, shown with dashed borders. Correct subclass predictions are depicted with cyan, dashed arrows, while red, dotted arrows indicate misclassifications.
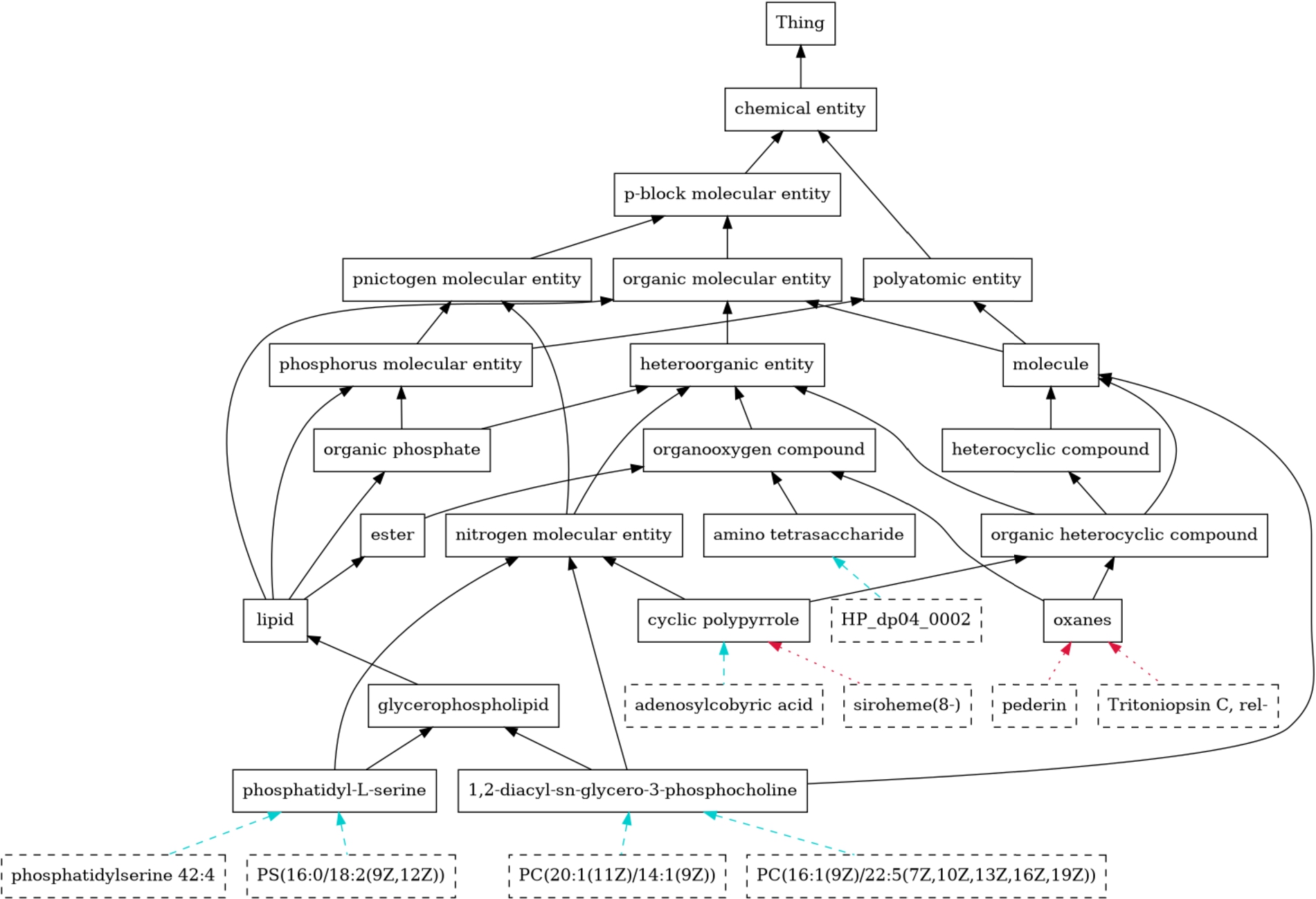
The presented model takes a given class of molecules, represented by a SMILES string, and assigns the corresponding superclasses from the CHEBI ontology. Figure 8 shows an illustrative example of the result of this process.
For our evaluations during and after training, we used the F1-score as the main measure. The F1 score is defined as the harmonic mean between precision and recall. These three metrics may be computed in different ways depending on the averaging scheme: (1) micro: collects the total number of true positives, false positives, and false negatives for each individual (i.e. each data row) and calculates the overall metric score as the average over all rows. (2) macro: calculates the score for each class and then computes their average. Table 2 and Fig. 9 compare the results of the current model with the previously obtained results for the LSTM model from [20] and the RoBERTa model from [36]. We saw an improvement in performance compared to the LSTM model both when we look at the distributions of values for the molecule-wise F1 scores (Fig. 9a) and for the class-wise F1 scores (Fig. 9b). A statistical comparison of the overall F1 score distributions shows that the difference in F1 scores is statistically significant (
The raw output values of our model are the probabilities of a sigmoid function. Therefore, a threshold value must be applied to these probabilities to produce a binary vector, indicating the final classifications. These results are based on the classification threshold value of 0.5. The precision – in our classification task – shows the ability of the model to not wrongly assign a label to a molecule, while the recall score reflects the model’s capability to discover all labels that were assigned to a molecule.
Table 2
The comparison of the scores achieved by two models on the
| Macro | Micro | |||||
| LSTM | RoBERTa | ELECTRA | LSTM | RoBERTa | ELECTRA | |
| F1 | 0.71 | 0.76 | 0.78 | 0.74 | 0.80 | 0.79 |
| Recall | 0.68 | 0.74 | 0.76 | 0.70 | 0.78 | 0.77 |
| Precision | 0.77 | 0.78 | 0.80 | 0.79 | 0.81 | 0.82 |
Fig. 9.
F1 scores on test fragment of the
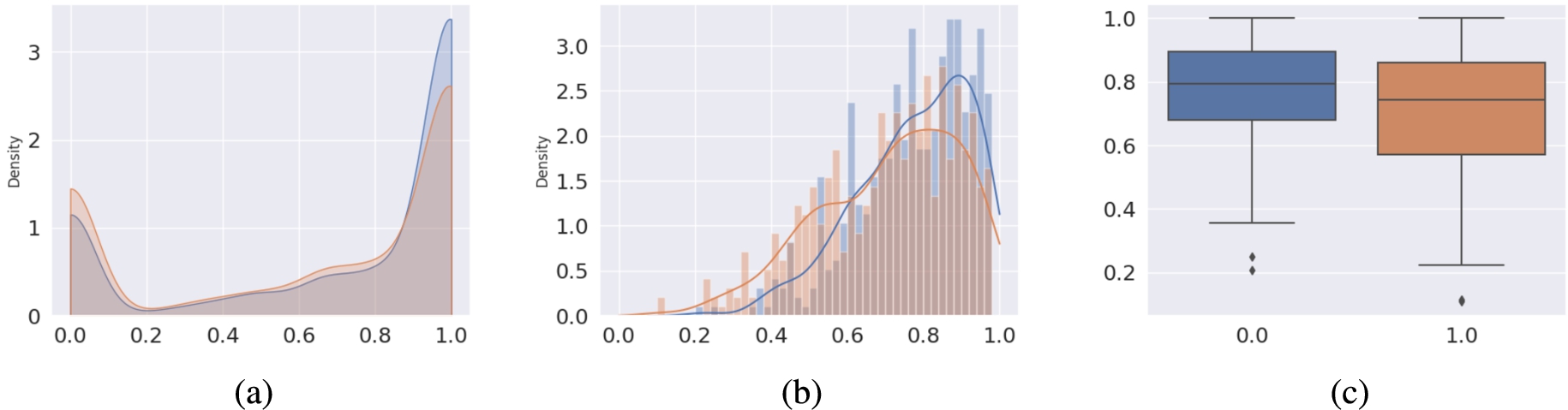
Fig. 10.
