
DFT, molecular docking and ADME prediction of tenofovir drug as a promising therapeutic inhibitor of SARS-CoV-2 Mpro
Abstract
In the present work, at first, DFT calculations were carried out to study the molecular structure of the tenofovir at B3LYP/MidiX level of theory and in the water as solvent. The HOMO/LUMO molecular orbitals, excitation energies and oscillator strengths of investigated drug were also calculated and presented. NBO analysis was performed to illustrate the intramolecular rehybridization and electron density delocalization. In the following, a molecular docking study was performed for screening of effective available tenofovir drug which may act as an efficient inhibitor for the SARS-CoV-2 Mpro. The binding energy value showed a good binding affinity between the tenofovir and SARS-CoV-2 Mpro with binding energy of-47.206 kcal/mol. Therefore, tenofovir can be used for possible application against the SARS-CoV-2 Mpro.
1Introduction
SARS-CoV-2 is a positive-sense single-stranded RNA virus closely related to SARS-CoV and Middle East respiratory syndrome coronavirus (MERS-CoV) that belong to the genus betacoronavirus in the Coronaviridae family [1]. Because of the emergence of severe acute respiratory syndrome coronavirus (SARS-CoV-2), a novel Coronavirus (2019-nCoV) has been developed [2–11], which causes more than 6 million deaths in the worldwide [12]. There is no clinical treatment for SARS-CoV-2. However, many efforts are being made to discover the treatment in preclinical and clinical trials. Protease inhibitors have been investigated for covid treatment [13–17]. Furthermore, ivermectin [18], nitazoxanide [19] and nafamostat [20] have remarkable activity in vitro.
(R)-(((1-(6-amino-9H-purin-9-yl)propan-2-yl)oxy)methyl)phosphonic acid known as tenofovir (Fig. 1) is used in the treatment of the HIV and the hepatitis B diseases in combination with other drugs [21–24]. Tenofovir is accompanied with a very low rate of antiviral resistance and low hepatotoxicity [25–28]. There are no persuasive reports about the attribution of liver injury of tenofovir. However, the tenofovir combination with didanosine leads to liver injury along with microvesicular fatty liver disease and lactic acidosis more commonly than didanosine with other antiretrovirals, which is due to drug-drug interactions [29, 30]. Liu et al. suggested many drugs as the candidates against 2019-nCoV [31]. According to their results, these drugs could form hydrogen bonds with Thr24, Asn28 and Asn119 residues, which are capable to bind to the pocket formed by these amino acids and causing major disruption of the function of 2019-nCoV.
Fig. 1
Chemical structure of the tenofovir.
The molecular docking is an important tool, which is used for modeling of the interaction between molecules and proteins [32–35]. Kumar et al. studied in silico based drug repurposing method using molecular docking studies on the spectrum of Food and Drug Administration (FDA)-approved antiviral drugs against SARS-CoV-2 [36]. Based on their results, three drugs called lopinavir-ritonavir, raltegravir, and tipranavir have shown the strongest binding to SARS-CoV-2 and MD simulation study confirmed the stability and conformational flexibility of these drugs in the enzyme active site. In another study, Elfiky [37] reported that ribavirin, remdesivir, sofosbuvir, galidesivir and tenofovir drugs can be used as potential drugs against SARS-CoV-2, which is related to their potential binding to its RNA-dependent RNA polymerase (RdRp). Peele et al. evaluated the efficacy of anti-viral drugs and plant-based antiviral drugs on the COVID-19 main protease viral protein of SARS-COV-2 using molecular docking and molecular dynamics simulations [38]. It was found that lopinavir, amodiaquine, and theaflavin digallate (TFDG) drugs have the best docking scores. Yu et al. studied the binding of chloroquine, remdesivir, ribavirin and luteolin drugs to the 2019-nCoV main proteins (3CLpro, PLpro, RdRp, and S) using computational methods [39]. According to their results, luteolin is the most potential antiviral drug.
In the present work the molecular structure of the tenofovir is studied using DFT method (B3LYP/MidiX) in water. Then, the interaction of tenofovir molecule with the SARS-CoV-2 Mpro is performed using molecular docking studies. The purpose of quantum chemical calculations is to prove that a strong complex is formed between the tenofovir molecule and Coronavirus, which leads to the destruction of old hydrogen bonds in the virus molecule and the formation of new ones. This leads to disrupt the viral protein information system.
2Computational methods
The 3D structure of the molecule was visualized using docking programs and various computational methods were developed to analyze the docking score [38]. Docking software calculates and predicts the interaction between the small molecules and the target proteins based on the binding energy, the number of Hydrogen and Pi bonds formed. The quantum chemical calculations have carried out for the tenfovir using DFT method (B3LYP/MidiX) and TD-DFT method with the Gaussian 09W program package [40]. The solvent effect has been incorporated using the Polarized Continuum Model (PCM) [41]. This method and basis set is used for calculation of organic compound containing benzene rings and P and N atoms [42]. The frontier molecular orbital (FMO) [43], molecular electrostatic potential (MEP) [44], natural bond orbital (NBO) [45] and charge analyses [46] were also studied. The optimized molecular structures, pictures of highest occupied molecular orbital (HOMO), lowest unoccupied orbital (LUMO) orbitals and MEP maps were visualized using GaussView 05 program [47].
Interaction between SARS-CoV-2 Mpro and tenfovir was investigated by HyperChem Professional 8.0.6 [48] (for windows, molecular modeling system, serial number 12-800-1501799999, 1995–2008 Hypercube, Inc. http://www.hyper.com/), PyMOL 2.3 (GL-, VERSION: 4.5.0 - Build 23.20.16.4973, License Expiry date: 01-jul-2020 https://pymol.org/2/#products) and Molegro Molecular Viewer 2.5 (MMW 2012.2.5.-2012-10-10- [WIN32], https://molegro-molecular-viewer.software.informer.com/2.5/) software programs. The protein sequences of 2019-nCoV were downloaded from GenBank (http://www.ncbi.nlm.nih.gov). Crystal structure of SARS-CoV-2 Mpro (PDB ID: 1UJ1) [49] was downloaded from Protein Data Bank (PDB, http://www.rcsb.org). As a preliminary step of docking, water molecules and the amino acid that does not belong to the protein should be removed by deleting the lines that start with “HETATM” and “CONNECT”. Then, the file structure was saved and it was ready for molecular docking analysis. The protein molecule was initialized by addition of hydrogen atoms and kolmen charges using the edit option and save the protein molecule as a written PDB. The following parameters were used for the calculations by HyperChem Professional 8.0.6: Algorithm: Steepest Descent, an RMS gradient of 0.1 kcal/Mol, maximum cycles: 32767.
The pharmacokinetic properties of tenofovir were calculated using Molinspiration software as well as validation of the Lipinski rule of 5 (RO5). Based on the rule, an orally acceptable drug-like molecule must have: 1. HBD < 5; 2. HBA < 10; 3. MW < 500 Dalton; 4. LogP < 5; 5. ROTB < 10 (added by Veber) [15]. However this rule is not able to predict whether a drug is active pharmacologically, it can use to get a deep insight about the pharmacokinetic properties such as Absorption, Distribution, Metabolism, and Excretion (ADME) of the investigated drug.
3Results and discussion
3.1Electronic structure of the tenofovir
The structure of the tenofovir was optimized by B3LYP/MidiX level of theory in the water as solvent (Fig. 2). The optimized geometrical parameters of tenofovir molecule are listed in Table 1, in accordance with the atom numbering scheme shown in Fig. 2. Table 1 indicates the calculated bond lengths of tenofovir molecule compared to experimentally available data [50]. It is found that in most cases, the calculated bond lengths are slightly greater than the available experimental values. The C = C and C-C bond lengths in the benzene ring are found to be 1.400 Å and 1.381 Å, respectively. This confirms C = C double-bond nature. Generally, the calculated geometrical parameters are in relatively good agreement with the experimental data, which provide the basis for FMO, MEP, NBO and molecular docking analyses.
Fig. 2
Optimized structure of the tenofovir by B3LYP/MidiX method.
