
Safety and Tolerability of Active Immunotherapy Targeting α-Synuclein with PD03A in Patients with Early Parkinson’s Disease: A Randomized, Placebo-Controlled, Phase 1 Study
Abstract
Background:
Immunotherapies targeting α-synuclein aim to limit its extracellular spread in the brain and prevent progression of pathology in Parkinson’s disease (PD). PD03A is a specific active immunotherapy (SAIT) involving immunization with a short peptide formulation.
Objective:
This phase 1 study characterized the safety and tolerability of PD03A in patients with early PD. A key secondary objective was to evaluate immunological activity following immunization.
Methods:
This was a phase 1 study of two different doses of PD03A versus placebo in PD patients. Patients were randomized (1:1:1) to receive four priming plus one booster vaccination of PD03A 15μg, PD03A 75μg or placebo and were followed for 52 weeks.
Results:
Overall, 36 patients were randomized, of which 35 received five immunizations and completed the study. All patients experienced at least one adverse event. Transient local injection site reactions affected all but two patients; otherwise most AEs were considered unrelated to study treatment. A substantial IgG antibody response against PD03 was observed with a maximum titer achieved at Week-12. Differences in titers between both active groups versus placebo were statistically significant from the second immunization at Week-8 until Week-52.
Conclusion:
The safety profile and positive antibody response of PD03A supports the further development of active immunotherapeutic approaches for the treatment of PD.
INTRODUCTION
Parkinson’s disease (PD), together with dementia with Lewy bodies (DLB) and multiple system atrophy (MSA), is classified as a synucleinopathy based on the shared pathogenic features of α-synuclein misfolding, oligomerization and formation of insoluble intracellular aggregates [1]. α-synuclein constitutes the major protein component of Lewy bodies [2], and the progressive loss of dopaminergic terminals (and subsequent motor impairment) is thought to be triggered by the aggregation and build-up of α-synuclein species that interfere with critical cell processes [3]. There is also strong experimental evidence for transsynaptic dissemination of aggregated α-synuclein through the extracellular space and a ‘prion-like’ spread of α-synuclein oligomers [4–6]. Studies in transgenic animal models have suggested that reducing oligomeric or aggregated forms of α-synuclein and their spread across the brain may have disease-modifying effects [7–10], providing a rationale to target pathological species of α-synuclein via immunotherapy [9, 11, 12].
To date, several monoclonal antibodies directed against α-synuclein have entered clinical development including two large double-blind phase 2 trials of passive immunotherapy in early PD [13–15]. One of them has recently reported preliminary signals of clinical efficacy after 52 weeks treatment [16]. These passive approaches currently require monthly intravenous infusions – a practical disadvantage that could be overcome through vaccination with specific active immunotherapy (SAIT), which aims to elicit a sustained, self-produced immune response against the target protein. The first results for an active immunization approach with the SAIT candidate, PD01A in PD patients have recently been published showing good safety and tolerability as well as substantial target engagement and a boostable immune response [17].
PD03A is a second SAIT candidate identified within the same preclinical screening program that identified PD01A. Like PD01A, it was selected for early clinical development based on a set of features including the specificity of the immune response and promising results from proof-of-concept studies in various models [18, 19]. The antigenic peptide (PD03) consists of a short ten amino acid long synthetic peptide that mimics an epitope in the C-terminal region of α-synuclein. This peptide is conjugated to the carrier protein keyhole limpet hemocyanin (KLH) and adjuvanted with aluminum hydroxide. The carrier protein provides the required T cell helper epitopes for the generation of a long-lasting, persistent, and boostable antigen-specific antibody response while the PD03 peptide antigen operates solely as a B cell epitope and is responsible for the specificity of the humoral immune response.
The primary objective of this phase 1, 52-week study was to characterize the safety, tolerability of two different doses of PD03A (given as five injections) in patients with early PD. A key secondary objective was to evaluate immunological activity following immunization, and we also included exploratory clinical assessments.
METHODS
Study design and participants
This was a phase 1, randomized, double-blind, placebo-controlled, 52-week study of two dosages of PD03A (15μg and 75μg) conducted in two Austrian centers between 18 November 2014 and 23 August 2016. The study was performed in accordance with Good Clinical Practice, the Declaration of Helsinki with amendments (2013), Austrian Drug Law and applicable international regulations. The protocol and associated documents were reviewed and approved by the institutional review boards of both centers, and all patients provided written informed consent. The study was registered at EudraCT (2014-000568-16).
Patients aged 45–68 years, with early stage PD [20] (Hoehn and Yahr stages 1 to 2), and a time from PD diagnosis of up to 4 years were eligible for inclusion. Patients had to be on stable symptomatic therapy with levodopa, dopamine agonists, MAO-B inhibitors or combinations; all antiparkinsonian drug classes were allowed if kept at a stable dose for≥3 months before study entry. During the study, adjustments to PD medications were made according to investigator judgement of clinical need. Baseline imaging results (dopamine transporter-SPECT and MRI) had to be consistent with a diagnosis of idiopathic PD. Patients were excluded if they had dementia, a history of relevant autoimmune disease, recent history of cancer, active infectious disease, relevant systemic illness, and history of relevant psychiatric illness. Patients who had previously received experimental immunotherapeutic treatment or immunosuppressive drugs were also excluded.
