
Parkinson’s Disease and Dementia with Lewy Bodies: One and the Same
Abstract
The question whether Parkinson’s disease dementia (PDD) and dementia with Lewy bodies (DLB) are expressions of the same underlying disease has been vigorously debated for decades. The recently proposed biological definitions of Lewy body disease, which do not assign any particular importance to the dopamine system over other degenerating neurotransmitter systems, has once more brought the discussion about different types of Lewy body disease to the forefront. Here, we briefly compare PDD and DLB in terms of their symptoms, imaging findings, and neuropathology, ultimately finding them to be indistinguishable. We then present a conceptual framework to demonstrate how one can view different clinical syndromes as manifestations of a shared underlying Lewy body disease. Early Parkinson’s disease, isolated RBD, pure autonomic failure and other autonomic symptoms, and perhaps even psychiatric symptoms, represent diverse manifestations of the initial clinical stages of Lewy body disease. They are characterized by heterogeneous and comparatively limited neuronal dysfunction and damage. In contrast, Lewy body dementia, an encompassing term for both PDD and DLB, represents a more uniform and advanced stage of the disease. Patients in this category display extensive and severe Lewy pathology, frequently accompanied by co-existing pathologies, as well as multi-system neuronal dysfunction and degeneration. Thus, we propose that Lewy body disease should be viewed as a single encompassing disease entity. Phenotypic variance is caused by the presence of individual risk factors, disease mechanisms, and co-pathologies. Distinct subtypes of Lewy body disease can therefore be defined by subtype-specific disease mechanisms or biomarkers.
INTRODUCTION
The question whether Parkinson’s disease (PD) and dementia with Lewy bodies (DLB) are expressions of the same underlying disease has been vigorously debated ever since consensus diagnostic criteria for DLB were first proposed in 1996 [1–7]. However, the need to resolve this discussion has not diminished in importance, given that the international research community is actively working towards a biological redefinition of various Lewy body diseases [8, 9].
Here, we review the literature on differences and similarities between DLB and PD dementia (PDD), including clinical symptoms, imaging findings, neuropathology, and the importance of co-pathologies. We find that PDD and DLB are, for all practical purposes, indistinguishable disease entities, reflective of an advanced stage of Lewy body disease often accompanied by Alzheimer disease (AD) co-pathology and widespread neurodegeneration.
To illustrate the problem of clinically-based PD and DLB diagnoses, Table 1 summarizes key information from three typical cases. Two of these patients, who are assigned the different diagnostic labels of DLB and PD, are virtually identical in every single aspect except for a slight difference in the motor-dementia interval. In contrast, both of these patients are strikingly different from the third patient, who nevertheless is also diagnosed with PD.
Table 1
Three typical patients with Lewy body disease
| Patient | 1 | 2 | 3 |
| Diagnosis | DLB | PD | PD |
| Age at diagnosis (y) | 75 | 73 | 50 |
| Age at dementia (y) | 75 | 75 | 75 |
| Motor-dementia interval (y) | 0 | 2 | 25 |
| Symptoms at diagnosis | |||
| Parkinsonism | yes | yes | yes |
| Hyposmia | yes | yes | yes |
| Autonomic | yes | yes | no |
| Depression | yes | yes | no |
| RBD | yes | yes | no |
| Major imaging pathology at diagnosis | |||
| Dopamine transporter | yes | yes | yes |
| MIBG cardiac scintigraphy | yes | yes | no |
| FDG PET | yes | yes | no |
| Cholinergic PET | yes | yes | no |
| Amyloid PET | yes | yes | no |
| Postmortem findings | |||
| Braak Lewy body stage | 6 | 6 | 5 |
| Significant AD co-pathology | yes | yes | no |
Characteristics of three patients with Lewy body disease. Patient 1 (DLB) and patient 2 (PD) show virtually identical findings but both are very different from patient 3 (PD).
Rather than upholding several distinct diagnoses based on separate clinical diagnostic criteria, we argue for the validity of a biological definition of Lewy body disease and its possible subtypes supported by biomarkers. The well-known clinical syndromes can thereby all be encompassed under an umbrella term of Lewy body disease. Some of these clinical syndromes, including early PD, isolated REM sleep behavior disorder (iRBD), pure autonomic failure (PAF), and idiopathic hyposmia, will reflect relatively restricted α-synuclein associated neurodegeneration. In contrast, clinical PDD and DLB will typically be evidence of more extensive and severe αSyn-associated neurodegeneration affecting multiple neurotransmitter systems. In the recently proposed staging systems, a patient will be diagnosed based on the presence of pathogenic, aggregated αSyn and neurodegeneration known to be specifically associated with this pathology [8, 9]. Other biomarkers may serve to define biological subtypes.
In the following, we shall briefly compare PDD and DLB with respect to symptoms, imaging findings, and neuropathology. We then propose a framework to illustrate how it is possible to think about different clinical syndromes as manifestations of the same underlying Lewy body disease. We also emphasize the importance of individual risk factors and co-pathologies, especially AD co-pathology, which seems to work synergistically with or additively to Lewy pathology to shape symptomatology in individual patients.
COGNITIVE AND PSYCHIATRIC SYMPTOMS
The cognitive and neuropsychiatric symptoms seen in DLB and PDD seem to be indistinguishable [10, 11]. Cognitive deficits in both disorders occur across all cognitive domains, including impairments in executive abilities, working memory, attention, visuospatial skills, processing speed and language [12–23]. It is controversial if memory is less affected in the early disease [24–26].
Neuropsychiatric symptoms are also very common in both PDD and DLB [27]. A systematic review [10] summarized the frequencies of neuropsychiatric symptoms in PDD and DLB as follows: depression (PDD 58%, DLB 49%), apathy (PDD 54%, DLB 57%), anxiety (PDD 49%, DLB 65%), hallucinations (PDD 54%, DLB 76%) and delusions (PDD 29%, DLB 57%) [27–29].
In general, these studies find that the frequency of cognitive and neuropsychiatric symptoms is very similar in PDD and DLB, or sometimes slightly higher in DLB. Thus, many individual PDD patients are indistinguishable from DLB patients. A recent large longitudinal study also demonstrated that diagnosed PDD and DLB patients show virtually identical rates of cognitive and motor decline when followed for up to five years [30]. Thus, based on the cognitive and neuropsychiatric profiles, there appears to be no justification to assume that PDD and DLB are different diseases.
IMAGING BIOMARKERS
Radiotracer and MRI studies generally report nearly identical findings in PDD and DLB. Very similar patterns of cerebral hypoperfusion and hypometabolism have been reported, with the most pronounced deficits seen in posterior parieto-occipital cortices [31–33].
Structural MRI studies of PDD and DLB have revealed comparable patterns of mostly posterior atrophy, occasionally slightly more pronounced in DLB [34, 35]. However, any statistical differences between PDD and DLB is only apparent when comparing large groups of patients, and many individual PDD and DLB patients are indistinguishable. Of note, atrophy will often be aggravated by the presence of co-morbid AD pathology, which is more common in DLB [36–38].
PET studies have shown significant depletion of cholinergic projections from the basal forebrain to the cortex in both PDD and DLB, with the most notable effects observed in the posterior parieto-occipital regions. Again, there is no obvious differences in the regional pattern or magnitude of loss in DLB compared with PDD [32, 39–43].
Patients with PD and DLB show similar loss of nigrostriatal dopaminergic innervation on dopamine transporter (DAT) striatal imaging [32, 44, 45]. When compared to non-demented PD patients, DLB shows considerably more symmetric loss and also more uniform loss in both the putamen and caudate nucleus [44, 45]. However, it is known that the dopaminergic deficit in PD becomes more symmetric at follow-up and also with progressive involvement of the caudate [46]. Thus, as non-demented PD patients progress, their dopamine scans look increasingly like DLB patients.
A small percentage (10–15%) of clinically-diagnosed DLB patients exhibit relatively normal DAT scans at diagnosis [47]. However, one study showed that when DLB patients with normal baseline DAT imaging were reexamined 3 years later, they had all progressed and showed pathologically low dopamine innervation [48]. A recent longitudinal study also demonstrated that patients with a baseline DLB diagnosis showed faster motor progression over an 8-year period compared to patients with a baseline PDD diagnosis [49].
Lewy body disease is characterized by profound loss of cardiac sympathetic innervation, which can be visualized with [123I]MIBG or [18F]fluorodopamine scans [50, 51]. At diagnosis, many RBD-negative PD patients display normal or near-normal MIBG parameters, whereas virtually all de novo PD patients with RBD show markedly pathological MIBG scans [52]. However, once PD patients have progressed to Hoehn & Yahr stage III, they almost always exhibit severe sympathetic denervation on MIBG [53, 54]. Likewise, nearly all DLB patients show pathological MIBG scans already at diagnosis [55–57]. Put together, virtually all patients with Lewy body disease will eventually develop severely pathological MIBG scans as the disease progresses, with no apparent differences between PDD and DLB.
In summary, many individual PDD and DLB patients have very similar, or identical, findings on all the aforementioned imaging modalities. At the group level, DLB patients sometimes show more severe pathology on imaging, but a large percentage of individual PDD and DLB patients are identical.
One notable exception relates to the frequency of AD co-pathology in PDD vs. DLB. A meta-analysis of amyloid PET scans concluded that 68% of DLB, 34% of PDD, and 5% of non-demented PD patients had pathological findings on amyloid PET [58, 59]. The implications of this important observation are discussed in more detail below.
NEUROPATHOLOGY
When comparing PDD and DLB, neuropathologists generally conclude that no pathologic substrate found at postmortem can reliably differentiate these clinically-defined disorders [6, 60–62]. Commonly, PDD patients show higher levels of neocortical Lewy pathology compared to non-demented PD patients, signifying that PDD represents a later, more advanced disease stage than non-demented PD [63, 64]. On the other hand, PDD and DLB patients show very similar global distribution and severity of Lewy pathology, and at autopsy the vast majority of cases are Braak Lewy body stage 5–6 [11, 65, 66]. Thus, with respect to Lewy body pathology there are no obvious differences between PDD and DLB.
Importantly, most studies report that intermediate-to-high levels of AD co-pathology (i.e., neurofibrillary tangles and beta-amyloid plaques), are more common in DLB than in PDD [60, 67, 68], although some have reported equal levels [66]. Many studies have reported a strong association between AD co-pathology and reduced cognitive status in autopsied PD cases [63, 64, 67, 69–73]. Generally, a combination of Lewy and AD pathologies shows a stronger correlation with dementia compared with patients with pure Lewy body disease [60, 67, 74]. As mentioned above, a similar relationship is seen with amyloid PET scans, which are positive in around 2/3 of DLB patients, 1/3 of PDD, but in only 5% of non-demented PD [58, 59].
Of note, at least two studies have clearly shown that the motor-dementia interval is strongly influenced by the amount of AD co-pathology (Fig. 1) [60, 67]. Demented patients with no or low-level AD co-pathology at postmortem usually develop parkinsonism many years before dementia. In contrast, patients who develop dementia within a few years of parkinsonism (irrespective of whether they had a PDD or DLB diagnosis clinically) more often have intermediate- or high-levels of AD pathology.
