
Effects of Helicobacter pylori on Levodopa Pharmacokinetics
Abstract
Background:
Infection with Helicobacter pylori seems overrepresented in Parkinson’s disease. Clinical observations suggest a suboptimal treatment effect of levodopa in Helicobacter positive patients.
Objective:
Describe and explain the connection between a Helicobacter pylori infection of the upper gut and changes in pharmacokinetics of oral levodopa treatment in Parkinson’s disease.
Methods:
PubMed, Google Scholar, and Cross Reference search was done using the key words and combined searches: Bioavailability, drug metabolism, dyskinesia, Helicobacter, L-dopa, levodopa, motor control, pharmacodynamics, pharmacokinetics, prevalence, unified Parkinson’s disease rating scale.
Results:
The prevalence of Helicobacter pylori in Parkinson’s disease patients is reported to be about 1.6-fold higher than in a control population in some studies. Helicobacter has therefore been assumed to be linked to Parkinson’s disease, but the mechanism is unclear. As regards symptoms and treatment, patients with Parkinson’s disease on levodopa therapy and with Helicobacter pylori infection display worse motor control than those without Helicobacter infection. Eradication of the infection improves levodopa response in Parkinson’s disease, likely as a consequence of an increased oral pre-systemic bioavailability of levodopa, likely to be explained by reduced Helicobacter-dependent levodopa consumption in the stomach. In addition, small intestinal bacterial overgrowth may also have an impact on the therapeutic setting for levodopa treatment but is less well established.
Conclusion:
Eradication of Helicobacter pylori improves levodopa bioavailability resulting in improved motor control. Eradication of Helicobacter should be considered in patients with poor symptomatic control and considerable motor fluctuations.
INTRODUCTION
Already in the 1960s, an association between gastric ulcer and Parkinson’s disease (PD) was reported. In a study of 200 PD patients and 200 age- and sex-matched controls, Strang [1] noted a statistically significant higher incidence of ulcers (diagnosed by X-ray or surgery) in PD patients compared to the controls, with ulcers preceding PD diagnosis by years. At first, the increased prevalence of gastric ulcer in PD patients was considered an independent feature of the disease with gastrointestinal (GI) symptoms as one of many clinical hallmarks experienced by all PD patients. Later, researchers found clear relationships between PD, levodopa treatment and Helicobacter pylori (HP), and many theories have been forwarded to explain the role of HP in PD [2– 5].
PD is a complex disease entity of unknown etiology [6]. The role of the GI system in PD has been much debated. The onset of PD is often preceded by a period of GI dysfunction, suggesting that the condition might originate in the gut and spread to the brain along the gut-brain axis. This hypothesis is, however, still debated [7, 8]. In this context, HP has been suggested to be involved in PD in several ways: First, epidemiological observations imply an association between HP and PD. Second, HP may interfere with the absorption of levodopa. Third, HP may affect the pharmacokinetic properties with subsequent effects on the levodopa efficacy.
In order to shed light on the importance of the gut microbiota in terms of HP infection and small intestinal bacterial overgrowth (SIBO) for the pharmacokinetics and pharmacodynamics of levodopa in PD treatment, a search for peer reviewed articles was carried out in PubMed, Google scholar and Cross Reference using the key words (alone and in combination): bioavailability, drug metabolism, dyskinesia, Helicobacter, l-dopa, levodopa, motor control, pharmacodynamics, pharmacokinetics, prevalence, unified Parkinson’s disease rating scale.
A CAUSATIVE ROLE OF HELICOBACTER PYLORI IN PARKINSON’S DISEASE?
There is today epidemiological data to suggest an increased prevalence of HP infection in PD patients (Table 1). As compared to healthy controls, one case-control study has shown HP antibodies to be 5-fold more common in PD patients over 80 years of age [9]; another study found HP to be 3-fold more common [10].
Table 1
Prevalence of Helicobacter pylori in Parkinson’s disease patients
| Author (year) [ref] | Helicobacter pylori prevalence in PD (%) | Helicobacter pylori prevalence in background population (%) |
| Dobbs et al. (2000) [2] | 48 | <40 |
| Pierantozzi et al. (2006) [11] | 36 | 50– 69 |
| Lee et al. (2008) [12] | 53 | 50– 69 |
| Dobbs et al. (2012) [13] | 70 | <40 |
| Tan et al. (2015) [14] | 32 | <40 |
| Mridula et al. (2017) [15] | 50 | 50– 69 |
Background population Helicobacter pylori prevalence according to [16].
A concluding meta-analysis of eight eligible studies comprising 33,125 patients showed the pooled odds ratio of PD in HP-infected persons to be 1.59 (1.37– 1.85). In subgroup analyses, the combined odds ratios of PD in HP-infected subjects were 1.96 (1.23– 3.12) in Asia, 1.55 (1.32– 1.82) in Europe, 1.59 (1.35– 1.88) in case-control studies, 1.56 (1.01– 2.39) in cross-sectional studies, 1.56 (1.32– 1.85) in studies with confounders adjusted, and 1.71 (1.21– 2.43) in studies with no confounder adjusted. Hence, HP infection might be associated with an increased risk of PD [17].
As regards a causative role of HP for development of PD, there is a hypothesis of HP generating a neurotoxic effect by elaborating cholesterol glucosides, ultimately leading to degeneration of dopaminergic neurons in the brain [5]. According to another hypothesis an HP infection which is not controlled by the immune system or not eradicated, permits immunologically activated molecules to pass the blood-brain barrier, leading to damage of dopaminergic cells in the brain and development of PD [18]. Hence, HP might be involved in the pathogenesis of PD by inducing apoptosis through an action nitric oxide on nerve cells in critical areas of the brain [19].
