
Astrocytes and Alpha-Synuclein: Friend or Foe?
Abstract
Despite its devastating disease burden and alarming prevalence, the etiology of Parkinson’s disease (PD) remains to be completely elucidated. PD is characterized by the degeneration of dopaminergic neurons in the substantia nigra pars compacta and this correlates with the accumulation of misfolded α-synuclein. While the aggregation of α-synuclein in the form of Lewy bodies or Lewy neurites is a well-established intraneuronal hallmark of the disease process, our understanding of the glial contribution to aberrant α-synuclein proteostasis is lacking. In this regard, restoring astrocyte function during early PD could offer a promising therapeutic avenue and understanding the involvement of astrocytes in handling/mishandling of α-synuclein is of particular interest. Here, we explore the growing body of scientific literature implicating aberrant astrocytic α-synuclein proteostasis with the seemingly inexorable pathological sequelae typifying PD. We also provide a perspective on how heterogeneity in the morphological relationship between astrocytes and neurons will need to be considered in the context of PD pathogenesis.
Parkinson’s disease (PD) is the most common synucleinopathy and second most common neurodegenerative disorder, affecting 2–3% of the population over 65 [1]. This progressive neurodegenerative disorder is clinically characterized by motor symptoms including rigidity, resting tremors, and postural instability as well as non-motor symptoms including autonomic and sensory disturbances, depression, and rapid eye movement sleep behavior disorder [2, 3]. Motor symptoms are underpinned by the degeneration of neurons in the substantia nigra pars compacta (SNc), causing dopamine deficiency in the striatum [4, 5]. While dopaminergic neuron loss is pathognomonic to PD, α-synuclein pathology affects neurons in multiple brain regions including monoaminergic neurons in the locus coeruleus [6, 7] and serotonergic neurons in the raphe nucleus [8–10]. The exact mechanisms mediating neurodegeneration in PD are currently unknown; however, α-synuclein aggregation in surviving neurons [a major component of Lewy bodies (LBs) and Lewy neurites (LNs)] is the most widely recognized pathological hallmark of the disease process [11]. These aggregates have been implicated in the disruption of a myriad of cellular processes, which culminate in declining neuronal function, thereby leading to neurodegeneration [12–14]. Given the urgent need for novel neuroprotective and disease modifying drugs to treat PD, this review focuses on the nascent, yet growing body of evidence suggesting a possible role for aberrant astrocytic proteostasis of α-synuclein in PD pathophysiology.
ALPHA-SYNUCLEIN AND PD
α-synuclein is a 140 amino acid protein predominantly localized to presynaptic terminals. It has three distinct regions which include: 1) a highly conserved N-terminal lipid-binding α-helix; 2) a hydrophobic non-amyloid-β component; and 3) an unstructured acidic C-terminus existing as a random coil [15, 16]. Under physiological conditions, α-synuclein has been shown to exist as a disordered monomer or more controversially as tetramers, while in its pathological form, it is mainly made up of insoluble oligomers and fibrils that have beta-sheet conformations [16, 17]. Despite being the focus of intensive research efforts, the normal physiological function of α-synuclein remains incompletely understood. However, it has been associated with multiple aspects of neuronal functionality, including the trafficking of synaptic vesicles and the formation of the SNARE complex in presynaptic terminals, the molecular chaperoning of targeted proteins, as well as the transport, synthesis, and storage of dopamine [18-20]. Contrastingly, unlike the neuronal roles so far discussed, there exists limited evidence with regards to a specific astrocytic role for endogenous α-synuclein.
Polymorphisms in the SNCA gene that encodes α-synuclein have been identified as risk factors for both sporadic and familial forms of PD [21]. Independent case-control association analyses of the 3-prime and 5-prime region of SNCA have identified strong associations with PD [22]. These findings have also been corroborated by large, unbiased genome-wide association studies and subsequent meta-analyses [23]. Additionally, multiplications (duplications and triplications) and point mutations (e.g., missense mutations including A53T, A30T, and E46K) in the SNCA gene have been strongly implicated in PD [24–27]. The profound genetic association between SNCA and PD is particularly pertinent given that transcriptomic analyses of neurons and astrocytes indicates a shared expression of numerous PD associated genes, including SNCA [28]. The translatability of this genetic redundancy to a specific astrocytic role in PD pathology remains a key unknown. Further research in this area could hold promise in explaining the differential susceptibilities of individuals or astrocytic populations in different brain regions to the aberrant proteostasis of α-synuclein.
NEUROPATHOLOGICAL ASSOCIATION BETWEEN ASTROCYTIC ALPHA-SYNUCLEIN AND PD
Analysis of postmortem brain tissue from PD patients yielded the first clear evidence for α-synuclein pathology in astrocytes. For example, the work of Wakabayashi et al. revealed that α-synuclein-immunoreactive inclusions are frequently found in SNc astrocytes of PD patients [29]. Furthermore, the number of α-synuclein-positive inclusions within SNc astrocytes positively correlated with the spatial severity of nigral dopaminergic neuron loss. These findings were supported by the seminal work of Braak et al. in which PD patient autopsies demonstrated that the distribution of astrocytic α-synuclein inclusions in prosencephalic brain regions closely topographically mirrored the cortical intraneuronal formation of LBs and LNs [30]. They used different anti-α-synuclein antibodies directed against its central domain and subsequently recorded immunoreactivity. These findings temporally paralleled ‘Braak staging’—a method proposed earlier by the group to classify the degree of pathology in PD [31]. Together, these studies indicate that astrocytes reflect intraneuronal LB/LN pathology from a spatiotemporal perspective, providing salience to the notion that there is a role for astrocytic α-synuclein aggregation in PD progression.
Recent studies have also identified an interesting link between the molecular circadian axis (BMAL1-BAG3 axis) in astrocytes and α-synuclein aggregation. Sheehan et al. showed that astrocyte-specific global deletion of the clock gene BMAL1 was sufficient to prevent α-synuclein pathology in vivo and induce astrocyte activation [32]. This was associated with increased astrocytic phagocytosis of α-synuclein by BAG3 (a macro-autophagy chaperone). These findings support previous work by McKee et al., which implicated an astrocytic role in the link between circadian clock function and neurodegeneration. They showed that deletion of astrocytic BMAL1 enhanced astrocyte activation and altered gene expression in mouse models of Alzheimer’s disease [33]. Together these findings highlight the capacity of astrocytic activation to mediate a neuroprotective state through mitigating aberrant α-synuclein accumulation.
ARE ASTROCYTIC AND NEURONAL ALPHA-SYNUCLEIN AGGREGATES DISTINCT?
When assessing astrocytic α-synuclein aggregates, the possible heterogeneity of α-synuclein between neurons and astrocytes must be considered. This is epitomized by the differential efficacies of the methods used to detect intraneuronal versus astrocytic α-synuclein inclusions. For example, unlike intraneuronal LBs, astrocytic LBs are not readily detectable with hematoxylin and eosin histology, silver staining, or phosphorylated serine 129 α-synuclein immunoreactivity [34]. Accessing specific α-synuclein species is also inherently difficult in formalin-fixed and paraffin-embedded human brain sections. Additionally, unlike neurons, astrocytic α-synuclein is not labelled or seldomly labelled when N or C terminal antibodies are used or when formic acid pre-treatment is excluded [30, 35].
Moreover, unlike neuronal LBs, astrocytic LBs do not colocalize with p62/SQSTM1 or ubiquitin. This indicates that astrocytic α-synuclein has undergone extensive modification to alter its ultrastructure, such as truncation. These findings are also in accordance with histological analyses showing that astrocytic inclusions of α-synuclein are more diffusely distributed through the cell body and processes, contrasting to the densely aggregated nature of neuronal LBs/LNs [30]. Given the differences in staining and localization of α-synuclein between astrocytes and neurons, it seems highly probable that astrocytes process α-synuclein differently to neurons. A systematic investigation of α-synuclein by Altay and colleagues has also identified that astrocytic α-synuclein has a unique sequence and post-translational modification signature. By using an expanded set of antibodies, the group revealed that astrocytic α-synuclein has both N- and C-terminal truncations and tyrosine 39 modifications [36]. These findings stress the diversity of α-synuclein pathology in the brain. Research specifically investigating the biochemical heterogeneity between astrocytic and neuronal α-synuclein is currently lacking in literature. However, these findings suggest that astrocytic α-synuclein has potentially been greatly underestimated by previous detection methods [37].
