
Ras signaling in aging and metabolic regulation
Abstract
Aberrant signal transduction downstream of the Ras GTPase has a well-established role in tumorigenesis. Mutations that result in hyperactivation of Ras are responsible for a third of all human cancers. Hence, small molecule inhibitors of the Ras signal transduction cascade have been under intense focus as potential cancer treatments. In both invertebrate and mammalian models, emerging evidence has also implicated components of the Ras signaling pathway in aging and metabolic regulation. Here, I review the current evidence for Ras signaling in these newly discovered roles highlighting the interactions between the Ras pathway and other longevity assurance mechanisms. Defining the role of Ras signaling in maintaining age-related health may have important implications for the development of interventions that could not only increase lifespan but also delay the onset and/or progression of age-related functional decline.
1Introduction
Increased age is the predominant risk factor for several major human pathologies, including cancer, diabetes, cardiovascular disease and neurodegeneration [1–5]. Long considered an inevitable consequence of life, recent advances have revealed that aging has an underlying biological process, influenced by both genetic and environmental factors [6]. Similar biological manipulations have been shown to extend lifespan and delay the adverse effects of aging across different organisms including yeast, worms, flies and mice [7]. Such striking evolutionary conservation suggests that understanding the molecular mechanisms that drive aging in these laboratory models are key to the development of pharmacological interventions to improve lifelong human health.
Among the most robust, evolutionary conserved longevity-assurance mechanisms identified to date are caloric or dietary restriction (DR), defined as reduced nutrient intake without malnutrition, and genetic down-regulation of nutrient signaling pathways, such as the insulin/insulin-like growth factor (IGF) signaling (IIS) and mechanistic target of rapamycin (mTOR) pathways [7]. Extensive cross-talk exists between these key regulators of organismal aging. Reciprocal regulation at multiple nodes between the IIS and mTOR signaling pathways results in a complex and highly-connected nutrient signaling network [8] while increasing evidence from genetic interaction studies suggests that signaling via the IIS/mTOR network plays an important role in mediating the longevity response to DR [9].The IIS/mTOR network may therefore provide prime targets for pharmacological interventions to delay aging. Indeed, recent studies have demonstrated that pharmacological modulation of mTOR signaling can similarly extend lifespan and protect against age-related pathologies in laboratory animals. For example, the mTOR kinase inhibitor, rapamycin, both extends lifespan in multiple organisms, including mammals, and improves several physiological outputs during aging [10–14]. Furthermore, inhibition of mTOR using rapamycin-like molecules or rapalogues in humans has been shown to improve age-related immunosenescence [15]. The underlying cellular mechanisms that elicit the longevity response upon exposure to chemical inhibition of mTOR are still not entirely understood but may include inhibition of protein synthesis and induction ofautophagy [10].
Lifespan extension via pharmacological inhibition of IIS has, until recently, proved somewhat more elusive. In mammals, a well-known signaling intermediary of the IIS pathway is the oncogenic Ras protein. Ras is a small GTPase, the activation of which initiates a signal transduction cascade via the extracellular signal-regulated kinase (ERK)/mitogen activated protein kinase (MAPK). Both Ras and its downstream signaling effector, ERK/MAPK, can modulate the activities of key regulatory molecules within the IIS/mTOR signaling network, including PI3K and mTOR, as well as cellular mediators of the response to DR such as AMPK. Thus, Ras or downstream components of its signal transduction pathway may offer novel targets for pharmacological manipulation of the IIS/mTOR nutrient signaling network.
Studies have recently demonstrated a role for Ras signal transduction itself as an important modulator of aging in both invertebrates and mammals [16, 17].Interestingly, studies in mammals have also highlighted a key role for components of the Ras/MAPK pathway in metabolic regulation, particularly in the regulation of fat metabolism. Further exploration of these functions of Ras signaling may therefore offer new avenues for therapeutic interventions for both aging and metabolic disease.
2The Ras signaling pathway
The Ras proteins are members of a superfamily of small GTPases that play a fundamental role in signal transduction from cell-surface receptor tyrosine kinases (RTKs) in response to a variety of extracellular stimuli including growth factors, hormones and cytokines [18]. Their primary function is to assemble intracellular signaling complexes thereby activating downstream cell signaling pathways (Fig. 1). As such, Ras proteins coordinate multiple cellularresponses including proliferation, differentiation, apoptosis, senescence and metabolism [19].
Fig.1
The Ras/MAPK signaling pathway. The Ras/MAPK signaling pathway responds to extracellular cues to control cell survival, proliferation and metabolism. Growth factor binding to receptor tyrosine kinases (RTK) activates autophosphorylation of the receptor which generates binding sites for the Grb2 and Shc adaptor proteins. These adaptor proteins recruit the Ras GTPase exchange factor (GEF), SOS, to the inner surface of the membrane. SOS catalyses the exchange of GDP to GTP on Ras and then the activated Ras-GTP recruits Raf to the complex. Raf then initiates a downstream phosphorylation cascade via MEK and ERK. Activated ERK phosphorylates multiple cytoplasmic and cytoskeletal proteins including ribosomal S6 kinase (RSK). In addition, activated ERK can translocate to the nucleus, where it phosphorylates and activates members of the the E-twenty-six (ETS) transcription factor family.