Translational studies in humans show that autoantibodies which are elevated in HP-positive PD patients recognize proteins that are essential for normal brain function, such as Nuclear factor I subtype A, platelet-derived growth factor B and eukaryotic translation initiation factor 4A3 [20]. This suggests that HP may cause the immune system to generate antibodies that are able to attack proteins within the brain that are required for normal brain function, thus making HP-infected people more vulnerable to development of PD. The presence of elevated autoantibodies against proteins essential for normal neurological functions suggests that immunomodulatory properties of HP may explain the association between the bacterium and greater PD motor dysfunction and disease severity. Today, the hypotheses that HP infection is a predisposing factor for PD, a disease progression modifier, or even a direct cause of PD remain largely unexplored. The GI pathology in PD seems multifactorial, involving HP, the intestinal microflora, inflammatory response, or misfolding of alpha-synuclein in the gut and brain, cholesterol and other metabolites, as well as potential neurotoxins directly from bacteria or dietary sources.
LEVODOPA THERAPY
The dopamine precursor levodopa (L-dopa, L-3,4-dihydroxyphenylalanine) is the hallmark of symptomatic treatment of PD. Peripherally acting DOPA decarboxylase inhibitors, such as carbidopa or ben-serazide, are always co-administered with levodopa in order to increase brain bioavailability of the drug. After gastric emptying, levodopa absorption in the small intestine is suggested to be mediated by the large neutral amino acids transport machinery, likely the L-type amino acid transporters (LATs) [21], which are obligatory exchangers and transport all common amino acids. Co-administration of levodopa and dietary amino acids is avoided in order to decrease the competition for the carrier-mediated transport the intestine. After uptake to the blood st-ream, levodopa crosses the blood-brain barrier by way of LATs, and is enzymatically decarboxylated in neuronal tissue into dopamine which exerts its pharmacological end-action on postsynaptic dopamine D2 (and possibly D3) receptors [22]. This pulsatile dopamine signaling is, however, short-lived and a rapid degradation takes place through the ac-tion of neuronal monoamine oxidase (MAO) and cathechol-O-methyl transferase (COMT). Therefore, administration of levodopa has to be repeated multiple times a day to achieve stable and acceptable body movements and motor control. Chronic levodopa treatment is limited by the shortening duration of the motor and non-motor improvement and fluctuating responses to the drug, resulting in ‘wearing-off’ phenomena and dyskinesia [23– 25], common problems in the clinical setting. The long-term treatment response gradually shortens along with disease progression and the pharmacokinetic behavior of levodopa is mirrored in the symptomatic relief as the patient has reached the phase of motor fluctuations. Several attempts to modify the pharmacokinetics of levodopa have been made. The most effective concept involves continuous drug delivery, especially duodenal infusion of levodopa including peripheral enzyme inhibitors [26]. Others are the use of peripheral DOPA decarboxylase inhibition, by e.g. carbidopa or benserazide, in order to increase brain bioavailability of levodopa, as well as MAO inhibition to prolong the effect duration of each dose.
Levodopa-induced dyskinesia is common in the treatment of PD. This complication is due to phasic stimulation of dopamine receptors, levodopa-to-dopamine conversion in serotonergic neurons, hyperactivity of glutamatergic transmission, and overstimulation of nicotinic autoreceptors enhancing dopamine release. Development of dyskinesia is a function of disease duration rather than cumulative levodopa medication [27]. Improvements of levodopa delivery and bioavailability have been suggested to reduce its prevalence [27].
Since the majority of PD patients are treated with oral levodopa formulations with rapid onset and turnover, any factor influencing the pharmacokinetics and pharmacodynamics of levodopa is of utmost interest for the optimization of therapy.
HELICOBACTER INTERFERENCE WITH LEVODOPA PHARMACOKINETICS AND EFFICACY
HP infection has been suggested to impair drug absorption. Diminished drug absorption was found in HP infected patients under replacement therapies with iron, thyroxin and levodopa. Following eradication therapy, an improvement of drug absorption was noticed along with an inverse correlation between severity of gastric inflammation and drug absorp-tion indices [28].
As regards the importance of HP for the levodopa treatment response in PD, pharmacokinetic studies of levodopa in HP-infected subjects have been carried out in a setting of an established PD diagnosis. In a first report, fluctuations in the levodopa absorption in six HP-infected patients with PD were observed, at difference with HP-negative counterparts. As an extension of this, unified Parkinson’s disease rating scale (UPDRS) scores were lower after HP eradication [29]. Furthermore, the ‘onset’ time of levodopa effects was longer, and its duration shorter in PD patients with concomitant HP infection compared to the HP-negative ones. It therefore seems that HP may disrupt levodopa absorption. To this end, eradication of HP decreased the onset time and increased the duration of the levodopa effect when compared to pretreatment measurements. It therefore appears that HP reversibly affects levodopa absorption. Examining the result of the plasma concentration-time graph of levodopa in detail shows that the absorption phase of a 250 mg oral single dose is similar between pre- and post-HP eradication conditions, whereas the elimination phase displays a greater bioavailability and higher drug exposure (area under the curve, AUC) after HP eradication over the remaining study period. Hence, data speaks for a prolonged and more efficient absorption process after HP eradication. This theor-etically would not be due to changes in rapid membrane passage or increased blood flow during the ab-sorption phase, but rather a slower effect possibly due to increased absorption of levodopa from the small intestine either through reduced bacterial metabolism of levodopa in the stomach and intestine, or a hastened gastric emptying. Furthermore, a study carried out in seven subjects showed similar results with increased bioavailability during the elimination phase of the plasma concentration-time graph (Fig. 1) [30].
