
Continuous Subcutaneous Levodopa Delivery for Parkinson’s Disease: A Randomized Study
Abstract
Background:
ND0612 is a continuous, subcutaneous levodopa/carbidopa delivery system in development for patients with Parkinson’s disease (PD) experiencing motor fluctuations
Objective:
Evaluate the efficacy and safety of two ND0612 dosing regimens in patients with PD.
Methods:
This was a 28-day open-label study (NCT02577523) in PD patients with ≥2.5 hours/day of OFF time despite optimized treatment. Patients were randomized to treatment with either a 24-hour infusion (levodopa/carbidopa dose of 720/90 mg) or a 14-hour ‘waking-day’ infusion (levodopa/carbidopa dose of 538/68 mg plus a morning oral dose of 150/15 mg). Supplemental oral doses of levodopa were permitted for patients in both groups if required. In-clinic assessments of OFF time (primary endpoint) and ON time with or without dyskinesia were determined by a blinded rater over 8 hours (normalized to 16 hours).
Results:
A total of 38 patients were randomized and 33 (87%) completed the study. Compared to baseline, OFF time for the overall population was reduced by a least squares (LS) mean[95% CI] of 2.0[– 3.3, – 0.7] hours (p = 0.003). ON time with no/mild dyskinesia (no troublesome dyskinesia) was increased from baseline by a LS mean of 3.3[2.0, 4.6] hours (p < 0.0001), and ON time with moderate/severe dyskinesia was reduced by a LS mean of 1.2[– 1.8, – 0.5] hours (p≤0.001). Reduction in OFF time was larger in the 24-hour group (– 2.8[– 4.6, – 0.9] hours; p = 0.004) than in the 14-hour group (– 1.3[– 3.1, 0.5] hours; p = 0.16). Complete resolution of OFF time was observed in 42% (n = 8) of patients in the 24-hour group. Infusion site reactions were the most common adverse event.
Conclusion:
This study demonstrates the feasibility and safety of continuous subcutaneous delivery of levodopa as a treatment for PD and provides preliminary evidence of efficacy.
INTRODUCTION
After more than 50 years, levodopa remains the most effective therapy for Parkinson’s disease (PD). However, chronic treatment is associated with motor complications in the majority of patients [1– 3], which can significantly impact activities of daily living and quality of life [4, 5]. Indeed, motor complications are the key reason PD patients undergo surgical therapies such as deep brain stimulation.
Increasing evidence indicates that motor complications are related to the non-physiologic restoration of brain dopamine with intermittent doses of standard oral levodopa [6, 7]. Under normal conditions, striatal dopamine concentrations are maintained at a relatively continuous level [8, 9]. However, in the dopamine-depleted state, intermittent levodopa doses result in marked fluctuations in striatal dopamine leading to molecular and neurophysiologic changes and ultimately to motor complications [6, 7]. These observations led to the hypothesis that continuous delivery of levodopa might restore dopamine to the brain in a more physiological manner and reduce the risk of motor complications. This concept has been supported by numerous animal studies [6, 7], and by a double-blind study in PD patients showing that continuous intrajejunal infusion of Levodopa/Carbidopa Intestinal Gel (LCIG) was associated with significantly reduced motor fluctuations compared to optimized oral levodopa [10]. However, continuous intestinal levodopa fusion is associated with potentially serious side effects related to the surgical procedure and the need for a persistent indwelling jejunal catheter [10, 11].
ND0612 (NeuroDerm, Rehovot Israel) is a drug-device combination consisting of a liquid formulation of levodopa/carbidopa continuously administered via a subcutaneous pump that provides plasma levodopa levels with less variability than standard oral levodopa [12]. The novel formulation is a sterile solution of levodopa and carbidopa with the addition of excipients that increase solubility and control stability and permit delivery at a low infusion rate. Here, we report the results of a proof-of-concept study designed to assess the efficacy, safety and tolerability of ND0612 in PD patients with motor fluctuations. An exploratory objective of this trial was to evaluate any potential benefits of a 14-hour (waking day) infusion compared to 24-hour (round the clock) infusion. No formal statistical comparison was planned between these two dosing regimens.
METHODS
Study design
The study was designed as a randomized, multicenter, 28-day trial with a blinded evaluator, and was conducted between December 2015 and January 2017 in 11 specialist PD sites in Europe, Israel and the United States. The study was registered in the database of the US National Library of Medicine (clinicaltrials.gov), record number NCT02577523, and conducted in accordance with the Declaration of Helsinki and the International Council for Harmonization tripartite guideline on the ethical principles of Good Clinical Practice (ICH E6) guidelines. The study protocol, informed consent forms, and other study-related documents were reviewed and approved by the local independent ethics committees and institutional review boards. Eligible participants who met entry criteria and signed an IRB-approved informed consent were enrolled into the study.
