
Early-Onset Parkinson’s Disease: Creating the Right Environment for a Genetic Disorder
Abstract
Parkinson’s disease (PD) by its common understanding is a late-onset sporadic movement disorder. However, there is a need to recognize not only the fact that PD pathogenesis expands beyond (or perhaps to) the brain but also that many early-onset patients develop motor signs before the age of 50 years. Indeed, studies have shown that it is likely the protein aggregation observed in the brains of patients with PD precedes the motor symptoms by perhaps a decade. Studies on early-onset forms of PD have shown it to be a heterogeneous disease with multiple genetic and environmental factors determining risk of different forms of disease. Genetic and neuropathological evidence suggests that there are α-synuclein centric forms (e.g., SNCA genomic triplication), and forms that are driven by a breakdown in mitochondrial function and specifically in the process of mitophagy and clearance of damaged mitochondria (e.g., PARKIN and PINK1 recessive loss-of-function mutations). Aligning genetic forms with recognized environmental influences will help better define patients, aid prognosis, and hopefully lead to more accurately targeted clinical trial design. Work is now needed to understand the cross-talk between these two pathomechanisms and determine a sense of independence, it is noted that autopsies studies for both have shown the presence or absence of α-synuclein aggregation. The integration of genetic and environmental data is critical to understand the etiology of early-onset forms of PD and determine how the different pathomechanisms crosstalk.
INTRODUCTION
Classically, Parkinson’s disease (PD) is considered a progressive, age-related neurodegenerative disorder of the elderly [1]. While this dogma is driven by the onset of motor symptoms, well-characterized prodromal clinical manifestations can precede motor deficits, and neuropathological protein aggregation likely begins decades before the onset of clinical symptoms given recent studies in Alzheimer’s disease (AD) and PD [2–4]. With the average age-at-onset of approximately 65 years being based on the appearance of motor symptoms, this raises the interesting question of whether prodromal clinical manifestations and early underlying molecular etiology justify the reclassification of PD as a ‘early-onset’ disease.
Defining early-onset Parkinson’s disease
Early-onset PD (EOPD), sometimes referred to as ‘young-onset Parkinson’s disease’, lacks a consensus definition, but generally refers to patients who develop the classical PD motor symptoms after 21 years old and before 50 years of age [5, 6]. Patients presenting with PD motor symptoms earlier than 21 years old are classified as ‘juvenile PD’ and usually present with atypical and more severe clinical features [7]. EOPD and juvenile PD cases together comprise between 5-15% of all patients with PD, and are more frequent in males than females (1.5-1.7 : 1) [8, 9]. Age of onset is not the only differential feature between EOPD and the more typical patients with late-onset PD (LOPD; age of onset > 60-65 years) as there are significant differences in disease progression, clinical features, response to medication, and postmortem pathology. Historically, EOPD cases have been reported to be more likely familial (∼20-30% of cases are familial) than sporadic LOPD (where 10-15% report family history) [10]. The discovery of novel EOPD genetic and environmental causes and their potential interplay may also still help to further elucidate the pathological mechanisms of the more common late-onset patients with PD. Like typical LOPD, EOPD is clinically and pathobiologically heterogeneous and the most current genetic and environmental etiologies of EOPD will be discussed herein (Fig. 1).
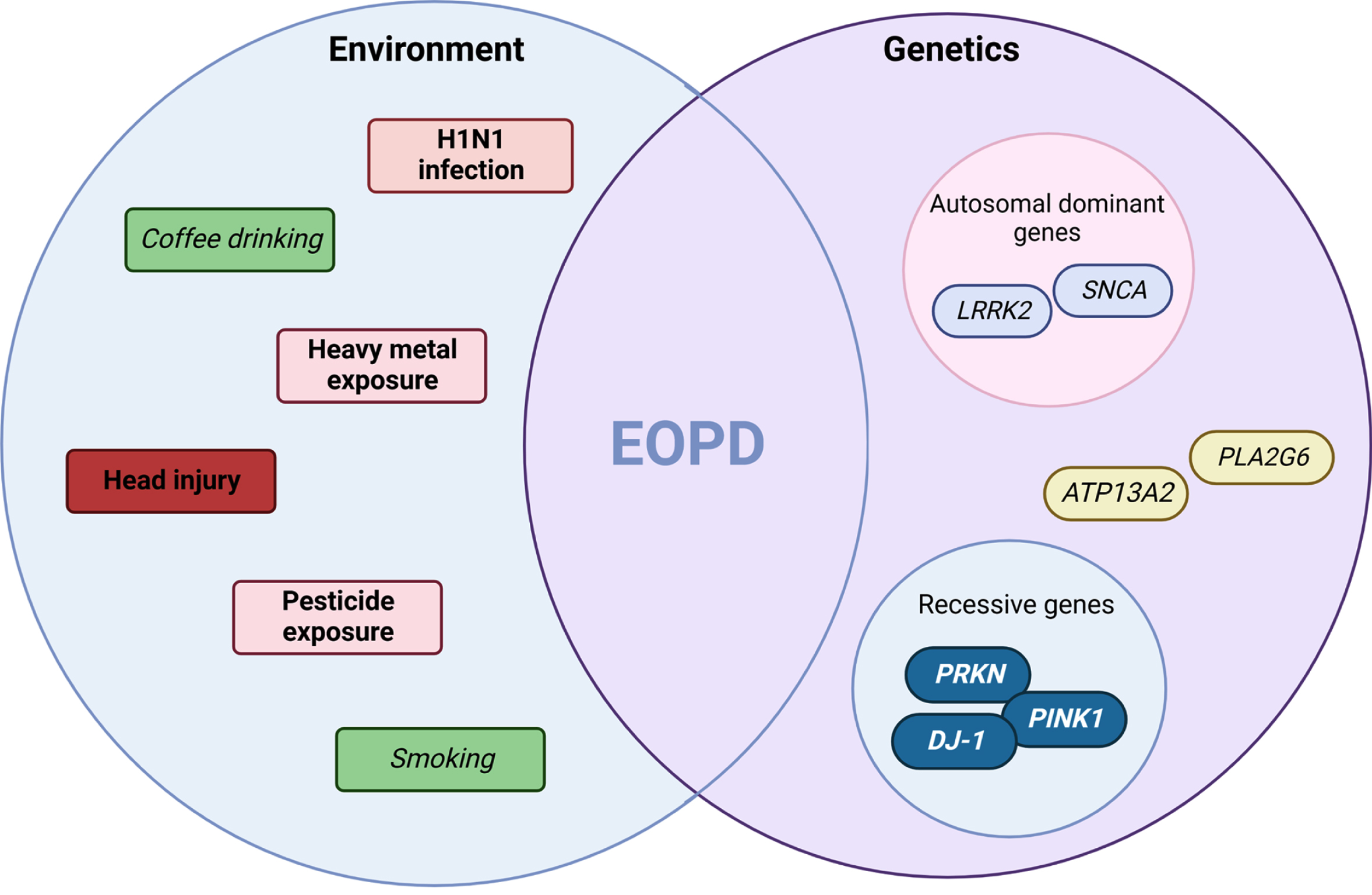
Fig. 1
Environment and genetic interplay in EOPD. An interpretation of the multi-hit hypothesis on early-onset Parkinson’s disease suggests it is the combination of environmental agents acting on the background of genetic determinants that pre-disposes the individual to disease. Highlighted are both risk (pink-red or bold) and protective (green or italics) factors. This concept in early-onset forms of disease may be due to stronger effects sizes from exposure or higher penetrant genetic mutations observed in specific genes (e.g., SNCA or PRKN) and molecular pathways (e.g., mitophagy). (Created by BioRender.com)
Clinical characteristics and neuropathology of EOPD
It is clear that for PD in general the clinical manifestation of symptoms is heterogeneous, and a number of clinical subtypes have been described [11, 12]. With this in mind, a diagnosis of EOPD is based on the presence of clinical manifestations that define parkinsonism before a predefined age, and it remains unclear whether using age at onset as a cut-off produces a more homogeneous patient group [13, 14]. A number of studies confirm no significant difference for tremor and bradykinesia between EOPD and LOPD; however, EOPD patients are less likely to present with gait disturbances [15], possibly due to a lower burden of comorbidities affecting gait either due to the younger age of onset, or a slower disease progression. Dystonic posture and post-exercise gait disorders have been reported frequently EOPD as the first symptom [16]. As EOPD and LOPD progress, differences can become more apparent with rigidity, dystonia, dyskinesia, painful cramps, and sexual dysfunctions being more frequent in EOPD than LOPD [17]. By contrast, EOPD patients have a lower risk of developing dementia compared to LOPD patients [17, 18], perhaps again reflecting the general overall health of the individual, e.g., better vascular health and potentially different mechanism of neuropathology progression between EOPD and LOPD. In addition, early studies suggested preservation of cardiac sympathetic innervation in patients with PRKN-related disease, these findings have not been observed consistently across EOPD or PRKN-patients [19–21].
The reasons for these clinical differences are not entirely clear, but age, environment, or underlying genetic variants likely play important roles. Additionally, diverse distribution of α-synuclein-positive Lewy body (LB) pathology may result in different clinical phenotypes. In LOPD, although remaining controversial, α-synuclein pathology is believed to play an important role in dopaminergic neuronal loss and the subsequent onset and progression of motor symptoms. However, patients diagnosed with EOPD can demonstrate heterogeneous neuropathology, which may or may not include α-synuclein deposits. Specifically, some patients with genetic forms of EOPD harboring familial mutations in PINK1 or PARKIN (PRKN) have been shown to mostly lack LB pathology [22–35]. The heterogeneity in α-synuclein pathology raises the question of whether the lack of LB pathology is due to the younger age or whether distinct pathologic mechanisms exist that alter buildup and distribution of the pathology. Interestingly, classical LB pathology in several PINK1 and PRKN cases was observed although confined to the brainstem [31, 36] suggesting the degenerative processes taking place in these forms of parkinsonism may differ from LOPD. The predominant nigral degeneration in the presence of few or no LB deposits led to these cases being defined as “pure nigropathies” with restricted pathology and underlying mitochondrial dysfunction as the primary substrate [37]. This hypothesis is further supported by the known role of environmental toxins (e.g., MPTP) in inducing mitochondrial-related parkinsonism which, together with the sporadic nature of disease, was responsible for PD being described as the archetypal non-genetic disease.
While PD is pathologically defined by the presence of LBs, the biological origin, mechanisms of formation, and role(s) in disease remain unclear to date. Interestingly, recent accumulating data suggest that membranous organelles together with α-synuclein are the main components of LB and that the process of LB formation itself could be one of the main drivers of neurodegeneration, rather than simple α-synuclein fibril formation [38–40]. Of note, alterations in lipid metabolism and trafficking as well as mitochondrial and autophagic-lysosomal function, emerge as common biological pathways that can contribute to LB formation [41], and may provide further links between the various genetic and environmental factors in EOPD and/or LOPD. These studies have raised more questions than answers in regard to whether there are specific subtypes of disease, and whether EOPD represents a distinct entity compared to LOPD.