Fig.1
Pharmacokinetic model of levodopa plasma concentrations after oral administration of 250 mg levodopa with or w/o antibiotic treatment directed against Helicobacter pylori in Parkinsons’s disease patients (after reference [30]).
![Pharmacokinetic model of levodopa plasma concentrations after oral administration of 250 mg levodopa with or w/o antibiotic treatment directed against Helicobacter pylori in Parkinsons’s disease patients (after reference [30]).](https://content.iospress.com:443/media/jpd/2021/11-1/jpd-11-1-jpd202298/jpd-11-jpd202298-g001.jpg)
Later, Lee and co-workers (2008) [12] reported an observational study of 30 uninfected versus 35 HP-infected patients. The levodopa ‘onset’ time was found to be longer and ‘on’-time duration shorter in HP-infected patients than in HP-noninfected patients (78.4±28.2 vs. 56.7±25.1 and 210.0±75.7 vs. 257.7±68.9 min). Upon HP eradication the delayed levodopa ‘onset’ time and short ‘on’-time duration was significantly improved to values similar to those in noninfected PD patients. In another short-term study over three weeks, the mean levodopa equivalent daily dose (824 mg vs. 707 mg) was significantly higher among HP-positive patients than in controls. Control subjects also had a significantly longer ‘on’- duration and daily ‘on’-time, and better ‘on’ UPDRS-III scores. The HP-positives had significantly longer time to turn ‘on’ after levodopa intake. At the 3-week follow-up, HP eradication had significantly improved the mean ‘on’ UPDRS-III score, as well as onset time, ‘on’-duration, and daily ‘on’-time [15].
In a comparison of 20 HP-infected with 20 age- and gender-matched non-infected PD patients, there were no differences in disease severity or daily levodopa dosage, although HP-infected patients had about 10% higher levodopa-equivalent dosage [31]. The HP-infected group had less motor complications, such as ‘wearing-off’ and dyskinesias. It was speculated that HP infection could be beneficial in terms of motor complications by keeping levodopa concentrations low and possibly less fluctuating than in noninfected patients. By eradicating HP, the increased levodopa bioavailability would then mimic an increased levodopa dosage or addition of a COMT inhibitor, factors that are associated with ‘wearing-off’ and dyskinesias.
LONG-TERM STUDIES OF HELICOBACTER ERADICATION ON LEVODOPA TREATMENT
As the increased bioavailability of levodopa might have been due to a short-term stimulating effect of clarithromycin on gastric emptying, data were further substantiated by long-term observations over a prolonged period of three months where HP-eradicated patients showed a significant increase of levodopa absorption, associated with a significant improvement of clinical disability and prolonged ‘on-time’ duration, whereas the gastritis/duodenitis scores significantly decreased in line with improved levodopa pharmacokinetics [11]. Again, data has been interpreted as a reversible HP-induced interference with the levodopa clinical response related to impaired drug absorption, probably due to an active gastroduodenitis which should hamper the LATs to operate in a functional manner for the absorption of levodopa. Similarly, a parallel study with HP eradication and de-blinding of data at one year found a markedly improved stride-length in patients on active treatment versus placebo was found [32].
In a 12-week study of 82 patients, 27 of which were HP-positive, showed significantly poorer total UPDRS and 39 item Parkinson’s disease question-naire (PDQ-39) scores as compared to the HP-neg-ative ones. After HP eradication, the mean levodopa onset time was improved, being shortened by 14 minutes with mean motor ‘on’-duration time increased by 56 minutes at week 6 and by 38 minutes at week 12. Total UPDRS scores were drastically improved, with separate scores for parts II, III and IV all being significantly better. Total PDQ-39 scores and subdomains mobility, activities of daily living, emotional well-being and stigma were also improved. Hence, HP eradication rectifies levodopa onset time, ‘on’-duration, motor control severity and quality of life parameters [33].
Recently, Liu and co-workers [34] reported a 1-year follow-up study of 48 patients among which the motor examination scores according to the UPDRS-III were significantly reduced both when comparing to baseline within the observation group and to a comparative HP-infected group. The main improvements within the observation group were finger taps, rapid alternating movements of hands and leg agility, whereas differences to the HP-infected group showed significant improvement in rapid alternating movements of the hands, leg agility and total motor examination according to UPDRS-III. Even if the subgroups in this study were small, the final conclusion of the study confirmed that eradication of HP primarily improves bradykinesia as typically represented by the subscores of the UPDRS-III.
CRITICISMS
The concept of HP in PD has received critici-sms as the cause-effect relationship between HP and PD has been considered unclear and overall poor. Further evidence is expected before clear conclusions can be drawn on this matter. Narożańska et al. [35] detected HP positivity in 18 of 37 PD patients, but found no significant difference between HP-positive and HP-negative patients in terms of scores on the UPDRS and Hoehn-Yahr scales. Levodopa/benserazide 100/25 mg was given in the fasted state and plasma samples were repeatedly collected until 4 hours after dose intake. AUC was 8% higher in the non-infected patients but the difference was not statistically significant. The authors note that there were large inter-individual differences in pharmacokinetics within both groups, which is a common experience in PD [36]. Some patients have worse pharmacokinetic excursions only in the afternoon indicating an interaction between HP and meals. Taken together, comparisons are difficult to make, unless plasma samples are intra-individually compared before and after eradication. In a letter, Fiddian-Green [37] disputed the specificity of the HP antibiotic treatment, with the understanding that any one bacterial strain or species in the GI tract may be involved in the treatment result. To our knowledge, the only bacteria compellingly shown to be harbored in the upper GI tract, and with the ability to affect uptake of levodopa, is HP. The eradication treatment against HP employed in the studies presented above, and in common use since at least two decades back with the combination of clarithromycin and amoxicillin along with proton pump inhibition, has not convincingly been shown to affect any other bacterial pathogen in the upper gut.