On study entry, patients slept in the clinic over-night and anti-parkinsonian medications were withheld after midnight. The following morning, they were randomized (1:1) using a computer-generated sequence (block size of 4) to ND0612 treatment administered either as a 24-hour ‘round the clock’ SC infusion delivering a total levodopa/carbidopa dose of 720/90 mg or as a 14-hour ‘waking day’ SC infusion delivering a levodopa/carbidopa dose of 537.6/67.2 mg plus a morning oral dose of levodopa/carbidopa 150/15 mg. Dose levels of ND0612 were based on the maximal amount of LD/CD that could be delivered with the current pump system as determined in a previous phase 1 study [12]. It should be noted that levodopa, and to a greater extent carbidopa have higher bioavailability when delivered subcutaneously, and that the 8:1 ratio of carbidopa:levodopa that is employed efficiently inhibits the dopa-decarboxylase enzyme [13]. The infusion was initiated on top of each patient’s anti-parkinsonian medication, but, oral levodopa/carbi-dopa could be rapidly downtitrated to account for the additional levodopa/carbidopa. The dose could not be changed once discharged from clinic. Other anti-parkinsonian drugs were permitted but had to be maintained at a constant dose throughout the study. On the fourth clinic day, patients were discharged to their homes on their assigned ND0612 dosing regimen, plus any additional levodopa/carbidopa required and other anti-parkinsonian medications as taken before the study.
Population
The trial enrolled male and female patients, aged 30– 80 years, with a diagnosis of PD consistent with the UK Brain Bank criteria [14], who were Hoehn & Yahr≤3 in the ON state, had≥2.5 hours of OFF time per day, had predictable and well-defined early morning OFF periods (patient report), and a ‘good’ early morning response to levodopa. Patients had to be taking≥4 levodopa doses per day (or≥3 doses/day of Rytary) and to have taken a therapeutic dose of≥2 other classes of PD medications. PD medications had to have been maintained at stable doses for≥30 days prior to the start of the study. Patients also had to have a caregiver able to provide support during the study period if required. Exclusion criteria included atypical parkinsonism, prior neurosurgical procedure for PD, prior treatment with LCIG or DBS, Mini-Mental State Examination score≤26, psychosis or hallucinations during the prior 6 months, and any clinically significant medical, surgical, or psychiatric condition in the judgment of the investigator. Prior exposure to SC apomorphine injection/infusion was permitted but patients must have discontinued administration≥4 weeks before the screening visit. Patients also had to be approved for study entry by an independent enrollment committee to confirm eligibility and that patient understood and was capable of performing all study-related activities. This methodology is now commonly used in clinical trials to avoid error in diagnosis, minimize variability, and avoid dropout.
Study treatment
A Home Nursing Service representative visited patients each day to supervise the administration of the infusion (administered via 2 cannulas inserted into the SC layer of the skin) and operation of the pump system. For this study, ND0612 was delivered using a modified Cane Crono Twin ambulatory infusion pump. Patients randomized to the 24-hour ND0612 treatment received the infusion at a rate of 0.64 mL/hour from 4AM to 10PM and 0.08 mL/hour between 10PM and 4AM. Night-time flow rates were kept at the lowest possible flow rate until approximately 4AM to conserve infused levodopa for daytime availability. The pump was then automatically switched to the high rate with the intention that plasma levodopa levels would be sufficiently high to permit the patient to experience an ON episode upon awakening. Patients randomized to the 14-hour ND0612 received the infusion at a rate of 0.64 mL/hour starting in the morning (when the nurse arrived to start the infusion) and discontinued prior to sleep. They also received a booster dose of oral levodopa/carbidopa (150/15 mg) in the morning at the time of the start of the infusion to facilitate reaching plasma levels that permit patient to turn ON. The infusion sites could be on the abdomen, flanks, or outer thighs, and were changed every morning in a rotating manner.
Evaluations
In-clinic assessments of OFF time, ON time without dyskinesia or with mild, moderate or severe dyskinesia were determined by a blinded rater at 30-minute intervals for a total of 8 hours upon waking. Evaluations were performed at Baseline, Day 3 (last full in-patient day) and Day 28 (end of study). Additional evaluations performed at baseline and final visit included patient and blinded-rater determination of when a ‘full’ ON had taken place, Clinician and Patient Global Impressions of improvement (CGI-I and PGI-I), Unified Parkinson’s Disease Rating Scale [15] motor score (UPDRS Part-III) assessed just before taking the morning oral dose of levodopa and/or the start/change of the infusion, UPDRS activities of daily living (UPDRS Part-II), Quality of Life in Parkinson’s Disease (PDQ-39) scale [16] and Parkinson’s Disease Sleep Scale (PDSS-2) [17].
Adverse events were recorded each day throughout the study. Pain at infusion site was scored using a Visual Analogue Scale (VAS) scale (0 mm=no pain; 100 mm=worst possible pain). Other safety assessments included standard laboratory assessments and vital signs, the Epworth Sleepiness Scale (ESS) [18], the Questionnaire for Impulsive-Compulsive Disorders in Parkinson’s Disease Rating Scale (QUIP-RS) [19], and the Columbia Suicide Severity Rating Scale (C-SSRS) [20].
Statistical analysis
A sample size of 15 completers per treatment group was sufficient to detect a within-group change from baseline to week 28 in daily OFF time of 0.78 hours with 80% power assuming a standard deviation of 1.0 hours and a 2-sided significance level of 0.05.
The primary endpoint was the within group change in OFF time between Baseline and Day 28 as assessed by the blinded rater, normalized to the 16-hour waking day. This is a standard manner of reporting OFF time in order to control for the numbers of hours during the day that are available for reporting motor state among different patients. Secondary endpoints included change from baseline to Day 28 in ON time with no or mild dyskinesia and in ON time with moderate or severe dyskinesia (normalized to 16 hours), percentage of patients who were fully ON at 8:00 AM and 9:00 AM (24-hour group only), CGI-I, PGI-I, UPDRS Part-III, UPDRS Part-II, PDQ-39 and PDSS-2. Exploratory endpoints included percentage of patients who achieved a 50% and 100% reduction in daily OFF time, and the amount of dyskinesia reduction in patients who had ≥1 hour of moderate to severe dyskinesia at baseline.