Environmental determinants
Exposure to environmental agents may modify the risk of developing PD with the same environmental variables affecting LOPD believed to also play a role in EOPD. Studies specifically investigating the association between environmental exposure and EOPD found that only a positive history of head injury seemed to increase the risk of EOPD compared to LOPD [42]. Interestingly, smoking was found to be protective against PD in general with a stronger association with a younger age at onset [43]. Coffee consumption has been reported as possible protective factors in the development of PD; however, the role of coffee intake has not been extensively explored in EOPD yet.
Historically, the H1N1 influenza pandemic of 1917-1918 has been considered a possible environmental cause of parkinsonism. In particular, the development of von Economo encephalitis was directly caused by H1N1 [44]. In addition, results of a study on trends of incidence and birth cohorts of PD reported a slight but significant increase of future risk of PD in men with a date of birth between 1915-1924 [4]. Recently, exposure to H5N1 has been considered a possible PD causative infection in experimental model of PD [45]. It is possible that systemic viral (or potentially bacterial) infection represents the first “hit” of the dual-hit theory of PD [46]. Additional definitive studies will be needed in the future to explore the role of the current SARS-CoV-2 (COVID-19) pandemic on PD risk although studies have shown that neurodegenerative processes are likely[47–49].
The discovery of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced parkinsonism dramatically advanced the understanding of the pathophysiology of parkinsonism and increased the understanding of possible risk factors for EOPD [50]. MPTP-induced mitochondrial damage raises the possibility that cases of non-drug induced EOPD have mitochondrial dysfunction as the underlying pathogenesis [51]. On the other hand, in the genetic and non-genetic forms of EOPD, the involvement of environmental agents seems to cause less acute and more long-lasting effect later in life. Paraquat and a number of pesticides have been identified as a risk for PD either as a direct exposure or via polluted water [52, 53]; however, the information on EOPD is limited. Certainly, the duration and amount of exposure has an important weight in the development of PD, together with an individual (genetic) predisposition.
Therefore, in EOPD the environmental risk factors (alone or acting in cluster) may need a stronger individual genetic predisposition to contribute to EOPD. In fact, a study reported an increase risk of developing PD and having a more severe disease in farmers that had head trauma, exposure to pesticides and a genetic susceptibility allele of the SNCA REP1 expansion (common variation at the SNCA locus is hypothesized to increase expression of α-synuclein protein) but not in farmers with head trauma, exposure to pesticides and no REP1 risk allele [54, 55]. If this is valid for LOPD, we may argue that a stronger individual (genetic) predisposition is needed in EOPD and/or a stronger exposure to environmental risk factors.
ENVIRONMENT-GENE INTERACTIONS
The majority of PD cases are idiopathic, with inherited genetic forms accounting for approximately 10% of PD cases [56]. Many have postulated that PD arises from an interaction between genetic susceptibility and environmental exposures [57]. One of the most studied interactions in PD is between smoking and monoamine oxidase B (MAO-B) enzyme. As noted earlier, smoking has been shown to be protective against PD; however, different polymorphisms in the gene encoding MAO-B may have opposite effects on PD risk in smokers [58]. Another example of gene-environment interaction comes from a genetically predetermined inability to metabolize organophosphates and pesticides that in turn may result in an increased risk of PD [59]. Chronic exposure to rotenone, a mitochondrial complex I inhibitor and pesticide, may alter the genetic expression and production of α-synuclein and DJ-1, leading to PD pathogenesis [60]. In addition, chronic exposure to heavy metals and pesticides is associated with a younger onset of PD [61]. It is also possible that genetic factors only become relevant when placed in the context of an environmental agent and these genes may be more complicated to identify than those that have been identified as monogenic drivers of disease.
Genetics determinants
PD is a genetically heterogeneous disease with family members who carry the same genetic mutation presenting with varying symptomatic manifestation (e.g., age of onset, severity of symptoms, and disease progression rate), suggesting that even less complex Mendelian and monogenic cases of the disease rely on environmental factors or additional genetic modifiers/risks.
Currently, pathogenic variants of variable penetrance have been identified in a handful of genes widely accepted as major determinants or strong risk factors for inherited forms of parkinsonism and PD including SNCA, PINK1, PRKN, DJ-1, LRRK2, VPS35, VPS13C, FBXO7, PLA2G6, SYNJ1, ATP13A2, RAB39B, and GBA [62]. Genes known to cause hereditary ataxias (ATXN2, ATXN3, and FMR1) and frontotemporal dementia (C9ORF72, GRN, MAPT, and TARDBP) have also been suggested to influence PD as a secondary phenotype [63]. Additional genes including UCHL1, HTRA2, GIGYF2, CHCHD2, DNAJC13, TMEM230, LRP10, PODXL, and NUS1 have been linked to isolated familial PD cases but are either too rare for proper validation/replication studies or lack definitive evidence of pathogenicity [62–65].
Some genetic mutations are associated with atypical presentations of EOPD, with extrapyramidal symptoms as part of a more complex phenotype. Various forms of variable dystonia-parkinsonism syndromes associated with early onset parkinsonian features have been described; mutations in the ATP13A2 (PARK9) gene can cause a constellation of symptoms including supranuclear up-gaze palsy, oculogyric dystonic spams, facial-faucial-finger mini-myoclonic and autonomic dysfunction along with more typical parkinsonian signs and symptoms [66]. Mutations in the PLA2G6 (PARK14) might be associated with levodopa-responsive EOPD along with marked truncal hypotonia, optic atrophy, cerebellar ataxia and mental retardation [67]. A similar pattern can be observed in those genetic disorders clinically defined as spinocerebellar ataxias (SCA) (e.g., SCA 2, SCA 3, SCA 8, and SCA 17) in which parkinsonism may occur as a prominent sign early in life, and signs of cerebellar dysfunction are the main findings [68, 69]. Genetic forms of EOPD usually show a good response to Levodopa, but a higher risk of developing dyskinesia, with a shorter latency between treatment initiation and motor fluctuations onset [70, 71]. Interestingly, EOPD patients may present a slower disease progression and longer survival compared to LOPD [72, 73].
The identification of genes related to PD has provided useful insights into specific biological pathways involved in PD pathobiology and progression. Mutations in confirmed PD genes mainly alter the dopaminergic neuron maintenance/homeostasis through various mechanisms including, synaptic vesicle protein defects, alterations of mitochondrial quality control, and endosome-autophagy-lysosome dysfunctions [74–76]. For EOPD, two genetic drivers of disease are currently considered: 1) the three recessive genes PINK1, PRKN, and DJ-1 and 2) the autosomal dominant genes SNCA and LRRK2.
SNCA and LRRK2: from early to late, from familial to sporadic
As the presence of LBs is considered the defining pathological confirmation of a PD diagnosis [77], the most relevant genetic cause of EOPD is the highly penetrant mutations observed in the SNCA gene encoding α-synuclein. SNCA point mutations and genomic multiplications cause autosomal dominantly inherited forms of EOPD with variable penetrance and severity [78]. SNCA coding variation is rare and to date only a handful of pathogenic autosomal dominant missense mutations have been discovered along with genomic multiplications of the entire gene [79]. Patients who carry additional copies of the SNCA gene (e.g., duplications and triplications) often have an earlier onset and more severe disease phenotypes that appear to be dose-dependent [78]. Interestingly, EOPD patients with the α-synuclein p.A30P substitution have a slightly later disease onset than patients carrying p.A53T, whereas patients with the highly penetrant p.E46K mutation are more likely to develop cognitive symptoms and dementia within two years after the PD diagnosis [80].
The normal function of α-synuclein remains unclear but it is thought to play several roles in synaptic regulation and neurotransmitter release [81]. Pathologically, overexpression of α-synuclein or folding defects/toxic gain of function mutations lead to the accumulation and aggregation of this protein in the pre-synaptic termini of neurons. This leads to a cascade of events that includes neurotransmission inhibition, inhibition of exocytosis, cell stress, and cell death, which then spreads in a prion-like manner through the synaptically connected brain regions at variable rates consistent with Braak staging [82]. However recent links to other genes related to PD also link α-synuclein levels to mitochondrial function (PINK1 and PRKN) and endosomal-lysosomal pathways (LRRK2) [74, 75].
Leucine-rich repeat kinase 2 (LRRK2) is a large protein with multiple domains and GTPase and kinase activity that is widely expressed in different tissues [83–85]. Mutations in LRRK2 are the most common genetic cause of autosomal dominant PD, where the most common disease-causing mutant is likely a gain of function, LRRK2 p.G2019S [86]. Even though LRRK2 variants are generally associated with LOPD, clinical presentation is heterogeneous with a number of reports of EOPD patients harboring LRRK2 mutations [87]. The clinical heterogeneity observed seems to be dependent on sex, ethnic background and familial history [88]. Reduced penetrance is becoming a better recognized feature of late-onset complex disease and is likely a combination of risk and protective determinants (both environmental and genetic). Interestingly, large genome-wide association studies have confirmed common risk variants with small effect size in both SNCA and LRRK2 that contribute to overall disease risk of PD [89].
PINK1 and PRKN: regulation of mitochondria quality control
The high energy demand of dopaminergic neurons means a large amount of reactive oxygen species (ROS) is produced in these neurons. As ROS are toxic to mitochondria and to the cell, it is essential that the mitochondrial quality control is efficient in these neurons [75, 90]. Mitochondrial damage and clearance defects are relevant for neurodegenerative disorders in general, and EOPD in particular has been hypothesized to result from a breakdown of these quality control pathways in some cases [91]. This hypothesis is in part driven by the identification of recessive loss-of-function mutations in two genes, PINK1 and PRKN [92, 93], and moreover their functional linkage into a stress-activated mitophagy pathway - the degradation of selectively damaged mitochondria via the autophagy-lysosome system (Fig. 2) [75].