WHAT ARE THE ORIGINS OF ASTROCYTIC ALPHA-SYNUCLEIN AGGREGATES?
While evident that astrocytic α-synuclein accumulation seems to be a part of the pathological sequelae of PD, the next question is with regards to the origin of this aggregated protein. The emerging evidence for this can be broadly categorized into three forms. The first is de novo induction of α-synuclein pathology, the second is phagocytic or pinocytic uptake of extracellular α-synuclein and the third and final is direct transfer from neurons to astrocytes through active processes including tunnelling nanotubules (TNTs) or exosomes. Evidence for each of these mechanisms will be considered in the following sections. Figure 1 summarizes these mechanisms.
Fig. 1
Postulated mechanisms of α-synuclein aggregation in astrocytes. This schematic summarizes four potential mechanisms for α-synuclein accumulation in astrocytes. (1) De novo aggregation of α-synuclein showing α-synuclein transcription, translation, and aggregation in astrocytes, independent of neuronal α-synuclein. (2) Tunnelling nanotubules (TNTs) are actin rich membranous connections between cytosols of individual cells that are capable of bidirectional α-synuclein transfer between astrocytes and neurons. (3) Secretion of exosomes from astrocytes and neurons have been postulated as a mechanism for bidirectional α-synuclein transfer between neurons and astrocytes. (4) Phagocytic or pinocytic uptake of α-synuclein from neurons to astrocytes is widely regarded as the most likely model by which α-synuclein accumulates in astrocytes.
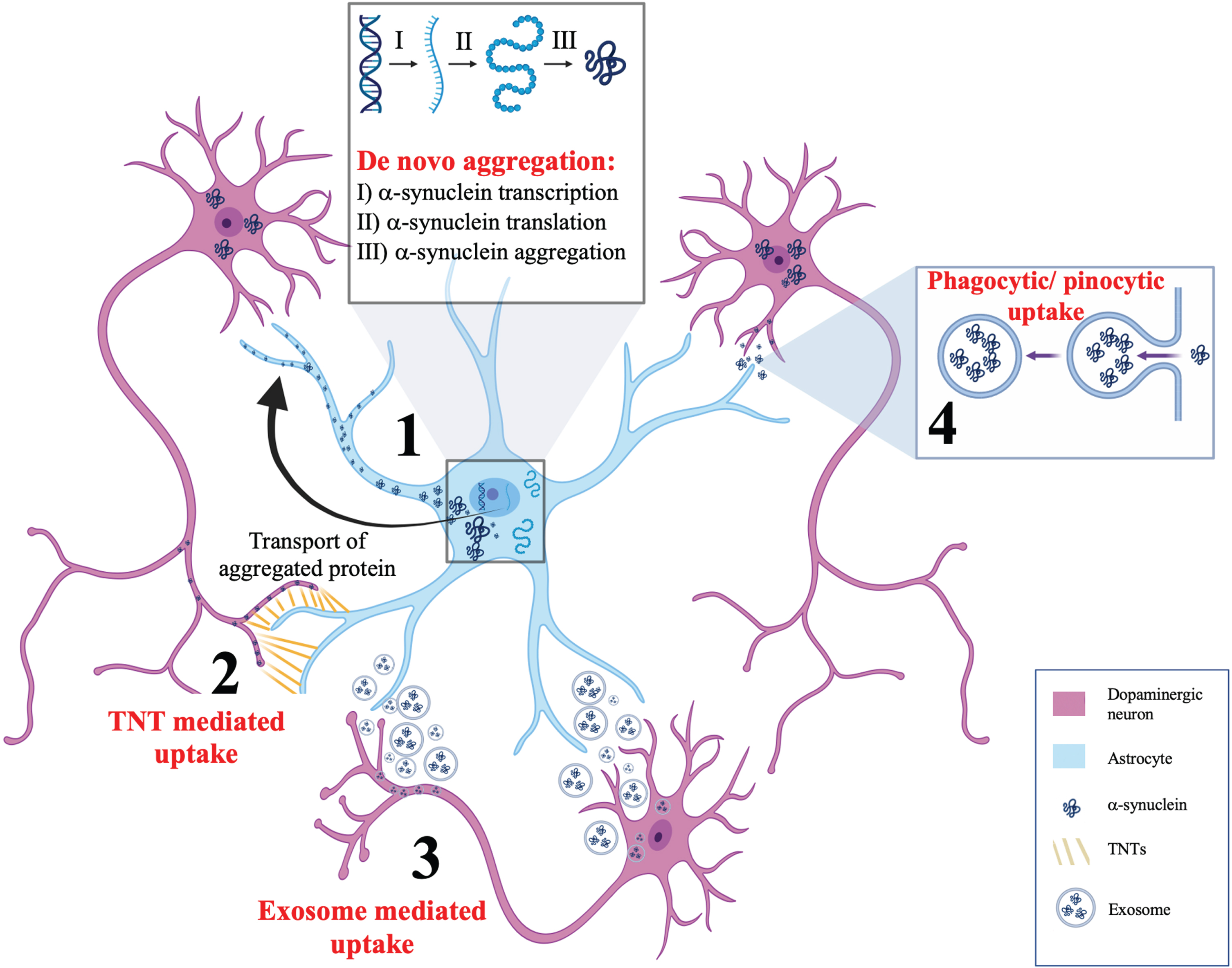
De novo aggregation of α-synuclein in astrocytes
The notion of reactive de novo aggregation of α-synuclein in astrocytes has limited support [28]. Studies supporting this phenomenon have identified α-synuclein immunoreactive protoplasmic astrocytes in regions of the human brain that do not have LBs such as the dorsal thalamus [38]. However, it must be noted that the endogenous expression of α-synuclein in astrocytes is often less than what would be required in order to facilitate de novo aggregation and subsequent transmission [39]. This has been verified by mRNA and protein assays showing that astrocytic cell lines only have small detectable amounts of α-synuclein [40–42]. Nonetheless, one must also consider that endogenous astrocytic α-synuclein levels are not static and instead dynamically vary depending upon the stimuli to which the astrocytes are exposed to. For example, exposure to inflammatory cytokines has been shown to mediate the upregulation of α-synuclein in cultured astrocytes [40]. Thus, it seems most probable that while the upregulation of endogenous α-synuclein is contributory to PD disease progression, it does not represent the leading cause of α-synuclein aggregation in astrocytes.
Phagocytic or pinocytic uptake
Phagocytic or pinocytic uptake of α-synuclein originating from neurons is widely regarded as the most likely means by which astrocytes accumulate α-synuclein. This neuron-to-astrocyte transmission model is exemplified by the seminal work of Lee et al. The group treated primary astrocytes with conditioned media containing α-synuclein aggregates from differentiated SH-SY5Y cells (a human neuroblastoma cell line) and demonstrated that endocytic uptake of α-synuclein occurred by astrocytes, resulting in the formation of aggregates that were resistant to degradation by proteinase K [43]. This work was corroborated by the findings of Tsunemi et al. in which induced pluripotent stem-derived dopaminergic neurons and astrocytes were investigated from healthy people and those with mutations in lysosomal ATP13A2 (a familial PD-associated mutation) [44]. The authors utilized ELISA-based detection of α-synuclein to show that under normal conditions, α-synuclein levels in astrocytes were low but increased when cultured in media containing α-synuclein monomers or oligomers/fibrils. Coculture of astrocytes and neurons led to decreased accumulation of α-synuclein in neurons in conjunction with decreased intraneuronal transfer of α-synuclein. Furthermore, astrocytes had a higher proteolytic capacity than neurons, associated with increased levels of both EEA1 (an early endosomal protein) and LAMP1 (a lysosomal protein) relative to neurons. Given these findings, it seems feasible that long-term exposure to pathological α-synuclein could impair astrocytic lysosomal function, culminating in the accumulation of α-synuclein in astrocytes.