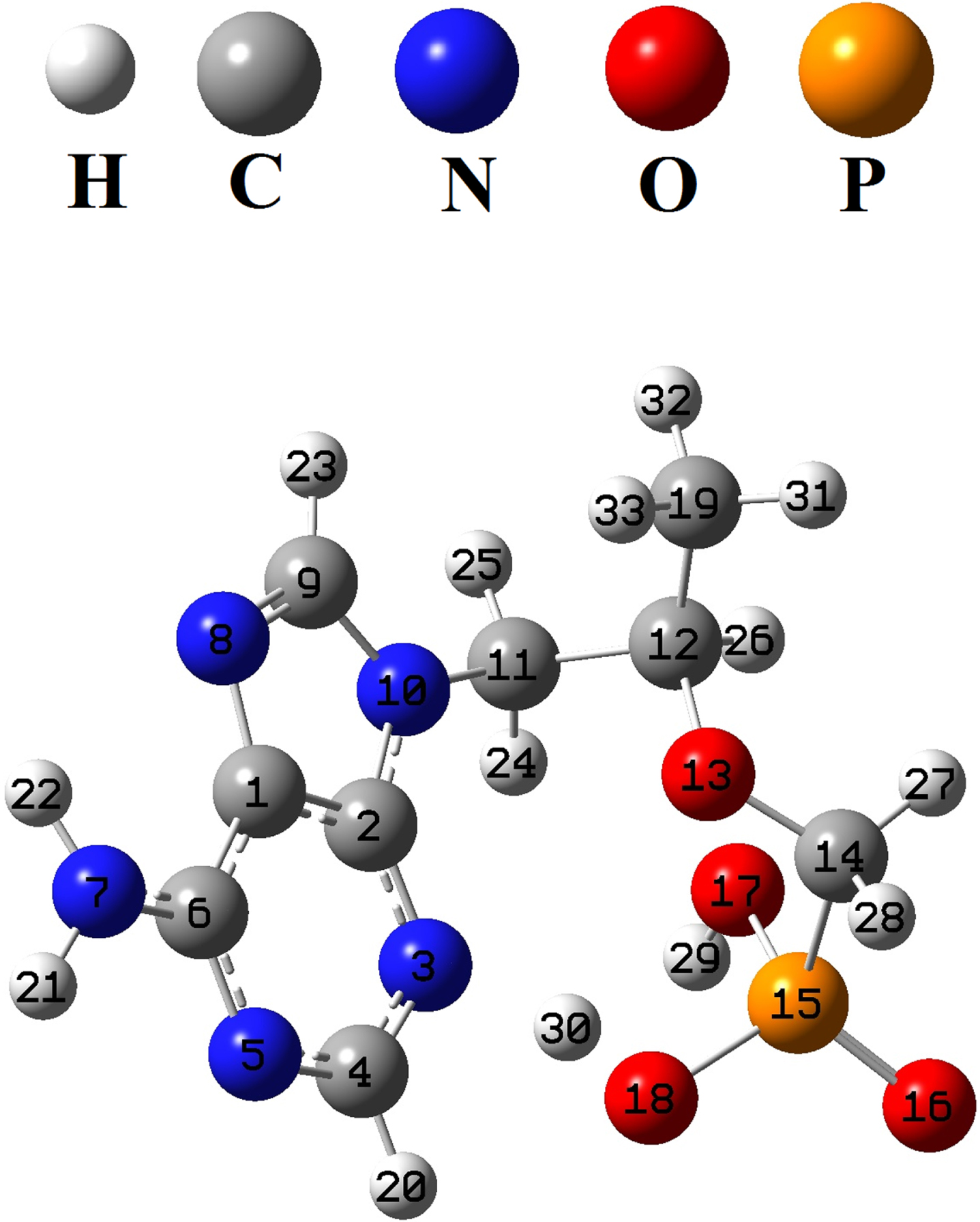
Table 1
Selected optimized geometrical parameters of the tenofovir molecule
| Bond | Bond length (°) | Experimental | Bond | Bond angle (Å) | Experimental |
| C1-C2 | 1.400 | 1.384 | C1-C2-N3 | 124.40 | 120.3 |
| C1-C6 | 1.381 | 1.376 | C1-C6-N5 | 119.14 | 117.6 |
| C1-N8 | 1.370 | 1.366 | C1-C6-N7 | 122.30 | 121.2 |
| C2-N3 | 1.353 | 1.345 | C1-N8-C9 | 103.87 | 107.5 |
| N3-C4 | 1.348 | 1.340 | C2-N3-C4 | 112.78 | 112.6 |
| C4-N5 | 1.363 | 1.352 | N3-C4-N5 | 128.71 | 128.1 |
| N5-C6 | 1.360 | 1.349 | C4-N5-C6 | 117.50 | 118.1 |
| C6-N7 | 1.357 | 1.266 | N5-C6-N7 | 118.50 | 121.2 |
| N8-C9 | 1.371 | 1.369 | C1-C2-N10 | 105.57 | 110.9 |
| C2-N10 | 1.378 | 1.369 | N3-C2-N10 | 129.98 | 125.7 |
| C9-N10 | 1.393 | 1.366 | N3-C4-H20 | 115.08 | 115.9 |
| N10-C11 | 1.465 | 1.470 | N5-C4-H20 | 116.20 | 115.9 |
| C11-C12 | 1.543 | 1.523 | C6-N7-H21 | 116.33 | 120.0 |
| C12-O13 | 1.455 | 1.402 | C6-N7-H22 | 117.09 | 120.0 |
| C12-C19 | 1.531 | 1.523 | N8-C9-H23 | 125.52 | 124.1 |
| O13-C14 | 1.431 | 1.402 | C2-N10-C11 | 127.01 | 127.5 |
| C14-P15 | 1.869 | 1.856 | C9-N10-C11 | 127.09 | 127.5 |
| P15-O16 | 1.620 | 1.615 | N10-C9-H23 | 120.87 | 124.1 |
| P15-O17 | 1.627 | 1.615 | N10-C11-C12 | 112.33 | 109.5 |
| P15-O18 | 1.625 | 1.615 | C11-C12-O13 | 106.97 | 109.5 |
| C4-H20 | 1.089 | 1.100 | C11-C12-C19 | 113.88 | 109.4 |
| N7-H21 | 1.024 | 1.050 | C12-O13-C14 | 114.56 | 120.0 |
| N7-H22 | 1.023 | 1.050 | C14-P15-O16 | 115.86 | 109.5 |
| C9-H23 | 1.083 | 1.100 | C14-P15-O17 | 99.05 | 109.5 |
| C11-H24 | 1.093 | 1.113 | C14-P15-O18 | 108.34 | 109.5 |
| C11-H25 | 1.096 | 1.113 | O16-P15-O17 | 114.02 | 109.5 |
| C12-H26 | 1.097 | 1.113 | O16-P15-O18 | 112.33 | 109.5 |
| C14-H27 | 1.100 | 1.113 | O17-P15-O18 | 106.05 | 109.5 |
| C14-H28 | 1.097 | 1.113 | P15-O17-H29 | 106.69 | 120.0 |
| O17-H29 | 0.992 | 0.942 | P15-O18-H30 | 112.22 | 120.0 |
| O18-H30 | 1.059 | 0.942 | H21-N7-H22 | 116.68 | 120.0 |
| C19-H31 | 1.098 | 1.113 | N10-C11-H24 | 108.60 | 109.4 |
| C19-H32 | 1.099 | 1.113 | N10-C11-H25 | 108.03 | 109.5 |
| C19-H33 | 1.098 | 1.113 | O13-C12-H26 | 110.63 | 109.5 |
3.2FMO analysis of the tenofovir
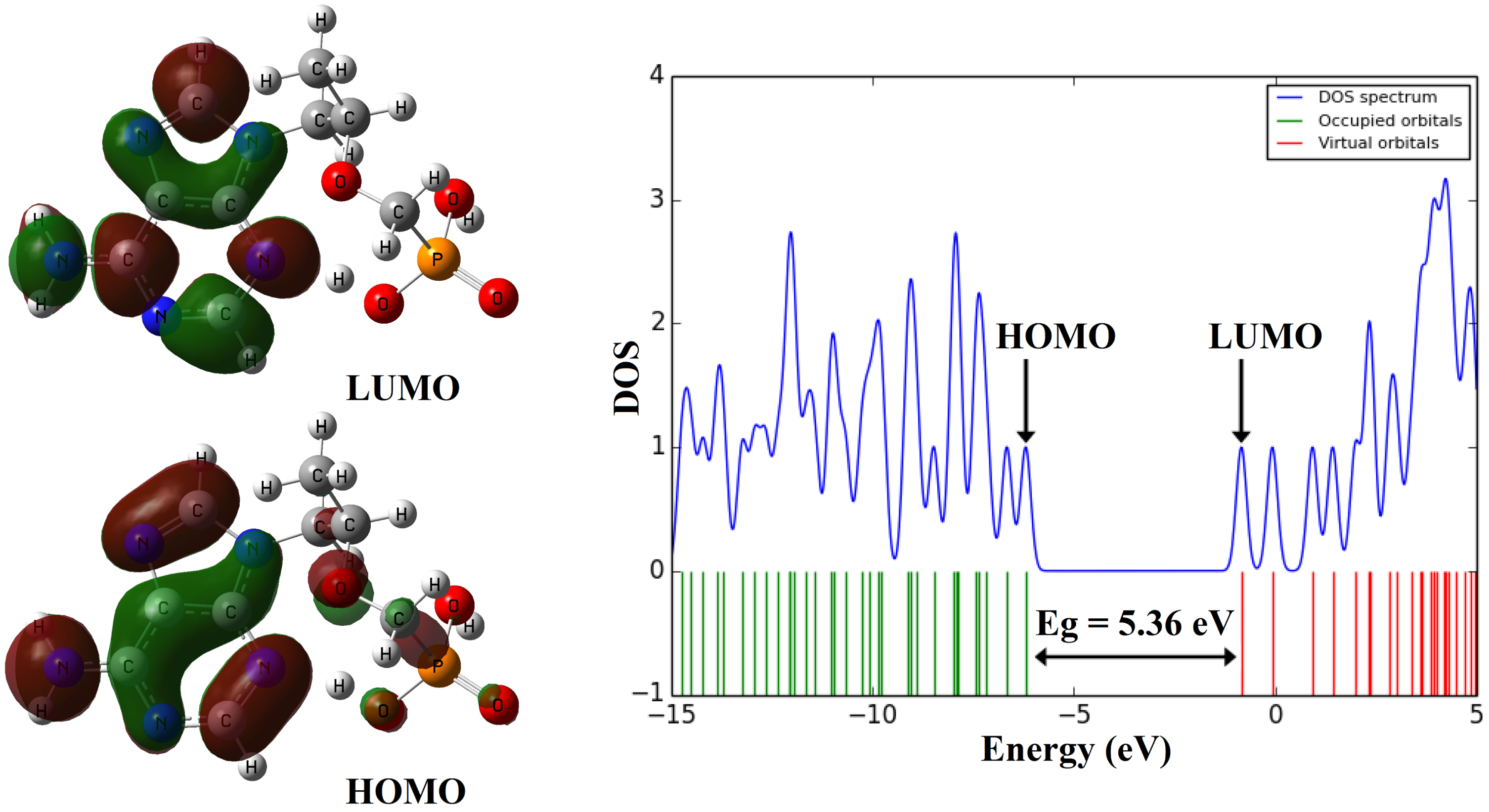
The results of FMO analysis and electronic properties are reported in Table 2. The picture of HOMO and LUMO orbitals are shown in Fig. 3. According to the results of FMO analysis, the maximum of the HOMO to LUMO charge transfer in tenofovir molecule is related to the contribution of pi (π) bonds and lone pairs (n⟶π* and π ⟶ π*). A detail of quantum molecular descriptors [51] of tenofovir are reported in Table 2. The density of states spectrum (DOS) [52] of the tenofovir is displayed in Fig. 2.