Fig. 1
Schematic illustration of the prevalence of significant AD co-pathology at postmortem in individuals with Lewy body dementia. Higher rates are observed in patients with short motor-dementia intervals. For example, a PDD patient with a 5-years motor-dementia may have a 60% probability of AD co-pathology at death, whereas another PDD patient with a 20-years motor-dementia may have only a 20% probability of AD pathology at death. No obvious distinction is evident between DLB and the PDD patients with the shortest motor-dementia interval, underscoring the arbitrary nature of “the 1-year rule” (based on data from [58, 60, 67]).
![Schematic illustration of the prevalence of significant AD co-pathology at postmortem in individuals with Lewy body dementia. Higher rates are observed in patients with short motor-dementia intervals. For example, a PDD patient with a 5-years motor-dementia may have a 60% probability of AD co-pathology at death, whereas another PDD patient with a 20-years motor-dementia may have only a 20% probability of AD pathology at death. No obvious distinction is evident between DLB and the PDD patients with the shortest motor-dementia interval, underscoring the arbitrary nature of “the 1-year rule” (based on data from [58, 60, 67]).](https://content.iospress.com:443/media/jpd/2024/14-3/jpd-14-3-jpd240002/jpd-14-jpd240002-g001.jpg)
These postmortem and PET imaging data strongly suggest that the presence of AD co-pathology accelerates cognitive decline. In short, relatively pure Lewy pathology most commonly results in a PD diagnosis, and the development of the advanced PDD stage typically takes many years. The few DLB cases without AD-pathology at postmortem frequently had GBA mutations or other co-pathologies such as cerebrovascular disease [60]. In contrast, significant amounts of AD co-pathology regularly lead to more rapid dementia, but again, there is no clear difference between PDD patients with a short motor-dementia interval and DLB patients with an even shorter motor-dementia interval (Fig. 1). Of note, PDD patients with considerable amounts of AD co-pathology seems to have clinical phenotype that in many respects is more similar to DLB [60, 63, 72, 75–77], and one study showed that the arbitrary “1-year rule” had very poor diagnostic accuracy with respect to predicting the amount of neocortical Lewy bodies or the amount of AD co-pathology in PDD vs. DLB [60].
In summary, postmortem studies show that PDD and DLB have highly similar burdens and distribution of Lewy pathology. Significant AD co-pathology is more commonly seen in DLB and in PDD patients with a short motor-dementia interval, but with no clear distinction between the two.
RELATIONSHIP BETWEEN LEWY BODY DISEASE AND AD CO-PATHOLOGY
Figure 2 provides a visual summary to illustrate how AD and Lewy body pathologies might theoretically interact to produce the different clinical syndromes of DLB, PD and PDD.
The rows in the figure depict Lewy body disease divided clinically into brain-first and body-first disease [52, 78, 79]. The latter is characterized by development of iRBD, autonomic symptoms, and pathological MIBG scintigraphy before diagnosis [52], and more often shows relatively symmetric dopamine degeneration [45, 78, 80]. These symptoms and signs are all risk factors for rapid dementia (summarized in [78]). Thus, body-first Lewy body disease in itself leads to faster cognitive decline compared to RBD-negative brain-first disease.
Fig. 2
Hypothetical interaction between Lewy body disease and AD-type co-pathology. Patients with clinical body-first Lewy body disease show faster progression towards dementia compared to brain-first disease. Prodromal iRBD, autonomic symptoms, pathological MIBG scintigraphy, and symmetric dopamine degeneration are all risk factors of accelerated cognitive decline and signify a body-first etiology. AD-type co-pathology is also strongly associated with fast dementia in Lewy body disease. Thus, body-first Lewy body disease with co-morbid AD-pathology will often result in a DLB diagnosis or sometimes a PD diagnosis with rapid dementia (dark blue quadrant; short arrow signifies fast progression to dementia). Brain-first Lewy body disease without concomitant AD-pathology is almost always diagnosed as PD and the evolution towards dementia is often much slower (light blue quadrant; long arrow signifies slow progression to dementia). The other remaining quadrants result in clinical syndromes with in-between rates of progression towards cognitive decline. Finally, the figure shows how different genetic risk factors could theoretically be distinctly associated with the different axes of Lewy body vs. AD pathology. LRRK2 mutations are associated with a benign, RBD-negative Lewy body disease, and thus not with dementia or DLB. GBA mutations associate with an RBD-positive, fast progressive Lewy body disease but not with AD. The association between GBA and dementia is therefore related mainly to the axis of Lewy body disease. In contrast, APOEɛ4 promotes the formation of AD co-pathology and is therefore also associated with dementia in Lewy body disease, due to the contribution from AD pathology.
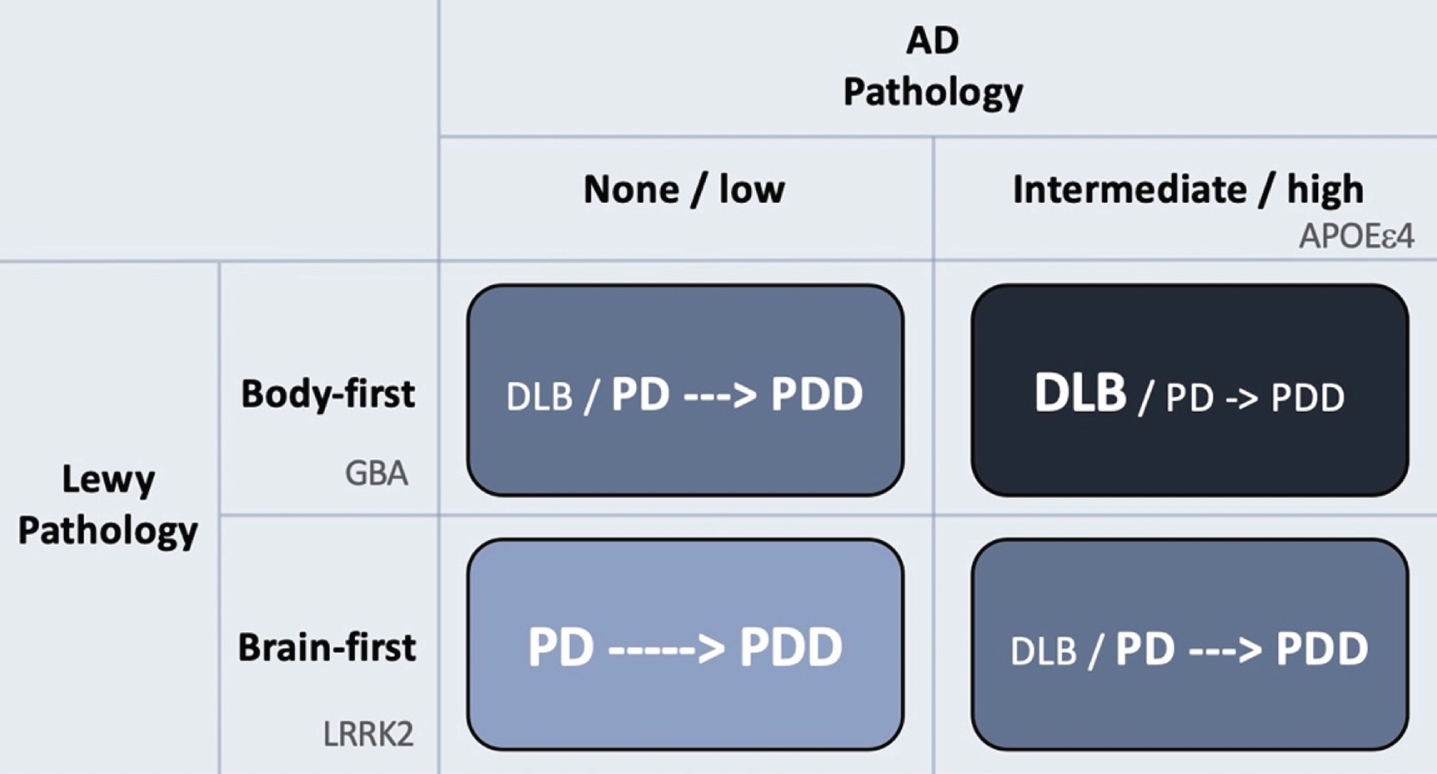
The columns of Fig. 2 depict the burden of AD co-pathology. As reviewed above, AD co-pathology is also strongly associated with faster dementia in Lewy body disease. Hence, it appears that patients diagnosed with DLB mainly consists of individuals, who have a combination of body-first Lewy body disease and co-morbid AD pathology. If such cases receive a PD diagnosis first, they will most likely progress rapidly to PDD.
In contrast, patients with brain-first Lewy body disease in the absence of AD co-pathology nearly always receive a PD diagnosis, and the evolution towards PDD is much slower. Such patients are generally younger, display initially more asymmetric dopamine loss, fewer non-motor symptoms, and are RBD-negative at diagnosis [81–84].
Patients in the remaining two quadrants (Fig. 2) will probably show in-between rates of progression towards cognitive decline.
Finally, it should be remembered that the relationship between AD and Lewy pathology is probably not simply additive. There is evidence to suggest that these pathologies can cross-seed each other [85–87]. Of note, patients with early brainstem-predominant Lewy body disease comprise almost half of all cases in postmortem series, and they do not exhibit more AD co-pathology than the background population. In contrast, patients with advanced Lewy body disease on average display much higher frequencies of AD pathology than expected [58, 59, 85, 88–90]. This observation implies that the gradual accumulation of Lewy pathology can trigger the formation of AD co-pathology.
Given the strong association between AD co-pathologies and cognitive decline in Lewy body disease, it is an exciting prospect that anti-amyloid treatments could potentially slow down cognitive decline in Lewy body disease.
GENETICS
A review of genetic risk factors in Lewy body disease is beyond the scope of this paper, so only a few interesting associations will be highlighted here. Several studies have demonstrated that mutations in the glucocerebrosidase (GBA) gene in PD are associated with a more severe phenotype of Lewy body disease, including faster progression, increased prevalence of RBD and other non-motor symptoms, accelerated cognitive decline, and reduced survival. Not surprisingly, GBA mutations are also associated with the clinical DLB diagnosis [91–96].
In contrast, mutations in the leucine-rich repeat kinase 2 (LRRK2) gene are generally associated with more benign type of Lewy body disease with slower progression, lower risk of RBD and other non-motor manifestations, and late dementia in PD, and are not associated with DLB [97–100].
The apolipoprotein E4 (APOE ɛ4) genotype strongly elevates the risk of AD [101, 102]. Some studies did not detect associations between APOE ɛ4 and elevated risk of either PD or iRBD [103, 104], but do show that this genotype predicts dementia in PD [61, 105–107]. In any case, the APOE ɛ4 genotype seems to be much more strongly associated with AD than with Lewy pathology.
Thus, it can be speculated that different genetic risk factors are mainly associated with independent components of protein pathologies (Fig. 2). LRRK2 mutations might be associated with a relatively benign olfactory-first or brain-first etiology of Lewy body disease and is therefore not associated with dementia or DLB. In contrast, GBA mutations, given their more rapid disease progression and high non-motor symptom comorbidity, may predispose to a body-first etiology of Lewy body disease leading to faster dementia and is therefore associated with DLB. Importantly, GBA mutations are not associated with AD per se, so its association with DLB seems to relate purely to the Lewy body component of DLB. Finally, the APOE ɛ4 genotype is also associated with DLB, not because of any particularly strong association with Lewy body disease, but rather because it promotes concomitant AD pathology, which in itself accelerates cognitive decline.
