
Do Bacterial Outer Membrane Vesicles Contribute to Chronic Inflammation in Parkinson’s Disease?
Abstract
Parkinson’s disease (PD) is an increasingly common neurodegenerative disease. It has been suggested that the etiology of idiopathic PD is complex and multifactorial involving environmental contributions, such as viral or bacterial infections and microbial dysbiosis, in genetically predisposed individuals. With advances in our understanding of the gut-brain axis, there is increasing evidence that the intestinal microbiota and the mammalian immune system functionally interact. Recent findings suggest that a shift in the gut microbiome to a pro-inflammatory phenotype may play a role in PD onset and progression. While there are links between gut bacteria, inflammation, and PD, the bacterial products involved and how they traverse the gut lumen and distribute systemically to trigger inflammation are ill-defined. Mechanisms emerging in other research fields point to a role for small, inherently stable vesicles released by Gram-negative bacteria, called outer membrane vesicles in disease pathogenesis. These vesicles facilitate communication between bacteria and the host and can shuttle bacterial toxins and virulence factors around the body to elicit an immune response in local and distant organs. In this perspective article, we hypothesize a role for bacterial outer membrane vesicles in PD pathogenesis. We present evidence suggesting that these outer membrane vesicles specifically from Gram-negative bacteria could potentially contribute to PD by traversing the gut lumen to trigger local, systemic, and neuroinflammation. This perspective aims to facilitate a discussion on outer membrane vesicles in PD and encourage research in the area, with the goal of developing strategies for the prevention and treatment of the disease.
INTRODUCTION
Parkinson’s disease (PD) is a multifactorial disease encompassing many body systems but is clinically recognized as a debilitating neurodegenerative disease that affects motor function [1]. The cardinal motor symptoms include tremor, bradykinesia, postural instability, and rigidity. Concurrently, there are significant non-motor symptoms, including constipation, loss of a sense of smell, and sleep disturbances [1]. There is no single definitive cause for idiopathic PD (∼90% of cases), instead, it is hypothesized that a mosaic of genetic and environmental risk factors coalesces to initiate disease [2]. These factors include, but are not limited to, exposure to environmental insults (such as neurotoxins, infection, and pollutants) and genetic susceptibility [3–7]. Ultimately, the consequences of these exposures accumulate in the aging body and are suggested to lead to neuroinflammation, the initial loss of synapses in the striatum, neuronal loss in the substantia nigra (SN), and the aggregation of insoluble alpha-synuclein (α-syn) in Lewy bodies, the pathological hallmark features of PD [8].
A conceptual framework has been proposed to characterize the contribution of known environmental and biological factors to disease pathogenesis [8]. This framework proposes there are three phases to consider: triggers, facilitators, and aggravators [8]. Triggers (such as bacterial infections or exposure to industrial toxins) enable disease initiation when present alongside other facilitators [8–10]. Facilitators (such as peripheral inflammation, aging, or genetic susceptibility) are proposed to spread the disease and impact the central nervous system (CNS) [8, 11]. Aggravators (such as neuroinflammation, oxidative stress, and impaired autophagy) are hypothesized to exacerbate and accelerate neuronal dysfunction and /or further the spread of already initiated pathological events [8, 12]. This framework incorporates the multifactorial nature of PD and proposes a continuum of stages whereby factors may differentially contribute to disease progression depending on the disease stage. Inflammation is a consistent feature throughout this framework [8, 13]. The gastrointestinal (GI) tract is considered a site of inflammation in PD [10, 14] with elevated levels of pro-inflammatory bacteria and reduced levels of certain anti-inflammatory bacteria observed in people with PD (PwP) [15]. Researchers have hypothesized that this dysbiosis contributes to the development of disease, however, the mechanism is unclear [10, 14, 16, 17].
Here, we propose outer membrane vesicles (OMVs) released by gut bacteria as facilitators and aggravators of PD. We suggest they are a key mechanism by which gut bacteria mediate intercellular communication and transfer of immunomodulatory compounds around the body, contributing to systemic and neuroinflammation, and consequently neurodegeneration.
INFLAMMATION AND ETIOLOGY OF PD
The etiological risk factors of PD are varied, but many converge in their involvement in inflammatory processes [13, 18]. In the brain reactive microgliosis and elevation of inflammatory markers are some of the most commonly described features alongside neurodegeneration and α-syn deposition [19–26]. Previously, neuroinflammation was explained away as a mere consequence of neurodegenerative processes; however, many research groups have demonstrated in animal models that inflammation itself can contribute to furthering disease processes especially when it becomes chronic [27, 28]. Compellingly proinflammatory cytokines such as tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), and IL-6, and important inflammatory pathway proteins like nuclear factor-kappa B (NF-κB) and inducible nitric oxide synthase (iNOS) are found to be elevated in the periphery and CNS of PwP [21, 24, 29–31]. Similar elevations of cytokines, other inflammation-associated proteins, and immune cell changes have also been recapitulated in Parkinsonian animal models [32–36]. A role in promoting inflammation is a commonality between many of the proposed etiological factors that are hypothesized to trigger or facilitate PD.
Advanced age is the greatest risk factor for PD, and during aging there is a dysregulation of inflammatory processes due to the life-long cumulative effects of immune activation from various self- and non-self-stimuli [11]. This can lead to the development of sterile, low-grade, and chronic inflammation as a person ages, which is delineated from normal physiological inflammation and termed inflammaging [37, 38]. Many of the common changes to immune function that occur with age overlap with PD-associated immune dysfunction, such as the development of gut dysbiosis, reduced macrophage activity, increased inflammasome expression, and increased blood-brain-barrier (BBB) permeability [38–40].
Genetic variations that are associated with PD are also known to regulate immune responses. Notably, the leucine-rich repeat kinase 2 (LRRK2) gene encodes a protein that is highly expressed in immune cells and is known to have a role in response to bacterial infections and the innate immune system [41, 42]. Mutations in LRRK2 are suggested to lead to chronic overactivation of microglia and neuroinflammation [43, 44]. The gene SCNA encodes for α-syn, a major component of Lewy bodies, and while the physiological roles of α-syn remain unclear, data is emerging that describes a role in immune regulation due to antimicrobial, antifungal, and antiviral effects [6, 44, 45]. PINK1, a protein kinase, and parkin, an E3 ubiquitin ligase, control the elimination of dysfunctional mitochondria from the cell in a process known as mitophagy. PINK1 and PRKN (the genes encoding PINK1 and parkin, respectively) deficient mice show minimal signs of neurodegeneration unless exposed to a pro-inflammatory trigger such as bacterial infection or lipopolysaccharide (LPS) administration suggesting PINK1 and PRKNs roles in protecting against inflammation-driven neurodegeneration [46, 47]. Both DJ-1 and ATP13A2 deficiency attenuate the anti-inflammatory effects of astrocytes [48, 49]. This suggests that these genetic changes may contribute to PD by increasing one’s susceptibility to pro-inflammatory stimuli. Furthermore, over 90 independent risk signals have been identified in recent genome-wide association studies, of which the risk factor gene’s functions are enriched in chemical signaling pathways involved with responding to stressors [50].
Environmental exposure to toxins, mainly pesticides, has been long linked with PD, with a meta-analysis finding a 66% increased risk of developing PD after pesticide exposure [51]. Pesticides like paraquat and rotenone, have direct toxic effects by interfering with mitochondrial function, but have also been shown to activate inflammatory pathways, like cytokine release and microgliosis [52, 53].
Conversely, smoking (nicotine), caffeine consumption, and the regular use of anti-inflammatory analgesics have been proposed to confer protection against the development of PD due to their anti-inflammatory effects [54–58].
Bacterial, viral, and fungal infections are associated with an increased risk of PD [10, 59, 60]. Specific examples of these infections include Helicobacter pylori, hepatitis C virus, Malassezia, and Chlamydophila pneumoniae [5]. The connection between H. pylori and PD has long been discussed, in a way even before the discovery of H. pylori, as a link between peptic ulcers (of which H. pylori is a major causal agent) and PD was discovered in the 1960 s [61]. In more recent years H. pylori infection has been found to be more prevalent in PD populations and can affect PD motor symptoms, with the eradication of H. pylori improving PD motor dysfunction [62, 63]. Some have posited that H. pylori infections could be causal or at the very least a promoter of the development of PD [64]. PwP have been shown to also be more likely to be seropositive for several other bacteria including Borrelia burgdorferi and C. pneumoniae than healthy controls [65]. Furthermore, individuals who have previously been hospitalized for CNS infection [60] are at increased risk of PD as are those who have experienced GI infections caused by viral or bacterial pathogens [10].
While inflammations’ influence and perpetuation of PD processes is generally well accepted, some believe there is merit to inflammation as a disease initiator. Garretti and colleagues demonstrated that α-syn immunization of a specific HLA genetic mouse model leads to many GI features of PD, including constipation and enteric inflammation. These effects were mediated by CD4+ T cells and therefore the immune response promoted a PD-like phenotype [66]. Matheoud and colleagues described similar autoimmune processes that could be initiating neuronal dysfunction. They demonstrated in vivo that infection triggers antigen presentation of mitochondrial self-proteins. This presentation leads to the development of mitochondria-specific cytotoxic T cells in the periphery and brain, loss of synapses in the striatum, and motor dysfunction [47]. These studies demonstrate how inflammation need not just play a role in perpetuating the disease, but could initiate PD.
THE MICROBIOTA-GUT-BRAIN AXIS IN PD
The gut has long been of interest in PD research, as several lines of clinical and preclinical evidence converge upon a theory that the gut is a critical site of PD etiopathogenesis for some PwP [16]. In the prodromal stages of the disease (before the emergence of motor symptoms) GI symptoms, in particular mild constipation, are common [67, 68]. PD is associated with inflammatory bowel diseases (IBD) as they share common genetic risk factors, like LRRK2, CARD15, and ABCB1; and some groups of people with IBD are at a greater risk of developing PD [69, 70]. GI inflammation has been shown to accelerate the onset and exacerbate PD motor symptoms, influence α-syn aggregation, promote neuroinflammation, and exacerbate dopaminergic cell death in the midbrain in PD animal models [71–73].
Similarly to the pathological features of the CNS in PD, inflammation and α-syn inclusions occur also in the enteric nervous system [17, 74–83]. α-syn is proposed to be capable of transsynaptic spreading, trafficking via the vagus nerve to seed α-syn pathology in the CNS, as demonstrated in several animal model experiments [84–87]. These findings have led to some researchers proposing that PD could begin in the gut. The hypothesis revolves around the notion that initial pathology is triggered by an infectious agent or toxin, resulting in an inflammation-driven process that promotes α-syn misfolding within the gut and subsequent spreading of pathogenic α-syn from the gut to the brain via the vagus nerve [85, 88, 89]. Other features of GI dysfunction are established including increased gut permeability (or “leaky gut”), increased bacterial invasion, and oxidative stress, which could be contributing factors and consequences of GI inflammation [83, 90]. While the gut is a critical site of etiopathogenesis in PD, whether disease begins in the gut is still a matter of contention.
An altered microbiome in PD
Since 2015, our understanding of the microbiota-gut-brain axis has evolved after several groups found that PwP exhibit an altered gut-microbiota compared to healthy controls [91–93]. Since then, at least 30 individual peer-reviewed studies have been published confirming these results [14, 94–126]. Recent meta-analyses taking into account many of these studies support PD gut dysbiosis as a robust finding, even when taking into account study design and geography [127, 128]. The most consistent findings in the PD gut microbiome are a lower abundance of protective anti-inflammatory bacteria genera, such as Roseburia, Blautia, and Faecalibacterium, and a greater relative abundance of potentially deleterious bacteria genera like Akkermansia. [14, 15, 127, 128]. Several of these studies have found significant correlations between elevated pro-inflammatory or opportunistic pathogen genera, such as Escherichia, Klebsiella, and Porphyromonas [124]. The cause of intestinal microbial dysbiosis in PD is not fully understood, but changes to the microbiota can be caused by aging, diet, heavy metals, toxins, and pesticides; which are also known environmental risk factors and triggers for PD [129–133].
An altered microbiome is not considered to be a simple consequence of the disease, but an active contributor to its progression [112]. Animal studies have demonstrated a role for the microbiota influencing gut permeability, local, systemic, and neural inflammation, enteric nervous system (ENS) and autonomic nervous system (ANS) signaling, microglial development, BBB integrity, and α-syn misfolding [134–137]. The reduction of anti-inflammatory bacteria involved in the maintenance of barrier integrity and immune regulation could lead to a greater susceptibility to pro-inflammatory insults, either from other harmful gut bacteria, pathogens, or environmental toxins [128]. Furthermore, specific changes to the gut microbiota have correlations with the severity of PD [98, 105, 112].
Evidence of early changes in the microbiome further supports a role for microbial dysbiosis as a functional contributor to PD. Several studies investigating recently diagnosed and drug-naïve PwP have confirmed that microbiotic dysbiosis is not a late phenomenon and is not due to antiparkinsonian drugs [95, 104, 121]. Most compellingly, studies that have explored the gut microbiota of people with rapid eye movement sleep behavior disorder (RBD), the most specific symptom predictor of future PD diagnosis, have shown that they also exhibit microbial dysbiosis that is more similar in composition to people diagnosed with PD that it is to healthy persons [101, 126]. This again supports that microbial dysbiosis is a feature of PD that manifests early.
