
A Case Report of Severe Type B Lactic Acidosis Following First Dose of Nivolumab in a VHL-Mutated Metastatic Renal Cell Carcinoma
Abstract
We report a case of severe type B lactic acidosis (LA) in a 51-year-old male, 12 days after he received his first dose of nivolumab for metastatic Von Hippel Lindau (VHL)-mutated, clear cell renal cell carcinoma. Throughout his hospital course, infection, hypoperfusion, and tissue necrosis were not identified. We propose that his LA may have resulted from either inherent tumor glycolysis or immune activation and enhanced metabolism. The patient’s course was complicated by acute renal failure, and his LA rose progressively, eventually necessitating daily hemodialysis (HD). After receiving five consecutive days of HD, the patient started everolimus daily with the intent of reducing glycolytic metabolism. Subsequently, the rate of lactic acid production slowed, and HD was no longer required after two doses of everolimus. To our knowledge, this is the first reported case of type B LA following nivolumab administration, and the use of everolimus to treat type B LA in a patient with renal cancer.
INTRODUCTION
We report a case of life threatening lactic acidosis (LA) following a first dose of nivolumab, a monoclonal antibody against the negative immunoregulatory human cell surface receptor, programmed death-1 (PD-1), in a Von Hippel-Lindau (VHL)-mutated renal cell carcinoma (RCC), and subsequent resolution of LA following everolimus, a mammalian Target of Rapamycin (mTOR) inhibitor, administration. To our knowledge, this case of severe type B LA highlights a previously unreported, adverse effect following nivolumab treatment, and proposes using the metabolic effects of everolimus to manage the acidosis.
CLINICAL HISTORY
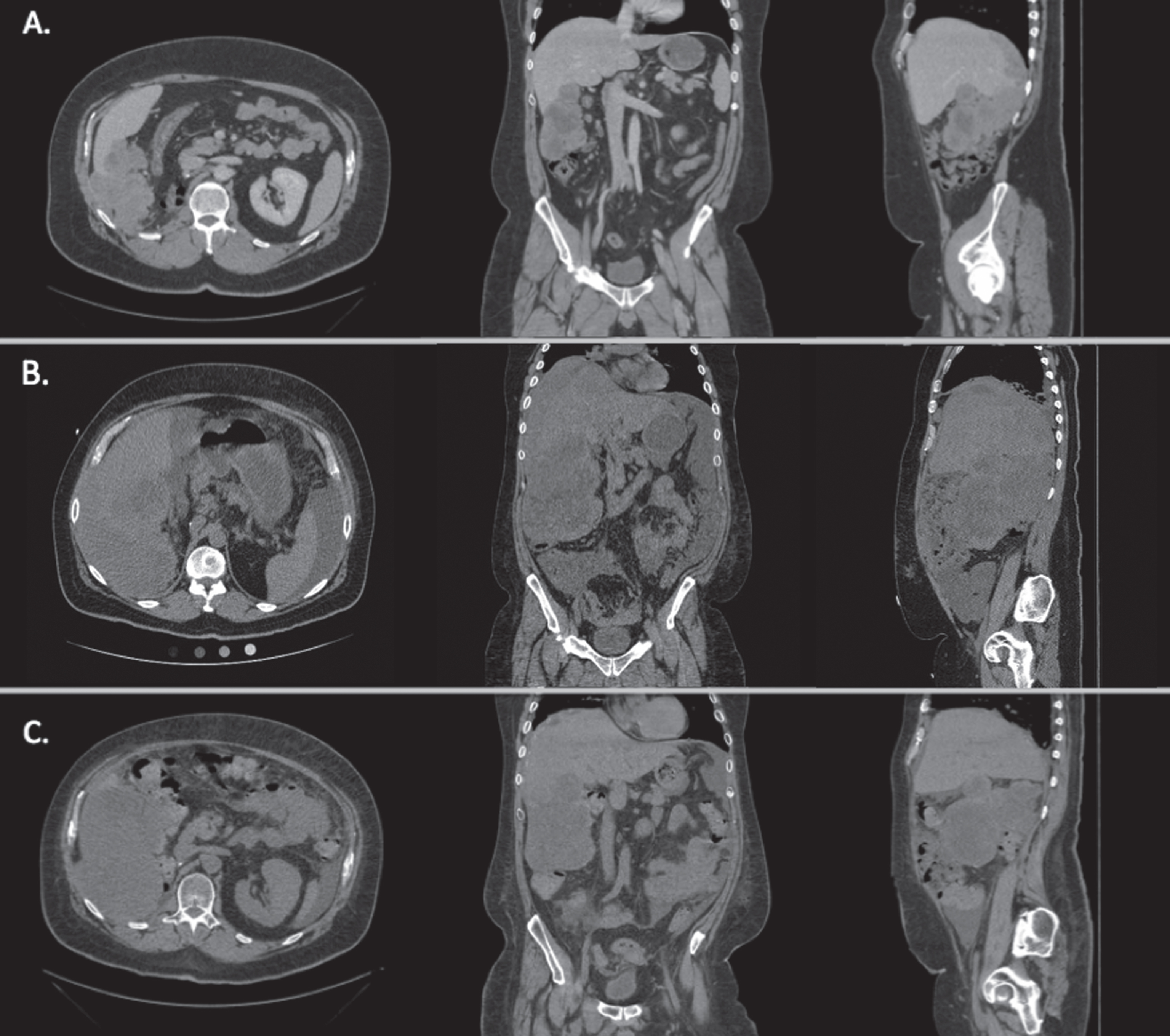
A 51-year-old male presented to the emergency department (ED) with abdominal distention, generalized weakness, and exertional dyspnea. He had been identified to have an 11.5 cm right kidney mass eight months previously. Pathology at the time of right nephrectomy, approximately seven months prior to admission, revealed clear cell renal cell carcinoma (RCC), Fuhrman grade 4 with sarcomatoid features, positive margins and intra-kidney venous involvement, T3a, N0, M0, stage III. Genetic testing of the tumor revealed mutations in