Table 2
The calculated electronic properties of the tenofovir
| Property | B3LYP |
| Electronic energy (a.u.) | –1260.545 |
| Dipole moment (Debye) | 9.40 |
| Point Group | C1 |
| EHOMO (eV) | –6.2 |
| ELUMO (eV) | –0.84 |
| Energy gap (eV) | 5.36 |
| Ionization potential, I (eV) | 6.2 |
| Electron affinity, A (eV) | 0.84 |
| Electronegativity, χ (eV) | 3.52 |
| Global hardness, η (eV) | 2.68 |
| Chemical potential, μ (eV) | –3.52 |
| Global electrophilicity, ω (eV) | 2.31 |
| Chemical softness, S (eV) | 0.18 |
Fig. 3
The shape of HOMO and LUMO orbitals and DOS plot of the molecule tenofovir.
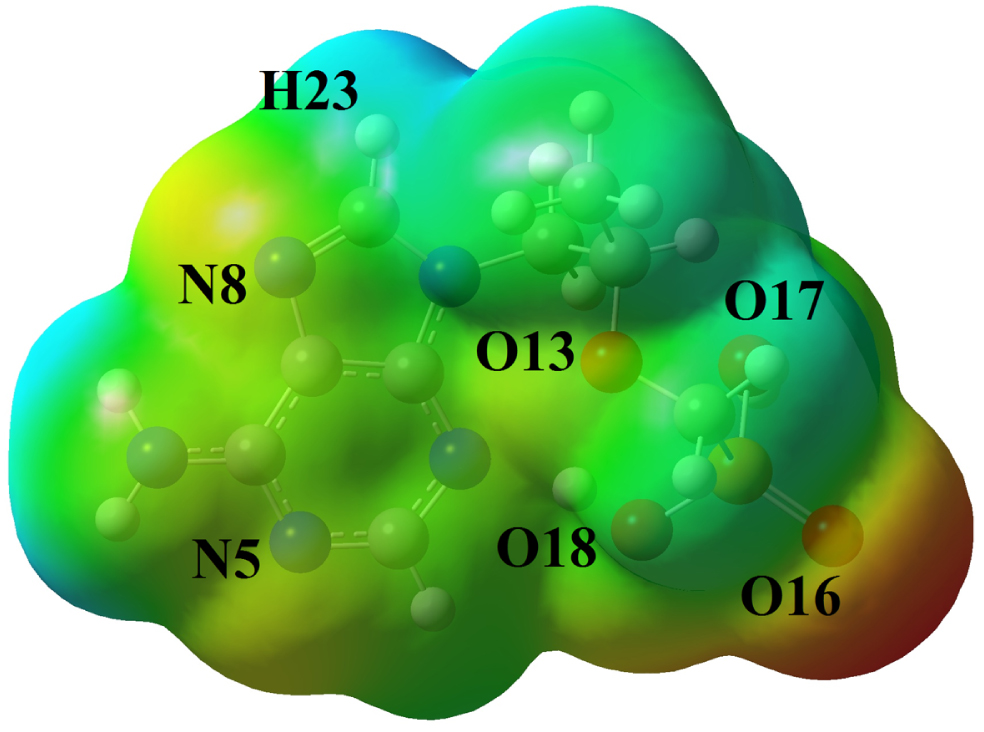
3.3MEP analysis of the tenofovir
The MEP analysis [53] was studied for optimized tenofovir molecule and the corresponding map was visualized in Fig. 4. According to Fig. 4, the oxygen atom (O16) of C = O group is found to be an electron donor (red color), which is due to the lone pair electrons of the oxygen atoms. Therefore, the O16 atom is nucleophilic region. Also, the O17 and O18 atoms in the O-H groups are shown slightly negative charge sites (orange color). The O13, N5 and N8 atoms with yellow color are partially electron-rich areas. The hydrogen atoms are shown electron-poor and electrophilic areas (blue color). The green color areas in the tenofovir molecule indicate the areas with zero potential and neutral. The electrophilic and nucleophilic areas represent the interaction between the tenofovir molecule and other biomolecules in living organisms.
Fig. 4
MEP map of the tenofovir molecule.
3.4Charge analysis of the tenofovir
The Mulliken atomic charges and natural charges [54] for the tenofovir molecule calculated are reported in Table S1. The carbon atom of methyl groups in the tenofovir molecule has the highest negative charge (Mulliken: –0.459e and NBO: –0.720e) rather than other carbon atoms due to the hyperconjugation effect. The phosphorus atom has a positive value (Mulliken charge: 0.371e, NBO: 2.093e). All the nitrogen atoms display a negative charge. The N10 atom has the highest negative Mulliken charge about –0.755e rather than other nitrogen, while the highest negative natural charge is observed for N7 atom (–0.750e). All oxygen atoms in the tenofovir molecule have the negative charge and O13 atom has the highest negative Mulliken charge about –0.621e, whereas O16 atom of carbonyl group has the highest negative natural charge about –1.005e. All the hydrogen atoms display a positive charge. The H29 and H30 atoms have the highest positive charge about 0.421e and 0.430e (Mulliken charge) and 0.486e and 0.482e (natural charge), respectively, rather than other hydrogen atoms due to the attachment to electron-withdrawing oxygen atoms.
3.5Electronic structure and excited states of the tenofovir molecule
Theoretical absorption spectrum [55] of the optimized tenofovir molecule is calculated using TDB3LYP/MidiX level of theory at 100–400 nm by the IEFPCM solvent model [56]. The absorption spectrum of the tenofovir (Fig. 5) has a wide absorption at about 100–400 nm, which characterized by maxima at 186.51 nm. The strong peak at 186.51 nm is due to the Charge-Transfer (CT) excited state S0⟶S12 and is described by a wave function corresponding to a superposition of six configurations for one-electron excitations (H-5 (L (31%), H (L + 2 (38%), H-6 (L + 1 (6%), H-5 (L + 1 (3%), H-3 (L + 1 (6%), H (L + 1 (4%)). Excitation of an electron from HOMO to LUMO+1 (38%) molecular orbital (MO) gives the main contribution to the formation of the absorption band at 186.51 nm (Fig. 5). Excitation of one electron at 179.44 nm belonged to the transition into the excited singlet state S0⟶S16 and is described by a wave function corresponding to a superposition of two configurations for one-electron excitations (H-3 (L + 1 (38%), H-1 (L + 2 (38%), H-5 (L (7%), H-5 (L + 1 (2%), H-4 (L + 1 (2%), H-1 (L + 3 (2%)). Excitation of an electron from HOMO-3 to LUMO+1 (38%) and HOMO-1 to LUMO+2 (38%) MO gives the main contribution to the formation of the absorption band at 179.44 nm (Table 3). The other excited states of the title compound at 248.69, 240.77, 234.75, 231.02, 210.75, 208.98, 207.92, 205.64, 204.92, 194.62, 190.47, 184.91, 182.50, 176.38, 175.11, 174.81, 172.53 nm have very small intensity (f ≈ 0) that is nearly forbidden by orbital symmetry considerations and not discussed.
Fig. 5
UV-vis spectrum of the tenofovir.