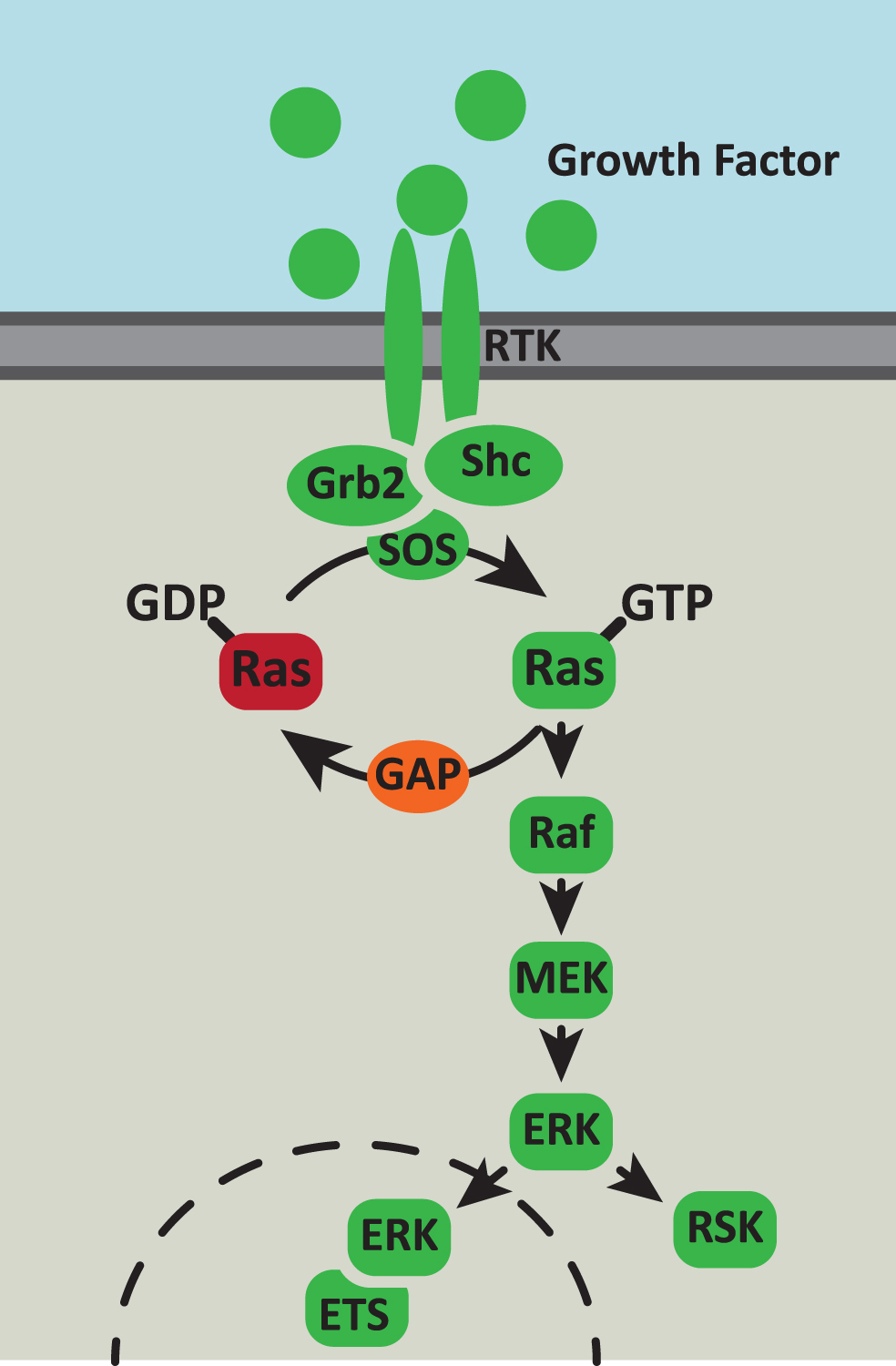
Ras proteins function as binary molecular switches, cycling between inactive GDP-bound and active GTP-bound states. The balance of active versus inactive Ras is determined by the competing activities of guanine nucleotide exchange factors (GEFs) and GTPase activating proteins (GAPs): Ras GEFs catalyse the replacement of GDP by GTP and Ras GAPs increase the rate of GTP hydrolysis [20, 21]. In the active GTP-bound conformation, two regions within the Ras protein, the Switch regions I and II, undergo significant structural changes, forming a GTP-dependent interface for binding to downstream effector molecules with high affinity and specificity [22]. Oncogenic mutations in Ras usually arise as a result of amino acid substitutions that prevent the GAP-dependent hydrolysis of GTP, thereby generating constitutively active Ras molecules [23].
Upon RTK activation, Ras facilitates the assembly of signaling complexes to the inner surface of the plasma membrane by specific protein-protein interactions [21] (Fig. 1). The cytoplasmic tail of the activated RTK recruits the Grb2 adaptor protein either via a direct SH2 domain-phosphotyrosine interaction or through association with the adaptor protein, Shc. Grb2 in turn then binds to the Ras-GEF, SOS, localising Ras to the activated RTK-bound complex. Active GTP-bound Ras then binds to and activates its effector molecules, including Raf, initiating a phosphorylation cascade via MEK and the extracellular signal-regulated kinase (ERK)/mitogen-activated protein kinase (MAPK). Activated ERK phosphorylates multiple cytoplasmic and cytoskeletal proteins, including MAPK-activated protein kinases and ribosomal S6 kinase [24]. In addition, activated ERK can translocate to the nucleus, where it phosphorylates and activates several transcription factors including members of the the E-twenty-six (ETS) transcription factor family [25]. The target specificity of activated ERK is therefore controlled by substrate availability, subcellular localisation and ERK scaffolding, in which scaffolding proteins tether ERK or its upstream kinase, MEK, to specific substrates and/or subcellular compartments [24]. Hence, signal transduction through Ras and its downstream effectors influences several cellular processes including proliferation, migration and differentiation. Increasing evidence suggests Ras signaling may also play an important role in animalaging.
3A role for Ras signaling in aging
3.1Yeast
Lifespan in the budding yeast, Saccharomyces cerevisiae, is measured by two metrics: replicative lifespan which corresponds to the number of daughter cells produced by an individual mother cell and chronological lifespan that measures the survival time of non-dividing cells in the stationary phase [26]. The first genes that were implicated in yeast longevity were the two Ras homologues, RAS1 and RAS2, which influence both replicative and chronological lifespan. Deletion of RAS1 extends replicative lifespan while deletion of RAS2 extends chronological lifespan [27, 28]. In yeast, the Ras proteins form part of a nutrient signaling pathway that includes cyclic AMP (cAMP) and protein kinase A (PKA). The effects of RAS1 or RAS2 deletion on yeast lifespan were shown to be mediated via altered signaling through this pathway [27]. Furthermore, direct deletion or inactivation of several components of the pathway were also found to increase both replicative and chronological lifespan [29–31]. The stress responsive transcription factors, Msn2 and Msn4, which are activated in response to RAS-cAMP-PKA signaling, were required for the effects of reduced RAS-cAMP-PKA signaling on chronological lifespan [30] and may also mediate the effects of RAS-cAMP-PKA inhibition on replicativelifespan [32].
The differential effects of RAS1 and RAS2 deletion on replicative versus chronological lifespan are interesting particularly as they both signal through cAMP-PKA to influence yeast aging. The relationship between replicative and chronological lifespan is not clear but increasing evidence suggests that the two are linked. For example, chronological aging results in a subsequent decrease in replicative lifespan [33]. The most obvious difference between replicative and chronological aging is the increased metabolic activity of actively replicating cells compared to cells in the stationary phase which themselves will be more subjected to stress. Thus, the divergent outcomes of RAS1 and RAS2 deletion on either replicative or chronological aging may reflect differential effects of RAS1 and RAS2 functions on metabolic and stress resistance pathways.
3.2Neurospora
Neurospora crassa also possesses two RAS genes: ras-1 [34] and ras-2 [35], and at least three genes predicted to encode Ras-GEFs [36]. The band (bd) mutation, a long known and widely used tool in the study of Neurospora circadian biology, was recently identified as a dominant mutation in ras-1 [37], resulting in an amino acid substitution within the switch region II, which mediates the interaction of Ras with its GEFs [38]. Mutation of similar residues in yeast may be important for proper activation of adenylyl cyclase [39]. Interestingly, the ras-1bd mutation has recently been shown to increase chronological lifespan in Neurospora [40]. The ras-1bdmutation does not seem to cause dramatic effects on Ras activity as there are no reproducible differences in the levels of active GTP-bound Ras in ras-1bd mutants compared to wild-type [37]. The signaling dynamics downstream of ras-1 in this organism are not well described but it is likely that the ras-1bd mutation elicits its effects by affecting the interaction of Ras with one or more GEFs and/or downstream effectors of signaling.