Fig. 2
Mitophagy pathway. Two of the genes that have been identified to drive early-onset recessive forms of Parkinson’s disease have been demonstrated to play a major role in mitochondrial quality control measures of healthy mitochondria (A). The recessive loss of function for either the PINK1 or “PARKIN (PRKN)” proteins disrupts the normal mitochondrial surveillance process which induces the process of mitophagy for clearance of the damaged organelle (B). This dysfunction is believed to lead to the accumulation of damaged mitochondria eventually resulting in neuronal death. (Created by BioRender.com)
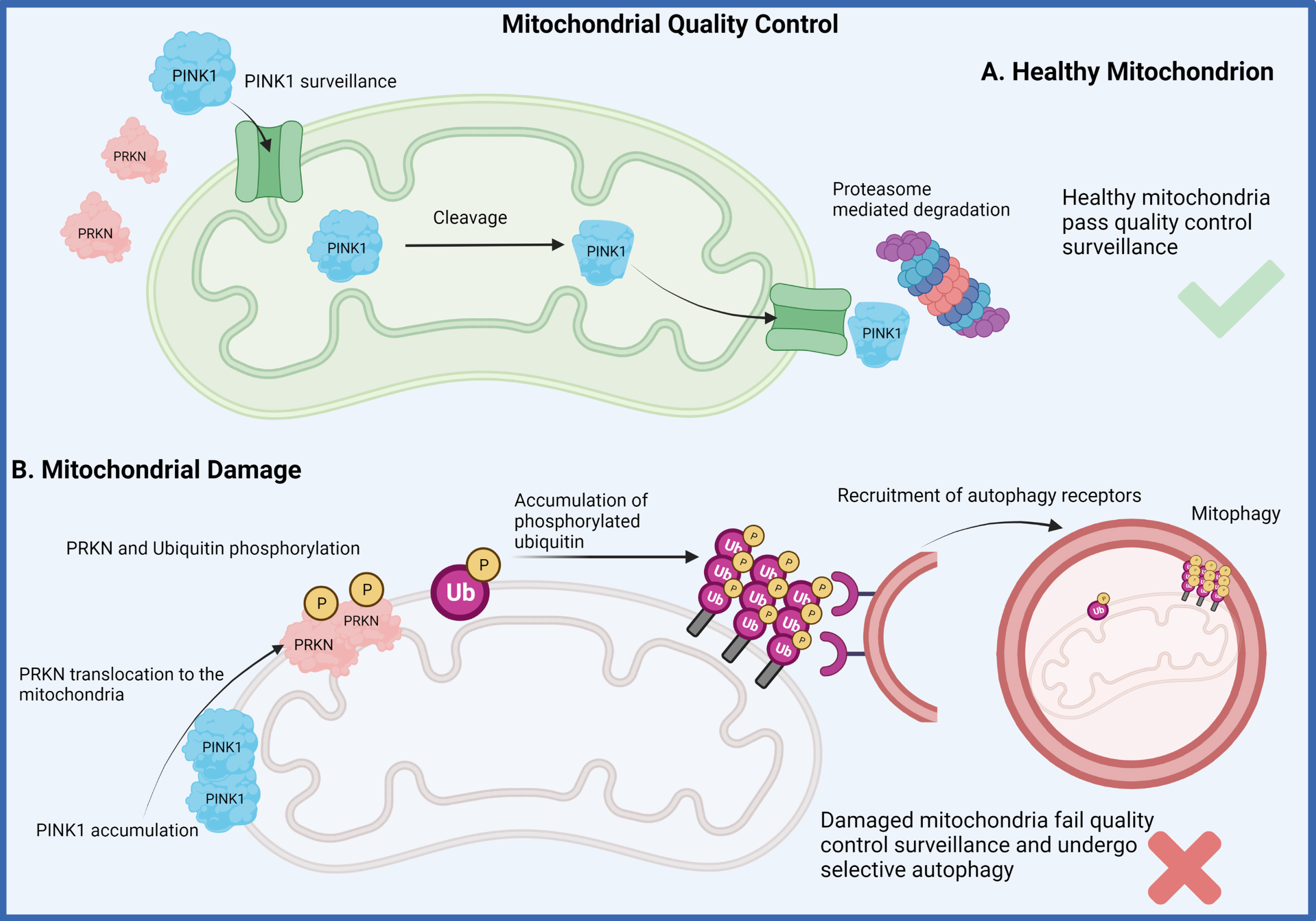
PINK1 is a mitochondrial ubiquitin (Ub) kinase that constantly surveilles organelle health through its regulated import into healthy organelles and rapid cleavage and degradation therefrom. When mitochondrial damage is present though, PINK1 is no longer imported but locally accumulates on the outer mitochondrial membrane where it recruits and activates the E3 Ub ligase PRKN from the cytosol to jointly label and target damaged mitochondria for degradation [75]. PRKN encodes for PARKIN, which was first shown to function in the Ub-proteasome degradation pathway in the early 2000 s [94, 95]. Experiments in drosophila were the first to genetically link PINK1 and PARKIN into a linear pathway associated with mitochondrial health and function [96, 97]. Yet, the biological link between PINK1 and PARKIN and their roles in mitophagy were only established later in human cell culture [98–101]. Homozygous loss of function mutations in PINK1 and PRKN cause EOPD, with mutations in PRKN representing the most common genetic defect in EOPD patients [93, 102]. Although the symptoms are indistinguishable from sporadic cases, multiple post-mortem brains with PRKN mutations do not harbor α-synuclein aggregation and LB pathology [103] and only one report suggests PINK1-linked Lewy pathology [31].
The mean age at onset of patients with PINK1 mutations is 33 years old; however, the onset and severity of the symptoms is highly variable. For example, the youngest patient diagnosed with PINK1-related disease was just 5 years old, whereas some PINK1 patients first present with symptoms at 61 years of age or older [104, 105]. This extreme example suggests that there is more to the picture than the PINK1 loss-of-function variants, with a possibility of population-specific phenotypic expressions of certain gene mutations. However, it is also clear that the extent and duration of mitochondrial stress and damage play critical roles in the activation of the PINK1-PARKIN mitophagy pathway. Moreover, proper flux through the autophagy-lysosome system is crucial for swift and effective degradation and recycling of the mitochondrial cargo.
Interestingly, even though the function of DJ-1 may not be directly involved in the PINK1-PARKIN mitophagy pathway, there is growing evidence that loss of function variants in DJ-1 do in fact interact in parallel with the mitophagy pathway during disease progression [106, 107]. DJ-1 loss of function variants were first described in 2 consanguineous families from geographically isolated regions in the Netherlands and Italy [108], and patients with DJ-1 loss of function variants are clinically similar to PINK1-PRKN patients [109]. Multiple functions have been attributed to DJ-1 since it was first identified as an oncogene, including as a chaperone with protease activity, a transcriptional regulator, and a role in the maintenance of mitochondrial homeostasis [110]. Most of those functions could not be demonstrated in vivo so far, until a recent study in DJ-1 knock-out rat and mice brains demonstrated that PTEN inhibition and AKT signaling seem to be the main functions of DJ-1 in the progression of neurodegeneration [107]. AKT signaling promotes the attachment of hexokinase1 and 2 (HK1 and HK2) to the mitochondrial outer membrane, which is an important step that aids the function of the PINK1-PARKIN pathway. DJ-1 absence in rat and mice brain cells have displayed delocalized HK1 from mitochondria to the cytosol, which in turn may disrupt the efficiency of mitophagy and increase oxidative stress and cell death.
There is controversy whether PINK1 and PRKN deficiency and genetic variation might also contribute to risk for development of PD later in life, so the targeting of this pathway might confer therapeutic value to both EOPD and LOPD [64, 75, 111–114]. With the rise in Next Generation Sequencing and characterization of gene expression regulators it will be possible to determine whether EOPD and LOPD patients who carry heterozygous PINK1-PRKN deleterious variants also carry additional regulatory variants that could lower the expression efficiency of these proteins under stress. However, it is also becoming clear now that not all mutations are alike and thus genetic and functional analyses must be performed on the single variant level in order to truly determine the pathogenicity [115]. Yet, the amount of mitochondrial damage and the critical threshold functions of PINK1 and PARKIN in that context may also vary from individual to individual and even from cell to cell, further impeding a pure genetic approach.
AGE-AT-ONSET MODIFIERS
Patients with EOPD have considerable age-at-onset (AAO) variation. Genetic variation and sex differences very likely contribute to AAO [116]. Multiple studies have attempted to identify genes or variants that modify the AAO of PD with very little success, likely due to the low sample size of their datasets. The largest aggregate of PD genome wide association studies (GWAS) to date includes a meta-analysis of 17 datasets, with 37,688 PD patients, 18,618 UK Biobank proxy-cases and 1.4 million controls. This study identified 90 independent common genetic risk factors in individuals of European ancestry [89]. Currently, there are efforts to generate a predictive genetic risk score (GRS) that is based on cumulative genetic risk, this may also help determine AAO prediction, as it has been shown that a subset of risk variants overlap with AAO modifiers [117]. A Mendelian randomization portal that uses thousands of GWAS data is now available for the research community in hopes to identify modifiers of disease progression (https://pdgenetics.shinyapps.io/MRportal/) [118].
GBA: lysosomal pathway central in PD pathobiology
Rare coding variants in GBA are one the best established risk and AAO modifiers of PD. Homozygous detrimental variants in GBA cause Gaucher’s disease (GD) which is the most common lysosomal storage disease worldwide [119]. Patients who are heterozygous for GD-causing variants have a 10-30% increased chance of developing PD by 80 years of age; this reflects a 20-fold higher risk compared to the general population [120]. PD patients with GBA mutations may exhibit typical manifestations of PD but have a higher risk for dementia with Lewy bodies than patients with LOPD [121]. Patients with homozygotic GBA mutations seem to be more susceptible to psychiatric symptoms such as psychosis, hallucinations, depression, anxiety, and apathy, although this has not been confirmed in all clinical studies [120]. Different GBA mutations result in different risks of developing PD [122]. It is proposed that patients with known disease-causing variants in other PD genes (e.g., SNCA, LRRK2, PINK1, and PRKN) that carry GBA mutations may develop an earlier onset and more severe disease [123, 124]. A recent study showed that PD and Lewy body dementia cases with GBA variants often carry a substantial number of other PD associated risk variants, which confirms the age of onset modifier hypothesis, and further highlights the importance of the lysosomal pathway in PD pathobiology (Fig. 3) [117, 125]. In addition, heterozygotic carries of GBA mutations, may also carry a number of variants of uncertain significance that can further provide heterogeneity to the clinical manifestations, but are likely contributing to the neurometabolic process responsible for the symptoms and disease progression. The interplay between AAO modifier genes with the rest of the genome, epigenome and environmental risk/protective factors is not understood yet, but likely it is one of the major contributors to the endophenotypic variation of LOPD and more specifically to EOPD.