Top: Stacked bar chart for the number of false negatives (orange) and true positives (teal), Bottom: Bar chart for the number of false positives. Both plots are based on Electra’s performance on the test portion of the
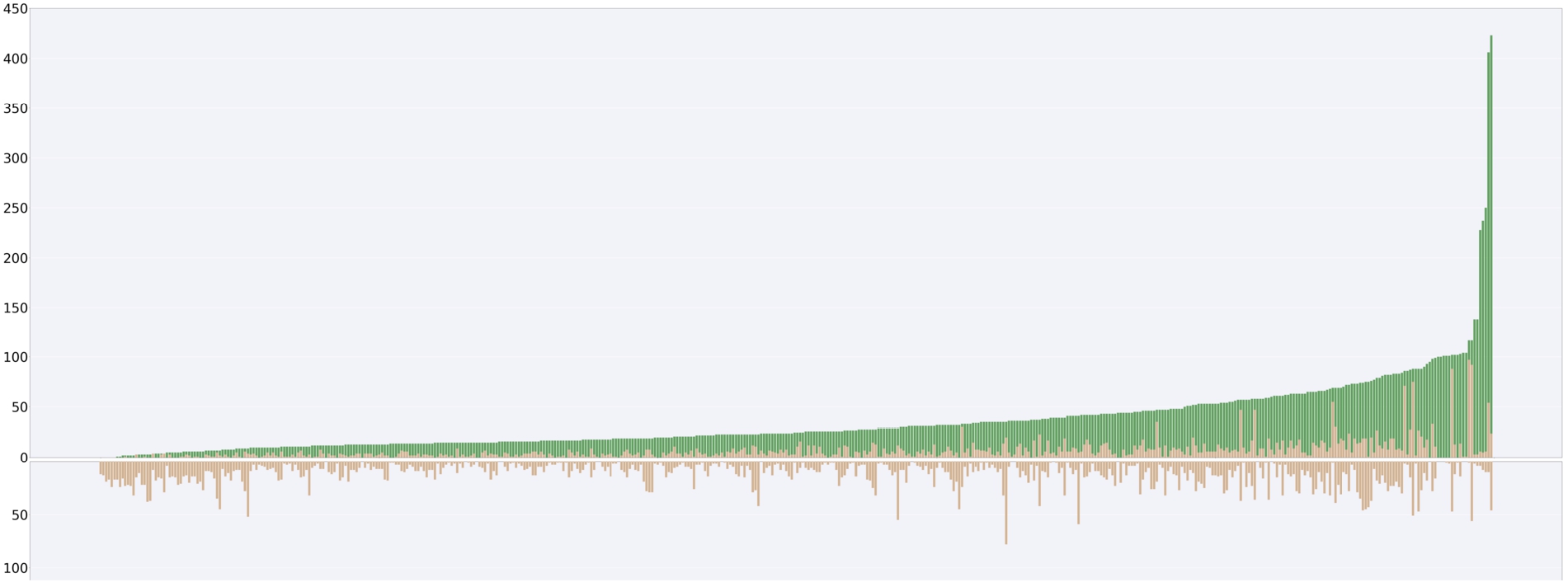
Table 3
Comparison of the scores achieved by ELECTRA on the
| Macro | Micro | |||
| BPE | Chemical tokens | BPE | Chemical tokens | |
| F1 | 0.75 | 0.77 | 0.84 | 0.82 |
| Recall | 0.75 | 0.74 | 0.86 | 0.79 |
| Precision | 0.75 | 0.80 | 0.82 | 0.84 |
As can be seen in Fig. 10, some of the predicted subclass relationships are correct according to ChEBI, while others are incorrect. Among the classes in the predictions, some have a larger number of false-positive classifications than others. This aligns with our analysis from previous work, discussed in detail in [20], that has shown that certain classes were harder to predict, often due to specific structural features such as cycles and aromaticity. The performance for smaller classes (those with fewer members in the underlying ontology) worsened compared to the
4.2.Interpretability
Self-attention in Transformer-based models enables the model to explore several locations in the input sequence to produce a better embedding for the tokens. As a result, the embeddings encode different contextual information for the same token in different positions (and different sequences). The architecture of the RoBERTa and ELECTRA models contain a stack of Transformers’ encoders, each consisting of multiple attention heads. Since the attention heads do not share parameters, each head learns a unique set of attention weights. Intuitively, attention weights determine the importance of each token for the embeddings of the next layers [50]. In this sense, visualizing the attention weights of Transformer-based models helps to interpret the model with respect to the relative importance of different input items for making classifications [49]. While the benefit of attention visualization may be limited in explaining particular predictions, depending on the task, attention can be quite useful in interpreting the model’s overall predictions [37,41,47]. In fact, attention heads can reveal a wide variety of model behaviors and some of these heads may be more significant for model interpretation than others [49].
We examined, for our ELECTRA model, how attention corresponds to different chemical structural elements, at both the token and molecule level. Figures 11 and 12 illustrates a selection of the attention weights within the model when processing the molecule naphthionic acid (illustrated). Darker lines in the network plot in Fig. 12 indicate a stronger attention relation between tokens. Similarly, darker green in the molecule plot indicates that higher attention was paid to that particular part of the molecule.
ChEBI specifies that this molecule is a subclass of arenesulfonic acid (CHEBI:33555), defined as a sulfonic acid that features one or more carbon rings. These defining structures are attended to by our model, when it classifies naphthionic acid correctly as a subclass of arenesulfonic acid. The left column in Fig. 12b shows the attention of the model from the ring index “2” at position 8 to the fused carbon ring structure. Those parts of the molecules to which the system pays high attention when visiting ring index “2” are also highlighted in 11a. The right column in Fig. 12b and, respectively, Fig. 11b show the attention of the model to the sulfonic acid group from the sulfur atom “S” at position 17. These structures are essential for the correct classification of the given molecule as arenesulfonic acid. Visualisations such as those in Fig. 11 provide a representation of the attention structure that is more intuitive for chemists, and provide a sort of visual explanation for the classification.
Fig. 11.
Some attentions for naphthionic acid, projected to the molecule.