3.3Flies
In the fruit fly, Drosophila melanogaster, key outputs of Ras activation during development are the two ETS transcription factors: Pointed (Pnt), a transcriptional activator which is stimulated in response to Ras activation, and Anterior open (AOP), a transcriptional repressor that is inhibited by Ras activation. Both Pointed and AOP regulate expression of the same genes by binding to the same regulatory elements but with opposing outcomes [41–43]. Expression of an activated form of AOP was previously shown to be sufficient for lifespan extension in flies [44], implicating the Ras signal transduction pathway in Drosophila aging. More recent studies have demonstrated a direct role for Ras signaling during aging in this animal model as genetic inhibition of either Ras itself or ERK were found to extend lifespan [17]. Furthermore, these effects on lifespan were dependent on activation of AOP. Importantly, pharmacological inhibition of ERK using trametinib, a small molecule inhibitor of the upstream kinase, MEK, also resulted in longevity [17], thereby identifying a direct role for inhibition of the canonical Ras/MAPK signaling pathway as a pro-longevity assurance mechanism in an animal model.
3.4Worms
The canonical Ras signaling pathway is conserved in the nematode worm, Caenorhabditis elegans [45]. Ras itself is encoded by the let-60 gene, mutation of which disrupts several developmental processes including development of the vulva and excretory systems and sex-myoblast migration [45]. Downstream effectors of Let-60 signaling in worms include the ETS transcription factor, Lin-1, that is potentially regulated by MAPK phosphorylation [46]. Similar to the Drosophila Ras protein, Let-60 has been implicated in signal transduction downstream of the insulin receptor, Daf-2, during aging [47]. However, in worms, extension of lifespan by mutation of daf-2 is associated with activation rather than inhibition of Let-60 activity [47]. An important distinction in the signal transduction cascade between flies and worms is the absence of an AOP orthologue in C. elegans. Differences in the transcriptional output downstream of Ras may therefore explain the differential effects of Ras inhibition on lifespan in these two animal models.
Interestingly, the ETS transcription factor, ETS-4, has recently been identified as a longevity determinant in worms [48]. Epistasis analysis of loss-of-function alleles places ETS-4 in a parallel pathway to Daf-2 during aging and in a similar manner to AOP, ETS-4 shares common transcriptional targets with the FOXO transcription factor, Daf-16 [48]. This suggests that despite the divergent roles of Ras signaling in lifespan, the ETS and FOXO transcription factors regulate the expression of common target genes that influence lifespan, a function that is conserved from worms to flies.
3.5Mammals
In mammals, there are four members of the Ras protein family, N-RAS, H-RAS, K-RAS4A andK-RAS4B, expressed from three genes. A role for Ras signaling in mammalian aging by direct genetic disruption of Ras has yet to be demonstrated. Mice deficient for H-RAS, N-RAS or K-RAS4A are viable and do not show any obvious health deficits in early adulthood [49–51]. Yet, with the exception of K-RAS4A which does not impact on lifespan [52], the long-term effects of RAS deficiency on animal lifespan is still to beaddressed.
Several studies have implicated downstream components of the Ras signaling pathway in mammalian aging. For example, fibroblasts isolated from long-lived species of mammals and birds as well as long-lived mouse mutants show altered kinetics of ERK phosphorylation in response to stress associated with increased stress resistance, linking ERK activation to longevity [53, 54].
Mice deficient for the p66 isoform of the adapter protein, Shc, which forms part of the intracellular signaling complex coupling activated Ras to activated RTKs, has also been implicated as a determinant of mammalian longevity. Mice carrying a targeted knockout of p66shc show a 30% increase in lifespan compared to control littermates [55]. However, while the p66 isoform of Shc binds to activated RTKs and forms stable complexes with Grb2, it does not appear to facilitate MAPK activation [56] suggesting that the effects of p66shc mutation on mouse lifespan were not mediated via inhibition of the canonical Ras/MAPK signal transduction pathway. It should be noted that the role of p66Shc as a longevity protein has been recently disputed as in a separate study, no significant differences in lifespan was observed between p66Shc deficient animals and theircontrols [57].
Recently, more direct evidence of a role for Ras signaling in mammalian aging was described as mice deficient for the tissue specific Ras-GEF, RasGrf1, which is predominantly expressed within the pancreatic islets and in certain regions of the brain including the hippocampus and hypothalamus [58], were not only long-lived but also exhibited better motor coordination in older animals compared to their control littermates [16]. The longer lifespan of RasGrf1 deficient mice was shown to not simply be caused by a protection against cancer because tumor-free survival was also increased in these animals. Instead, old RasGrf1 mutant animals showed enhanced protection against oxidative stress, lower circulating IGF-1 levels and increased SIRT1 expression compared to controls, all of which may have contributed to their longer lifespan [16]. Furthermore, RasGrf1 lies downstream of both the insulin and IGF-1 receptors, supporting a role for reduced Insulin/IGF-1 signaling in the beneficial effects of RasGrf1 deficiency on lifespan [59]. Functional analysis has demonstrated that the RasGrf1 protein stimulates the dissociation of GDP from Ras [59]. However, RasGrf1 also shows affinity for other ligands in addition to Ras, including Rac, Rho, microtubules, PI[4,5]P2, and fasfatidic acid [60] and so it remains unclear whether RasGrf1-dependent longevity is a direct result of specific inhibition of Ras and its downstream signalingcascade.
In humans, mutations in HRAS are associated with Costello Syndrome, a rare multi-systemic disorder characterised by a failure to thrive, short stature, developmental delay or intellectual disability, soft skin and distinctive facial features [61]. As the disorder progresses with age, patients often show signs of premature aging including osteoporosis and osteopenia, potentially linking Ras activity with human aging [61]. Genome-wide association studies have also identified genetic variants of HRAS1 that are associated with longevity in humans [62]. Furthermore, genetic variants of HRAS1 and APOE have been shown to interact synergistically and are associated with both exceptional longevity and healthy aging [63]. In addition, chronological aging in humans as well as progeria are associated with activation of ERK signaling [64]. Thus, both HRAS and downstream ERK activity may play important roles in humanaging.
4Ras signaling interacts with other aging pathways
4.1Caloric restriction
Caloric or dietary restriction, defined as a reduction in nutrient intake without malnutrition, has been shown to extend lifespan in multiple species. In yeast, caloric restriction by glucose deprivation results in extension of chronological lifespan and is associated with down-regulation of the RAS-cAMP-PKA signaling pathway [65]. Moreover, the transcriptional activities of the RAS-cAMP-PKA responsive transcription factors, Msn2 and Msn4, were increased in response to caloric restriction and their deletion was sufficient to block the effects of caloric restriction on yeast lifespan [32]. Thus, at least part of the transcriptional response to caloric restriction in yeast is mediated via inhibition of Ras signaling.
Several studies in mammals have also linked caloric restriction to Ras and/or ERK activity. Cells isolated from the skin of calorically restricted rats had reduced HRAS expression and increased DNA methylation at the HRAS locus [66, 67]. Hyper-methylated promoters will often be recognised by transcriptional repressor complexes and so may lead to HRAS silencing which could contribute to the reduced cancer incidence in calorically restricted animals [66]. Calorically restricted rodents also show reduced levels of both activated Ras-GTP and activated ERK[66, 68] suggesting that signal transduction downstream of Ras is also impaired upon caloric restriction but whether these changes in Ras signal transduction contribute to lifespan extension remains to be determined.