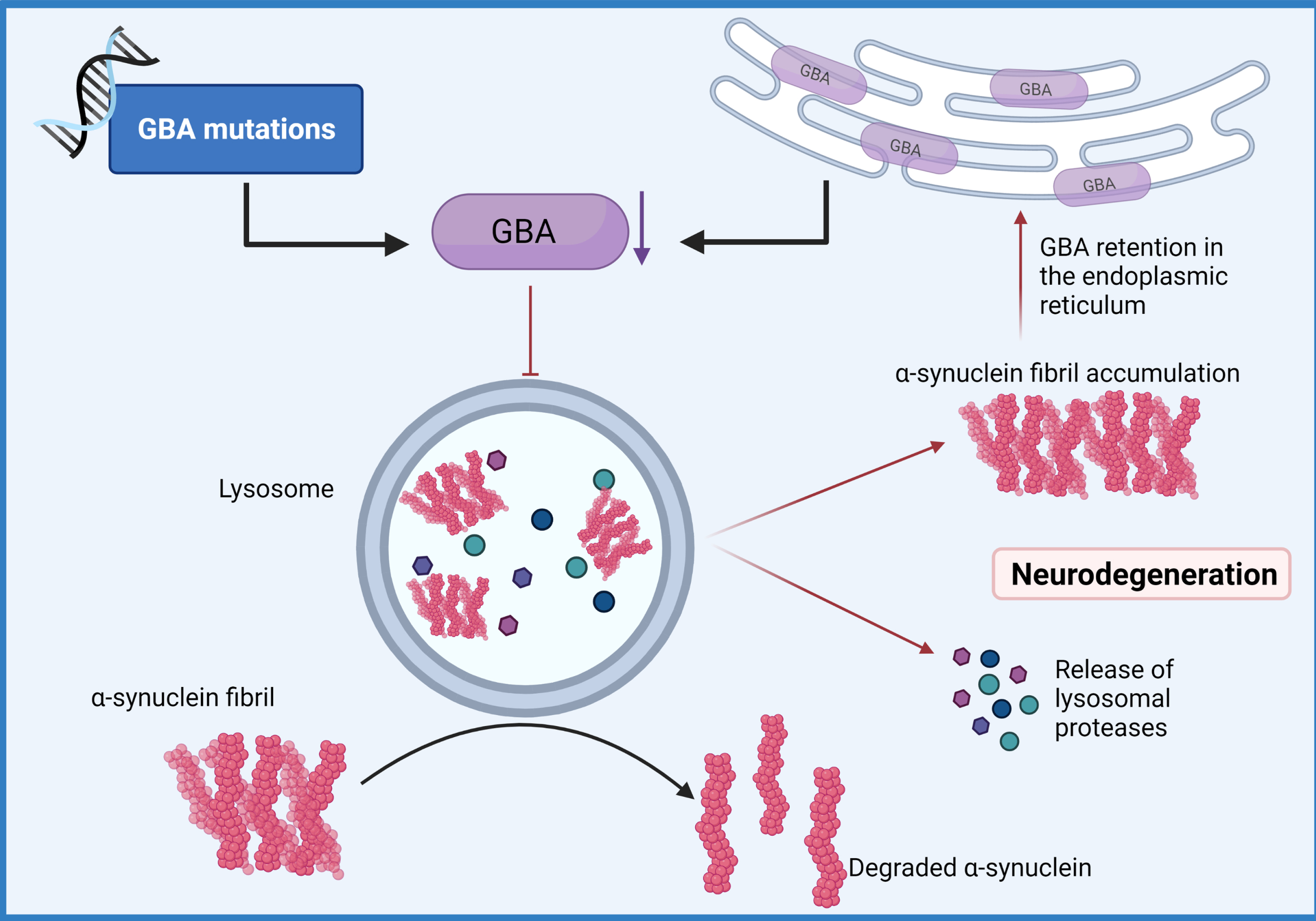
Fig. 3
GBA-mediated Lysosomal dysfunction. Mutations of the GBA gene that result in haploinsufficiency have been shown to result in loss of lysosomal homeostasis, a reduction of glucocerebrosidase activity, the build-up of lysosomal glucosylceramides and impairs α-synuclein degradation enhancing aggregation. (Created by BioRender.com)
CONCLUSIONS
Over the last two decades it has become clear that up to 10% of patients with EOPD harbor a major genetic determinant [126]. Nonetheless, in most patients, disease cannot be explained by a monogenetic form of inheritance, rather a genetic-environment interaction appears to play a pivotal role in the onset and progression of the disease [57]. GWAS have identified susceptible gene loci for many diseases, including neurodegenerative disorders, however, the variability in common alleles can’t explain heritability of common disorders by itself. Moreover, GWAS cannot detect those genes that influence disease onset/progression dependent upon an interaction with other genes or with the environment [127].
Functional analyses of lipid metabolism, mitochondrial health, neurometabolic cellular activity, and autophagic-lysosome capacity will be critical to place these proposed gene-environment interactions into context. Specific readouts such as phosphorylated ubiquitin, the joint PINK1-PARKIN product and so-called mitophagy tag, may not only be helpful to reveal cross-talk between and potential convergence of PD pathways in cells, tissues, and biofluids, but may also aid in better stratification of patients [128–130]. Likewise, further approaches to identify and compare specific molecular signatures, could be critical to re-categorize forms of PD based on biological pathways, in addition to clinical and pathological definitions, rather than simply age at onset [131].
After the initial discovery of SNCA gene mutations as a cause of PD more than 20 years ago, focused research has led to the identification of multiple genes linked to PD onset and progression [132–135]. Gene-environment interactions might be necessary to initiate the cascade of events leading to the onset of PD, and in some cases modify the risk of developing PD as is the case for caffeine exposure and specific polymorphisms that reduce PD risk [136] or smoking and decreased risk of PD that can be reversed in the presence of specific genetic variants [58]. Advances in research and technology have enabled genetic testing to become widely accessible and allowed consumers to access their genetic information without a healthcare professional intermediate. Genetic testing companies such as 23andme empower customers, not only with the ability to access information regarding their ancestry, but also their genetic predisposition to develop certain diseases, or their response to a specific drug, all without having to see a physician. A number of PD risk loci have been identified or confirmed using a genetic database based on the data collected through 23andme DNA samples [137].
A major question that remains unanswered by current genetic-environment studies is whether EOPD and LOPD are distinct disease entities or different staging on the same spectrum of disease. As discussed herein, there are clinical and neuropathologic differences in the phenotypic manifestation of disease signs, but it is not clear whether these are a simply a reflection of overall health (biological age) in the younger patients. Age is recognized as the major risk factor for LOPD, but this is absent in patients with EOPD and may reflect a stronger genetic/environmental component in the latter. However, given the presented genetic and environmental data it may be the scenario whereby there are distinct forms of PD, e.g., α-synuclein-centric versus mitophagy-driven, that may produce/influence both EOPD and LOPD. If this is the case, then whether it is a benefit to split patients by an arbitrary age cut-off is up for debate. What we really need as a field is an unbiased biologic readout that categorizes patients into subtypes and allows a more accurate definition of disease for clinical trial participation. A good example of this is the application of seed assays, such as RT-QuIC (real-time quaking induced conversion), which will facilitate the identification of patients with toxic α-synuclein protein driven forms of disease [138, 139]. Also, the recent work on phospho-ubiquitin (Serine-65) as a marker of mitochondrial health and readout for the mitophagy/mitochondrial quality control pathway, is another example of a potential biologic marker of a specific disease process [128]. Finally, the exposure to toxins may also in part define these subtypes, creating an environment on which the genetic determinants will act to drive phenotypic heterogeneity. Whether we can use these combinations to define specific patients’ subtypes, beyond the simple dichotomy of EOPD or LOPD, will be critical for drug development and successful clinical trials. As we start using machine-learning approaches to integrate large scale multimodal datasets, we can perhaps identify these key interactions, tease out cause versus effect questions, and redefine early-onset forms of PD.
ACKNOWLEDGMENTS
We acknowledge the support of the many people with Parkinson’s disease, both early and late-onset forms, and their families and caregivers who have driven many of the studies described in this review. In addition, we note the support of many clinical neurologists, clinical study coordinators, research technicians, fellows and students who have contributed to the studies on early-onset PD. Mayo Clinic is an American Parkinson Disease Association (APDA) Mayo Clinic Information and Referral Center, an APDA Center for Advanced Research, and the Mayo Clinic Lewy Body Dementia Association (LBDA) Research Center of Excellence. Mayo Clinic is also an LBD Center without Walls (U54-NS110435). OAR is supported by National Institute of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) [R01 NS085070 and U54 NS110435], the Department of Defense Congressionally Directed Medical Research Programs (CDMRP) [W81XWH-17-1-0249], the Michael J. Fox Foundation for Parkinson’s Research (MJFF), the American Parkinson Disease Association (APDA), and American Brain Foundation. PJM is supported by NIH/NINDS R01 NS085070, R01 NS110085 and U54 NS110435, the APDA Center for Advanced Research, the Mayo Clinic Foundation, and the American Brain Foundation. WS is supported by National Institute on Aging (NIH/NIA) [R56 AG062556], National Institute of Neurological Disorders and Stroke (NIH/NINDS) [R01 NS085070, R01 NS110085 and U54 NS110435], the Department of Defense Congressionally Directed Medical Research Programs (CDMRP) [W81XWH-17-1-0248], the Florida Department of Health - Ed and Ethel Moore Alzheimer’s Disease Research Program [9AZ10], the Michael J. Fox Foundation for Parkinson’s Research (MJFF), the American Parkinson Disease Association (APDA), Mayo Clinic Foundation, the Center for Biomedical Discovery (CBD) and the Robert and Arlene Kogod Center on Aging, and a sponsored research agreement with Amazentis SA.
CONFLICT OF INTEREST
PJM: Support from NIH/NINDS R01 NS085070, R01 NS110085 and U54 NS110435, the APDA Center for Advanced Research, the Mayo Clinic Foundation, and the American Brain Foundation.
WS: Support from National Institute on Aging (NIH/NIA) [R56 AG062556], National Institute of Neurological Disorders and Stroke (NIH/NINDS) [R01 NS085070, R01 NS110085 and U54 NS110435], the Department of Defense Congressionally Directed Medical Research Programs (CDMRP) [W81XWH-17-1-0248], the Michael J. Fox Foundation for Parkinson’s Research (MJFF), the American Parkinson Disease Association (APDA),
RS: Support from the National Institute on Aging, the National Institute of Neurological Disorders and Stroke, the Parkinson’s Disease Foundation, and Acadia Pharmaceuticals.
OAR: Support from National Institute of Health/National Institute of Neurological Disorders and Stroke (NIH/NINDS) [R01 NS085070 and U54 NS110435], the Department of Defense Congressionally Directed Medical Research Programs (CDMRP) [W81XWH-17-1-0249], the Michael J. Fox Foundation for Parkinson’s Research (MJFF), the American Parkinson Disease Association (APDA), and American Brain Foundation.
The other authors have no relevant disclosures to report.