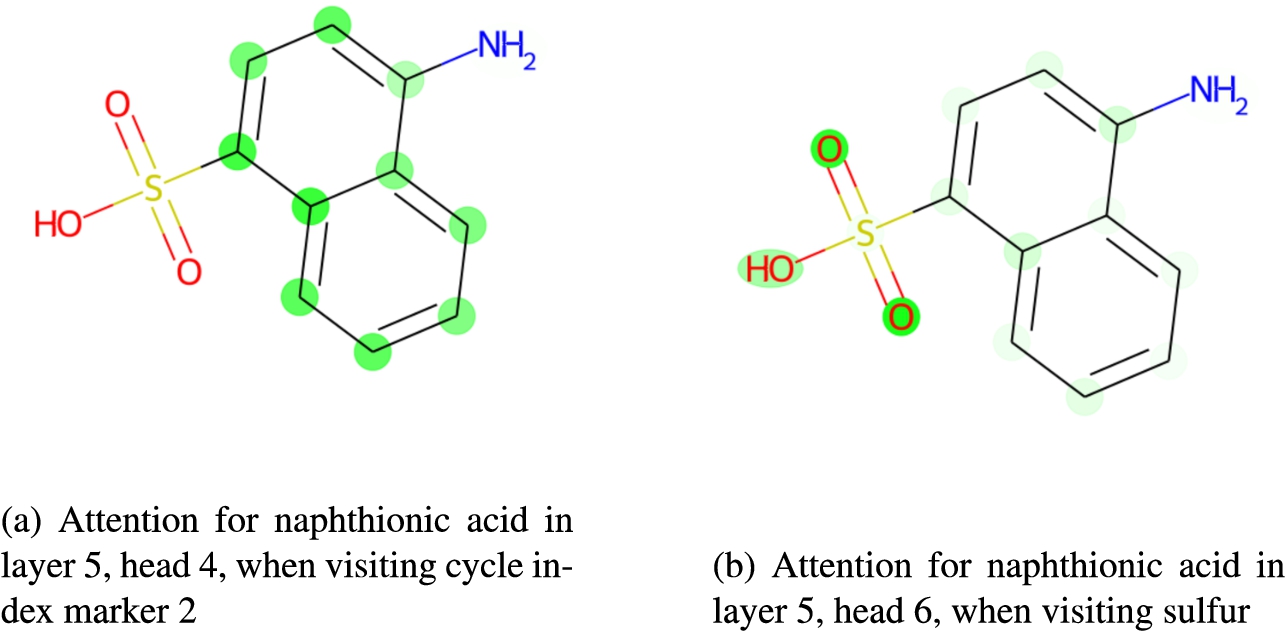
Fig. 12.
Attention relations for naphthionic acid.
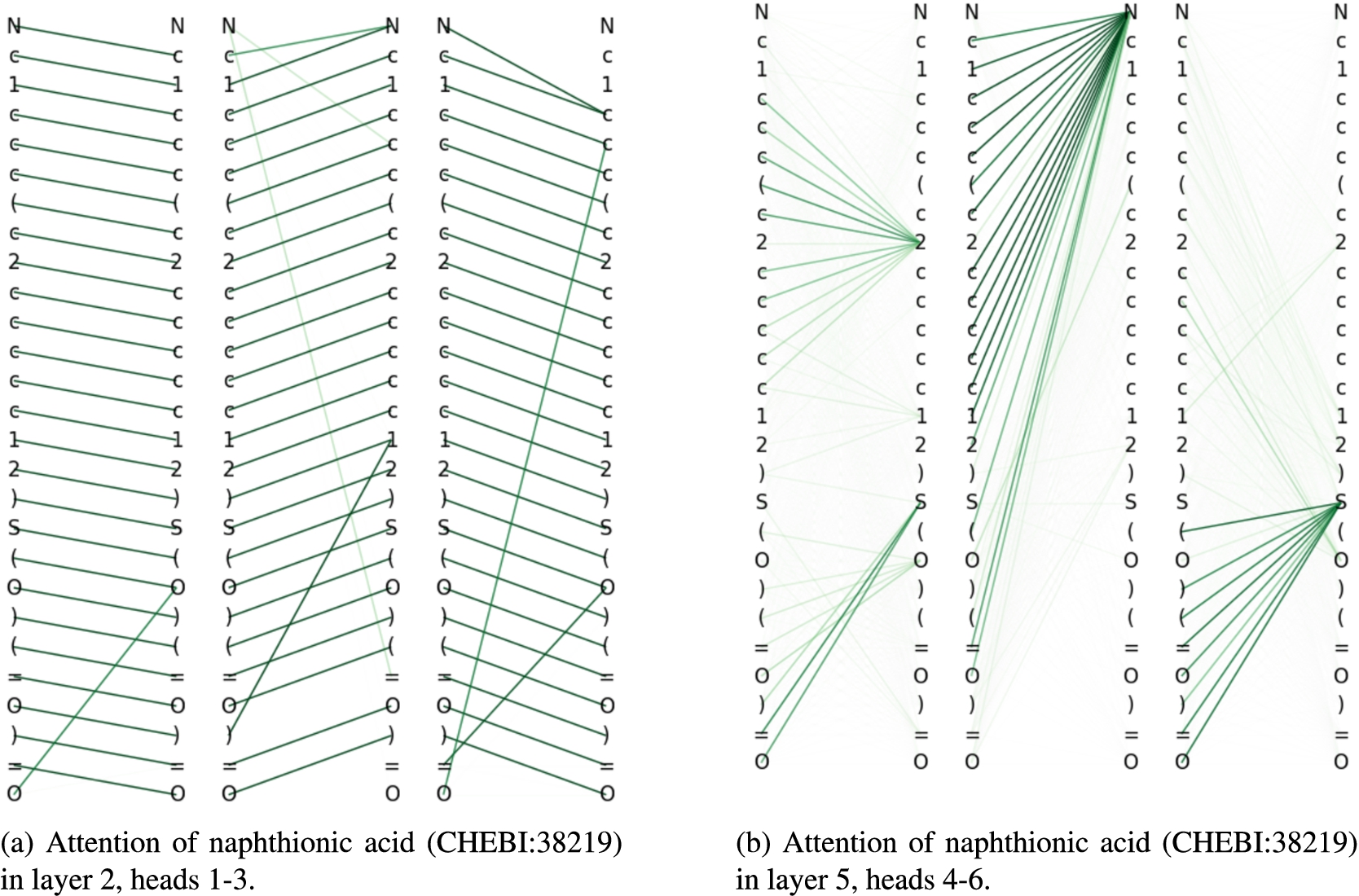
The middle column of attention weights in Fig. 12b shows that the model also paid attention to a substantial portion of the molecular structure from the nitrogen atom at position 0. Nitrogen is the essential element in amino compounds. As there were other amino classes that were part of the 500 selected classes, such as amino monosaccharide (CHEBI:60926), glutamic acid derivative (CHEBI:22693) or ethanolamines (CHEBI:23981), it is possible that the model needs to analyse larger parts of the molecule in order to rule out those possible candidates for superclasses.
We also found that some universal patterns emerged during classification. The attention distribution in the first layer was almost homogenous. The second and third layer focused mostly on direct neighbourhoods (preceding and following tokens) within the SMILES string, as depicted in Fig. 12a. Attention to neighbourhood tokens is important for understanding how general chemical connectivity within molecules is specified in the SMILES language. The fourth, fifth and sixth layer focused on larger structures, as shown in Fig. 12b.
The visualisations that we presented in this section provide a useful way to interpret the attention mechanism of the model, because in the case of true positives or true negatives, they enable a chemist to check whether the classification by the model was indeed based on the chemically salient substructures of a molecule. If that is the case, it provides some validation for the assumption that the model learned some chemically meaningful distinctions. Analogously, if a molecule is classified wrongly, because the model fails to pay attention to the presence of one of the relevant substructures (e.g., a false negative classification), then the visualisation of the attention mechanism is helpful to understand which of the salient substructures the model fails to recognise. A presentation of these predictions and visualisations to a group of chemists informally received enthusiastic confirmation that these visualisations provide meaningful insights into the operation of the system accessible to domain experts. We therefore expect the visualisations presented here to improve the ontology development process. However, our approach is limited to the visualisation of attention to the presence of substructures. This is a significant limitation because some chemical classes are defined by the absence of certain structures, which our approach cannot visualise.