MECHANISM OF HELICOBACTER-INDUCED LEVODOPA MALABSORPTION
In a speculative article by Lyte in 2010 [38] a mechanism with direct utilization of levodopa by HP to maintain its ecological niche within the GI tract was suggested. The ability of levodopa to influence the in vitro growth of HP in an iron-restricted minimal medium, as well as its utilization of levodopa for its own growth requirement was presented. This mechanism would then reduce the amount of orally administered levodopa available for absorption in the treatment of PD (Table 2).
Table 2
Colony-forming units of Helicobacter pylori incubated in a microaerophilic environment in the presence or absence of levodopa (5×10–4 moles/L) in a serum-based minimal medium (after Lyte [38])
| Condition | 0 hours | 36 hours |
| Non-supplemented | 1.6×102 | 1.2×103 |
| Levodopa-supplemented | 1.6×102 | 2.7×109 |
The fact that levodopa can directly stimulate HP growth strongly suggests that microbiota-based interactions play a role in PD and specifically the need to progressively increase drug dosage over the course of the disease. More to the point, it demonstrates that HP has some kind of receptor for lev-odopa, perhaps metabolizing it, thus reducing bio-availability and further suggests a positive feedback loop. As a theoretical background, the growth of Helicobacter strains require som the specific aminoacids such as arginine, histidine, isoleucine, leucine, methionine, valine and phenylalanine. This need for phenylalanine opens the possibility for a bacterial consumption of levodopa in the stomach [39]. Time- and concentration-dependent incubation experiments show a significant decrease in levodopa concentrations when in contact with HP. The levodopa concentration has been estimated to 12– 47% of initial concentrations, dependening on the HP density. In addition, fluorescein isothiocyanate (FITC)-labelled HP were shown to have markedly reduced bacterial adhesion to gastric epithelial cells. These results demonstrate a direct interaction of levodopa with membrane proteins of HP responsible for the adhesion to gastric epithelial cells, while the concentration of levodopa in a bacterial suspension was strongly reduced [40]. Thus, the interaction between levodopa and HP provides a mechanism for the purported benefit of HP-directed antibiotic therapy.
8PREVIOUS OVERVIEWS
A Cochrane database systematic review [41] carried out in 2011 failed to confirm any outcome of HP eradication in PD (cf. Table 1). That review was, however, based on only two completed trials: Bjarnason et al. [32] and Pierantozzi et al. [11]. An unfinished study by Bronstein and Fox from 2008 was included, even though the study was terminated early with no end results because of low prevalence of HP in the study population (cf. Table 1). Only 13 patients were included in that study, which did not permit any conclusion to be drawn [42]. The authors of the Cochrane review concluded that more clinical trials are needed using standard measures of motor symptoms. It should also be important to look at the cost of both screening and treatment of HP in PD patients for further considerations. In 2016, Camci and Oguz [43] presented an overview including 11 papers on HP in PD patients. They concluded that HP can prevent the absorption of certain drugs, one of which is levodopa. Infection with this bacterium causes motor fluctuations by impairing levodopa absorption. HP eradication in PD patients by applying a short course of antibiotics was found to increase the absorption of levodopa and thereby decrease motor fluctuations. Another overview of late by McGee et al [44] again concluded that not only are people with PD 1.5– 3 fold more likely to be infected with HP than normal subjects, but also that HP-infected PD patients display worse motor functions compared with HP-negative patients. These observations suggest that eradication treatment can increase levodopa absorption.
WHAT IS NEW?
Not only HP, but also SIBO has been studied in PD. A large study in 103 subjects using the lactulose and glucose hydrogen breath tests found SIBO to be present in 25– 33% of PD patients, along with poor motor control [45, 46], and in other studies up to 67% [47, 48], but without association to GI symptoms [44]. Another study showed increased urinary excretion of indoxyl sulfate (indican), indicating SIBO to be significantly more common in PD than in controls [49]. Fasano and collaborators studied the impact of SIBO as well as HP in patients with PD. SIBO was found in 55% and HP in 33% of the patients. The prevalence of unpredictable motor fluctuations was markedly higher in patients with both infections (87.5% vs. 8.3%). Patients with isolated SIBO had longer ‘off’ time daily and more episodes of delayed-‘on’ and no-‘on’. Antibacterial treatment with rifaximin improved motor fluctuations without affecting the levodopa pharmacokinetics. HP positives were also found to have more ‘off’ time, ‘wearing-off’ and ‘no’-on time than HP negatives [50]. It has been argued that SIBO may play a role in the development of motor dysfunction by increasing intestinal permeability and bacterial translocation, hence promoting a pro-inflammatory environment with consequences on levodopa absorption and enteric neuroinflammatory alpha-synuclein aggregates [51]. The presence of SIBO might be under-recognized in PD, yet to have important clinical implications along the lines of HP infection with malabsorption of levodopa. Today, there is consensus on the diagnosis using a glucose or lactulose breath test [52]. Based on theoretical grounds the most attractive treatment would be rifaximin as a poorly absorbable antibiotic which acts against Gram-positive and Gram-negative aerobic and anaerobic bacteria, given at a dose of 550 mg B.I.D. for 14 days with a follow-up clinical control within two weeks [53].