4.2Insulin/IGF-1 signaling
Ras is a well-established signaling intermediary of the mammalian insulin/IGF-1-signaling (IIS) pathway [69], which plays an evolutionary conserved role in the modulation of animal lifespan [6, 7, 70]. A central factor in the ability of reduced IIS to extend lifespan is activation of the Forkhead boxO (FOXO) transcription factors via inhibition of the lipid kinase PI3K and its downstream target AKT [7]. The activated insulin receptor also recruits activated Ras via insulin receptor substrate (IRS) proteins that couple the receptor to the Ras-GEF, SOS, by binding to the Grb2 adaptor protein [69]. In Drosophila, selective mutation of the insulin receptor substrate, chico, that disrupts signaling via the Ras/MAPK pathway was found to extend lifespan [17]. Moreover, the Ras-responsive transcription factor, AOP, was required for lifespan extension by chico mutation [17]. Thus, in flies at least part of the longevity response to reduced IIS occurs via inhibition of Ras/MAPK signal transduction.
Extensive cross-talk occurs between Ras/MAPK signaling and PI3K/AKT signaling via both stimulatory and inhibitory mechanisms (Fig. 2). For example, activated Ras-GTP can bind directly to and allosterically activate the catalytic subunit of PI3K [71]. During growth, such an interaction is required for maximal PI3K activation [72]. Also, ERK phosphorylation of FOXO3A directs the transcription factor for degradation via the ubiquitin-proteasome system [73]. Meanwhile, AKT can negatively regulate ERK activation by phosphorylating inhibitory sites in Raf, sequestering it within the cytosol away from Ras and MEK [74]. Thus, the two signaling branches downstream of the insulin receptor are intricately connected.
Fig.2
Integration of Ras/MAPK signaling with other aging pathways. The Ras/MAPK signaling pathway is intricately connected to other cellular pathways that impact on aging. Ras signaling is activated downstream of the activated insulin receptor. Ras can directly bind to and allosterically activate PI3K. Activated ERK phosphorylates an inhibitory site on AMPK negatively regulating its activation. Phosphorylation of the TSC by ERK increases mTORC1 activity. Phosphorylation by ERK inhibits the TSC’s GAP function [76] thereby increasing mTORC1 activity. ERK also activates mTORC1 via phosphorylation of RAPTOR. ERK phosphorylation of the FOXO3A transcription factor leads to FOXO3A degradation via the ubiquitin proteasome system. Dietary restriction (DR) inhibits Ras-GTP and ERK activity, activates AMPK, inhibits mTOR and may inhibit insulin signaling via PI3K and AKT. Positive regulatory interactions are indicated by arrows. Negative regulatory interactions are shown as blunt-ended lines.
![Integration of Ras/MAPK signaling with other aging pathways. The Ras/MAPK signaling pathway is intricately connected to other cellular pathways that impact on aging. Ras signaling is activated downstream of the activated insulin receptor. Ras can directly bind to and allosterically activate PI3K. Activated ERK phosphorylates an inhibitory site on AMPK negatively regulating its activation. Phosphorylation of the TSC by ERK increases mTORC1 activity. Phosphorylation by ERK inhibits the TSC’s GAP function [76] thereby increasing mTORC1 activity. ERK also activates mTORC1 via phosphorylation of RAPTOR. ERK phosphorylation of the FOXO3A transcription factor leads to FOXO3A degradation via the ubiquitin proteasome system. Dietary restriction (DR) inhibits Ras-GTP and ERK activity, activates AMPK, inhibits mTOR and may inhibit insulin signaling via PI3K and AKT. Positive regulatory interactions are indicated by arrows. Negative regulatory interactions are shown as blunt-ended lines.](https://content.iospress.com:443/media/nha/2017/4-3/nha-4-3-nha160021/nha-4-nha160021-g002.jpg)
4.3mTOR
Both genetic and pharmacological inhibition of the mechanistic target of rapamycin (mTOR) kinase results in longevity in both invertebrates and mammals [10, 12, 75]. mTOR resides within two distinct cellular complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). Each complex contains a different repertoire of protein components and targets different downstream substrates for phosphorylation. Thus, mTORC1 responds to growth factors, energy status, amino acid levels, and cellular stress and is acutely inhibited by rapamycin. mTORC2 is activated in response to insulin/IGF-1 signaling via PI3K.
The mTORC1 complex comprises the mTOR kinase coupled with the RAPTOR scaffold protein, PRAS40 and the mLST8/GbL protein, which is involved in mTORC1 assembly and stability [76]. Activation of canonical Ras/MAPK signaling has been shown to increase mTORC1 activity. Activated ERK phosphorylates RAPTOR promoting mTORC1 phosphorylation of its downstream target, 4E-BP [77]. mTORC1 activation by the tuberous sclerosis complex (TSC) is also influenced by ERK activity. The TSC functions as a GAP for the small GTPase, RHEB, that binds to and activates mTORC1. Phosphorylation by ERK functions to inhibit the TSC’s GAP function [78] thereby increasing mTORC1 activity (Fig. 2).
4.4AMPK
The AMP-activated protein kinase (AMPK) is a major regulator of energy metabolism, stress resistance and proteostasis. Several studies have shown that AMPK signaling declines during aging suggesting that AMPK inhibition may contribute to the aging process [79]. Moreover, activation of AMPK is sufficient to extend lifespan across different species [80–82]. AMPK activity is regulated both positively and negatively by phosphorylation. Activated ERK can directly phosphorylate AMPK on Ser485, an inhibitory AMPK site [83] (Fig. 2) and stress-induced ERK activation can promote the translocation of AMPK from the cytoplasm to the nucleus [84]. In addition, the kinase suppressor of Ras (KSR), which acts as a scaffold for ERK at the cell membrane to co-localise ERK with its upstream kinases, also positively regulates AMPK activation [85].
5Ras, cellular senescence and aging
Cellular senescence describes the process whereby proliferating cells cease dividing and enter an irreversible state of growth arrest associated with several distinctive phenotypic changes. These include widespread changes in chromatin and gene expression, the secretion of pro-inflammatory chemokines and cytokines, growth factors, and proteases, a characteristic feature of senescent cells called the senescence-associated secretory phenotype (SASP), and the activation of tumor suppression pathways [86]. Several factors act as triggers to induce cells to senesce including telomere shortening, DNA damage and activation of oncogenic pathways including activation of Ras and chronic stimulation of MAPK signal transduction [87]. Together, cellular senescence and the SASP are thought to provide an effective safeguard against tumorigenesis by preventing the proliferation of damaged and potentially cancerous cells.
Cellular senescence has long been linked to aging and the development of age-related pathologies. In mammals, including primates, senescent cells accumulate in mitotically active tissues in association with both increased age [88] and age-associated diseases such as osteoarthritis and Alzheimer’s Disease [89]. Several mechanisms for how senescent cells may promote age-related pathology have been proposed. The accumulation of senescent cells may negatively impact on the regenerative potential of aging tissues by depleting the number of actively dividing cells. Alternatively, senescent cells may contribute to the disruption of the aging stem cell niche via the SASP [86]. Importantly, the clearance of senescent cells within a progeroid murine model was able to delay the progression of age-related pathologies but did not reverse their effects [90].