Aside from changes to the gut microbiota as measured from fecal samples, small intestine bacterial overgrowth (SIBO) is another form of dysbiosis that is commonly diagnosed in PD [138]. SIBO is known to impact on the absorption of levodopa, subsequently impacting on motor function, and is associated with greater disease severity [139, 140]. SIBO can promote GI inflammation and weaken barrier integrity, which can expose the local and peripheral immune system to harmful bacterial and non-bacterial agents [141, 142]. The connections between PD and SIBO highlight another way in which bacteria may be relevant to facilitating disease processes.
in vivo studies have demonstrated the importance of the gut flora to the development of disease, showing that PD mouse models that would normally develop motor deficits, α-syn aggregation, and microglial activation do not develop these phenotypes when raised in a germ-free environment [135]. The same group also showed that germ-free PD model mice have restoration of the PD phenotype after colonization of their GI tract with a microbiota derived from PwP or healthy controls, however, the severity of the PD phenotype was greater after the colonization with PwP-derived microbes [135].
Inflammation and the microbiota-gut-brain axis in PD
Although it is difficult to dispute that the microbiota plays a role in PD, the question remains: how does a shift in intestinal microbial diversity facilitate the progression of disease? In good gut health, most gut bacteria act indirectly on host cells as they remain in the gut lumen, separated from the epithelium by a thick mucosal layer that protects against their invasion [143, 144]. Being predominantly restricted to the gut lumen, in order to communicate with host cells, such as epithelial and immune cells, bacteria must act indirectly via the release of secreted factors including OMVs.
Bacterial metabolites and secreted factors such as short-chain fatty acids (SCFAs) and toxins (such as lipopolysaccharide (LPS)) have been investigated as mechanisms by which microbiota affect global changes in disease, in particular in PD [94, 113, 145–147]. LPS, the major component of the outer membrane of Gram-negative bacteria and a potent activator of the innate immune response, has been shown to be increased in the blood of PwP [148, 149]. This finding is substantiated by research indicating higher levels of the LPS binding protein (LBP) in serum during the prodromal phase of the disease [150]. This connection extends beyond systematic effects, as demonstrated by increased expression of the microbial-associated molecular pattern (MAMPs) receptors, specifically toll-like receptor 4 (TLR4) and toll-like receptor 2 (TLR2), in postmortem PD brain tissue [151–153]. For many years LPS has been used to model neurodegenerative disease [147]. PD researchers use LPS (typically from E. coli strains) to elicit a neuroinflammatory response that ultimately leads to neurodegeneration of the dopaminergic neurons of the SN [154–156]. This response is mediated by the activation of microglia and the subsequent release of pro-inflammatory cytokines and destructive reactive oxygen species [32, 154, 155, 157]. LPS injections directly into the SN (and not other sites in the brain) elicit this response, indicating that LPS can direct PD-specific neurodegenerative processes [155, 156]. LPS administration has been shown to increase α-syn expression in wild-type animal ENS and SN, exacerbate α-syn aggregation in PD models, and make animals more susceptible to neurotoxins [158–161]. When administered systemically at a low dose, LPS models the neuronal features of PD, as well as GI features such as increased intestinal permeability and increased α-syn expression in the colon [160].
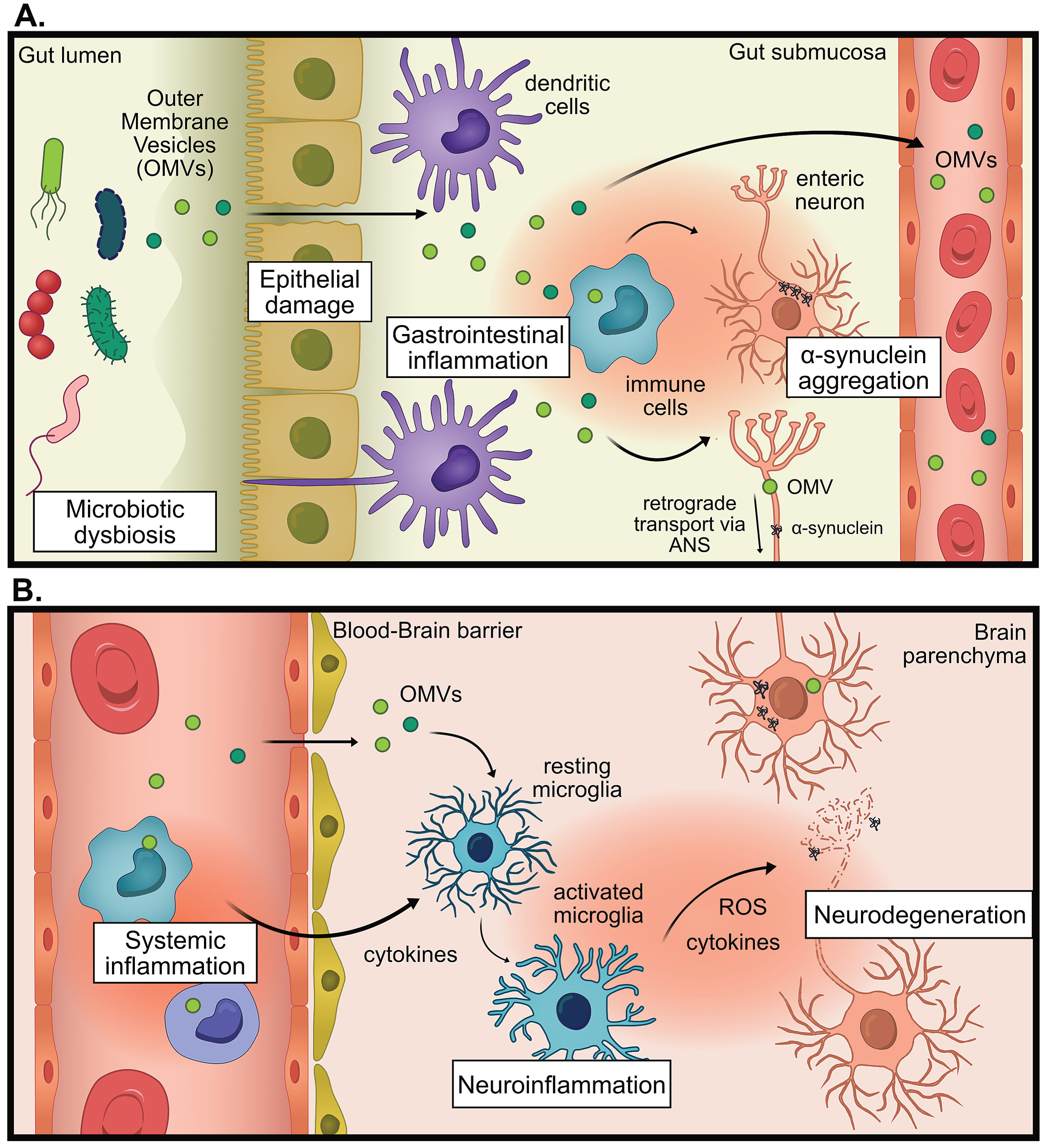
LPS found on E. coli strains and other Enterobacteriaceae members, are considered to be the most immunogenic forms of LPS [162, 163]. Notably, Enterobacteriaceae have an increased presence in the gut microbiota of PwP along with other Gram-negative LPS-producing families Veruccomicrobioaceae and Christensenellaceae [93, 94, 98, 102, 106, 110, 118, 123] and Klebsiella species, and Porphyromonas asaccharolytica [124]. In the field of neurodegenerative disease, there has been a prevailing assumption that secreted, non-vesicle-associated factors, such as free LPS, breach the intestinal barrier and trigger an immune response that contributes to disease progression. We propose an expansion of this hypothesis to include a crucial role for OMVs in this process. Bacterial OMVs have yet to be investigated in the context of PD despite being a major secretion and communication pathway for bacteria, being highly immunogenic, and able to deliver cargo across biological barriers like the gut and BBB [164, 165]. For visual hypothesis see Fig. 1.
Fig. 1
The proposed hypothesis for the pro-inflammatory role of outer membrane vesicles (OMVs) in Parkinson’s disease (PD). A) In states of microbial dysbiosis, OMVs and other microbial factors can cause damage to epithelial barriers, causing the translocation of OMVs past the epithelium, where immune cells can be activated to promote gastrointestinal inflammation which increases intestinal permeability. Information about gastrointestinal inflammation can be directly communicated to the brain via the autonomic nervous system (ANS), by altering neural signaling or retrograde transport of pathogenic proteins (for example α-synuclein) or OMVs. B) OMVs reach the systemic circulation whereby they promote systemic inflammation. OMVs could promote neuroinflammation indirectly via increasing systemic inflammation, leading to weakening of the blood-brain barrier, entry of immune cells into the brain and increased cytokine signaling. OMVs could also promote neuroinflammation directly by infiltrating the brain and activating microglia. This chronic inflammation can contribute to neurodegeneration.
BACTERIAL OUTER MEMBRANE VESICLES
OMVs are nanosized membranous structures that range from 20–200 nm and have known roles in bacterial communication and survival [166]. These vesicles are secreted as distinct entities to mediate intercellular communication between bacteria and their host cell tissues [167]. OMVs are complex structures that are comprised of many different antigenic bacterial proteins, lipids, and nucleic acids on their membrane surfaces and also as cargo within their lumen. The extracellular vesicles released from Gram-negative and Gram-positive bacteria are generally termed OMVs and membrane vesicles (MVs), respectively; however, nomenclature variation exists in the literature [166]. For the purpose of this review, we will focus on the extracellular vesicles released from Gram-negative bacteria, which we will refer to as OMVs; however, we do acknowledge the potential relevance of Gram-positive MVs in disease also (for a review, see [168]).
There are multiple mechanisms by which Gram-negative bacteria shed OMVs, such as envelope crosslink modulation, envelope component accumulation, and insertion of particular lipids into the outer membrane, the importance of this being that OMV release is not a stochastic event, and highly regulated by the cell (reviewed in [169]). Depending on their bacterial source, OMVs have varied functions that are related to their composition, for example, delivery of toxins, horizontal gene transfer, and biofilm formation (see review by Gilmore et al. for further information [170]). Unlike the parental bacterium, OMVs are not so restricted to the gut lumen and can traverse tight junctions to deliver their payload over long distances within the host, while protecting vesicular contents from the external environment [171, 172]. Bacterial vesicles have been detected in a multitude of tissues and fluids including blood and plasma [173–175], cerebrospinal fluid [176, 177], gastric mucosa [178], urine [179–181], saliva [179] and feces [174, 175, 182–184], indicating their widespread biodistribution, including in the brain [185, 186].
The role of OMVs in immune modulation
One major function attributed to gut-derived OMVs that is relevant to PD is host immune system modulation. OMVs, released by bacteria in the gut, can trigger local and distant proinflammatory immune activation by interaction with both innate and adaptive immune cell types including, macrophages, dendritic cells, neutrophils, and B cells [187–194]. Enclosed by a membrane, OMVs harbor many types of inflammogens such as bacterial DNAs, RNAs, peptidoglycan, lipoproteins, LPSs, and toxins, that can result in a complex multimodal mechanism of immune activation [195]. Porphyromonus gingivalis and Aggregatibacter actinomycetemcomitans OMVs have been demonstrated to interact with neutrophils by coating their plasma membrane, and are also internalized by macrophages and epithelial cells [190]. Once bound to neutrophils P. gingivalis OMVs induce cell activation but also degrade neutrophil effector protein myeloperoxidase, which results in greater survival of the P. gingivalis parent bacterium [190]. OMVs from E. coli as well as total bacterial extracellular vesicles from feces have been demonstrated to cause sepsis-like inflammation in rodents after intraperitoneal injection or intravenous infusion [183, 196–198]. In high enough doses, the sepsis-like effects caused by OMVs can be lethal, in contrast, there is no lethality when the animals are administered a dose of pure LPS that is more than double the LPS dose contained in the OMV preparation [197].
While OMVs contain many MAMPs, a major component is their LPS-rich outer membrane [164, 199]. OMVs, studded with LPS on their surface are able to deliver LPS to many types of cells [200], including microglia [28]. LPS predominantly activates immune cells by binding extracellularly to TLR4/CD14/MD2 receptor complex, which results in intracellular NF-κB signaling and thus the release of pro-inflammatory cytokines, TNF-α, IL-1β, and free radicals, nitric oxide and superoxide [201]. However, OMVs have been identified as a method of delivering LPS intracellularly where they activate caspases and the NLRP3 inflammasome [200]. Several E. coli strains (enterotoxigenic and avirulent strains) release OMVs that can be genotoxic, contain a heat-labile toxin, and can confer protection to the producer bacteria and others against some antibiotics [195, 202, 203]. E. coli OMVs have been shown to induce mitochondrial dysfunction, mitochondrial apoptosis, and activate inflammation, which is particularly relevant in PD, as mitochondrial impairment is characteristic of the disease [204].
The finding of elevated pro-inflammatory Gram-negative bacteria and elevated synthesis of LPS in PwP may indicate that the gut microbiome in PwP produces a greater amount of OMVs which contain highly potent LPS. LPS has been shown to be increased in the blood of PwP [148, 149], however the methods employed in these studies did not differentiate free monomeric LPS from LPS contained on OMVs, suggesting LPS found in the bloodstream of PwP could be OMV-associated.