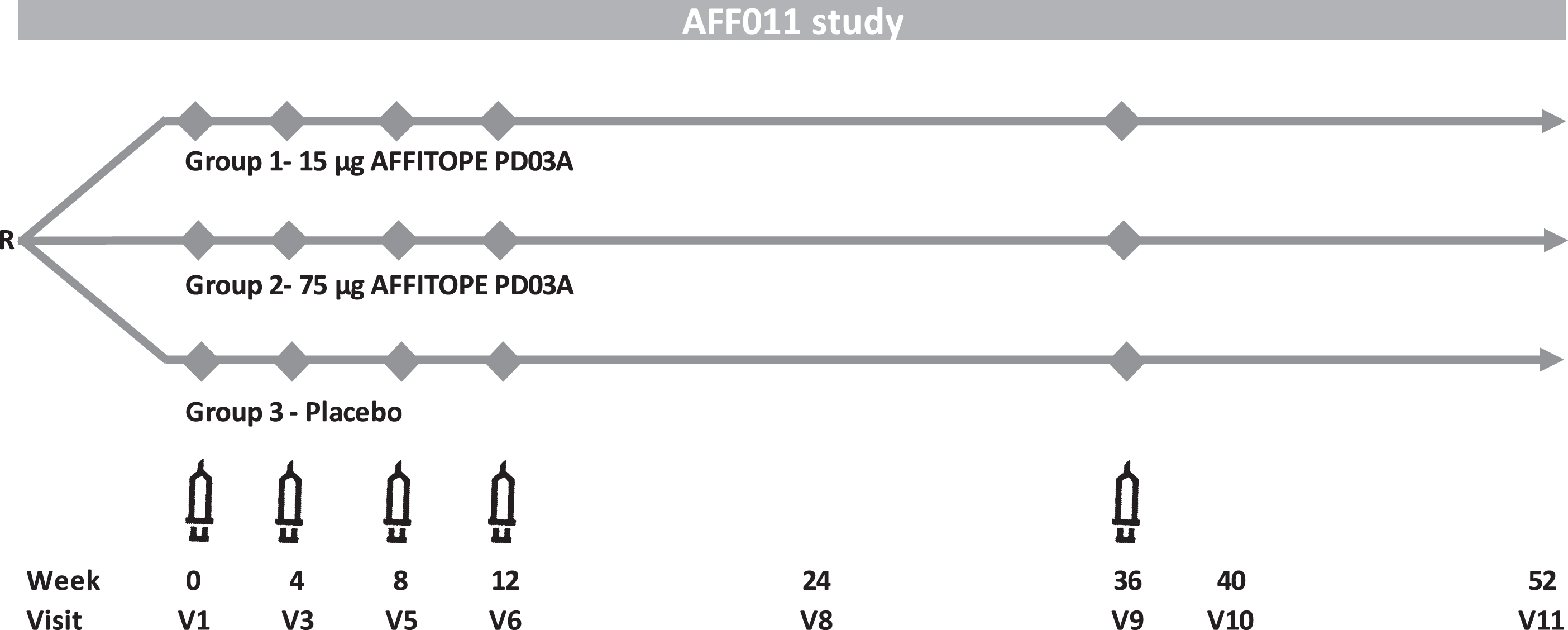
Eligible patients were randomized (1:1:1) to receive subcutaneous injections of either PD03A 15μg, PD03A 75μg or matching placebo (i.e., aluminum hydroxide in PBS). Doses refer to the net peptide amount of the applied drug product. The randomization sequence was computer-generated using the permuted blocks method with fixed block size. Priming immunizations were given at Weeks 0, 4, 8, 12, and a single booster immunization was given at Week 36 (Fig. 1). Following each immunization, patients were closely monitored for 1 hour and potential late-phase allergic reactions were assessed via telephone interview on the following day. Patients were followed for 52 weeks after the first vaccination (16 weeks after the last vaccination). Serum samples were collected at screening, baseline and Weeks 4, 8, 12, 24, 36, 40, and 52. A lumbar puncture with CSF sampling was performed during the screening period and at Week 40 or early discontinuation.
Fig. 1
PD03A phase 1 study scheme.
Safety and tolerability assessments
Safety and tolerability were assessed as primary endpoints through the recording of local or systemic treatment emergent adverse events (TEAEs) including serious and/or non-serious TEAEs possibly related to the study drug as well as TEAE-related study discontinuations. Patients completed diaries to record any injection site reaction and/or systemic reactions, on a daily basis, over a period of 7 consecutive days following each vaccination, starting about eight hours after the first vaccination. Injection site reactions (erythema, edema, induration) were classified as ‘severe’ if they were > 10 cm in diameter and pain was classified as severe if preventing daily activity or requiring use of narcotic pain reliever. In addition, patients had a complete physical examination, including vital signs, standard hematology and clinical chemistry assessments, urinalysis and serology at each study visit. Cranial magnetic resonance imaging (MRI) was performed at baseline, visit 8 (Week 24) and end of study and assessed for volumetric changes as well as any changes suggestive of encephalitic reactions (e.g., vasogenic edema, meningoencephalitis, meningioma, and microhemorrhage).
Key secondary outcomes
The key secondary objective of the study was to assess immunological activity following the immunizations. Serum samples were serially diluted (1:3 dilution steps) and evaluated by an external provider (eBioscience, Vienna, Austria) using an ELISA validated to specifically detect IgG antibodies. IgG reactivity was tested against the immunizing peptide PD03 and against the PD03 related naturally occurring α-synuclein target epitope, which represents a surrogate for the target protein structure. Titers were immune characterized for reactivity with PD03 and the native epitope on the target α-synuclein protein. A serial dilution of a human IgG pool coated to the ELISA plate was used as a calibration curve and results are presented as geometric mean end-titers (defined as last serum dilution which gave a signal that was higher than the signal of the calibration curve at penultimate dilution). Titers were also determined to the carrier protein KLH (to confirm patients’ immune competence); these were reported as half max titers. In addition, the presence of treatment-induced α-synuclein specific antibodies in the cerebrospinal fluid (CSF) was also evaluated.
Exploratory outcomes
Clinical parameters including the Movement Disorder Society-Unified Parkinson's Disease Rating Scale (MDS-UPDRS) [21], Non-Motor Symptoms Scale (NMSS) [22], PD Quality of life questionnaire (PDQ39) [23], Investigator’s global evaluation [24], and a cognitive test battery (Wisconsin Card Sorting Test, Hopkins Verbal Learning Test, Benton Judgement of line orientation, Letter Number Sequencing Test, Symbol Digit Modalities Test, Paced Auditory Serial Addition Test and Montreal Cognitive Assessment) were assessed by blinded-raters during screening and at week 24 and 52. DaT-SPECT scans (DaTscan [GE Healthcare, Chicago, IL, USA] with Siemens, Dual Head Nuclear Camera, Erlangen, Germany) were performed at baseline and Week 52 or early discontinuation visit (EDV). Changes in striatal DAT binding ratios were calculated using the occipital cortex as the reference, and analyses were performed in the native acquisition space.