In addition to the in vitro studies above demonstrating directional spread of α-synuclein from neurons to astrocytes, in vivo studies have also demonstrated the ability of astrocytes to transfer α-synuclein to neurons. Gu et al. used inducible transgenic mouse lines that overexpressed human A53T α-synuclein under the astrocyte-specific glial fibrillary acid protein (GFAP) promoter [45]. The mice had diffuse intraneuronal LBs throughout the CNS, demonstrating that astrocyte-to-neuronal spread of pathologic α-synuclein had occurred. Additionally, dopaminergic neuron loss was profound, with the relative susceptibilities of the SNc and ventral tegmental area (VTA) paralleling that of human PD. While highly informative, this study does have caveats. Firstly, the inducible transgenic approach used cannot be considered 100% astrocyte-selective since GFAP can also be expressed in neuronal stem cells [46, 47], thus making it difficult to conclusively discern whether the effects seen are attributable solely to astrocytic overexpression of α-synuclein. Additionally, not all astrocytes express GFAP and it is primarily upregulated during astrogliosis, limiting the translatability of these findings to all astrocytes [48]. Furthermore, A53T α-synuclein forms more protofibrils than wildtype protein, causing one to posit whether the effects seen are specific to the particular form of overexpressed α-synuclein.
TNTs and exosomes
While the bidirectional spread of α-synuclein between neurons and astrocytes is a feasible mechanism for its spread in the brain, the predictable manner by which transmission seems to occur in PD brains [31] is indicative of a mechanism involving α-synuclein transfer between cells via physical conduits. This has been supported by the discovery of TNTs. These thin actin-rich membranous channels connect the cytosols of individual cells and are postulated to bidirectionally transfer α-synuclein between neurons and astrocytes [49]. Rostami et al. showed that astrocytes derived from human embryonic stem cells are able to transfer α-synuclein to healthy astrocytes by TNTs, leading to its subsequent storage in the trans-Golgi region. A limitation of this study was the fluorescent tagging of α-synuclein (Cy3-labeled α-synuclein oligomer) which increased its molecular mass and potentially altered the ability and/or mechanisms by which the protein was transferred between cells. Indeed, tagging of α-synuclein with variants of green-fluorescent protein has been shown to reduce its secretion in vitro [50]. Additionally, the authors show that treatment with latrunculin B pharmacologically inhibit actin polymerization and thus TNT formation. This suggests that the roles of TNT-mediated transmission may have been underestimated, as the inhibitory agent used likely has widespread effects on cell functionality, especially considering the diverse role of actin more generally as an integral component of the cytoskeleton. Rostami and colleagues have since used live cell imaging data to highlight a synergistic effect of astrocytic and microglial α-synuclein processing [51]. Co-cultured astrocytes and microglia were shown to be in constant contact with each other via TNTs. Additionally, microglia were able to attract and clear intracellular protein deposits from astrocytes when attached to their cell membranes. Overall, these findings demonstrate a key role for astrocytic interactions with glia and neurons in the transfer and clearance of α-synuclein.
Exosomes are another means by which α-synuclein is believed to be transmitted between neurons and astrocytes. In vitro studies have demonstrated that exosomes carrying α-synuclein are able to cause neuronal cell death [52]. Emmanouilidou et al. generated wildtype α-synuclein-Tet-off inducible SH-SY5Y cells and showed that α-synuclein is secreted by externalized vesicles in a calcium dependent process. Despite the experiment being performed on neuron-like cells and not on astrocytes, this still indicates a feasible mechanism by which astrocytic α-synuclein pathology could spread. Interneuronal transfer of α-synuclein in PD by means of exosomes is an active field of research, however relatively few studies have provided direct evidence for the transmission of α-synuclein from neurons to astrocytes or vice versa. Venturini et al. demonstrated that exosomes can be released from astrocytic processes prepared from the adult rat cerebral cortex and that they are able to selectively target neurons [53]. While the subcellular origins of the exosomes identified could not be ascertained, given that these extracellular vesicles were able to transport the neuroprotective molecule neuroglobin, their possible role in the transport α-synuclein warrants further investigation.
Overall, while highly informative in highlighting the bidirectional transfer of α-synuclein between neurons and astrocytes, the in vitro and in vivo work discussed demonstrates a significant heterogeneity in both the type of α-synuclein and astrocytes investigated. This is compounded when one notes that PD is known to cause degeneration primarily at the level of the SNc, possibly limiting the translatability of findings from other brain regions.
HOW IS ALPHA-SYNUCLEIN INTERNALIZED BY ASTROCYTES?
The specific mechanisms underlying the internalization of α-synuclein by astrocytes are unclear; however, astrocytic and neuronal mechanisms are widely believed to be dissimilar. The internalization of α-synuclein by neurons can occur through a number of means, including the interaction with cell surface heparin sulphates [54], receptors such as Lag3 [55], or the sodium-potassium transporting ATPase subunit α3 [56]. The relevance of these findings to astrocytes remains uncertain since mass spectroscopy has indicated that astrocytes possess a unique interactome and thus likely process and internalize α-synuclein differently to neurons [57]. Currently no astrocytic receptors that specifically bind α-synuclein assemblies have been identified thereby thwarting development of a therapeutic intervention based off making these receptors unavailable for binding.
ASTROCYTE HETEROGENEITY AND ASTROCYTE-NEURON COUPLING
Despite being the most abundant glial cell, astrocytes have classically been considered as a homogenous cell type. More recently, however, a growing body of literature has supported the notion of astrocyte heterogeneity being indicative of functional diversity [58]. Advancements in techniques to label and modulate astrocyte activity have highlighted not only their vast pleiomorphism but also their varied interactions with a myriad of neuronal types [59]. As such, compartmentalizing this ubiquitous glial cell into subcategories comes with challenges, not least because a single molecular marker linking astrocytic phenotype, morphology and function remains elusive. Nevertheless, it is clear that the traditional delineation of astrocytes into two main classes: protoplasmic (found in grey matter) and fibrous (found in white matter) is entirely insufficient when considering their potential role in neurodegenerative disease.
Interestingly, a robust study by Chai et al. showed that morphological differences were associated with astrocyte specialization within the mouse cortex. Mouse hippocampal astrocytes were shown to occupy a smaller tissue area than striatal astrocytes, with the latter typically occupying territories with more neuronal cell bodies [60]. Fascinatingly, the degree of morphological astrocyte-neuron coupling has also shown to be variable in different brain regions [61, 62]. Furthermore, advancements in sequencing-based approaches have highlighted the transcriptomic heterogeneity between astrocytes in different brain regions [60, 62]. Subregional specialization has already been implicated in the selective degeneration or protection of neurons in PD [63]. Kostuk et al. showed that in vitro elimination of astrocytes exacerbated SN dopaminergic neuron vulnerability to MPP+ (a PD mimetic toxin) and induced a novel neuronal vulnerability in the VTA (which is normally relatively spared in PD). Additionally, VTA and SN dopaminergic neurons were both protected from MPP+ by VTA astrocytes in a dose dependent manner via a secreted molecule. Batiuk et al. have since corroborated these findings and identified five transcriptomically distinct astrocyte subtypes using single-cell RNA sequencing of astrocytes in the cortex and hippocampus of adult mice [64]. Taken together these studies emphasize that subregional astrocytic heterogeneity could underly the variable propensity of neurons in different brain regions to undergo degeneration. Further inter- and intra-regional astrocytic heterogeneity will be necessary to fully appreciate the role of different astrocyte populations in the PD brain.
Astrocytes have already been shown to secrete mediators controlling dendrite growth locally [65]. Indeed, the unique morphological astrocyte-neuron coupling in different brain regions, could underlie the variable propensity of astrocytes to uptake and/or exocytose α-synuclein. Since astrocytic processes directly abut synapses, subcellular compartmentalization (influenced by reciprocal astrocyte-neuron communication) may be key in influencing their functional role within local and wider brain circuits. As such, astrocyte heterogeneity cannot only be considered in the context of isolated glial cells but must also include their varied interactions with neurons in local circuits.