OMVs interact with cells either by ligand binding to externally expressed pattern recognition receptors (PRRs) such as toll-like receptors (TLRs) or intracellular receptors like nucleotide-binding-oligomerization domain-containing proteins (NOD) [188, 199, 205–207]. In the context of PD, the major PRR families that are involved in sensing MAMPs, (TLRs, and Nod-like receptors (NLR)), are implicated in the disease [208]. Leukocyte TLR2 responses are lowered in PD compared to controls. This could indicate a reduced ability to respond to bacterial lipoproteins and other TLR2 ligands [209]. Conversely, leukocyte TLR4 responses are increased in young PwP. When stimulated with LPS, leukocytes from these individuals have a more robust TNFα cytokine release than healthy controls, indicating that in PD there is an overactive proinflammatory response early in the disease [209]. The expression of TLRs is increased in the brain, (particularly in the caudate putamen), blood, and GI of PwP [83, 153]. One study found that TLR2 expression is found to be increased in the neurons of PwP, and in particular, neurons harboring aggregated α-syn [151]. Another study demonstrated that the SN, a brain region highly susceptible to neuroinflammation, is one of the regions with the highest TLR4 expression [152]. NLR family proteins, such as NOD2 which detects peptidoglycan, and the NLRP3 inflammasome complex which detects a wide variety of MAMPs (as well as damage-associated molecular patterns) have also been implicated in PD [39, 210, 211]. Levels of NLRP3 are increased in the serum and peripheral blood mononuclear cells of PwP and these levels positively correlate with the level of serum α-syn [39, 210]. Variant alleles in the gene that encodes NOD2 (CARD15) are associated with an increased risk of PD [211]. Both NLRP3 and NOD2 detect OMVs and their components, for example, NLRP3 detects E. coli OMV-delivered LPS and NOD2 detects OMVs from A. actinomycetemcomitans [200, 205].
The changes to TLR expression and other PRRs could indicate that PwP may be differentially able to detect bacterial components, like OMVs, and that they may produce aberrant immune responses which could contribute to disease as a result. TLRs are mostly expressed in immune cells, including microglia, but neurons, in both the gut epithelium and brain, also express TLRs [212–214]. This naturally leads to the implication that OMVs may be capable of directly interacting with neurons (should they come in contact), however, this has not yet been investigated to the best of our knowledge.
OMVs and the epithelium
OMVs released in the gut lumen can pass through the thick mucosal layer and associate with epithelial cells, where they can initiate cytokine release, affect cell growth, and affect tight junction protein expression [178]. OMVs are known to pass through the epithelium either paracellularly or transcellularly to access the lamina propria, where they can interact with local immune cells [164, 178, 215]. OMVs from bacteria such as Desulfovibrio fairfieldensis, Campylobacter jejuni, H. pylori, and P. gingivalis have been found to reduce the expression of important tight junction proteins, like occludin, zonulin-1, and E-cadherin, which results in the weakening of the epithelial barrier and could potentially promote their own translocation from the lumen into deeper layers of the gut and beyond [175, 216–219]. Tulkens and colleagues demonstrated that there were increased bacterial extracellular vesicles in the plasma of people with intestinal barrier dysfunction compared to healthy controls and that the bacterial extracellular vesicle concentration was associated with greater plasma zonulin-1 levels (indicating intestinal barrier dysfunction). They also demonstrated that in an in vitro colitis model that bacterial extracellular vesicle concentration was increased in the basal side as zonulin-1 levels decreased, indicating paracellular translocation from the apical side where they were applied [175]. This role in affecting epithelial permeability is particularly relevant in PD as elevated intestinal permeability and the translocation of bacterial products occur in PwP.
OMVs beyond the gut
It is unlikely that major OMV translocation beyond the gut lumen is an innocuous event as OMV’s ability to influence the function of many cell types could have flow-on effects. The ability of OMVs to travel throughout the body allows for bacteria restricted to the gut to elicit long-range effects indirectly, permitting them to functionally interact with the important sites of PD pathology, mainly the gut and potentially the brain. A study investigating OMV biodistribution demonstrated that labeled OMVs administered orally to mice are predominantly found in the GI tract and liver and OMVs delivered intraperitoneally are predominantly found in the liver, and less so in the spleen, kidneys, and lungs (this study did not investigate the brain) [171]. This study was performed in healthy mice and therefore supports that OMVs can pass through the gut even without gut impairments [171].
As described above OMVs are highly effective immune activators and their presence in the circulation and distant organs has major implications for the facilitation of inflammation systemically. We acknowledge the possibility that OMV translocation from the gut lumen may not occur in sufficient quantities to affect systemic and neural inflammation directly, however, it is plausible that infiltration of OMVs into the lamina propria and submucosa may promote local inflammation and alterations to ENS signaling to an extent that results in a modulation of the peripheral immune system and CNS signaling. OMVs, in this case, need not traverse into the vasculature or brain, while still contributing to PD pathogenesis.
The presence of OMVs in the bloodstream and urine has been established in other conditions and biodistribution studies and this supports that OMVs can pass through epithelial layers to traverse into the vasculature [171, 173, 180, 181, 220]. It has been demonstrated that individuals with GI barrier disruption have a greater number of bacterial extracellular vesicles in their circulation compared to healthy controls [175]. Though this has yet to be tested in PwP, it stands to reason that bacterial extracellular vesicles could similarly be elevated in the circulation, because of the evidence of increased intestinal permeability or reduced gut tight-junction proteins in PwP [90, 221, 222].
OMVs and the brain
Several research groups have hypothesized that OMVs can cross the BBB, especially in cases where the BBB is impaired, theorizing based on the nanosize of OMVs and citing evidence of bacterial components such as bacterial nucleic acids and LPS being found in brains, particularly of those with neurological dysfunction [165, 223]. It has been demonstrated that OMVs and LPSs, from species like E. coli and P. gingivalis, can impair endothelial barriers including the BBB, which would perhaps promote uptake of OMVs or other toxic materials, allow bacterial infiltration into the brain and also promote neuroinflammation via peripheral immune cell infiltration [224–230]. The BBB in PD has been suggested to be dysfunctional, as evidenced by positron electron tomography hyperpermeability experiments, investigations of cerebrospinal fluid-serum albumin ratios, and also histological investigation of BBB integrity markers [231–233]. While the cause of the BBB dysfunction is unknown in PD, these observations imply that the BBB may be susceptible to OMV penetration.
There is the potential that OMVs could cross an intact BBB, as eukaryotic EVs have been suggested to do [234], or in a similar manner to how they transcellularly cross epithelial and endothelial cells [171]. Evidence for OMVs crossing the BBB in animal models is beginning to emerge [185, 186, 226, 235–237]. A. actinomycetemcomitans OMVs can cross the BBB, deliver bacterial RNA, and induce the release of cytokines following intracardial administration [235]. In a follow-up study, OMVs were shown to be taken up by microglia and meningeal macrophages [186]. Orally-gavaged OMVs from Paenalcaligenes hominis are delivered to the hippocampus via the autonomic nervous system resulting in cognitive impairment more potently than orally-gavaged LPS [238]. To the best of our knowledge, this study is the first to experimentally link OMVs and neuronal dysfunction in the CNS, showing retrograde delivery from the gut to the brain via the vagus nerve, which is believed to be an important conduit between the gut and the brain in PD and links the gut to key sites of PD pathology, such as the dorsal motor nucleus [238–240]. It also highlights the possibility of a route for gut-to-brain trafficking of OMVs that bypasses the circulation and thus avoids the challenges of evading the peripheral immune system and crossing the BBB.
In another study, which provided evidence of OMV passage into the brain, H. pylori-derived OMVs were shown to pass into the brain through the transcellular method [185]. This study also demonstrated that by orally delivering OMVs to Alzheimer’s model mice, the OMVs are taken up by astrocytes. Notably, OMVs increased plaque load, inflammation, neuronal dysfunction, and also accelerated cognitive decline [185]. H. pylori-derived OMVs have also been demonstrated to traffic to the mouse brain following venous and oral administration, where they activate astrocytes and cause neuronal damage [237]. As previously discussed, there is are connection between H. pylori infection and PD, therefore an investigation into H. pylori-specific OMVs in PD has sufficient merit. A separate study investigating Alzheimer’s disease demonstrated that after oral gavage of P. gingivalis OMVs into middle-aged wild-type mice, the OMVs reached the brain, impaired expression of tight junction proteins, were able to induce inflammation, and impaired learning and memory [241].
While the studies described above use non-physiological quantities of OMVs in acute animal models, throughout the lifetime of a person, minute insults from gut-derived OMVs could contribute to PD by fueling a state of chronic inflammation, ultimately contributing to neurodegeneration over the course of decades.
CONCLUDING REMARKS
The etiology and pathogenesis of PD are multifactorial, and the interaction between age, genetic susceptibility, environmental influence, and microbial function all appear to contribute to PD pathogenesis. There is increasing appreciation of the importance of the microbiome in health and disease. A greater understanding of bidirectional gut-brain axis functions will allow for more sophisticated examinations of the interaction between gut and brain health in systemic diseases such as PD.
A multitude of studies demonstrate that models using inflammogens, like LPS, recapitulate the neuroinflammation and neurodegeneration seen in PwP. There is an abundance of evidence supporting changes in the GI tract of PwP including increased intestinal permeability, microbial dysbiosis, and evidence of increased LPS in blood. Thus, a hypothesis has formed that people with GI dysfunction have greater amounts of LPS reaching their circulation, which promotes systemic inflammation and promotes neurodegeneration. OMVs are a physiological mechanism by which MAMPs are released from bacteria to promote immune responses. Considering their presence in the bloodstream and potential ability to cross the BBB, we propose a more likely hypothesis, that OMVs released from bacteria in the gut trigger inflammation to contribute to the progression of PD.
The implication of this hypothesis opens new avenues for therapeutic interventions in PD and potentially other diseases where OMV-induced inflammation plays a role. This is because OMVs elicit distinct host immune responses so therapeutic approaches specifically tailored for soluble LPS for example, may prove inadequate in addressing inflammation triggered by OMVs [242]. Re-establishing a healthy microbiome and strengthening gut barrier integrity could be one mechanism for reducing OMV-induced inflammation. TLR4 and other OMV-detecting PRR antagonists could prevent increases in inflammatory processes. Lastly, OMVs could provide a new avenue for biomarker development with the difference in the relative abundance of plasma OMVs used as a complementary diagnostic tool for PD.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
The Florey Institute of Neuroscience and Mental Health acknowledges the support from the Victorian Government and in particular funding from the Operational Infrastructure Support Grant. We acknowledge an Australian Government Research Training Program Scholarship (TK).
CONFLICT OF INTEREST
The authors have no conflicts of interest to report.