EVOLUTION OF LEWY BODY DISEASE
Figure 3 depicts a hypothetical framework to illustrate how Lewy body disease may evolve from the preclinical stage, over several heterogenous early- and moderate-stage subtypes, to finally converge on a more homogenous, advanced stage, during which the patient typically becomes demented. The advanced stage can be termed Lewy body dementia and includes both PDD and DLB. Such patients are characterized by widespread deposition of Lewy body pathology in both subcortical and most commonly (but not always) cortical regions. Moreover, multiple neurotransmitter systems, including autonomic, noradrenergic, dopaminergic, serotonergic, and cholinergic, are typically significantly damaged at this clinical stage, and many patients display considerable amounts of comorbid AD pathology. Patients with DLB and PDD have similarly poor prognosis with a median survival from onset of dementia of 4–6 years [108, 109].
Fig. 3
A hypothetical framework to illustrate how Lewy body disease develops from the pre-clinical stage over several possible well-defined early- and moderate clinical disease stages to converge on a more homogenous advanced disease stage, in which patients are typically demented. Rare exceptions to the shown trajectories may exist. The figure lists some neuroanatomical structures, which typically show damage during that particular clinical stage. Based on postmortem data, imaging, and clinical phenotypes, most patients can be categorized into two overall types: brain-first (olfactory/limbic-first) or a body-first (gut/autonomic-first). A small group with mixed (multi-focal) origin of αSyn-pathology may lead to equally mixed clinical symptom subtypes. Irrespective of the origin of αSyn-pathology, and of the heterogeneity seen in symptoms, neurodegeneration, and αSyn deposition during earlier clinical subtypes, patients will tend to converge on a homogenous advanced stage characterized by widespread αSyn pathology and neurodegeneration, parkinsonism, RBD, non-motor symptoms, and cognitive decline. The figure also depicts approximate amounts of Lewy pathology (LB) deposition, and neurodegeneration (N). Pre-clinically, patients are by definition LB-positive but the level of neurodegeneration N, if any, is insufficient to cause symptoms. Conversely, in the advanced stage, widespread LB pathology in peripheral and CNS structures are commonly seen accompanied by marked multi-system neurodegeneration. Amyg, amygdala; DMV, dorsal motor nucleus of vagus; LC, locus coeruleus; MCI, mild cognitive impairment; NBM, nucleus basalis of Meynert; SY, sympathetic ganglia and fibers.
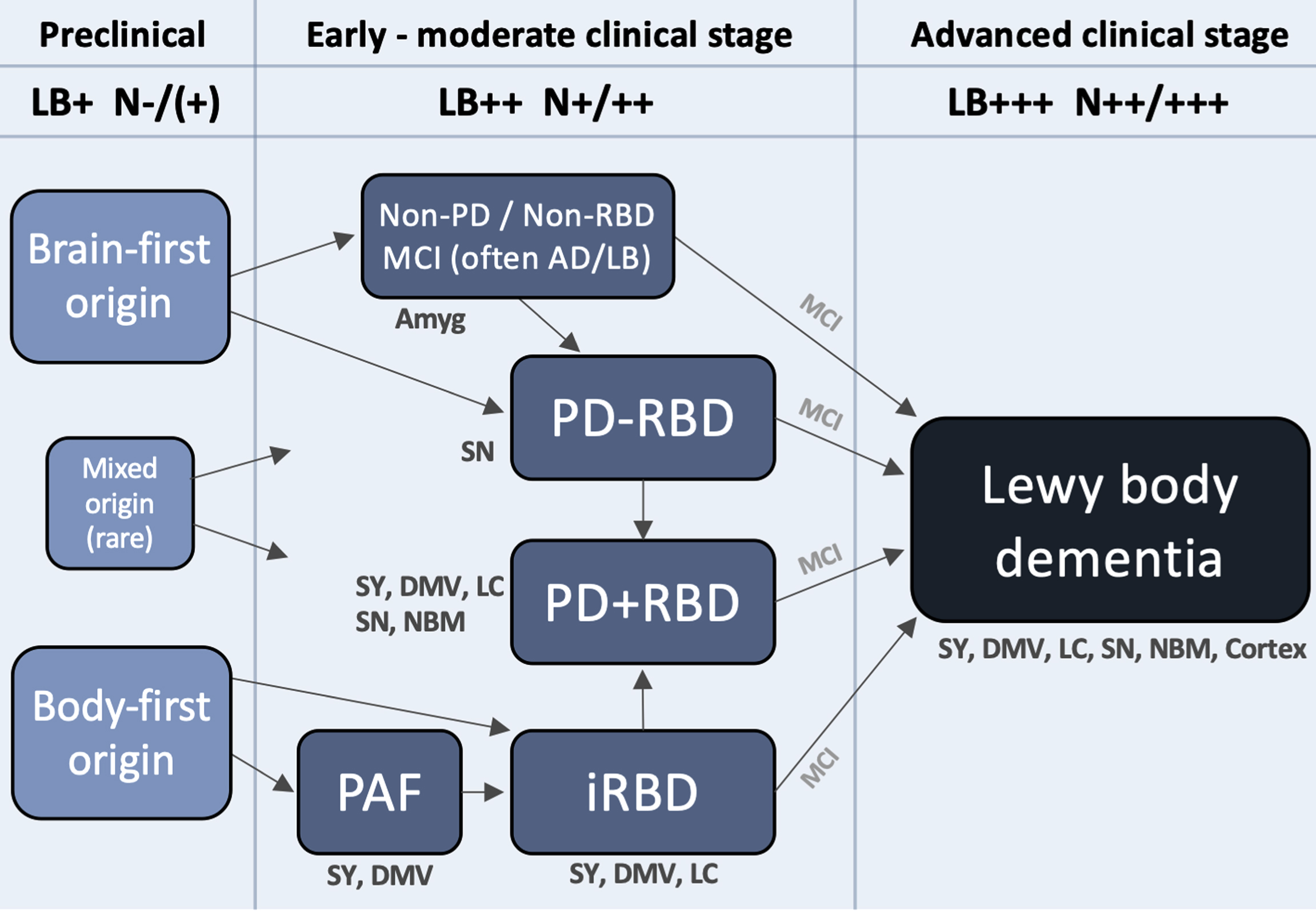
Initially there is a preclinical disease stage in which pathological αSyn may be detectable in CSF, blood, or other tissues, but before significant neurodegeneration has occurred. This is followed by several well-known clinical syndromes, characterized by limited symptoms, and relatively restricted neuronal damage. These syndromes include parkinsonism (including early PD), iRBD, PAF, and MCI-LB, but in principle also some less specific syndromes and symptoms, such as hyposmia, constipation, depression, and anxiety. Finally, there is advanced disease.
The preclinical stage is characterized by formation and initial propagation of aggregated αSyn species. By analyzing data from two postmortem cohorts, we have previously shown that the majority of brains with very restricted incidental Lewy body disease can be divided into two major categories [85, 88, 90]: (i) a brain-first group with Lewy pathology in the olfactory bulb, limbic system and top of the brainstem, but without pathology in the lower brainstem, spinal cord, or peripheral autonomic nervous system, and (ii) a body-first group with Lewy pathology in the peripheral autonomic nervous system, lower brainstem, and spinal cord, but without pathology in the olfactory bulb, limbic system, or upper brainstem. Additionally, a small mixed group can be defined, exhibiting pathology in olfactory/limbic structures, as well as in autonomic and lower brainstem structures. This group, however, lacks pathology in the intervening structures, including the upper brainstem. These mixed cases were relatively rare in our analysis (<10%), and thus the majority of cases are compatible with the pathology having started in a single, restricted region, either the olfactory bulb or enteric nervous system, with secondary spreading from those two originating sites [85, 88, 110].
Pure autonomic failure (PAF) is a rare clinical diagnosis characterized by predominantly peripheral autonomic symptoms with few or no CNS symptoms [111]. Patients universally show severe autonomic denervation on MIBG scintigraphy, but their dopamine and other CNS systems are typically much more normal [112, 113]. Only few PAF cases have come to autopsy, but these typically showed Lewy pathology restricted to the lower brainstem or in some cases higher Braak stages [114–119]. At follow-up, many PAF patients convert to PD or DLB, and those who do are almost always RBD-positive [112, 120]. This points to a relatively stereotypical trajectory from PAF to iRBD and finally to PD or DLB. Of note, PAF is rarely diagnosed. The majority of individuals with iRBD are not typically diagnosed with PAF before receiving an iRBD diagnosis, although a large percentage of iRBD patients do exhibit orthostatic hypotension, which in some cases may have preceded the emergence of RBD itself [121]. Such iRBD patients could therefore have been eligible for the PAF diagnosis.
Patients with Lewy body-positive iRBD convert to PD (54%) or DLB (46%) [122]. Although postmortem data on iRBD cases are rare, the published cases showed Lewy pathology in the brain, typically Braak stages 3-5 [123–126]. In vivo, most iRBD cases show Lewy pathology in peripheral autonomic nervous fibers, i.e., in salivary glands, skin, and colon although limited reproducibility has been reported for colon biopsies [127–130]. Patients with iRBD exhibit almost universal loss of the cardiac sympathetic system on MIBG imaging [131–133], decreased cholinergic innervation of the colon signifying parasympathetic denervation [131], low neuromelanin signal of the locus coeruleus [131, 134], but commonly show normal or near-normal dopamine transporter imaging [131, 132]. Thus, patients with iRBD show neuronal degeneration in the lower brainstem and autonomic system and progresses to either PD or DLB in almost equal proportions.
According to the Brain- vs. Body-first model, an olfactory-first or limbic-first type of Lewy body disease leads to clinical brain-first PD subtype, characterized by fairly asymmetric dopamine degeneration [78]. At diagnosis, such patients have fewer non-motor symptoms and are typically RBD-negative [52, 84]. On imaging, these de novo brain-first PD patients show limited or no damage to the locus coeruleus, parasympathetic, or sympathetic systems [52, 135, 136]. However, on follow-up most of these initially RBD-negative PD patients will eventually develop RBD and an increasing burden of non-motor symptoms. In support, it has been shown that 87% of late-stage PD and more than 75% of DLB patients are RBD-positive, whereas only about 1/3 of PD patients developed RBD before diagnosis [55, 57, 137, 138].
Although follow-up studies of well-characterized brain-first PD patients are lacking, it is known that virtually all PD patients at H&Y stage III show severe cardiac denervation on MIBG scintigraphy [53, 54]. Thus, it seems likely that most brain-first patients will eventually converge on the same advanced Lewy body stage, characterized by neurodegeneration across all vulnerable peripheral and CNS nuclei and accompanied by RBD and clinical dementia. In support, studies have estimated that at least 80% of all PD patients develop dementia during life [72, 108, 139, 140], and it remains possible that indeed all patients with clinical Lewy body disease will eventually dement, if they survive long enough.