VHL, p53, and CHEK2, consistent with a clear cell primary histology. Three months prior to admission, CT chest, abdomen and pelvis with IV contrast demonstrated the occurance of extensive metastatic disease with a large heterogeneous mass in the right abdomen, multiple peritoneal nodules and right-sided pulmonary nodules (Fig. 1A). The patient was treated with sunitinib (a vascular endothelial growth factor receptor 2 [VEGFR2], platelet-derived growth factor receptor b [PDGFRb], and c-kit inhibitor) in combination with infusional gemcitabine, as a part of standard protocol [1]. Repeat CT scan one month prior to admission showed disease progression (Fig. 1B). This prompted the initiation of second line treatment with nivolumab, given intravenously at a standard dose of 3 mg/kg (408 mg total). 12 days following the first dose of nivolumab, the patient presented to the emergency department.
Fig.1
Axial, coronal, and sagittal CT images of the patient’s abdomen and pelvis revealing metastatic disease at different time points during his therapy. A. Prior to sunitinib and gemcitabine therapy. B. After sunitinib and gemcitabine therapy, prior to nivolumab therapy and one month prior to admission. C. 47 days after admission. Image shown after nivolumab and during everolimus therapy. Overall response was mixed as compared to prior to everolimus therapy.
Upon presentation to the emergency department, he complained of extreme fatigue, dyspnea, and diarrhea in the preceding week. He was afebrile (37C), tachycardic (137 bpm), hypotensive (81/66 mm Hg), tachypneic (respiratory rate 30) with an oxygen saturation of 100% while breathing ambient air. Initial work-up was notable for a leukocytosis (23×103/mcL) with 86% neutrophils, anemia with Hct 33% (previous baseline was 30%), acute kidney injury with creatinine of 2.23 mg/dL (baseline 1.1), mild hyperkalemia (5.3 mmol/L). He had an anion gap metabolic acidosis (serum bicarbonate 15, anion gap: 21, venous pH 7.29) with significant LA (lactic acid 10.1 mEq/L). LDH had risen from 683 units/L six weeks prior to 2798 units/L. Additional admission labs may be found in Table 1. A chest X-ray revealed a new right-sided pleural effusion, and CT of the abdomen and pelvis revealed increased size of his known mesenteric and hepatic metastases, but no evidence of bowel ischemia or infection.