The primary endpoint was analyzed using a Mixed Model for Repeated Measures (MMRM) with no imputation for missing data. The MMRM assumed an unstructured covariance and included baseline values as a covariate and the randomized treatment regimen, scheduled study day, interaction between study visit and treatment, and region as fixed factors. Continuous secondary efficacy outcome measures were analyzed using MMRM with baseline value as covariate and the randomized treatment regimen and region as fixed factors, or an ANCOVA (for endpoints that were assessed only at baseline and final visits). Categorical secondary endpoints were compared using the Cochran-Mantel-Haenszel test. No corrections were made for multiple endpoints; P values are provided for descriptive purposes only.
RESULTS
Patient disposition and baseline characteristics
Between 29 December 2015 and 04 November 2016, 49 patients were screened, 38 met eligibility criteria and were randomized (19 to each group), and 33 patients (87%) completed the study (Fig. 1). Two patients discontinued due to adverse events (one due to an infusion site reaction with abscess; one due to worsening of PD symptoms), two due to lack of efficacy, and one withdrew consent. Patient baseline characteristics are provided in Table 1. All patients took supplemental oral levodopa therapy, and baseline and final doses in the two groups were comparable (Table 3). The two dosing groups were also reasonably well balanced for use of concomitant anti-PD medications and these were not changed during the study.
Fig. 1
Study flow.
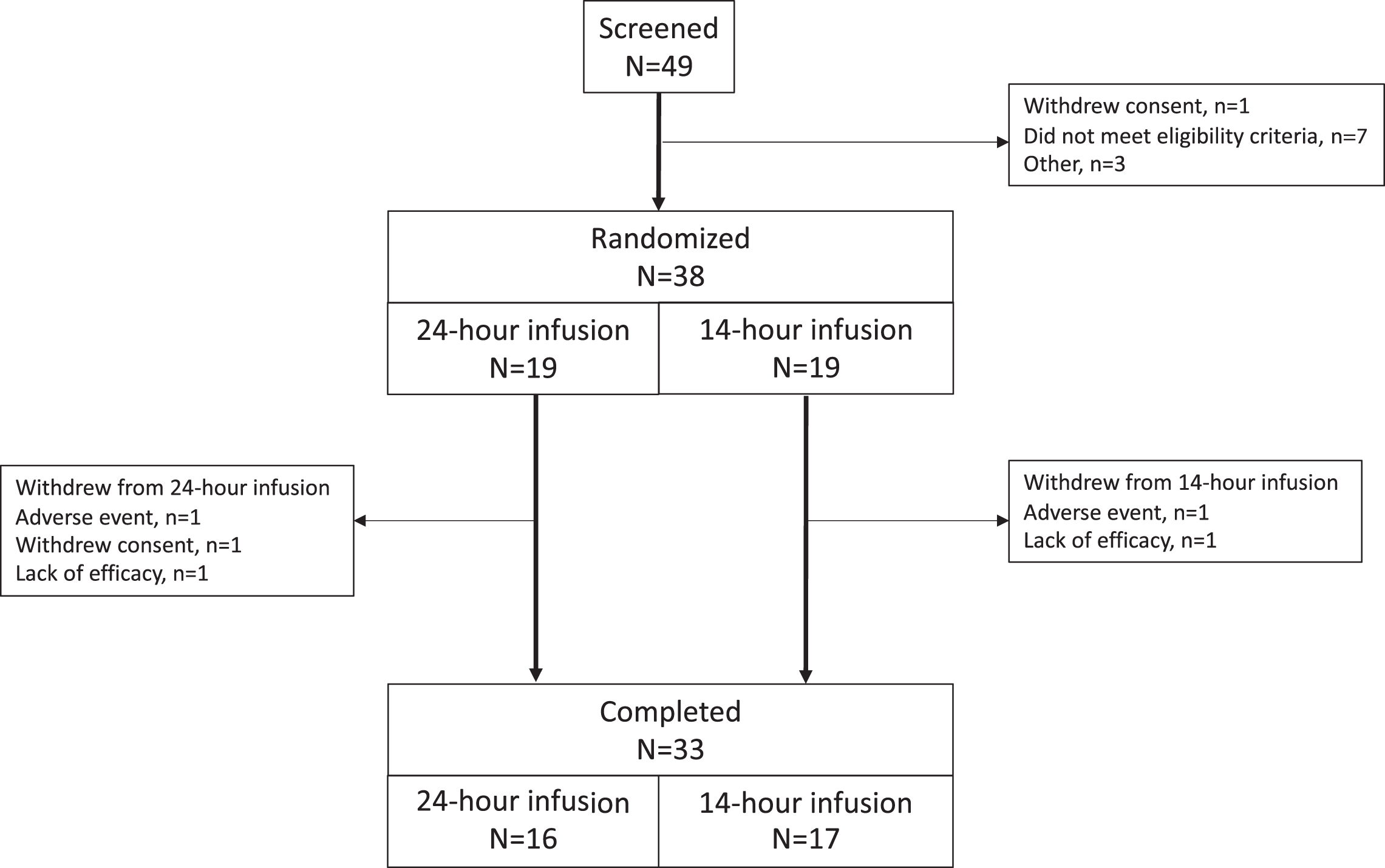
Table 1
Baseline characteristics
| 24-hour infusion | 14-hour infusion | Overall | |
| (N = 19) | (N = 19) | (N = 38) | |
| Age (y); mean±SD | 63.0±10.1 | 64.0±8.5 | 63.5±9.2 |
| Sex; n (%) male | 12 (63.2) | 14 (73.7) | 26 (68.4) |
| Modified Hoehn and Yahr Stage; n (%) | |||
| 1 | 1 (5.3) | – | 1 (2.6) |
| 1.5 | – | 1 (5.3) | 1 (2.6) |
| 2 | 13 (68.4) | 11 (57.9) | 24 (63.2) |
| 2.5 | 4 (21.1) | 5 (26.3) | 9 (23.7) |
| 3 | 1 (5.3) | 2 (10.5) | 3 (7.9) |
| Years since PD diagnosis; mean±SD | 10.7±5.5 | 12.2±5.0 | 11.5±5.2 |
| Years since motor fluctuation onset; mean±SD | 5.7±6.9 | 5.5±4.8 | 5.6±5.9 |
| Years since dyskinesia onset | 3.1±2.7 | 4.2±3.4 | 3.7±3.1 |
| Patients with≥1 h of moderate-severe dyskinesia at baseline; n (%) | 6 (31.6) | 8 (42.1) | 14 (36.8) |
| Daily OFF time (h); mean±SD | 5.6±2.1 | 5.0±2.4 | 5.3±2.2 |
| Daily ON time with no or mild dyskinesia (h) | 9.2±3.3 | 8.5±3.3 | 8.9±3.2 |
| Daily ON time with moderate or severe dyskinesia (h) | 1.2±2.8 | 2.5±3.7 | 1.9±3.3 |
| Concomitant PD medications; n (%) | |||
| Levodopa | 996±552 | 948±305 | |
| Dopamine agonists | 9 (47.4) | 13 (68.4) | 22 (57.9) |
| Monoamine oxidase inhibitors | 11 (57.9) | 10 (52.6) | 21 (55.3) |
| Amantadine | 5 (26.3) | 7 (36.8) | 12 (31.6) |
| Entacapone | 2 (10.5) | 4 (21.1) | 6 (15.8) |