For completeness, Fig. 3 includes a clinical category termed “non-PD/non-RBD”. Among patients in the advanced Lewy body dementia stage, the majority had developed parkinsonism, RBD, or both prior to onset of dementia. Such patients are captured by the PD and iRBD categories in the framework, but that still leaves a small group of pre-demented symptomatic patients without parkinsonism or RBD. Plausibly, such patients might have hyposmia, neuropsychiatric symptoms including anxiety, depression, and hallucinations, and MCI [57]. It is possible that mixed AD-Lewy body pathology could be particularly common in this patient group, leading to accelerated psychiatric symptoms and cognitive decline due to significant co-morbid AD pathology. The proposed ‘psychiatric-onset’ form of DLB, for which there is currently insufficient evidence to support distinct clinical criteria, could be more prevalent in this group, but it is unknown whether this type of DLB exhibits higher loads of AD co-pathology [141]. In any case, some of these patients reach the advanced dementia stage before parkinsonism or RBD has appeared, but these symptoms are likely to appear during the dementia stage. In support, it has been shown that a small percentage (10–15%) of DLB patients show normal dopamine imaging at diagnosis, but these will eventually develop dopamine degeneration upon follow-up [48]. It is also important to note that some individuals in the “non-PD / non-RBD” category may develop parkinsonism before dementia and thus fulfil the criteria for PD before they reach the advanced stage.
LIMITATIONS
The framework suggested here (Fig. 3) is based on integrating the brain- vs. body-first model with other concepts and observations from the literature. This model is still hypothetical, and its limitations have been thoroughly reviewed previously [52, 78, 79, 85, 110]. While the model proposes that the specific origin site of initial Lewy pathology and its subsequent propagation through the connectome is a major determining factor for the evolution of different clinical phenotypes, we want to emphasize that many other pathogenic factors, such as genetic variants, inflammation, and selective neuronal vulnerability factors, are important for the shaping the variable disease course in individual patients.
Our proposed framework is meant to explain the relationship between progressive Lewy pathology, co-pathologies, and different clinical syndromes. It is not intended as a staging or diagnostic system in its current form. Several obstacles remain with regards to the validity of both this framework and recently published biological staging systems [8, 9], such as determining the sensitivity and specificity of synuclein seed amplification assays to diagnose prodromal and preclinical Lewy body disease [130, 142, 143].
More work is certainly needed with respect to understanding different clinical phenotypes and their underlying pathophysiological causes, including PAF and psychiatric-onset DLB. Such distinct clinical phenotypes may be caused by particular individual vulnerability factors interacting with pathological α-synuclein and other aggregated protein pathologies.
Finally, we must always consider carefully how changes in diagnostic criteria or syndromic conceptualization may adversely impact patient care and advocacy efforts.
SUMMARY
In the framework presented here, we propose that the different clinical syndromes, such as iRBD, PAF, PD, PDD, and DLB, should be viewed as manifestations of a common underlying Lewy body disease, but which can be modified by genetic variants, selective neuronal vulnerability factors, and the presence of various co-pathologies, with the most significant being AD pathology.
Patients in the earliest clinical disease stages will show the greatest degree of heterogeneity, since the pathogenic process can originate in different locations of the neuroaxis and the associated neurodegeneration is still relatively localized. However, the majority of patients with Lewy body disease appears to progress towards a more homogenous advanced disease stage. In this stage, currently diagnosed as PDD or DLB, most patients will exhibit a full-spectrum clinical phenotype, including dementia, psychiatric manifestations, parkinsonism, RBD, autonomic issues, and other non-motor symptoms. The imaging literature also supports that patients with advanced disease commonly show evidence of neurodegeneration across all major neurotransmitter systems known to be vulnerable to Lewy pathology, which is not the case for early-stage disease.
Future research should focus on understanding how different clinical signs and symptoms map to underlying neuronal dysfunction and damage, including brain region-specific changes, with the goal of facilitating personalized treatments. This may potentially lead to biological subtypes of Lewy body disease, if it can be demonstrated that they have a specific biological-clinical “fingerprint”. But the best starting point is to acknowledge that this disease does not conform to our current clinical diagnostic criteria and can manifest itself quite variably, in terms of age of onset, organ and neurotransmitter systems affected, symptom presentation and evolution over time, and disease progression over many years.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The authors have no funding to report.
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.
Daniel Weintraub is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.
REFERENCES
[1] | McKeith IG , Dickson DW , Lowe J , Emre M , O’Brien JT , Feldman H , Cummings J , Duda JE , Lippa C , Perry EK , Aarsland D , Arai H , Ballard CG , Boeve B , Burn DJ , Costa D , Del Ser T , Dubois B , Galasko D , Gauthier S , Goetz CG , Gomez-Tortosa E , Halliday G , Hansen LA , Hardy J , Iwatsubo T , Kalaria RN , Kaufer D , Kenny RA , Korczyn A , Kosaka K , Lee VM , Lees A , Litvan I , Londos E , Lopez OL , Minoshima S , Mizuno Y , Molina JA , Mukaetova-Ladinska EB , Pasquier F , Perry RH , Schulz JB , Trojanowski JQ , Yamada M , Consortium on DLB ((2005) ) Diagnosis and management of dementia with Lewy bodies: Third report of the DLB Consortium. Neurology 65: , 1863–1872. |
[2] | Kosaka K , Yoshimura M , Ikeda K , Budka H ((1984) ) Diffuse type of Lewy body disease: Progressive dementia with abundant cortical Lewy bodies and senile changes of varying degree–a new disease? Clin Neuropathol 3: , 185–192. |
[3] | Friedman JH ((2018) ) Dementia with Lewy bodies and Parkinson disease dementia: It is the same disease! Parkinsonism Relat Disord Suppl 1: , S6–S9. |
[4] | Weintraub D ((2023) ) What’s in a name? The time has come to unify Parkinson’s disease and dementia with Lewy bodies. Mov Disord 38: , 1977–1981. |
[5] | Mensikova K , Matej R , Colosimo C , Rosales R , Tuckova L , Ehrmann J , Hrabos D , Kolarikova K , Vodicka R , Vrtel R , Prochazka M , Nevrly M , Kaiserova M , Kurcova S , Otruba P , Kanovsky P ((2022) ) Lewy body disease or diseases with Lewy bodies? NPJ Parkinsons Dis 8: , 3. |
[6] | Jellinger KA , Korczyn AD ((2018) ) Are dementia with Lewy bodies and Parkinson’s disease dementia the same disease? BMC Med 16: , 34. |
[7] | Aarsland D , Ballard CG , Halliday G ((2004) ) Are Parkinson’s disease with dementia and dementia with Lewy bodies the same entity? J Geriatr Psychiatry Neurol 17: , 137–145. |
[8] | Simuni T , Chahine LM , Poston K , Brumm M , Buracchio T , Campbell M , Chowdhury S , Coffey C , Concha-Marambio L , Dam T , DiBiaso P , Foroud T , Frasier M , Gochanour C , Jennings D , Kieburtz K , Kopil CM , Merchant K , Mollenhauer B , Montine T , Nudelman K , Pagano G , Seibyl J , Sherer T , Singleton A , Stephenson D , Stern M , Soto C , Tanner CM , Tolosa E , Weintraub D , Xiao Y , Siderowf A , Dunn B , Marek K ((2024) ) A biological definition of neuronal alpha-synuclein disease: Towards an integrated staging system for research. Lancet Neurol 23: , 178–190. |
[9] | Hoglinger GU , Adler CH , Berg D , Klein C , Outeiro TF , Poewe W , Postuma R , Stoessl AJ , Lang AE ((2024) ) A biological classification of Parkinson’s disease: The SynNeurGe research diagnostic criteria. Lancet Neurol 23: , 191–204. |
[10] | Fields JA ((2017) ) Cognitive and Neuropsychiatric Features in Parkinson’s and Lewy Body Dementias. Arch Clin Neuropsychol 32: , 786–801. |
[11] | Galvin JE , Pollack J , Morris JC ((2006) ) Clinical phenotype of Parkinson disease dementia. Neurology 67: , 1605–1611. |
[12] | Caviness JN , Driver-Dunckley E , Connor DJ , Sabbagh MN , Hentz JG , Noble B , Evidente VG , Shill HA , Adler CH ((2007) ) Defining mild cognitive impairment in Parkinson’s disease. Mov Disord 22: , 1272–1277. |
[13] | Dubois B , Pillon B ((1997) ) Cognitive deficits in Parkinson’s disease. J Neurol 244: , 2–8. |
[14] | Kehagia AA , Barker RA , Robbins TW ((2010) ) Neuropsychological and clinical heterogeneity of cognitive impairment and dementia in patients with Parkinson’s disease. Lancet Neurol 9: , 1200–1213. |
[15] | Muslimovic D , Post B , Speelman JD , Schmand B ((2005) ) Cognitive profile of patients with newly diagnosed Parkinson disease. Neurology 65: , 1239–1245. |
[16] | Raskin SA , Borod JC , Tweedy J ((1990) ) Neuropsychological aspects of Parkinson’s disease. Neuropsychol Rev 1: , 185–221. |
[17] | Stella F , Gobbi LT , Gobbi S , Oliani MM , Tanaka K , Pieruccini-Faria F ((2007) ) Early impairment of cognitive functions in Parkinson’s disease. Arq Neuropsiquiatr 65: , 406–410. |