Table 1
Admission lab values
| Variable | Reference Range | Admission Value |
| Sodium (mmol/liter) | 136–144 | 135 |
| Potassium (mmol/liter) | 3.3–4.8 | 5.3 |
| Chloride (mmol/liter) | 98–107 | 99 |
| Carbon dioxide (mmol/liter) | 21–29 | 15 |
| Urea nitrogen (mg/dl) | 8–26 | 48 |
| Creatinine (mg/dl) | 0.72–1.25 | 2.23 |
| Glucose (mg/dl) | 70–99 | 110 |
| Calcium (mg/dl) | 8.4–10.5 | 11.3 |
| White cell count (×103/mcl) | 3.9–10.7 | 23.1 |
| Hemoglobin (g/dl) | 14.0–18.1 | 10.3 |
| Hematocrit (%) | 41–49 | 33 |
| Platelet count (×103/mcl) | 135–371 | 340 |
| Lactic acid (mEq/liter) | 0.5–2.2 | 10.1 |
| Venous pH | 7.29 | |
| Venous pCO2 (mmHg) | 38–50 | 35 |
| Venous pO2 (mmHg) | 30–40 | 53 |
| Venous O2 saturation(%) | 90 |
He was admitted to the medical intensive care unit due to his persistent hypotension, concerning for sepsis. Vancomycin and piperacillin/tazobactam were administered. Blood cultures were negative after 48 hours. Despite normalization of his blood pressure with aggressive IV hydration, his LA increased dramatically (Fig. 2) with persistent acidosis despite serial IV bicarbonate treatments. Differential diagnoses at that stage included severe sepsis despite negative cultures and ongoing broad spectrum antibiotic coverage, ischemic bowel despite negative exam and imaging, type B LA from tumor progression, RCC pseudoprogression secondary to nivolumab, nivolumab-induced immune activation, and adrenal insufficiency. High-dose methylprednisolone (125 mg IV daily) was initiated on hospital day three to treat systemic inflammation and suspected nivolumab-induced immune activation.
Fig.2
Lactic acid and bicarbonate levels during hospitalization. Lactic acid trend is shown in red, while bicarbonate levels are shown in black. Time zero represents time of admission. * represents a four-hour hemodialysis session. + represents administration of daily everolimus 10 mg.
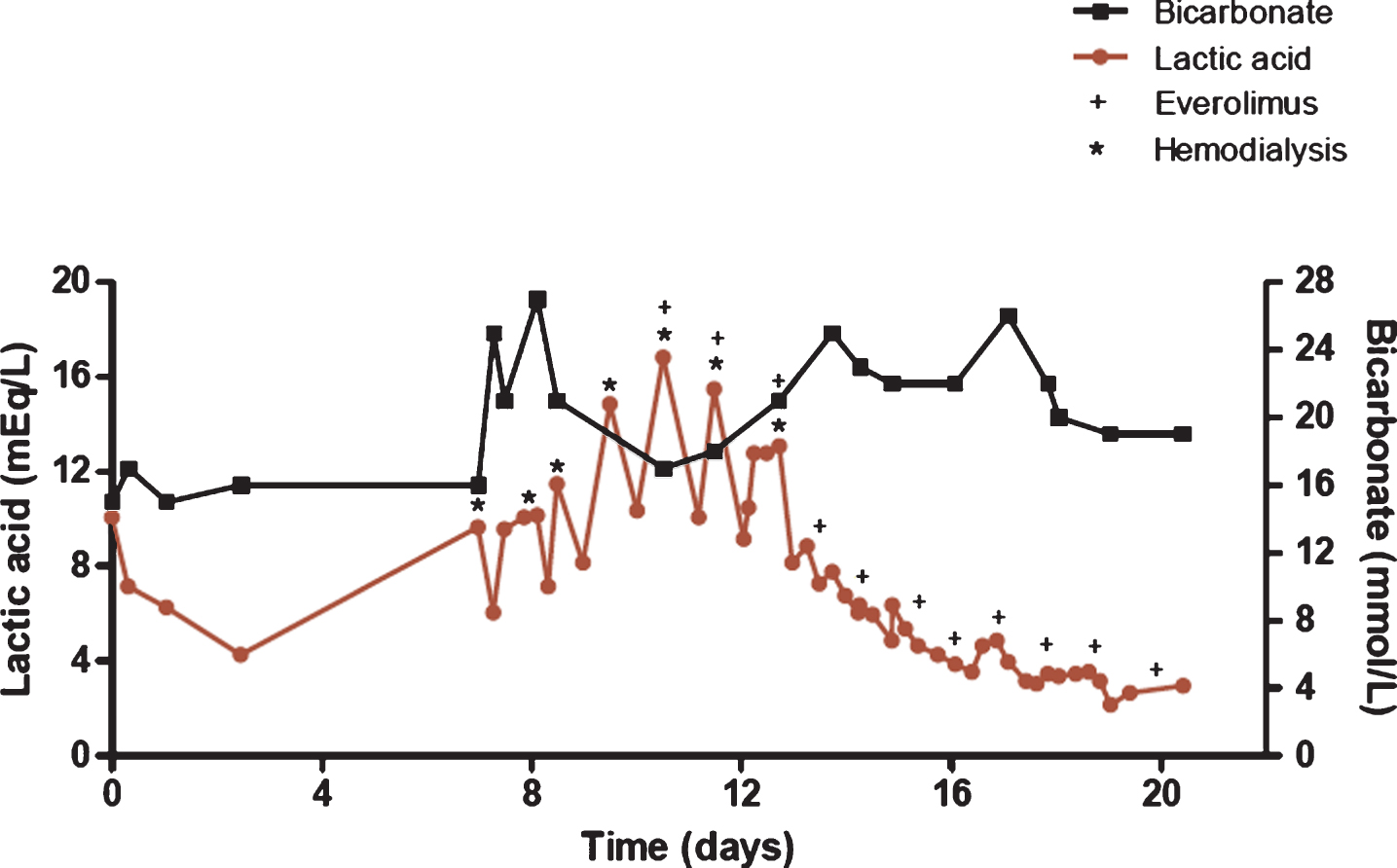