REFERENCES
[1] | Lees AJ , Hardy J , Revesz T ((2009) ) Parkinson’s disease. Lancet 373: , 2055–2066. |
[2] | Bateman RJ , Xiong C , Benzinger TL , Fagan AM , Goate A , Fox NC , Marcus DS , Cairns NJ , Xie X , Blazey TM , Holtzman DM , Santacruz A , Buckles V , Oliver A , Moulder K , Aisen PS , Ghetti B , Klunk WE , McDade E , Martins RN , Masters CL , Mayeux R , Ringman JM , Rossor MN , Schofield PR , Sperling RA , Salloway S , Morris JC , Dominantly Inherited Alzheimer Network ((2012) ) Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med 367: , 795–804. |
[3] | Savica R , Boeve BF , Mielke MM ((2018) ) When do alpha-synucleinopathiesstart? An epidemiological timeline: A review. JAMA Neurol 75: , 503–509. |
[4] | Savica R , Grossardt BR , Bower JH , Ahlskog JE , Rocca WA ((2016) ) Time trends in the incidence of Parkinson disease. JAMA Neurol 73: , 981–989. |
[5] | Quinn N , Critchley P , Marsden CD ((1987) ) Young onset Parkinson’s disease. Mov Disord 2: , 73–91. |
[6] | Mehanna R , Smilowska K , Fleisher J , Post B , Hatano T , Pimentel Piemonte ME , Kumar KR , McConvey V , Zhang B , Tan EK , Savica R , International P , Movement Disorder Society Task Force on Early OnsetParkinson’s Disease ((2022) ) Age cutoff for early-onset Parkinson’sdisease: Recommendations from the International Parkinson andMovement Disorder Society Task Force on Early Onset Parkinson’sDisease. Mov Disord Clin Pract 9: , 869–878. |
[7] | Paviour DC , Surtees RA , Lees AJ ((2004) ) Diagnostic considerations in juvenile parkinsonism. Mov Disord 19: , 123–135. |
[8] | Ascherio A , Schwarzschild MA ((2016) ) The epidemiology of Parkinson’sdisease: Risk factors and prevention.. Lancet Neurol 15: , 1257–1272. |
[9] | Marras C , Beck JC , Bower JH , Roberts E , Ritz B , Ross GW , Abbott RD , Savica R , Van Den Eeden SK , Willis AW , Tanner CM ((2018) ) Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis 4: , 21. |
[10] | Schrag A , Schott JM ((2006) ) Epidemiological, clinical, and genetic characteristics of early-onset parkinsonism. Lancet Neurol 5: , 355–363. |
[11] | Konno T , Deutschlander A , Heckman MG , Ossi M , Vargas ER , Strongosky AJ , van Gerpen JA , Uitti RJ , Ross OA , Wszolek ZK ((2018) ) Comparison of clinical features among Parkinson’s disease subtypes: A large retrospective study in a single center. J Neurol Sci 386: , 39–45. |
[12] | Dulski J , Uitti R , Ross OA , Wszolek ZK ((2022) ) Genetic architecture of Parkinson’s disease subtypes - Review of the literature. Front Aging Neurosci 14: , 1023574. |
[13] | Bower JH , Maraganore DM , McDonnell SK , Rocca WA ((1999) ) Incidence and distribution of parkinsonism in Olmsted County, Minnesota, 1976-1990. Neurology 52: , 1214–1220. |
[14] | Hughes AJ , Daniel SE , Kilford L , Lees AJ ((1992) ) Accuracy of clinicaldiagnosis of idiopathic Parkinson’s disease: A clinico-pathologicalstudy of 100 cases. J Neurol Neurosurg Psychiatry 55: , 181–184. |
[15] | Mehanna R , Moore S , Hou JG , Sarwar AI , Lai EC ((2014) ) Comparing clinical features of young onset, middle onset and late onset Parkinson’s disease. Parkinsonism Relat Disord 20: , 530–534. |
[16] | Armstrong MJ , Okun MS ((2020) ) Time for a new image of Parkinson disease. JAMA Neurol 77: , 1345–1346. |
[17] | Mehanna R , Jankovic J ((2019) ) Young-onset Parkinson’s disease: Its unique features and their impact on quality of life. Parkinsonism Relat Disord 65: , 39–48. |
[18] | Chaudhary S , Joshi D , Pathak A , Mishra VN , Chaurasia RN , Gupta G ((2018) ) Comparison of cognitive profile in young- and late-onset Parkinson’s disease patients. Ann Indian Acad Neurol 21: , 130–132. |
[19] | De Rosa A , Pellegrino T , Pappata S , Pellecchia MT , Peluso S , Sacca F , Barone P , Cuocolo A , De Michele G ((2017) ) Myocardial (123)I-metaiodobenzylguanidine scintigraphy in patients with homozygous and heterozygous parkin mutations. J Nucl Cardiol 24: , 103–107. |
[20] | Orimo S , Amino T , Yokochi M , Kojo T , Uchihara T , Takahashi A , Wakabayashi K , Takahashi H , Hattori N , Mizuno Y ((2005) ) Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord 20: , 1350–1353. |
[21] | Suzuki M , Hattori N , Orimo S , Fukumitsu N , Abo M , Kono Y , Sengoku R , Kurita A , Honda H , Inoue K ((2005) ) Preserved myocardial[123I]metaiodobenzylguanidine uptake in autosomal recessive juvenileparkinsonism: First case report. Mov Disord 20: , 634–636. |
[22] | Santpere G , Ferrer I ((2009) ) LRRK2 and neurodegeneration. Acta Neuropathol 117: , 227–246. |
[23] | Hayashi S , Wakabayashi K , Ishikawa A , Nagai H , Saito M , Maruyama M , Takahashi T , Ozawa T , Tsuji S , Takahashi H ((2000) ) An autopsy case of autosomal-recessive juvenile parkinsonism with a homozygous exon 4 deletion in the parkin gene. Mov Disord 15: , 884–888. |
[24] | Gouider-Khouja N , Larnaout A , Amouri R , Sfar S , Belal S , Ben Hamida C , Ben Hamida M , Hattori N , Mizuno Y , Hentati F ((2003) ) Autosomal recessive parkinsonism linked to parkin gene in a Tunisian family. Clinical, genetic and pathological study. Parkinsonism Relat Disord 9: , 247–251. |
[25] | Farrer M , Chan P , Chen R , Tan L , Lincoln S , Hernandez D , Forno L , Gwinn-Hardy K , Petrucelli L , Hussey J , Singleton A , Tanner C , Hardy J , Langston JW ((2001) ) Lewy bodies and parkinsonism in families with parkin mutations. Ann Neurol 50: , 293–300. |
[26] | Johansen KK , Torp SH , Farrer MJ , Gustavsson EK , Aasly JO ((2018) ) A case of Parkinson’s disease with no Lewy body pathology due to a homozygous exon deletion in parkin. Case Rep Neurol Med 2018: , 6838965. |
[27] | Miyakawa S , Ogino M , Funabe S , Uchino A , Shimo Y , Hattori N , Ichinoe M , Mikami T , Saegusa M , Nishiyama K , Mori H , Mizuno Y , Murayama S , Mochizuki H ((2013) ) Lewy body pathology in a patient with a homozygous parkin deletion. Mov Disord 28: , 388–391. |
[28] | Nybo CJ , Gustavsson EK , Farrer MJ , Aasly JO ((2020) ) Neuropathological findings in PINK1-associated Parkinson’s disease. Parkinsonism Relat Disord 78: , 105–108. |
[29] | Pramstaller PP , Schlossmacher MG , Jacques TS , Scaravilli F , Eskelson C , Pepivani I , Hedrich K , Adel S , Gonzales-McNeal M , Hilker R , Kramer PL , Klein C ((2005) ) Lewy body Parkinson’s disease in a large pedigree with 77 Parkin mutation carriers. Ann Neurol 58: , 411–422. |
[30] | Sakuwa M , Adachi T , Yoshida K , Adachi Y , Nakano T , Hanajima R ((2021) ) An autopsy case of PARK2 due to a homozygous exon 2 deletion of parkin and associated with alpha-synucleinopathy. Neuropathology 41: , 293–300. |
[31] | Samaranch L , Lorenzo-Betancor O , Arbelo JM , Ferrer I , Lorenzo E , Irigoyen J , Pastor MA , Marrero C , Isla C , Herrera-Henriquez J , Pastor P ((2010) ) PINK1-linked parkinsonism is associated with Lewy body pathology. Brain 133: , 1128–1142. |
[32] | Sasaki S , Shirata A , Yamane K , Iwata M ((2004) ) Parkin-positive autosomal recessive juvenile Parkinsonism with alpha-synuclein-positive inclusions. Neurology 63: , 678–682. |
[33] | Seike N , Yokoseki A , Takeuchi R , Saito K , Miyahara H , Miyashita A , Ikeda T , Aida I , Nakajima T , Kanazawa M , Wakabayashi M , Toyoshima Y , Takahashi H , Matsumoto R , Toda T , Onodera O , Ishikawa A , Ikeuchi T , Kakita A ((2021) ) Genetic variations and neuropathologic features of patients with PRKN mutations. Mov Disord 36: , 1634–1643. |
[34] | Steele JC , Guella I , Szu-Tu C , Lin MK , Thompson C , Evans DM , Sherman HE , Vilarino-Guell C , Gwinn K , Morris H , Dickson DW , Farrer MJ ((2015) ) Defining neurodegeneration on Guam by targeted genomic sequencing. Ann Neurol 77: , 458–468. |
[35] | Takanashi M , Li Y , Hattori N ((2016) ) Absence of Lewy pathology associated with PINK1 homozygous mutation. Neurology 86: , 2212–2213. |
[36] | Poulopoulos M , Levy OA , Alcalay RN ((2012) ) The neuropathology of genetic Parkinson’s disease. Mov Disord 27: , 831–842. |
[37] | Ahlskog JE ((2009) ) Parkin and PINK1 parkinsonism may represent nigral mitochondrial cytopathies distinct from Lewy body Parkinson’s disease. Parkinsonism Relat Disord 15: , 721–727. |
[38] | Shahmoradian SH , Lewis AJ , Genoud C , Hench J , Moors TE , Navarro PP , Castano-Diez D , Schweighauser G , Graff-Meyer A , Goldie KN , Sutterlin R , Huisman E , Ingrassia A , Gier Y , Rozemuller AJM , Wang J , Paepe A , Erny J , Staempfli A , Hoernschemeyer J , Grosseruschkamp F , Niedieker D , El-Mashtoly SF , Quadri M , Van IWFJ , Bonifati V , Gerwert K , Bohrmann B , Frank S , Britschgi M , Stahlberg H , Van de Berg WDJ , Lauer ME ((2019) ) Lewy pathology in Parkinson’s disease consists of crowded organelles and lipid membranes. Nat Neurosci 22: , 1099–1109. |
[39] | Trinkaus VA , Riera-Tur I , Martinez-Sanchez A , Bauerlein FJB , Guo Q , Arzberger T , Baumeister W , Dudanova I , Hipp MS , Hartl FU , Fernandez-Busnadiego R ((2021) ) In situ architecture of neuronal alpha-synuclein inclusions. Nat Commun 12: , 2110. |