4.3.Classification of never-seen chemicals
To evaluate the performance of the model on never-seen chemicals, we applied it to a set of 140,913 SMILES extracted from PubChem that had ‘hazard class’ annotations and were not present in ChEBI.
Fig. 13.
Left: Bar plot indicating how many molecules were assigned a particular number of class predictions, for example the bar at 0 indicates that 41,097 molecules were assigned no class prediction. (right) Scatter plot indicating the number of molecules predicted to belong to each of the 500 ChEBI classes in the prediction task (ordered), with the rolling average indicated by a line.
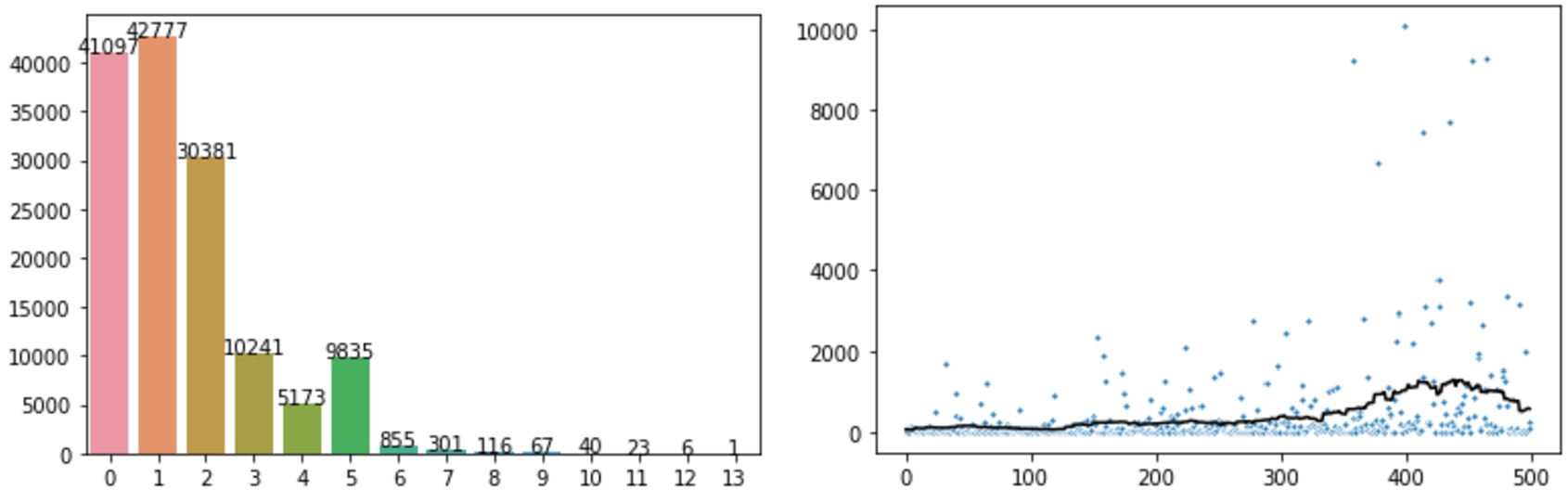
Among these, we found that 29% (41,097) – almost a third – of the input molecules were not assigned any class at all (Fig. 13, left), which can be regarded as a ‘don’t know’ response from the model. Assessing these molecules reveals that many contain isotopic SMILES with unusual atoms such as 13C Carbon isotopes or 2H Hydrogen isotopes, not present in the training dataset. Others had explicit charges, complex branching structures and explicit stereochemistry, all attributes that are poorly represented in the training data. Although a high percentage, this result is consistent with our earlier findings with similar models and offers a good motivation to connect deep learning-based models together with other approaches in ensembles so that if the learning-based model offers a ‘don’t know’ response, the system as a whole can still offer a reasonable prediction for every input molecule.
The remaining input molecules were assigned to at least one class, and amongst those 57,039 were assigned to multiple classes, including one molecule that was assigned to 13 distinct classes (Fig. 13, left). The distribution of molecules into classes is spread quite well across the 500 classes with which the model is trained (Fig. 13, right). Figure 14 shows a subset of the ChEBI hierarchy illustrating the assignment of molecules to classes, with colours indicating how many molecules were assigned to each class (red = fewer, green = more). In general, the classes lower down in the hierarchy received fewer molecule assignments, as would be expected. The full visualisation of all the 500 selected classes is available at Zenodo.55
Fig. 14.
Zooming in on a subset of the predicted classes, coloured by how many molecules are assigned to that class with red = fewer and green = more. Dark red = fewer than 10, Dark Green = more than 100, Orange = 10–100, Grey = not included in the 500 predicted classes but included in the visualisation for hierarchical completeness.
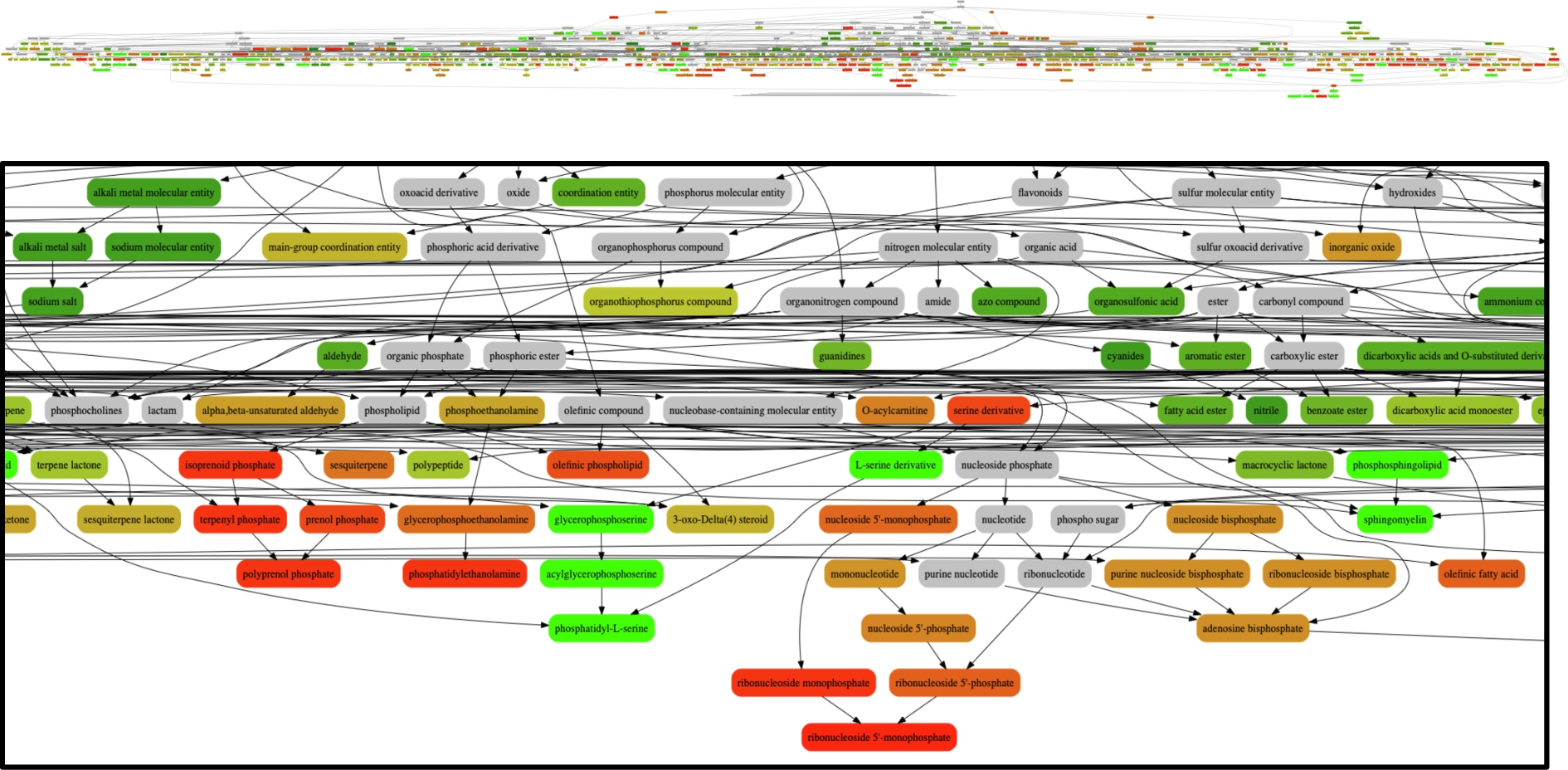
We then created an ontology extension from the subset of ChEBI classes included in our model with newly added classes based on the classified input molecules. Each input molecule is assigned a class in the extended ontology and the predicted classes are asserted as superclasses. The extended ontology is available at Zenodo.66
We combined this extended ontology with the disjointness axioms between chemical classes in ChEBI [16], and then tested for consistency using the HermiT reasoner in Protégé. Most of the predicted classifications were consistent, but we found that 5 of the input molecules’ predicted classifications resulted in a disjointness violation. These were caused by:
– Classification as both carbohydrate acid and carbohydrate acid ‘derivative’. In these cases, the derivative class shares the same core structural features as the class from which it is derived; however, the class definition reflects not only presence but also absence of features, which may be harder to learn.