Oncogenic mutations in Ras that result in its hyperactivation can trigger senescence [91]. Similarly, chronic activation of ERK/MAPK signaling, which has been observed in aging tissues [64, 92], can also induce senescence. Inhibition of the Ras/MAPK signal transduction pathway may therefore promote longevity by preventing cellular senescence. In support of this, down-regulation of RasGrf1 in VSELs (very small embryonic-like stem cells) prevents senescence and age-related depletion of these cells in adult tissues which may contribute to the longevity of RasGrf1 deficient mice [58].
6Ras signaling and metabolic regulation
Identification of the oncogenic Ras signaling pathway as a conserved modulator of animal aging joins an emerging theme that manipulating the activity of cancer-promoting pathways, either by increasing the activity of tumor suppressor proteins or by inhibition of oncogenes, can promote longevity. Other examples include increasing the activity of the tumor suppressor PTEN or inhibition of PI3K, a direct target for PTEN activation, increasing expression of the tumor suppressor proteins encoded in the Ink4/Arf locus, or inhibition of the Myc oncogene, all of which lead to longevity in murine models [93–96]. Interestingly, lifespan extension in these models could not simply be attributed to their protective effects against cancer as cancer-free individuals were still longer-lived. Similarly, mice deficient for RasGrf1 showed decreased cancer incidence but lifespan extension was also observed in cancer-free animals [16]. Also, pharmacological inhibition of MEK in Drosophila did not prevent hyperplasia of the adult intestinal epithelium [17], a cancer-like pathology caused by over-proliferation of the intestinal stem cells. Ras signaling may therefore, at least to some extent, influence lifespan independently of its well-established oncogenic functions.
One intriguing potential mechanism by which inhibition of Ras signal transduction could promote longevity is through changes in metabolic regulation, particularly fat metabolism. Aging in several animal models is often accompanied by metabolic dysfunction, especially aberrant fat metabolism. In humans, increased fat deposition, particularly in visceral fat stores, commonly leads to metabolic disease including type 2 diabetes and cardiovascular disease. Surgical removal of visceral fat in rodents extends lifespan and improves several metabolic outcomes with age including insulin sensitivity [97]. Similarly, long-lived dietary restricted mouse models and mutants with impaired IIS or mTOR signaling are often lean and show improved age-related metabolic profiles [98]. However, other long-lived mouse mutants, such as the growth hormone deficient mouse, show increased fat mass [99] suggesting that total lipid levels themselves may not be a key determinant of aging. Instead, age-related deterioration in tissue function may result from lipotoxicity, the ectopic deposition of fat that occurs when the adipocyte fat storage capacity is exceeded or impaired.
Multiple studies have linked activation of ERK to aberrant fat metabolism or accumulation. Mice lacking ERK1 have fewer adipocytes and decreased adiposity compared to their controls [100]. Conversely, in several models of obesity, including high-fat feeding or genetic interventions such as in ob/ob mice, activated ERK levels were elevated in metabolically active tissues such as the liver and white adipose tissue [101–103]. Reducing the levels of activated ERK can also provide beneficial outcomes in obesitymodels. Thus, ERK1 deficient mice were more resistant to obesity when challenged with a high-fat diet [100]. Similarly, mice exposed to a small molecule inhibitor of MEK not only showed lowered levels of activated ERK but improved metabolic profiles in response to high-fat feeding [102]. Interestingly, these interactions between ERK activity and metabolism appear to be conserved. In Drosophila, activities of the Ras-responsive ETS transcription factors, Pnt and AOP, are also associated with metabolic functions [44].
7Conclusions and future perspectives
The assignment of new roles for Ras signaling in animal aging and metabolic regulation offers substantial opportunity for new therapies to target aging and age-related disease. An effective strategy for developing new treatments for aging is the repurposing of existing drugs that have already been approved for human use. Such drugs have known mechanisms of action and have been well screened for both safety and toxicity effects. The Ras signaling pathway has been intensively studied in the context of cancer. Thus, Ras and its downstream signaling effectors have been the focus of extensive screening to identify compounds that inhibit the pathway for use as cancer treatments. Such strategies have already isolated several small molecule inhibitors of the Ras/MAPK pathway that have entered clinical trials or are already in clinical use. Interestingly, one of these small molecule inhibitors, trametinib, which targets the MEK kinase with high specificity, extends lifespan in Drosophila [17] and offers protection against deleterious metabolic effects of both diet- and genetically-induced obesity in mice [102]. Moreover, the therapeutic concentration in which trametinib elicited these metabolic effects was much lower than that required to inhibit tumor growth in xenograft models.
A direct role for Ras/MAPK signaling in mammalian aging has yet to be demonstrated. However, the high degree of evolutionary conservation within this signal transduction cascade suggests that the function of reduced Ras/MAPK signaling as a longevity assurance mechanism is also likely to be conserved. Determining the extent to which the metabolic effects of Ras/MAPK inhibition are relevant for aging and their contribution to longevity will be a major challenge for the future. Nevertheless, such studies could identify important new targets for interventions to diminish the detrimental effects of aging, moving towards direct clinical applications to improve lifelong health.
Acknowledgments
The author would like to thank Nazif Alic for critical reading of the manuscript. The author is currently supported by an internal start-up award given by the School of Life and Health Sciences, AstonUniversity.