Statistics
Due to the explorative nature of this first-in-human study, no formal statistical sample size calculation was performed. The study was designed to provide a precision of estimation for frequent AEs and side effects (35%occurrence) of±30%for n = 10 and±10%for n = 30 (two-sided 95%confidence intervals). The safety set included all patients who entered the study and received at least one dose of study medication. Immunological and clinical analyses were performed on the ITT population. Immune responders (i.e., patients with seroconversion) were defined as PD03A immunized patients with PD03 peptide titer ratio≥4 fold relative to baseline. Longitudinal MRI volumetric analysis was performed using a mixed models procedure including age and sex as covariates; all volumetric measures were adjusted for total intracranial volume.
Patients were analyzed according to the treatment received. Data are primarily descriptive, with no imputation for missing data. Between group differences were assessed by t-test (normally distributed data) or non-parametric Wilcoxon rank sum test (if non-normal distribution). The baseline value was defined as value of the last assessment before the first vaccination. If the baseline assessment was not available, a value from assessment at Screening was used as a baseline. All analyses were performed using SAS® software (Version 9.4).
RESULTS
The study was conducted between November 2014 and August 2016, 36 patients with early stage idiopathic PD were randomized and 35 completed the study and received all injections of study drug (Fig. 2). One patient in the placebo group discontinued after two injections due to a new diagnosis of polymyalgia rheumatica which was an exclusion criterion for the trial. Baseline characteristics were similar between study groups (Table 1).
Fig. 2
Patient disposition.
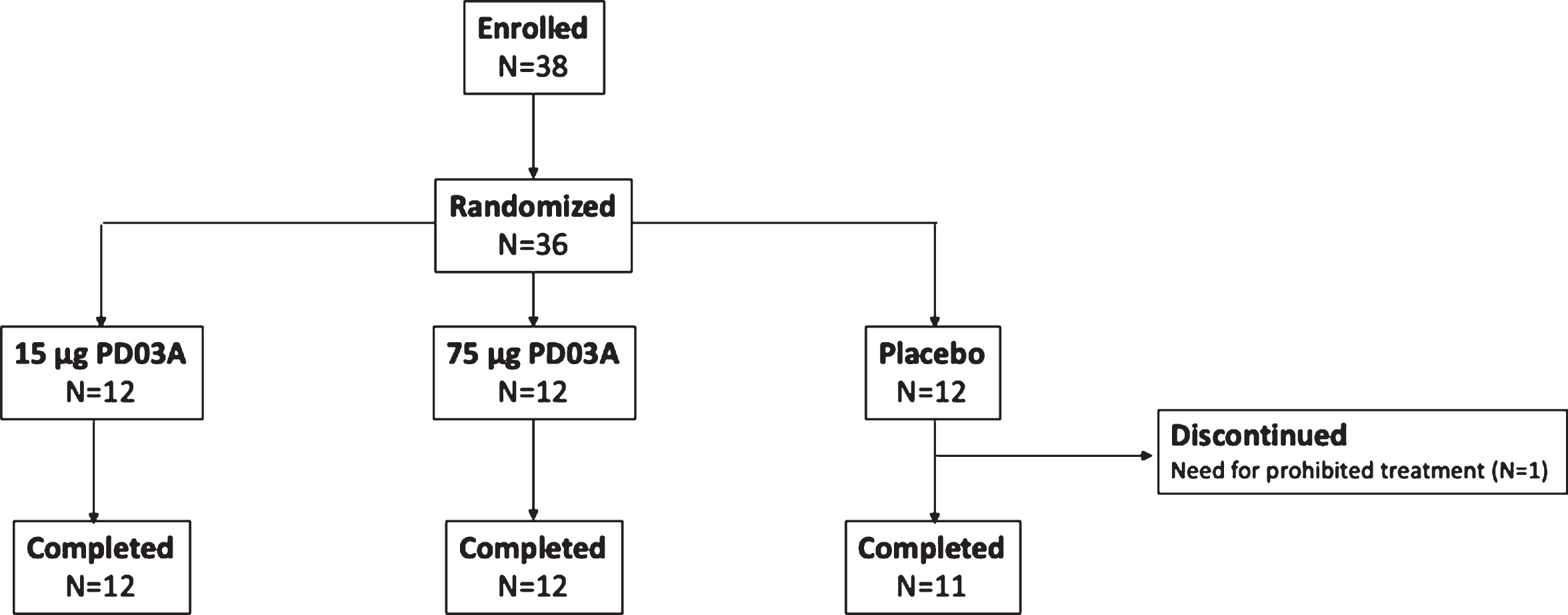
Table 1
Baseline characteristics
| Parameter | 15μg PD03A (N = 12) | 75μg PD03A (N = 12) | Placebo (N = 12) |
| Age (y); mean (SD) | 58.7 (7.4) | 54.9 (6.5) | 61.4 (4.6) |
| Sex (Female/Male); n (%) | 7 (58.3)/5 (41.7) | 2 (16.7)/10 (83.3) | 5 (41.7)/7 (58.3) |
| Mean duration of PD (y); (SD) | 1.7 (1.2) | 2.3 (1.3) | 2.0 (1.2) |
| Anti-Parkinson drugs; n (%) | 11 (91.7) | 12 (100) | 12 (100) |
| Dopamine agonists | 9 (75.0) | 11 (91.7) | 8 (66.7) |
| MAO-B inhibitors | 6 (50.0) | 6 (50.0) | 8 (66.7) |
| Levodopa | 4 (33.3) | 8 (66.7) | 5 (41.7) |
Safety and tolerability
All patients of all study groups experienced treatment-emergent adverse events (TEAEs, total of 805 events) (Table 2). Systemic AEs were infrequent and transient, while local injection site reactions (erythema, swelling, induration, warmth, pain, pruritus, granuloma) affected all but two male patients. In general, local injection site reactions were reported within 7 days post-vaccination, and most of them were of mild or moderate intensity and all of them transient. Twelve injection site reactions were rated as severe and were recorded for 4 patients receiving active treatment: injection site erythema (n = 1 receiving PD03A 15μ g and n = 2 patients receiving PD03A 75μg) and injection site swelling (n = 1 patient receiving PD03A 15μg and n = 1 patient receiving PD03A 75μg). All of these resolved within a week and none led to study withdrawal. There was no trend for increased severity of skin reactions with successive injections.