The functional significance of this unique morphological astrocyte-neuron coupling remains unknown. It is likely that the degree of coupling is relevant in the wider spatiotemporal context of computational brain function. A key question that remains is whether morphological astrocyte-neuron coupling is plastic and if so, whether aberrant remodeling leads to neurodegeneration within the nigrostriatal pathway. Disease states such as PD and the aberrant associated accumulation of α-synuclein could contribute, at least to a degree, to this relationship.
CAN ALPHA-SYNUCLEIN ALTER CALCIUM SIGNALS IN ASTROCYTES?
Astrocyte calcium signaling, suggestive of cellular communication, prompted considerable interest within the field. Importantly, these signals are able to occur in a manner both dependent and independent of neuronal activity [66–68]. Given the aforementioned astrocyte-neuron morphological coupling, astrocyte calcium signals appear optimally positioned to modulate feedback and activity within the tripartite synapse and hence local circuits. While discussion of the numerous mechanisms of calcium signal generation are beyond the scope of this review, we instead question the role of these signals in the neurodegenerative PD brain. α-synuclein uptake by astrocytes has been shown to disrupt calcium homeostasis [68, 69]. Importantly, the ramifications of such altered calcium signals must not be underestimated due to coupling of astrocytes via gap junctions [70]. This raises the question of whether α-synuclein uptake by astrocytes could induce aberrant calcium signaling that disrupts local circuitry through widespread adverse effects on astrocyte-neuron coupling in susceptible brain regions. Nanclares et al. have indeed showed that astrocytes in transgenic mice over-expressing human A53T mutant α-synuclein displayed a calcium hyper-excitability, independent of neurotransmitter receptor activation. Furthermore, mice expressing human A53T mutant α-synuclein selectively in neurons did not demonstrate astrocyte calcium hyper-excitability, implicating a cell-autonomous process [71].
The mechanisms underlying astrocytic calcium signal generation are only beginning to be unraveled [72]. Extracellular influx [73] and release from subcellular compartments including the mitochondria [74] and the endoplasmic reticulum [75] have been implicated. Furthermore, different astrocytic regions have shown varied mechanisms of inducing calcium signals [76]. For example, the astrocytic soma has a greater reliance upon metabotropic receptor activity relative to the fine astrocytic processes which rely on mitochondrial calcium fluxes to a greater degree [74, 77]. α-synuclein uptake by astrocytes has already been shown to damage tethering between mitochondria and ER [69] resulting in adverse effects on calcium signaling. Further, aberrant calcium homeostasis in astrocytes has been linked to microglial activation, a well-documented phenomenon in human PD [17, 78]. We postulate that there exists an association between abnormal α-synuclein uptake by astrocytes, the disruption of calcium signaling and microglial activation [72] and believe this will be of increasing research interest in the coming decades.
ASTROCYTIC ALPHA-SYNUCLEIN: THE BALANCE BETWEEN NEUROPROTECTION AND NEURODEGENERATION
Given the in vitro and in vivo findings discussed so far, astrocytes appear to be intrinsically linked to α-synuclein mediated pathology and the progression of PD [79]. The full range of functional consequences of α-synuclein accumulation diverges on a multitude of cellular functions including mitochondrial dysfunction [80], endoplasmic reticulum stress [81], and iron metabolism disturbance [82]. However, one particularly notable aspect of astrocytic α-synuclein accumulation that has been implicated by many of the studies previously mentioned is the induction of neuroinflammation. Figure 2 depicts a neuroinflammatory model for the role of α-synuclein and astrocytes in determining the balance between neuroprotection and neurodegeneration during PD.
Fig. 2
A neuroinflammatory model for astrocytic α-synuclein determining the balance between neuroprotection and neurodegeneration. Model schematic depicting a neuroinflammatory role for astrocytes in maintaining the balance between neuroprotection and neurodegeneration during PD. (1) α-synuclein is secreted by the soma of neurons and internalized by astrocytes. This somatodendritic neuron-astrocyte coupling may be seen in the midbrain. (2) α-synuclein is externalized by the axon of neurons and internalized by astrocytes. This morphological coupling may be seen in the striatum. (3) Bidirectional, efficient transfer of α-synuclein between astrocytes and neurons facilitates neuroprotection. (4) Aberrant α-synuclein accumulation induces astrocyte reactivity. Depending on the load of α-synuclein in the astrocyte, astrocyte reactivity can range from mild (blue astrocyte) to moderate (astrocyte with red process tips) to severe reactivity (red astrocyte). (5) Highly reactive astrocytes will secrete cytokines (red vesicles) resulting in a pro-inflammatory microenvironment and consequent neurodegeneration. In addition, transfer of α-synuclein from astrocytes to neurons can induce neurodegeneration.
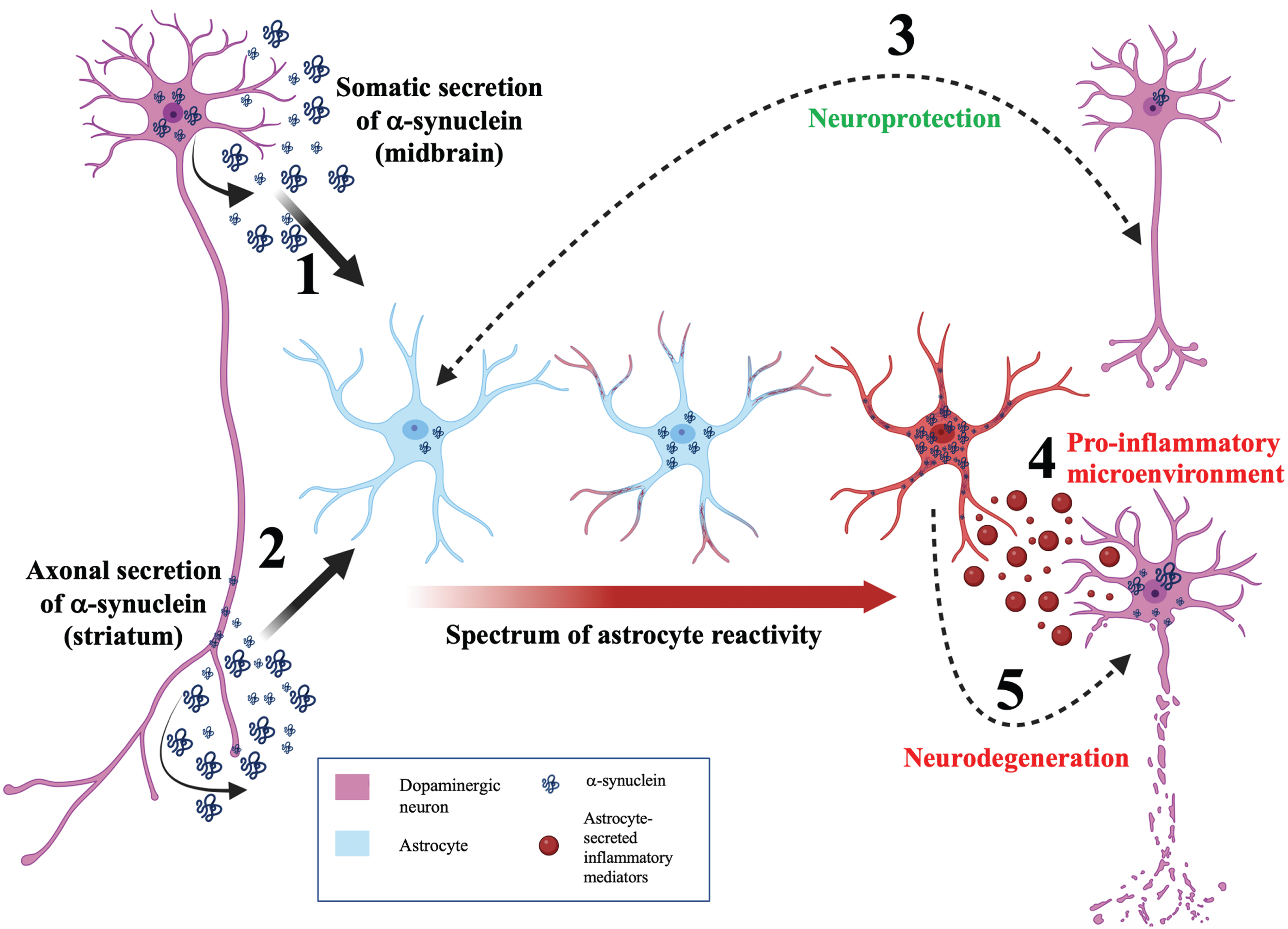
Astrogliosis describes the process of astrocyte activation and is associated with characteristic shift between a spectrum of phenotypes [83]. This is evidenced by astrocytes from induced pluripotent stem cells of PD patients secreting more proinflammatory cytokines (such as IL-6) upon inflammatory stimulation [84]. Furthermore, blocking A1 astrocyte formation has been shown to prolong life in human (A53T) α-synuclein mouse models of PD [85] and to be protective against the loss of dopaminergic neurons. However, the demarcation of astrocytes into discrete A1 and A2 subgroups is highly simplistic and highlights the requirement for further transcriptomic and functional studies to identify the continuous spectrum of astrocytic disease-associated profiles [86]. This raises the fascinating notion that astrocyte heterogeneity may alter PD progression and pathophysiology depending on the spatiotemporal context of the aberrant α-synuclein proteostasis.