Current research [54] verifies that the human GI microbiota can metabolize levodopa thereby reducing the bioavailability of the drug. The conversion of levodopa to dopamine can take place via a pyridoxal phosphate-dependent tyrosine decarboxylase elaborated by Enterococcus faecalis and is directly followed by the transformation of dopamine to tyramine by a molybdenum-dependent dehydroxylase from another bacterium, the Eggerthella lenta. To this end, a compound (S-α-fluoromethyltyrosine; AFMT) was developed that inhibits this breakdown activity in the parkinsonian microbiota, which should lead to increased bioavailability of levodopa. Luminal levels of levodopa in the GI tract are compromised by a high abundance of gut bacterial tyrosine decarboxylase in patients with Parkinson’s disease. A high abundance of bacterial tyrosine decarboxylases at the site of levodopa absorption, i.e. the proximal small intestine where SIBO prevails, may reduce the bioavailability of levodopa [55]. The data highlights the possibility of therapeutically targeting the gut microbial levodopa decarboxylation in order to increase absorption of the drug, increasing efficacy.
CONCLUSION
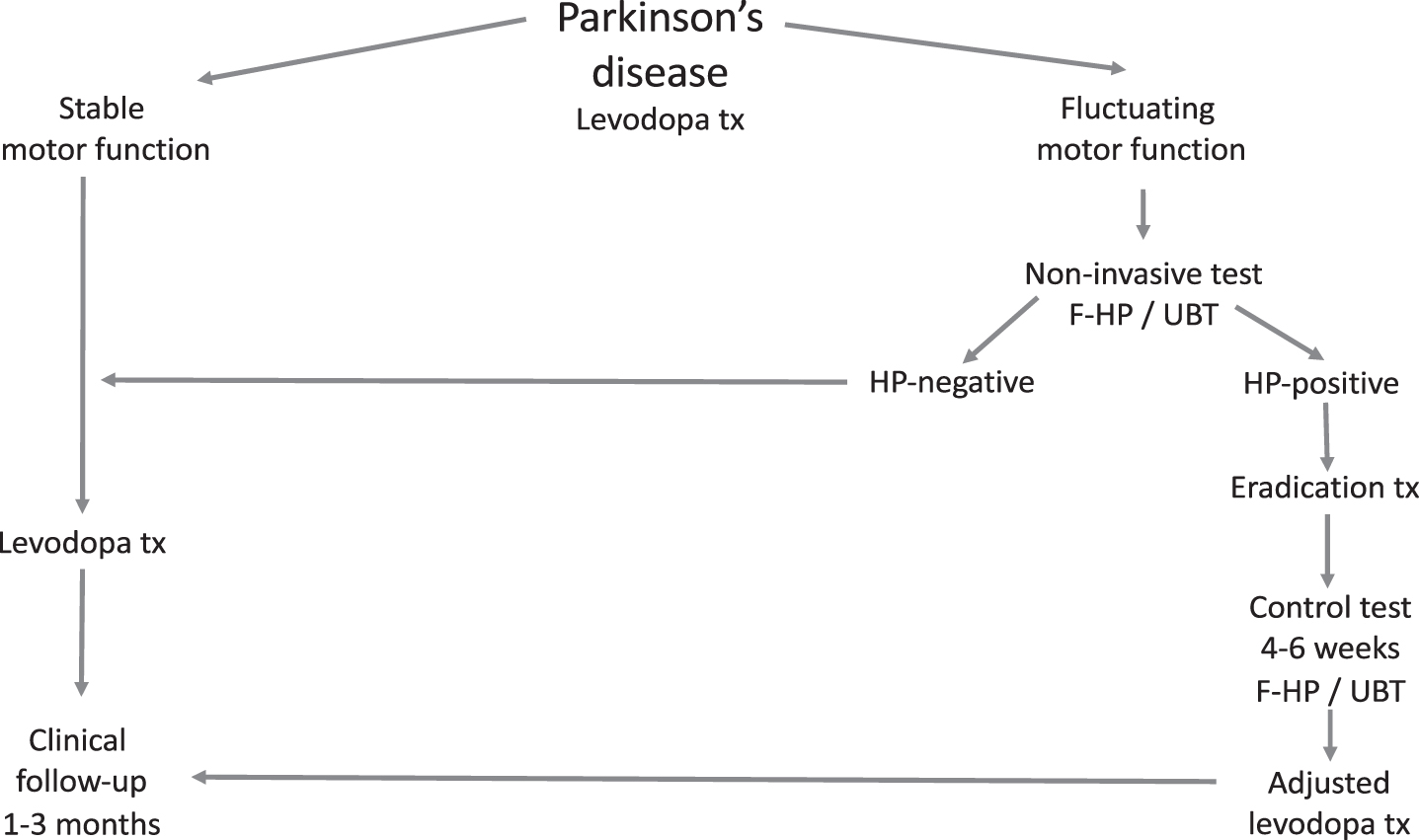
After reviewing the literature the medical community remains in suspense. Data speaks in favor of an interaction between levodopa treatment and the presence of HP in the upper GI tract. By eradication of HP, improved levodopa bioavailability and treatment efficacy is achievable. Whether or not obligatory screening and treatment of all patients with concurrent PD and HP should be pursued is a matter of debate. In cases having poor symptomatic control and considerable motor fluctuations, measures aimed at HP diagnosis and eradication seem justified. A suggested flowchart for the work-up and treatment of HP in PD is given in Fig. 2.