Table 2
Treatment-emergent adverse events
| 15 μg PD03A (N = 12) | 75 μg PD03A (N = 12) | Placebo (N = 12) | |
| Patients with≥1 TEAE; n (%) | 12 (100) | 12 (100) | 12 (100) |
| Patients with≥1 serious TEAE; n (%) | 2 (16.7) | 0 | 3 (25.0) |
| Patients with any TEAE leading to treatment discontinuation; n | 0 | 0 | 0 |
| Patients with≥1 local site reaction; n (%) | 12 (100) | 10 (83.3) | 12 (100) |
| Treatment emergent, treatment-related* systemic AEs occurring in ≥1 patient in any group* | |||
| Headache; n (%) | 0 | 1 (8.3) | 0 |
| Fatigue; n (%) | 0 | 0 | 1 (8.3) |
| Myalgia; n (%) | 1 (8.3) | 1 (8.3) | 0 |
*considered probably or certainly related to study treatment.
Eight serious TEAEs were reported in five patients (n = 3 in the placebo group, n = 2 in the 15μg group, and none in the 75μg group); all were considered unlikely or unrelated to study treatment (placebo: unstable angina, n = 1; mitral valve incompetence n = 1; syncope & lumbar fracture n = 1; PD03A 15μg: inguinal hernia n = 1, traumatic brain injury with subdural hematoma following a fall n = 1). There was no consistent trend for a dose-effect relationship with respect to TEAEs and no TEAE led to study treatment discontinuation.
On MRI, the active-treatment arms showed stable whole brain, ventricular and hippocampal volumes; trends to decreasing putaminal and pallidal volumes were observed in all three groups (Supplementary Figure 1). No new MRI abnormalities occurred after the baseline scan, with the exception of one patient in the 15μg group who developed a subdural hematoma related to a fall. No significant changes were reported for laboratory parameters or vital signs and there were no clinically significant abnormalities in any parameter reflecting activation of the immune system.
Immunogenicity results
Both the low dose and the high dose immunization with PD03A induced a sustained IgG antibody response against the immunizing peptide PD03 (Fig. 3A). Titers peaked at week 12 (4 weeks after the third immunization) and subsequently declined with a half-life of approximately 12-14 weeks. The geometric group mean titer of antibodies against the immunizing peptide PD03 increased from 1:73 at baseline to 1:2653 at week 12 in the 15μg dose group, and from 1:94 to 1:3240 in the 75μg dose group. Booster immunization at week 36 reactivated the specific antibody response with peak titers achieved 4 weeks after the injection resulting in geometric group mean titers of 1:2401 in the 15μg group and 1:2247 in the 75μg dose group. Twenty one out of 24 patients (88%) from the active groups showed immunological responses towards the PD03 peptide indicating that most of the patients met the predefined cutoff for serum conversion against PD03 at a factor level of at least or higher than 4 times baseline. Differences in titers between both active groups compared to placebo were statistically significant (p > 0.050) from the second immunization at Week 8 until Week 52. No statistically significant difference was seen between immune responses to the 15μg and 75μg dosages.
Fig. 3
Geometric mean antibody titers over time for (A) PD03A peptide and (B) native target sequence on α-synuclein.
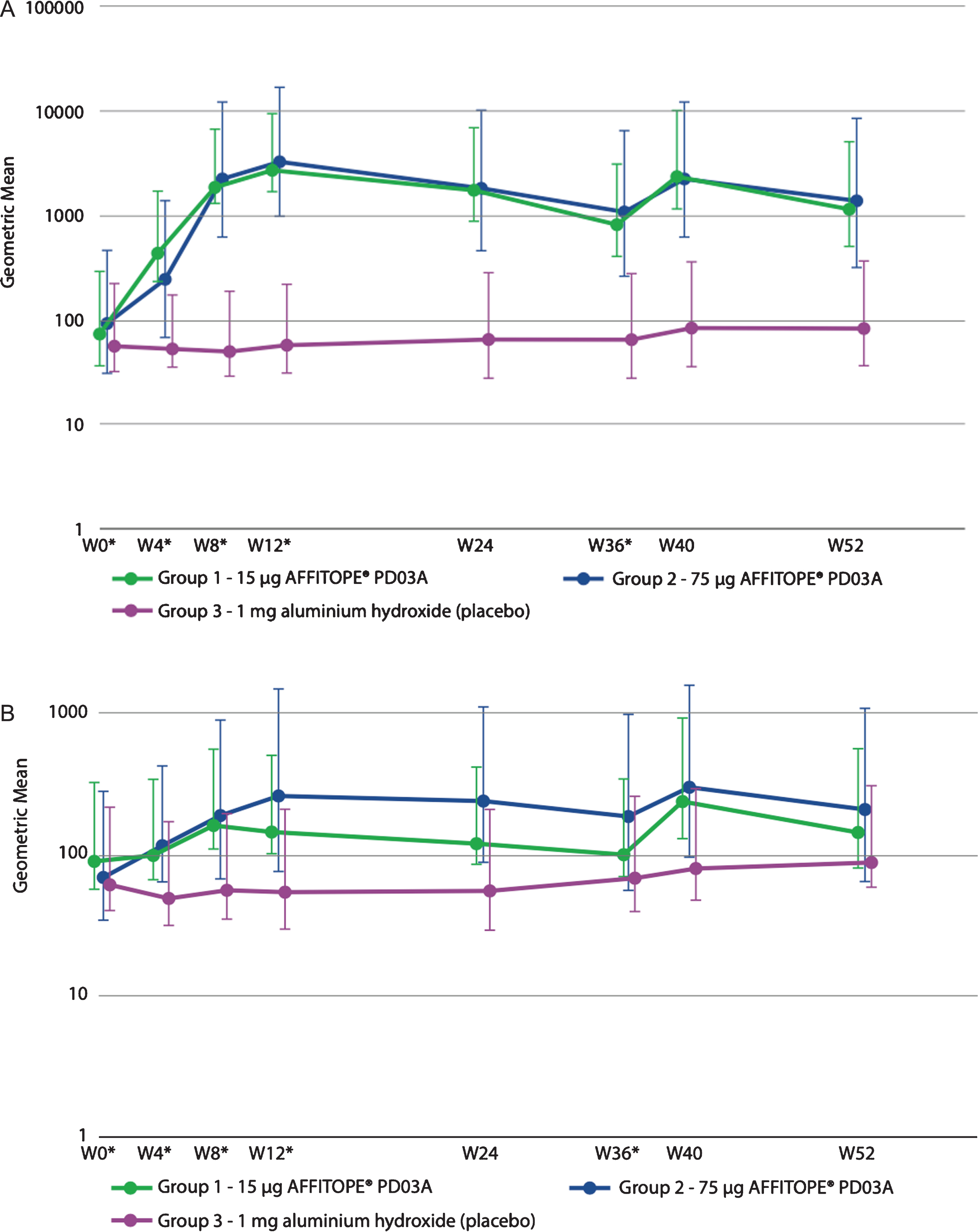