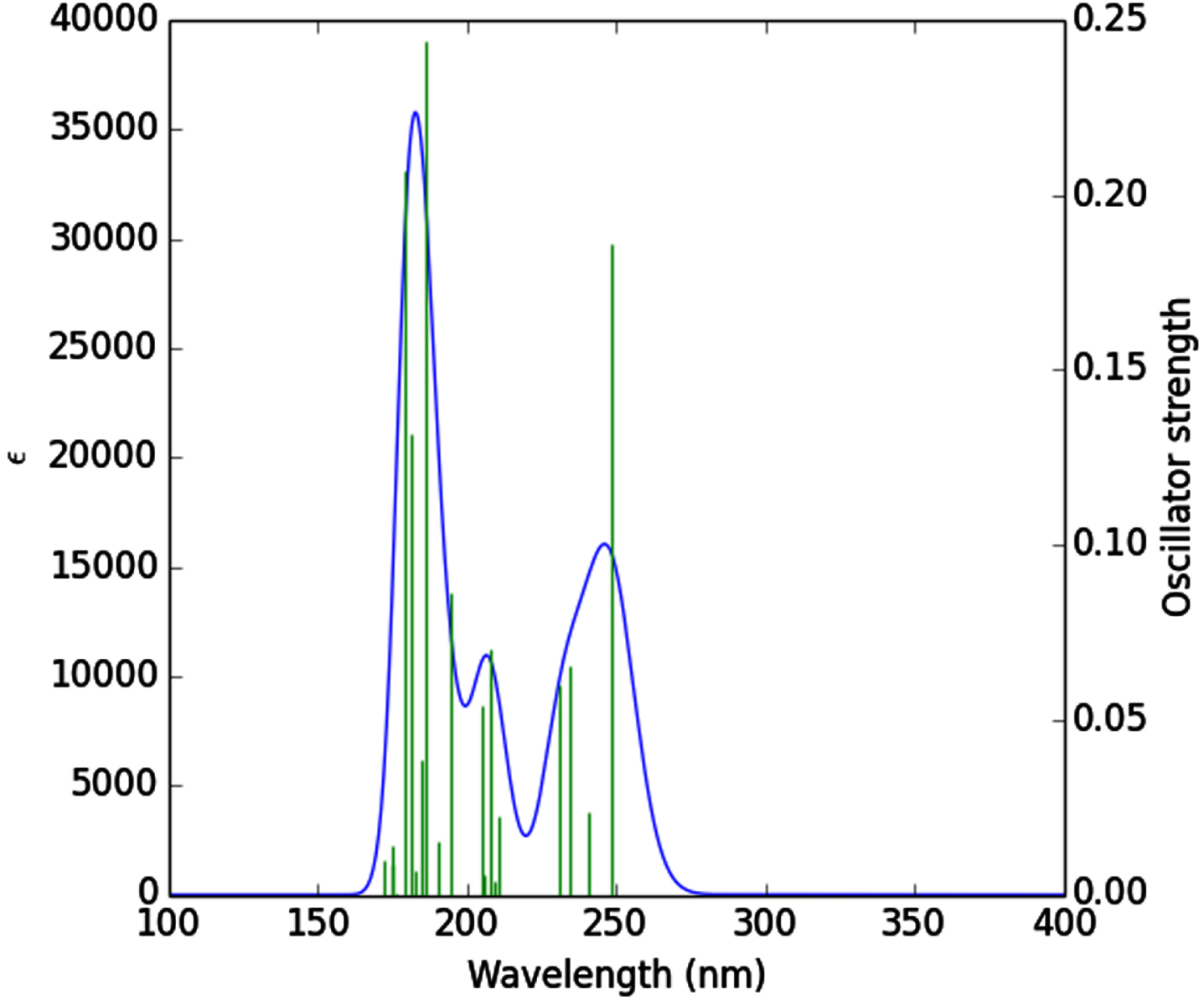
Table 3
Calculated electronic absorption parameters of the tenofovir
| Excited state | Wavelength (nm) | Excitation energy (eV) | Configurations composition (corresponding transition orbitals) | Oscillator strength (f) |
| S0⟶S12 | 186.51 | 6.64 | H-5⟶L (31%), H⟶L+2 (38%), H-6⟶L+1 (6%), H-5⟶L+1 (3%), H-3⟶L+1 (6%), H⟶L+1 (4%) | 0.24 |
| S0⟶S15 | 181.25 | 6.84 | H-3⟶L+1 (33%), H-1⟶L+2 (48%), H-5⟶L+1 (7%), H⟶L+3 (3%) | 0.13 |
| S0⟶S16 | 179.44 | 6.90 | H-3⟶L+1 (38%), H-1⟶L+2 (38%), H-5⟶L (7%), H-5⟶L+1 (2%), H-4⟶L+1 (2%), H-1⟶L+3 (2%) | 0.20 |
3.6NBO analysis of the tenofovir
The stabilization energy (E (2)) displays the value of participation of electrons in the resonance between atoms, which is calculated according to equation 1 [57, 58].
(1)
The results of NBO analysis are summarized in Table S2. According to the results of NBO, the σ(C1-N8) orbital in the purine ring acts as an electron donor and the σ*(C1-C6), σ*(C2-N3), σ*(C9-H23) anti-bonding orbitals participate as electron acceptors with resonance energies (E(2)) about 2.36 kcal/mol, 3.14 kcal/mol, 4.69 kcal/mol, respectively. These values show σ(C1-N8)⟶σ*(C9-H23) interaction has the higher stabilization energy (4.69 kcal/mol) rather than σ(C1-N8)⟶σ*(C1-C6) and σ(C1-N8)⟶σ*(C2-N3) interactions. The σ(C14-P15) orbital acts as an electron donor and the σ*(P15-O16), σ*(P15-O17), σ*(P15-O18), σ*(O17-H29) anti-bonding orbitals participate as electron acceptors with resonance energies (E(2)) of 4.29 kcal/mol, 4.60 kcal/mol, 6.11 kcal/mol, 1.47 kcal/mol, respectively. These values display σ(C14-P15)⟶σ*(P15-O18) transition has the higher resonance energy (6.11 kcal/mol) comparing with σ(C14-P15)⟶σ*(P15-O16), σ(C14-P15)⟶σ*(P15-O17) and σ(C14-P15)⟶σ*(O17-H29) interactions. The π ⟶ π* interactions at the tenofovir are such as C1-C2⟶N3-C4, C1-C2⟶N5-C6, C1-C2⟶N8-C9, N3-C4⟶C1-C2, N3-C4⟶N5-C6, N5-C6⟶C1-C2, N8-C9⟶C1-C2 transitions with stabilization energies (E(2)) of 10.83, 30.16, 14.54, 24.87, 6.61, 9.30, 42.95, 17.10 kcal/mol, respectively. The π(N5-C6)⟶π*(C1-C2) interaction has the higher resonance energy (42.95 kcal/mol) rather than other π ⟶ π* interactions. The n2(O16) orbital acts as an electron donor and σ*(C14-P15), σ*(P15-O18) anti-bonding orbitals participate as electron acceptors with resonance energies (E(2)) about 14.37 kcal/mol, 26.18 kcal/mol, respectively. The n2(O16)⟶σ*(P15-O18) interaction has the higher stabilization energy (26.18 kcal/mol) comparing with n2(O16)⟶σ*(C14-P15) interaction. The highest resonance energies of the tenofovir are observed for n1(N5)⟶π*(N5-C6), n1(N10)⟶π*(N8-C9) and n1(N10)⟶π*(N8-C9) transitions with resonance energies (E(2)) of 51.58 kcal/mol, 45.31 kcal/mol, 46.83 kcal/mol, respectively.