| Trihexyphenidyl | 1 (5.3) | 0 | 1 (2.6) |
Efficacy results
Efficacy results are provided in Table 2. The change from baseline to final visit (Day 28) in OFF time (primary endpoint) for the entire population was reduced by a least squares (LS) mean of 2.0 hours [95% CI: – 3.3, – 0.7; p = 0.003) (Fig. 2). The LS mean reduction in OFF time was larger in the 24-hour ND0612 infusion group (– 2.8 hours – 4.6, – 0.9]; p = 0.004) than in the 14-hour infusion group (– 1.3 hours [– 3.1, 0.5]; p = 0.16). At Day 28, 63% (n = 12) of patients who received the 24-hour ND0612 infusion and 42% (n = 8) in the 14-hour infusion group had a reduction of≥50% in OFF time compared to baseline. A reduction in OFF time to zero hours was observed in 42% (n = 8) of patients in the 24-hour infusion group. However, this could not be expected in the 14-hour infusion group since levodopa treatment was only started when the home nurse representative arrived (i.e. morning OFF time prior to start of oral and infusion levodopa was unavoidable). For patients in the 24-hour infusion group, the proportion of patients who achieved full ON by 8:00 AM increased from 15.8% on Day 1 (oral levodopa/carbidopa only) to 43.8% at Day 28, and the proportion of patients achieving full ON by 9:00AM increased from 36.8% at Day 1 to 81.3% at Day 28.
Table 2
Efficacy endpoints
| Outcome | 24-hour infusion | 14-hour infusion | Overall |
| (N = 19) | (N = 19) | (N = 38) | |
| OFF time (h) | |||
| Baseline; mean±SD | 5.6±2.1 | 5.0±2.4 | 5.3±2.2 |
| LS Mean [95% CI] change from baseline to Day 28 | – 2.8 [– 4.6, – 0.9] | – 1.3 [– 3.1, 0.5] | – 2.0 [– 3.3, – 0.7] |
| p value vs. baseline | 0.004 | 0.16 | 0.003 |
| ON time with no or mild dyskinesia (h) | |||
| Baseline; mean±SD | 9.2±3.3 | 8.5±3.3 | 8.9±3.2 |
| LS Mean [95% CI] change from baseline to Day 28 | 3.7 [1.9, 5.6] | 2.8 [1.0, 4.6] | 3.3 [2.0, 4.6] |
| p value vs. baseline | <0.001 | 0.003 | <0.0001 |
| ON time with moderate or severe dyskinesia (h) | |||
| Baseline; mean±SD | 1.2±2.8 | 2.5±3.7 | 1.9±3.3 |
| LS Mean [95% CI] change from baseline to Day 28 | – 1.2 [– 2.1, – 0.3] | – 1.2 [– 2.1, – 0.3] | – 1.2 [– 1.8, – 0.5] |
| p value vs. baseline | 0.01 | 0.01 | <0.001 |
| Proportion of patients achieving ON at 8:00 AM; n (%) | |||
| Baseline | 3 (15.8) | NA | NA |
| Day 28 | 7 (43.8) | NA | NA |
| Proportion of patients achieving ON at 9:00 AM; n (%) | |||
| Baseline | 7 (36.8) | NA | NA |
| Day 28 | 13 (81.3) | NA | NA |
| Clinical Global Impression of Improvement | |||
| N (%) improved at Day 3 | 10 (52.6) | 9 (47.4) | 19 (50.0) |
| N (%) improved at Day 28 | 14 (77.8) | 13 (68.4) | 27 (73.0) |
| Patient Global Impression of Improvement | |||
| N (%) improved at Day 3 | 14 (73.7) | 10 (52.6) | 24 (55.2) |
| N (%) improved at Day 28 | 14 (77.8) | 15 (78.9) | 29 (80.6) |
| UPDRS Part III scores (8:00 AM) | |||
| Baseline; mean±SD | 37.4±14.5 | 37.3±13.3 | 37.3±13.7 |
