
Combining Biomarkers with Genetics In Prodromal/Earliest Phase Parkinson’s Disease
Abstract
Family studies have linked several rare genetic variants to hereditary forms of Parkinson’s disease (PD). In addition to these monogenic forms, many PD cases are associated with genetic risk factors. Asymptomatic individuals carrying pathogenic variants linked to PD are at risk of developing the disease later in life, thereby providing a unique opportunity for the detection of the earliest pathophysiological and later clinical changes and, importantly, also of protective and compensatory features and mechanisms. However, the rarity of monogenic PD-causing variants is a major challenge of this approach. In this review, we discuss recent advances in the search for biomarkers in the prodromal/earliest phase of genetically linked PD.
Plain Language Summary
While the cause of most cases of Parkinson’s disease (PD) is still unknown, and age is considered the greatest risk factor, a combination of environmental influences and genetics are thought to affect disease risk and progression. The identification of carriers of pathogenic genetic changes, who have not yet developed motor symptoms of PD, offers the chance to closely monitor developing signs of PD. Some of these signs may be suitable biomarkers and could be used to predict early stages of the disease. In this review, we discuss recent advances in the search for biomarkers in the prodromal/earliest phase of genetically linked PD.
INTRODUCTION
Genetic variants of the germline are inherited or arise de novo and are present from conception. There are several hereditary forms of Parkinson’s disease (PD).1,2 When including GBA1 variants as the strongest known risk factor for PD with age-dependent “penetrance” of pathogenic variants, genetic forms of PD explain the etiology in about ∼14% of all PD patients.3
The pathology of PD—this largely applies to both “idiopathic” and genetic PD—is characterized in most cases by the presence of aggregated α-synuclein in the form of Lewy bodies and Lewy neurites in the brain. Current evidence suggests that the deposition and spread of pathological forms of misfolded α-synuclein drive the development and progression of the disease.4 By the time a clinical diagnosis is confirmed, it is estimated that 40–60% of dopaminergic neurons have degenerated and synaptic function has reduced by up to 80%.5 Therefore, neuroprotective interventions are required early on i.e. during the prodromal phase of PD. In this period, a clinical diagnosis based on fully evolved motor parkinsonism is not yet possible.6–8 This latent phase can vary from 5 to more than 20 years and is characterized by a variety of non-motor symptoms, in isolation or combined, including autonomic dysfunction, rapid eye movement sleep behavior disorder, olfaction, and depression.9–12 As indicated above, the prodromal phase is an important stage of the course of PD for investigating the pathophysiological cascade and, when neuroprotective treatment becomes available, to possibly slow or prevent the onset of motor symptoms in PD.6 In order to identify individuals during this phase and thus provide an early diagnosis of PD, biomarkers are urgently needed. However, the identification of suitable biomarkers is challenging as the type of symptoms, prevalence, time of occurrence, and rate of progression of prodromal symptoms vary considerably between patients.13
While the cause of most cases of PD is still unknown, and age is considered the greatest risk factor, a combination of environmental influences and genetics are thought to affect disease risk and progression. The identification of carriers of pathogenic variants or polygenic risk scores, who have not yet developed motor symptoms of PD, offers the chance to closely monitor developing signs of PD that may be suitable as biomarkers. In this review, we discuss recent advances in the search for biomarkers in the prodromal/earliest phase of genetically linked PD.