A key question that remains in the field is whether neuroinflammation is induced by LBs or neurodegeneration, as both events occur concurrently in the diseased PD brain. The pre-formed fibril (PFF) model has shed some light on this matter [87, 88]. Sorrentino and colleagues characterized the prionoid transmission of α-synuclein by injecting pre-formed exogenous α-synuclein intramuscularly into mice [89]. Neurodegeneration, immune changes, and survival were analyzed spatiotemporally with varying doses of α-synuclein seeds. A clear temporal delineation between the induction of α-synuclein proteostasis (and subsequent motor neuron degeneration) and the induction of immune changes (including astrogliosis) was seen, with the former preceding the latter. Importantly the group observed that astrocytes colocalize with α-synuclein inclusions implicating their role in α-synuclein transfer across the neuroaxis. Furthermore, immune reactivity has been shown in astrocytes derived from induced pluripotent stem cells exposed to α-synuclein fibrillar polymorphs. Russ et al. [90] found that astrocytes exposed to α-synuclein fibrils become antigen presenting cells, associated with changes in expression of HLA genes encoding MHC class I and II proteins. Similarly, Schaser and colleagues used a transgenic mouse PFF model to show that somatic α-synuclein pathology begins rapidly in neurons [91]. However, as time progressed since PFF injection, α-synuclein inclusions were observed in non-neuronal cells (mainly astrocytes). Together these studies provide key insight into the immune reactive role astrocytes play in neurodegeneration and the neuronal origin of α-synuclein in astrocytes.
Based on the mechanisms discussed thus far, a feasible model is that α-synuclein accumulation in astrocytes occurs following a combination of passive uptake by astrocytes and active extrusion from neurons. Initially, astrocytes may internalize and degrade this α-synuclein in a neuroprotective manner, however, a threshold seems to be eventually reached whereby uptake is limited by astrocytic impairment, thereby facilitating the formation of a LB pathology. Hence, the astrocytic response to α-synuclein release from neurons can be likened to a ‘double-edged sword’, which can be either neuroprotective or neurotoxic depending on the capacity of astrocytes to internalize and degrade α-synuclein. If this threshold is reached, induction of inflammatory cytokine production by astrocytes can occur (possibly manifesting as aberrant calcium signaling), establishing an inflammatory microenvironment and leading to sustained neuronal damage. Concomitantly, propagation and prion-like transmission of α-synuclein bidirectionally between neurons and astrocytes exacerbates this process. Notably the multitude of factors defining this ‘threshold’ are variable depending on a vast interplay of numerous poorly understood factors. For example, the ability of astrocytes to mediate a neuroprotective role through α-synuclein clearance is critically defined by their capacity for α-synuclein internalization and susceptibility to undergo astrogliosis, both of which exist on a spectrum determined by the context of the glial microenvironment and their overall functional and computational requirements within the brain. Thus, the ability of an astrocyte to subserve a protective role depends not only on the remit of its particular cell lineage but also on the context within which it functions in the brain.
CONCLUSION
In summary, the targeting of astrocytic α-synuclein inclusions and/or the prevention of their consequences by astrocyte-targeted therapies offers an attractive therapeutic avenue. While intraneuronal approaches provide proof of concept, for example by promoting intracellular degradation of α-synuclein by enhancing autophagic processes [92, 93] the specific targeting of astrocytic α-synuclein aggregates offers a new, relatively unexplored therapeutic approach.
Research investigating the potential contribution of astrocytes to aberrant α-synuclein proteostasis has yielded novel insight into the potential role of these cells in PD pathology. The findings discussed highlight the need for α-synuclein research to extend its remit beyond the traditional neuron-focused scope that has dominated the field for over two decades, so as to encompass the integral role of neuron-astrocyte interactions. Astrocytic functionality seems to operate over a spectrum of neuroprotective and neurotoxic roles, the dynamics of which are determined by their capacity to scavenge and degrade neuron-derived α-synuclein. Outstanding questions and future directions include determining: the trigger for astrocytes to mediate this neurotoxic role, whether astrocyte dysfunction initiates or exacerbates PD pathology and whether the restoration of astrocytic function as a therapeutic approach will be sufficient to preclude aberrant α-synuclein proteostasis. Understanding these ongoing questions requires further research into astrocyte heterogeneity, correlates of α-synuclein internalization capacity, the heterogeneity, and functional differences of different α-synuclein conformations, the astrocytic-specific effects of different physiological states such as cell-activation status and changes in calcium signals, and finally, the ability of current detection methods to accurately identify and characterize astrocytic α-synuclein. Further elucidation of the mechanisms underpinning α-synuclein misprocessing and the neuron-astrocyte interactions involved could reveal a much-needed novel therapeutic target.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This work was partially funded by a research grant from the National Institutes of Health (NIH) research grant, R01NS115809 to RS.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Poewe W , Seppi K , Tanner CM , Halliday GM , Brundin P , Volkmann J , Schrag A-E , Lang AE ((2017) ) Parkinson disease. Nat Rev Dis Primers 3: , 17013. |
[2] | Sveinbjornsdottir S ((2016) ) The clinical symptoms of Parkinson’s disease. J Neurochem 139: Suppl 1, 318–324. |
[3] | Moustafa AA , Chakravarthy S , Phillips JR , Gupta A , Keri S , Polner B , Frank MJ , Jahanshahi M ((2016) ) Motor symptoms in Parkinson’s disease: A unified framework. Neurosci Biobehav Rev 68: , 727–740. |
[4] | Zhai S , Tanimura A , Graves SM , Shen W , Surmeier DJ ((2018) ) Striatal synapses, circuits, and Parkinson’s disease. Curr Opin Neurobiol 48: , 9–16. |
[5] | Chung SJ , Lee HS , Yoo HS , Lee YH , Lee PH , Sohn YH ((2020) ) Patterns of striatal dopamine depletion in early Parkinson disease: Prognostic relevance. Neurology 95: , e280–e290. |
[6] | Madelung CF , Meder D , Fuglsang SA , Marques MM , Boer VO , Madsen KH , Petersen ET , Hejl AM , Løkkegaard A , Siebner HR ((2022) ) Locus coeruleus shows a spatial pattern of structural disintegration in Parkinson’s disease. Mov Disord 37: , 479–489. |