[40] | Mahul-Mellier AL , Burtscher J , Maharjan N , Weerens L , Croisier M , Kuttler F , Leleu M , Knott GW , Lashuel HA ((2020) ) The process of Lewy body formation, rather than simply alpha-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci U S A 117: , 4971–4982. |
[41] | Erskine D , Koss D , Korolchuk VI , Outeiro TF , Attems J , McKeith I ((2021) ) Lipids, lysosomes and mitochondria: Insights into Lewy bodyformation from rare monogenic disorders.. Acta Neuropathol 141: , 511–526. |
[42] | Tsai CH , Lo SK , See LC , Chen HZ , Chen RS , Weng YH , Chang FC , Lu CS ((2002) ) Environmental risk factors of young onset Parkinson’s disease: A case-control study.. Clin Neurol Neurosurg 104: , 328–333. |
[43] | Ritz B , Ascherio A , Checkoway H , Marder KS , Nelson LM , Rocca WA , Ross GW , Strickland D , Van Den Eeden SK , Gorell J ((2007) ) Pooled analysis of tobacco use and risk of Parkinson disease. Arch Neurol 64: , 990–997. |
[44] | Henry J , Smeyne RJ , Jang H , Miller B , Okun MS ((2010) ) Parkinsonism and neurological manifestations of influenza throughout the 20th and 21st centuries. Parkinsonism Relat Disord 16: , 566–571. |
[45] | Jang H , Boltz D , Sturm-Ramirez K , Shepherd KR , Jiao Y , Webster R , Smeyne RJ ((2009) ) Highly pathogenic H5N1 influenza virus can enter the central nervous system and induce neuroinflammation and neurodegeneration. Proc Natl Acad Sci U S A 106: , 14063–14068. |
[46] | Hawkes CH , Del Tredici K , Braak H ((2007) ) Parkinson’s disease: Adual-hit hypothesis. Neuropathol Appl Neurobiol 33: , 599–614. |
[47] | Smeyne RJ , Noyce AJ , Byrne M , Savica R , Marras C ((2021) ) Infection and risk of Parkinson’s disease. J Parkinsons Dis 11: , 31–43. |
[48] | Douaud G , Lee S , Alfaro-Almagro F , Arthofer C , Wang C , McCarthy P , Lange F , Andersson JLR , Griffanti L , Duff E , Jbabdi S , Taschler B , Keating P , Winkler AM , Collins R , Matthews PM , Allen N , Miller KL , Nichols TE , Smith SM ((2022) ) SARS-CoV-2 is associated with changes in brain structure in UK Biobank. Nature 604: , 697–707. |
[49] | Prudencio M , Erben Y , Marquez CP , Jansen-West KR , Franco-Mesa C , Heckman MG , White LJ , Dunmore JA , Cook CN , Lilley MT , Song Y , Harlow CF , Oskarsson B , Nicholson KA , Wszolek ZK , Hickson LJ , O’Horo JC , Hoyne JB , Gendron TF , Meschia JF , Petrucelli L ((2021) ) Serum neurofilament light protein correlates with unfavorable clinical outcomes in hospitalized patients with COVID-19. Sci Transl Med 13: , eabi7643. |
[50] | Ballard PA , Tetrud JW , Langston JW ((1985) ) Permanent humanparkinsonism due to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine(MPTP): Seven cases. Neurology 35: , 949–956. |
[51] | Schapira AH , Holt IJ , Sweeney M , Harding AE , Jenner P , Marsden CD ((1990) ) Mitochondrial DNA analysis in Parkinson’s disease. Mov Disord 5: , 294–297. |
[52] | Gatto NM , Cockburn M , Bronstein J , Manthripragada AD , Ritz B ((2009) ) Well-water consumption and Parkinson’s disease in rural California. Environ Health Perspect 117: , 1912–1918. |
[53] | Berry C , La Vecchia C , Nicotera P ((2010) ) Paraquat and Parkinson’s disease. Cell Death Differ 17: , 1115–1125. |
[54] | Brighina L , Frigerio R , Schneider NK , Lesnick TG , de Andrade M , Cunningham JM , Farrer MJ , Lincoln SJ , Checkoway H , Rocca WA , Maraganore DM ((2008) ) Alpha-synuclein, pesticides, and Parkinsondisease: A case-control study. Neurology 70: , 1461–1469. |
[55] | Gatto NM , Rhodes SL , Manthripragada AD , Bronstein J , Cockburn M , Farrer M , Ritz B ((2010) ) alpha-Synuclein gene may interact with environmental factors in increasing risk of Parkinson’s disease. Neuroepidemiology 35: , 191–195. |
[56] | Elbaz A , Grigoletto F , Baldereschi M , Breteler MM , Manubens-Bertran JM , Lopez-Pousa S , Dartigues JF , Alperovitch A , Tzourio C , Rocca WA ((1999) ) Familial aggregation of Parkinson’s disease: Apopulation-based case-control study in Europe. EUROPARKINSON StudyGroup. Neurology 52: , 1876–1882. |
[57] | Gao HM , Hong JS ((2011) ) Gene-environment interactions: Key tounraveling the mystery of Parkinson’s disease. Prog Neurobiol 94: , 1–19. |
[58] | Checkoway H , Franklin GM , Costa-Mallen P , Smith-Weller T , Dilley J , Swanson PD , Costa LG ((1998) ) A genetic polymorphism of MAO-B modifies the association of cigarette smoking and Parkinson’s disease. Neurology 50: , 1458–1461. |
[59] | Benmoyal-Segal L , Vander T , Shifman S , Bryk B , Ebstein RP , Marcus E-L , Stessman J , Darvasi A , Herishanu Y , Friedman A , Soreq H ((2005) ) Acetylcholinesterase/paraoxonase interactions increase the risk of insecticide-induced Parkinson’s disease. FASEB J 19: , 452–454. |
[60] | Betarbet R , Canet-Aviles RM , Sherer TB , Mastroberardino PG , McLendon C , Kim JH , Lund S , Na HM , Taylor G , Bence NF , Kopito R , Seo BB , Yagi T , Yagi A , Klinefelter G , Cookson MR , Greenamyre JT ((2006) ) Intersecting pathways to neurodegeneration in Parkinson’s disease: Effects of the pesticide rotenone on DJ-1, alpha-synuclein, and theubiquitin-proteasome system. Neurobiol Dis 22: , 404–420. |
[61] | Ratner MH , Farb DH , Ozer J , Feldman RG , Durso R ((2014) ) Younger age at onset of sporadic Parkinson’s disease among subjects occupationally exposed to metals and pesticides. Interdiscip Toxicol 7: , 123–133. |
[62] | Lunati A , Lesage S , Brice A ((2018) ) The genetic landscape of Parkinson’s disease. Rev Neurol (Paris) 174: , 628–643. |
[63] | Cardona F , Perez-Tur J ((2017) ) Other proteins involved in Parkinson’s disease and related disorders. Curr Protein Pept Sci 18: , 765–778. |
[64] | Puschmann A ((2013) ) Monogenic Parkinson’s disease and parkinsonism:Clinical phenotypes and frequencies of known mutations. Parkinsonism Relat Disord 19: , 407–415. |
[65] | Lanoue V , Chai YJ , Brouillet JZ , Weckhuysen S , Palmer EE , Collins BM , Meunier FA ((2019) ) STXBP1 encephalopathy: Connecting neurodevelopmental disorders with alpha-synucleinopathies? . Neurology 93: , 114–123. |
[66] | Williams DR , Hadeed A , al-Din AS , Wreikat AL , Lees AJ ((2005) ) KuforRakeb disease: Autosomal recessive, levodopa-responsive parkinsonismwith pyramidal degeneration, supranuclear gaze palsy, and dementia. Mov Disord 20: , 1264–1271. |
[67] | Schneider SA , Bhatia KP , Hardy J ((2009) ) Complicated recessive dystonia parkinsonism syndromes. Mov Disord 24: , 490–499. |
[68] | Schneider SA , Alcalay RN ((2017) ) Neuropathology of genetic synucleinopathies with parkinsonism: Review of the literature. Mov Disord 32: , 1504–1523. |
[69] | Klein C , Schneider SA , Lang AE ((2009) ) Hereditary parkinsonism: Parkinson disease look-alikes–an algorithm for clinicians to “PARK” genes and beyond. Mov Disord 24: , 2042–2058. |
[70] | Kostic V , Przedborski S , Flaster E , Sternic N ((1991) ) Early development of levodopa-induced dyskinesias and response fluctuations in young-onset Parkinson’s disease. Neurology 41: , 202–205. |
[71] | Pantelatos A , Fornadi F ((1993) ) Clinical features and medical treatment of Parkinson’s disease in patient groups selected in accordance with age at onset. Adv Neurol 60: , 690–697. |
[72] | Alves G , Wentzel-Larsen T , Aarsland D , Larsen JP ((2005) ) Progressionof motor impairment and disability in Parkinson disease: Apopulation-based study. Neurology 65: , 1436–1441. |
[73] | Camerucci E , Stang CD , Hajeb M , Turcano P , Mullan AF , Martin P , Ross OA , Bower JH , Mielke MM , Savica R ((2021) ) Early-onset parkinsonismand early-onset Parkinson’s disease: A population-based study(2010-2015). J Parkinsons Dis 11: , 1197–1207. |
[74] | Hou X , Watzlawik JO , Fiesel FC , Springer W ((2020) ) Autophagy in Parkinson’s disease. J Mol Biol 432: , 2651–2672. |
[75] | Truban D , Hou X , Caulfield TR , Fiesel FC , Springer W ((2017) ) PINK1,Parkin, and mitochondrial quality control: What can we learn aboutParkinson’s disease pathobiology? J Parkinsons Dis 7: , 13–29. |
[76] | Schulte C , Gasser T ((2011) ) Genetic basis of Parkinson’s disease:Inheritance, penetrance, and expression. Appl Clin Genet 4: , 67–80. |
[77] | Tolmasov M , Djaldetti R , Lev N , Gilgun-Sherki Y ((2016) ) Pathological and clinical aspects of alpha/beta synuclein in Parkinson’s disease and related disorders. Expert Rev Neurother 16: , 505–513. |
[78] | Konno T , Ross OA , Puschmann A , Dickson DW , Wszolek ZK ((2016) ) Autosomal dominant Parkinson’s disease caused by SNCA duplications. , S. Parkinsonism Relat Disord 22: Suppl 1, 1–6. |
[79] | Houlden H , Singleton AB ((2012) ) The genetics and neuropathology of Parkinson’s disease. Acta Neuropathol 124: , 325–338. |
[80] | Hernandez DG , Reed X , Singleton AB ((2016) ) Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J Neurochem 139: Suppl 1, 59–74. |
[81] | Butler B , Saha K , Rana T , Becker JP , Sambo D , Davari P , Goodwin JS , Khoshbouei H ((2015) ) Dopamine transporter activity is modulated by alpha-synuclein. J Biol Chem 290: , 29542–29554. |