GENETICS OF PARKINSON’S DISEASE
Family studies of PD reported rare variants in more than 20 genes.2 However, the relevance of some of these genes and variants is unclear, and replication or functional validation studies are missing. Over the past three decades, seven genes have unequivocally been linked to monogenic PD, including four autosomal dominantly inherited ones (α-synuclein (SNCA), leucine-rich repeat kinase 2 (LRRK2), vacuolar protein sorting-associated protein 35 (VPS35), and Coiled-Coil-Helix-Coiled-Coil-Helix Domain Containing 2 (CHCHD2)) and three recessively transmitted ones (E3 ubiquitin ligase Parkin (PRKN), PTEN-induced putative kinase 1 (PINK1), and PARK7 (DJ-1)).14 The G2019S mutation in the LRRK2 gene is the most common form of monogenic PD and accounts for up to 1–6% of sporadic and 3–19% of familial PD.6 It is inherited with reduced penetrance that ranges between 30 and 80% at age 80. Recently, a large family study has revealed a new pathogenic variant in RAB32 that causes autosomal dominant PD suggesting that there are additional monogenic forms to be detected.15 Molecular studies of these genes provide unique opportunities to investigate the mechanisms of neuronal degeneration in models of PD. Collectively, these studies demonstrated the importance of failure of proteostasis, defects in vesicular transport and endolysosomal function, and failure of mitochondrial quality control/oxidative stress responses in PD.16
In addition to the monogenic forms, many PD cases are associated with genetic risk factors. Seventy-eight independent genome-wide significant loci were identified in a large-scale multi-ancestry meta-analysis of PD including 49,049 cases, 18,785 proxy cases, and 2,458,063 controls.17 Variants at the SNCA locus and in the beta-glucocerebrosidase 1 (GBA1) gene have been discovered as the most important genetic risk factors for idiopathic PD. Notably, more than 100 variants in GBA1 have been associated with an increased risk of developing the disease.13 However, there is substantial variability in the pathogenicity of different variants. For example, GBA1 variants classified as severe (e.g., L483P) show an odds ratio of 10–15, while mild variants (e.g., N409S) have an odds ratio of less than five.13 This illustrates the heterogeneous character of PD even within the group of patients harboring GBA1 risk variants. Therefore, GBA1-subgroup classification based on variant severity is important for subsequent studies. Notably, variability in variant distribution is also reported among different ethnicities. For example, a novel GBA1 variant, associated with an increased risk of developing PD, has recently been identified in the African population.18 This highlights the importance of understanding ancestry-specific genetic risk in PD, which is still understudied in non-European populations.19 Studying genetic risk in underrepresented populations may not only be relevant to enhancing our knowledge of underlying pathological mechanisms in PD but is also important when it comes to targeted treatments in clinical trials.18
COMBINING BIOMARKERS AND GENETICS IN PRODROMAL PARKINSON’S DISEASE
PD is a complex disorder comprised of various clinical phenotypes, pathological findings, and genetic predispositions.20 Recent cohort studies have confirmed the high phenotypic variability and the numerous genetic variants associated with PD risk contribute to the heterogeneity of multiple motor and nonmotor aspects.21,22 For example, the timescale over which PD with dementia may develop ranges from months to decades and neurodegeneration is not limited to the nigrostriatal dopaminergic denervation.23,24 This heterogeneity in PD makes it challenging to find reliable biomarkers and intensive research efforts have been performed to detect biomarkers in cerebrospinal fluid (CSF), blood, and through imaging modalities. Within the realm of biofluidic biomarkers, α-synuclein, glial fibrillary acidic protein (GFAP), amyloid-beta, tau, p-tau, as well as inflammatory markers, mitochondrial DNA (mtDNA), and microRNAs have been discussed.25,26 Of these, α-synuclein seed amplification assays (SAAs) have shown promise for the biochemical diagnosis of PD.27–29 GFAP could be of use mainly in tracking PD progression, the Alzheimer’s disease biomarkers amyloid-beta and (p)-tau are relevant to evaluating cognitive decline and motor function in PD.30,31 Significantly increased levels of inflammatory markers (blood: IL-6, TNF-α, IL-1β, STNFR1, CRP, CCL2, CX3CL1, and CXCL12; CSF: IL-6, TNF-α, IL-1β, CRP and CCL2) were recently verified in a meta-analysis of PD patients and control groups confirming that neuroinflammation plays a role in PD.32 The use of microRNAs and mtDNA as biomarkers for PD is new and promising but still in a developing phase.26,33 Imaging modalities to test for PD include magnetic resonance imaging (MRI), dopamine transporter (DAT) single-photon emission computed tomography (SPECT), fluoro-3,4-dihydroxyphenylalanine (FDOPA) positron emission tomography (PET), and [18F]fluorodeoxyglucose (FDG) PET.25,34