Fig.2
Suggested flow chart for the diagnosis, treatment (tx) and follow-up of Helicobacter pylori (HP) infection in Parkinson’s disease. F-HP, fecal Helicobacter pylori antigen analysis; UBT, urea breath test.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
ACKNOWLEDGMENTS
The present work was supported by Parkinsonfonden, Neuroförbundet, Uppsala University and ALF 2019. We thank Assoc Prof Dominic-Luc Webb, Department of Medical Sciences, Uppsala University, for comments and language revision.
REFERENCES
[1] | Strang RR ((1965) ) The association of gastro-duodenal ulceration and Parkinson’s disease. Med J Aust 1: , 842–843. |
[2] | Dobbs RJ , Charlett A , Dobbs SM , Weller C , Peterson DW ((2000) ) Parkinsonism: Differential age-trend in Helicobacter pylori antibody. Aliment Pharmacol Ther 14: , 1199–1205. |
[3] | Blaecher C , Smet A , Flahou B , Pasmans F , Ducatelle R , Taylor D , Weller C , Bjarnason I , Charlett A , Lawson AJ , Dobbs RJ , Dobbs SM , Haesebrouck F ((2013) ) Significantly higher frequency of Helicobacter suis in patients with idiopathic parkinsonism than in control patients. Aliment Pharmacol Ther 38: , 1347–1353. |
[4] | Dobbs SM , Dobbs RJ , Weller C , Charlett A ((2000) ) Link between Helicobacter pylori infection and idiopathic parkinsonism. Med Hypotheses 55: , 93–98. |
[5] | Schulz JD , Hawkes EL , Shaw CA ((2006) ) Cycad toxins, Helicobacter pylori and parkinsonism: Cholesterol glucosides as the common denomenator. Med Hypotheses 66: , 1222–1226. |
[6] | Barnum CJ , Tansey MG ((2012) ) Neuroinflammation and non-motor symptoms: The dark passenger of Parkinson’s disease?. Curr Neurol Neurosci Rep 12: , 350–358. |
[7] | Breen DP , Halliday GM , Lang AE ((2019) ) Gut-brain axis and the spread of α-synuclein pathology: Vagal highway or dead end?. Mov Disord 34: , 307–316. |
[8] | Keshavarzian A , Engen P , Bonvegna S , Cilia R ((2020) ) The gut microbiome in Parkinson’s disease: A culprit or a bystander?. Prog Brain Res 252: , 357–450. |
[9] | Weller C , Charlett A , Oxlade NL , Dobbs SM , Dobbs RJ , Peterson DW , Bjarnason IT ((2005) ) Role of chronic infection and inflammation in the gastrointestinal tract in the etiology and pathogenesis of idiopathic parkinsonism. Part 3: Predicted probability and gradients of severity of idiopathic parkinsonism based on H. pylori antibody profile. Helicobacter 10: , 288–297. |
[10] | Charlett A , Dobbs RJ , Dobbs SM , Weller C , Brady P , Peterson DW ((1999) ) Parkinsonism: Siblings share Helicobacter pylori seropositivity and facets of syndrome. Acta Neurol Scand 99: , 26–35. |
[11] | Pierantozzi M , Pietroiusti A , Brusa L , Galati S , Stefani A , Lunardi G , Fedele E , Sancesario G , Bernardi G , Bergamaschi A , Magrini A , Stanzione P , Galante A ((2006) ) Helicobacter pylori eradication and l-dopa absorption in patients with PD and motor fluctuations. Neurology 66: , 1824–1829. |
[12] | Lee WY , Yoon WT , Shin HY , Jeon SH , Rhee PL ((2008) ) Helicobacter pylori infection and motor fluctuations in patients with Parkinson’s disease. Mov Disord 23: , 1696–1700. |
[13] | Dobbs RJ , Charlett A , Dobbs SM , Weller C , A Ibrahim MA , Iguodala O , Smee C , Plant JM , Lawson AJ , Taylor D , Bjarnason I ((2012) ) Leukocyte-subset counts in idiopathic parkinsonism provide clues to a pathogenic pathway involving small intestinal bacterial overgrowth. A surveillance study. Gut Pathog 4: , 12. |
[14] | Tan AH , Mahadeva S , Marras C , Thalha AM , Kiew CK , Yeat CM , Ng SW , Ang SP , Chow SK , Loke MF , Vadivelu JS , Ibrahim N , Yong HS , Tan CT , Fox SH , Lang AE , Lim SY ((2015) ) Helicobacter pylori infection is associated with worse severity of Parkinson’s disease. Parkinsonism Relat Disord 21: , 221–225. |
[15] | Mridula KR , Borgohain R , Chandrasekhar Reddy V , Bandaru VCh , Suryaprabha T ((2017) ) Association of Helicobacter pylori with Parkinson’s disease. J Clin Neurol 13: , 181–186. |
[16] | Hooi JKY , Lai WY , Ng WK , Suen MMY , Underwood FE , Tanyingoh D , Malfertheiner P , Graham DY , Wong VWS , Wu JCY , Chan FKL , Sung JJY , Kaplan GG , Ng SC ((2017) ) . Global prevalence of Helicobacter pylori infection: Systematic review and meta-analysis. Gastroenterology 153: , 420–429. |
[17] | Shen X , Yang H , Wu Y , Zhang D , Jiang H ((2017) ) Meta-analysis: Association of Helicobacter pylori infection with Parkinson’s diseases. Helicobacter 22: , 1–6. |