[18] | Azuma T , Cruz RF , Bayles KA , Tomoeda CK , Montgomery EB Jr ((2003) ) A longitudinal study of neuropsychological change in individuals with Parkinson’s disease. Int J Geriatr Psychiatry 18: , 1043–1049. |
[19] | Mosimann UP , Mather G , Wesnes KA , O’Brien JT , Burn DJ , McKeith IG ((2004) ) Visual perception in Parkinson disease dementia and dementia with Lewy bodies. Neurology 63: , 2091–2096. |
[20] | Benito-Leon J , Louis ED , Posada IJ , Sanchez-Ferro A , Trincado R , Villarejo A , Mitchell AJ , Bermejo-Pareja F ((2011) ) Population-based case-control study of cognitive function in early Parkinson’s disease (NEDICES). J Neurol Sci 310: , 176–182. |
[21] | Mondon K , Gochard A , Marque A , Armand A , Beauchamp D , Prunier C , Jacobi D , de Toffol B , Autret A , Camus V , Hommet C ((2007) ) Visual recognition memory differentiates dementia with Lewy bodies and Parkinson’s disease dementia. J Neurol Neurosurg Psychiatry 78: , 738–741. |
[22] | Filoteo JV , Salmon DP , Schiehser DM , Kane AE , Hamilton JM , Rilling LM , Lucas JA , Zizak V , Galasko DR ((2009) ) Verbal learning and memory in patients with dementia with Lewy bodies or Parkinson’s disease with dementia. J Clin Exp Neuropsychol 31: , 823–834. |
[23] | Park KW , Kim HS , Cheon SM , Cha JK , Kim SH , Kim JW ((2011) ) Dementia with Lewy bodies versus Alzheimer’s disease and Parkinson’s disease dementia: A comparison of cognitive profiles. J Clin Neurol 7: , 19–24. |
[24] | Martinez-Horta S , Kulisevsky J ((2011) ) Is all cognitive impairment in Parkinson’s disease “mild cognitive impairment”? J Neural Transm (Vienna) 118: , 1185–1190. |
[25] | Goldman JG , Litvan I ((2011) ) Mild cognitive impairment in Parkinson’s disease. Minerva Med 102: , 441–459. |
[26] | Dalrymple-Alford JC , Livingston L , MacAskill MR , Graham C , Melzer TR , Porter RJ , Watts R , Anderson TJ ((2011) ) Characterizing mild cognitive impairment in Parkinson’s disease. Mov Disord 26: , 629–636. |
[27] | Aarsland D , Bronnick K , Ehrt U , De Deyn PP , Tekin S , Emre M , Cummings JL ((2007) ) Neuropsychiatric symptoms in patients with Parkinson’s disease and dementia: Frequency, profile and associated care giver stress. J Neurol Neurosurg Psychiatry 78: , 36–42. |
[28] | Borroni B , Agosti C , Padovani A ((2008) ) Behavioral and psychological symptoms in dementia with Lewy-bodies (DLB): Frequency and relationship with disease severity and motor impairment. Arch Gerontol Geriatr 46: , 101–106. |
[29] | Sadak TI , Katon J , Beck C , Cochrane BB , Borson S ((2014) ) Key neuropsychiatric symptoms in common dementias: Prevalence and implications for caregivers, clinicians, and health systems. Res Gerontol Nurs 7: , 44–52. |
[30] | Gonzalez MC , Tovar-Rios DA , Alves G , Dalen I , Williams-Gray CH , Camacho M , Forsgren L , Backstrom D , Lawson RA , Macleod AD , Counsell CE , Paquet C , DeLena C , D’Antonio F , Pilotto A , Padovani A , Blanc F , Falup-Pecurariu C , Lewis SJG , Rejdak K , Papuc E , Hort J , Nedelska Z , O’Brien J , Bonanni L , Marquie M , Boada M , Pytel V , Abdelnour C , Alcolea D , Beyer K , Tysnes OB , Aarsland D , Maple-Grodem J ((2023) ) Cognitive and motor decline in dementia with Lewy bodies and Parkinson’s disease dementia. Mov Disord Clin Pract 10: , 980–986. |
[31] | Kasama S , Tachibana H , Kawabata K , Yoshikawa H ((2005) ) Cerebral blood flow in Parkinson’s disease, dementia with Lewy bodies, and Alzheimer’s disease according to three-dimensional stereotactic surface projection imaging. Dement Geriatr Cogn Disord 19: , 266–275. |
[32] | Klein JC , Eggers C , Kalbe E , Weisenbach S , Hohmann C , Vollmar S , Baudrexel S , Diederich NJ , Heiss WD , Hilker R ((2010) ) Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology 74: , 885–892. |
[33] | Yong SW , Yoon JK , An YS , Lee PH ((2007) ) A comparison of cerebral glucose metabolism in Parkinson’s disease, Parkinson’s disease dementia and dementia with Lewy bodies. Eur J Neurol 14: , 1357–1362. |
[34] | Beyer MK , Janvin CC , Larsen JP , Aarsland D ((2007) ) A magnetic resonance imaging study of patients with Parkinson’s disease with mild cognitive impairment and dementia using voxel-based morphometry. J Neurol Neurosurg Psychiatry 78: , 254–259. |
[35] | Whitwell JL , Weigand SD , Shiung MM , Boeve BF , Ferman TJ , Smith GE , Knopman DS , Petersen RC , Benarroch EE , Josephs KA , Jack CR Jr. ((2007) ) Focal atrophy in dementia with Lewy bodies on MRI: A distinct pattern from Alzheimer’s disease. Brain 130: , 708–719. |
[36] | Nemoto K , Sakaguchi H , Kasai W , Hotta M , Kamei R , Noguchi T , Minamimoto R , Arai T , Asada T ((2021) ) Differentiating dementia with Lewy bodies and Alzheimer’s disease by deep learning to structural MRI. J Neuroimaging 31: , 579–587. |
[37] | Zhu H , Lu H , Wang F , Liu S , Shi Z , Gan J , Du X , Yang Y , Li D , Wang L , Ji Y ((2021) ) Characteristics of cortical atrophy and white matter lesions between dementia with Lewy bodies and Alzheimer’s disease: A case-control study. Front Neurol 12: , 779344. |
[38] | Caso F , Agosta F , Scamarcia PG , Basaia S , Canu E , Magnani G , Volonte MA , Filippi M ((2021) ) A multiparametric MRI study of structural brain damage in dementia with lewy bodies: A comparison with Alzheimer’s disease. Parkinsonism Relat Disord 91: , 154–161. |
[39] | Shimada H , Hirano S , Shinotoh H , Aotsuka A , Sato K , Tanaka N , Ota T , Asahina M , Fukushi K , Kuwabara S , Hattori T , Suhara T , Irie T ((2009) ) Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology 73: , 273–278. |
[40] | Okkels N , Horsager J , Labrador-Espinosa M , Kjeldsen PL , Damholdt MF , Mortensen J , Vestergard K , Knudsen K , Andersen KB , Fedorova TD , Skjaerbaek C , Gottrup H , Hansen AK , Grothe MJ , Borghammer P ((2023) ) Severe cholinergic terminal loss in newly diagnosed dementia with Lewy bodies. Brain 146: , 3690–3704. |
[41] | Hiraoka K , Okamura N , Funaki Y , Hayashi A , Tashiro M , Hisanaga K , Fujii T , Takeda A , Yanai K , Iwata R , Mori E ((2012) ) Cholinergic deficit and response to donepezil therapy in Parkinson’s disease with dementia. Eur Neurol 68: , 137–143. |
[42] | Nejad-Davarani S , Koeppe RA , Albin RL , Frey KA , Muller M , Bohnen NI ((2019) ) Quantification of brain cholinergic denervation in dementia with Lewy bodies using PET imaging with [(18)F]-FEOBV. Mol Psychiatry 24: , 322–327. |
[43] | Bohnen NI , Kaufer DI , Hendrickson R , Ivanco LS , Lopresti BJ , Constantine GM , Mathis Ch A , Davis JG , Moore RY , Dekosky ST ((2006) ) Cognitive correlates of cortical cholinergic denervation in Parkinson’s disease and parkinsonian dementia. J Neurol 253: , 242–247. |
[44] | Walker Z , Costa DC , Walker RW , Lee L , Livingston G , Jaros E , Perry R , McKeith I , Katona CL ((2004) ) Striatal dopamine transporter in dementia with Lewy bodies and Parkinson disease: A comparison. Neurology 62: , 1568–1572. |
[45] | Fedorova TD , Knudsen K , Horsager J , Hansen A , Okkels N , Gottrup H , Vang K , Borghammer P ((2023) ) Dopaminergic dysfunction is more symmetric in Dementia with Lewy bodies compared to Parkinson’s disease. J Parkinsons Dis 13: , 515–523. |
[46] | Cao R , Chen X , Xie C , Hu P , Wang K ((2020) ) Serial dopamine transporter imaging of nigrostriatal function in Parkinson’s disease with probable REM sleep behavior disorder. Front Neurosci 14: , 349. |
[47] | McKeith I , O’Brien J , Walker Z , Tatsch K , Booij J , Darcourt J , Padovani A , Giubbini R , Bonuccelli U , Volterrani D , Holmes C , Kemp P , Tabet N , Meyer I , Reininger C , Group DLBS ((2007) ) Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: A phase III, multicentre study. Lancet Neurol 6: , 305–313. |
[48] | van der Zande JJ , Booij J , Scheltens P , Raijmakers PG , Lemstra AW ((2016) ) [(123)]FP-CIT SPECT scans initially rated as normal became abnormal over time in patients with probable dementia with Lewy bodies. Eur J Nucl Med Mol Imaging 43: , 1060–1066. |
[49] | Choudhury P , Zhang N , Adler CH , Chen K , Belden C , Driver-Dunckley E , Mehta SH , Shprecher DR , Serrano GE , Shill HA , Beach TG , Atri A ((2023) ) Longitudinal motor decline in dementia with Lewy bodies, Parkinson disease dementia, and Alzheimer’s dementia in a community autopsy cohort. Alzheimers Dement 19: , 4377–4387. |
[50] | Orimo S , Yogo M , Nakamura T , Suzuki M , Watanabe H ((2016) ) Brain imaging in Aging Special Issue of Ageing Research Reviews I-meta-iodobenzylguanidine (MIBG) cardiac scintigraphy in alpha-synucleinopathies. Ageing Res Rev 30: , 122–133. |
[51] | Goldstein DS , Sewell L , Sharabi Y ((2011) ) Autonomic dysfunction in PD: A window to early detection? J Neurol Sci 310: , 118–122. |
[52] | Horsager J , Andersen KB , Knudsen K , Skjaerbaek C , Fedorova TD , Okkels N , Schaeffer E , Bonkat SK , Geday J , Otto M , Sommerauer M , Danielsen EH , Bech E , Kraft J , Munk OL , Hansen SD , Pavese N , Goder R , Brooks DJ , Berg D , Borghammer P ((2020) ) Brain-first versus body-first Parkinson’s disease: A multimodal imaging case-control study. Brain 143: , 3077–3088. |
[53] | Nagayama H , Hamamoto M , Ueda M , Nagashima J , Katayama Y ((2005) ) Reliability of MIBG myocardial scintigraphy in the diagnosis of Parkinson’s disease. J Neurol Neurosurg Psychiatry 76: , 249–251. |
[54] | Kashihara K , Imamura T , Shinya T ((2010) ) Cardiac 123I-MIBG uptake is reduced more markedly in patients with REM sleep behavior disorder than in those with early stage Parkinson’s disease. Parkinsonism Relat Disord 16: , 252–255. |