[82] | Vidyadhara DJ , Lee JE , Chandra SS ((2019) ) Role of the endolysosomal system in Parkinson’s disease. J Neurochem 150: , 487–506. |
[83] | Mamais A , Kluss JH , Bonet-Ponce L , Landeck N , Langston RG , Smith N , Beilina A , Kaganovich A , Ghosh MC , Pellegrini L , Kumaran R , Papazoglou I , Heaton GR , Bandopadhyay R , Maio N , Kim C , LaVoie MJ , Gershlick DC , Cookson MR ((2021) ) Mutations in LRRK2 linked to Parkinson disease sequester Rab8a to damaged lysosomes and regulate transferrin-mediated iron uptake in microglia. PLoS Biol 19: , e3001480. |
[84] | Melrose HL , Kent CB , Taylor JP , Dachsel JC , Hinkle KM , Lincoln SJ , Mok SS , Culvenor JG , Masters CL , Tyndall GM , Bass DI , Ahmed Z , Andorfer CA , Ross OA , Wszolek ZK , Delldonne A , Dickson DW , Farrer MJ ((2007) ) A comparative analysis of leucine-rich repeat kinase 2 (Lrrk2) expression in mouse brain and Lewy body disease. Neuroscience 147: , 1047–1058. |
[85] | Bonet-Ponce L , Cookson MR ((2022) ) LRRK2 recruitment, activity, and function in organelles. FEBS J 289: , 6871–6890. |
[86] | Rui Q , Ni H , Li D , Gao R , Chen G ((2018) ) The role of LRRK2 in neurodegeneration of Parkinson disease. Curr Neuropharmacol 16: , 1348–1357. |
[87] | Peng F , Sun YM , Chen C , Luo SS , Li DK , Wang YX , Yang K , Liu FT , Zuo CT , Ding ZT , An Y , Wu JJ , Wang J ((2017) ) The heterozygous R1441C mutation of leucine-rich repeat kinase 2 gene in a Chinese patient with Parkinson disease: A five-year follow-up and literatures review. J Neurol Sci 373: , 23–26. |
[88] | Shu L , Zhang Y , Pan H , Xu Q , Guo J , Tang B , Sun Q ((2018) ) Clinicalheterogeneity among LRRK2 variants in Parkinson’s disease: Ameta-analysis. Front Aging Neurosci 10: , 283. |
[89] | Nalls MA , Blauwendraat C , Vallerga CL , Heilbron K , Bandres-Ciga S , Chang D , Tan M , Kia DA , Noyce AJ , Xue A , Bras J , Young E , von Coelln R , Simón-Sáanchez J , Schulte C , Sharma M , Krohn L , Pihlstrøm L , Siitonen A , Iwaki H , Leonard H , Faghri F , Gibbs JR , Hernandez DG , Scholz SW , Botia JA , Martinez M , Corvol JC , Lesage S , Jankovic J , Shulman LM , Sutherland M , Tienari P , Majamaa K , Toft M , Andreassen OA , Bangale T , Brice A , Yang J , Gan-Or Z , Gasser T , Heutink P , Shulman JM , Wood NW , Hinds DA , Hardy JA , Morris HR , Gratten J , Visscher PM , Graham RR , Singleton AB; 23andMe ResearchTeam; System Genomics of Parkinson’s Disease Consortium;International Parkinson’s Disease Genomics Consortium ((2019) ) Identification of novel risk loci, causal insights, and heritablerisk for Parkinson’s disease: A meta-analysis of genome-wideassociation studies. Lancet Neurol 18: , 1091–1102. |
[90] | Liu J , Liu W , Li R , Yang H ((2019) ) Mitophagy in Parkinson’s disease:From pathogenesis to treatment. Cells 8: , 712. |
[91] | Liu XL , Wang YD , Yu XM , Li DW , Li GR ((2018) ) Mitochondria-mediated damage to dopaminergic neurons in Parkinson’s disease (Review). Int J Mol Med 41: , 615–623. |
[92] | Valente EM , Salvi S , Ialongo T , Marongiu R , Elia AE , Caputo V , Romito L , Albanese A , Dallapiccola B , Bentivoglio AR ((2004) ) PINK1 mutations are associated with sporadic early-onset parkinsonism. Ann Neurol 56: , 336–341. |
[93] | Kitada T , Asakawa S , Hattori N , Matsumine H , Yamamura Y , Minoshima S , Yokochi M , Mizuno Y , Shimizu N ((1998) ) Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: , 605–608. |
[94] | Tanaka K , Suzuki T , Chiba T , Shimura H , Hattori N , Mizuno Y ((2001) ) Parkin is linked to the ubiquitin pathway. J Mol Med (Berl) 79: , 482–494. |
[95] | Shimura H , Hattori N , Kubo S , Mizuno Y , Asakawa S , Minoshima S , Shimizu N , Iwai K , Chiba T , Tanaka K , Suzuki T ((2000) ) Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25: , 302–305. |
[96] | Park J , Lee SB , Lee S , Kim Y , Song S , Kim S , Bae E , Kim J , Shong M , Kim JM , Chung J ((2006) ) Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441: , 1157–1161. |
[97] | Clark IE , Dodson MW , Jiang C , Cao JH , Huh JR , Seol JH , Yoo SJ , Hay BA , Guo M ((2006) ) Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441: , 1162–1166. |
[98] | Geisler S , Holmstrom KM , Skujat D , Fiesel FC , Rothfuss OC , Kahle PJ , Springer W ((2010) ) PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12: , 119–131. |
[99] | Geisler S , Holmstrom KM , Treis A , Skujat D , Weber SS , Fiesel FC , Kahle PJ , Springer W ((2010) ) The PINK1/Parkin-mediated mitophagy is compromised by PD-associated mutations. Autophagy 6: , 871–878. |
[100] | Narendra D , Tanaka A , Suen DF , Youle RJ ((2008) ) Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: , 795–803. |
[101] | Narendra DP , Jin SM , Tanaka A , Suen DF , Gautier CA , Shen J , Cookson MR , Youle RJ ((2010) ) PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: , e1000298. |
[102] | Valente EM , Abou-Sleiman PM , Caputo V , Muqit MM , Harvey K , Gispert S , Ali Z , Del Turco D , Bentivoglio AR , Healy DG , Albanese A , Nussbaum R , Gonzalez-Maldonado R , Deller T , Salvi S , Cortelli P , Gilks WP , Latchman DS , Harvey RJ , Dallapiccola B , Auburger G , Wood NW ((2004) ) Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304: , 1158–1160. |
[103] | Doherty KM , Hardy J ((2013) ) Parkin disease and the Lewy body conundrum. Mov Disord 28: , 702–704. |
[104] | Al-Rumayyan A , Klein C , Alfadhel M ((2017) ) Early-onset parkinsonism:Case report and review of the literature. Pediatr Neurol 67: , 102–106 e101. |
[105] | Ferraris A , Valente EM , Bentivoglio AR ((2009) ) Prevalence and phenotypic of PINK1 mutations in Parkinson’s spectrum disease. US Neurology 5: , 34–37. |
[106] | Thomas KJ , McCoy MK , Blackinton J , Beilina A , van der Brug M , Sandebring A , Miller D , Maric D , Cedazo-Minguez A , Cookson MR ((2011) ) DJ-1 acts in parallel to the PINK1/parkin pathway to control mitochondrial function and autophagy. Hum Mol Genet 20: , 40–50. |
[107] | Hauser DN , Mamais A , Conti MM , Primiani CT , Kumaran R , Dillman AA , Langston RG , Beilina A , Garcia JH , Diaz-Ruiz A , Bernier M , Fiesel FC , Hou X , Springer W , Li Y , de Cabo R , Cookson MR ((2017) ) Hexokinases link DJ-1 to the PINK1/parkin pathway. Mol Neurodegener 12: , 70. |
[108] | Bonifati V , Rizzu P , van Baren MJ , Schaap O , Breedveld GJ , Krieger E , Dekker MC , Squitieri F , Ibanez P , Joosse M , van Dongen JW , Vanacore N , van Swieten JC , Brice A , Meco G , van Duijn CM , Oostra BA , Heutink P ((2003) ) Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 299: , 256–259. |
[109] | Cookson MR ((2012) ) Parkinsonism due to mutations in PINK1, parkin, and DJ-1 and oxidative stress and mitochondrial pathways. , a. Cold Spring Harb Perspect Med 2: , 009415. |
[110] | Ottolini D , Cali T , Negro A , Brini M ((2013) ) The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum Mol Genet 22: , 2152–2168. |
[111] | Newman LE , Shadel GS ((2018) ) Pink1/Parkin link inflammation, mitochondrial stress, and neurodegeneration. J Cell Biol 217: , 3327–3329. |
[112] | Castelo Rueda MP , Raftopoulou A , Gogele M , Borsche M , Emmert D , Fuchsberger C , Hantikainen EM , Vukovic V , Klein C , Pramstaller PP , Pichler I , Hicks AA ((2021) ) Frequency of heterozygous Parkin (PRKN) variants and penetrance of Parkinson’s disease risk markers in the population-based CHRIS Cohort. Front Neurol 12: , 706145. |
[113] | Krohn L , Grenn FP , Makarious MB , Kim JJ , Bandres-Ciga S , Roosen DA , Gan-Or Z , Nalls MA , Singleton AB , Blauwendraat C , International Parkinson’s Disease Genomics Consortium (IPDGC) ((2020) ) Comprehensive assessment of PINK1 variants in Parkinson’s disease. Neurobiol Aging 91: , 168 e161–168 e165. |
[114] | Lubbe SJ , Bustos BI , Hu J , Krainc D , Joseph T , Hehir J , Tan M , Zhang W , Escott-Price V , Williams NM , Blauwendraat C , Singleton AB , Morris HR , for International Parkinson’s Disease Genomics Consortium (IPDGC) ((2021) ) Assessing the relationship between monoallelic PRKN mutations and Parkinson’s risk. Hum Mol Genet 30: , 78–86. |
[115] | Puschmann A , Fiesel FC , Caulfield TR , Hudec R , Ando M , Truban D , Hou X , Ogaki K , Heckman MG , James ED , Swanberg M , Jimenez-Ferrer I , Hansson O , Opala G , Siuda J , Boczarska-Jedynak M , Friedman A , Koziorowski D , Rudzinska-Bar M , Aasly JO , Lynch T , Mellick GD , Mohan M , Silburn PA , Sanotsky Y , Vilarino-Guell C , Farrer MJ , Chen L , Dawson VL , Dawson TM , Wszolek ZK , Ross OA , Springer W ((2017) ) Heterozygous PINK1 p.G411S increases risk of Parkinson’s disease via a dominant-negative mechanism. Brain 140: , 98–117. |
[116] | Nalls MA , Escott-Price V , Williams NM , Lubbe S , Keller MF , Morris HR , Singleton AB , International Parkinson’s Disease Genomics Consortium (IPDGC) ((2015) ) Genetic risk and age in Parkinson’s disease: Continuum not stratum. Mov Disord 30: , 850–854. |