[18] | Dobbs RJ , Dobbs SM , Weller C , Charlett A , Bjarnason IT , Curry A , Ellis DS , Ibrahim MA , McCrossan MV , O’Donohue J , Owen RJ , Oxlade NL , Price AB , Sanderson JD , Sudhanva M , Williams J ((2008) ) Helicobacter hypothesis for idiopathic parkinsonism: Before and beyond. Helicobacter 13: , 309–322. |
[19] | Kountouras J , Zavos C , Polyzos SA , Deretzi G , Vardaka E , Giartza-Taxidou E , Katsinelos P , Rapti E , Chatzopoulos D , Tzilves D , Stergiopoulos C , Christodoulou K ((2012) ) Helicobacter pylori infection and Parkinson’s disease: Apoptosis as an underlying common contributor. Eur J Neurol 19: , e56. |
[20] | Suwarnalata G , Tan AH , Isa H , Gudimella R , Anwar A , Loke MF , Mahadeva SY , Vadivelu J ((2016) ) Augmentation of autoantibodies by Helicobacter pylori in Parkinson’s disease patients may be linked to greater severity. PLoS One 11: , e0153725. |
[21] | Fotiadis D , Kanai Y , Palacín M ((2013) ) The SLC3 and SLC7 families of amino acid transporters. Mol Asp Med 34: , 139–158. |
[22] | Xu Y , Yan J , Zhou P , Li J , Gao H , Xia Y , Wang Q ((2012) ) Neurotransmitter receptors and cognitive dysfunction in Alzheimer’s disease and Parkinson’s disease. Prog Neurobiol 97: , 1–13. |
[23] | Marsden CD , Parkes JD ((1977) ) Success and problems of long-term levodopa therapy in Parkinson’s disease. Lancet 1: , 345–349. |
[24] | Peppe A , Dambrosia JM , Chase TN ((1993) ) Risk factors for motor response complications in L-dopa-treated parkinsonian patients. Adv Neurol 60: , 698–702. |
[25] | Marsden CD ((1994) ) Parkinson’s disease. J Neurol Neurosurg Psychiatry 57: , 672–681. |
[26] | Senek M , Nyholm D ((2014) ) Continuous drug delivery in Parkinson’s disease. CNS Drugs 28: , 19–27. |
[27] | Espay AJ , Morgante F , Merola A , Fasano A , Marsili L , Fox SH , Bezard E , Picconi B , Calabresi P , Lang AE ((2018) ) Levodopa-induced dyskinesia in Parkinson disease: Current and evolving concepts. Ann Neurol 84: , 797–811. |
[28] | Fiorini G , Bland JM , Hughes E , Castelli V , Vaira D ((2015) ) A systematic review on drugs absorption modifications after eradication in} positive patients undergoing replacement therapy. J Gastrointest Liver Dis 24: , 95–100. |
[29] | Pierantozzi M , Pietroiusti A , Sancesario G , Lunardi G , Fedele E , Giacomini P , Frasca S , Galante A , Marciani MG , Stanzione P ((2001) ) Reduced L-dopa absorption and increased clinical fluctuations in Helicobacter pylori-infected Parkinson’s disease patients. Neurol Sci 22: , 89–91. |
[30] | Pierantozzi M , Pietroiusti A , Galante A , Sancesario G , Lunardi G , Fedele E , Giacomini P , Stanzione P ((2001) ) Helicobacter pylori-induced reduction of acute levodopa absorption in Parkinson’s disease patients. Ann Neurol 50: , 686–687. |
[31] | Rahne KE , Tagesson C , Nyholm D ((2013) ) Motor fluctuations and Helicobacter pylori in Parkinson’s disease. J Neurol 260: , 2974–2980. |
[32] | Bjarnason IT , Charlett A , Dobbs RJ , Dobbs SM , Ibrahim MA , Kerwin RW , Mahler RF , Oxlade NL , Peterson DW , Plant JM , Price AB , Weller C ((2005) ) Role of chronic infection and inflammation in the gastrointestinal tract in the etiology and pathogenesis of idiopathic parkinsonism. Part 2: Response of facets of clinical idiopathic parkinsonism to Helicobacter pylori eradication. A randomized, double-blind, placebo-controlled efficacy study. Helicobacter 10: , 276–287. |
[33] | Hashim H , Azmin S , Razlan H , Yahya NW , Tan HJ , Manaf MR , Ibrahim NM ((2014) ) Eradication of Helicobacter pylori infection improves levodopa action, clinical symptoms and quality of life in patients with Parkinson’s disease. PLoS One 9: , e112330. |
[34] | Liu H , Su W , Li S , Du W , Ma X , Jin Y , Li K , Chen H ((2017) ) Eradication of Helicobacter pylori infection might improve clinical status of patients with Parkinson’s disease, especially on bradykinesia. Clin Neurol Neurosurg 160: , 101–104. |
[35] | Narożańska E , Białecka M , Adamiak-Giera U , Gawrońska-Szklarz B , Sołtan W , Schinwelski M , Robowski P , Madaliński MH , Sławek J ((2014) ) Pharmacokinetics of levodopa in patients with Parkinson disease and motor fluctuations depending on the presence of Helicobacter pylori infection. Clin Neuropharmacol 37: , 96–99. |
[36] | Nyholm D , Lennernäs H , Gomes-Trolin C , Aquilonius SM ((2002) ) Levodopa pharmacokinetics and motor performance during activities of daily living in patients with Parkinson’s disease on individual drug combinations. Clin Neuropharmacol 25: , 89–96. |