References
[1] | Blokh D , Stambler I . Information theoretical analysis of aging as a risk factor for heart disease. Aging and Disease. (2015) ;6: (3):196–207. |
[2] | Niccoli T , Partridge L . Ageing as a risk factor for disease. Current Biology. (2012) ;22: (17):R741–52. |
[3] | Pike CJ . Sex and the development of Alzheimer’s disease. Journal of Neuroscience Research. (2017) ;95: (1-2):671–80. |
[4] | van der Ende MY , Hartman MH , Hagemeijer Y , Meems LM , de Vries HS , Stolk RP , et al. The lifelines cohort study: Prevalence and treatment of cardiovascular disease and risk factors. International Journal of Cardiology. (2016) ;228: :495–500. |
[5] | White MC , Holman DM , Boehm JE , Peipins LA , Grossman M , Henley SJ . Age and cancer risk: A potentially modifiable relationshiAmerican Journal of Preventive Medicine. S. (2014) ;46: :7–15. |
[6] | Lopez-Otin C , Blasco MA , Partridge L , Serrano M , Kroemer G . The hallmarks of aging. Cell. (2013) ;153: (6):1194–217. |
[7] | Fontana L , Partridge L , Longo VD . Extending healthy life span–from yeast to humans. Science. (2010) ;328: (5976):321–6. |
[8] | Partridge L , Alic N , Bjedov I , Piper MD . Ageing in Drosophila: The role of the insulin/Igf and TOR signaling network. Exp Gerontol. (2011) ;46: (5):376–81. |
[9] | Santos J , Leitao-Correia F , Sousa MJ , Leao C . Dietary restriction and nutrient balance in aging. Oxidative Medicine and Cellular Longevity. (2016) ;2016: :10. |
[10] | Bjedov I , Toivonen JM , Kerr F , Slack C , Jacobson J , Foley A , et al. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster. Cell Metabolism. (2010) ;11: (1):35–46. |
[11] | Flynn JM , O’Leary MN , Zambataro CA , Academia EC , Presley MP , Garrett BJ , et al. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell. (2013) ;12: (5):851–62. |
[12] | Harrison DE , Strong R , Sharp ZD , Nelson JF , Astle CM , Flurkey K , et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. (2009) ;460: (7253):392–5. |
[13] | Majumder S , Caccamo A , Medina DX , Benavides AD , Javors MA , Kraig E , et al. Lifelong rapamycin administration ameliorates age-dependent cognitive deficits by reducing IL-1beta and enhancing NMDA signaling. Aging Cell. (2012) ;11: (2):326–35. |
[14] | Wilkinson JE , Burmeister L , Brooks SV , Chan CC , Friedline S , Harrison DE , et al. Rapamycin slows aging in mice. Aging Cell. (2012) ;11: (4):675–82. |
[15] | Mannick JB , Del Giudice G , Lattanzi M , Valiante NM , Praestgaard J , Huang B , et al. mTOR inhibition improves immune function in the elderly. Sci Transl Med. (2014) ;6: (268):268ra179. |
[16] | Borras C , Monleon D , Lopez-Grueso R , Gambini J , Orlando L , Pallardo FV , et al. RasGrf1 deficiency delays aging in mice. Aging. (2011) ;3: (3):262–76. |
[17] | Slack C , Alic N , Foley A , Cabecinha M , Hoddinott MP , Partridge L . The Ras-ERK-ETS-signaling pathway is a drug target for longevity. Cell. (2015) ;162: (1):72–83. |
[18] | McKay MM , Morrison DK . Integrating signals from RTKs to ERK/MAPK. Oncogene. (2007) ;26: (22):3113–21. |
[19] | Schlessinger J . Cell signaling by receptor tyrosine kinases. Cell. (2000) ;103: (2):211–25. |
[20] | Goitre L , Trapani E , Trabalzini L , Retta SF . The Ras superfamily of small GTPases: The unlocked secrets. Methods in Molecular Biology. (2014) ;1120: :1–18. |
[21] | Rajalingam K , Schreck R , Rapp UR , Albert S . Ras oncogenes and their downstream targets. Biochimica et Biphysica Acta. (2007) ;1773: (8):1177–95. |
[22] | Wittinghofer A , Nassar N . How Ras-related proteins talk to their effectors. Trends in Biochemical Sciences. (1996) ;21: (12):488–91. |
[23] | Barbacid M . ras genes. Annual Review of Biochemistry. (1987) ;56: :779–827. |
[24] | Mendoza MC , Er EE , Blenis J . The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends in Biochemical Sciences. (2011) ;36: (6):320–8. |
[25] | McCubrey JA , Steelman LS , Chappell WH , Abrams SL , Wong EWT , Chang F , et al. Roles of the RAF/MEK/ERK pathway in cell growth, malignant transformation and drug resistance. Biochimica et Biophysica Acta. (2007) ;1773: (8):1263–84. |
[26] | Jazwinski SM . The RAS genes: A homeostatic device in Saccharomyces cerevisiae longevity. Neurobiology of Aging. (1999) ;20: (5):471–8. |
[27] | Fabrizio P , Liou LL , Moy VN , Diaspro A , Valentine JS , Gralla EB , et al. SOD2 functions downstream of Sch9 to extend longevity in yeast. Genetics. (2003) ;163: (1):35–46. |
[28] | Sun J , Kale SP , Childress AM , Pinswasdi C , Jazwinski SM . Divergent roles of RAS1 and RAS2 in yeast longevity. The Journal of Biological Chemistry. (1994) ;269: (28):18638–45. |
[29] | Fabrizio P , Pozza F , Pletcher SD , Gendron CM , Longo VD . Regulation of longevity and stress resistance by Sch9 in yeast. Science. (2001) ;292: (5515):288–90. |
[30] | Fabrizio P , Pletcher SD , Minois N , Vaupel JW , Longo VD . Chronological aging-independent replicative life span regulation by Msn2/Msn4 and Sod2 in Saccharomyces cerevisiae. FEBS Lett. (2004) ;557: (1-3):136–42. |
[31] | Lin SJ , Defossez PA , Guarente L . Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science. (2000) ;289: (5487):2126–8. |
[32] | Medvedik O , Lamming DW , Kim KD , Sinclair DA . MSN2 and MSN4 link calorie restriction and TOR to sirtuin-mediated lifespan extension in Saccharomyces cerevisiae. PLoS Biology. (2007) ;5: (10):e261. |
[33] | Delaney JR , Murakami C , Chou A , Carr D , Schleit J , Sutphin GL , et al. Dietary restriction and mitochondrial function link replicative and chronological aging in Saccharomyces cerevisiae. Exp Gerontol. (2013) ;48: (10):1006–13. |
[34] | Altschuler DL , Muro A , Schijman A , Almonacid FB , Torres HN . Neurospora crassa cDNA clones coding for a new member of the ras protein family. FEBS Lett. (1990) ;273: (1-2):103–6. |
[35] | Kana-uchi A , Yamashiro CT , Tanabe S , Murayama T . A ras homologue of Neurospora crassa regulates morphology. Molecular & General Genetics. (1997) ;254: (4):427–32. |
[36] | Borkovich KA , Alex LA , Yarden O , Freitag M , Turner GE , Read ND , et al. Lessons from the genome sequence of Neurospora crassa: Tracing the path from genomic blueprint to multicellular organism. Microbiology and Molecular Biology Reviews 68: (1):1–108. |
[37] | Belden WJ , Larrondo LF , Froehlich AC , Shi M , Chen CH , Loros JJ , et al. The band mutation in Neurospora crassa is a dominant allele of ras-1 implicating RAS signaling in circadian output. Genes Dev. (2007) ;21: (12):1494–505. |