Antibody titers against the α-synuclein target epitope were lower than those detected against PD03, but the profile of antibody development was very similar (Fig. 3B). Geometric mean titers increased from 1:90 at baseline to 1:148 after three immunizations in the 15μg dose group and from 1:71 to 1:270 in the 75μg dose group, respectively. In both groups the booster injection induced higher geometric mean titers than those overserved with the priming injections reaching titers of 1:244 in the 15μg dose group and 1:305 in the 75μg dose group. Thus, the primary immunization produced a substantial memory effect that was reactivated and augmented during the booster immunization. Statistically significant differences compared to placebo were detected for the 15μg PD03A group after the second immunization at week 8 (p = 0.0189) and at after the booster immunization at week 40 (p = 0.0258). For the 75μg PD03A group, significant differences were detected after the second immunization at week 8 (p = 0.0175) until week 24 and after the booster immunization at week 40 (p = 0.0175). There were no significant differences between the two dose levels.
All patients (both active groups) demonstrated an immune response towards KLH with a titer profile very similar to that observed with the immunizing PD03 peptide and the α-synuclein target epitope, thus demonstrating a general responsiveness of the patients’ immune system (Supplementary Figure 2). As expected, antibody reactivity to the immunizing peptide PD03, to the α-synuclein target epitope and to the carrier protein did not change over time in the placebo group. No α-synuclein-specific antibody titers could be measured in the CSF-derived samples from the active immunization groups.
Exploratory outcomes
MDS-UPDRS scores (subscales 1 to 3) showed little change from baseline to end of study and there were no significant differences between treatment groups (Supplementary Table 1). At the last visit, mean±SD MDS-UPDRS Part 3 (motor) scores change from baseline was 1.2±7.1 points in the placebo group versus 3.0±6.5 in the 15μg group and 0.2±7.0 in the 75μg group. At the same time there was a small to moderate increase in the mean Levodopa Equivalent Dose (LED) over the course of the study (Supplementary Table 2). Across all groups, there were little or no changes from baseline in non-motor symptoms (MDS-UPDRS Part 1, non-motor experiences of daily living scores and NMSS scores), cognitive assessments and PDQ-39 scores. There were no differences in change of DAT-SPECT striatal binding ratios from baseline to end-of-study in any study group (Supplementary Figure 3).
Discussion
The results of this phase 1, first-in-human study indicate that immunization with PD03A is safe and well-tolerated in patients with early PD. PD03A immunotherapy triggered the induction of a specific IgG response to the injected PD03 peptide as well as to the α-synuclein target epitope, which could be rapidly reactivated upon a booster injection. Overall, 88%of patients from the active groups showed an immunological response towards the PD03 peptide and differences in immune response were statistically significant between active groups and placebo. The specific immune response could also be rapidly reactivated after a booster application.
Overall, PD03A demonstrated a good safety and tolerability profile, with transient local injection site reactions being the main treatment-emergent adverse events. The most common TEAEs with PD03A were injection site erythema, swelling, induration, pruritus, or injection-site pain. These were transient, mild to moderate in severity and consistent with known vaccine-associated hypersensitivity [25]. Four patients receiving active immunization experienced severe local reactions, but all resolved in less than a week and none led to study withdrawal. Most systemic TEAEs were unspecific and considered unlikely to be related to study drug, while headache, fatigue and myalgia reported by four patients following an injection would appear as plausible vaccination-related AEs. There was no evidence for CNS inflammatory responses on MRI, and there was also no indication of accelerated brain atrophy with active treatment as has been described in a recent passive immunotherapy trial targeting Aβ in Alzheimer’s disease [26].