Based on the numerous studies of neuropathological findings, genetic contributions, and biomarker research, a novel biologically based classification of PD has been proposed. This classification takes the complexity and heterogeneity of the disease into account and defines disease status by the presence or absence of pathological α-synuclein in tissue or CSF, by the evidence of underlying neurodegeneration described by neuroimaging procedures, and by the occurrence of gene variants that cause or strongly predispose to PD.14 This biological classification of PD could improve objective preclinical and clinical diagnosis, disease staging, and enable the accurate subdivision of PD according to disease mechanisms. Moreover, it could also advance research in multiple fields, such as epidemiology, biomarker discovery, and patient-specific development of disease-modifying therapies. To assess biomarkers of prodromal PD in a longitudinal and multimodal fashion, a homogeneous group of non-manifesting carriers of gene variants, which cause or strongly predispose to PD, need to be recruited. However, the rarity of monogenic PD-causing variants is a major challenge of this approach. Therefore, only a few studies combining biomarker analysis and genetic PD have been performed so far. These studies focused mainly on LRRK2-PD (G2019S), PRKN-PD, and GBA1-PD variant carriers.28,29,35,36
α-SYNUCLEIN SEED AMPLIFICATION ASSAYS
α-synuclein-SAAs are based on the self-replicative nature of misfolded α-synuclein aggregates (seeds). In these assays, α-synuclein seeds in biological fluids are amplified by a cyclical process and detected by fluorescent dyes specific for amyloids.37
α-synuclein-SAAs have been performed using CSF and blood samples from patients with genetic PD (Table 1). Analysis of CSF samples revealed that GBA1-PD variant carriers show an overall high percentage of positive seeding (more than 90%).29,36,38 Patient cohorts of LRRK2-PD mutation carriers are reported with 40%, 67%, and 78% of positive SAA results suggesting a heterogeneous phenotype among LRRK2 mutation carriers.29,36,39 However, the performed SAAs may not be sensitive enough to detect low amounts of pathological α-synuclein aggregates. Interestingly, within the group of non-manifesting GBA1 (N409S) and LRRK2 (G2019S) mutation carriers, 8% had positive α-synuclein SAA results, of whom 9% were LRRK2 and 7% were GBA1 carriers.29 This result correlates with the lifetime penetrance of PD among GBA1 N409S carriers but does not reflect estimates of PD penetrance among LRRK2 G2019S carriers. It remains to be seen whether the non-manifesting mutation carriers with a positive SAA result will develop PD in the future. Patients with bi-allelic mutations in recessively inherited genes (PRKN, PINK1) did not show CSF α-synuclein seeding, whereas heterozygous mutation carriers showed α-synuclein seeding in 59% of the individuals.36
Table 1
α-synuclein seed amplification assays in patients with genetic Parkinson’s disease
| Tissue | Genetic/disease status | Positive seeding+/n (%) | Reference |
| CSF | LRRK2-PD (G2019S) | 6/15 (40) | 39 |
| LRRK2 NMC (G2019S) | 3/16 (19) | ||
| Controls | 2/10 (20) | ||
| GBA1-PD | 7/7 (100) | 38 | |
| LRRK2-PD | 0/1 (0) | ||
| Controls | 1/62 (2) | ||
| GBA1-PD (risk variants) | 48/53 (91) | 36 | |
| GBA1-PD (mild variants) | 11/17 (65) | ||
| GBA1-PD (severe variants) | 27/29 (93) | ||
| LRRK2-PD | 7/9 (78) | ||
| PRKN-PD (n = 15), PINK1-PD (n = 1), DJ-1-PD (n = 1) (heterozygous) | 10/17 (59) | ||
| PRKN-PD (n = 1), PINK1-PD (n = 2) (bi-allelic) | 0/3 (0) | ||
| GBA1, LRRK2 NMC | 2/14 (14) | ||
| Controls | 2/26 (8) | ||
| SNCA-PD (triplication) | 1/1 (100) | 40 | |
| Controls | 2/26 (8) | ||
| GBA1-PD (N409S; mild variant) | 47/49 (96) | 29 | |
| LRRK2-PD (G2019S) | 83/123 (67) | ||
| GBA1 NMC | 11/151 (7) | ||
| LRRK2 NMC | 14/159 (9) | ||
| Controls | 6/163 (4) | ||
| Blood | PRKN-PD (bi-allelic) | 0/17 (0) | 28 |
| Controls | 11/128 (9) | ||
| PRKN-PD (bi-allelic) | 8/13 (62) | 41 | |
| Controls | 0/10 (0) |
NMC, non-manifesting carrier.