3.7Molecular docking analysis
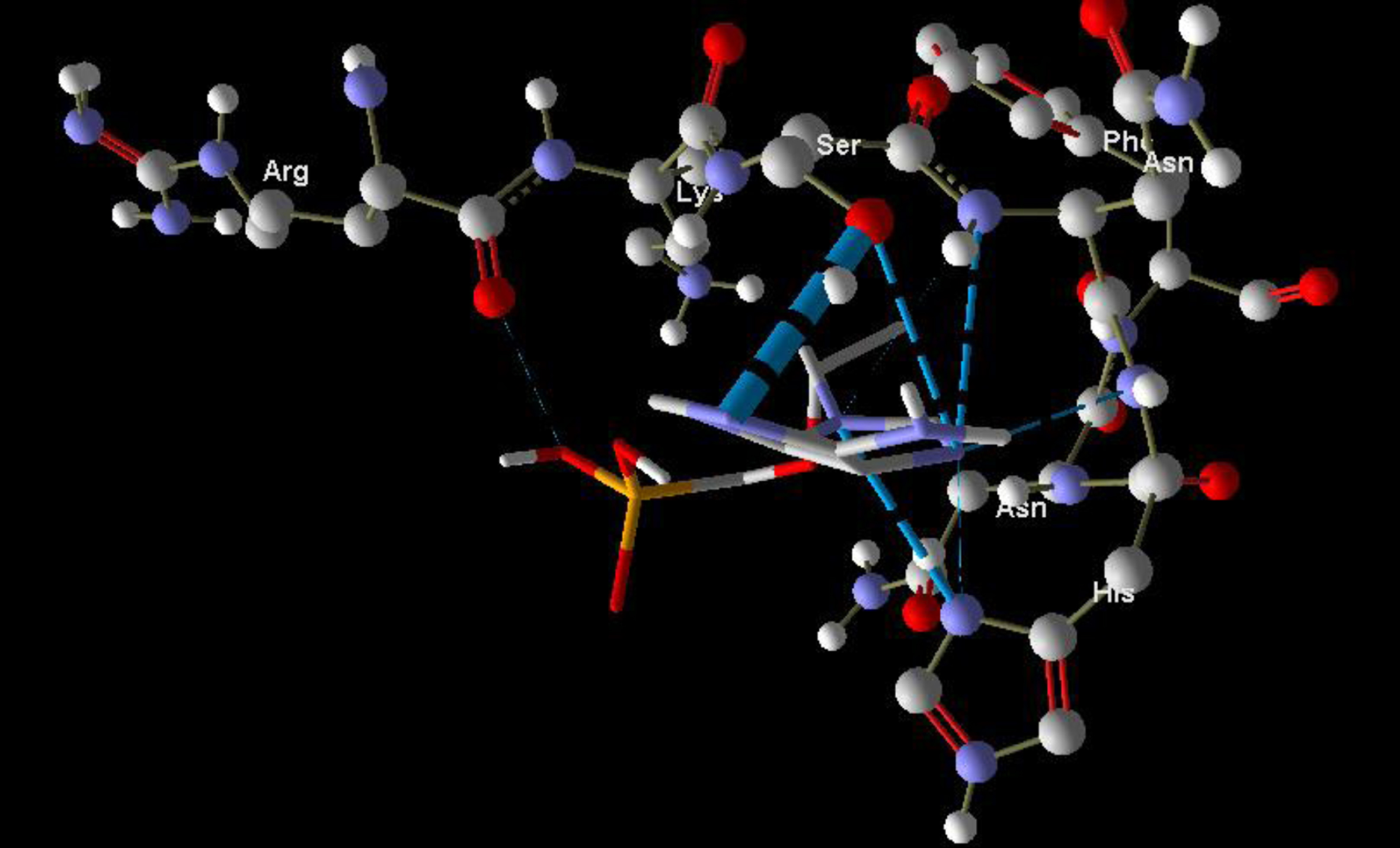
Molecular basis of interactions between SARS-CoV-2 Mpro molecule and the tenofovir can be understood with the help of docking analysis and interactions as observed in Fig. 6. There are 6 positions with a strong interaction between the tenofovir drug molecule and the SARS-CoV-2 Mpro, which leads to the degradation of the protein structure. The best position is illustrated here. Molecular docking energy data for mentioned ligand as well as hydrogen bonds are listed in the Table 4. The binding energy for SARS-CoV-2 Mpro and tenofovir is -77.63 kcal/mol in which shows good binding affinity between the tenofovir and SARS-CoV-2 Mpro compared to the results obtained by Kumar et al. [36], which have been reported the binding affinity about –8.1 kcal/mol for tenofovir-disoproxil and SARS-CoV-2 Mpro. As seen from Fig. 7 and Table 2 eight hydrogen bonding formation are observed between reduces of Arg60 bonded with O atom, Ser62 bonded with N atoms of the tenofovir. Also, Ser62, His64, Asn63 are connected to negatively and positively charged in the tenofovir binding environment (Figs. 8, 9). According to obtained results, it is found that the tenofovir ligand has the best affinity for the SARS-CoV-2 Mpro compared to colistin, valrubicin, icatibant, bepotastine, epirubicin, epoprostenol, vapreotide and aprepitant antiviral drugs with the binding energy of –11.206, –10.934, –9.607, –10.273, –9.091, 10.582, –9.892 and –11.376 kcal/mol, respectively [31].
Fig. 6
Interaction of the tenofovir with SARS-CoV-2 Mpro.
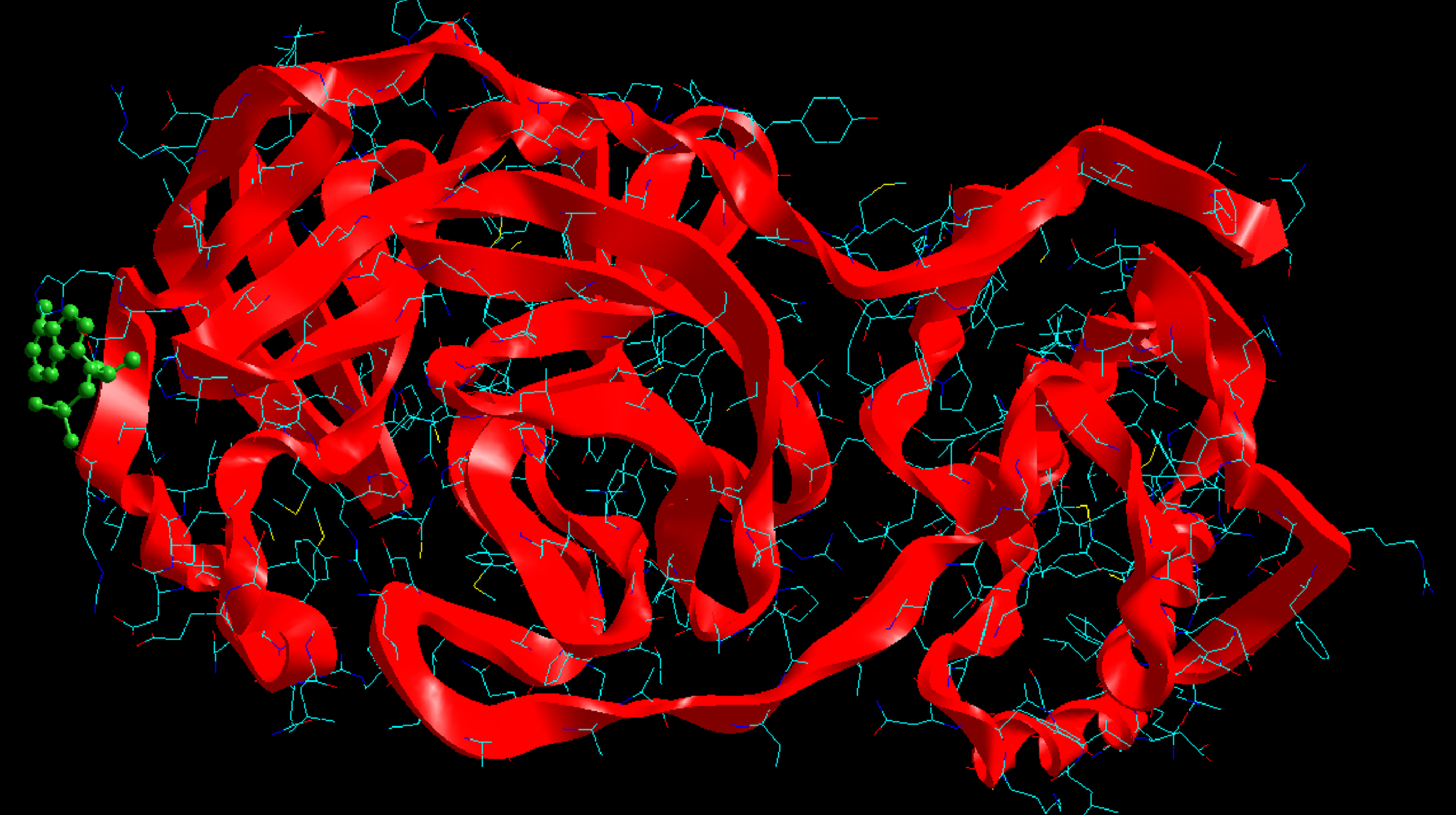
Table 4
Molecular docking energy data for mentioned ligand and Hydrogen bonding
| Protein | Bonded residues | ID | Hydrogen bond | Bond distance ( Å) | Binding energy (kcal/mol) |
| 2019-nCoV | Arg | 60 | 1 | 1.5028 | –5.2473 |
| 2019-nCoV | Ser | 62 | 1 | 1.6358 | –6.2907 |
| 2019-nCoV | Ser | 62 | 1 | 1.9311 | –3.4003 |
| 2019-nCoV | Asn | 63 | 1 | 1.4703 | –2.9054 |
| 2019-nCoV | Asn | 63 | 1 | 1.5554 | –4.5941 |
| 2019-nCoV | His | 64 | 1 | 1.9901 | –3.0399 |
| 2019-nCoV | His | 64 | 1 | 2.1143 | –5.0754 |
| 2019-nCoV | His | 64 | 1 | 2.3487 | –7.2362 |
Fig. 7
Hydrogen bonds between the tenofovir and SARS-CoV-2 Mpro.