REFERENCES
[1] | Kalia LV , Lang AE ((2015) ) Parkinson’s disease. Lancet 386: , 896–912. |
[2] | Pang SY , Ho PW , Liu HF , Leung CT , Li L , Chang EES , Ramsden DB , Ho SL ((2019) ) The interplay of aging, genetics and environmental factors in the pathogenesis of Parkinson’s disease. Transl Neurodegener 8: , 23. |
[3] | Kieburtz K , Wunderle KB ((2013) ) Parkinson’s disease: Evidence for environmental risk factors. Mov Disord 28: , 8–13. |
[4] | Ascherio A , Schwarzschild MA ((2016) ) The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol 15: , 1257–1272. |
[5] | Wang H , Liu X , Tan C , Zhou W , Jiang J , Peng W , Zhou X , Mo L , Chen L ((2020) ) Bacterial, viral, and fungal infection-related risk of Parkinson’s disease: Meta-analysis of cohort and case-control studies. Brain Behav 10: , e01549. |
[6] | Caggiu E , Arru G , Hosseini S , Niegowska M , Sechi G , Zarbo IR , Sechi LA ((2019) ) Inflammation, infectious triggers, and Parkinson’s disease. Front Neurol 10: , 122. |
[7] | Limphaibool N , Iwanowski P , Holstad MJV , Kobylarek D , Kozubski W ((2019) ) Infectious etiologies of parkinsonism: Pathomechanisms and clinical implications. Front Neurol 10: , 652. |
[8] | Johnson ME , Stecher B , Labrie V , Brundin L , Brundin P ((2019) ) Triggers, facilitators, and aggravators: Redefining Parkinson’s disease pathogenesis. Trends Neurosci 42: , 4–13. |
[9] | Nandipati S , Litvan I ((2016) ) Environmental exposures and Parkinson’s disease. Int J Environ Res Public Health 13: . |
[10] | Nerius M , Doblhammer G , Tamgüney G ((2020) ) GI infections are associated with an increased risk of Parkinson’s disease. Gut 69: , 1154. |
[11] | Calabrese V , Santoro A , Monti D , Crupi R , Di Paola R , Latteri S , Cuzzocrea S , Zappia M , Giordano J , Calabrese EJ , Franceschi C ((2018) ) Aging and Parkinson’s disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic Biol Med 115: , 80–91. |
[12] | Menza M , DeFronzo Dobkin R , Marin H , Mark MH , Gara M , Bienfait K , Dicke A , Kusnekov A ((2010) ) The role of inflammatory cytokines in cognition and other non-motor symptoms of Parkinson’s disease. Psychosomatics 51: , 474–479. |
[13] | Tansey MG , Wallings RL , Houser MC , Herrick MK , Keating CE , Joers V ((2022) ) Inflammation and immune dysfunction in Parkinson disease. Nat Rev Immunol 22: , 657–673. |
[14] | Wallen ZD , Appah M , Dean MN , Sesler CL , Factor SA , Molho E , Zabetian CP , Standaert DG , Payami H ((2020) ) Characterizing dysbiosis of gut microbiome in PD: Evidence for overabundance of opportunistic pathogens. NPJ Parkinsons Dis 6: , 11. |
[15] | Romano S , Savva GM , Bedarf JR , Charles IG , Hildebrand F , Narbad A ((2021) ) Meta-analysis of the Parkinson’s disease gut microbiome suggests alterations linked to intestinal inflammation. NPJ Parkinsons Dis 7: , 27. |
[16] | Shannon K , Vanden Berghe P ((2018) ) The enteric nervous system in PD: Gateway, bystander victim, or source of solutions. Cell Tissue Res 373: , 313–326. |
[17] | Umamahesan C , Augustin AD , Hayee BH , Ibrahim MAA , Taylor D , Weller C , Charlett A , Dobbs RJ , Dobbs SM ((2021) ) Intestinal inflammation and compromised barrier function in idiopathic parkinsonism: Scenario captured by systematic review. Neuroimmunol Neuroinflammation 8: , 313. |
[18] | Troncoso-Escudero P , Parra A , Nassif M , Vidal RL ((2018) ) Outside in: Unraveling the role of neuroinflammation in the progression of Parkinson’s disease. Front Neurol 9: , 860. |
[19] | McGeer PL , Itagaki S , Akiyama H , McGeer EG ((1988) ) Rate of cell death in parkinsonism indicates active neuropathological process. Anna Neurol 24: , 574–576. |
[20] | McGeer PL , Itagaki S , Boyes BE , McGeer EG ((1988) ) Reactive microglia are positive for HLA-DR in the: Substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38: , 1285–1291. |
[21] | Mogi M , Harada M , Kondo T , Riederer P , Inagaki H , Minami M , Nagatsu T ((1994) ) Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci Lett 180: , 147–150. |
[22] | Blum-Degena D , Müller T , Kuhn W , Gerlach M , Przuntek H , Riederer P ((1995) ) Interleukin-1β and interleukin-6 are elevated in the cerebrospinal fluid of Alzheimer’s and de novo Parkinson’s disease patients. Neurosci Lett 202: , 17–20. |
[23] | Hunot S , Boissière F , Faucheux B , Brugg B , Mouatt-Prigent A , Agid Y , Hirsch EC ((1996) ) Nitric oxide synthase and neuronal vulnerability in Parkinson’s disease. Neuroscience 72: , 355–363. |
[24] | Hunot S , Brugg B , Ricard D , Michel PP , Muriel MP , Ruberg M , Faucheux BA , Agid Y , Hirsch EC ((1997) ) Nuclear translocation of NF-κb is increased in dopaminergic neurons of patients with Parkinson disease. Proc Natl Acad Sci U S A 94: , 7531–7536. |
[25] | Banati RB , Daniel SE , Blunt SB ((1998) ) Glial pathology but absence of apoptotic nigral neurons in long-standing Parkinson’s disease. Mov Disord 13: , 221–227. |
[26] | Mirza B , Hadberg H , Thomsen P , Moos T ((1999) ) The absence of reactive astrocytosis is indicative of a unique inflammatory process in Parkinson’s disease. Neuroscience 95: , 425–432. |
[27] | Hunter RL , Dragicevic N , Seifert K , Choi DY , Liu M , Kim HC , Cass WA , Sullivan PG , Bing G ((2007) ) Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J Neurochem 100: , 1375–1386. |
[28] | Qin L , Wu X , Block ML , Liu Y , Breese GR , Hong JS , Knapp DJ , Crews FT ((2007) ) Systemic LPS causes chronic neuroinflammation and progressive neurodegeneration. Glia 55: , 453–462. |
[29] | Mogi M , Harada M , Riederer P , Narabayashi H , Fujita K , Nagatsu T ((1994) ) Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett 165: , 208–210. |
[30] | Knott C , Stern G , Wilkin GP ((2000) ) Inflammatory regulators in Parkinson’s disease: iNOS, lipocortin-1, and cyclooxygenases-1 and -2. Mol Cell Neurosci 16: , 724–739. |
[31] | Qin XY , Zhang SP , Cao C , Loh YP , Cheng Y ((2016) ) Aberrations in peripheral inflammatory cytokine levels in Parkinson disease: A systematic review and meta-analysis. JAMA Neurol 73: , 1316–1324. |
[32] | Gao HM , Hong JS , Zhang W , Liu B ((2002) ) Distinct role for microglia in rotenone-induced degeneration of dopaminergic neurons. J Neurosci 22: , 782–790. |
[33] | Gao HM , Jiang J , Wilson B , Zhang W , Hong JS , Liu B ((2002) ) Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: Relevance to Parkinson’s disease. J Neurochem 81: , 1285–1297. |
[34] | Liu B , Du L , Hong JS ((2000) ) Naloxone protects rat dopaminergic neurons against inflammatory damage through inhibition of microglia activation and superoxide generation. J Pharmacol Exp Ther 293: , 607–617. |
[35] | Liu B , Jiang JW , Wilson BC , Du L , Yang SN , Wang JY , Wu GC , Cao XD , Hong JS ((2000) ) Systemic infusion of naloxonereduces degeneration of rat substantia nigral dopaminergic neurons induced by intranigral injection oflipopolysaccharide. J Pharmacol Exp Ther 295: , 125–132. |
[36] | Arimoto T , Bing G ((2003) ) Up-regulation of inducible nitric oxide synthase in the substantia nigra by lipopolysaccharide causes microglial activation and neurodegeneration. Neurobiol Dise 12: , 35–45. |
[37] | Franceschi C , Garagnani P , Parini P , Giuliani C , Santoro A ((2018) ) Inflammaging: A new immune–metabolic viewpoint for age-related diseases. Nat Rev Endocrinol 14: , 576–590. |
[38] | Collier TJ , Kanaan NM , Kordower JH ((2017) ) Aging and Parkinson’s disease: Different sides of the same coin? . Mov Disord 32: , 983–990. |
[39] | Fan Z , Pan YT , Zhang ZY , Yang H , Yu SY , Zheng Y , Ma JH , Wang XM ((2020) ) Systemic activation of NLRP3 inflammasome and plasma α-synuclein levels are correlated with motor severity and progression in Parkinson’s disease. J Neuroinflammation 17: , 11. |
[40] | Gritsenko A , Green JP , Brough D , Lopez-Castejon G ((2020) ) Mechanisms of NLRP3 priming in inflammaging and age related diseases. Cytokine Growth Factor Rev 55: , 15–25. |
[41] | Rui Q , Ni H , Li D , Gao R , Chen G ((2018) ) The role of LRRK2 in neurodegeneration of Parkinson disease. Curr Neuropharmacol 16: , 1348–1357. |
[42] | Shutinoski B , Hakimi M , Harmsen IE , Lunn M , Rocha J , Lengacher N , Zhou YY , Khan J , Nguyen A , Hake-Volling Q , El-Kodsi D , Li J , Alikashani A , Beauchamp C , Majithia J , Coombs K , Shimshek D , Marcogliese PC , Park DS , Rioux JD , Philpott DJ , Woulfe JM , Hayley S , Sad S , Tomlinson JJ , Brown EG , Schlossmacher MG ((2019) ) Lrrk2 alleles modulate inflammation during microbial infection of mice in a sex-dependent manner. Sci Transl Med 11: , eaas9292. |
[43] | Kim B , Yang MS , Choi D , Kim JH , Kim HS , Seol W , Choi S , Jou I , Kim EY , Joe EH ((2012) ) Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS One 7: , e34693. |
[44] | Beatman EL , Massey A , Shives KD , Burrack KS , Chamanian M , Morrison TE , Beckham JD ((2015) ) Alpha-synuclein expression restricts RNA viral infections in the brain. J Virol 90: , 2767–2782. |
[45] | Park SC , Moon JC , Shin SY , Son H , Jung YJ , Kim NH , Kim YM , Jang MK , Lee JR ((2016) ) Functional characterization ofalpha-synuclein protein with antimicrobial activity. Biochem Biophys Res Commun 478: , 924–928. |
[46] | Frank-Cannon TC , Tran T , Ruhn KA , Martinez TN , Hong J , Marvin M , Hartley M , Trevino I , O’Brien DE , Casey B , Goldberg MS , Tansey MG ((2008) ) Parkin deficiency increases vulnerability to inflammation-related nigraldegeneration. J Neurosci 28: , 10825–10834. |
[47] | Matheoud D , Cannon T , Voisin A , Penttinen AM , Ramet L , Fahmy AM , Ducrot C , Laplante A , Bourque MJ , Zhu L , Cayrol R , Le Campion A , McBride HM , Gruenheid S , Trudeau LE , Desjardins M ((2019) ) Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1(-/-) mice. Nature 571: , 565–569. |
[48] | Choi DJ , An J , Jou I , Park SM , Joe EH ((2019) ) A Parkinson’s disease gene, DJ-1, regulates anti-inflammatory roles of astrocytes through prostaglandin D(2) synthase expression. Neurobiol Dis 127: , 482–491. |
[49] | Qiao C , Yin N , Gu HY , Zhu JL , Ding JH , Lu M , Hu G ((2016) ) Atp13a2 deficiency aggravates astrocyte-mediated neuroinflammation via NLRP3 inflammasome activation. CNS Neurosci Ther 22: , 451–460. |
[50] | Nalls MA , Blauwendraat C , Vallerga CL , Heilbron K , Bandres-Ciga S , Chang D , Tan M , Kia DA , Noyce AJ , Xue A , BrasJ , Young E , von Coelln R , Simón-Sánchez J , Schulte C , Sharma M , Krohn L , Pihlstrøm L , Siitonen A , Iwaki H , Leonard H , Faghri F , Gibbs JR , Hernandez DG , Scholz SW , Botia JA , Martinez M , Corvol JC , Lesage S , Jankovic J , Shulman LM , Sutherland M , Tienari P , Majamaa K , Toft M , Andreassen OA , Bangale T , Brice A , Yang J , Gan-Or Z , Gasser T , Heutink P , Shulman JM , Wood NW , Hinds DA , Hardy JA , Morris HR , Gratten J , Visscher PM , Graham RR , Singleton AB; 23andMe Research Team; System Genomics of Parkinson’s Disease Consortium; InternationalParkinson’s Disease Genomics Consortium ((2019) ) Identification of novel risk loci, causal insights, and heritablerisk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol 18: , 1091–1102. |
[51] | Gunnarsson LG , Bodin L ((2019) ) Occupational exposures and neurodegenerative diseases-a systematic literature review and meta-analyses. Int J Environ Res Public Health 16: . |