| LS Mean [95% CI] change from baseline to Day 28 | – 19.1 [– 25.6, – 12.5] | – 10.7 [– 16.8, – 4.6] | – 14.6 [– 19.4, – 9.7] |
| p value vs. baseline | <0.0001 | 0.001 | <0.0001 |
| UPDRS Part II scores | |||
| Baseline; mean±SD | 12.5±6.4 | 14.5±6.7 | 13.5±6.6 |
| LS Mean [95% CI] change from baseline to Day 28 | – 2.9 [– 5.4, – 0.5] | – 1.9 [– 4.2, 0.4] | – 2.4 [– 4.1, – 0.7] |
| p value vs. baseline | 0.02 | 0.11 | 0.008 |
| PDSS-2 scores | |||
| Baseline; mean±SD | 20.6±7.9 | 21.5±7.8 | 21.1±7.8 |
| LS Mean [95% CI] change from baseline to Day 28 | – 4.1 [– 8.0, – 0.2] | – 0.8 [– 4.4, 2.9] | – 2.3 [– 5.1, 0.5] |
| p value vs. baseline | 0.04 | 0.68 | 0.10 |
| PDQ-39 Summary Index scores | |||
| Baseline; mean±SD | 24.8±10.7 | 36.8±14.6 | 30.8±14.0 |
| LS Mean [95% CI] change from baseline to Day 27 | – 7.5 [– 12.9, – 2.1] | – 3.7 [– 8.9, 1.5] | – 5.5 [– 9.2, – 1.8] |
| p value vs. baseline | 0.008 | 0.16 | 0.005 |
| Oral levodopa dose (mg) | |||
| Baseline; mean±SD | 996±552 | 948±305 | 972±441 |
| Change from baseline to Day 28; mean±SD | – 648±314 | – 592±371 | – 615±344 |
| Frequency oral levodopa dosing; mean±SD | |||
| Baseline; mean±SD | 6.8±3.2 | 6.9±2.2 | 6.9±2.7 |
| Day 28; mean±SD | 2.3±1.3 | 2.3±2.1 | 2.3±1.9 |
Fig. 2
Changes in ON and OFF time after 28 days ND0612 infusion.
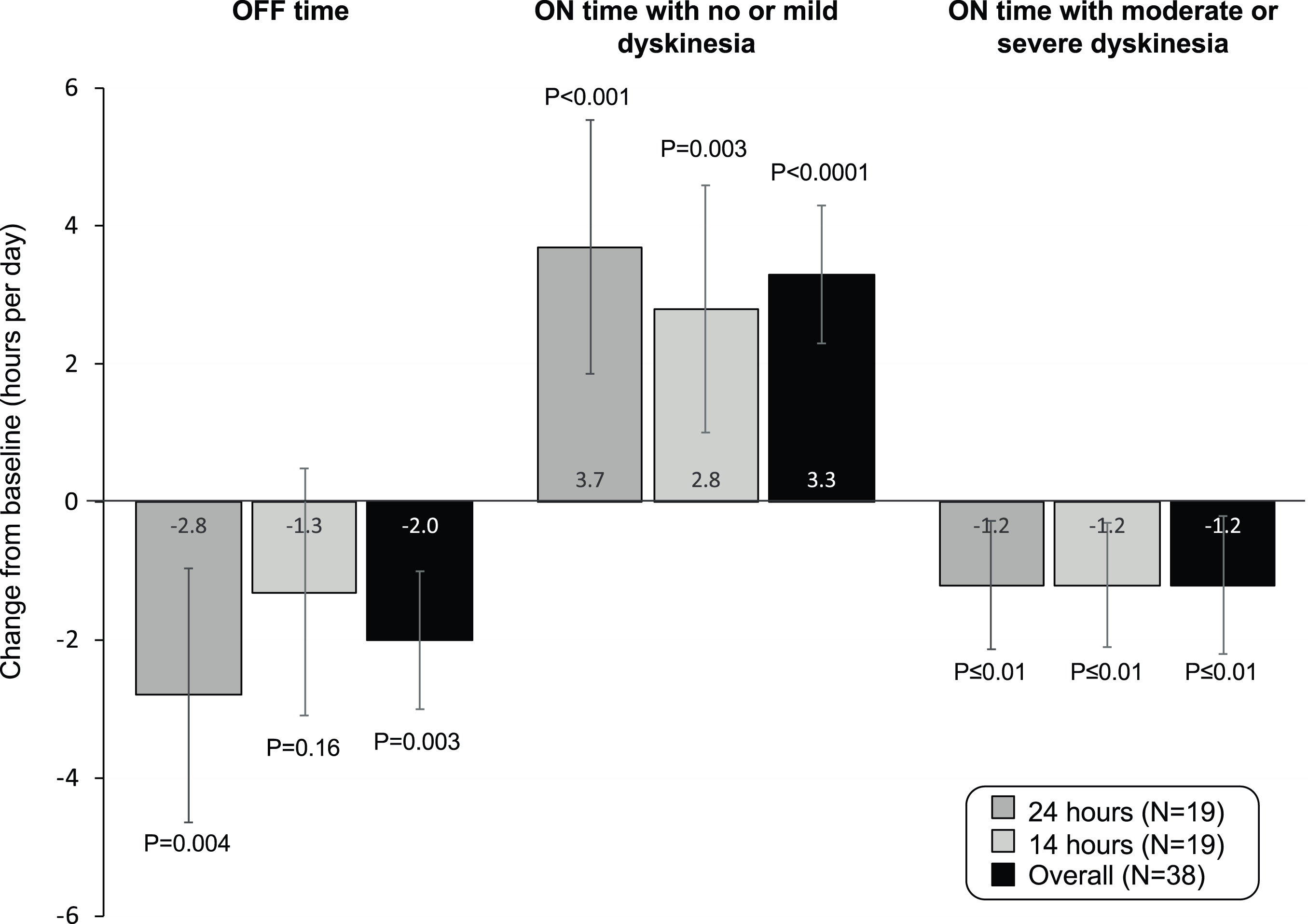
ON time with no or mild dyskinesia increased from baseline for the entire cohort by a LS mean of 3.3 [2.0, 4.6] hours (p < 0.0001). LS mean increases were 3.7 [1.9, 5.6] hours in the 24-hour infusion group (p < 0.001) and 2.8 [1.0, 4.6] hours in the 14-hour infusion group (p = 0.003). ON time with moderate or severe dyskinesia was reduced by a LS mean of 1.2 [– 1.8, – 0.5] hours for the overall cohort (p < 0.001) and was similar in both groups. Fourteen patients had≥1 hour of moderate to severe dyskinesia at baseline; in this subgroup, treatment with ND0612 reduced ON time with moderate-severe dyskinesia from 5.1 hours at baseline to 1.8 hours at Day 28 (LS mean reduction of 3.5 [– 6.0, – 1.0] hours; p = 0.011).
Overall, half (n = 19) of the patients were improved on CGI-I at Day 3, and this proportion increased to 73% at Day 28. Similar benefits were seen with the PGI-I. UPDRS assessments (UPDRS Part II and III) showed nominally significant improvement from baseline, particularly with 24-hour ND0612 infusion (Table 2). PDQ-39 measures of quality of life improved in the overall group by least mean square of – 5.5 [– 9.2, – 1.8] (p = 0.005), with greater improvements in the 24-hour infusion group and in the items corresponding to activities in daily living, bodily discomfort and mobility (Supplementary Table 1).
Oral levodopa dosing frequency was reduced from a mean of 6.9 times at baseline to 2.3 times per day (including the required morning dose) at Day 28 and mean total daily oral levodopa dose was reduced by a mean of – 615±344 mg (– 648±314 mg in the 24-hour group and – 592±371 mg in the 14-hour group).