We have recently reported the results for the SAIT candidate, PD01A which also targets the α-synuclein protein [17]. Neither SAIT has shown signs of dose-dependent safety patterns and there has been no suggestion of cumulative toxicity over time or with booster injections. Also, similar to the findings with PD01A [17], active immunization with PD03A resulted in a significant increase in titers against the immunizing peptide and the specific immune response could also be rapidly reactivated after booster application. Neither of the two SAIT candidates showed significant dose-dependent immune responses after the priming injections. However, whereas PD01A clearly induced higher titers after the 75μg booster application compared to the 15μg booster dose, this ‘boost’ effect was not observed with PD03A, suggesting that the two SAIT candidates differ in their potential to reactivate antibody responses. In parallel to this study, PD01A and PD03A have also been tested in patients with MSA [27]. In that study, PD03A was also shown to induce a specific antibody response and to be generally well tolerated, but the detected antibody response triggered by PD01A was higher than that of PD03A.
The finding that the immune response to the α-synuclein target epitope was approximately one order of magnitude lower than that seen to the immunizing peptide can partly be explained by the binding of product-induced antibodies to the target structure. Since PD03-induced antibodies bind to oligomeric forms of α-synuclein it is likely that both arms of the antibodies are bound to the target structure and thus become ‘masked’ for ELISA readouts using α-synuclein target epitope as substrate. It is also possible that the surrogate substrate for oligo-α-synuclein used in the ELISA assay does not fully reproduce the target structure and the antibody titers measured in this assay underestimate the level of antibodies generated against the oligomeric forms of α-synuclein. It is interesting to note that the baseline titers for PD03 (1:73 and 1:94 in the 15μg and 75μg dose groups, respectively) and the related α-synuclein target epitope (1:90 and 1:71, in the 15μg and 75μg dose groups, respectively) were already somewhat elevated at baseline, potentially indicating that there might be an IgG antibody fraction present in plasma samples of PD patients that reacted with PD03 and α-synuclein. Naturally occurring autoantibodies (NAbs) towards α-synuclein have been detected in human plasma and are assumed to be involved in the maintenance of physiological and immune homeostasis (e.g., by removing aging cells, cellular debris and even aggregated α-synuclein) [28]. However, it has been reported that the repertoire of IgG autoantibodies against α-synuclein is significantly reduced in patients with PD [29], and the biological meaning and relevance of anti-α-synuclein antibodies in PD requires further study.
The antigenic peptides PD01 and PD03 were designed to mimic the amino acid sequence of a critical epitope in the C-terminal region of the α-synuclein protein, with the introduction of targeted amino acid substitutions in the original sequence. This is done with the aim of breaking immune tolerance to this self-protein and generating high titer antibody responses to the immunizing peptide which cross react with the native protein, without induction of harmful off-target auto immune responses [19]. This aim was realized with the PD01 candidate product [17], and the approach has been confirmed in this study with PD03 with respect to safety and immunogenicity. We did not detect vaccine-induced antibodies against monomeric or filamentous α-synuclein in the CSF at week 40. Little is known about the level of α-synuclein-specific antibodies in the brain required to achieve a therapeutic clinical effect, although prior studies with passive immunotherapy approaches have shown that antibody levels in CSF are dependent on the antibody concentration in the circulation [13, 14], and that plasma antibody concentrations should be between 40–400μg/ml [13]. While the exact plasma antibody concentration induced by PD03 in this study has not been evaluated, it is considered likely that the immunogenicity of SAIT products has to be increased. With this in mind, the formulation of the SAIT product being developed for the planned phase 2 clinical trial has been optimized with the aim of inducing antibody levels in humans that are one order of magnitude higher compared to the levels achieved in this study.
Limitations of this small phase 1 study include the fact that it was not powered to detect signals of clinical efficacy and, indeed, there were no statistically significant differences observed in striatal DAT-SPECT binding ratios nor the clinical scales used in this study. In addition, investigators were allowed to adapt symptomatic medications when clinically needed during the trial and the group which demonstrated the least worsening in motor symptoms (the 75μg dose group) also received the highest mean increase in levodopa dose equivalents from baseline to week 52. We performed an exploratory analysis of correlations between changes in clinical parameters and antibody response and observed signals of possible benefit some clinical outcomes at last visit in individual patients with high values of α-synuclein specific antibody. However, there was no consistent trend or correlation. This is perhaps not surprising given the small sample size and short duration of the study. Larger studies with a longer observation period will be required to evaluate such correlations.
In summary, the safety profile and positive antibody response of PD03A as evidenced in this study further supports the development of the SAIT approach for the treatment of PD in a phase 2 clinical trial. The lead SAIT candidate PD01A, has shown higher immunogenicity compared to the current results with PD03A in a similar patient population [17] and also in a recent study in MSA patients [27], arguing for its preferential use in future clinical trials. Based on the data from these studies, a larger 18-month, phase 2 clinical trial is planned to investigate the clinical efficacy of PD01. Further studies will also be needed to determine the persistence of the immune response and the best interval for booster immunizations, but available data from the PD01A trial [17] indicate that a yearly booster immunization could be sufficient to maintain high titers of therapeutic antibodies.
ACKNOWLEDGMENTS
We thank the patients and site staff involved in the study. The authors would like to thank the AFF011 DSMB board for their feedback and Carsten Schwenke for the statistical oversight. Medical writing support was provided by Anita Chadha-Patel (ACP Clinical Communications, funded by AFFiRiS AG).
The study was part of an EU-funded program (FP7, SYMPATH grant agreement 602999)
CONFLICT OF INTEREST
This study was part of an EU funded program (FP7, SYMPATH Consortium). Werner Poewe was an investigator in the study and reports receiving personal fees from AFFiRiS. Dieter Volc and Caroline Thun-Hohenstein were investigators in the study and have received funding from AFFiRiS AG. Rossella Medori has received consultancy fees from AFFiRiS; Petra Lührs and Alexandra Kutzelnigg are employed by AFFiRiS AG and Achim Schneeberger was employed by AFFiRiS AG at the time of study. Klaus Seppi and Atbin Djamshidian were investigators in the study. Wassilios Meissner and Olivier Rascol were advisors to the study as part of the SYMPATH Consortium.