Analysis of blood samples from patients with genetic PD has only been performed for biallelic PRKN-PD in two independent studies so far. The first study showed no positive seeding in 17 tested patients,28 whereas the second study found an α-synuclein seeding rate of 62% in 13 patients.41 Even though biallelic PRKN mutation carriers have only been tested in a small subset of patients, the different SAA methodologies used in these studies might have led to the discrepant results.
In summary, while data suggest that α-synuclein-SAAs represent very promising biomarkers for PD diagnosis, it remains unclear whether this method will be suitable for detecting prodromal stages of genetically linked PD. Notably, in patients with rapid eye movement sleep behavior disorder, α-synuclein positivity detected by SAA in CSF was associated with an increased risk of subsequent diagnosis of PD or dementia with Lewy bodies.42
NEUROIMAGING MARKERS
DAT-SPECT has been established as a method to detect the presence of nigrostriatal deficit not only in clinical PD but also in the early or pre-symptomatic stages of the disease and to monitor DAT loss with the progression of neurodegeneration.43 Therefore, DAT-SPECT has also been preferentially used to study genetic PD. Overall, GBA1 and LRRK2 mutations present a relatively asymmetric striatal dopaminergic neuronal loss, similar to idiopathic PD. Most cases with PRKN, PINK1, and SNCA mutations show a symmetrical loss.35
Decreased DAT binding was shown in 43.7% of 49 asymptomatic G2019S carriers.44 In contrast, in a large cross-sectional study investigating non-manifesting LRRK2 and GBA1 mutation carriers (208 and 184 individuals, respectively), only a minority had a DAT deficit (11% of LRRK2 and 3% of GBA1 non-manifesting carriers).45 Compared with healthy controls, both LRRK2 and GBA1 non-manifesting carriers had significantly increased mean scores on the Movement Disorders Society Unified Parkinson’s Disease Rating Scale.45 These data suggest that subtle motor and non-motor signs of PD in non-manifesting carriers can precede a DAT deficit. Notably, dopamine turnover analyzed via FDOPA-PET was elevated in asymptomatic LRRK2 mutation carriers compared to age-matched control values.46
In an attempt to assess whether a combination of systematic clinical testing and different imaging techniques in familial PD patients could detect subclinical signs in the preclinical and prodromal stages of PD, a retrospective evaluation was reported of a cohort of 39 participants, which included LRRK2 mutation carriers at different disease stages (preclinical, prodromal, clinical), who underwent DAT-SPECT.6 In the preclinical group, 28% of the participants had normal and 72% had abnormal DAT-SPECT scans. In the prodromal group, all the participants showed abnormal DAT-SPECT scans, albeit with different patterns of severity. In the clinical group of LRRK2 mutation carriers, all participants had severely abnormal DAT-SPECT scans.
CONCLUSIONS AND PERSPECTIVES
α-synuclein SAAs show clear potential to detect idiopathic PD and to distinguish patients from healthy controls. However, genetic PD has currently been studied only in a very limited number of patients and genetic conditions. DAT-SPECT has been established as a reliable biomarker in idiopathic and genetic PD, but the findings for prodromal PD of genetic subgroups are not yet conclusive. We are still at the beginning of our ability to understand and diagnose the early stages of the disease, mainly due to the lack of data that reflect these stages at the molecular level.