Safety and tolerability
Adverse events are described in Table 3. Overall, 76.3% (n = 29) of patients experienced≥1 AE, with a similar incidence reported with: the two infusion regimens. The most commonly reported AEs were infusion site reactions (60.5% of all patients). These included nodules (47.4%), bruising (18.4%), and erythema (18.4%). Twenty-seven patients (71.1%) had a skin nodule >1 cm (nine of these patients did not report nodules as an AE). The mean±SD VAS score for pain [range 0– 100] at Day 28 was 6.2±10.9; six patients reported no pain. Four patients had SAEs. In the 24-hour group, one patient had an abscess at the infusion site and orthostatic hypotension and one patient had suspected panniculitis, which was eventually ruled out. In the 14-hour group, one patient fell and fractured his nose and one patient had worsened PD symptoms. There were no clinically significant changes in laboratory values, suicidal ideation, ESS, or QUIP scores. Ten patients receiving 24-hour ND0612 infusion and five receiving 14-hour infusion experienced a pump malfunction; of these three patients required a replacement pump and the remainder were resolved through a telephone helpline system.
Table 3
Adverse Events
| Patients, n (%) | 24-hour infusion | 14-hour infusion | Overall |
| (N = 19) | (N = 19) | (N = 38) | |
| Overall summary | |||
| ≥1 adverse event | 15 (78.9) | 14 (73.7) | 29 (76.3) |
| Treatment-related adverse event | 13 (68.4) | 12 (63.2) | 25 (65.8) |
| Serious adverse event | 2 (10.5) | 2 (10.5) | 4 (10.5) |
| Adverse event leading to study drug discontinuation | 2 (10.5) | 1 (5.3) | 3 (7.9) |
| Adverse events occurring in ≥ 2 patients | |||
| Infusion site nodule | 11 (57.9) | 7 (36.8) | 18 (47.4) |
| Infusion site bruising | 4 (21.1) | 3 (15.8) | 7 (18.4) |
| Infusion site erythema | 5 (26.3) | 2 (10.5) | 7 (18.4) |
| Infusion site hemorrhage | 2 (10.5) | 3 (15.8) | 5 (13.2) |
| Infusion site hematoma | 3 (15.8) | 1 (5.3) | 4 (10.5) |
| Infusion site pain | 1 (5.3) | 3 (15.8) | 4 (10.5) |
| Parkinson’s disease (worsening) | 1 (5.3) | 2 (10.5) | 3 (7.9) |
| Infusion site edema | 1 (5.3) | 1 (5.3) | 2 (5.3) |
| Infusion site pruritus | 2 (10.5) | 0 | 2 (5.3) |
| Headache | 2 (10.5) | 0 | 2 (5.3) |
| Depression | 0 | 2 (10.5) | 2 (5.3) |
| Orthostatic hypotension | 2 (10.5) | 0 | 2 (5.3) |
DISCUSSION
We present the results of the first clinical trial examining the efficacy, safety and tolerability of continuous SC levodopa/carbidopa infusion with ND0612 as a therapy for PD patients with motor fluctuations. While this preliminary study was partially unblinded, and lacked a placebo control, the study demonstrates the feasibility of this treatment approach and suggests that continuous SC delivery of levodopa/carbidopa can safely reduce OFF time and simultaneously improve dyskinesia.