– Classifications into multiple parent classes involving specific counts of groups, e.g. as both a diglyceride and a triglyceride. In these cases, perhaps larger targeted training sets would enable the network to better distinguish the features associated with these classes.
– Classifications into classes from both the inorganic and organic branches of the ontology, e.g. inorganic ion and steroid ester, or inorganic ion and dibenzopyrans.
As a final step, we then removed those 5 molecules which had conflicting classifications from the automated ontology extension, resulting in a consistent ontology extension. The extended ontology was manually inspected for correctness and no further problems were detected.
5.Discussion
5.1.Ontology extension and interpretability
ChEBI already makes use of an automated tool to extend its coverage beyond the manually curated core, namely ClassyFire, a rules-based system. This approach has limitations, notably that ClassyFire is structured around a different chemical ontology with only a partial mapping to ChEBI, and ClassyFire’s rules are manually maintained.
The deep-learning-based approach that we presented can overcome the limitations of rules-based approaches by allowing dynamic creation of classifiers based on a given existing ontology structure, and can be integrated into the ChEBI development process in the same way. The resulting system can then be used to integrate the given class into the ontology and translate the classification results into subsumption relations. By applying this approach to all classes in the dataset of hazardous chemicals, we were able to extend ChEBI automatically with an additional 99,816 classes. Yet, for optimal applicability, the approach must meet certain quality criteria. Ozaki [39] defined six goals for ontology learning, which we use to structure our discussion of our results.
Handling of inconsistencies and noise Our model is trained on information that originated solely from the ontology itself. This design decision eliminates external sources of inconsistencies and noise compared to approaches that utilise text corpora. The comparison of the F1 scores in the table in Fig. 9 shows that this classification outperforms the current state-of-the-art approaches – including the formerly leading LSTM-based model. In particular, for those chemical classes that were the most challenging in the previous approach, the current approach performed almost twice as well, as illustrated in Fig. 9 (b). It should be noted that there nevertheless remain some chemical classes that perform worse than others. For example, classes that are based on cyclic structures pose challenges, as their information may be scattered around the respective SMILES strings. Alternative input formats and network architectures may be explored in the future to better handle these structures. The model may also benefit from a larger amount of data. The distribution of class memberships depicted in Fig. 5 indicates that the dataset features some classes far more often than others. These classes are more prominent, often by virtue of being higher in the ontology subclass hierarchy and, therefore, represent broader classes of chemicals that may share members with other classes. Such an imbalance can skew the training in favour of those classes. Different sampling and regularisation techniques may be explored in the future to address this issue.
Unsupervised The presented approach is a variant of ontology extension. The ontology is therefore a mandatory input, from which the information that is needed for the ontology extension is extracted. The resulting dataset includes labels for each molecule. Strictly speaking, it is thus a supervised learning approach. However, these labels are extracted fully automatically from the input – the ontology. Therefore, no additional annotation by experts or other manual data pre-processing is necessary.
Human interaction As the ontology is extended automatically, no interaction is required.
Expressivity The system extends the given ontology using the same ontology language that has been used to build it. ChEBI is developed as an OWL ontology, which comes with expressive OWL-DL semantics. The extension adds classes and additional subclass relationships, which only uses a small element of the OWL-DL language, but the final extended ontology includes the axioms from the original as well and is verified for consistency accordingly.
Efficiency In [39] ‘efficiency’ is defined as the time it takes to build the ontology. This notion of efficiency does not translate well to a multi-level task that can be split into training and classification. This is due to differences in the times required for training and for classification. While the former task is often time consuming and elaborate, the same does not hold for the often quick classification. We, therefore, distinguish between two different types of efficiency.
Training efficiency describes the time that has to be invested in training the system for its specific task. Training of our model is divided in pre-training and fine-tuning. The pre-training with 100 epochs took around 20 hours for the
Classification efficiency refers to the time that is invested during the actual classification, i.e. the time that it would take to extend an existing ontology. Classification of 6,569 chemical entities in our test dataset took around 2 minutes. The classification of more than 140,000 molecules from the hazardous dataset can be done in under 10 minutes. This lays the foundation for a classification system that can be used to classify unseen chemical in near real-time, and represents a significant performance improvement over classification of never-seen chemicals using rule-based approaches.
Interpretability The formerly best classifier was based on an LSTM architecture. This approach outperformed ClassyFire, but this performance came with a disadvantage: The reason for a specific classification was not transparent. This is problematic, because the experts that check the ontology extension need insights into the system’s decision processes in order to evaluate the classifications. An interpretable approach is therefore crucial. The attention mechanism of the RoBERTa and ELECTRA architectures that have been used in the present work helps to address this issue. Attention weights can be seen as a measure of how much focus is put on an individual token. A homogeneous distribution of attention shows that nothing has been focused on in particular, whilst a high attention weight shows that a particular token had a high impact. Figure 12b shows that structures that are important for chemical classification are attended to during the classification process. Yet, not all attention heads show the same level of interpretability. On the one hand there were clear patterns in the attention within the first three layers, but these patterns were universal and did not differ between different chemical classes. This limits the use for interpretability of these layers. Attention heads in layers four and five, on the other hand, showed a tendency to focus on distinct structures within the molecules. Visualisations such as those in Figs 11 and 12b can be used to help interpret decisions made by the model, and aid the experts during the ontology extension process. While it is a limitation of the present work that we did not conduct a formal evaluation with users of the interpretability of these visualisations, we informally presented the visualisations to two chemists who are familiar with the ontology, and received positive feedback. It should, however, be noted that not all attention can be seen as a positive indicator for the classifications that have been made. Our analysis has shown that the model is also attending to substructures that are a contraindication for certain subsumption relations. Yet, these attentions can also be valuable for human experts, as they indicate the reason for the decision of the model for why certain subsumptions were not annotated.