However, there are promising findings, especially in carriers of the GBA1 N409S variant, who show evidence of abnormal CSF α-synuclein aggregation prior to other detectable clinical or biomarker changes, including alterations in DAT imaging.29 Nevertheless, further studies on more easily accessible biomarkers, such as a blood-based diagnosis are needed. Notably, increased blood-derived mtDNA damage was found not only in LRRK2-linked PD patients but also in asymptomatic LRRK2 mutation carriers suggesting that mtDNA damage may precede a clinical PD diagnosis and is detectable in prodromal stages.26 It remains to be investigated whether the non-manifesting LRRK2 mutation carriers with elevated mtDNA damage will develop clinical PD.
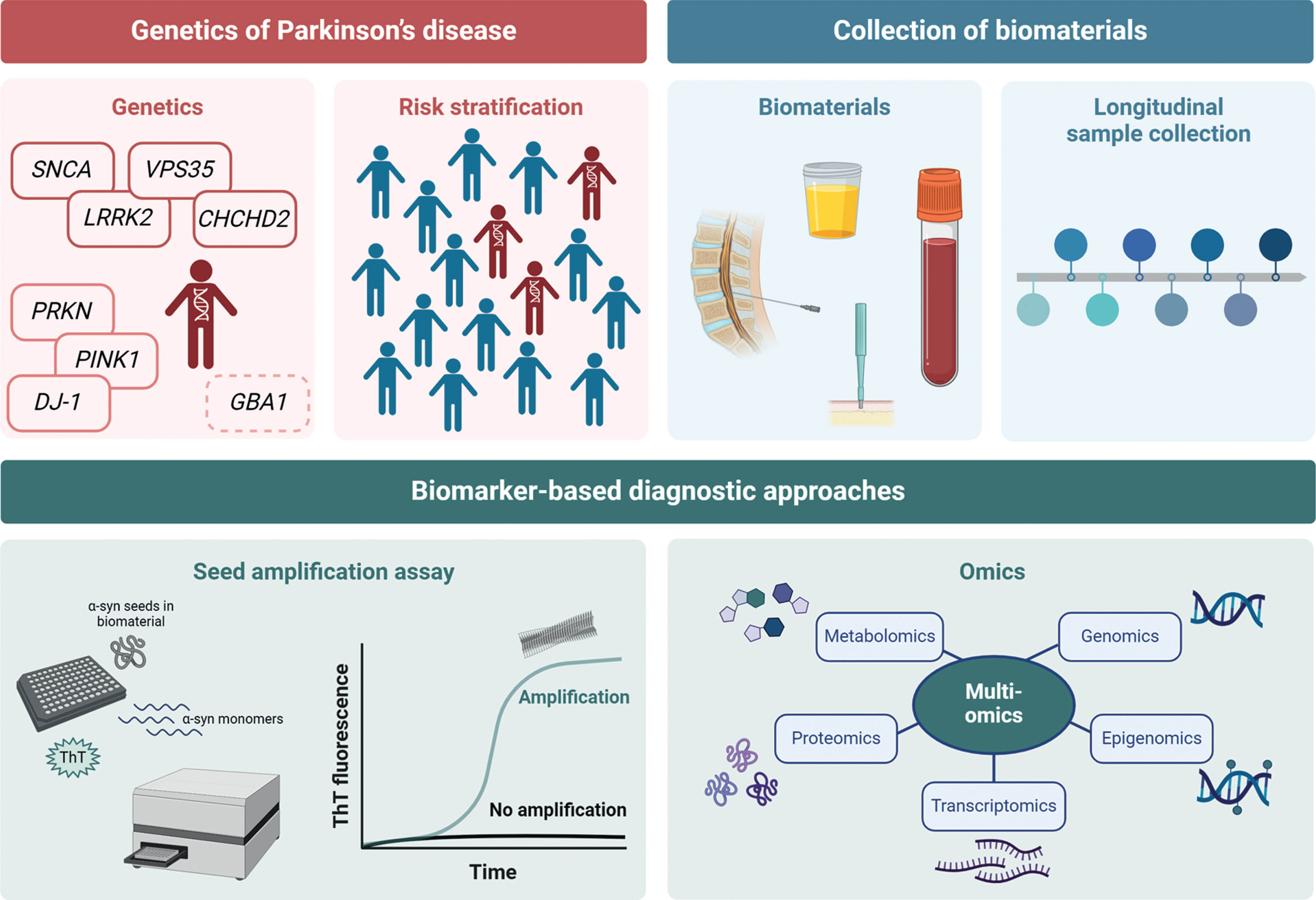
For future studies, it will be important to identify genetically defined subgroups to establish at-risk cohorts (Fig. 1). Patients should be stratified according to genetic variant severity and those carrying severe mutations to be preferentially included. Longitudinal research is needed to examine the predictive value of α-synuclein SAA, mtDNA damage, and DAT-SPECT over time. In this context, repeated collections of biomaterials, starting at the prodromal disease stage, are required, and samples will have to be analyzed in a multimodal fashion, including multi-omics as well as patient-derived experimental disease models to detect new biomarkers specific for PD. In a recent approach, mass spectrometry-based proteomic phenotyping was used to identify a panel of blood biomarkers in early PD.47 Furthermore, a combination of systems biology and data integration methods was employed to identify differences in transcriptomic and metabolic signatures between PD patient and healthy individual-derived midbrain neural precursor cells.48 These and other approaches will provide new single biomarkers or biomarker panels that form a distinct signature of PD. Moreover, performing timely studies during the progression from health to disease might inform us about biomarkers that reflect the underlying pathological processes and present possible read-outs for target engagement.
Fig. 1
Scheme on the search for new biomarkers. (i) Stratification of risk groups of asymptomatic mutation carriers, (ii) longitudinal collection of biomaterials, (iii) verification of biomarker search by using the established α-synuclein seed amplification assays, and (iv) search for new biomarkers using a multi-omics approach. Created with BioRender.com.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
C.K. serves as a medical advisor to Centogene, Takeda, Retromer Therapeutics, and Lundbeck, and received speakers’ honoraria from Bial and Desitin and royalties from Oxford University Press.