[7] | Guo YJ , Xiong H , Chen K , Zou JJ , Lei P ((2022) ) Brain regions susceptible to alpha-synuclein spreading. Mol Psychiatry 27: , 758–770. |
[8] | Qamhawi Z , Towey D , Shah B , Pagano G , Seibyl J , Marek K , Borghammer P , Brooks DJ , Pavese N ((2015) ) Clinical correlates of raphe serotonergic dysfunction in early Parkinson’s disease. Brain 138: , 2964–2973. |
[9] | Pasquini J , Ceravolo R , Brooks DJ , Bonuccelli U , Pavese N ((2020) ) Progressive loss of raphe nuclei serotonin transporter in early Parkinson’s disease: A longitudinal (123)I-FP-CIT SPECT study. Parkinsonism Relat Disord 77: , 170–175. |
[10] | Giguère N , Burke Nanni S , Trudeau LE ((2018) ) On cell loss and selective vulnerability of neuronal populations in Parkinson’s disease. Front Neurol 9: , 455. |
[11] | Spillantini MG , Schmidt ML , Lee VMY , Trojanowski JQ , Jakes R , Goedert M ((1997) ) α-Synuclein in Lewy bodies. Nature 388: , 839–840. |
[12] | Bennett MC ((2005) ) The role of alpha-synuclein in neurodegenerative diseases. Pharmacol Ther 105: , 311–331. |
[13] | Meade RM , Fairlie DP , Mason JM ((2019) ) Alpha-synuclein structure and Parkinson’s disease –lessons and emerging principles. Mol Neurodegener 14: , 29. |
[14] | Miyazaki I , Asanuma M ((2020) ) Neuron-astrocyte interactions in Parkinson’s disease. Cells 9: , 2623. |
[15] | Jellinger K ((2012) ) The role of α-synuclein in neurodegeneration –An update. Transl Neurosci 3: , 75–122. |
[16] | Eliezer D , Kutluay E , Bussell R Jr. , Browne G ((2001) ) Conformational properties of alpha-synuclein in its free and lipid-associated states. J Mol Biol 307: , 1061–1073. |
[17] | Bartels T , Choi JG , Selkoe DJ ((2011) ) α-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 477: , 107–110. |
[18] | Burré J , Sharma M , Südhof TC ((2014) ) α-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A 111: , E4274–4283. |
[19] | Abeliovich A , Schmitz Y , Fariñas I , Choi-Lundberg D , Ho WH , Castillo PE , Shinsky N , Verdugo JM , Armanini M , Ryan A , Hynes M , Phillips H , Sulzer D , Rosenthal A ((2000) ) Mice lacking alpha-synuclein display functional deficits in the nigrostriatal dopamine system. Neuron 25: , 239–252. |
[20] | Rekas A , Ahn KJ , Kim J , Carver JA ((2012) ) The chaperone activity of α-synuclein: Utilizing deletion mutants to maits interaction with target proteins.Proteins 80: , 1316–1325. |
[21] | Zhang Y , Shu L , Sun Q , Pan H , Guo J , Tang B ((2018) ) A comprehensive analysis of the association between SNCA polymorphisms and the risk of Parkinson’s disease. Front Mol Neurosci 11: , 391. |
[22] | Mueller JC , Fuchs J , Hofer A , Zimprich A , Lichtner P , Illig T , Berg D , Wüllner U , Meitinger T , Gasser T ((2005) ) Multiple regions of alpha-synuclein are associated with Parkinson’s disease. Ann Neurol 57: , 535–541. |
[23] | Nalls MA , Plagnol V , Hernandez DG , Sharma M , Sheerin UM , Saad M , Simón-Sánchez J , Schulte C , Lesage S , Sveinbjörnsdóttir S , Stefánsson K , Martinez M , Hardy J , Heutink P , Brice A , Gasser T , Singleton AB , Wood NW ((2011) ) Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet 377: , 641–649. |
[24] | Polymeropoulos MH , Lavedan C , Leroy E , Ide SE , Dehejia A , Dutra A , Pike B , Root H , Rubenstein J , Boyer R , Stenroos ES , Chandrasekharappa S , Athanassiadou A , Papapetropoulos T , Johnson WG , Lazzarini AM , Duvoisin RC , Di Iorio G , Golbe LI , Nussbaum RL ((1997) ) Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 276: , 2045–2047. |
[25] | Ibáñez P , Bonnet AM , Débarges B , Lohmann E , Tison F , Pollak P , Agid Y , Dürr A , Brice A ((2004) ) Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet 364: , 1169–1171. |
[26] | Ibáñez P , Lesage S , Janin S , Lohmann E , Durif F , Destée A , Bonnet AM , Brefel-Courbon C , Heath S , Zelenika D , Agid Y , Dürr A , Brice A ((2009) ) Alpha-synuclein gene rearrangements in dominantly inherited parkinsonism: Frequency, phenotype, and mechanisms. Arch Neurol 66: , 102–108. |
[27] | Singleton AB , Farrer M , Johnson J , Singleton A , Hague S , Kachergus J , Hulihan M , Peuralinna T , Dutra A , Nussbaum R , Lincoln S , Crawley A , Hanson M , Maraganore D , Adler C , Cookson MR , Muenter M , Baptista M , Miller D , Blancato J , Hardy J , Gwinn-Hardy K ((2003) ) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302: , 841. |
[28] | Zhang Y , Chen K , Sloan SA , Bennett ML , Scholze AR , O’Keeffe S , Phatnani HP , Guarnieri P , Caneda C , Ruderisch N , Deng S , Liddelow SA , Zhang C , Daneman R , Maniatis T , Barres BA , Wu JQ ((2014) ) An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci 34: , 11929–11947. |
[29] | Wakabayashi K , Hayashi S , Yoshimoto M , Kudo H , Takahashi H ((2000) ) NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol 99: , 14–20. |
[30] | Braak H , Sastre M , Del Tredici K ((2007) ) Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol 114: , 231–241. |
[31] | Braak H , Del Tredici K , Rüb U , de Vos RA , Jansen Steur EN , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[32] | Sheehan PW , Nadarajah CJ , Kanan MF , Patterson JN , Novotny B , Lawrence JH , King MW , Brase L , Inman CE , Yuede CM , Lee J , Patel TK , Harari O , Benitez BA , Davis AA , Musiek ES ((2023) ) An astrocyte BMAL1-BAG3 axis protects against alpha-synuclein and tau pathology. Neuron 111: , 2383–2398.e2387. |
[33] | McKee CA , Lee J , Cai Y , Saito T , Saido T , Musiek ES ((2022) ) Astrocytes deficient in circadian clock gene Bmal1 show enhanced activation responses to amyloid-beta pathology without changing plaque burden. Sci Rep 12: , 1796. |
[34] | Sorrentino ZA , Giasson BI , Chakrabarty P ((2019) ) α-Synuclein and astrocytes: Tracing the pathways from homeostasis to neurodegeneration in Lewy body disease. Acta Neuropathol 138: , 1–21. |
[35] | Kovacs GG , Lee VM , Trojanowski JQ ((2017) ) Protein astrogliopathies in human neurodegenerative diseases and aging. Brain Pathol 27: , 675–690. |
[36] | Altay MF , Liu AKL , Holton JL , Parkkinen L , Lashuel HA ((2022) ) Prominent astrocytic alpha-synuclein pathology with unique post-translational modification signatures unveiled across Lewy body disorders. Acta Neuropathol Commun 10: , 163. |
[37] | Hausmann L , Schulz JB ((2021) ) Preface to an editorial controversy on α-synuclein: How good are cellular models?: This is areface for an Editorial controversy between Hilal A. Lashuel “Monitoring alpha-synuclein oligomerization and aggregation using bimolecular fluorescence complementation assays: What you see is not always what you get” and Tiago F. Outeiro “Alpha-synuclein oligomerization and aggregation: A model will always be a model” Read Hilal A. Lashuel’s reply “Alpha-Synuclein oligomerization and aggregation: All models are useful but only if we know what they model”. J Neurochem 157: , 869–871. |