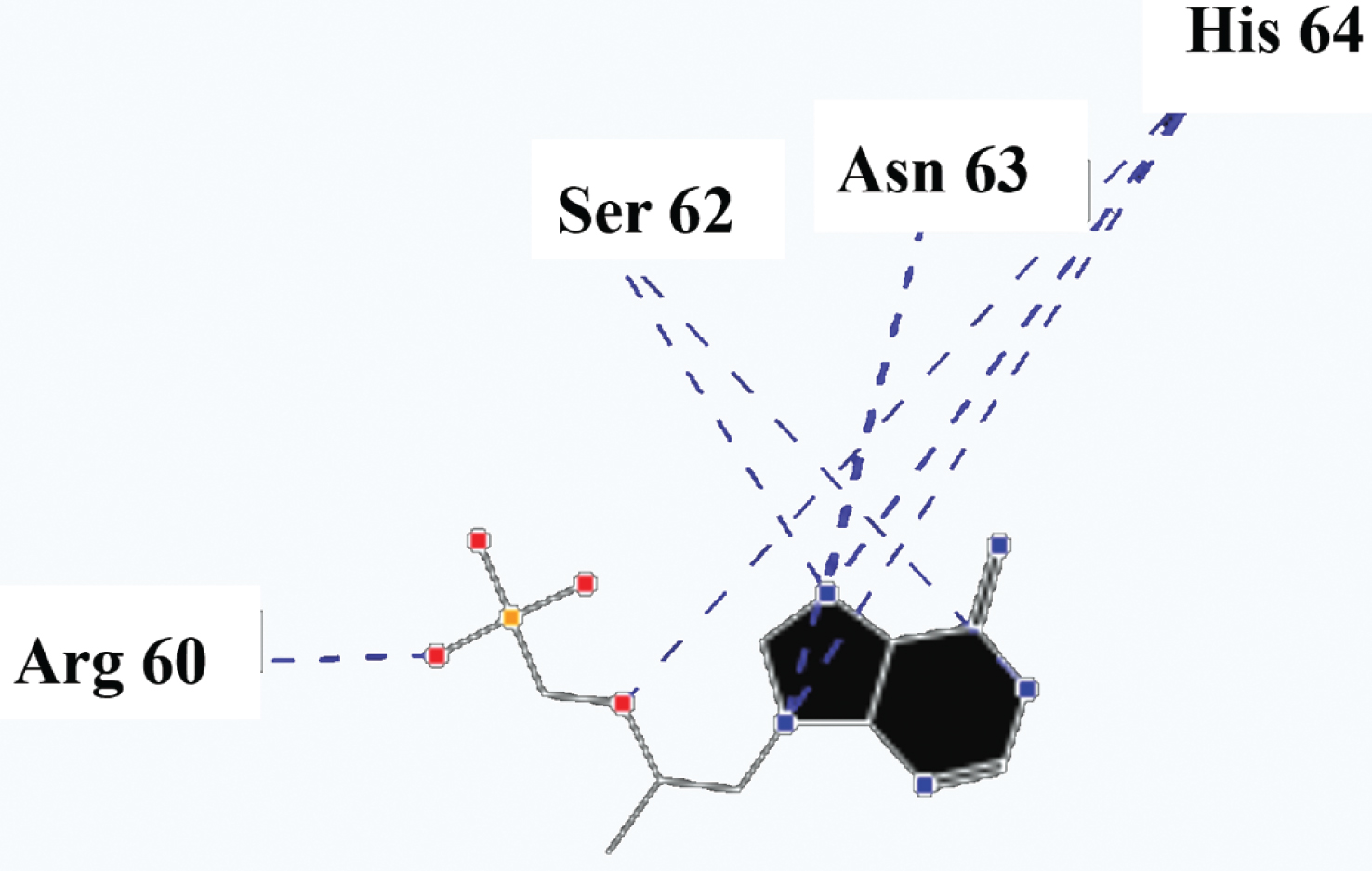
Fig. 8
Steric interactions between the tenofovir and SARS-CoV-2 Mpro.
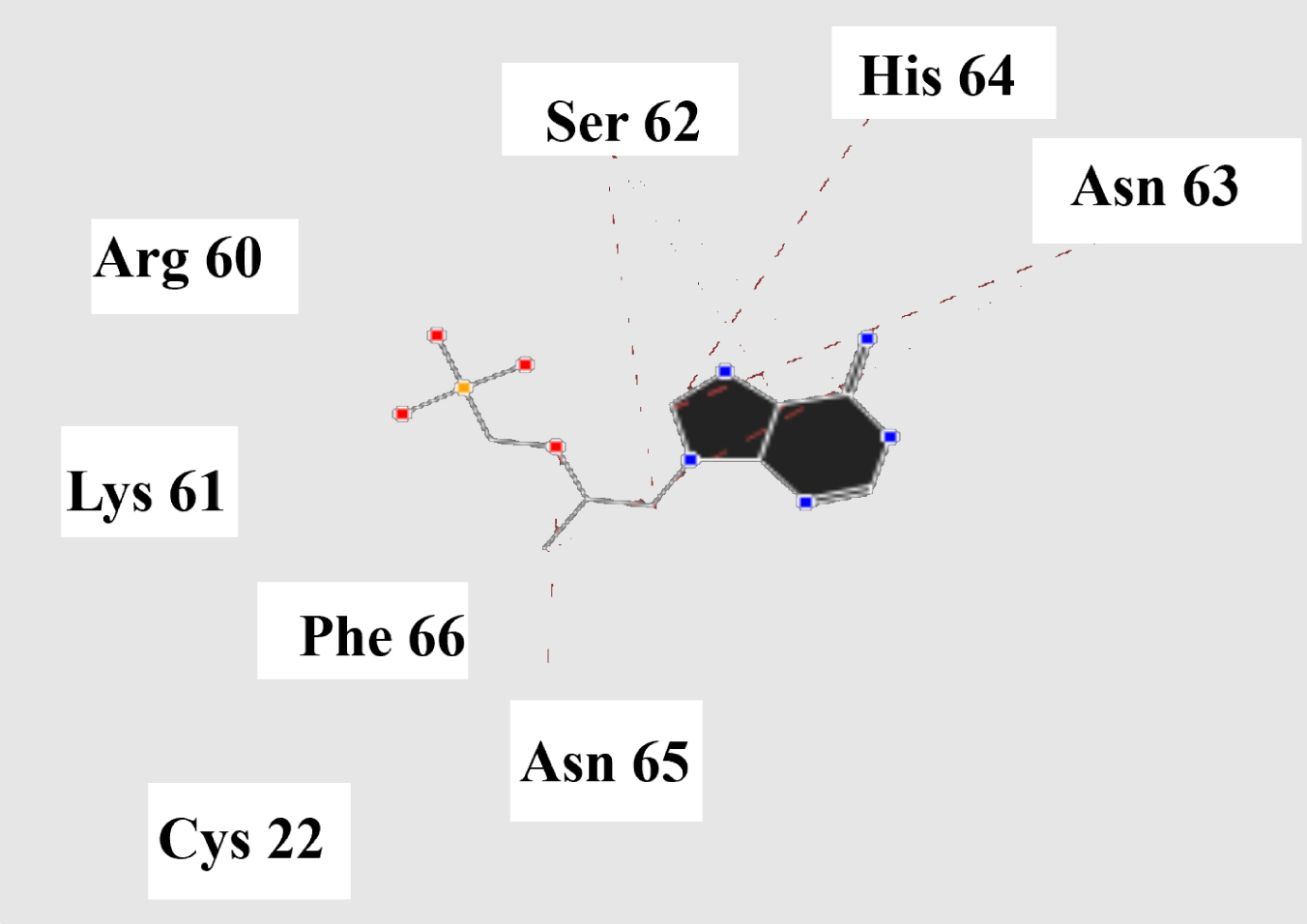
Fig. 9
Molecular docking of the tenofovir to SARS-CoV-2 Mpro.
3.8Bioactivity and pharmacokinetic properties
Drug-likeness evaluated by the Lipinski rule of five deals four simple physicochemical parameter ranges (MWT≤500, log P≤5, H-bond donors≤5, H-bond acceptors≤10) accompanied with 90% of orally active drugs, which have passed phase II clinical trial. MiLog P values of these compounds are observed to be < 5 (–2.63–3.19) showed their good permeability across the cell membrane. These compounds were observed to have TPSA will be below 140Å2 (229.46 for tenofovir triphosphate), molecular weight < 500 (the best structure is tenofovir), No. of hydrogen bond donors≤5 (the best structure is tenofovir), No. of hydrogen acceptor≤10, n-violations 0 (for tenofovir and Tucatinib), number of rotatable flexible bonds > 5. The results of calculations are presented in Table 5.
Table 5
Pharmacokinetic properties of the compounds tenofovir, selpercatinib [59], tucatinib [59] and tenofovir triphosphate
| Compound | miLogP | TPSA | natoms | MW | nON | nOHNH | nviolations | nrotb | volume |
| Tenofovir | –0.62 | 136.39 | 19 | 287.22 | 9 | 4 | 0 | 5 | 232.58 |
| Selpercatinib | 1.35 | 112.04 | – | 525.60 | 8 | 1 | 1 | 8 | – |
| Tucatinib | 3.19 | 110.85 | – | 480.52 | 7 | 2 | 0 | 6 | – |
| Tenofovir triphosphate | –2.63 | 229.46 | 27 | 447.17 | 15 | 6 | 2 | 9 | 323.28 |
*_ miLogP (the octanol/water partition coefficient), TPSA (the molecular polar surface area), natoms (the number of atoms of the molecule), nON (the number of hydrogen bond acceptors), nOHNH (the number of hydrogen bond donors), nviolations (the number of violations of the Ro5), nrotb (the number of rotatable bonds), volume (the molecular volume), and MW (the molecular weight).
4Conclusion
In Summary, density functional theory and molecular docking investigations were carried out for the binding of tenofovir to SARS-CoV-2 Mpro. The most of the charge transfer from the HOMO to LUMO in tenofovir drug molecule is due to n⟶π* and π ⟶ π* transitions. The energy gap (Eg) value of tenofovir was estimated about 5.36 eV. All oxygen atoms in the tenofovir molecule have the negative charge and O16 atom has the highest negative natural charge about –1.005e. The absorption spectrum of the tenofovir has a wide absorption at about 100–400 nm which characterized by maxima at 186.51 and 179.44 nm. Docking study was performed to analyze the binding interaction of the tenofovir ligand with the target protein. It was found that the tenofovir shows the most binding energy (–47.206 kcal/Mol) towards of the SARS-CoV-2 Mproin comparison with other known antiviral drugs, including colistin, valrubicin, icatibant, bepotastine, epirubicin, epoprostenol, vapreotide, aprepitant. Thus, this compound can be considered in the future clinical trials of drugs against 2019-nCoV. The molecular properties related to drug-likeness have been predicted based on the bioavailability using methodologies defined in the previous literatures as well as the descriptors used to quantify the bioactivity allowed the characterization of the studied tenofovir molecule.