REFERENCES
1. | Morris HR , Spillantini MG , Sue CM , et al. The pathogenesis of Parkinson’s disease. Lancet (2024) ; 403: : 293–304. |
2. | Blauwendraat C , Nalls MA and Singleton AB. The genetic architecture of Parkinson’s disease. Lancet Neurol (2020) ; 19: : 170–178. |
3. | Skrahina V , Gaber H , Vollstedt E , et al. The Rostock International Parkinson’s Disease (ROPAD) Study: Protocol and initial findings. Mov Disord (2021) ; 36: : 1005–1010. |
4. | Brás IC and Outeiro TF. Alpha-synuclein: Mechanisms of release and pathology progression in synucleinopathies. Cells (2021) ; 10: : 1–19. |
5. | Mahlknecht P , Seppi K and Poewe W. The concept of prodromal Parkinson’s disease. J Parkinsons Dis (2015) ; 5: : 681–697. |
6. | Hustad E and Aasly JO. Clinical and imaging markers of prodromal Parkinson’s disease. Front Neurol (2020) ; 11: : 395. |
7. | Postuma RB and Berg D. Prodromal Parkinson’s disease: The decade past, the decade to come. Mov Disord (2019) ; 34: : 665–675. |
8. | Gan-Or Z , Girard SL , Noreau A , et al. Parkinson’s disease genetic loci in rapid eye movement sleep behavior disorder. J Mol Neurosci (2015) ; 56: : 617–622. |
9. | Bloem BR , Okun MS and Klein C. Parkinson’s disease. Lancet (2021) ; 397: : 2284–2303. |
10. | Postuma RB and Berg D. Advances in markers of prodromal Parkinson disease. Nat Rev Neurol (2016) ; 12: : 622–634. |
11. | Kalia LV and Lang AE. Parkinson’s disease. Lancet (2015) ; 386: : 896–912. |
12. | Yilmaz R , Hopfner F , van Eimeren T , et al. Biomarkers of Parkinson’s disease: 20 years later. J Neural Transm (2019) ; 126: : 803–813. |
13. | Höglinger G , Schulte C , Jost WH , et al. GBA-associated PD: Chances and obstacles for targeted treatment strategies. J Neural Transm (2022) ; 129: : 1219–1233. |
14. | Höglinger GU , Adler CH , Berg D , et al. A biological classification of Parkinson’s disease: The SynNeurGe research diagnostic criteria. Lancet Neurol (2024) ; 23: : 191–204. |
15. | Gustavsson EK , Follett J , Trinh J , et al. RAB32 Ser71Arg in autosomal dominant Parkinson’s disease: Linkage, association, and functional analyses. Lancet Neurol (2024) ; 23: : 603–614. |
16. | Panicker N , Ge P , Dawson VL , et al. The cell biology of Parkinson’s disease. J Cell Biol (2021) ; 220: : e202012095. |
17. | Kim JJ , Vitale D , Otani DV , et al. Multi-ancestry genome-wide association meta-analysis of Parkinson’s disease. Nat Genet (2024) ; 56: : 27–36. |
18. | Rizig M , Bandres-Ciga S , Makarious MB , et al. Identification of genetic risk loci and causal insights associated with Parkinson’s disease in African and African admixed populations: A genome-wide association study. Lancet Neurol (2023) ; 22: : 1015–1025. |