Our analysis shows that the presented approach achieves the goals of ontology learning stipulated in [39]. One additional issue that needs to be addressed is applicability. At the heart of the presented approach is a neural network that is trained based on the annotations of the ontology. In the same way as any text analysis approach to ontology generation is dependent on the existence of suitable text corpora, our approach requires that the ontology contains enough information to train a model to predict the superclasses of a new class. ChEBI is an ideal use case, because SMILES annotations provide rich, structured information that we could harness for training the model. Another potential application domain for our approach in biology are proteins, which are also classified based on their structures, features of which can be annotated in relevant ontologies. Moreover, our approach is not limited to ontologies with structural information represented in annotations. E.g., for ontologies in material science one could consider training the model based on the physical properties (e.g., density, hardness, thermal conductivity), which are typically represented using data properties. In short, our approach to ontology extension is applicable to reference ontologies that associate classes with sufficient information that a neural network may learn the classification criteria that the ontology developers are using.
5.2.Data sets and learning methods
The current approach has been pre-trained on a combination of different data sources. The dataset that could be used for fine-tuning, however, is still just a subset of the ontology corresponding to 500 classes, thus falling short of the full scope of the entire ontology. In principle, with a longer training time corresponding to a larger dataset, the approach could be extended to encompass all classes in ChEBI which include a sufficient number of structurally defined member molecules for training purposes. While we do not want to speculate as to what this minimum number of examples for successful training might be, as this is an empirical question, also considering that the model may require fewer examples for classes that have very unusual or paradigmatic structural features, we can nevertheless estimate that at least 50 members per class would be required, which is met by around 1,200 classes in ChEBI currently. This could be seen as a limitation of applicability of this method, as many of the classes that chemists may be interested in may have fewer members. However, targeted curation efforts could be used to add examples to important classes for any given use case. In future work we will explore the benefit of up-sampling smaller classes to expand the applicability of our approach, and we will also explore ensemble-based architectures in which more specific classes are predicted by other methods that require smaller training datasets.
It may be worth observing that the performance of the deep learning model may be evaluated differently depending on the ontology extension use case. The model gives a substantial number of ‘don’t know’ results (i.e. no predicted classification for a given input molecule) which under the current evaluation metric (F1) count negatively alongside the incorrect classifications, as they affect recall. However, for some use cases these may be treated differently: For the use case of ontology extension it may be preferable for the model to preferentially avoid incorrect classifications by returning no classification for challenging input structures, however, if the use case involves automated curation of a set of measured molecules – for example in a metabolomics assay – then this behaviour would be more detrimental. Thus, future work may explore different weightings of training metrics suitable for these different use cases.
6.Conclusion and future work
We have presented a novel approach to the problem of ontology extension, applied to the chemical domain. Instead of extending the ontology using external resources, we created a model using the ontology’s own structured annotations. This Transformer-based model can not only classify previously unseen chemical entities (such as molecules) into the appropriate classes, but also provides information about relevant aspects of its internal structure on which the decision is based. At the same time, it was able to outperform previous approaches to ontology-based chemical classification in terms of predictive performance.
However, the trained model still struggles with several chemical classes that depend on specific structural features. E.g, classes that exhibit cyclic structures are often found in the lower quantile of classification quality. This behaviour can be traced back to the way molecules are encoded into the SMILES notation. This weakness might be addressed by using architectures that operate directly on the molecular structures, such as Graph Neural Networks [44].
Another limitation of our current work is that the system only extends the taxonomy of the ontology with subclass axioms between atomic classes. As one of our next steps we are planning to use ChEBI’s partonomy to learn new axioms of the form
We have illustrated our approach within the chemical domain, but as we discussed in Section 5, the approach is applicable to any ontology that contains classes that are annotated with information that is relevant to their position in the class hierarchy.
While our approach supports an automatic extension of an ontology, it can also be used in a semi-automated fashion to help ontology developers in their manual curation of the ontology. Since the model is trained based on the content of a manually curated ontology, improving and extending this ontology will lead to better quality training data and, thus, enable better predictions. Hence, there is a potential for a positive feedback loop between manual development and the AI-based extension.
An additional limitation of our current approach is that it does not use most of the logical axioms of the ontology during the learning process. Our analysis has shown that the classification is still violating some ontological axioms. This highlights the need for a stronger combination of neural and symbolic approaches. A logic-based framework could be employed to detect results that were likely to be mis-classifications – analogously to how we use ChEBI’s disjointness axioms, but generalised. Another strategy to address this gap would be to represent logical axioms in the form of Logical Neural Networks [43] in order to detect possible inconsistencies already in the learning process and to penalise them accordingly. Overall, there is still a pressing need for research in the field of (semi-)automatic ontology extension. Here, the growing field of neuro-symbolic integration can serve as the interface between formal ontologies and the power of deep learning. The possibility of incorporating explanations may further the understanding of the inner workings of learned models and, therefore, raise trust in these systems.
Alternative encodings for the input molecules may also yield benefits. For example, Xinhao Li et al. [28] introduced SMILES Pair Encoding (SPE) as a new data-driven tokenization approach that recognizes common SMILES sub-strings as unique tokens. Inspired by the BPE algorithm, the SPE is capable of learning a vocabulary of frequently occurring SMILES substrings from available large datasets of chemical compounds. This may tokenize the molecules into substructures with more chemical information, better expressing the molecular functions. The SPE may also contribute to better interpretability for the model, by providing tokens that are more chemically interpretable. In addition, the tokenized input sequence from SPE has fewer tokens than our current tokenizers – deep learning models with shorter inputs have a lower computational cost.
The masked language modeling paradigm used in the RoBERTa model requires a significant amount of computational power. The ELECTRA model uses a more efficient pretraining method, namely as replaced token detection. Moreover, due to the fact that this new approach is defined over all input tokens instead of a set of masked ones, it learns a better contextual representation of tokens. Hence, given the same amount of computing resources, the ELECTRA model converges faster than RoBERTa [11]. This enabled us to employ a smaller ELECTRA model (in terms of the hidden size of encoder blocks and the number of attention heads) while still achieving a comparable result to our RoBERTa model. Furthermore, findings from ChemBERTa [10] showed that increasing the amount of data used for pretraining leads to learning more robust representations of the molecules and therefore the performance of the final classification task improves with more pertaining data. In this paper, we only employed a small part of the PubChem dataset and a subset of the ChEBI ontology for the pretraining.
Our future research will include using SPE as featurization strategy, employing faster models while using more data from larger datasets for pretraining, and exploring approaches to systematically extract explanations for model predictions from the associated attention weights.
Finally, the visualisations that we used to interpret classifications (e.g., those shown in Figs 11 and 12b) were only exemplariliy extracted from our Transformer model. An important task for future work is to develop a tool that is able to automatically show these kind of visualisations for all classifications, which we can then use to evaluate them systematically with human experts. Based on the evaluation, the tool could learn to focus on the most helpful visualisations and to distinguish attentions that are positive indicators for a certain classification from those that are negative indicators.
Notes
1 This paper is an extended and revised version of a publication at the 3rd International Workshop on Data meets Applied Ontologies in Explainable AI (DAO-XAI 2021) [36].
2 [2,27,29,30] only generate subclass axioms. [5,45,54] can detect relations between classes, but they only generate some kind of triples, and apparently do not generate ontology axioms, which would involve e.g. the decision between an existential and a universal restriction. [40] can generate complex concept definitions also involving existential restrictions.