Despite the high dose steroids and resolution of hypotension, the serum lactate continued to rise. Nephrology was consulted given his acute renal failure (eGFR 29 mL/min/1.73m2) with severe acidosis, and hemodialysis (HD) was initiated on hospital day seven. The patient’s lactic acid continued to rise at such a rate that daily HD was required (Fig. 2). On hospital day 11, the patient was administered everolimus 10 mg daily by mouth with the intention of both inhibiting glycolytic activity to reduce lactic acid production, and as an approved targeted agent for treating RCC. As shown in Fig. 2, the patient required HD for two consecutive days following the initiation of everolimus, however the rate of lactic acid production decreased immediately after the first dose. The patient was discharged home on hospital day 22 with a lactic acid level of 3.0 mEq/L, and did not require further HD.
The patient continued to receive everolimus at 10 mg daily as palliative treatment, demonstrating a mixed response to treatment on the first six week interval CT-scan, approximately two months after receiving nivolumab (Fig. 1C). Unfortunately, his disease progressed thereafter, and he died of complications from his metastatic disease in the fifth month after initiation of everolimus.
DISCUSSION
This case documents dramatic metabolic derangement subsequent to nivolumab administration, with resolution after initiation of the mTOR inhibitor, everolimus, in a patient with sustained life-threatening LA and metastatic RCC. Based on literature review, this is the first case report of type B LA in RCC following nivolumab administration.
Throughout this patient’s course, there was no evidence that his LA was a consequence of tissue necrosis or inadequate perfusion or oxygenation, classified as a type A LA. Type B LA is a diagnosis of exclusion, and may be a consequence of medications such as metformin, linezolid, nucleoside reverse transcriptase inhibitors, metabolic enzyme deficiencies, thiamine deficiency, renal or hepatic failure, and malignancy. Type B LA has been reported most commonly in the setting of lymphomas and leukemias [2], but has also been reported in cases of small cell lung cancer, breast cancer, and metastasis from unknown primaries [3]. No cases in renal cell carcinoma have been reported previously.