19. | Khani M , Cerquera-Cleves C , Kekenadze M , et al. Towards a global view of Parkinson’s disease genetics. Ann Neurol (2024) ; 95: : 831–842. |
20. | Wüllner U , Borghammer P , Choe C un , et al. The heterogeneity of Parkinson’s disease. J Neural Transm (2023) ; 130: : 827–838. |
21. | Iwaki H , Blauwendraat C , Leonard HL , et al. Genetic risk of Parkinson disease and progression: An analysis of 13 longitudinal cohorts. Neurol Genet (2019) ; 5: : e348. |
22. | Bartl M , Dakna M , Schade S , et al. Longitudinal change and progression indicators using the Movement Disorder Society-Unified Parkinson’s Disease Rating Scale in two independent cohorts with early Parkinson’s disease. J Parkinsons Dis (2022) ; 12: : 437–452. |
23. | Dong-Chen X , Yong C , Yang X , et al. Signaling pathways in Parkinson’s disease: Molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther (2023) ; 8: : 73. |
24. | Carceles-Cordon M , Weintraub D and Chen-Plotkin AS. Cognitive heterogeneity in Parkinson’s disease: A mechanistic view. Neuron (2023) ; 111: : 1531–1546. |
25. | Yamashita KY , Bhoopatiraju S , Silverglate BD , et al. Biomarkers in Parkinson’s disease: A state of the art review. Biomark Neuropsychiatry (2023) ; 9: : 100074. |
26. | Qi R , Sammler E , Gonzalez-Hunt CP , et al. A blood-based marker of mitochondrial DNA damage in Parkinson’s disease. Sci Transl Med (2023) ; 15: : eabo1557. |
27. | Kluge A , Bunk J , Schaeffer E , et al. Detection of neuron-derived pathological α-synuclein in blood. Brain (2022) ; 145: : 3058–3071. |
28. | Okuzumi A , Hatano T , Matsumoto G , et al. Propagative α-synuclein seeds as serum biomarkers for synucleinopathies. Nat Med (2023) ; 29: : 1448–1455. |
29. | Siderowf A , Concha-Marambio L , Lafontant DE , et al. Assessment of heterogeneity among participants in the Parkinson’s Progression Markers Initiative cohort using α-synuclein seed amplification: A cross-sectional study. Lancet Neurol (2023) ; 22: : 407–417. |
30. | Lin CH and Wu RM. Biomarkers of cognitive decline in Parkinson’s disease. Parkinsonism Relat Disord (2015) ; 21: : 431–443. |
31. | McGrowder DA , Miller F , Vaz K , et al. Cerebrospinal fluid biomarkers of Alzheimer’s disease: Current evidence and future perspectives. Brain Sci (2021) ; 11: : 215. |
32. | Qu Y , Li J , Qin Q , et al. A systematic review and meta-analysis of inflammatory biomarkers in Parkinson’s disease. NPJ Parkinsons Dis (2023) ; 9: : 18. |
33. | Zago E , Dal Molin A , Dimitri GM , et al. Early downregulation of hsa-miR-144-3p in serum from drug-naíve Parkinson’s disease patients. Sci Rep (2022) ; 12: : 1330. |
34. | Tang CC , Poston KL , Eckert T , et al. Differential diagnosis of parkinsonism: A metabolic imaging study using pattern analysis. Lancet Neurol (2010) ; 9: : 149–158. |