Werner Poewe reports receiving personal fees from AFFiRiS, AbbVie, AstraZeneca, BIAL, Boston Scientific, Britannia, Intec, Ipsen, Lundbeck, NeuroDerm, Neurocrine, Denali Pharmaceuticals, Novartis, Orion Pharma, Prexton, Teva, UCB and Zambon. He receives royalties from Thieme, Wiley Blackwell, Oxford University Press and Cambridge University Press and grant support from the Michael J Fox Foundation, EU FP7 and Horizon 2020. Dieter Volc and Caroline Thun-Hohenstein were investigators in the study and received funding from AFFiRiS AG. Klaus Seppi reports personal fees from Teva, UCB, Lundbeck, AOP Orphan Pharmaceuticals AG, Roche, Grünenthal, Stada, Licher Pharma, Biogen and Abbvie, honoraria from the International Parkinson and Movement Disorders Society, research grants from FWF Austrian Science Fund, Michael J. Fox Foundation, and AOP Orphan Pharmaceuticals AG, outside the submitted work. Rossella Medori has received consultancy fees from AFFiRiS. Petra Lührs, Alexandra Kutzelnigg, Achim Schneeberger and Günther Staffler are currently or were in the past employed by AFFiRiS AG.
Atbin Djamshidian reports receiving honoraria from Abbvie and Biogen.
Wassilios Meissner reports fees for editorial activities with Springer Nature and Elsevier, consultancy fees from Lundbeck and Biohaven, and teaching honoraria from UCB.
Olivier Rascol has acted as a scientific advisor for drug companies developing antiparkinsonian medications (AbbVie, Adamas, Acorda, Addex, Aguettant, Alkahest, AlzProtect, Apopharma, Astrazeneca, Bial, Biogen, Britannia, Buckwang, Cerevel, Clevexel, Irlab, Eli-Lilly, Lundbeck, Neuroderm, ONO Pharma, Orion Pharma, Osmotica, Oxford Biomedica, Pfizer, Prexton Therapeutics, Sanofi, Servier, Sunovion, Théranexus, Takeda, Teva, UCB, Watermark Research, XenoPort, XO, Zambon) and has received unrestricted scientific grants from academic non-profit entities (Agence Nationale de la Recherche (ANR), CHU de Toulouse, France-Parkinson, INSERM-DHOS Recherche Clinique Translationnelle, MJ Fox Foundation, Programme Hospitalier de Recherche Clinique du Ministère de la Santé, European Commission (FP7, H2020).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-212594.
REFERENCES
[1] | Golde TE , Borchelt DR , Giasson BI , Lewis J ((2013) ) Thinking laterally about neurodegenerative proteinopathies. J Clin Invest 123: , 1847–1855. |
[2] | Poewe W , Seppi K , Tanner CM , Halliday GM , Brundin P , Volkmann J , Schrag AE , Lang AE ((2017) ) Parkinson disease. Nat Rev Dis Primers 3: , 17013. |
[3] | Alam P , Bousset L , Melki R , Otzen DE ((2019) ) alpha-synuclein oligomers and fibrils: A of species, a spectrum of toxicities. J Neurochem 150: , 522–534. |
[4] | Chu Y , Muller S , Tavares A , Barret O , Alagille D , Seibyl J , Tamagnan G , Marek K , Luk KC , Trojanowski JQ , Lee VMY , Kordower JH ((2019) ) Intrastriatal alpha-synuclein fibrils in monkeys: Spreading, imaging and neuropathological changes. Brain 142: , 3565–3579. |
[5] | Prusiner SB , Woerman AL , Mordes DA , Watts JC , Rampersaud R , Berry DB , Patel S , Oehler A , Lowe JK , Kravitz SN , Geschwind DH , Glidden DV , Halliday GM , Middleton LT , Gentleman SM , Grinberg LT , Giles K ((2015) ) Evidence for alpha-synuclein prions causing multiple system atrophy in humans with parkinsonism. Proc Natl Acad Sci U S A 112: , E5308–5317. |
[6] | Visanji NP , Lang AE , Kovacs GG ((2019) ) Beyond the synucleinopathies: Alpha synuclein as a driving force in neurodegenerative comorbidities. Transl Neurodegener 8: , 28. |
[7] | Games D , Seubert P , Rockenstein E , Patrick C , Trejo M , Ubhi K , Ettle B , Ghassemiam M , Barbour R , Schenk D , Nuber S , Masliah E ((2013) ) Axonopathy in an alpha-synuclein transgenic model of Lewy body disease is associated with extensive accumulation of C-terminal-truncated alpha-synuclein. Am J Pathol 182: , 940–953. |
[8] | Kim C , Lee SJ ((2008) ) Controlling the mass action of alpha-synuclein in Parkinson’s disease. J Neurochem 107: , 303–316. |
[9] | Valera E , Masliah E ((2013) ) Immunotherapy for neurodegenerative diseases: Focus on alpha-synucleinopathies. Pharmacol Ther 138: , 311–322. |
[10] | Brundin P , Dave KD , Kordower JH ((2017) ) Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol 298: , 225–235. |
[11] | Schwartz M ((2017) ) Can immunotherapy treat neurodegeneration? Science 357: , 254–255. |
[12] | Antonini A , Bravi D , Sandre M , Bubacco L ((2020) ) Immunization therapies for Parkinson’s disease: State of the art and considerations for future clinical trials. Expert Opin Invest Drugs 29: , 685–695. |
[13] | Jankovic J , Goodman I , Safirstein B , Marmon TK , Schenk DB , Koller M , Zago W , Ness DK , Griffith SG , Grundman M , Soto J , Ostrowitzki S , Boess FG , Martin-Facklam M , Quinn JF , Isaacson SH , Omidvar O , Ellenbogen A , Kinney GG ((2018) ) Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-alpha-synuclein monoclonal antibody, in patients with Parkinson disease: A randomized clinical trial. JAMA Neurol 75: , 1206–1214. |