Nivolumab is a monoclonal antibody (IgG4) that inhibits PD-1 signaling by binding to the PD-1 receptor on activated T-cells and blocking the interaction between PD-1 and the ligands PD-L1 and PD-L2 expressed on tumor cells and immune cells. As a result, the negative signal that suppresses the activation and proliferation of T-cells is removed, and activated T-cells can execute an anti-tumor immune response [4, 5]. PD-1 inhibitors have been used extensively in the treatment of several malignancies including malignant melanoma, lung cancer, renal cell carcinoma, recurrent glioblastoma, and lymphoma [4, 6–8].
The non-selective activation of cellular immunity by PD-1 inhibitors has been associated with severe pneumonitis, colitis, hypophysitis, nephritis, hepatitis, thyroiditis, and myocarditis [9–15]. An alternative explanation for the patient’s type B LA is tremendous immune activation following nivolumab therapy. Activated T cells are known to be highly glycolytic [16], and in this case the patient had a persistent leukocytosis, although this represented a neutrophilia. Repeated imaging taken during his hospitalization showed gross disease stability, thus massive tumor lysis in response to immune attack or rapid disease progression were less likely explanations. Ultimately, we concluded that this case of tremendous type B LA most likely represented aerobic glycolysis by RCC cells (Warburg effect) or by T-cells activated by nivolumab. It is possible that the reduction in tumor burden was in part a delayed response of nivolumab-induced T cell activation and tumor infiltration. It is also possible that the mixed response observed post-hospitalization was due to the effect of everolimus; however, in the absence of mTOR pathway activating mutations, this would be less commonly expected. The patient’s renal failure likely contributed to the rapid rise in lactate due to impaired lactate metabolism and excretion [17].
The strict correlation of the decline in LA upon the administration of everolimus is also notable. The rapid time course of LA reduction does not support the clearance of nivolumab alone. The LA resolved to a more physiologic level within four days of administering everolimus, while seven days of HD resulted in only brief reductions in lactic acid levels at the end of an HD session followed by even higher concentrations each following day.
HYPOTHETICAL MECHANISM