[117] | Blauwendraat C , Heilbron K , Vallerga CL , Bandres-Ciga S , von Coelln R , Pihlstrom L , Simon-Sanchez J , Schulte C , Sharma M , Krohn L , Siitonen A , Iwaki H , Leonard H , Noyce AJ , Tan M , Gibbs JR , Hernandez DG , Scholz SW , Jankovic J , Shulman LM , Lesage S , Corvol JC , Brice A , van Hilten JJ , Marinus J , andMe Research T , Eerola-Rautio J , Tienari P , Majamaa K , Toft M , Grosset DG , Gasser T , Heutink P , Shulman JM , Wood N , Hardy J , Morris HR , Hinds DA , Gratten J , Visscher PM , Gan-Or Z , Nalls MA , Singleton AB , International Parkinson’s Disease Genomics Consortium (IPDGC) ((2019) ) Parkinson’s disease age at onset genome-wide association study: Defining heritability, genetic loci, and alpha-synuclein mechanisms. Mov Disord 34: , 866–875. |
[118] | Noyce AJ , Bandres-Ciga S , Kim J , Heilbron K , Kia D , Hemani G , Xue A , Lawlor DA , Smith GD , Duran R , Gan-Or Z , Blauwendraat C , Gibbs JR , andMe Research Team IPsDGC, Hinds DA , Yang J , Visscher P , Cuzick J , Morris H , Hardy J , Wood NW , Nalls MA , Singleton AB ((2019) ) The Parkinson’s Disease Mendelian Randomization Research Portal. Mov Disord 34: , 1864–1872. |
[119] | Sidransky E ((2012) ) Gaucher disease: Insights from a rare Mendeliandisorder. Discov Med 14: , 273–281. |
[120] | Balestrino R , Schapira AHV ((2018) ) Glucocerebrosidase and Parkinsondisease: Molecular, clinical, and therapeutic implications. Neuroscientist 24: , 540–559. |
[121] | Sato S , Li Y , Hattori N ((2017) ) Lysosomal defects in ATP13A2 and GBA associated familial Parkinson’s disease. J Neural Transm (Vienna) 124: , 1395–1400. |
[122] | Gan-Or Z , Alcalay RN , Makarious MB , Scholz SW , Blauwendraat C , International Parkinson’s Disease Genomics Consortium (IPDGC) ((2019) ) Classification of GBA Variants and Their Effects in Synucleinopathies. Mov Disord 34: , 1581–1582. |
[123] | Iwaki H , Blauwendraat C , Leonard HL , Liu G , Maple-Grodem J , Corvol JC , Pihlstrom L , van Nimwegen M , Hutten SJ , Nguyen KH , Rick J , Eberly S , Faghri F , Auinger P , Scott KM , Wijeyekoon R , Van Deerlin VM , Hernandez DG , Day-Williams AG , Brice A , Alves G , Noyce AJ , Tysnes OB , Evans JR , Breen DP , Estrada K , Wegel CE , Danjou F , Simon DK , Ravina B , Toft M , Heutink P , Bloem BR , Weintraub D , Barker RA , Williams-Gray CH , van de Warrenburg BP , Van Hilten JJ , Scherzer CR , Singleton AB , Nalls MA ((2019) ) Genetic risk of Parkinson disease andprogression: An analysis of 13 longitudinal cohorts. NeurolGenet 5: , e348. |
[124] | Iwaki H , Blauwendraat C , Leonard HL , Kim JJ , Liu G , Maple-Grødem J , Corvol JC , Pihlstrøm L , van Nimwegen M , Hutten SJ , Nguyen KH , Rick J , Eberly S , Faghri F , Auinger P , Scott KM , Wijeyekoon R , Van Deerlin VM , Hernandez DG , Gibbs JR , International Parkinson’s Disease Genomics Consortium Chitrala KN , Day-Williams AG , Brice A , Alves G , Noyce AJ , Tysnes OB , Evans JR , Breen DP , Estrada K , Wegel CE , Danjou F , Simon DK , Andreassen O , Ravina B , Toft M , Heutink P , Bloem BR , Weintraub D , Barker RA , Williams-Gray CH , van de Warrenburg BP , Van Hilten JJ , Scherzer CR , Singleton AB , Nalls MA ((2019) ) Genomewide association study of Parkinson’s disease clinical biomarkers in 12 longitudinal patients’ cohorts. Mov Disord 34: , 1839–1850. |
[125] | Blauwendraat C , Reed X , Krohn L , Heilbron K , Bandres-Ciga S , Tan M , Gibbs JR , Hernandez DG , Kumaran R , Langston R , Bonet-Ponce L , Alcalay RN , Hassin-Baer S , Greenbaum L , Iwaki H , Leonard HL , Grenn FP , Ruskey JA , Sabir M , Ahmed S , Makarious MB , Pihlstrom L , Toft M , van Hilten JJ , Marinus J , Schulte C , Brockmann K , Sharma M , Siitonen A , Majamaa K , Eerola-Rautio J , Tienari PJ , andMe Research T, Pantelyat A , Hillis AE , Dawson TM , Rosenthal LS , Albert MS , Resnick SM , Ferrucci L , Morris CM , Pletnikova O , Troncoso J , Grosset D , Lesage S , Corvol JC , Brice A , Noyce AJ , Masliah E , Wood N , Hardy J , Shulman LM , Jankovic J , Shulman JM , Heutink P , Gasser T , Cannon P , Scholz SW , Morris H , Cookson MR , Nalls MA , Gan-Or Z , Singleton AB ((2020) ) Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 143: , 234–248. |
[126] | Larsen SB , Hanss Z , Kruger R ((2018) ) The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res 373: , 21–37. |
[127] | Hamza TH , Chen H , Hill-Burns EM , Rhodes SL , Montimurro J , Kay DM , Tenesa A , Kusel VI , Sheehan P , Eaaswarkhanth M , Yearout D , Samii A , Roberts JW , Agarwal P , Bordelon Y , Park Y , Wang L , Gao J , Vance JM , Kendler KS , Bacanu SA , Scott WK , Ritz B , Nutt J , Factor SA , Zabetian CP , Payami H ((2011) ) Genome-wide gene-environment study identifies glutamate receptor gene GRIN2A as a Parkinson’s disease modifier gene via interaction with coffee. PLoS Genet 7: , e1002237. |
[128] | Fiesel FC , Ando M , Hudec R , Hill AR , Castanedes-Casey M , Caulfield TR , Moussaud-Lamodiere EL , Stankowski JN , Bauer PO , Lorenzo-Betancor O , Ferrer I , Arbelo JM , Siuda J , Chen L , Dawson VL , Dawson TM , Wszolek ZK , Ross OA , Dickson DW , Springer W ((2015) ) (Patho-)physiological relevance of PINK1-dependent ubiquitinphosphorylation. EMBO Rep 16: , 1114–1130. |
[129] | Hou X , Fiesel FC , Truban D , Castanedes Casey M , Lin WL , Soto AI , Tacik P , Rousseau LG , Diehl NN , Heckman MG , Lorenzo-Betancor O , Ferrer I , Arbelo JM , Steele JC , Farrer MJ , Cornejo-Olivas M , Torres L , Mata IF , Graff-Radford NR , Wszolek ZK , Ross OA , Murray ME , Dickson DW , Springer W ((2018) ) Age- and disease-dependent increase of the mitophagy marker phospho-ubiquitin in normal aging and Lewy body disease. Autophagy 14: , 1404–1418. |
[130] | Watzlawik JO , Hou X , Fricova D , Ramnarine C , Barodia SK , Gendron TF , Heckman MG , DeTure M , Siuda J , Wszolek ZK , Scherzer CR , Ross OA , Bu G , Dickson DW , Goldberg MS , Fiesel FC , Springer W ((2021) ) Sensitive ELISA-based detection method for the mitophagy marker p-S65-Ub in human cells, autopsy brain, and blood samples. Autophagy 17: , 2613–2628. |
[131] | Laperle AH , Sances S , Yucer N , Dardov VJ , Garcia VJ , Ho R , Fulton AN , Jones MR , Roxas KM , Avalos P , West D , Banuelos MG , Shu Z , Murali R , Maidment NT , Van Eyk JE , Tagliati M , Svendsen CN ((2020) ) iPSC modeling of young-onset Parkinson’s disease reveals a molecular signature of disease and novel therapeutic candidates. Nat Med 26: , 289–299. |
[132] | Hardy J ((2010) ) Genetic analysis of pathways to Parkinson disease. Neuron 68: , 201–206. |
[133] | Satake W , Nakabayashi Y , Mizuta I , Hirota Y , Ito C , Kubo M , Kawaguchi T , Tsunoda T , Watanabe M , Takeda A , Tomiyama H , Nakashima K , Hasegawa K , Obata F , Yoshikawa T , Kawakami H , Sakoda S , Yamamoto M , Hattori N , Murata M , Nakamura Y , Toda T ((2009) ) Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat Genet 41: , 1303–1307. |
[134] | Simon-Sanchez J , Schulte C , Bras JM , Sharma M , Gibbs JR , Berg D , Paisan-Ruiz C , Lichtner P , Scholz SW , Hernandez DG , Kruger R , Federoff M , Klein C , Goate A , Perlmutter J , Bonin M , Nalls MA , Illig T , Gieger C , Houlden H , Steffens M , Okun MS , Racette BA , Cookson MR , Foote KD , Fernandez HH , Traynor BJ , Schreiber S , Arepalli S , Zonozi R , Gwinn K , van der Brug M , Lopez G , Chanock SJ , Schatzkin A , Park Y , Hollenbeck A , Gao J , Huang X , Wood NW , Lorenz D , Deuschl G , Chen H , Riess O , Hardy JA , Singleton AB , Gasser T ((2009) ) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41: , 1308–1312. |
[135] | Nalls MA , Plagnol V , Hernandez DG , Sharma M , Sheerin UM , Saad M , Simon-Sanchez J , Schulte C , Lesage S , Sveinbjornsdottir S , Stefansson K , Martinez M , Hardy J , Heutink P , Brice A , Gasser T , Singleton AB , Wood NW ((2011) ) Imputation of sequence variants foridentification of genetic risks for Parkinson’s disease: Ameta-analysis of genome-wide association studies.. Lancet 377: , 641–649. |
[136] | Popat RA , Van Den Eeden SK , Tanner CM , Kamel F , Umbach DM , Marder K , Mayeux R , Ritz B , Ross GW , Petrovitch H , Topol B , McGuire V , Costello S , Manthripragada AD , Southwick A , Myers RM , Nelson LM ((2011) ) Coffee, ADORA2A, and CYP1A2: The caffeine connection inParkinson’s disease. Eur J Neurol 18: , 756–765. |
[137] | Chang D , Nalls MA , Hallgrimsdottir IB , Hunkapiller J , van der Brug M , Cai F , Kerchner GA , Ayalon G , Bingol B , Sheng M , Hinds D , Behrens TW , Singleton AB , Bhangale TR , Graham RR ((2017) ) A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet 49: , 1511–1516. |
[138] | Manne S , Kondru N , Jin H , Serrano GE , Anantharam V , Kanthasamy A , Adler CH , Beach TG , Kanthasamy AG ((2020) ) Blinded RT-QuIC analysis of alpha-synuclein biomarker in skin tissue from Parkinson’s disease patients. Mov Disord 35: , 2230–2239. |
[139] | Shahnawaz M , Mukherjee A , Pritzkow S , Mendez N , Rabadia P , Liu X , Hu B , Schmeichel A , Singer W , Wu G , Tsai AL , Shirani H , Nilsson KPR , Low PA , Soto C ((2020) ) Discriminating alpha-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 578: , 273–277. |