Supplementary material
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/MGC-220046.
References
[1] | Hassan A.O. , Case J.B. , Winkler E.S. , Thackray L.B. , Kafai’ N.M. , Bailey A.L. , McCune B.T. , Fox J.M. , Chen R.E. , Alsoussi W.B. , Turner J.S. , Schmitz A.J. , Lei T. , Shrihari S. , Keeler Sh.P. , Fremont D.H. , Greco S. , McCray P.B. Jr, Perlman S. , Holtzman M.J. , Ellebedy A.H. and Diamond M.S. , A SARS-CoV-2 infection model in mice demonstrates protection by neutralizing antibodies, Cell 182: ((2020) ), 744–753. |
[2] | Li G. , Fan Y. , Lai Y. , Han T. , Li Z. , Zhou P. , Pan P. , Wang W. , Hu D. , Liu X. , Zhang Q. and Wu J. , Coronavirus infections and immune responses, J Med Virol 92: ((2020) ), 424–432. |
[3] | Shahab S. and Sheikhi M. , Triazavirin-Potential inhibitor for 2019-nCoV coronavirus M protease: A DFT study, Curr Mol Med 20: ((2020) ), 1–13. |
[4] | Liang J. , Karagiannis C. , Pitsillou E. , Darmawan K.K. , Ng K. , Hung A. and Karagiannis T.C. , Site mapping and small molecule blind docking reveal a possible target site on the SARS-CoV-2 main protease dimer interface, ComputBiol Chem 89: ((2020) ), 107372. |
[5] | Marinho E.M. , de Andrade Neto J.B. , Silva J. , da Silva C.R. , Cavalcanti B.C. , Marinho E.S. and Júnior H.V.N. , Virtual screeningbased on molecular docking of possible inhibitors of Covid-19 mainprotease. , Microb Pathog 148: ((2020) ), 104365. |
[6] | Jiménez-Alberto A. , Ribas-Aparicio R.M. , Aparicio-Ozores G. and Castelán-Vega J.A. , Virtual screening of approved drugs as potential SARS-CoV-2 main protease inhibitors, Comput Biol Chem 88: ((2020) ), 107325. |
[7] | Meyer-Almes F.J. , Repurposing approved drugs as potential inhibitors of 3CL-protease of SARS-CoV-2: Virtual screening and structure based drug design, Comput Biol Chem 88: ((2020) ), 107351. |
[8] | Enmozhi S.K. , Raja K. , Sebastine I. and Joseph J. , Andrographolide as a potential inhibitor of SARS-CoV-2 main protease: an in silico approach, J Biomol Struct Dyn 39: ((2020) ), 6317–6323. |
[9] | Khan M.A. , Mahmud S. , Alam A.S.M.R.U. , Rahman M.E. , Ahmed F. and Rahmatullah M. , Comparative molecular investigation of the potential inhibitors against SARS-CoV-2 main protease: a molecular docking study, J Biomol Struct Dyn 39: ((2020) ), 6317–6323. |
[10] | Das S. , Sarmah S. , Lyndem S. and Singha R.A. , An investigation into the identification of potential inhibitors of SARS-CoV-2 main protease using molecular docking study, J Biomol Struct Dyn 39: ((2020) ), 3347–3357. |
[11] | Rasool N. , Akhtar A. and Hussain W. , Insights into the inhibitory potential of selective phytochemicals against Mpro of 2019-nCoV: a computer-aided study, Struct Chem 31: ((2020) ), 1777–1783. |
[12] | https://www.statista.com/statistics/1093256/novel-coronavirus-2019ncov-deaths-worldwide-by-country/. |
[13] | Ahn D.G. , Shin H.J. , Kim M.H. , Lee S. , Kim H.S. , Myoung J. , Kim B.T. and Kim S.J. , Current status of epidemiology, diagnosis, therapeutics, and vaccines for novel coronavirus disease (COVID-19), J Mol Biol 30: ((2020) ), 313–324. |
[14] | Sanders J.M. , Monogue M.L. , Jodlowski T.Z. and Cutrell J.B. , Pharmacologic treatments for coronavirus disease (COVID-19): A review, Jama 323: ((2020) ), 1824–1836. |
[15] | Liu C. , Zhou Q. , Garner L.Y. , Watkins S.P. , Carter L.J. , Smoot J. , Gregg A.C. , Daniels A.D. and Jervey S. , Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases, ACS Cent Sci 6: ((2020) ), 315–331. |
[16] | Wang L.S. , Wang Y.R. , Ye D.W. and Liu Q.Q. , A review of the 2019 novel Coronavirus (COVID-19) based on current evidence, Int J Antimicrob Agents 55: ((2020) ), 105948. |
[17] | Wang M. , Cao. R. , Zhang L. , Yang X. , Liu J. , Xu M. , Shi Z. , Hu Z. , Zhong W. and Xiao G. , Remdesivir and chloroquine effectively inhibit therecently emerged novel coronavirus 2019-nCoV) in vitro, Cell Res 30: ((2020) ), 269–271. |
[18] | Caly L. , Druce J.D. , Catton M.G. , Jans D.A. and Wagstaff K.M. , The FDA-approved drug ivermectin inhibits the replication of SARS-CoV-2in vitro, Antivir Res 178: ((2020) ), 104787. |
[19] | Martins-Filho P.R. , Barreto-Alves J.A. and Fakhouri R. , Potential role for nitazoxanide in treating SARS-CoV-2 infection, Am J Physiol Lung Cell Mol Physiol 319: ((2020) ), 35–36. |
[20] | Jang S. and Rhee J.Y. , Three cases of treatment with Nafamostat in elderly patients with COVID-19 pneumonia who need oxygen therapy, Int J Infect Dis 96: ((2020) ), 500–502. |
[21] | Gallant J.E. and Deresinski S. , Tenofovir Disoproxil Fumarate, Clin Infect Dis 37: ((2003) ), 944–950. |
[22] | Derstine B.P. , Tomlin J.W. , Peck C. , Dietz J.P. , Herrera B.T. , Cardoso F. , Paymode D.J. , Yue A.C. , Arduengo A.J. and Opatz T. , An Efficient Synthesis of Tenofovir (PMPA): A 24. Key Intermediate Leading to Tenofovir-based HIV Medicines, Org Process Res Dev 24: ((2020) ), 1420–1427. |
[23] | Byrne R. , Carey I. and Agarwal K. , Tenofovir alafenamide in the treatment of chronic hepatitis B virus infection: rationale and clinical trial evidence, Therap Adv Gastroenterol 11: ((2018) ), 1756284818786108. |
[24] | Ray A.S. , Fordyce M.W. and Hitchcock M.J.M. , Tenofovir alafenamide: a novel prodrug of tenofovir for the treatment of human immunodeficiency virus, Antivir Res 125: ((2016) ), 63–70. |
[25] | Seo H.Y. , Lee H.A. , Ko S.Y. , Wang J.H. , Kim J.H. , Choe W.H. and Kwon S.K. , Clinical impact of the early alanine amininotransferase flareduring tenofovir monotherapy in treatment-naïve patients withchronic hepatitis B, Clin Mol Hepatol 23: ((2017) ), 154–159. |
[26] | Buti M. , Tsai N. , Petersen J. , Flisiak R. , Gurel S. , Krastev Z. , Aguilar S. , Flaherty J.F. , Martins E.B. , Charuworn P. , Kitrinos K.M. , Subramanian G.M. , Gane E. and Marcellin P. , Seven-year efficacy and safety of treatment with tenofovir disoproxil fuma-rate for chronic hepatitis B virus infection, Dig Dis Sci 60: ((2015) ), 1457–1464. |
[27] | Reijnders J.G.P. and Janssen H.L.A. , Potency of tenofovir in chronic hepatitis B: Mono or combination therapy? J Hepatol 48: ((2008) ), 383–386. |
[28] | De Clercq E. , Ferir G. , Kaptein S. and Neyts J. , Antiviral treatment of chronic hepatitis B virus (HBV) infections, Viruses 2: ((2010) ), 1279–1305. |
[29] | Sulkowski M.S. , Management of hepatic complications in HIV-infected persons, Int J Infect Dis 197: ((2008) ), 279–293. |
[30] | Kang M.K. and Park J.G. , Tenofovir disoproxil fumarate-induced severe liver injury in a patient with chronic hepatitis B virus infection, Digest Liver Dis 50: ((2018) ), 621–632. |
[31] | Liu X. and Wang X.J. , Potential inhibitors against -nCoV coronavirus M protease from clinically approved medicines, J Genet Genomics 47: ((2020) ), 119–121. |
[32] | Meng X.Y. , Zhang H.X. , Mezei M. and Cui M. , Molecular Docking: A powerful approach for structure-based drug discovery, Curr. Comput. Aided Drug Des 7: ((2011) ), 146–157. |
[33] | De Ruyck J. , Brysbaert G. , Blossey R. and Lensink M.F. , Molecular docking as a popular tool in drug design, an in silico travel, Adv Appl Bioinforma Chem 9: ((2016) ), 1–11. |