[38] | Song YJ , Halliday GM , Holton JL , Lashley T , O’Sullivan SS , McCann H , Lees AJ , Ozawa T , Williams DR , Lockhart PJ , Revesz TR ((2009) ) Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J Neuropathol Exp Neurol 68: , 1073–1083. |
[39] | Mori F , Tanji K , Yoshimoto M , Takahashi H , Wakabayashi K ((2002) ) Demonstration of alpha-synuclein immunoreactivity in neuronal and glial cytoplasm in normal human brain tissue using proteinase K and formic acid pretreatment. Exp Neurol 176: , 98–104. |
[40] | Tanji K , Imaizumi T , Yoshida H , Mori F , Yoshimoto M , Satoh K , Wakabayashi K ((2001) ) Expression of alpha-synuclein in a human glioma cell line and its up-regulation by interleukin-1beta. Neuroreport 12: , 1909–1912. |
[41] | Booth HDE , Hirst WD , Wade-Martins R ((2017) ) The role of astrocyte dysfunction in Parkinson’s disease pathogenesis. Trends Neurosci 40: , 358–370. |
[42] | Zhang Y , Sloan SA , Clarke LE , Caneda C , Plaza CA , Blumenthal PD , Vogel H , Steinberg GK , Edwards MS , Li G , Duncan JA , 3rd, Cheshier SH , Shuer LM , Chang EF , Grant GA , Gephart MG , Barres BA ((2016) ) Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89: , 37–53. |
[43] | Lee HJ , Suk JE , Patrick C , Bae EJ , Cho JH , Rho S , Hwang D , Masliah E , Lee SJ ((2010) ) Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem 285: , 9262–9272. |
[44] | Tsunemi T , Ishiguro Y , Yoroisaka A , Valdez C , Miyamoto K , Ishikawa K , Saiki S , Akamatsu W , Hattori N , Krainc D ((2020) ) Astrocytes protect human dopaminergic neurons from α-synuclein accumulation and propagation. J Neurosci 40: , 8618–8628. |
[45] | Gu XL , Long CX , Sun L , Xie C , Lin X , Cai H ((2010) ) Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol Brain 3: , 12. |
[46] | Liu Y , Namba T , Liu J , Suzuki R , Shioda S , Seki T ((2010) ) Glial fibrillary acidic protein-expressing neural progenitors give rise to immature neurons via early intermediate progenitors expressing both glial fibrillary acidic protein and neuronal markers in the adult hippocampus. Neuroscience 166: , 241–251. |
[47] | Jurga AM , Paleczna M , Kadluczka J , Kuter KZ ((2021) ) Beyond the GFAP-astrocyte protein markers in the brain. Biomolecules 11: , 1361. |
[48] | Brahmachari S , Fung YK , Pahan K ((2006) ) Induction of glial fibrillary acidic protein expression in astrocytes by nitric oxide. J Neurosci 26: , 4930–4939. |
[49] | Rostami J , Holmqvist S , Lindström V , Sigvardson J , Westermark GT , Ingelsson M , Bergström J , Roybon L , Erlandsson A ((2017) ) Human astrocytes transfer aggregated alpha-synuclein via tunneling nanotubes. J Neurosci 37: , 11835–11853. |
[50] | Lee HJ , Patel S , Lee SJ ((2005) ) Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J Neurosci 25: , 6016–6024. |
[51] | Rostami J , Mothes T , Kolahdouzan M , Eriksson O , Moslem M , Bergström J , Ingelsson M , O’Callaghan P , Healy LM , Falk A , Erlandsson A ((2021) ) Crosstalk between astrocytes and microglia results in increased degradation of α-synuclein and amyloid-β aggregates. J Neuroinflammation 18: , 124. |
[52] | Emmanouilidou E , Melachroinou K , Roumeliotis T , Garbis SD , Ntzouni M , Margaritis LH , Stefanis L , Vekrellis K ((2010) ) Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J Neurosci 30: , 6838–6851. |
[53] | Venturini A , Passalacqua M , Pelassa S , Pastorino F , Tedesco M , Cortese K , Gagliani MC , Leo G , Maura G , Guidolin D , Agnati LF , Marcoli M , Cervetto C ((2019) ) Exosomes from astrocyte processes: Signaling to neurons. Front Pharmacol 10: , 1452. |
[54] | Ihse E , Yamakado H , van Wijk XM , Lawrence R , Esko JD , Masliah E ((2017) ) Cellular internalization of alpha-synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci Rep 7: , 9008. |
[55] | Mao X , Ou MT , Karuppagounder SS , Kam TI , Yin X , Xiong Y , Ge P , Umanah GE , Brahmachari S , Shin JH , Kang HC , Zhang J , Xu J , Chen R , Park H , Andrabi SA , Kang SU , Gonçalves RA , Liang Y , Zhang S , Qi C , Lam S , Keiler JA , Tyson J , Kim D , Panicker N , Yun SP , Workman CJ , Vignali DA , Dawson VL , Ko HS , Dawson TM ((2016) ) Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 353: , aah3374. |
[56] | Shrivastava AN , Redeker V , Fritz N , Pieri L , Almeida LG , Spolidoro M , Liebmann T , Bousset L , Renner M , Léna C , Aperia A , Melki R , Triller A ((2015) ) α-synuclein assemblies sequester neuronal α3-Na+/K+-ATPase and impair Na+gradient. EMBO J 34: , 2408–2423. |
[57] | Shrivastava AN , Redeker V , Fritz N , Pieri L , Almeida LG , Spolidoro M , Liebmann T , Bousset L , Renner M , Léna C , Aperia A , Melki R , Triller A ((2016) ) Data in support of the identification of neuronal and astrocyte proteins interacting with extracellularly applied oligomeric and fibrillar α-synuclein assemblies by mass spectrometry. Data Brief 7: , 221–228. |
[58] | Holt MG ((2023) ) Astrocyte heterogeneity and interactions with local neural circuits. Essays Biochem 67: , 93–106. |
[59] | Yu X , Nagai J , Khakh BS ((2020) ) Improved tools to study astrocytes. Nat Rev Neurosci 21: , 121–138. |
[60] | Chai H , Diaz-Castro B , Shigetomi E , Monte E , Octeau JC , Yu X , Cohn W , Rajendran PS , Vondriska TM , Whitelegge JP , Coppola G , Khakh BS ((2017) ) Neural circuit-specialized astrocytes: Transcriptomic, proteomic, morphological, and functional evidence. , . Neuron 95: , 531–549.e539. |
[61] | Genoud C , Quairiaux C , Steiner P , Hirling H , Welker E , Knott GW ((2006) ) Plasticity of astrocytic coverage and glutamate transporter expression in adult mouse cortex. PLoS Biol 4: , e343. |
[62] | Doyle JP , Dougherty JD , Heiman M , Schmidt EF , Stevens TR , Ma G , Bupp S , Shrestha P , Shah RD , Doughty ML , Gong S , Greengard P , Heintz N ((2008) ) Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell 135: , 749–762. |
[63] | Kostuk EW , Cai J , Iacovitti L ((2019) ) Subregional differences in astrocytes underlie selective neurodegeneration or protection in Parkinson’s disease models in culture. Glia 67: , 1542–1557. |
[64] | Batiuk MY , Martirosyan A , Wahis J , de Vin F , Marneffe C , Kusserow C , Koeppen J , Viana JF , Oliveira JF , Voet T , Ponting CP , Belgard TG , Holt MG ((2020) ) Identification of region-specific astrocyte subtypes at single cell resolution. Nat Commun 11: , 1220. |
[65] | Miller SJ , Philips T , Kim N , Dastgheyb R , Chen Z , Hsieh YC , Daigle JG , Datta M , Chew J , Vidensky S , Pham JT , Hughes EG , Robinson MB , Sattler R , Tomer R , Suk JS , Bergles DE , Haughey N , Pletnikov M , Hanes J , Rothstein JD ((2019) ) Molecularly defined cortical astroglia subpopulation modulates neurons via secretion of Norrin. Nat Neurosci 22: , 741–752. |