[52] | Zhang D , Li S , Hou L , Jing L , Ruan Z , Peng B , Zhang X , Hong JS , Zhao J , Wang Q ((2021) ) Microglial activation contributes to cognitive impairments in rotenone-induced mouse Parkinson’s disease model. J Neuroinflammation 18: , 4. |
[53] | Tong T , Duan W , Xu Y , Hong H , Xu J , Fu G , Wang X , Yang L , Deng P , Zhang J , He H , Mao G , Lu Y , Lin X , Yu Z , Pi H , Cheng Y , Xu S , Zhou Z ((2022) ) Paraquat exposure induces Parkinsonism by altering lipid profile and evoking neuroinflammation in the midbrain. Environ Int 169: , 107512. |
[54] | Ross GW , Petrovitch H ((2001) ) Current evidence for neuroprotective effects of nicotine and caffeine against Parkinson’s disease. Drugs Aging 18: , 797–806. |
[55] | Piao WH , Campagnolo D , Dayao C , Lukas RJ , Wu J , Shi FD ((2009) ) Nicotine and inflammatory neurological disorders. Acta Pharmacol Sin 30: , 715–722. |
[56] | Madeira MH , Boia R , Ambrosio AF , Santiago AR ((2017) ) Having a coffee break: The impact of caffeine consumption on microglia-mediated inflammation in neurodegenerative diseases. Mediators Inflamm 2017: , 4761081. |
[57] | Gao X , Chen H , Schwarzschild MA , Ascherio A ((2011) ) Use of ibuprofen and risk of Parkinson disease. Neurology 76: , 863–869. |
[58] | Gagne JJ , Power MC ((2010) ) Anti-inflammatory drugs and risk of Parkinson disease: A meta-analysis. Neurology 74: , 995–1002. |
[59] | Vlajinac H , Dzoljic E , Maksimovic J , Marinkovic J , Sipetic S , Kostic V ((2013) ) Infections as a risk factor for Parkinson’s disease: A case-control study. Int J Neurosci 123: , 329–332. |
[60] | Fang F , Wirdefeldt K , Jacks A , Kamel F , Ye W , Chen H ((2012) ) CNS infections, sepsis and risk of Parkinson’s disease. Int J Epidemiol 41: , 1042–1049. |
[61] | Strang RR ((1966) ) The occurrence of peptic ulceration in patients with parkinsonism. Acta Neurol Scand 42: , 124–127. |
[62] | Hashim H , Azmin S , Razlan H , Yahya NW , Tan HJ , Manaf MR , Ibrahim NM ((2014) ) Eradication of Helicobacter pylori infection improves levodopa action, clinical symptoms and quality of life in patients with Parkinson’s disease. PLoS One 9: , e112330. |
[63] | Dardiotis E , Tsouris Z , Mentis AA , Siokas V , Michalopoulou A , Sokratous M , Dastamani M , Bogdanos DP , Deretzi G , Kountouras J ((2018) ) H. pylori and Parkinson’s disease: Meta-analyses including clinical severity. Clin Neurol Neurosurg 175: , 16–24. |
[64] | Dobbs RJ , Dobbs SM , Weller C , Charlett A , Bjarnason IT , Curry A , Ellis DS , Ibrahim MA , McCrossan MV , O’Donohue J ((2008) ) Helicobacter hypothesis for idiopathic parkinsonism: Before and beyond. Helicobacter 13: , 309–322. |
[65] | Bu X-L , Wang X , Xiang Y , Shen L-L , Wang Q-H , Liu Y-H , Jiao S-S , Wang Y-R , Cao H-Y , Yi X , Liu C-H , Deng B , Yao X-Q , Xu Z-Q , Zhou H-D , Wang Y-J ((2015) ) The association between infectious burden and Parkinson’s disease: A case-control study. Parkinsonism Relat Disord 21: , 877–881. |
[66] | Garretti F , Monahan C , Sloan N , Bergen J , Shahriar S , Kim SW , Sette A , Cutforth T , Kanter E , Agalliu D , Sulzer D ((2023) ) Interaction of an α-synuclein epitope with HLA-DRB1(*)15:01 triggers enteric features in mice reminiscent of prodromal Parkinson’s disease. Neuron 111: , 3397–3413.e3395. |
[67] | Knudsen K , Fedorova T , Bekker A , Iversen P , Østergaard K , Krogh K , Borghammer P ((2017) ) Objective colonic dysfunction is far more prevalent than subjective constipation in Parkinson’s disease: A colon transit and volume study. J Parkinsons Dis 7: , 359–367. |
[68] | Durcan R , Wiblin L , Lawson RA , Khoo TK , Yarnall AJ , Duncan GW , Brooks DJ , Pavese N , Burn DJ , Group I-PS ((2019) ) Prevalence and duration of non-motor symptoms in prodromal Parkinson’s disease. Eur J Neurol 26: , 979–985. |
[69] | Peter I , Dubinsky M , Bressman S , Park A , Lu C , Chen N , Wang A ((2018) ) Anti–tumor necrosis factor therapy and incidence of Parkinson disease among patients with inflammatory bowel disease. JAMA Neurol 75: , 939–946. |
[70] | Lin JC , Lin CS , Hsu CW , Lin CL , Kao CH ((2016) ) Association between Parkinson’s disease and inflammatory bowel disease: A nationwide Taiwanese retrospective cohort study. Inflamm Bowel Dis 22: , 1049–1055. |
[71] | Garrido-Gil P , Rodriguez-Perez AI , Dominguez-Meijide A , Guerra MJ , Labandeira-Garcia JL ((2018) ) Bidirectional neural interaction between central dopaminergic and gut lesions in Parkinson’s disease models. Mol Neurobiol 55: , 7297–7316. |
[72] | Kishimoto Y , Zhu W , Hosoda W , Sen JM , Mattson MP ((2019) ) Chronic mild gut inflammation accelerates brain neuropathology and motor dysfunction in alpha-synuclein mutant mice. Neuromolecular Med 21: , 239–249. |
[73] | Ugalde-Muniz P , Fetter-Pruneda I , Navarro L , Garcia E , Chavarria A ((2020) ) Chronic systemic inflammation exacerbates neurotoxicity in a Parkinson’s disease model. Oxid Med Cell Longev 2020: , 4807179. |
[74] | Braak H , Del Tredici K , Rüb U , De Vos RAI , Jansen Steur ENH , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[75] | Braak H , de Vos RA , Bohl J , Del Tredici K ((2006) ) Gastric alpha-synuclein immunoreactive inclusions in Meissner’sand Auerbach’s plexuses in cases staged for Parkinson’s disease-related brain pathology. Neurosci Lett 396: , 67–72. |
[76] | Wakabayashi K , Takahashi H , Takeda S , Ohama E , Ikuta F ((1988) ) Parkinson’s disease: The presence of Lewy bodies in Auerbach’s and Meissner’s plexuses. Acta Neuropathol 76: , 217–221. |
[77] | Wakabayashi K , Takahashi H , Ohama E , Ikuta F ((1990) ) Parkinson’s disease: An immunohistochemical study of Lewy body-containing neurons in the enteric nervous system. Acta Neuropathol 79: , 581–583. |
[78] | Beach TG , Adler CH , Sue LI , Vedders L , Lue L , White Iii CL , Akiyama H , Caviness JN , Shill HA , Sabbagh MN , Walker DG , Arizona Parkinson’s Disease Consortium ((2010) ) Multi-organ distribution of phosphorylated alpha-synuclein histopathology in subjects with Lewy body disorders. Acta Neuropathol 119: , 689–702. |
[79] | Shannon KM , Keshavarzian A , Dodiya HB , Jakate S , Kordower JH ((2012) ) Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s Disease? Evidence from 3 cases. Mov Disord 27: , 716–719. |
[80] | Shannon KM , Keshavarzian A , Mutlu E , Dodiya HB , Daian D , Jaglin JA , Kordower JH ((2012) ) Alpha-synuclein in colonic submucosa in early untreated Parkinson’s disease. Mov Disord 27: , 709–715. |
[81] | Visanji NP , Marras C , Kern DS , Al Dakheel A , Gao A , Liu LWC , Lang AE , Hazrati LN ((2015) ) Colonic mucosal a-synuclein lacks specificity as a biomarker for Parkinson disease. Neurology 84: , 609–616. |
[82] | Stokholm MG , Danielsen EH , Hamilton-Dutoit SJ , Borghammer P ((2016) ) Pathological alpha-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann Neurol 79: , 940–949. |
[83] | Perez-Pardo P , Dodiya HB , Engen PA , Forsyth CB , Huschens AM , Shaikh M , Voigt RM , Naqib A , Green SJ , Kordower JH , Shannon KM , Garssen J , Kraneveld AD , Keshavarzian A ((2019) ) Role of TLR4 in the gut-brain axis in Parkinson’s disease: A translational study from men to mice. Gut 68: , 829–843. |
[84] | Uemura N , Yagi H , Uemura MT , Hatanaka Y , Yamakado H , Takahashi R ((2018) ) Inoculation of alpha-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol Neurodegener 13: , 21. |
[85] | Kim S , Kwon SH , Kam TI , Panicker N , Karuppagounder SS , Lee S , Lee JH , Kim WR , Kook M , Foss CA , Shen C , Lee H , Kulkarni S , Pasricha PJ , Lee G , Pomper MG , Dawson VL , Dawson TM , Ko HS ((2019) ) Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron 103: , 627–641 e627. |
[86] | Walker LC , LeVine H , 3rd ((2012) ) Corruption and spread of pathogenic proteins in neurodegenerative diseases. J Biol Chem 287: , 33109–33115. |
[87] | Jucker M , Walker LC ((2013) ) Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 501: , 45–51. |
[88] | Braak H , Rüb U , Gai WP , Del Tredici K ((2003) ) Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm 110: , 517–536. |
[89] | Houser MC , Tansey MG ((2017) ) The gut-brain axis: Is intestinal inflammation a silent driver of Parkinson’s disease pathogenesis? NPJ Parkinsons Dis 3: , 3. |
[90] | Forsyth CB , Shannon KM , Kordower JH , Voigt RM , Shaikh M , Jaglin JA , Estes JD , Dodiya HB , Keshavarzian A ((2011) ) Increased intestinal permeability correlates with sigmoid mucosa alpha-synuclein staining and endotoxin exposure markers in early Parkinson’s disease. , e. PLoS One 6: , 28032. |
[91] | Hasegawa S , Goto S , Tsuji H , Okuno T , Asahara T , Nomoto K , Shibata A , Fujisawa Y , Minato T , Okamoto A , Ohno K , Hirayama M ((2015) ) Intestinal dysbiosis and lowered serum lipopolysaccharide-binding protein in Parkinson’s disease. PLoS One 10: , e0142164. |
[92] | Keshavarzian A , Green SJ , Engen PA , Voigt RM , Naqib A , Forsyth CB , Mutlu E , Shannon KM ((2015) ) Colonic bacterial composition in Parkinson’s disease. Mov Disord 30: , 1351–1360. |
[93] | Scheperjans F , Aho V , Pereira PAB , Koskinen K , Paulin L , Pekkonen E , Haapaniemi E , Kaakkola S , Eerola-Rautio J , Pohja M , Kinnunen E , Murros K , Auvinen P ((2015) ) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord 30: , 350–358. |
[94] | Unger MM , Spiegel J , Dillmann KU , Grundmann D , Philippeit H , Bürmann J , Faßbender K , Schwiertz A , Schäfer KH ((2016) ) Short chain fatty acids and gut microbiota differ between patients with Parkinson’s disease and age-matched controls. Parkinsonism Relat Disord 32: , 66–72. |
[95] | Bedarf JR , Hildebrand F , Coelho LP , Sunagawa S , Bahram M , Goeser F , Bork P , Wullner U ((2017) ) Functional implications of microbial and viral gut metagenome changes in early stage L-DOPA-naive Parkinson’s disease patients. Genome Med 9: , 39. |
[96] | Hill-Burns EM , Debelius JW , Morton JT , Wissemann WT , Lewis MR , Wallen ZD , Peddada SD , Factor SA , Molho E , Zabetian CP , Knight R , Payami H ((2017) ) Parkinson’s disease and Parkinson’s disease medications have distinct signatures of the gut microbiome. Mov Disord 32: , 739–749. |
[97] | Hopfner F , Kunstner A , Muller SH , Kunzel S , Zeuner KE , Margraf NG , Deuschl G , Baines JF , Kuhlenbaumer G ((2017) ) Gut microbiota in Parkinson disease in a northern German cohort. Brain Res 1667: , 41–45. |
[98] | Li W , Wu X , Hu X , Wang T , Liang S , Duan Y , Jin F , Qin B ((2017) ) Structural changes of gut microbiota in Parkinson’s disease and its correlation with clinical features. Sci China Life Sci 60: , 1223–1233. |
[99] | Minato T , Maeda T , Fujisawa Y , Tsuji H , Nomoto K , Ohno K , Hirayama M ((2017) ) Progression of Parkinson’s disease is associated with gut dysbiosis: Two-year follow-up study. PLoS One 12: , e0187307. |
[100] | Petrov VA , Saltykova IV , Zhukova IA , Alifirova VM , Zhukova NG , Dorofeeva YB , Tyakht AV , Kovarsky BA , Alekseev DG , Kostryukova ES , Mironova YS , Izhboldina OP , Nikitina MA , Perevozchikova TV , Fait EA , Babenko VV , Vakhitova MT , Govorun VM , Sazonov AE ((2017) ) Analysis of gut microbiota in patients with Parkinson’s disease. Bull Exp Biol Med 162: , 734–737. |
[101] | Heintz-Buschart A , Pandey U , Wicke T , Sixel-Doring F , Janzen A , Sittig-Wiegand E , Trenkwalder C , Oertel WH , Mollenhauer B , Wilmes P ((2018) ) The nasal and gut microbiome in Parkinson’s disease and idiopathic rapid eyemovement sleep behavior disorder. Mov Disord 33: , 88–98. |