[55] | Baumann-Vogel H , Hor H , Poryazova R , Valko P , Werth E , Baumann CR ((2020) ) REM sleep behavior in Parkinson disease: Frequent, particularly with higher age. PLoS One 15: , e0243454. |
[56] | Pao WC , Boeve BF , Ferman TJ , Lin SC , Smith GE , Knopman DS , Graff-Radford NR , Petersen RC , Parisi JE , Dickson DW , Silber MH ((2013) ) Polysomnographic findings in dementia with Lewy bodies. Neurologist 19: , 1–6. |
[57] | van de Beek M , van Steenoven I , van der Zande JJ , Barkhof F , Teunissen CE , van der Flier WM , Lemstra AW ((2020) ) Prodromal dementia with Lewy bodies: Clinical characterization and predictors of progression. Mov Disord 35: , 859–867. |
[58] | Petrou M , Dwamena BA , Foerster BR , MacEachern MP , Bohnen NI , Muller ML , Albin RL , Frey KA ((2015) ) Amyloid deposition in Parkinson’s disease and cognitive impairment: A systematic review. Mov Disord 30: , 928–935. |
[59] | Diaz-Galvan P , Przybelski SA , Lesnick TG , Schwarz CG , Senjem ML , Gunter JL , Jack CR , Min HP , Jain M , Miyagawa T , Forsberg LK , Fields JA , Savica R , Graff-Radford J , Jones DT , Botha H , St Louis EK , Knopman DS , Ramanan VK , Ross O , Graff-Radford N , Day GS , Dickson DW , Ferman TJ , Petersen RC , Lowe VJ , Boeve BF , Kantarci K ((2023) ) beta-amyloid load on PET along the continuum of dementia with Lewy bodies. Neurology 101: , e178–e188. |
[60] | Irwin DJ , Grossman M , Weintraub D , Hurtig HI , Duda JE , Xie SX , Lee EB , Van Deerlin VM , Lopez OL , Kofler JK , Nelson PT , Jicha GA , Woltjer R , Quinn JF , Kaye J , Leverenz JB , Tsuang D , Longfellow K , Yearout D , Kukull W , Keene CD , Montine TJ , Zabetian CP , Trojanowski JQ ((2017) ) Neuropathological and genetic correlates of survival and dementia onset in synucleinopathies: A retrospective analysis. Lancet Neurol 16: , 55–65. |
[61] | Irwin DJ , Hurtig HI ((2018) ) The contribution of tau, amyloid-beta and alpha-synuclein pathology to dementia in Lewy body disorders. J Alzheimers Dis Parkinsonism 8: , 444. |
[62] | Ballard C , Ziabreva I , Perry R , Larsen JP , O’Brien J , McKeith I , Perry E , Aarsland D ((2006) ) Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology 67: , 1931–1934. |
[63] | Irwin DJ , White MT , Toledo JB , Xie SX , Robinson JL , Van Deerlin V , Lee VM , Leverenz JB , Montine TJ , Duda JE , Hurtig HI , Trojanowski JQ ((2012) ) Neuropathologic substrates of Parkinson disease dementia. Ann Neurol 72: , 587–598. |
[64] | Compta Y , Parkkinen L , O’Sullivan SS , Vandrovcova J , Holton JL , Collins C , Lashley T , Kallis C , Williams DR , de Silva R , Lees AJ , Revesz T ((2011) ) Lewy- and Alzheimer-type pathologies in Parkinson’s disease dementia: Which is more important? Brain 134: , 1493–1505. |
[65] | Beach TG , Adler CH , Lue L , Sue LI , Bachalakuri J , Henry-Watson J , Sasse J , Boyer S , Shirohi S , Brooks R , Eschbacher J , White CL 3rd Akiyama H , Caviness J , Shill HA , Connor DJ , Sabbagh MN , Walker DG Arizona Parkinson’s Disease Consortium ((2009) ) Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117: , 613–634. |
[66] | Martin WRW , Younce JR , Campbell MC , Racette BA , Norris SA , Ushe M , Criswell S , Davis AA , Alfradique-Dunham I , Maiti B , Cairns NJ , Perrin RJ , Kotzbauer PT , Perlmutter JS ((2023) ) Neocortical Lewy body pathology parallels Parkinson’s dementia, but not always. Ann Neurol 93: , 184–195. |
[67] | Halliday G , Hely M , Reid W , Morris J ((2008) ) The progression of pathology in longitudinally followed patients with Parkinson’s disease. Acta Neuropathol 115: , 409–415. |
[68] | Jellinger KA ((2022) ) Are there morphological differences between Parkinson’s disease-dementia and dementia with Lewy bodies? Parkinsonism Relat Disord 100: , 24–32. |
[69] | Jellinger KA , Seppi K , Wenning GK , Poewe W ((2002) ) Impact of coexistent Alzheimer pathology on the natural history of Parkinson’s disease. J Neural Transm (Vienna) 109: , 329–339. |
[70] | Reid WG , Hely MA , Morris JG , Loy C , Halliday GM ((2011) ) Dementia in Parkinson’s disease: A 20-year neuropsychological study (Sydney Multicentre Study). J Neurol Neurosurg Psychiatry 82: , 1033–1037. |
[71] | Howlett DR , Whitfield D , Johnson M , Attems J , O’Brien JT , Aarsland D , Lai MK , Lee JH , Chen C , Ballard C , Hortobagyi T , Francis PT ((2015) ) Regional multiple pathology scores are associated with cognitive decline in Lewy body dementias. Brain Pathol 25: , 401–408. |
[72] | Hely MA , Reid WG , Adena MA , Halliday GM , Morris JG ((2008) ) The Sydney multicenter study of Parkinson’s disease: The inevitability of dementia at 20 years. Mov Disord 23: , 837–844. |
[73] | Jellinger KA , Attems J ((2008) ) Prevalence and impact of vascular and Alzheimer pathologies in Lewy body disease. Acta Neuropathol 115: , 427–436. |
[74] | Quadalti C , Palmqvist S , Hall S , Rossi M , Mammana A , Janelidze S , Dellavalle S , Mattsson-Carlgren N , Baiardi S , Stomrud E , Hansson O , Parchi P ((2023) ) Clinical effects of Lewy body pathology in cognitively impaired individuals. Nat Med 29: , 1964–1970. |
[75] | Selikhova M , Williams DR , Kempster PA , Holton JL , Revesz T , Lees AJ ((2009) ) A clinico-pathological study of subtypes in Parkinson’s disease. Brain 132: , 2947–2957. |
[76] | Coughlin D , Xie SX , Liang M , Williams A , Peterson C , Weintraub D , McMillan CT , Wolk DA , Akhtar RS , Hurtig HI , Branch Coslett H , Hamilton RH , Siderowf AD , Duda JE , Rascovsky K , Lee EB , Lee VM , Grossman M , Trojanowski JQ , Irwin DJ ((2019) ) Cognitive and pathological influences of tau pathology in Lewy body disorders. Ann Neurol 85: , 259–271. |
[77] | Irwin DJ , Lee VM , Trojanowski JQ ((2013) ) Parkinson’s disease dementia: Convergence of alpha-synuclein, tau and amyloid-beta pathologies. Nat Rev Neurosci 14: , 626–636. |
[78] | Borghammer P ((2021) ) The alpha-Synuclein Origin and Connectome Model (SOC Model) of Parkinson’s disease: Explaining motor asymmetry, non-motor phenotypes, and cognitive decline. J Parkinsons Dis 11: , 455–474. |
[79] | Borghammer P , Van Den Berge N ((2019) ) Brain-first versus gut-first Parkinson’s disease: A hypothesis. , S281-S. J Parkinsons Dis 9: , 295. |
[80] | Knudsen K , Fedorova TD , Horsager J , Andersen KB , Skjaerbaek C , Berg D , Schaeffer E , Brooks DJ , Pavese N , Van Den Berge N , Borghammer P ((2021) ) Asymmetric dopaminergic dysfunction in brain-first versus body-first Parkinson’s disease subtypes. J Parkinsons Dis 11: , 1677–1687. |
[81] | Fereshtehnejad SM , Romenets SR , Anang JB , Latreille V , Gagnon JF , Postuma RB ((2015) ) New clinical subtypes of Parkinson disease and their longitudinal progression: A prospective cohort comparison with other phenotypes. JAMA Neurol 72: , 863–873. |
[82] | Merola A , Romagnolo A , Dwivedi AK , Padovani A , Berg D , Garcia-Ruiz PJ , Fabbri M , Artusi CA , Zibetti M , Lopiano L , Pilotto A , Bonacina S , Morgante F , Zeuner K , Griewing C , Schaeffer E , Rodriguez-Porcel F , Kauffman M , Turcano P , de Oliveira LM , Palermo G , Shanks E , Del Sorbo F , Bonvegna S , Savica R , Munhoz RP , Ceravolo R , Cilia R , Espay AJ ((2020) ) Benign versus malignant Parkinson disease: The unexpected silver lining of motor complications. J Neurol 267: , 2949–2960. |
[83] | Kim JS , Park HE , Park IS , Oh YS , Ryu DW , Song IU , Jung YA , Yoo IR , Choi HS , Lee PH , Lee KS ((2017) ) Normal ‘heart’ in Parkinson’s disease: Is this a distinct clinical phenotype? Eur J Neurol 24: , 349–356. |
[84] | Horsager J , Knudsen K , Sommerauer M ((2022) ) Clinical and imaging evidence of brain-first and body-first Parkinson’s disease. Neurobiol Dis 164: , 105626. |
[85] | Borghammer P , Just MK , Horsager J , Skjaerbaek C , Raunio A , Kok EH , Savola S , Murayama S , Saito Y , Myllykangas L , Van Den Berge N ((2022) ) A postmortem study suggests a revision of the dual-hit hypothesis of Parkinson’s disease. NPJ Parkinsons Dis 8: , 166. |
[86] | Bassil F , Brown HJ , Pattabhiraman S , Iwasyk JE , Maghames CM , Meymand ES , Cox TO , Riddle DM , Zhang B , Trojanowski JQ , Lee VM ((2020) ) Amyloid-beta (Abeta) plaques promote seeding and spreading of alpha-synuclein and tau in a mouse model of Lewy body disorders with Abeta pathology. Neuron 105: , 260–275e266. |
[87] | Williams T , Sorrentino Z , Weinrich M , Giasson BI , Chakrabarty P ((2020) ) Differential cross-seeding properties of tau and alpha-synuclein in mouse models of tauopathy and synucleinopathy. Brain Commun 2: , fcaa090. |
[88] | Borghammer P , Horsager J , Andersen K , Van Den Berge N , Raunio A , Murayama S , Parkkinen L , Myllykangas L ((2021) ) Neuropathological evidence of body-first vs. brain-first Lewy body disease. Neurobiol Dis 161: , 105557. |
[89] | Tanei ZI , Saito Y , Ito S , Matsubara T , Motoda A , Yamazaki M , Sakashita Y , Kawakami I , Ikemura M , Tanaka S , Sengoku R , Arai T , Murayama S ((2021) ) Lewy pathology of the esophagus correlates with the progression of Lewy body disease: A Japanese cohort study of autopsy cases. Acta Neuropathol 141: , 25–37. |
[90] | Raunio A , Kaivola K , Tuimala J , Kero M , Oinas M , Polvikoski T , Paetau A , Tienari PJ , Myllykangas L ((2019) ) Lewy-related pathology exhibits two anatomically and genetically distinct progression patterns: A population-based study of Finns aged 85. Acta Neuropathol 138: , 771–782. |