35. | Varrone A and Pellecchia MT. SPECT molecular imaging in familial Parkinson’s disease. Int Rev Neurobiol (2018) ; 142: : 225–260. |
36. | Brockmann K , Quadalti C , Lerche S , et al. Association between CSF alpha-synuclein seeding activity and genetic status in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol Commun (2021) ; 9: : 175. |
37. | Concha-Marambio L , Pritzkow S , Shahnawaz M , et al. Seed amplification assay for the detection of pathologic alpha-synuclein aggregates in cerebrospinal fluid. Nat Protoc (2023) ; 18: : 1179–1196. |
38. | Rossi M , Candelise N , Baiardi S , et al. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol (2020) ; 140: : 49–62. |
39. | Garrido A , Fairfoul G , Tolosa ES , et al. α-synuclein RT-QuIC in cerebrospinal fluid of LRRK2-linked Parkinson’s disease. Ann Clin Transl Neurol (2019) ; 6: : 1024–1032. |
40. | Wurster I , Quadalti C , Rossi M , et al. Linking the phenotype of SNCA triplication with PET-MRI imaging pattern and alpha-synuclein CSF seeding. NPJ Parkinsons Dis (2022) ; 8: : 117. |
41. | Kluge A , Borsche M , Streubel-Gallasch L , et al. α-synuclein pathology in PRKN-linked Parkinson’s disease – new insights from a blood-based seed amplification assay. Ann Neurol (2024) ; 95: : 1173–1177. |
42. | Iranzo A , Fairfoul G , Ayudhaya ACN , et al. Detection of α-synuclein in CSF by RT-QuIC in patients with isolated rapid-eye-movement sleep behaviour disorder: A longitudinal observational study. Lancet Neurol (2021) ; 20: : 203–212. |
43. | Noyce AJ , Lees AJ and Schrag AE. The prediagnostic phase of Parkinson’s disease. J Neurol Neurosurg Psychiatry (2016) ; 87: : 871–878. |
44. | Sierra M , Sánchez-Juan P , Martínez-Rodríguez MI , et al. Olfaction and imaging biomarkers in premotor LRRK2 G2019S-associated Parkinson disease. Neurology (2013) ; 80: : 621–626. |
45. | Simuni T , Uribe L , Cho HR , et al. Clinical and dopamine transporter imaging characteristics of non-manifest LRRK2 and GBA mutation carriers in the Parkinson’s Progression Markers Initiative (PPMI): A cross-sectional study. Lancet Neurol (2020) ; 19: : 71–80. |
46. | Sossi V , De La Fuente-Fernández R , Nandhagopal R , et al. Dopamine turnover increases in asymptomatic LRRK2 mutations carriers. Mov Disord (2010) ; 25: : 2717–2723. |
47. | Hällqvist J , Bartl M , Dakna M , et al. Plasma proteomics identify biomarkers predicting Parkinson’s disease up to 7 years before symptom onset. Nat Commun (2024) ; 15: : 4759. |
48. | Zagare A , Preciat G , Nickels SL , et al. Omics data integration suggests a potential idiopathic Parkinson’s disease signature. Commun Biol (2023) ; 6: : 1179. |