References
[1] | D. Allemang, P. Garbacz, P. Grądzki, E. Kendall and R. Trypuz, An analysis of the debate over structural universals, in: Formal Ontology in Information Systems – Proceedings of the 11th International Conference, FOIS 2021, Bozen-Bolzano, Italy, F. Neuhaus and B. Brodaric, eds, Frontiers in Artificial Intelligence and Applications. |
[2] | S. Althubaiti, Ş. Kafkas, M. Abdelhakim and R. Hoehndorf, Combining lexical and context features for automatic ontology extension, Journal of Biomedical Semantics 11: (1) ((2020) ), 1–13. doi:10.1186/s13326-019-0218-0. |
[3] | M.N. Asim, M. Wasim, M.U.G. Khan, W. Mahmood and H.M. Abbasi, A survey of ontology learning techniques and applications, Database 2018: ((2018) ), bay101. doi:10.1093/database/bax101. |
[4] | H. Assadi, Construction of a regional ontology from text and its use within a documentary system, in: FOIS’98 – 1st International Conference on Formal Ontology in Information Systems, Frontiers in Artificial Intelligence and Applications, Vol. 46: , IOS Press, Trento, Italy, (1998) , pp. 236–252, https://hal.archives-ouvertes.fr/hal-01617868. |
[5] | P.H. Barchi and E.R. Hruschka, Never-ending ontology extension through machine reading, in: 2014 14th International Conference on Hybrid Intelligent Systems, IEEE, (2014) , pp. 266–272. doi:10.1109/HIS.2014.7086210. |
[6] | C. Biemann, Ontology learning from text: A survey of methods, LDV Forum, 20: ((2005) ), 75–93. |
[7] | C. Bobach, T. Böhme, U. Laube, A. Püschel and L. Weber, Automated compound classification using a chemical ontology, Journal of Cheminformatics 4: (1) ((2012) ), 1–12. doi:10.1186/1758-2946-4-40. |
[8] | M. Booshehri, L. Emele, S. Flügel, H. Förster, J. Frey, U. Frey, M. Glauer, J. Hastings, C. Hofmann, C. Hoyer-Klick et al., Introducing the open energy ontology: Enhancing data interpretation and interfacing in energy systems analysis, Energy and AI 5: ((2021) ), 100074. doi:10.1016/j.egyai.2021.100074. |
[9] | L.L. Chepelev, J. Hastings, M. Ennis, C. Steinbeck and M. Dumontier, Self-organizing ontology of biochemically relevant small molecules, BMC Bioinformatics 13: (3) ((2012) ). doi:10.1186/1471-2105-13-3. |
[10] | S. Chithrananda, G. Grand and B. Ramsundar, ChemBERTa: Large-scale self-supervised pretraining for molecular property prediction, 2020, arXiv preprint arXiv:2010.09885. |
[11] | K. Clark, M.-T. Luong, Q.V. Le and C.D. Manning, Electra: Pre-training text encoders as discriminators rather than generators, 2020, arXiv preprint arXiv:2003.10555. |
[12] | T.F.G.G. Cova and A.A.C.C. Pais, Deep learning for deep chemistry: Optimizing the prediction of chemical patterns, Frontiers in Chemistry 7: ((2019) ), 809. doi:10.3389/fchem.2019.00809. |
[13] | Y. Djoumbou Feunang, R. Eisner, C. Knox, L. Chepelev, J. Hastings, G. Owen, E. Fahy, C. Steinbeck, S. Subramanian, E. Bolton, R. Greiner and D.S. Wishart, ClassyFire: Automated chemical classification with a comprehensive, computable taxonomy, Journal of Cheminformatics 8: (1) ((2016) ), 61. doi:10.1186/s13321-016-0174-y. |
[14] | K. Dührkop et al., Systematic classification of unknown metabolites using high-resolution fragmentation mass spectra, Nature Biotechnology ((2020) ), 1–10. doi:10.1038/s41587-020-0740-8. |
[15] | H.J. Feldman et al., CO: A chemical ontology for identification of functional groups and semantic comparison of small molecules, FEBS Letters 579: ((2005) ), 4685–4691. doi:10.1016/j.febslet.2005.07.039. |
[16] | J.D. Ferreira, J. Hastings and F.M. Couto, Exploiting disjointness axioms to improve semantic similarity measures, Bioinformatics 29: ((2013) ), 2781–2787. Publisher: Oxford University Press. |
[17] | G. Fu, C. Batchelor, M. Dumontier, J. Hastings, E. Willighagen and E. Bolton, PubChemRDF: Towards the semantic annotation of PubChem compound and substance databases, Journal of Cheminformatics 7: ((2015) ), 34. doi:10.1186/s13321-015-0084-4. |
[18] | L.H. Gilpin, D. Bau, B.Z. Yuan, A. Bajwa, M. Specter and L. Kagal, Explaining explanations: An overview of interpretability of machine learning, in: 2018 IEEE 5th International Conference on Data Science and Advanced Analytics (DSAA), IEEE, (2018) , pp. 80–89. doi:10.1109/DSAA.2018.00018. |
[19] | J. Hastings, M. Dumontier, D. Hull, M. Horridge, C. Steinbeck, U. Sattler, R. Stevens, T. Hörne and K. Britz, Representing chemicals using OWL, description graphs and rules, in: Proc. of OWL: Experiences and Directions (OWLED 2010), (2010) . |
[20] | J. Hastings, M. Glauer, A. Memariani, F. Neuhaus and T. Mossakowski, Learning chemistry: Exploring the suitability of machine learning for the task of structure-based chemical ontology classification, Journal of Cheminformatics 13: (1) ((2021) ), 1–20. doi:10.1186/s13321-020-00477-w. |
[21] | J. Hastings, D. Magka, C. Batchelor, L. Duan, R. Stevens, M. Ennis and C. Steinbeck, Structure-based classification and ontology in chemistry, Journal of Cheminformatics 4: ((2012) ), 8. doi:10.1186/1758-2946-4-8. |
[22] | J. Hastings, G. Owen, A. Dekker, M. Ennis, N. Kale, V. Muthukrishnan, S. Turner, N. Swainston, P. Mendes and C. Steinbeck, ChEBI in 2016: Improved services and an expanding collection of metabolites, Nucleic Acids Research 44: (D1) ((2016) ), D1214–D1219. doi:10.1093/nar/gkv1031. |
[23] | M. Herrero-Zazo, I. Segura-Bedmar, J. Hastings and P. Martínez, DINTO: Using OWL ontologies and SWRL rules to infer drug-drug interactions and their mechanisms, Journal of Chemical Information and Modeling 55: (8) ((2015) ), 1698–1707. doi:10.1021/acs.jcim.5b00119. |
[24] | D.P. Hill, N. Adams, M. Bada, C. Batchelor, T.Z. Berardini, H. Dietze, H.J. Drabkin, M. Ennis, R.E. Foulger, M.A. Harris, J. Hastings, N.S. Kale, P. de Matos, C.J. Mungall, G. Owen, P. Roncaglia, C. Steinbeck, S. Turner and J. Lomax, Dovetailing biology and chemistry: Integrating the gene ontology with the ChEBI chemical ontology, BMC Genomics 14: ((2013) ), 513. doi:10.1186/1471-2164-14-513. |
[25] | H.W. Kim et al., NPClassifier: A deep neural network-based structural classification tool for natural products, 2020. doi:10.26434/chemrxiv.12885494. |