[91] | Nalls MA , Duran R , Lopez G , Kurzawa-Akanbi M , McKeith IG , Chinnery PF , Morris CM , Theuns J , Crosiers D , Cras P , Engelborghs S , De Deyn PP , Van Broeckhoven C , Mann DM , Snowden J , Pickering-Brown S , Halliwell N , Davidson Y , Gibbons L , Harris J , Sheerin UM , Bras J , Hardy J , Clark L , Marder K , Honig LS , Berg D , Maetzler W , Brockmann K , Gasser T , Novellino F , Quattrone A , Annesi G , De Marco EV , Rogaeva E , Masellis M , Black SE , Bilbao JM , Foroud T , Ghetti B , Nichols WC , Pankratz N , Halliday G , Lesage S , Klebe S , Durr A , Duyckaerts C , Brice A , Giasson BI , Trojanowski JQ , Hurtig HI , Tayebi N , Landazabal C , Knight MA , Keller M , Singleton AB , Wolfsberg TG , Sidransky E ((2013) ) A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol 70: , 727–735. |
[92] | Cilia R , Tunesi S , Marotta G , Cereda E , Siri C , Tesei S , Zecchinelli AL , Canesi M , Mariani CB , Meucci N , Sacilotto G , Zini M , Barichella M , Magnani C , Duga S , Asselta R , Solda G , Seresini A , Seia M , Pezzoli G , Goldwurm S ((2016) ) Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann Neurol 80: , 662–673. |
[93] | Blauwendraat C , Reed X , Krohn L , Heilbron K , Bandres-Ciga S , Tan M , Gibbs JR , Hernandez DG , Kumaran R , Langston R , Bonet-Ponce L , Alcalay RN , Hassin-Baer S , Greenbaum L , Iwaki H , Leonard HL , Grenn FP , Ruskey JA , Sabir M , Ahmed S , Makarious MB , Pihlstrøm L , Toft M , van Hilten JJ , Marinus J , Schulte C , Brockmann K , Sharma M , Siitonen A , Majamaa K , Eerola-Rautio J , Tienari PJ , 23andMe Research Team; Pantelyat A , Hillis AE , Dawson TM , Rosenthal LS , Albert MS , Resnick SM , Ferrucci L , Morris CM , Pletnikova O , Troncoso J , Grosset D , Lesage S , Corvol JC , Brice A , Noyce AJ , Masliah E , Wood N , Hardy J , Shulman LM , Jankovic J , Shulman JM , Heutink P , Gasser T , Cannon P , Scholz SW , Morris H , Cookson MR , Nalls MA , Gan-Or Z , Singleton AB ((2020) ) Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 143: , 234–248. |
[94] | Stoker TB , Camacho M , Winder-Rhodes S , Liu G , Scherzer CR , Foltynie T , Evans J , Breen DP , Barker RA , Williams-Gray CH ((2020) ) Impact of GBA1 variants on long-term clinical progression and mortality in incident Parkinson’s disease. J Neurol Neurosurg Psychiatry 91: , 695–702. |
[95] | Yahalom G , Greenbaum L , Israeli-Korn S , Fay-Karmon T , Livneh V , Ruskey JA , Ronciere L , Alam A , Gan-Or Z , Hassin-Baer S ((2019) ) Carriers of both GBA and LRRK2 mutations, compared to carriers of either, in Parkinson’s disease: Risk estimates and genotype-phenotype correlations. Parkinsonism Relat Disord 62: , 179–184. |
[96] | Thaler A , Kozlovski T , Gurevich T , Bar-Shira A , Gana-Weisz M , Orr-Urtreger A , Giladi N , Mirelman A ((2018) ) Survival rates among Parkinson’s disease patients who carry mutations in the LRRK2 and GBA genes. Mov Disord 33: , 1656–1660. |
[97] | Marras C , Alcalay RN , Caspell-Garcia C , Coffey C , Chan P , Duda JE , Facheris MF , Fernández-Santiago R , Ruíz-Martínez J , Mestre T , Saunders-Pullman R , Pont-Sunyer C , Tolosa E , Waro B LRRK2 Cohort Consortium ((2016) ) Motor and nonmotor heterogeneity of LRRK2-related and idiopathic Parkinson’s disease. Mov Disord 31: , 1192–1202. |
[98] | Kestenbaum M , Alcalay RN ((2017) ) Clinical features of LRRK2 carriers with Parkinson’s disease. Adv Neurobiol 14: , 31–48. |
[99] | Heckman MG , Soto-Ortolaza AI , Contreras MYS , Murray ME , Pedraza O , Diehl NN , Walton R , Labbe C , Lorenzo-Betancor O , Uitti RJ , van Gerpen J , Ertekin-Taner N , Smith GE , Kantarci K , Savica R , Jones DT , Graff-Radford J , Knopman DS , Lowe VJ , Jack CR Jr , Petersen RC , Parisi JE , Rademakers R , Wszolek ZK , Graff-Radford NR , Ferman TJ , Dickson DW , Boeve BF , Ross OA ((2016) ) LRRK2 variation and dementia with Lewy bodies. Parkinsonism Relat Disord 31: , 98–103. |
[100] | Saunders-Pullman R , Mirelman A , Alcalay RN , Wang C , Ortega RA , Raymond D , Mejia-Santana H , Orbe-Reilly M , Johannes BA , Thaler A , Ozelius L , Orr-Urtreger A , Marder KS , Giladi N , Bressman SB , Consortium LAJ ((2018) ) Progression in the LRRK2-asssociated Parkinson disease population. JAMA Neurol 75: , 312–319. |
[101] | Jansen WJ , Ossenkoppele R , Knol DL , Tijms BM , Scheltens P , Verhey FR , Visser PJ Amyloid Biomarker Study G Aalten P , Aarsland D , Alcolea D , Alexander M , Almdahl IS , Arnold SE , Baldeiras I , Barthel H , van Berckel BN , Bibeau K , Blennow K , Brooks DJ , van Buchem MA , Camus V , Cavedo E , Chen K , Chetelat G , Cohen AD , Drzezga A , Engelborghs S , Fagan AM , Fladby T , Fleisher AS , van der Flier WM , Ford L , Forster S , Fortea J , Foskett N , Frederiksen KS , Freund-Levi Y , Frisoni GB , Froelich L , Gabryelewicz T , Gill KD , Gkatzima O , Gomez-Tortosa E , Gordon MF , Grimmer T , Hampel H , Hausner L , Hellwig S , Herukka SK , Hildebrandt H , Ishihara L , Ivanoiu A , Jagust WJ , Johannsen P , Kandimalla R , Kapaki E , Klimkowicz-Mrowiec A , Klunk WE , Kohler S , Koglin N , Kornhuber J , Kramberger MG , Van Laere K , Landau SM , Lee DY , de Leon M , Lisetti V , Lleo A , Madsen K , Maier W , Marcusson J , Mattsson N , de Mendonca A , Meulenbroek O , Meyer PT , Mintun MA , Mok V , Molinuevo JL , Mollergard HM , Morris JC , Mroczko B , Van der Mussele S , Na DL , Newberg A , Nordberg A , Nordlund A , Novak GP , Paraskevas GP , Parnetti L , Perera G , Peters O , Popp J , Prabhakar S , Rabinovici GD , Ramakers IH , Rami L , Resende de Oliveira C , Rinne JO , Rodrigue KM , Rodriguez-Rodriguez E , Roe CM , Rot U , Rowe CC , Ruther E , Sabri O , Sanchez-Juan P , Santana I , Sarazin M , Schroder J , Schutte C , Seo SW , Soetewey F , Soininen H , Spiru L , Struyfs H , Teunissen CE , Tsolaki M , Vandenberghe R , Verbeek MM , Villemagne VL , Vos SJ , van Waalwijk van Doorn LJ , Waldemar G , Wallin A , Wallin AK , Wiltfang J , Wolk DA , Zboch M , Zetterberg H ((2015) ) Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 313: , 1924–1938. |
[102] | Ossenkoppele R , Jansen WJ , Rabinovici GD , Knol DL , van der Flier WM , van Berckel BN , Scheltens P , Visser PJ , Amyloid PETSG , Verfaillie SC , Zwan MD , Adriaanse SM , Lammertsma AA , Barkhof F , Jagust WJ , Miller BL , Rosen HJ , Landau SM , Villemagne VL , Rowe CC , Lee DY , Na DL , Seo SW , Sarazin M , Roe CM , Sabri O , Barthel H , Koglin N , Hodges J , Leyton CE , Vandenberghe R , van Laere K , Drzezga A , Forster S , Grimmer T , Sanchez-Juan P , Carril JM , Mok V , Camus V , Klunk WE , Cohen AD , Meyer PT , Hellwig S , Newberg A , Frederiksen KS , Fleisher AS , Mintun MA , Wolk DA , Nordberg A , Rinne JO , Chetelat G , Lleo A , Blesa R , Fortea J , Madsen K , Rodrigue KM , Brooks DJ ((2015) ) Prevalence of amyloid PET positivity in dementia syndromes: A meta-analysis. JAMA 313: , 1939–1949. |
[103] | Talyansky S , Le Guen Y , Kasireddy N , Belloy ME , Greicius MD ((2023) ) APOE-epsilon4 and BIN1 increase risk of Alzheimer’s disease pathology but not specifically of Lewy body pathology. Acta Neuropathol Commun 11: , 149. |
[104] | Pavelka L , Rauschenberger A , Landoulsi Z , Pachchek S , Marques T , Gomes CPC , Glaab E , May P , Krüger R NCER-PD Consortium ((2022) ) Body-first subtype of Parkinson’s disease with probable REM-sleep behavior disorder is associated with non-motor dominant phenotype. J Parkinsons Dis 12: , 2561–2573. |
[105] | Tsuang D , Leverenz JB , Lopez OL , Hamilton RL , Bennett DA , Schneider JA , Buchman AS , Larson EB , Crane PK , Kaye JA , Kramer P , Woltjer R , Trojanowski JQ , Weintraub D , Chen-Plotkin AS , Irwin DJ , Rick J , Schellenberg GD , Watson GS , Kukull W , Nelson PT , Jicha GA , Neltner JH , Galasko D , Masliah E , Quinn JF , Chung KA , Yearout D , Mata IF , Wan JY , Edwards KL , Montine TJ , Zabetian CP ((2013) ) APOE epsilon4 increases risk for dementia in pure synucleinopathies. JAMA Neurol 70: , 223–228. |
[106] | Ruffmann C , Calboli FC , Bravi I , Gveric D , Curry LK , de Smith A , Pavlou S , Buxton JL , Blakemore AI , Takousis P , Molloy S , Piccini P , Dexter DT , Roncaroli F , Gentleman SM , Middleton LT ((2016) ) Cortical Lewy bodies and Abeta burden are associated with prevalence and timing of dementia in Lewy body diseases. Neuropathol Appl Neurobiol 42: , 436–450. |
[107] | Tsuang DW , Wilson RK , Lopez OL , Luedecking-Zimmer EK , Leverenz JB , DeKosky ST , Kamboh MI , Hamilton RL ((2005) ) Genetic association between the APOE*4 allele and Lewy bodies in Alzheimer disease. Neurology 64: , 509–513. |
[108] | De Pablo-Fernandez E , Lees AJ , Holton JL , Warner TT ((2019) ) Prognosis and neuropathologic correlation of clinical subtypes of Parkinson disease. JAMA Neurol 76: , 470–479. |
[109] | Armstrong MJ , Alliance S , Corsentino P , DeKosky ST , Taylor A ((2019) ) Cause of death and end-of-life experiences in individuals with dementia with Lewy bodies. J Am Geriatr Soc 67: , 67–73. |
[110] | Borghammer P ((2023) ) The brain-first vs. body-first model of Parkinson’s disease with comparison to alternative models. J Neural Transm (Vienna) 130: , 737–753. |
[111] | Kaufmann H ((1996) ) Consensus statement on the definition of orthostatic hypotension, pure autonomic failure and multiple system atrophy. Clin Auton Res 6: , 125–126. |
[112] | Kaufmann H , Norcliffe-Kaufmann L , Palma JA , Biaggioni I , Low PA , Singer W , Goldstein DS , Peltier AC , Shibao CA , Gibbons CH , Freeman R , Robertson D , Autonomic Disorders C ((2017) ) Natural history of pure autonomic failure: A United States prospective cohort. Ann Neurol 81: , 287–297. |
[113] | Donadio V , Incensi A , Piccinini C , Cortelli P , Giannoccaro MP , Baruzzi A , Liguori R ((2016) ) Skin nerve misfolded alpha-synuclein in pure autonomic failure and Parkinson disease. Ann Neurol 79: , 306–316. |