[102] | Lin A , Zheng W , He Y , Tang W , Wei X , He R , Huang W , Su Y , Huang Y , Zhou H , Xie H ((2018) ) Gut microbiota in patients with Parkinson’s disease in southern China. Parkinsonism Relat Disord 53: , 82–88. |
[103] | Qian Y , Yang X , Xu S , Wu C , Song Y , Qin N , Chen SD , Xiao Q ((2018) ) Alteration of the fecal microbiota in Chinese patients with Parkinson’s disease. Brain Behav Immun 70: , 194–202. |
[104] | Tetz G , Brown SM , Hao Y , Tetz V ((2018) ) Parkinson’s disease and bacteriophages as its overlooked contributors. Sci Rep 8: , 10812. |
[105] | Aho VTE , Pereira PAB , Voutilainen S , Paulin L , Pekkonen E , Auvinen P , Scheperjans F ((2019) ) Gut microbiota in Parkinson’s disease: Temporal stability and relations to disease progression. EBioMedicine 44: , 691–707. |
[106] | Barichella M , Severgnini M , Cilia R , Cassani E , Bolliri C , Caronni S , Ferri V , Cancello R , Ceccarani C , Faierman S , Pinelli G , De Bellis G , Zecca L , Cereda E , Consolandi C , Pezzoli G ((2019) ) Unraveling gut microbiota in Parkinson’s disease and atypical parkinsonism. Mov Disord 34: , 396–405. |
[107] | Jin M , Li J , Liu F , Lyu N , Wang K , Wang L , Liang S , Tao H , Zhu B , Alkasir R ((2019) ) Analysis of the gut microflora in patients with Parkinson’s disease. Front Neurosci 13: , 1184. |
[108] | Li C , Cui L , Yang Y , Miao J , Zhao X , Zhang J , Cui G , Zhang Y ((2019) ) Gut microbiota differs between Parkinson’s disease patients and healthy controls in Northeast China. Front Mol Neurosci 12: , 171. |
[109] | Lin CH , Chen CC , Chiang HL , Liou JM , Chang CM , Lu TP , Chuang EY , Tai YC , Cheng C , Lin HY , Wu MS ((2019) ) Altered gut microbiota and inflammatory cytokine responses in patients with Parkinson’s disease. J Neuroinflammation 16: , 129. |
[110] | Pietrucci D , Cerroni R , Unida V , Farcomeni A , Pierantozzi M , Mercuri NB , Biocca S , Stefani A , Desideri A ((2019) ) Dysbiosis of gut microbiota in a selected population of Parkinson’s patients. Parkinsonism Relat Disord 65: , 124–130. |
[111] | Weis S , Schwiertz A , Unger MM , Becker A , Fassbender K , Ratering S , Kohl M , Schnell S , Schafer KH , Egert M ((2019) ) Effect of Parkinson’s disease and related medications on the composition of the fecal bacterial microbiota. NPJ Parkinsons Dis 5: , 28. |
[112] | Cirstea MS , Yu AC , Golz E , Sundvick K , Kliger D , Radisavljevic N , Foulger LH , Mackenzie M , Huan T , Finlay BB , Appel-Cresswell S ((2020) ) Microbiota composition and metabolism are associated with gut function in Parkinson’s disease. Mov Disord 35: , 1208–1217. |
[113] | Grun D , Zimmer VC , Kauffmann J , Spiegel J , Dillmann U , Schwiertz A , Fassbender K , Fousse M , Unger MM ((2020) ) Impact of oral COMT-inhibitors on gut microbiota and short chain fatty acids in Parkinson’s disease. Parkinsonism Relat Disord 70: , 20–22. |
[114] | Ren T , Gao Y , Qiu Y , Jiang S , Zhang Q , Zhang J , Wang L , Zhang Y , Wang L , Nie K ((2020) ) Gut microbiota altered in mild cognitive impairment compared with normal cognition in sporadic Parkinson’s disease. Front Neurol 11: , 137. |
[115] | Khedr EM , Ali AM , Deaf E , Hassan HM , Alaa A , Gamea A ((2021) ) Gut microbiota in Parkinson’s disease patients: Hospital-based study. Egypt J Neurol Psychiatry Neurosurg 57: , 153. |
[116] | Li P , Killinger BA , Ensink E , Beddows I , Yilmaz A , Lubben N , Lamp J , Schilthuis M , Vega IE , Woltjer R , Pospisilik JA , Brundin P , Brundin L , Graham SF , Labrie V ((2021) ) Gut microbiota dysbiosis is associated with elevated bile acids in Parkinson’s disease. Metabolites 11: , 29. |
[117] | Li Z , Lu G , Luo E , Wu B , Li Z , Guo J , Xia Z , Zheng C , Su Q , Zeng Y , Yee Chan W , Su X , Qiu X , Zheng X , Cai Q , Xu Y , Chen Y , Fan Y , Chen W , Yu Z , Chen X , Zheng C , Wang M , Sang Poon W , Luo X ((2022) ) Oral, nasal, and gut microbiota in Parkinson’s disease. Neuroscience 480: , 65–78. |
[118] | Rosario D , Bidkhori G , Lee S , Bedarf J , Hildebrand F , Le Chatelier E , Uhlen M , Ehrlich SD , Proctor G , Wullner U , Mardinoglu A , Shoaie S ((2021) ) Systematic analysis of gut microbiome reveals the role of bacterial folate and homocysteine metabolism in Parkinson’s disease. Cell Rep 34: , 108807. |
[119] | Vascellari S , Melis M , Palmas V , Pisanu S , Serra A , Perra D , Santoru ML , Oppo V , Cusano R , Uva P , Atzori L , Morelli M , Cossu G , Manzin A ((2021) ) Clinical phenotypes of Parkinson’s disease associate with distinct gutmicrobiota and metabolome enterotypes. Biomolecules 11: , 144. |
[120] | Cerroni R , Pietrucci D , Teofani A , Chillemi G , Liguori C , Pierantozzi M , Unida V , Selmani S , Mercuri NB , Stefani A ((2022) ) Not just a snapshot: An Italian longitudinal evaluation of stability of gut microbiota findings in Parkinson’s disease. Brain Sci 12: , 739. |
[121] | Zhang P , Huang P , Du J , He Y , Liu J , He G , Cui S , Zhang W , Li G , Chen S ((2022) ) Specific gut microbiota alterations in essential tremor and its difference from Parkinson’s disease. NPJ Parkinsons Dis 8: , 98. |
[122] | Zhang Y , He X , Qian Y , Xu S , Mo C , Yan Z , Yang X , Xiao Q ((2022) ) Plasma branched-chain and aromatic amino acids correlate with the gut microbiota and severity of Parkinson’s disease. NPJ Parkinsons Dis 8: , 48. |
[123] | Nakahara K , Nakane S , Ishii K , Ikeda T , Ando Y ((2023) ) Gut microbiota of Parkinson’s disease in an appendectomy cohort: A preliminary study. Sci Rep 13: , 2210. |
[124] | Wallen ZD , Demirkan A , Twa G , Cohen G , Dean MN , Standaert DG , Sampson TR , Payami H ((2022) ) Metagenomics of Parkinson’s disease implicates the gut microbiome in multiple disease mechanisms. Nat Commun 13: , 6958. |
[125] | Palacios N , Wilkinson J , Bjornevik K , Schwarzschild MA , McIver L , Ascherio A , Huttenhower C ((2023) ) Metagenomics of the gut microbiome in Parkinson’s disease: Prodromal changes. Ann Neurol 94: , 486–501. |
[126] | Huang B , Chau SWH , Liu Y , Chan JWY , Wang J , Ma SL , Zhang J , Chan PKS , Yeoh YK , Chen Z , Zhou L , Wong SH , Mok VCT , To KF , Lai HM , Ng S , Trenkwalder C , Chan FKL , Wing YK ((2023) ) Gut microbiome dysbiosis across early Parkinson’s disease, REM sleep behavior disorder and their first-degree relatives. Nat Commun 14: , 2501. |
[127] | Toh TS , Chong CW , Lim SY , Bowman J , Cirstea M , Lin CH , Chen CC , Appel-Cresswell S , Finlay BB , Tan AH ((2022) ) Gut microbiome in Parkinson’s disease: New insights from meta-analysis. Parkinsonism Relat Disord 94: , 1–9. |
[128] | Shen T , Yue Y , He T , Huang C , Qu B , Lv W , Lai HY ((2021) ) The association between the gut microbiota and Parkinson’s disease, a meta-analysis. Front Aging Neurosci 13: , 636545. |
[129] | Yuan X , Pan Z , Jin C , Ni Y , Fu Z , Jin Y ((2019) ) Gut microbiota: An underestimated and unintended recipient for pesticide-induced toxicity. Chemosphere 227: , 425–434. |
[130] | Xie W , Gao J , Jiang R , Liu X , Lai F , Tang Y , Xiao H , Jia Y , Bai Q ((2020) ) Twice subacute MPTP administrations induced time-dependent dopaminergic neurodegeneration and inflammation in midbrain and ileum, as well as gut microbiota disorders in PD mice. Neurotoxicology 76: , 200–212. |
[131] | Swarte JC , Eelderink C , Douwes RM , Said MY , Hu S , Post A , Westerhuis R , Bakker SJL , Harmsen HJM ((2020) ) Effect of high versus low dairy consumption on the gut microbiome: Results of a randomized, cross-over study. Nutrients 12: , 2129. |
[132] | Wang H , Zhang S , Yang F , Xin R , Wang S , Cui D , Sun Y ((2020) ) The gut microbiota confers protection in the CNS against neurodegeneration induced by manganism. Biomed Pharmacother 127: , 110150. |
[133] | Shao M , Zhu Y ((2020) ) Long-term metal exposure changes gut microbiota of residents surrounding a mining and smelting area. Sci Rep 10: , 4453–4453. |
[134] | Sun MF , Zhu YL , Zhou ZL , Jia XB , Xu YD , Yang Q , Cui C , Shen YQ ((2018) ) Neuroprotective effects of fecal microbiota transplantation on MPTP-induced Parkinson’s disease mice: Gut microbiota, glial reaction and TLR4/TNF-alpha signaling pathway. Brain Behav Immun 70: , 48–60. |
[135] | Sampson TR , Debelius JW , Thron T , Janssen S , Shastri GG , Ilhan ZE , Challis C , Schretter CE , Rocha S , Gradinaru V , Chesselet MF , Keshavarzian A , Shannon KM , Krajmalnik-Brown R , Wittung-Stafshede P , Knight R , Mazmanian SK ((2016) ) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. , . Cell 167: , 1469–1480.e12. |
[136] | Braniste V , Al-Asmakh M , Kowal C , Anuar F , Abbaspour A , Tóth M , Korecka A , Bakocevic N , Guan NL , Kundu P , Gulyás B , Halldin C , Hultenby K , Nilsson H , Hebert H , Volpe BT , Diamond B , Pettersson S ((2014) ) The gutmicrobiota influences blood-brain barrier permeability in mice. Sci Transl Med 6: , 263ra158. |
[137] | Erny D , De Angelis ALH , Jaitin D , Wieghofer P , Staszewski O , David E , Keren-Shaul H , Mahlakoiv T , Jakobshagen K , Buch T , Schwierzeck V , Utermöhlen O , Chun E , Garrett WS , McCoy KD , Diefenbach A , Staeheli P , Stecher B , Amit I , Prinz M ((2015) ) Host microbiota constantly control maturation and function of microglia in the CNS. Nat Neurosci 18: , 965–977. |
[138] | Li X , Feng X , Jiang Z , Jiang Z ((2021) ) Association of small intestinal bacterial overgrowth with Parkinson’s disease: A systematic review and meta-analysis. Gut Pathog 13: , 25. |
[139] | Niu XL , Liu L , Song ZX , Li Q , Wang ZH , Zhang JL , Li HH ((2016) ) Prevalence of small intestinal bacterial overgrowth in Chinese patients with Parkinson’s disease. J Neural Transm (Vienna) 123: , 1381–1386. |
[140] | Fasano A , Bove F , Gabrielli M , Petracca M , Zocco MA , Ragazzoni E , Barbaro F , Piano C , Fortuna S , Tortora A , Di Giacopo R , Campanale M , Gigante G , Lauritano EC , Navarra P , Marconi S , Gasbarrini A , Bentivoglio AR ((2013) ) The role of small intestinal bacterial overgrowth in Parkinson’s disease. Mov Disord 28: , 1241–1249. |
[141] | Lauritano EC , Valenza V , Sparano L , Scarpellini E , Gabrielli M , Cazzato A , Ferraro PM , Gasbarrini A ((2010) ) Small intestinal bacterial overgrowth and intestinal permeability. Scand J Gastroenterol 45: , 1131–1132. |
[142] | Ricci JE , Chebli LA , Ribeiro TC , Castro A , Gaburri PD , Pace FH , Barbosa KV , Ferreira LE , Malaguti C , DelgadoÁ H ((2018) ) Small-intestinal bacterial overgrowth is associated with concurrent intestinal inflammation but notwith systemic inflammation in Crohn’s disease patients. J Clin Gastroenterol 52: , 530–536. |
[143] | Donaldson GP , Lee SM , Mazmanian SK ((2016) ) Gut biogeography of the bacterial microbiota, nature reviews. Microbiology 14: , 20–32. |
[144] | Johansson ME , Phillipson M , Petersson J , Velcich A , Holm L , Hansson GC ((2008) ) The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc Natl Acad Sci U S A 105: , 15064–15069. |
[145] | Chen SJ , Chen CC , Liao HY , Lin YT , Wu YW , Liou JM , Wu MS , Kuo CH , Lin CH ((2022) ) Association of fecal and plasma levels of short-chain fatty acids with gut microbiota and clinical severity in Parkinson disease patients. Neurology 98: , e848–e858. |
[146] | Tufekci KU , Genc S , Genc K ((2011) ) The endotoxin-induced neuroinflammation model of Parkinson’s disease. Parkinsons Dis 2011: , 487450. |
[147] | Brown GC ((2019) ) The endotoxin hypothesis of neurodegeneration. J Neuroinflammation 16: , 180. |