The mutations involved in renal cell carcinoma lead to dysregulation of the metabolic pathways involved oxygen, iron, nutrient utilization [18]. Next generation sequencing (NGS) testing of the reported patient’s renal tumor showed mutation of the Von Hippel-Lindau gene (VHL) on the short arm of chromosome 3, which is a common sporadic mutation found in clear cell RCC [19–24].
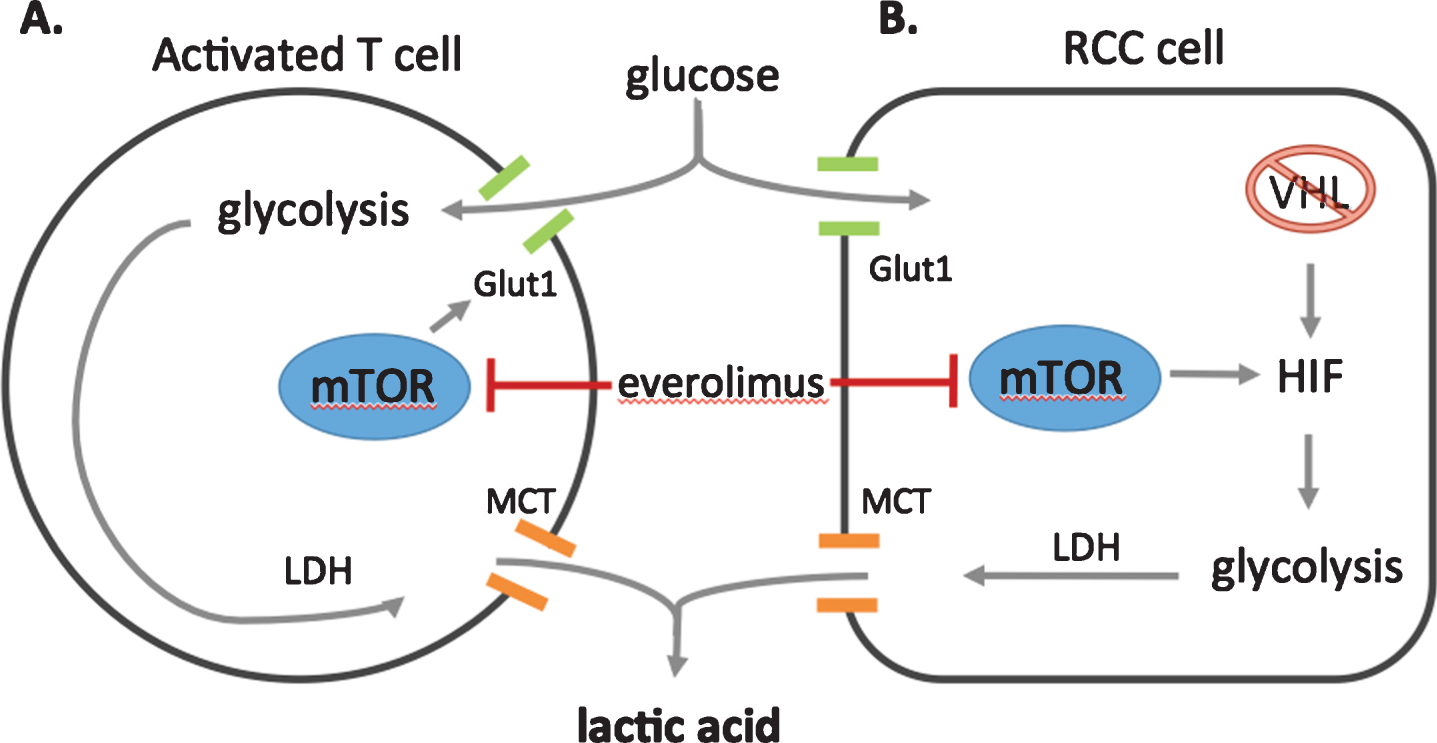
A disrupted VHL complex cannot degrade hypoxia inducible factor (HIF), which accumulates and leads to transcription of genes, including those that promote anaerobic glycolysis [18]. Most VHL mutated tumors display modest or variable glucose uptake, however more aggressive molecular features correspond with increased glucose uptake. In the context of HIF accumulation and severely impaired oxidative phosphorylation, the kidney cancer cell relies primarily on glycolysis for energy production, which can lead to the production of lactic acid as shown in Fig. 3.
Fig.3
Potentials sources of lactic acid production. A. Activated T-cells, such as those stimulated an anti-PD-1 antibody such as nivolumab, are known to have increased glycolysis, leading to lactic acid production via lactate dehydrogenase (LDH). Lactic acid is exported from the cell via a monocarboxylase transporter (MCT) B. Alternatively, in clear cell RCC cells, Von Hippel Lindau (VHL) mutated tumor cells lose their ability to degrade hypoxia induced factor (HIF). The accumulation of HIF promotes anaerobic glycolysis, which produces lactic acid as an end product. mTOR signaling enhances anaerobic glycolysis further, increasing the localization of glucose transporters (Glut1) to the cell membrane for more efficient glucose uptake. Everolimus, the mammalian target of rapamycin (mTOR) inhibitor, blocks HIF translation, promotes the degradation of HIF via the proteasome, and limits glucose importation from glucose transporters, culminating in decreased glycolysis.
A better understanding of the effects of disrupted VHL function has led to the development of targeted therapies such as sunitinib [25], that inhibit VEGF/VEGF receptor signaling. The mammalian target of rapamycin (mTOR) signaling also plays a role in the pathogenesis of many renal cancers, owing to common mutations in regulatory signaling factors (phosphatase and tensin homolog (PTEN), tuberous sclerosis (TSC1/2)) [26–28], and evidence of activated mTOR signaling is present in many high risk tumors [29]. Everolimus, an mTOR inhibitor, was approved in the second or further line setting based on the RECORD1 study [30], where it demonstrated a 30% reduction in disease progression and a median increase in progression free survival of 4.0 [95% CI 3.7–5.5] vs 1.9 [95% CI 1.8–1.9] months.