[38] | Milburn MV , Tong L , deVos AM , Brunger A , Yamaizumi Z , Nishimura S , et al. Molecular switch for signal transduction: Structural differences between active and inactive forms of protooncogenic Ras proteins. Science. (1990) ;247: (4945):939–45. |
[39] | Segal M , Marbach I , Willumsen BM , Levitzki A . Two distinct regions of Ras participate in functional interaction with GDP-GTP exchangers. European Journal of Biochemistry. (1995) ;228: (1):96–101. |
[40] | Case ME , Griffith J , Dong W , Tigner IL , Gaines K , Jiang JC , et al. The aging biological clock in Neurospora crassa. Ecology and evolution. (2014) ;4: (17):3494–507. |
[41] | Brunner D , Ducker K , Oellers N , Hafen E , Scholz H , Klambt C . The ETS domain protein pointed-P2 is a target of MAP kinase in the sevenless signal transduction pathway. Nature. (1994) ;370: (6488):386–9. |
[42] | Halfon MS , Carmena A , Gisselbrecht S , Sackerson CM , Jimenez F , Baylies MK , et al. Ras pathway specificity is determined by the integration of multiple signal-activated and tissue-restricted transcription factors. Cell. (2000) ;103: (1):63–74. |
[43] | O’Neill EM , Rebay I , Tjian R , Rubin GM . The activities of two Ets-related transcription factors required for Drosophila eye development are modulated by the Ras/MAPK pathway. Cell. (1994) ;78: (1):137–47. |
[44] | Alic N , Giannakou ME , Papatheodorou I , Hoddinott MP , Andrews TD , Bolukbasi E , et al. Interplay of dFOXO and two ETS-family transcription factors determines lifespan in Drosophila melanogaster. PLoS Genetics. (2014) ;10: (9):e1004619. |
[45] | Sternberg PW , Han M . Genetics of RAS signaling in C. elegans. Trends Genet. (1998) ;14: (11):466–72. |
[46] | Tan PB , Lackner MR , Kim SK . MAP kinase signaling specificity mediated by the LIN-1 Ets/LIN-31 WH transcription factor complex during C. elegans vulval induction. Cell. (1998) ;93: (4):569–80. |
[47] | Nanji M , Hopper NA , Gems D . LET-60 RAS modulates effects of insulin/IGF-1 signaling on development and aging in Caenorhabditis elegans. Aging Cell. (2005) ;4: (5):235–45. |
[48] | Thyagarajan B , Blaszczak AG , Chandler KJ , Watts JL , Johnson WE , Graves BJ . ETS-4 is a transcriptional regulator of life span in Caenorhabditis elegans. PLoS Genetics. (2010) ;6: (9):e1001125. |
[49] | Umanoff H , Edelmann W , Pellicer A , Kucherlapati R . The murine N-RAS gene is not essential for growth and development. Proceedings of the National Academy of Sciences. (1995) ;92: (5):1709–13. |
[50] | Ise K , Nakamura K , Nakao K , Shimizu S , Harada H , Ichise T , et al. Targeted deletion of the H-ras gene decreases tumor formation in mouse skin carcinogenesis. Oncogene. (2000) ;19: (26):2951–6. |
[51] | Plowman SJ , Williamson DJ , O’Sullivan MJ , Doig J , Ritchie AM , Harrison DJ , et al. While K-ras is essential for mouse development, expression of the K-ras 4A splice variant is dispensable. Molecular and Cellular Biology. (2003) ;23: (24):9245–50. |
[52] | Plowman SJ , Arends MJ , Brownstein DG , Luo F , Devenney PS , Rose L , et al. The K-Ras 4A isoform promotes apoptosis but does not affect either lifespan or spontaneous tumor incidence in aging mice. Experimental Cell Research. (2006) ;312: (1):16–26. |
[53] | Elbourkadi N , Austad SN , Miller RA . Fibroblasts from long-lived species of mammals and birds show delayed, but prolonged, phosphorylation of ERK. Aging Cell. (2014) ;13: (2):283–91. |
[54] | Sun LY , Steinbaugh MJ , Masternak MM , Bartke A , Miller RA . Fibroblasts from long-lived mutant mice show diminished ERK1/2 phosphorylation but exaggerated induction of immediate early genes. Free Radical Biology & Medicine. (2009) ;47: (12):1753–61. |
[55] | Migliaccio E , Giorgio M , Mele S , Pelicci G , Reboldi P , Pandolfi PP , et al. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. (1999) ;402: (6759):309–13. |
[56] | Migliaccio E , Mele S , Salcini AE , Pelicci G , Lai KM , Superti-Furga G , et al. Opposite effects of the p52shc/p46shc and p66shc splicing isoforms on the EGF receptor-MAP kinase-fos signaling pathway. The EMBO Journal. (1997) ;16: (4):706–16. |
[57] | Ramsey JJ , Tran D , Giorgio M , Griffey SM , Koehne A , Laing ST , et al. The influence of Shc proteins on life span in mice. The Journals of Gerontology Series A, Biological Sciences and Medical Sciences. (2014) ;69: (10):1177–85. |
[58] | Ratajczak MZ , Kucia M , Liu R , Shin DM , Bryndza E , Masternak MM , et al. RasGrf Genomic imprinting, VSELs, and aging. Aging. (2011) ;3: (7):692–7. |
[59] | Fernandez-Medarde A , Santos E . The RasGrf family of mammalian guanine nucleotide exchange factors. Biochimica et Biophysica Acta. (2011) ;1815: (2):170–88. |
[60] | Mirisola MG , Longo VD . Conserved role of Ras-GEFsin promoting aging: From yeast to mice. Aging. (2011) ;3: (4):340–3. |
[61] | Rauen KA . HRAS and the Costello syndrome. Clinical Genetics. (2007) ;71: (2):101–8. |
[62] | Bonafe M , Barbi C , Olivieri F , Yashin A , Andreev KF , Vaupel JW , et al. An allele of HRAS1 3’variable number of tandem repeats is a frailty allele: Implication for an evolutionarily-conserved pathway involved in longevity. Gene. (2002) ;286: (1):121–6. |
[63] | Jazwinski SM , Kim S , Dai J , Li L , Bi X , Jiang JC , et al. HRAS1 and LASS1 with APOE are associated with human longevity and healthy aging. Aging Cell. (2010) ;9: (5):698–708. |
[64] | Aliper AM, Csoka AB, Buzdin A, Jetka T, Roumiantsev S, Moskalev A and Zhavoronkov A. Signaling pathway activation drift during aging: Hutchinson-Gilford Progeria Syndrome fibroblasts are comparable to normal middle-age and old-age cells. Aging. (2015) ;7: (1):26–37. |
[65] | Wei M , Fabrizio P , Hu J , Ge H , Cheng C , Li L , et al. Life span extension by calorie restriction depends on rim15 and transcription factors downstream of Ras/PKA, tor, and Sch9. PLoS Genetics. (2008) ;4: (1):e13. |
[66] | Xie L , Jiang Y , Ouyang P , Chen J , Doan H , Herndon B , et al. Effects of dietary calorie restriction or exercise on the PI3K and Ras signaling pathways in the skin of mice. The Journal of Biological Chemistry. (2007) ;282: (38):28025–35. |
[67] | Li Y , Daniel M , Tollefsbol TO . Epigenetic regulation of caloric restriction in aging. BMC Medicine. (2011) ;9: (1):98. |
[68] | Liu Y , Duysen E , Yaktine AL , Au A , Wang W , Birt DF . Dietary energy restriction inhibits ERK but not JNK or p38 activity in the epidermis of SENCAR mice. Carcinogenesis. (2001) ;22: (4):607–12. |
[69] | White MF . The insulin signaling system and the IRS proteins. Diabetologia. (1997) ;40: (2):S2–S17. |