[26] | O. Kutz, J. Hastings and T. Mossakowski, Modelling highly symmetrical molecules: Linking ontologies and graphs artificial intelligence: Methodology, systems, and applications, in: Artificial Intelligence: Methodology, Systems, and Applications, A. Ramsay and G. Agre, eds, Lecture Notes in Computer Science, Vol. 7557: , Springer Berlin / Heidelberg, Berlin, Heidelberg, (2012) , pp. 103–111, Section: 11. ISBN 978-3-642-33184-8. doi:10.1007/978-3-642-33185-5_11. |
[27] | H. Li, R. Armiento and P. Lambrix, A method for extending ontologies with application to the materials science domain, Data Science Journal 18: (1) ((2019) ), 1–21. |
[28] | X. Li and D. Fourches, SMILES pair encoding: A data-driven substructure tokenization algorithm for deep learning, Journal of Chemical Information and Modeling 61: (4) ((2021) ), 1560–1569. doi:10.1021/acs.jcim.0c01127. |
[29] | F. Liu and G. Li, The extension of domain ontology based on text clustering, in: IHMSC 2018, Vol. 1: , (2018) , pp. 301–304. doi:10.1109/IHMSC.2018.00076. |
[30] | W. Liu, A. Weichselbraun, A. Scharl and E. Chang, Semi-automatic ontology extension using spreading activation, Journal of Universal Knowledge Management ((2005) ), 50–58. |
[31] | Y. Liu, M. Ott, N. Goyal, J. Du, M. Joshi, D. Chen, O. Levy, M. Lewis, L. Zettlemoyer and V. Stoyanov, Roberta: A robustly optimized bert pretraining approach, 2019, arXiv preprint arXiv:1907.11692. |
[32] | A. Maedche and S. Staab, Ontology learning for the semantic web, IEEE Intelligent Systems 16: (2) ((2001) ), 72–79. doi:10.1109/5254.920602. |
[33] | D. Magka, M. Krötzsch and I. Horrocks, A rule-based ontological framework for the classification of molecules, Journal of Biomedical Semantics 5: (1) ((2014) ), 17. doi:10.1186/2041-1480-5-17. |
[34] | D. Magka, B. Motik and I. Horrocks, Modelling structured domains using description graphs and logic programming, in: The Semantic Web: Research and Applications, D. Hutchison et al., eds, Lecture Notes in Computer Science, Vol. 7295: , Springer Berlin Heidelberg, Berlin, Heidelberg, (2012) , pp. 330–344. ISBN 978-3-642-30283-1. doi:10.1007/978-3-642-30284-8_29. |
[35] | A.C. Mater and M.L. Coote, Deep learning in chemistry, Journal of Chemical Information and Modeling 59: (6) ((2019) ), 2545–2559. doi:10.1021/acs.jcim.9b00266. |
[36] | A. Memariani, M. Glauer, F. Neuhaus, T. Mossakowski and J. Hastings, Automated and explainable ontology extension based on deep learning: A case study in the chemicaldomain, in: Proceedings of the 3rd Workshop on Data Meets Applied Ontologies in XAI, Bratislava, September 18–19, 2021, R.C. et al., ed., CEUR Workshop Proceedings, Vol. 2998: , (2021) , http://ceur-ws.org/Vol-2998/. |
[37] | P. Moradi, N. Kambhatla and A. Sarkar, Interrogating the explanatory power of attention in neural machine translation, 2019, arXiv preprint arXiv:1910.00139. |
[38] | P. Moreno, S. Beisken, B. Harsha, V. Muthukrishnan, I. Tudose, A. Dekker, S. Dornfeldt, F. Taruttis, I. Grosse, J. Hastings, S. Neumann and C. Steinbeck, BiNChE: A web tool and library for chemical enrichment analysis based on the ChEBI ontology, BMC Bioinformatics 16: ((2015) ), 56. doi:10.1186/s12859-015-0486-3. |
[39] | A. Ozaki, Learning description logic ontologies: Five approaches. Where do they stand?, KI-Künstliche Intelligenz 34: (3) ((2020) ), 317–327. doi:10.1007/s13218-020-00656-9. |
[40] | A. Petrova, Y. Ma, G. Tsatsaronis, M. Kissa, F. Distel, F. Baader and M. Schroeder, Formalizing biomedical concepts from textual definitions, Journal of Biomedical Semantics 6: (1) ((2015) ), 1–17. doi:10.1186/2041-1480-6-1. |
[41] | D. Pruthi, M. Gupta, B. Dhingra, G. Neubig and Z.C. Lipton, Learning to deceive with attention-based explanations, 2019, arXiv preprint arXiv:1909.07913. |
[42] | B. Ramsundar, Molecular machine learning with DeepChem, PhD thesis, Stanford University, 2018. |
[43] | R. Riegel, A. Gray, F. Luus, N. Khan, N. Makondo, I.Y. Akhalwaya, H. Qian, R. Fagin, F. Barahona, U. Sharma et al., Logical neural networks, 2020, arXiv preprint arXiv:2006.13155. |
[44] | F. Scarselli, M. Gori, A.C. Tsoi, M. Hagenbuchner and G. Monfardini, The graph neural network model, IEEE Transactions on Neural Networks 20: (1) ((2008) ), 61–80. doi:10.1109/TNN.2008.2005605. |
[45] | A. Schutz and P. Buitelaar, Relext: A tool for relation extraction from text in ontology extension, in: International Semantic Web Conference, Springer, (2005) , pp. 593–606. |
[46] | P. Schwaller, T. Gaudin, D. Lanyi, C. Bekas and T. Laino, “Found in translation”: Predicting outcomes of complex organic chemistry reactions using neural sequence-to-sequence models, Chemical Science 9: (28) ((2018) ), 6091–6098. doi:10.1039/C8SC02339E. |
[47] | S. Serrano and N.A. Smith, Is attention interpretable? 2019, arXiv preprint arXiv:1906.03731. |
[48] | B. Smith, M. Ashburner, C. Rosse, J. Bard, W. Bug, W. Ceusters, L.J. Goldberg, K. Eilbeck, A. Ireland, C.J. Mungall et al., The OBO foundry: Coordinated evolution of ontologies to support biomedical data integration, Nature Biotechnology 25: (11) ((2007) ), 1251–1255. doi:10.1038/nbt1346. |
[49] | J. Vig, A multiscale visualization of attention in the transformer model, 2019, arXiv preprint arXiv:1906.05714. |
[50] | J. Vig, A. Madani, L. Varshney, C. Xiong and N. Rajani, BERTology meets biology: Interpreting attention in protein language models, 2020, arXiv preprint arXiv:2006.15222. |
[51] | Y. Wang, J. Xiao, T. Suzek, J. Zhang, J. Wang and S. Bryant, PubChem: A public information system for analyzing bioactivities of small molecules, Nucl Acids Res 37: ((2009) ), W623–W633. doi:10.1093/nar/gkp456. |
[52] | D. Weininger and Daylight Inc, The SMiles ARbitrary Target Specification (SMARTS) manual, 2020, http://www.daylight.com/dayhtml/doc/theory/theory.smarts.html. |
[53] | Z. Wu, B. Ramsundar, E.N. Feinberg, J. Gomes, C. Geniesse, A.S. Pappu, K. Leswing and V. Pande, MoleculeNet: A benchmark for molecular machine learning, Chemical Science 9: (2) ((2018) ), 513–530. doi:10.1039/C7SC02664A. |
[54] | Y. Zhou, L. Zhang and S. Niu, The research of concept extraction in ontology extension based on extended association rules, in: 2016 IEEE International Conference of Online Analysis and Computing Science (ICOACS), IEEE, (2016) , pp. 111–114. doi:10.1109/ICOACS.2016.7563059. |