[34] | Chaudhary M.K. , Srivastava A. , Singh K.K. , Tandon P. and Joshi B.D. , Computational evaluation on molecular stability, reactivity, and drug potential of frovatriptan from DFT and molecular docking approach, Comput Theor Chem 1191: ((2020) ), 113031. |
[35] | Ferreira L.G. , Dos Santos R.N. , Oliva G. and Andricopulo A.D. , Molecular docking and structure-based drug design strategies, Molecules 20: ((2015) ), 13384–13421. |
[36] | Kumar Y. , Singh H. and Patel C.N. , In silico prediction of potential inhibitors for the Main protease of SARS-CoV-2 using molecular docking and dynamics simulation based drug-repurposing, J Infect Public Heal 13: ((2020) ), 1210–1223. |
[37] | Elfiky A.A. , Ribavirin, Remdesivir and Sofosbuvir, Galidesivir, Tenofovir against SARS-CoV-2 RNA dependent RNA polymerase (RdRp): A molecular docking study, Life Sci 253: ((2020) ), 117592. |
[38] | Peele K.A. , Durthi C.P. , Srihansa T. , Krupanidhi S. , Sai A.V. , Babu D.J. , Indira M. , Reddy A.R. and Venkateswarulu T.C. , Molecular docking and dynamic simulations for antiviral compounds against SARS-CoV-2: A computational study, Inform Med Unlocked 19: ((2020) ), 100345. |
[39] | Yu. R. , Chen L. , Lan R. , Shen R. and Li P. , Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking, Int J Antimicrob Agents 56: ((2020) ), 106012. |
[40] | Frisch M.J. , Trucks G.W. , Schlegel H.B. , Scuseria G.E. , Robb M.A. , Cheeseman J.R. , Scalmani G. , Barone V. , Mennucci B. , Petersson G.A. , Nakatsuji H. , Caricato M. , Li X. , Hratchian H.P. , Izmaylov A.F. , Bloino J. , Zheng G. , Sonnenberg J.L. , Hada M. , Ehara M. , Toyota K. , Fukuda R. , Hasegawa J. , Ishida M. , Nakajima T. , Honda Y. , Kita O. , Nakai H. , Vreven T. , Montgomery J. , Peralta J.E. , Ogliaro F. , Bearpark M.J. , Heyd J. , Brothers E.N. , Kudin K.N. , Staroverov V.N. , Kobayashi R. , Normand J. , Raghavachari K. , Rendell A.P. , Burant J.C. , Iyengar S.S. , Tomasi J. , Cossi M. , Rega N. , Millam N.J. , Klene M. , Knox J.E. , Cross J.B. , Bakken V. , Adamo C. , Jaramillo J. , Gomperts R. , Stratmann R.E. , Yazyev O. , Austin A.J. , Cammi R. , Pomelli C. , Ochterski J.W. , Martin R.L. , Morokuma K. , Zakrzewski V.G. , Voth G.A. , Salvador P. , Dannenberg J.J. , Dapprich S. , Daniels A.D. , Farkas O. , Foresman J.B. , Ortiz J.V. , Cioslowski J. and Fox D.J. , Gaussian 09, in, Gaussian, Inc., Wallingford, CT, USA, (2009) . |
[41] | Tomasi J. , Mennucci B. and Cammi R. , Quantum mechanical continuum solvation models, Chem Rev 105: ((2005) ), 2999–3093. |
[42] | Shahab. S. , Sheikhi. M. , Filippovich L. , Khaleghian M. , Dikusar E. , Yahyaei H. and Yousefzadeh B.M. , Spectroscopic studies (geometryoptimization, E⟶Z isomerization, UV/Vis, excitedstates, FT-IR, HOMO-LUMO, FMO, MEP, NBO, polarization) andanisotropy of thermal and electrical conductivity of new azomethinedyes in stretched polymer matrix, Silicon 10: ((2018) ), 2361–2365. |
[43] | Bulat F.A. , Chamorro E. , Fuentealba P. and Alejandro T.L. , Condensation of frontier molecular orbital Fukui functions, J Phys Chem A 108: ((2004) ), 342–349. |
[44] | Shahab. S. , Filippovich L. , Sheikhi M. , Kumar R. , Dikusar E. , Yahyaei H. and Muravsky A. , Polarization, excited states, trans-cis properties and anisotropy of thermal and electrical conductivity of the 4-(phenyldiazenyl) aniline in PVA matrix, J Mol Struct 1141: ((2017) ), 703–709. |
[45] | Weinhold F. and Landis C.R. , Neutral bond orbitals and extensions of localized, Chem Educ Res Pract 10: ((2001) ), 91–104. |
[46] | Akman F. , A comparative study based on molecular structure, spectroscopic, electronic, thermodynamic and NBO analysis of some nitrogen-containing monomers, Polym Bull 68: ((2020) ), 1–31. |
[47] | Frisch A. , Nielson A.B. and Holder A.J. , GAUSSVIEW User Manual, Gaussian Inc., Pittsburgh, PA, (2000) . |
[48] | Singh R. , Kumar D. , Goswami Y.C. and Sharma R. , Synthesis, spectral studies and quantum-chemical investigations on S-benzyl β-N-(4-NN biscynodiethylaminophenylmethylene) dithiocarbazate, Arab J Chem 12: ((2019) ), 1537–1544. |
[49] | Yang H. , Yang M. , Ding Y. , Liu Y. , Lou Z. , Zhou Z. , Sun L. , Mo L. , Ye S. , Pang H. , Gao G.F. , Anand K. , Bartlam M. , Hilgenfeld R. and Rao Z. , The crystal structures of severe acute respiratory syndrome virus main protease and its complex with an inhibitor, Proc Natl Acad Sci U S A 100: ((2003) ), 13190–13195. |
[50] | Ramkumaar G.R. , Srinivasan S. , Bhoopathy T.J. and Gunasekaran S. , Vibrational spectroscopic studies of tenofovir using density functional theory method, J Chem 2013: ((2013) ), 1–13. |
[51] | Shahab S. , Sheikhi M. , Filippovich L. , Dikusar E. , Rouhani M. , Pazniak A. and Kumar R. , Molecular investigations of the new synthesized azomethines as antioxidants: Theoretical and experimental studies, Curr Mol Med 19: ((2019) ), 419–433. |
[52] | Shahab S. , Sheikhi M. , Filippovich L. , Ignatovich Z. , Muravsky A. , Alnajjar R. , Kaviani S. and Strogova A. , Optimization, Spectroscopic (FT-IR, Excited States, UV/Vis) Studies, FMO, ELF, LOL, QTAIM and NBO Analyses and Electronic Properties of Two New Pyrimidine Derivatives, Chinese, J Struct Chem 38: ((2019) ), 1615–1639. |
[53] | Shahab S. , Sheikhi M. , Filippovich L , Ihnatovich Z. , Koroleva E. , Drachilovskaya M. , Atroshko M. and Pazniak A. , Spectroscopic (FT-IR, excited states, UV/Vis, polarization) properties, synthesis and quantum chemical studies of new azomethine derivatives, Dyes Pigm 170: ((2019) ), 107647. |
[54] | Sheikhi. M. , Shahab. S. , Filippovich L. , Yahyaei H. , Dikusar E. and Khaleghian M. , New derivatives of (E,E)-azomethines: Design, quantum chemical modeling, spectroscopic (FT-IR, UV/Vis, polarization) studies, synthesis and their applications: Experimental and theoretical investigations, J Mol Struct 1152: ((2018) ), 368–385. |
[55] | Shahab S. , Sheikhi M. , Khaleghian M. , Sharifi S. and Kaviani S. , Theoretical study of interaction between apalutamide anticancer drug and thymine by DFT method, Chinese J Struct Chem 38: ((2019) ), 1645–1663. |
[56] | Amovilli C. , Barone V. , Cammi R. , Cancés E. , Cossi M. , Mennucci B. , Pomelli C.S. and Tomasi J. , Recent advances in the description of solvent effects with the polarizable continuum model, Adv Quantum Chem 32: ((1998) ), 27–261. |
[57] | Shahab S. , Sheikhi M. , Filippovich L. , Dikusar E. , Yahyaei H. , Kumar R. and Khaleghian M. , Design of geometry, synthesis, spectroscopic (FT-IR, UV/Vis, excited state, polarization) and anisotropy (thermal conductivity and electrical) properties of new synthesized derivatives of (E,E)-azomethines in colored stretched poly (vinyl alcohol) matrix, J Mol Struct 1157: ((2018) ), 536–550. |
[58] | Yahyaei H. , Shahab S. , Sheikhi M. , Filippovich L. , Almodarresiyeh H.A. , Kumar R. , Dikusar E. , Yousefzadeh B.M. and Alnajjar R. , Anisotropy (optical, electrical and thermal conductivity) in thin polarizing films for UV/Vis regions of spectrum: Experimental and theoretical investigations, Spectrochim Acta A Mol Biomol Spectrosc 192: ((2018) ), 343–360. |
[59] | Parveen S. and Alnoman R.B. , Potential Exploration of Recent FDA-Approved Anticancer Drugs Against Models of SARS-CoV-2’s Main Protease and Spike Glycoprotein: A Computational Study, Res Appl Chem 11: ((2019) ), 10059–10073. |