For the entire population, there was a 2-hour reduction in OFF time and a 3.3-hour improvement in ON time with no/mild dyskinesia using an in-clinic 8-hour rating scale (normalized to 16 hours) based on assessments performed by blinded investigators. These changes were matched by a reduction from baseline in moderate-severe dyskinesia by an average of 1.2 hours which was also nominally significant versus baseline (p < 0.001). I Benefits were also noted in patient and physician rated CGI-C, as well as the UPDRS Part-III, UPDRS Part-II, and PDQ-39. The findings with ND0612 are in line with those observed with continuous intrajejunal infusion of LCIG [10], while avoiding the need for, and the risks associated with, a surgical procedure.
Although the study was not designed for a formal statistical comparison between the two regimens, benefits were nominally superior with the 24-hour infusion. In the 24-hour group, 12 patients (63%) had a 50% reduction in OFF time, and eight patients (42%) noted a complete resolution of OFF time. Reduction in early morning akinesia and improved sleep quality were only observed with 24-hour infusion. The study was, however, biased against the 14-hour infusion group as these patients received less infused levodopa, and had to wait for the nurse to arrive in the morning in order to set up the infusion system and take the morning levodopa dose, and thus may have remained in the OFF state for as many as 2 hours before initiating treatment. In contrast, those in the 24-hour infusion group were already receiving levodopa at the time of waking.
Local infusion site reactions were the most common AE. One patient in the 24-hour group developed an abscess at an infusion site and withdrew from the study. Infusion site reactions were otherwise not bothersome to patients and tended to resolve spontaneously over time. More nodules were seen with the 24-hour infusion, but it remains to be determined if duration of the infusion is a risk factor. It should be noted that infusion site reactions, particularly nodules, are commonly seen with subcutaneous infusion of drugs [21– 24]. The development of nodules can generally be minimized with diligent hygiene, daily rotation of sites, massage, and possibly the use of therapeutic ultrasound [24]. Aside from the infusion site reactions, AEs were consistent with the well-established safety profile of levodopa and there were no other clinically significant, unexpected AEs.
There are important limitations to this study. These include the open label design, lack of a placebo-control group, and the relatively small sample size. It should be noted, however, that rather than using the traditional patient-reported home diary, evaluations of motor state were made in clinic over 8 hours by a movement disorder specialist blinded to treatment group. This approach has the advantage of motor state being assessed by an expert, and avoids the problems of delay in form completion, recall errors, and diary fatigue associated with patient self-report [25]. The current infusion formulation only permits a limited amount of levodopa/carbidopa to be administered subcutaneously per day (approximately 700 mg which is bioequivalent to approximately 900 mg of oral levodopa). Thus, patients frequently require supplemental oral levodopa treatment. Greater benefits were seen with the 24-hour treatment group, but the study was not designed to formally compare this approach with the 14-hour waking day infusion as noted above. Potential advantages to a 24-hour infusion include reduced morning OFF periods and improved sleep, but it remains to be determined in a larger sample if there is more nighttime discomfort, more frequent infusion site reactions, or a greater risk of developing tolerance with this approach. On the other hand, a daytime infusion regimen allows more levodopa to be administered subcutaneously during the waking day, reduces infusion site exposure time, and may be preferred by patients for comfort during the night. Of note, most patients (n = 24) chose to continue treatment and rolled over to the ongoing long-term safety study.
In summary, this phase 2 study demonstrates the feasibility of continuously delivering levodopa/carbidopa via a subcutaneous infusion. The results are promising, and a well-controlled, randomized, double-blind, double-dummy study (NCT04006210) comparing continuous subcutaneous administration of levodopa/carbidopa with ND0612 versus optimized oral levodopa therapy has been initiated.
006 Study group
C. Warren Olanow, Alberto J. Espay, Fabrizio Stocchi, Aaron Ellenbogen, Ruth Djaldetti, Tanya Gurevitch, Gilad Yahalom, Sharon Hassin, Marco Onofrj, Ubaldo Bonucelli, Angelo Antonini, Tanya Simuni, Karl Kieburtz.
CONFLICTS OF INTEREST
This study was sponsored by NeuroDerm Ltd. C. Warren Olanow has stock ownership in Clintrex which was contracted by NeuroDerm to provide services for this study. Alberto J. Espay, Fabrizio Stocchi, Aaron Ellenbogen and Werner Poewe were all investigators in the 006 study and report fees for consultancy from NeuroDerm Ltd. Mika Leinonen reports consultancy for Clintrex LLC. Liat Adar, Ryan Case, Shir Fuchs Orenbach, Tami Yardeni and Sheila Oren are employed by NeuroDerm Ltd.