[148] | Loffredo L , Ettorre E , Zicari AM , Inghilleri M , Nocella C , Perri L , Spalice A , Fossati C , De Lucia MC , Pigozzi F , Cacciafesta M , Violi F , Carnevale R , Neurodegenerative Disease study group ((2020) ) Oxidative stress andgut-derived lipopolysaccharides in neurodegenerative disease: Role of NOX2. Oxid Med Cell Longev 2020: , 8630275. |
[149] | Wijeyekoon RS , Kronenberg-Versteeg D , Scott KM , Hayat S , Kuan WL , Evans JR , Breen DP , Cummins G , Jones JL , Clatworthy MR , Floto RA , Barker RA , Williams-Gray CH ((2020) ) Peripheral innate immune and bacterial signals relate to clinical heterogeneity in Parkinson’s disease. Brain Behav Immun 87: , 473–488. |
[150] | Zhao Y , Walker DI , Lill CM , Bloem BR , Darweesh SKL , Pinto-Pacheco B , McNeil B , Miller GW , Heath AK , Frissen M , Petrova D , Sánchez M-J , Chirlaque M-D , Guevara M , Zibetti M , Panico S , Middleton L , Katzke V , Kaaks R , Riboli E , Masala G , Sieri S , Zamora-Ros R , Amiano P , Jenab M , Peters S , Vermeulen R ((2023) ) Lipopolysaccharide-bindingprotein and future Parkinson’s disease risk: A European prospective cohort. J Neuroinflammation 20: , 170. |
[151] | Dzamko N , Gysbers A , Perera G , Bahar A , Shankar A , Gao J , Fu Y , Halliday GM ((2017) ) Toll-like receptor 2 is increased in neurons in Parkinson’s disease brain and may contribute to alpha-synuclein pathology. Acta Neuropathol 133: , 303–319. |
[152] | Hughes CD , Choi ML , Ryten M , Hopkins L , Drews A , Botia JA , Iljina M , Rodrigues M , Gagliano SA , Gandhi S , Bryant C , Klenerman D ((2019) ) Picomolar concentrations of oligomeric alpha-synuclein sensitizes TLR4 to play an initiating role in Parkinson’s disease pathogenesis. Acta Neuropathol 137: , 103–120. |
[153] | Drouin-Ouellet J , St-Amour I , Saint-Pierre M , Lamontagne-Proulx J , Kriz J , Barker RA , Cicchetti F ((2014) ) Toll-like receptor expression in the blood and brain of patients and a mouse model of Parkinson’s disease. Int J Neuropsychopharmacol 18: , pyu103. |
[154] | Castaño A , Herrera AJ , Cano J , Machado A ((1998) ) Lipopolysaccharide intranigral injection induces inflammatoryreaction and damage in nigrostriatal dopaminergic system. J Neurochem 70: , 1584–1592. |
[155] | Herrera AJ , Castaño A , Venero JL , Cano J , Machado A ((2000) ) The single intranigral injection of LPS as a newmodel for studying the selective effects of inflammatory reactions on dopaminergic system. Neurobiol Dis 7: , 429–447. |
[156] | Kim WG , Mohney RP , Wilson B , Jeohn GH , Liu B , Hong JS ((2000) ) Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain: Role of microglia. J Neurosci 20: , 6309–6316. |
[157] | De Pablos RM , Hererra AJ , Villaran RF , Cano J , Machado A ((2004) ) Dopamine-dependent neurotoxicity of lipopolysaccharide in substantia nigra. FASEB J 1096: , 1–22. |
[158] | Tanaka S , Ishii A , Ohtaki H , Shioda S , Yoshida T , Numazawa S ((2013) ) Activation of microglia induces symptoms of Parkinson’s disease in wild-type, but not in IL-1 knockout mice. J Neuroinflammation 10: , 907. |
[159] | Gao HM , Zhang F , Zhou H , Kam W , Wilson B , Hong JS ((2011) ) Neuroinflammation and alpha-synuclein dysfunction potentiate each other, driving chronic progression of neurodegeneration in a mouse model of Parkinson’s disease. Environ Health Perspect 119: , 807–814. |
[160] | Kelly LP , Carvey PM , Keshavarzian A , Shannon KM , Shaikh M , Bakay RA , Kordower JH ((2014) ) Progression of intestinal permeability changes and alpha-synuclein expression in a mouse model of Parkinson’s disease. Mov Disord 29: , 999–1009. |
[161] | He Q , Yu W , Wu J , Chen C , Lou Z , Zhang Q , Zhao J , Wang J , Xiao B ((2013) ) Intral LPS-mediated Parkinson’s model challenges the pathogenesis of nasal cavity and environmental toxins. , . PLoS One 8: , e78418. |
[162] | Alexander C , Rietschel ET ((2001) ) Bacterial lipopolysaccharides and innate immunity. J Endotoxin Res 7: , 167–202. |
[163] | Di Lorenzo F , De Castro C , Silipo A , Molinaro A ((2019) ) Lipopolysaccharide structures of Gram-negative populations in the gut microbiota and effects on host interactions. FEMS Microbiol Rev 43: , 257–272. |
[164] | Kaparakis-Liaskos M , Ferrero RL ((2015) ) Immune modulation by bacterial outer membrane vesicles. Nat Rev Immunol 15: , 375–387. |
[165] | Stentz R , Carvalho AL , Jones EJ , Carding SR ((2018) ) Fantastic voyage: The journey of intestinal microbiota-derived microvesicles through the body. Biochem Soc Trans 46: , 1021–1027. |
[166] | Kim JH , Lee J , Park J , Gho YS ((2015) ) Gram-negative and Gram-positive bacterial extracellular vesicles. Semin Cell Dev Biol 40: , 97–104. |
[167] | Knox K , Vesk M , Work E ((1966) ) Relation between excreted lipopolysaccharide complexes and surface structures of a lysine-limited culture of Escherichia coli. J Bacteriol 92: , 1206–1217. |
[168] | Liu Y , Defourny KAY , Smid EJ , Abee T ((2018) ) Gram-positive bacterial extracellular vesicles and their impact on health and disease. Front Microbiol 9: , 1502–1502. |
[169] | Schwechheimer C , Kuehn MJ ((2015) ) Outer-membrane vesicles from Gram-negative bacteria: Biogenesis and functions. Nat Rev Microbiol 13: , 605–619. |
[170] | Gilmore WJ , Johnston EL , Zavan L , Bitto NJ , Kaparakis-Liaskos M ((2021) ) Immunomodulatory roles and novel applications of bacterial membrane vesicles. Mol Immunol 134: , 72–85. |
[171] | Jones EJ , Booth C , Fonseca S , Parker A , Cross K , Miquel-Clopes A , Hautefort I , Mayer U , Wileman T , Stentz R , Carding SR ((2020) ) The uptake, trafficking, and biodistribution of bacteroides thetaiotaomicron generated outer membrane vesicles. Front Microbiol 11: , 57. |
[172] | Cecil JD , Sirisaengtaksin N , O’Brien-Simpson NM , Krachler AM ((2019) ) Outer membrane vesicle-host cellinteractions. Microbiol Spectr 7: , 10.1128/microbiolspec.PSIB-0001-2018. |
[173] | Park JY , Choi J , Lee Y , Lee JE , Lee EH , Kwon HJ , Yang J , Jeong BR , Kim YK , Han PL ((2017) ) Metagenome analysis of bodily microbiota in a mouse model of Alzheimer disease using bacteria-derived membrane vesicles in Blood. Exp Neurobiol 26: , 369–379. |
[174] | Tulkens J , De Wever O , Hendrix A ((2020) ) Analyzing bacterial extracellular vesicles in human body fluids by orthogonal biophysical separation and biochemical characterization. Nat Protoc 5: , 40–67. |
[175] | Tulkens J , Vergauwen G , Van Deun J , Geeurickx E , Dhondt B , Lippens L , De Scheerder M-A , Miinalainen I , Rappu P , De Geest BG , Vandecasteele K , Laukens D , Vandekerckhove L , Denys H , Vandesompele J , De Wever O , Hendrix A ((2020) ) Increased levels of systemic LPS-positive bacterial extracellular vesicles in patients with intestinal barrier dysfunction. Gut 69: , 191. |
[176] | Stephens DS , Edwards KM , Morris F , McGee ZA ((1982) ) Pili and outer membrane appendages on Neisseria meningitidis in the cerebrospinal fluid of an infant. J Infect Dis 146: , 568–568. |
[177] | Namork E , Brandtzaeg P ((2002) ) Fatal meningococcal septicaemia with “blebbing” meningococcus. Lancet 360: , 1741. |
[178] | Fiocca R , Necchi V , Sommi P , Ricci V , Telford J , Cover TL , Solcia E ((1999) ) Release of Helicobacter pylori vacuolating cytotoxin by both a specific secretion pathway and budding of outer membrane vesicles. Uptake of released toxin and vesicles by gastric epithelium. J Pathol 188: , 220–226. |
[179] | Kim Y-k (2019) Nano-vesicles derived from genus cupriavidus bacteria and use thereof. US Patent App. 16/563,594. http://www.freepatentsonline.com/y2019/0390284.html |
[180] | Yoo JY , Rho M , You YA , Kwon EJ , Kim MH , Kym S , Jee YK , Kim YK , Kim YJ ((2016) ) 16S rRNA gene-based metagenomic analysis reveals differences in bacteria-derived extracellular vesicles in the urine of pregnant and non-pregnant women. Exp Mol Med 48: , e208. |
[181] | Lee Y , Park JY , Lee EH , Yang J , Jeong BR , Kim YK , Seoh JY , Lee S , Han PL , Kim EJ ((2017) ) Rapid assessment of microbiota changes in individuals with autism spectrum disorder using bacteria-derived membrane vesicles in urine. Exp Neurobiol 26: , 307–317. |
[182] | Wu J , An M , Zhu J , Tan Z , Chen Y.G , Stidham R , Lubman D ((2019) ) A method for isolation and proteomic analysis of outer membrane vesicles from fecal samples by LC-MS/MS. J Proteomics Bioinform 12: , 38–42. |
[183] | Park KS , Lee J , Lee C , Park HT , Kim JW , Kim OY , Kim SR , Radinger M , Jung HY , Park J , Lotvall J , Gho YS ((2018) ) Sepsis-like systemic inflammation induced by nano-sized extracellular vesicles from feces. Front Microbiol 9: , 1735. |
[184] | Kameli N , Becker HEF , Welbers T , Jonkers D , Penders J , Savelkoul P , Stassen FR ((2021) ) Metagenomic profiling of fecal-derived bacterial membrane vesicles in Crohn’s disease patients. Cells 10: , 2795. |
[185] | Xie J , Cools L , Van Imschoot G , Van Wonterghem E , Pauwels MJ , Vlaeminck I , De Witte C , El Andaloussi S , Wierda K , De Groef L , Haesebrouck F , Van Hoecke L , Vandenbroucke RE ((2023) ) Helicobacter pylori-derived outer membrane vesicles contribute to Alzheimer’s disease pathogenesis via C3-C3aR signalling. J Extracell Vesicles 12: , e12306. |
[186] | Ha JY , Choi S-Y , Lee JH , Hong S-H , Lee H-J ((2020) ) Delivery of periodontopathogenic extracellular vesicles to brain monocytes and microglial IL-6 promotion by RNA cargo. Front Mol Biosci 7: , 596366. |
[187] | Tiku V , Tan MW ((2021) ) Host immunity and cellular responses to bacterial outer membrane vesicles. Trends Immunol 42: , 1024–1036. |
[188] | Ellis TN , Leiman SA , Kuehn MJ ((2010) ) Naturally produced outer membrane vesicles from Pseudomonas aeruginosa elicit a potent innate immune response via combined sensing of both lipopolysaccharide and protein components. Infect Immun 78: , 3822–3831. |
[189] | Al-Nedawi K , Mian MF , Hossain N , Karimi K , Mao YK , Forsythe P , Min KK , Stanisz AM , Kunze WA , Bienenstock J ((2015) ) Gut commensal microvesicles reproduce parent bacterial signals to host immune and enteric nervous systems. FASEB J 29: , 684–695. |
[190] | du Teil Espina M , Fu Y , van der Horst D , Hirschfeld C , López-Álvarez M , Mulder Lianne M , Gscheider C , Haider Rubio A , Huitema M , Becher D , Heeringa P , van Dijl Jan M ((2022) ) Coating and corruption of humanneutrophils by bacterial outer membrane vesicles. Microbiol Spectr 10: , e00753–00722. |
[191] | Vidakovics ML , Jendholm J , Morgelin M , Mansson A , Larsson C , Cardell LO , Riesbeck K ((2010) ) B cell activation by outer membrane vesicles–a novel virulence mechanism. PLoS Pathog 6: , e1000724. |
[192] | Lee EY , Bang JY , Park GW , Choi DS , Kang JS , Kim HJ , Park KS , Lee JO , Kim YK , Kwon KH , Kim KP , Gho YS ((2007) ) Global proteomic profiling of native outer membrane vesicles derived from Escherichia coli. Proteomics 7: , 3143–3153. |
[193] | Sartorio Mariana G , Valguarnera E , Hsu F-F , Feldman Mario F ((2022) ) Lipidomics analysis of outer membrane vesicles and elucidation of the inositol phosphoceramide biosynthetic pathway in Bacteroides thetaiotaomicron. Microbiol Spectr 10: , e00634–00621. |
[194] | Sjostrom AE , Sandblad L , Uhlin BE , Wai SN ((2015) ) Membrane vesicle-mediated release of bacterial RNA. Sci Rep 5: , 15329. |
[195] | Horstman AL , Kuehn MJ ((2000) ) Enterotoxigenic Escherichia coli secretes active heat-labile enterotoxin via outer membrane vesicles. J Biol Chem 275: , 12489–12496. |