Using everolimus to manage type B lactic acidosis in acute cancer syndromes has not been reported previously to our knowledge. The downstream effects of mTOR activity include translational activation of hypoxia-inducible factors (HIF), activation of glucose transport [31], and insulin-receptor signaling [32], both of which have potential to lead to increased glycolysis. In fact, Chen et al. [33] found that everolimus decreased standardized uptake values (SUVs) on follow up 18F fluorodeoxyglucose positron emission tomography (18F-FDG PET) scans in metastatic RCC patients, indicating that everolimus suppressed glycolysis within these tumors.
In addition to the tumor metabolic response, this patient underwent an immune checkpoint inhibitor therapy immediately prior to the development of lactic acidosis. In the absence of sepsis, tumor lysis, or accelerated tumor growth, it remains possible that immune cell activation drove the LA. The metabolic features of immune cells are also essential components of the immune response, and mTOR mediated glycolysis has been implicated in CD4 + effector T cell activation [34] and activated CD8 + T cells [35]. Thus, although the case is complicated by the use of high dose steroids, it remains possible that the enhanced lactic acid production was driven by an enabled T cell response. Figure 3 demonstrates the dual sources of lactic acid in this patient, and the proposed mechanism by which mTOR inhibitor therapy effectively disrupted this process.
CONCLUSION
PD-1 inhibitors have produced dramatic responses in treating several solid malignancies such as lung cancer, renal cell carcinoma, melanoma [4, 6, 7]. We are still discovering potential side effects from PD-1 inhibition, as this case of type B LA following a first dose of nivolumab demonstrates. Additional case reports of similar outcomes or other novel side effects will be beneficial in defining the full spectrum of responses to nivolumab and other immune checkpoint inhibitors. In addition to palliative treatment for kidney cancer, everolimus may be an effective transient treatment for type B LA. The use of an mTOR inhibitor may be helpful in other patients with RCC-associated type B LA.
REFERENCES
[1] | Michaelson MD , et al. Phase 2 trial of sunitinib and gemcitabine in patients with sarcomatoid and/or poor-risk metastatic renal cell carcinoma. Cancer (2015) ;121: (19):3435–43. |
[2] | Sia P , Plumb TJ , Fillaus JA . Type B lactic acidosis associated with multiple myeloma. Am J Kidney Dis (2013) ;62: (3):633–7. |
[3] | de Groot R , et al. Type B lactic acidosis in solid malignancies. Neth J Med (2011) ;69: (3):120–3. |
[4] | Motzer RJ , et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med (2015) ;373: (19):1803–13. |
[5] | Siska PJ , Rathmell JC . T cell metabolic fitness in antitumor immunity. Trends Immunol (2015) ;36: (4):257–64. |
[6] | Larkin J , et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med (2015) ;373: (1):23–34. |
[7] | Borghaei H , et al. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med (2015) ;373: (17):1627–39. |
[8] | Ansell SM , et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med (2015) ;372: (4):311–9. |
[9] | Nishino M , et al. Anti-PD-1-Related Pneumonitis during Cancer Immunotherapy. N Engl J Med (2015) ;373: (3):288–90. |
[10] | Postow MW . Toxicities associated with checkpoint inhibitor immunotherapy. UpToDate. (2016) . |
[11] | Okano Y , et al. Nivolumab-induced hypophysitis in a patient with advanced malignant melanoma. Endocr J. (2016) ;63: (10):905–12. |
[12] | Vandiver JW , Singer Z , Harshberger C . Severe Hyponatremia and Immune Nephritis Following an Initial Infusion of Nivolumab. Target Oncol (2016) ;11: (4):553–6. |