[70] | Kenyon C . The first long-lived mutants: Discovery of the insulin/IGF-1 pathway for ageing. Philosophical Transactions of the Royal Society of London Series B, Biological Sciences. (2011) ;366: (1561):9–16. |
[71] | Castellano E , Downward J . RAS interaction with PI3K: More than just another effector pathway. Genes & Cancer. (2011) ;2: (3):261–74. |
[72] | Orme MH , Alrubaie S , Bradley GL , Walker CD , Leevers SJ . Input from Ras is required for maximal PI(3)K signaling in Drosophila. Nature Cell Biology. (2006) ;8: (11):1298–302. |
[73] | Yang J-Y , Zong CS , Xia W , Yamaguchi H , Ding Q , Xie X , et al. ERK promotes tumorigenesis by inhibiting FOXO3a via MDM2-mediated degradation. Nature Cell Biology. (2008) ;10: (2):138–48. |
[74] | Zimmermann S , Moelling K . Phosphorylation and regulation of Raf by Akt (protein kinase B). Science. (1999) ;286: (5445):1741–4. |
[75] | Miller RA , Harrison DE , Astle CM , Baur JA , Boyd AR , de Cabo R , et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. The Journals of Gerontology Series A, Biological Sciences and Medical Sciences. (2011) ;66: (2):191–201. |
[76] | Kennedy BK , Lamming DW . The mechanistic target of rapamycin: The grand conducTOR of metabolism and aging. Cell metabolism. (2016) ;23: (6):990–1003 . |
[77] | Carriere A , Romeo Y , Acosta-Jaquez HA , Moreau J , Bonneil E , Thibault P , et al. ERK1/2 phosphorylate raptor to promote Ras-dependent activation of mTOR complex 1 (mTORC1). Journal of Biological Chemistry. (2011) ;286: (1):567–77. |
[78] | Ma L , Chen Z , Erdjument-Bromage H , Tempst P , Pandolfi PP . Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell. (2005) ;121: (2):179–93. |
[79] | Salminen A , Kaarniranta K . AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Research Reviews. (2012) ;11: (2):230–41. |
[80] | Apfeld J , O’Connor G , McDonagh T , DiStefano PS , Curtis R . The AMP-activated protein kinase AAK-2 links energy levels and insulin-like signals to lifespan in C. elegans. Genes & Development. (2004) ;18: (24):3004–9. |
[81] | Curtis R , O’Connor G , DiStefano PS . Aging networks in Caenorhabditis elegans: AMP-activated protein kinase (aak-2) links multiple aging and metabolism pathways. Aging Cell. (2006) ;5: (2):119–26. |
[82] | Funakoshi M , Tsuda M , Muramatsu K , Hatsuda H , Morishita S , Aigaki T . A gain-of-function screen identifies wdb and lkb1 as lifespan-extending genes in Drosophila. Biochemical and Biophysical Research Communications. (2011) ;405: (4):667–72. |
[83] | Lopez-Cotarelo P , Escribano-Diaz C , Gonzalez-Bethencourt IL , Gomez-Moreira C , Deguiz ML , Torres-Bacete J , et al. A novel MEK-ERK-AMPK signaling axis controls chemokine receptor CCR7-dependent survival in human mature dendritic cells. The Journal of Biological Chemistry. (2015) ;290: (2):827–40. |
[84] | Kodiha M , Rassi JG , Brown CM , Stochaj U . Localization of AMP kinase is regulated by stress, cell density, and signaling through the MEK-ERK1/2 pathway. American Journal of Physiology –Cell Physiology. (2007) ;293: (5):C1427–36. |
[85] | Costanzo-Garvey DL , Pfluger PT , Dougherty MK , Stock JL , Boehm M , Chaika O , et al. KSR2 Is an Essential Regulator of AMP Kinase, Energy Expenditure, and Insulin Sensitivity. Cell Metabolism. (2009) ;10: (5):366–78. |
[86] | van Deursen JM . The role of senescent cells in ageing. Nature. (2014) ;509: (7501):439–46. |
[87] | Campisi J . Aging, cellular senescence, and cancer. Annu Rev Physiol. (2013) ;75: :685–705. |
[88] | Jeyapalan JC , Sedivy JM . Cellular senescence and organismal aging. Mech Ageing Dev. (2008) ;129: (7-8):467–74. |
[89] | Naylor RM , Baker DJ , van Deursen JM . Senescent cells: A novel therapeutic target for aging and age-related diseases. Clin Pharmacol Ther. (2013) ;93: (1):105–16. |
[90] | Baker DJ , Wijshake T , Tchkonia T , LeBrasseur NK , Childs BG , van de Sluis B , et al. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature. (2011) ;479: (7372):232–6. |
[91] | Dimauro T , David G . Ras-induced senescence and its physiological relevance in cancer. Curr Cancer Drug Targets. (2010) ;10: (8):869–76. |
[92] | Williamson D , Gallagher P , Harber M , Hollon C , Trappe S . Mitogen-activated protein kinase (MAPK) pathway activation: Effects of age and acute exercise on human skeletal muscle. J Physiol. (2003) ;547: (Pt 3):977–87. |
[93] | Foukas LC , Bilanges B , Bettedi L , Pearce W , Ali K , Sancho S , et al. Long-term p110alpha PI3K inactivation exerts a beneficial effect on metabolism. EMBO Mol Med. (2013) ;5: (4):563–71. |
[94] | Ortega-Molina A , Efeyan A , Lopez-Guadamillas E , Munoz-Martin M , Gomez-Lopez G , Canamero M , et al. Pten positively regulates brown adipose function, energy expenditure, and longevity. Cell metabolism. (2012) ;15: (3):382–94. |
[95] | Matheu A , Maraver A , Collado M , Garcia-Cao I , Canamero M , Borras C , et al. Anti-aging activity of the Ink4/Arf locus. Aging Cell. (2009) ;8: (2):152–61. |
[96] | Hofmann JW , Zhao X , De Cecco M , Peterson AL , Pagliaroli L , Manivannan J , et al. Reduced expression of MYC increases longevity and enhances healthspan. Cell. (2015) ;160: (3):477–88. |
[97] | Muzumdar R , Allison DB , Huffman DM , Ma X , Atzmon G , Einstein FH , et al. Visceral adipose tissue modulates mammalian longevity. Aging Cell. (2008) ;7: (3):438–40. |
[98] | Selman C , Withers DJ . Mammalian models of extended healthy lifespan. Philosophical Transactions of the Royal Society B: Biological Sciences. (2011) ;366: (1561):99–107. |
[99] | Liang H , Masoro EJ , Nelson JF , Strong R , McMahan CA , Richardson A . Genetic mouse models of extended lifespan. Exp Gerontol. (2003) ;38: (11-12):1353–64. |
[100] | Bost F , Aouadi M , Caron L , Even P , Belmonte N , Prot M , et al. The extracellular signal-regulated kinase isoform ERK1 is specifically required for in vitro and in vivo adipogenesis. Diabetes. (2005) ;54: (2):402–11. |
[101] | Jiao P , Feng B , Li Y , He Q , Xu H . Hepatic ERK activity plays a role in energy metabolism. Molecular and Cellular Endocrinology. (2013) ;375: (1-2):157–66. |
[102] | Banks AS , McAllister FE , Camporez JP , Zushin PJ , Jurczak MJ , Laznik-Bogoslavski D , et al. An ERK/Cdk5 axis controls the diabetogenic actions of PPARgamma. Nature. (2015) ;517: (7534):391–5. |
[103] | Bi L , Chiang JY , Ding WX , Dunn W , Roberts B , Li T . Saturated fatty acids activate ERK signaling to downregulate hepatic sortilin 1 in obese and diabetic mice. Journal of Lipid Research. (2013) ;54: (10):2754–62. |