[196] | Shah B , Sullivan CJ , Lonergan NE , Stanley S , Soult MC , Britt LD ((2012) ) Circulating bacterial membrane vesicles cause sepsis in rats. Shock 37: , 621–628. |
[197] | Park K-S , Choi K-H , Kim Y-S , Hong BS , Kim OY , Kim JH , Yoon CM , Koh G-Y , Kim Y-K , Gho YS ((2010) ) Outer membrane vesicles derived from Escherichia coli induce systemic inflammatory response syndrome. PLoS One 5: , e11334. |
[198] | Svennerholm K , Park KS , Wikstrom J , Lasser C , Crescitelli R , Shelke GV , Jang SC , Suzuki S , Bandeira E , Olofsson CS , Lotvall J ((2017) ) Escherichia coli outer membrane vesicles can contribute to sepsis induced cardiac dysfunction. Sci Rep 7: , 17434. |
[199] | Kaparakis M , Turnbull L , Carneiro L , Firth S , Coleman HA , Parkington HC , Le Bourhis L , Karrar A , Viala J , Mak J , Hutton ML , Davies JK , Crack PJ , Hertzog PJ , Philpott DJ , Girardin SE , Whitchurch CB , Ferrero RL ((2010) ) Bacterialmembrane vesicles deliver peptidoglycan to NOD1 in epithelial cells. Cell Microbiol 12: , 372–385. |
[200] | Vanaja SK , Russo AJ , Behl B , Banerjee I , Yankova M , Deshmukh SD , Rathinam VAK ((2016) ) Bacterial outer membrane vesicles mediate cytosolic localization of LPS and caspase-11 activation. Cell 165: , 1106–1119. |
[201] | Ciesielska A , Matyjek M , Kwiatkowska K ((2020) ) TLR4 and CD14 trafficking and its influence on LPS-induced pro-inflammatory signaling. Cell Mol Life Sci 78: , 1233–1261. |
[202] | Tyrer PC , Frizelle FA , Keenan JI ((2014) ) Escherichia coli-derived outer membrane vesicles are genotoxic to human enterocyte-like cells. Infect Agent Cancer 9: , 2. |
[203] | Kulkarni HM , Nagaraj R , Jagannadham MV ((2015) ) Protective role of E. coli outer membrane vesicles against antibiotics. Microbiol Res 181: , 1–7. |
[204] | Deo P , Chow SH , Han M-L , Speir M , Huang C , Schittenhelm RB , Dhital S , Emery J , Li J , Kile BT , Vince JE , Lawlor KE , Naderer T ((2020) ) Mitochondrial dysfunction caused by outer membrane vesicles from Gram-negative bacteria activates intrinsic apoptosis and inflammation. Nat Microbiol 5: , 1418–1427. |
[205] | Thay B , Damm A , Kufer TA , Wai SN , Oscarsson J ((2014) ) Aggregatibacter actinomycetemcomitans outer membrane vesicles are internalized in human host cells and trigger NOD1- and NOD2-dependent NF-kappaB activation. Infect Immun 82: , 4034–4046. |
[206] | Marion Chad R , Lee J , Sharma L , Park K-S , Lee C , Liu W , Liu P , Feng J , Gho Yong S , Dela Cruz Charles S ((2019) ) Toll-like receptors 2 and 4 modulate pulmonary inflammation and host factors mediated by outer membrane vesicles derived from Acinetobacter baumannii. Infect Immun 87: , e00243–19. |
[207] | Jun SH , Lee JH , Kim BR , Kim SI , Park TI , Lee JC , Lee YC ((2013) ) Acinetobacter baumannii outer membrane vesicles elicit a potent innate immune response via membrane proteins. PLoS One 8: , e71751. |
[208] | Yu L , Wang L , Chen S ((2010) ) Endogenous toll-like receptor ligands and their biological significance. J Cell Mol Med 14: , 2592–2603. |
[209] | Rocha Sobrinho HMd , Silva DJd , Gomides LF , Dorta ML , Oliveira MAPd , Ribeiro-Dias F ((2018) ) TLR4 and TLR2 activation is differentially associated with age during Parkinson’s disease. Immunol Invest 47: , 71–88. |
[210] | Chatterjee K , Roy A , Banerjee R , Choudhury S , Mondal B , Halder S , Basu P , Shubham S , Dey S , Kumar H ((2020) ) Inflammasome and alpha-synuclein in Parkinson’s disease: A cross-sectional study. J Neuroimmunol 338: , 577089. |
[211] | Bialecka M , Kurzawski M , Klodowska-Duda G , Opala G , Juzwiak S , Kurzawski G , Tan E-K , Drozdzik M ((2007) ) CARD15 variants in patients with sporadic Parkinson’s disease. Neurosci Res 57: , 473–476. |
[212] | Barajon I , Serrao G , Arnaboldi F , Opizzi E , Ripamonti G , Balsari A , Rumio C ((2009) ) Toll-like receptors 3, 4, and 7 are expressed in the enteric nervous system and dorsal root ganglia. J Histochem Cytochem 57: , 1013–1023. |
[213] | Burgueno JF , Barba A , Eyre E , Romero C , Neunlist M , Fernandez E ((2016) ) TLR2 and TLR9 modulate enteric nervous system inflammatory responses to lipopolysaccharide. J Neuroinflammation 13: , 187. |
[214] | Heidari A , Yazdanpanah N , Rezaei N ((2022) ) The role of Toll-like receptors and neuroinflammation in Parkinson’s disease. J Neuroinflammation 19: , 135. |
[215] | Krsek D , Yara DA , Hrbácková H , Daniel O , Mancíková A , Schüller S , Bielaszewska M ((2023) ) Translocation of outer membrane vesicles from enterohemorrhagic Escherichia coli O157 across theintestinal epithelial barrier. Front Microbiol 14: , 1198945. |
[216] | Elmi A , Nasher F , Jagatia H , Gundogdu O , Bajaj-Elliott M , Wren B , Dorrell N ((2016) ) Campylobacter jejuni outer membrane vesicle-associated proteolytic activity promotes bacterial invasion by mediating cleavage of intestinal epithelial cell E-cadherin and occludin. Cell Microbiol 18: , 561–572. |
[217] | Turkina MV , Olofsson A , Magnusson KE , Arnqvist A , Vikstrom E ((2015) ) Helicobacter pylori vesicles carrying CagA localize in the vicinity of cell-cell contacts and induce histone H1 binding to ATP in epithelial cells. FEMS Microbiol Lett 362: . |
[218] | He Y , Shiotsu N , Uchida-Fukuhara Y , Guo J , Weng Y , Ikegame M , Wang Z , Ono K , Kamioka H , Torii Y , Sasaki A , Yoshida K , Okamura H ((2020) ) Outer membrane vesicles derived from Porphyromonas gingivalis induced cell death with disruption of tight junctions in human lung epithelial cells. Arch Oral Biol 118: , 104841. |
[219] | Nie Y , Xie XQ , Zhou L , Guan Q , Ren Y , Mao Y , Shi JS , Xu ZH , Geng Y ((2022) ) Desulfovibrio fairfieldensis-derived outer membrane vesicles damage epithelial barrier and induce inflammation and pyroptosis in macrophages. Cells 12: , 89. |
[220] | Jones E , Stentz R , Telatin A , Savva GM , Booth C , Baker D , Rudder S , Knight SC , Noble A , Carding SR ((2021) ) The origin of plasma-derived bacterial extracellular vesicles in healthy individuals and patients with inflammatory bowel disease: A pilot study. Genes (Basel) 12: , 1636. |
[221] | Clairembault T , Leclair-Visonneau L , Coron E , Bourreille A , Le Dily S , Vavasseur F , Heymann M-F , Neunlist M , Derkinderen P ((2015) ) Structural alterations of the intestinal epithelial barrier in Parkinson’s disease. Acta Neuropathol Commun 3: , 12. |
[222] | Schwiertz A , Spiegel J , Dillmann U , Grundmann D , Bürmann J , Faßbender K , Schäfer KH , Unger MM ((2018) ) Fecal markers of intestinal inflammation and intestinal permeability are elevated in Parkinson’s disease. Parkinsonism Relat Disord 50: , 104–107. |
[223] | Singhrao SK , Olsen I ((2018) ) Are Porphyromonas gingivalis outer membrane vesicles microbullets for sporadic Alzheimer’s disease manifestation? . J Alzheimers Dis Rep 2: , 219–228. |
[224] | Wispelwey B , Hansen EJ , Scheld WM ((1989) ) Haemophilus influenzae outer membrane vesicle-induced blood-brain barrier permeability during experimental meningitis. Infect Immun 57: , 2559–2562. |
[225] | Yu H , Kim KS ((2012) ) YgfZ contributes to secretion of cytotoxic necrotizing factor 1 into outer-membrane vesicles in Escherichia coli. Microbiology (Reading) 158: , 612–621. |
[226] | Wei S , Peng W , Mai Y , Li K , Wei W , Hu L , Zhu S , Zhou H , Jie W , Wei Z , Kang C , Li R , Liu Z , Zhao B , Cai Z ((2020) ) Outer membrane vesicles enhance tau phosphorylation and contribute to cognitive impairment. J Cell Physiol 235: , 4843–4855. |
[227] | Tomás-Camardiel M , Rite I , Herrera AJ , De Pablos RM , Cano J , Machado A , Venero JL ((2004) ) Minocycline reducesthe lipopolysaccharide-induced inflammatory reaction, peroxynitrite-mediated nitration of proteins, disruption ofthe blood-brain barrier, and damage in the nigral dopaminergic system. Neurobiol Dis 16: , 190–201. |
[228] | Varatharaj A , Galea I ((2017) ) The blood-brain barrier in systemic inflammation. Brain Behav Immun 60: , 1–12. |
[229] | Pritchard AB , Fabian Z , Lawrence CL , Morton G , Crean S , Alder JE ((2022) ) An investigation into the effectsof outer membrane vesicles and lipopolysaccharide of Porphyromonas gingivalis onblood-brain barrier integrity, permeability, and disruption of scaffolding proteins in a human in vitro model. J Alzheimers Dis 86: , 343–364. |
[230] | Farrugia C , Stafford GP , Murdoch C ((2020) ) Porphyromonas gingivalis outer membrane vesicles increase vascular permeability. J Dent Res 99: , 1494–1501. |
[231] | Pisani V , Stefani A , Pierantozzi M , Natoli S , Stanzione P , Franciotta D , Pisani A ((2012) ) Increased blood-cerebrospinal fluid transfer of albumin in advanced Parkinson’s disease. J Neuroinflammation 9: , 188. |
[232] | Gray MT , Woulfe JM ((2015) ) Striatal blood-brain barrier permeability in Parkinson’s disease. J Cereb Blood Flow Metab 35: , 747–750. |
[233] | Kortekaas R , Leenders KL , Van Oostrom JCH , Vaalburg W , Bart J , Willemsen ATM , Hendrikse NH ((2005) ) Blood-brain barrier dysfunction in Parkinsonian midbrain in vivo. Ann Neurol 57: , 176–179. |
[234] | Banks WA , Sharma P , Bullock KM , Hansen KM , Ludwig N , Whiteside TL ((2020) ) Transport of extracellular vesicles across the blood-brain barrier: Brain pharmacokinetics and effects of inflammation. Int J Mol Sci 21: , 4407. |
[235] | Han EC , Choi SY , Lee Y , Park JW , Hong SH , Lee HJ ((2019) ) Extracellular RNAs in periodontopathogenic outer membrane vesicles promote TNF-α production in human macrophages and cross the blood-brain barrier in mice. FASEB J 33: , 13412–13422. |
[236] | Bittel M , Reichert P , Sarfati I , Dressel A , Leikam S , Uderhardt S , Stolzer I , Phu TA , Ng M , Vu NK , Tenzer S , Distler U , Wirtz S , Rothhammer V , Neurath MF , Raffai RL , Gunther C , Momma S ((2021) ) Visualizing transfer of microbial biomolecules by outer membrane vesicles in microbe-host-communication in vivo. J Extracell Vesicles 10: , e12159. |
[237] | Palacios E , Lobos-Gonzalez L , Guerrero S , Kogan MJ , Shao B , Heinecke JW , Quest AFG , Leyton L , Valenzuela-Valderrama M ((2023) ) Helicobacter pylori outer membrane vesicles induce astrocyte reactivity through nuclear factor-kappaappa B activation and cause neuronal damage in vivo in a murine model. J Neuroinflammation 20: , 66. |
[238] | Lee KE , Kim JK , Han SK , Lee DY , Lee HJ , Yim SV , Kim DH ((2020) ) The extracellular vesicle of gut microbial Paenalcaligenes hominis is a risk factor for vagus nerve-mediated cognitive impairment. Microbiome 8: , 107. |
[239] | Tsukita K , Taguchi T , Sakamaki-Tsukita H , Tanaka K , Suenaga T ((2018) ) The vagus nerve becomes smaller in patients with Parkinson’s disease: A preliminary cross-sectional study using ultrasonography. Parkinsonism Relat Disord 55: , 148–149. |
[240] | Walter U , Tsiberidou P , Kersten M , Storch A , Lohle M ((2018) ) Atrophy of the vagus nerve in Parkinson’s disease revealed by high-resolution ultrasonography. Front Neurol 9: , 805. |
[241] | Gong T , Chen Q , Mao H , Zhang Y , Ren H , Xu M , Chen H , Yang D ((2022) ) Outer membrane vesicles of Porphyromonas gingivalis trigger NLRP3 inflammasome and induce neuroinflammation, tau phosphorylation, and memory dysfunction in mice. Front Cell Infect Microbiol 12: , 925435. |
[242] | Dhital S , Deo P , Stuart I , Naderer T ((2021) ) Bacterial outer membrane vesicles and host cell death signaling. Trends Microbiol 29: , 1106–1116. |