[66] | Shigetomi E , Bushong EA , Haustein MD , Tong X , Jackson-Weaver O , Kracun S , Xu J , Sofroniew MV , Ellisman MH , Khakh BS ((2013) ) Imaging calcium microdomains within entire astrocyte territories and endfeet with GCaMPs expressed using adeno-associated viruses. J Gen Physiol 141: , 633–647. |
[67] | Panatier A , Vallée J , Haber M , Murai KK , Lacaille JC , Robitaille R ((2011) ) Astrocytes are endogenous regulators of basal transmission at central synapses. Cell 146: , 785–798. |
[68] | Lia A , Henriques VJ , Zonta M , Chiavegato A , Carmignoto G , Gómez-Gonzalo M , Losi G ((2021) ) Calcium signals in astrocyte microdomains, a decade of great advances. Front Cell Neurosci 15: , 673433. |
[69] | Paillusson S , Gomez-Suaga P , Stoica R , Little D , Gissen P , Devine MJ , Noble W , Hanger DP , Miller CCJ ((2017) ) α-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol 134: , 129–149. |
[70] | Fujii Y , Maekawa S , Morita M ((2017) ) Astrocyte calcium waves propagate proximally by gap junction and distally by extracellular diffusion of ATP released from volume-regulated anion channels. Sci Rep 7: , 13115. |
[71] | Nanclares C , Poynter J , Martell-Martinez HA , Vermilyea S , Araque A , Kofuji P , Lee MK , Covelo A ((2023) ) Dysregulation of astrocytic Ca(2+) signaling and gliotransmitter release in mouse models of α-synucleinopathies. Acta Neuropathol 145: , 597–610. |
[72] | Bancroft EA , Srinivasan R ((2021) ) Emerging roles for aberrant astrocytic calcium signals in Parkinson’s disease. Front Physiol 12: , 812212. |
[73] | Srinivasan R , Huang BS , Venugopal S , Johnston AD , Chai H , Zeng H , Golshani P , Khakh BS ((2015) ) Ca(2+) signaling in astrocytes from Ip3r2(-/-) mice in brain slices and during startle responses in vivo. Nat Neurosci 18: , 708–717. |
[74] | Huntington TE , Srinivasan R ((2021) ) Astrocytic mitochondria in adult mouse brain slices show spontaneous calcium influx events with unique properties. Cell Calcium 96: , 102383. |
[75] | Okubo Y , Kanemaru K , Suzuki J , Kobayashi K , Hirose K , Iino M ((2019) ) Inositol 1,4,5-trisphosphate receptor type 2-independent Ca(2+) release from the endoplasmic reticulum in astrocytes. Glia 67: , 113–124. |
[76] | Verkhratsky A , Semyanov A , Zorec R ((2020) ) Physiology of astroglial excitability. , zqaa. Function (Oxf) 1: , 016. |
[77] | Agarwal A , Wu PH , Hughes EG , Fukaya M , Tischfield MA , Langseth AJ , Wirtz D , Bergles DE ((2017) ) Transient opening of the mitochondrial permeability transition pore induces microdomain calcium transients in astrocyte processes. Neuron 93: , 587–605.e587. |
[78] | McGeer PL , Itagaki S , Boyes BE , McGeer EG ((1988) ) Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38: , 1285–1291. |
[79] | Erustes AG , Stefani FY , Terashima JY , Stilhano RS , Monteforte PT , da Silva Pereira GJ , Han SW , Calgarotto AK , Hsu YT , Ureshino RP , Bincoletto C , Smaili SS ((2018) ) Overexpression of α-synuclein in an astrocyte cell line promotes autophagy inhibition and apoptosis. J Neurosci Res 96: , 160–171. |
[80] | Braidy N , Gai WP , Xu YH , Sachdev P , Guillemin GJ , Jiang XM , Ballard JW , Horan MP , Fang ZM , Chong BH , Chan DK ((2013) ) Uptake and mitochondrial dysfunction of alpha-synuclein in human astrocytes, cortical neurons and fibroblasts. Transl Neurodegener 2: , 20. |
[81] | Jiang P , Gan M , Ebrahim AS , Lin WL , Melrose HL , Yen SH ((2010) ) ER stress response plays an important role in aggregation of α-synuclein. Mol Neurodegener 5: , 56. |
[82] | Cui J , Guo X , Li Q , Song N , Xie J ((2020) ) Hepcidin-to-ferritin ratio is decreased in astrocytes with extracellular alpha-synuclein and iron exposure. Front Cell Neurosci 14: , 47. |
[83] | Liddelow SA , Guttenplan KA , Clarke LE , Bennett FC , Bohlen CJ , Schirmer L , Bennett ML , Münch AE , Chung WS , Peterson TC , Wilton DK , Frouin A , Napier BA , Panicker N , Kumar M , Buckwalter MS , Rowitch DH , Dawson VL , Dawson TM , Stevens B , Barres BA ((2017) ) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541: , 481–487. |
[84] | Sonninen TM , Hämäläinen RH , Koskuvi M , Oksanen M , Shakirzyanova A , Wojciechowski S , Puttonen K , Naumenko N , Goldsteins G , Laham-Karam N , Lehtonen M , Tavi P , Koistinaho J , Lehtonen Š ((2020) ) Metabolic alterations in Parkinson’s disease astrocytes. Sci Rep 10: , 14474. |
[85] | Yun SP , Kam TI , Panicker N , Kim S , Oh Y , Park JS , Kwon SH , Park YJ , Karuppagounder SS , Park H , Kim S , Oh N , Kim NA , Lee S , Brahmachari S , Mao X , Lee JH , Kumar M , An D , Kang SU , Lee Y , Lee KC , Na DH , Kim D , Lee SH , Roschke VV , Liddelow SA , Mari Z , Barres BA , Dawson VL , Lee S , Dawson TM , Ko HS ((2018) ) Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med 24: , 931–938. |
[86] | Khakh BS , Sofroniew MV ((2015) ) Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci 18: , 942–952. |
[87] | Stoll AC , Sortwell CE ((2022) ) Leveraging the preformed fibril model to distinguish between alpha-synuclein inclusion- and nigrostriatal degeneration-associated immunogenicity. Neurobiol Dis 171: , 105804. |
[88] | Gonzalez De La Cruz E , Vo Q , Moon K , McFarland KN , Weinrich M , Williams T , Giasson BI , Chakrabarty P ((2022) ) MhcII regulates transmission of α-synuclein-seeded pathology in mice. Int J Mol Sci 23: , 8175. |
[89] | Sorrentino ZA , Xia Y , Funk C , Riffe CJ , Rutherford NJ , Ceballos Diaz C , Sacino AN , Price ND , Golde TE , Giasson BI , Chakrabarty P ((2018) ) Motor neuron loss and neuroinflammation in a model of α-synuclein-induced neurodegeneration. Neurobiol Dis 120: , 98–106. |
[90] | Russ K , Teku G , Bousset L , Redeker V , Piel S , Savchenko E , Pomeshchik Y , Savistchenko J , Stummann TC , Azevedo C , Collin A , Goldwurm S , Fog K , Elmer E , Vihinen M , Melki R , Roybon L ((2021) ) TNF-α and α-synuclein fibrils differently regulate human astrocyte immune reactivity and impair mitochondrial respiration. Cell Rep 34: , 108895. |
[91] | Schaser AJ , Stackhouse TL , Weston LJ , Kerstein PC , Osterberg VR , López CS , Dickson DW , Luk KC , Meshul CK , Woltjer RL , Unni VK ((2020) ) Trans-synaptic and retrograde axonal spread of Lewy pathology following pre-formed fibril injection in an A53T alpha-synuclein mouse model of synucleinopathy. Acta Neuropathol Commun 8: , 150. |
[92] | Hebron ML , Lonskaya I , Moussa CE ((2013) ) Nilotinib reverses loss of dopamine neurons and improves motor behavior via autophagic degradation of α-synuclein in Parkinson’s disease models. Hum Mol Genet 22: , 3315–3328. |
[93] | Pagan F , Hebron M , Valadez EH , Torres-Yaghi Y , Huang X , Mills RR , Wilmarth BM , Howard H , Dunn C , Carlson A , Lawler A , Rogers SL , Falconer RA , Ahn J , Li Z , Moussa C ((2016) ) Nilotinib effects in Parkinson’s disease and dementia with Lewy bodies. J Parkinsons Dis 6: , 503–517. |