[37] | Fiddian-Green RG ((2007) ) Helicobacter pylori eradication and l-dopa absorption in patients with PD and motor fluctuations. Neurology 68: , 1085. |
[38] | Lyte M ((2010) ) Microbial endocrinology as a basis for improved L-DOPA bioavailability in Parkinson’s patients treated for Helicobacter pylori. Med Hypotheses 74: , 895–897. |
[39] | Reynolds DJ , Penn CW ((1994) ) Characteristics of Helicobacter pylori growth in a defined medium and determination of its amino acid requirements. Microbiology 140: , 2649–2656. |
[40] | Niehues M , Hensel A ((2009) ) } interaction of L-dopa with bacterial adhesins of Helicobacter pylori: An explanation for clinicial differences in bioavailability?. J Pharm Pharmacol 61: , 1303–1307. |
[41] | Rees K , Stowe R , Patel S , Ives N , Breen K , Clarke CE , Ben-Shlomo Y ((2011) ) . Helicobacter pylori eradication for Parkinson’s disease. Cochrane Database Syst Rev 11: , CD008453. |
[42] | Bronstein J , Fox MJ ((2008) ) Treating H. pylori in Parkinson’s patients with motor fluctuations. ClinTrials.gov, NCT00664209. Posted April 22, 2008, Updated December 2, 2017, Accessed June 12, 2019. |
[43] | Camci G , Oguz S ((2016) ) Association between Parkinson’s disease and Helicobacter pylori. J Clin Neurol 12: , 147–150. |
[44] | McGee DJ , Lu X-H , Disbrow EA ((2018) ) Stomaching the possibility of a pathogenic role for Helicobacter pylori in Parkinson’s disease. J Parkinsons Dis 8: , 367–374. |
[45] | Tan AH , Mahadeva S , Thalha AM , Gibson PR , Kiew CK , Yeat CM , Ng SW , Ang SP , Chow SK , Tan CT , Yong HS , Marras C , Fox SH , Lim SY ((2014) ) Small intestinal bacterial overgrowth in Parkinson’s disease. Parkinsonism Relat Disord 20: , 535–540. |
[46] | Niu XL , Liu L , Song ZX , Li Q , Wang ZH , Zhang JL , Li HH ((2016) ) Prevalence of small intestinal bacterial overgrowth in Chinese patients with Parkinson’s disease. J Neural Transm (Vienna) 123: , 1381–1386. |
[47] | Fasano A , Bove F , Gabrielli M , Petracca M , Zocco MA , Ragazzoni E , Barbaro F , Piano C , Fortuna S , Tortora A , Di Giacopo R , Campanale M , Gigante G , Lauritano EC , Navarra P , Marconi S , Gasbarrini A , Bentivoglio AR ((2013) ) The role of small intestinal bacterial overgrowth in Parkinson’s disease. Mov Disord 28: , 1241–1249. |
[48] | Dobbs RJ , Charlett A , Dobbs SM , Weller C , Ibrahim MA , Iguodala O , Smee C , Plant JM , Lawson AJ , Taylor D , Bjarnason I ((2012) ) Leukocyte-subset counts in idiopathic parkinsonism provide clues to a pathogenic pathway involving small intestinal bacterial overgrowth. A surveillance study. Gut Pathog 4: , 12. |
[49] | Cassani E , Barichella M , Cancello R , Cavanna F , Iorio L , Cereda E , Bolliri C , Zampella Maria P , Bianchi F , Cestaro B , Pezzoli G ((2015) ) Increased urinary indoxyl sulfate (indican): New insights into gut dysbiosis in Parkinson’s disease. Parkinsonism Relat Disord 21: , 389–393. |
[50] | Fasano A , Bove F , Gabrieli M , Petracca M , Zocco MA , Ragazzoni E , Barbaro F , Piano C , Fortuna S , Tortora A , Di Giacopo A , Campanale M , Gigante G , Lauritano EC , Navarra P , Marconi S , Gasparini A , Bentivoglio AR ((2013) ) The role of small intestinal bacterial overgrowth in Parkinson’s disease. Mov Disord 28: , 1241–1249. |
[51] | Losurdo G , Salvatore D’Abramo F , Indellicati G , Lillo C , Ierardi E , Di Leo A ((2020) ) The influence of small intestinal bacterial overgrowth in digestive and extra-intestinal disorders. Int J Mol Sci 21: , 3531. |
[52] | Rezaie A , Buresi M , Lembo A , Lin H , McCallum R , Rao S , Schmulson M , Valdovinos M , Zakko S , Pimentel M ((2017) ) Hydrogen and methane-based breath testing in gastrointestinal disorders: The North American consensus. Am J Gastroenterol 112: , 775–784. |
[53] | Gatta L , Scarpignato C ((2017) ) Systematic review with meta-analysis: Rifaximin is effective and safe for the treatment of small intestine bacterial overgrowth. Aliment Pharmacol Ther 45: , 604–616. |
[54] | Rekdal VM , Bess EN , Bisanz JE , Turnbaugh PJ , Balskus EP ((2019) ) Discovery and inhibition of an interspecies gut bacterial pathway for levodopa metabolism. Science 364: , 1–7. |
[55] | van Kessel SP , Frye AK , El-Gendy AO , Castejon M , Keshavarzian A , van Dijk G , El Aidy S ((2019) ) Gut bacterial tyrosine decarboxylases restrict levels of levodopa in the treatment of Parkinson’s disease. Nat Commun 10: , 310. |