[14] | Brys M , Fanning L , Hung S , Ellenbogen A , Penner N , Yang M , Welch M , Koenig E , David E , Fox T , Makh S , Aldred J , Goodman I , Pepinsky B , Liu Y , Graham D , Weihofen A , Cedarbaum JM ((2019) ) Randomized phase I clinical trial of anti-alpha-synuclein antibody BIIB054. Mov Disord 34: , 1154–1163. |
[15] | Chatterjee D , Kordower JH ((2019) ) Immunotherapy in Parkinson’s disease: Current status and future directions. Neurobiol Dis 132: , 104587. |
[16] | Pagano G , Taylor K , Cabrera J , Marchesi M , Zago W , Tripuraneni R , Boulay A , Vogt A , Boess F , Nikolcheva T , Svoboda H , Britschgi M , Lipsmeier F , Lindemann M , Dziadek S , Azulay J , Mollenhauer B , Manzanares L , Russell D , Boyd J , Nicholas A , Luquin M , Hauser RA , Simuni T , Gasser T , Poewe W , Kinney G , Doody R , Fontoura P , Umbricht D , Bonni A ((2020) ) PASADENA: A Phase 2 study to evaluate the safety and efficacy of prasinezumab in early Parkinson’s disease; Part 1 Week-52 results [abstract]. Mov Disord 35 (Suppl 1): , https://www.mdsabstracts.org/abstract/pasadena-a-phase-2-study-to-evaluate-the-safety-and-efficacy-of-prasinezumab-in-early-parkinsons-disease-part-1-week-52-results/. Accessed April 23, 2021. |
[17] | Volc D , Poewe W , Kutzelnigg A , Luhrs P , Thun-Hohenstein C , Schneeberger A , Galabova G , Majbour N , Vaikath N , El-Agnaf O , Winter D , Mihailovska E , Mairhofer A , Schwenke C , Staffler G , Medori R ((2020) ) Safety and immunogenicity of the alpha-synuclein active immunotherapeutic PD01A in patients with Parkinson’s disease: A randomised, single-blinded, phase 1 trial. Lancet Neurol 19: , 591–600. |
[18] | Mandler M , Valera E , Rockenstein E , Weninger H , Patrick C , Adame A , Santic R , Meindl S , Vigl B , Smrzka O , Schneeberger A , Mattner F , Masliah E ((2014) ) Next-generation active immunization approach for synucleinopathies: Implications for Parkinson’s disease clinical trials. Acta Neuropathol 127: , 861–879. |
[19] | Schneeberger A , Mandler M , Mattner F , Schmidt W ((2010) ) AFFITOME(R) technology in neurodegenerative diseases: The doubling advantage. Hum Vaccin 6: , 948–952. |
[20] | Hughes AJ , Daniel SE , Kilford L , Lees AJ ((1992) ) Accuracy of clinical diagnosis of idiopathic Parkinson’s disease: A clinico-pathological study of 100 cases. J Neurol Neurosurg Psychiatry 55: , 181–184. |
[21] | Goetz CG , Tilley BC , Shaftman SR , Stebbins GT , Fahn S , Martinez-Martin P , Poewe W , Sampaio C , Stern MB , Dodel R , Dubois B , Holloway R , Jankovic J , Kulisevsky J , Lang AE , Lees A , Leurgans S , LeWitt PA , Nyenhuis D , Olanow CW , Rascol O , Schrag A , Teresi JA , van Hilten JJ , LaPelle N ; Movement Disorder Society UPDRS Revision Task Force ((2008) ) Movement Disorder Society-sponsored revision of the Unified Parkinson’s Disease Rating Scale (MDS-UPDRS): Scale presentation and clinimetric testing results. Mov Disord 23: , 2129–2170. |
[22] | Chaudhuri KR , Martinez-Martin P , Brown RG , Sethi K , Stocchi F , Odin P , Ondo W , Abe K , Macphee G , Macmahon D , Barone P , Rabey M , Forbes A , Breen K , Tluk S , Naidu Y , Olanow W , Williams AJ , Thomas S , Rye D , Tsuboi Y , Hand A , Schapira AH ((2007) ) The metric properties of a novel non-motor symptoms scale for Parkinson’s disease: Results from an international pilot study. Mov Disord 22: , 1901–1911. |
[23] | Peto V , Jenkinson C , Fitzpatrick R , Greenhall R (1995) The development and validation of a short measure of functioning and well being for individuals with Parkinson’s disease. Qual Life Res 241–248. |
[24] | Guy W ((1976) ) Clinical global impressions. In ECDEU Assessment Manual for Psychopharmacology Department of Health, Education, and Welfare, Washington, DC., Rockville, MD, pp. 218–222. |
[25] | McNeil MM , DeStefano F ((2018) ) Vaccine-associated hypersensitivity. J Allergy Clin Immunol 141: , 463–472. |
[26] | Mintun MA , Lo AC , Duggan Evans C , Wessels AM , Ardayfio PA , Andersen SW , Shcherbinin S , Sparks J , Sims JR , Brys M , Apostolova LG , Salloway SP , Skovronsky DM ((2021) ) Donanemab in early Alzheimer’s disease. New Engl J Med 384: , 1691–1704. |
[27] | Meissner WG , Traon AP-L , Foubert-Samier A , Galabova G , Galitzky M , Kutzelnigg A , Laurens B , Lührs P , Medori R , Péran P , Sabatini U , Vergnet S , Volc D , Poewe W , Schneeberger A , Staffler G , Rascol O ((2020) ) A phase 1 randomized trial of specific active α-synuclein immunotherapies PD01A and PD03A in multiple system atrophy. Mov Disord 11: , 1957–1965. |
[28] | Lutz HU ((2007) ) Homeostatic roles of naturally occurring antibodies: An overview. J Autoimmun 29: , 287–294. |
[29] | Brudek T , Winge K , Folke J , Christensen S , Fog K , Pakkenberg B , Pedersen L ((2017) ) Autoimmune antibody decline in Parkinson’s disease and Multiple System Atrophy; a step towards immunotherapeutic strategies. Mol Neurodegener 12: , 44. |