[13] | Laubli H , et al. Acute heart failure due to autoimmune myocarditis under pembrolizumab treatment for metastatic melanoma. J Immunother Cancer (2015) ;3: :11. |
[14] | Semper H , et al. Drug-induced myocarditis after nivolumab treatment in a patient with PDL1- negative squamous cell carcinoma of the lung. Lung Cancer (2016) ;99: :117–9. |
[15] | Tanaka R , et al. Nivolumab-induced thyroid dysfunction. Jpn J Clin Oncol (2016) ;46: (6):575–9. |
[16] | Donnelly RP , Finlay DK . Glucose, glycolysis and lymphocyte responses. Mol Immunol (2015) ;68: (2 Pt C):513–9. |
[17] | Bellomo R . Bench-to-bedside review: Lactate and the kidney. Crit Care (2002) ;6: (4):322–6. |
[18] | Linehan WM , Srinivasan R , Schmidt LS . The genetic basis of kidney cancer: A metabolic disease. Nat Rev Urol (2010) ;7: (5):277–85. |
[19] | Gnarra JR , et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet (1994) ;7: (1):85–90. |
[20] | Nickerson ML , et al. Improved identification of von Hippel-Lindau gene alterations in clear cell renal tumors. Clin Cancer Res (2008) ;14: (15):4726–34. |
[21] | Shuin T , et al. Frequent somatic mutations and loss of heterozygosity of the von Hippel-Lindau tumor suppressor gene in primary human renal cell carcinomas. Cancer Res (1994) ;54: (11):2852–5. |
[22] | Duan DR , et al. Inhibition of transcription elongation by the VHL tumor suppressor protein. Science (1995) ;269: (5229):1402–6. |
[23] | Pause A , et al. The von Hippel-Lindau tumor-suppressor gene product forms a stable complex with human CUL-2, a member of the Cdc53 family of proteins. Proc Natl Acad Sci U S A (1997) ;94: (6):2156–61. |
[24] | Kibel A , et al. Binding of the von Hippel-Lindau tumor suppressor protein to Elongin B and C. Science (1995) ;269: (5229):1444–6. |
[25] | Motzer RJ , et al. Activity of SU11248, a multitargeted inhibitor of vascular endothelial growth factor receptor and platelet-derived growth factor receptor, in patients with metastatic renal cell carcinoma. J Clin Oncol (2006) ;24: (1):16–24. |
[26] | Hakimi AA , et al. Adverse outcomes in clear cell renal cell carcinoma with mutations of 3p21 epigenetic regulators BAP1 and SETD A report by MSKCC and the KIRC TCGA research network. Clin Cancer Res (2013) ;19: (12):3259–67. |
[27] | Chism DD , Rathmell WK . Seeing the forest for the trees: Kidney oncogenomes in relation to therapeutic outcomes. Clin Cancer Res (2014) ;20: (7):1721–3. |
[28] | Cancer Genome Atlas Research N, et al. The Cancer Genome Atlas Pan-Cancer analysis project. Nat Genet (2013) ;45: (10):1113–20. |
[29] | Hudes GR . Targeting mTOR in renal cell carcinoma. Cancer (2009) ;115: (Suppl 10):2313–20. |
[30] | Motzer RJ , et al. Efficacy of everolimus in advanced renal cell carcinoma: A double-blind, randomised, placebo-controlled phase III trial. Lancet (2008) ;372: (9637):449–56. |
[31] | Wieman HL , Wofford JA , Rathmell JC . Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell (2007) ;18: (4):1437–46. |
[32] | Gibbons JJ , Abraham RT , Yu K . Mammalian target of rapamycin: Discovery of rapamycin reveals a signaling pathway important for normal and cancer cell growth. Semin Oncol (2009) ;36: (Suppl 3):S3–S17. |
[33] | Chen JL , et al. FDG-PET as a predictive biomarker for therapy with everolimus in metastatic renal cell cancer. Cancer Med (2013) ;2: (4):545–52. |
[34] | Michalek RD , et al. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J Immunol (2011) ;186: (6):3299–303. |
[35] | Cammann C , et al. Early changes in the metabolic profile of activated CD(8+) T cells. BMC Cell Biol. (2016) ;17: (1):28. |