[114] | Hague K , Lento P , Morgello S , Caro S , Kaufmann H ((1997) ) The distribution of Lewy bodies in pure autonomic failure: Autopsy findings and review of the literature. Acta Neuropathol 94: , 192–196. |
[115] | Terao Y , Takeda K , Sakuta M , Nemoto T , Takemura T , Kawai M ((1993) ) Pure progressive autonomic failure: A clinicopathological study. Eur Neurol 33: , 409–415. |
[116] | Murayama S , Saito Y ((2022) ) [Pathological study of pure autonomic failure]. Brain Nerve 74: , 231–240. |
[117] | Arai K , Kato N , Kashiwado K , Hattori T ((2000) ) Pure autonomic failure in association with human alpha-synucleinopathy. Neurosci Lett 296: , 171–173. |
[118] | Roessmann U , Van den Noort S , McFarland DE ((1971) ) Idiopathic orthostatic hypotension. Arch Neurol 24: , 503–510. |
[119] | Vanderhaeghen JJ , Perier O , Sternon JE ((1970) ) Pathological findings in idiopathic orthostatic hypotension. Its relationship with Parkinson’s disease. Arch Neurol 22: , 207–214. |
[120] | Giannini G , Calandra-Buonaura G , Asioli GM , Cecere A , Barletta G , Mignani F , Ratti S , Guaraldi P , Provini F , Cortelli P ((2018) ) The natural history of idiopathic autonomic failure: The IAF-BO cohort study. Neurology 91: , e1245–e1254. |
[121] | Elliott JE , Lim MM , Keil AT , Postuma RB , Pelletier A , Gagnon JF , St Louis EK , Forsberg LK , Fields JA , Huddleston DE , Bliwise DL , Avidan AY , Howell MJ , Schenck CH , McLeland J , Criswell SR , Videnovic A , During EH , Miglis MG , Shprecher DR , Lee-Iannotti JK , Boeve BF , Ju YS North American Prodromal Synucleinopathy (NAPS) Consortium ((2023) ) Baseline characteristics of the North American prodromal Synucleinopathy cohort. Ann Clin Transl Neurol 10: , 520–535. |
[122] | Postuma RB , Iranzo A , Hu M , Hogl B , Boeve BF , Manni R , Oertel WH , Arnulf I , Ferini-Strambi L , Puligheddu M , Antelmi E , Cochen De Cock V , Arnaldi D , Mollenhauer B , Videnovic A , Sonka K , Jung KY , Kunz D , Dauvilliers Y , Provini F , Lewis SJ , Buskova J , Pavlova M , Heidbreder A , Montplaisir JY , Santamaria J , Barber TR , Stefani A , St Louis EK , Terzaghi M , Janzen A , Leu-Semenescu S , Plazzi G , Nobili F , Sixel-Doering F , Dusek P , Bes F , Cortelli P , Ehgoetz Martens K , Gagnon JF , Gaig C , Zucconi M , Trenkwalder C , Gan-Or Z , Lo C , Rolinski M , Mahlknecht P , Holzknecht E , Boeve AR , Teigen LN , Toscano G , Mayer G , Morbelli S , Dawson B , Pelletier A ((2019) ) Risk and predictors of dementia and parkinsonism in idiopathic REM sleep behaviour disorder: A multicentre study. Brain 142: , 744–759. |
[123] | Iranzo A , Gelpi E , Tolosa E , Molinuevo JL , Serradell M , Gaig C , Santamaria J ((2014) ) Neuropathology of prodromal Lewy body disease. Mov Disord 29: , 410–415. |
[124] | Boeve BF , Dickson DW , Olson EJ , Shepard JW , Silber MH , Ferman TJ , Ahlskog JE , Benarroch EE ((2007) ) Insights into REM sleep behavior disorder pathophysiology in brainstem-predominant Lewy body disease. Sleep Med 8: , 60–64. |
[125] | Uchiyama M , Isse K , Tanaka K , Yokota N , Hamamoto M , Aida S , Ito Y , Yoshimura M , Okawa M ((1995) ) Incidental Lewy body disease in a patient with REM sleep behavior disorder. Neurology 45: , 709–712. |
[126] | Boeve BF , Silber MH , Ferman TJ , Lin SC , Benarroch EE , Schmeichel AM , Ahlskog JE , Caselli RJ , Jacobson S , Sabbagh M , Adler C , Woodruff B , Beach TG , Iranzo A , Gelpi E , Santamaria J , Tolosa E , Singer C , Mash DC , Luca C , Arnulf I , Duyckaerts C , Schenck CH , Mahowald MW , Dauvilliers Y , Graff-Radford NR , Wszolek ZK , Parisi JE , Dugger B , Murray ME , Dickson DW ((2013) ) Clinicopathologic correlations in 172 cases of rapid eye movement sleep behavior disorder with or without a coexisting neurologic disorder. Sleep Med 14: , 754–762. |
[127] | Doppler K , Jentschke HM , Schulmeyer L , Vadasz D , Janzen A , Luster M , Hoffken H , Mayer G , Brumberg J , Booij J , Musacchio T , Klebe S , Sittig-Wiegand E , Volkmann J , Sommer C , Oertel WH ((2017) ) Dermal phospho-alpha-synuclein deposits confirm REM sleep behaviour disorder as prodromal Parkinson’s disease. Acta Neuropathol 133: , 535–545. |
[128] | Sprenger FS , Stefanova N , Gelpi E , Seppi K , Navarro-Otano J , Offner F , Vilas D , Valldeoriola F , Pont-Sunyer C , Aldecoa I , Gaig C , Gines A , Cuatrecasas M , Hogl B , Frauscher B , Iranzo A , Wenning GK , Vogel W , Tolosa E , Poewe W ((2015) ) Enteric nervous system alpha-synuclein immunoreactivity in idiopathic REM sleep behavior disorder. Neurology 85: , 1761–1768. |
[129] | Vilas D , Iranzo A , Tolosa E , Aldecoa I , Berenguer J , Vilaseca I , Marti C , Serradell M , Lomena F , Alos L , Gaig C , Santamaria J , Gelpi E ((2016) ) Assessment of alpha-synuclein in submandibular glands of patients with idiopathic rapid-eye-movement sleep behaviour disorder: A case-control study. Lancet Neurol 15: , 708–718. |
[130] | Kuzkina A , Rossle J , Seger A , Panzer C , Kohl A , Maltese V , Musacchio T , Blaschke SJ , Tamguney G , Kaulitz S , Rak K , Scherzad A , Zimmermann PH , Klussmann JP , Hackenberg S , Volkmann J , Sommer C , Sommerauer M , Doppler K ((2023) ) Combining skin and olfactory alpha-synuclein seed amplification assays (SAA)-towards biomarker-driven phenotyping in synucleinopathies. NPJ Parkinsons Dis 9: , 79. |
[131] | Knudsen K , Fedorova TD , Hansen AK , Sommerauer M , Otto M , Svendsen KB , Nahimi A , Stokholm MG , Pavese N , Beier CP , Brooks DJ , Borghammer P ((2018) ) staging of pathology in REM sleep behaviour disorder: A multimodality imaging case-control study. Lancet Neurol 17: , 618–628. |
[132] | Nishikawa N , Murata M , Hatano T , Mukai Y , Saitoh Y , Sakamoto T , Hanakawa T , Kamei Y , Tachimori H , Hatano K , Matsuda H , Taruno Y , Sawamoto N , Kajiyama Y , Ikenaka K , Kawabata K , Nakamura T , Iwaki H , Kadotani H , Sumi Y , Inoue Y , Hayashi T , Ikeuchi T , Shimo Y , Mochizuki H , Watanabe H , Hattori N , Takahashi Y , Takahashi R Japan Parkinson’s Progression Markers Initiative (J-PPMI) study group ((2022) ) Idiopathic rapid eye movement sleep behavior disorder in Japan: An observational study. Parkinsonism Relat Disord 103: , 129–135. |
[133] | Miyamoto T , Miyamoto M , Inoue Y , Usui Y , Suzuki K , Hirata K ((2006) ) Reduced cardiac 123I-MIBG scintigraphy in idiopathic REM sleep behavior disorder. Neurology 67: , 2236–2238. |
[134] | Ehrminger M , Latimier A , Pyatigorskaya N , Garcia-Lorenzo D , Leu-Semenescu S , Vidailhet M , Lehericy S , Arnulf I ((2016) ) The coeruleus/subcoeruleus complex in idiopathic rapid eye movement sleep behaviour disorder. Brain 139: , 1180–1188. |
[135] | Garcia-Lorenzo D , Longo-Dos Santos C , Ewenczyk C , Leu-Semenescu S , Gallea C , Quattrocchi G , Pita Lobo P , Poupon C , Benali H , Arnulf I , Vidailhet M , Lehericy S ((2013) ) The coeruleus/subcoeruleus complex in rapid eye movement sleep behaviour disorders in Parkinson’s disease. Brain 136: , 2120–2129. |
[136] | Sommerauer M , Fedorova TD , Hansen AK , Knudsen K , Otto M , Jeppesen J , Frederiksen Y , Blicher JU , Geday J , Nahimi A , Damholdt MF , Brooks DJ , Borghammer P ((2018) ) Evaluation of the noradrenergic system in Parkinson’s disease: An 11C-MeNER PET and neuromelanin MRI study. Brain 141: , 496–504. |
[137] | Sixel-Doering F , Muntean ML , Petersone D , Leha A , Lang E , Mollenhauer B , Trenkwalder C ((2023) ) The increasing prevalence of REM sleep behavior disorder with Parkinson’s disease progression: A polysomnography-supported study. Mov Disord Clin Pract 10: , 1769–1776. |
[138] | Choudhury P , Graff-Radford J , Aakre JA , Wurtz L , Knopman DS , Graff-Radford NR , Kantarci K , Forsberg LK , Fields JA , Pedraza O , Chen Q , Miyagawa T , Day GS , Tipton P , Savica R , Botha H , Lachner C , Dredla B , Reichard RR , Petersen RC , Dickson DW , Boeve BF , Ferman TJ ((2022) ) The temporal onset of the core features in dementia with Lewy bodies. Alzheimers Dement 18: , 591–601. |
[139] | Aarsland D , Andersen K , Larsen JP , Lolk A , Kragh-Sorensen P ((2003) ) Prevalence and characteristics of dementia in Parkinson disease: An 8-year prospective study. Arch Neurol 60: , 387–392. |
[140] | Emre M , Aarsland D , Brown R , Burn DJ , Duyckaerts C , Mizuno Y , Broe GA , Cummings J , Dickson DW , Gauthier S , Goldman J , Goetz C , Korczyn A , Lees A , Levy R , Litvan I , McKeith I , Olanow W , Poewe W , Quinn N , Sampaio C , Tolosa E , Dubois B ((2007) ) Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Mov Disord 22: , 1689-1707; quiz 1837. |
[141] | Gunawardana CW , Matar E , Lewis SJG ((2024) ) The clinical phenotype of psychiatric-onset prodromal dementia with Lewy bodies: A scoping review. J Neurol 271: , 606–617. |
[142] | Siderowf A , Concha-Marambio L , Lafontant DE , Farris CM , Ma Y , Urenia PA , Nguyen H , Alcalay RN , Chahine LM , Foroud T , Galasko D , Kieburtz K , Merchant K , Mollenhauer B , Poston KL , Seibyl J , Simuni T , Tanner CM , Weintraub D , Videnovic A , Choi SH , Kurth R , Caspell-Garcia C , Coffey CS , Frasier M , Oliveira LMA , Hutten SJ , Sherer T , Marek K , Soto C Parkinson’s Progression Markers Initiative ((2023) ) Assessment of heterogeneity among participants in the Parkinson’s Progression Markers Initiative cohort using alpha-synuclein seed amplification: A cross-sectional study. Lancet Neurol 22: , 407–417. |
[143] | Liguori R , Donadio V , Wang Z , Incensi A , Rizzo G , Antelmi E , Biscarini F , Pizza F , Zou W , Plazzi G ((2023) ) A comparative blind study between skin biopsy and seed amplification assay to disclose pathological alpha-synuclein in RBD. NPJ Parkinsons Dis 9: , 34. |