AUTHOR DISCLOSURES
Professor Olanow reports owning shares in Clintrex which provides services for multiple pharmaceutical and biotech companies. Dr Espay has received grant support from the NIH and the Michael J Fox Foundation; personal compensation as a consultant/scientific advisory board member for Abbvie, Neurocrine, Amneal, Adamas, Acadia, Acorda, InTrance, Sunovion, Lundbeck, and US WorldMeds; publishing royalties from Lippincott Williams & Wilkins, Cambridge University Press, and Springer; and honoraria from US WorldMeds, Acadia, Sunovion, the American Academy of Neurology, and the Movement Disorders Society. Professor Stocchi reports honoraria and consulting fees from Britannia Pharmaceuticals, GlaxoSmithKline, Boehringer Ingelheim, Lundbeck, Orion, Novartis, Teva, Pfizer and Zambon. Dr Ellenbogen reports honoraria and consulting fees from Allergan, Ipsen, Revance, Lundbeck, Affiris, Acorda, Kyowa, Biohaven, Teva, and Adamas. Professor Poewe reports receiving personal fees from AbbVie, AFFiRiS, AstraZeneca, BIAL, Boston Scientific, Britannia, Intec, Ipsen, Lundbeck, NeuroDerm, Neurocrine, Denali Pharmaceuticals, Novartis, Orion Pharma, Prexton, Teva, UCB and Zambon. He receives royalties from Thieme, Wiley Blackwell, Oxford University Press and Cambridge University Press and grant support from the Michael J Fox Foundation, EU FP7 and Horizon 2020.
ACKNOWLEDGMENTS
We thank the patients and site staff involved in the study. For their valuable contributions to this work, the authors would like to acknowledge Cecile Durlach, Olivia Rosenfeld and Yael Ullman (NeuroDerm Ltd) as well as Cheryl Fitzer-Attas and Katrina Paumier (Mitsubishi Tanabe Mitsubishi Tanabe Pharma America, Inc). We also thank Anita Chadha-Patel PhD (ACP Clinical Communications Ltd, funded by NeuroDerm, Ltd) for medical writing support (literature searching, referencing and editing).
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JPD-202285.
REFERENCES
[1] | Ahlskog JE , Muenter MD ((2001) ) Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 16: , 448–458. |
[2] | Hely MA , Morris JG , Traficante R , Reid WG , O’Sullivan DJ , Williamson PM ((1999) ) The Sydney multicentre study of Parkinson’s disease: Progression and mortality at 10 years. J Neurol Neurosurg Psychiatry 67: , 300448–307. |
[3] | Olanow CW , Kieburtz K , Rascol O , Poewe W , Schapira AH , Emre M , Nissinen H , Leinonen M , Stocchi F , Stalevo Reduction in Dyskinesia Evaluation in Parkinson’s Disease (STRIDE-PD) Investigators ((2013) ) Factors predictive of the development of Levodopa-induced dyskinesia and wearing-off in Parkinson’s disease. Mov Disord 28: , 1064–1071. |
[4] | Chapuis S , Ouchchane L , Metz O , Gerbaud L , Durif F ((2005) ) Impact of the motor complications of Parkinson’s disease on the quality of life. Mov Disord 20: , 224–230. |
[5] | Hechtner MC , Vogt T , Zollner Y , Schroder S , Sauer JB , Binder H , Singer S , Mikolajczyk R ((2014) ) Quality of life in Parkinson’s disease patients with motor fluctuations and dyskinesias in five European countries. Parkinsonism Relat Disord 20: , 969–974. |
[6] | Olanow CW , Obeso JA , Stocchi F ((2006) ) Continuous dopamine-receptor treatment of Parkinson’s disease: Scientific rationale and clinical implications. Lancet Neurol 5: , 677–687. |
[7] | Olanow CW , Calabrese P , Obeso JA ((2020) ) The concept of continuous dopaminergic delivery – current status. Mov Disord, doi: 10.1002/mds.28215 |
[8] | Abercrombie ED , Bonatz AE , Zigmond MJ ((1990) ) Effects of L-dopa on extracellular dopamine in striatum of normal and 6-hydroxydopamine-treated rats. Brain Res 525: , 36–44. |
[9] | Miller DW , Abercrombie ED ((1999) ) Role of high-affinity dopamine uptake and impulse activity in the appearance of extracellular dopamine in striatum after administration of exogenous L-DOPA: Studies in intact and 6-hydroxydopamine-treated rats. J Neurochem 72: , 1516–1522. |
[10] | Olanow CW , Kieburtz K , Odin P , Espay AJ , Standaert DG , Fernandez HH , Vanagunas A , Othman AA , Widnell KL , Robieson WZ , Pritchett Y , Chatamra K , Benesh J , Lenz RA , Antonini A , LCIG Horizon Study Group ((2014) ) Continuous intrajejunal infusion of levodopa-carbidopa intestinal gel for patients with advanced Parkinson’s disease: A randomised, controlled, double-blind, double-dummy study. Lancet Neurol 13: , 141–149. |
[11] | Fernandez HH , Boyd JT , Fung VSC , Lew MF , Rodriguez RL , Slevin JT , Standaert DG , Zadikoff C , Vanagunas AD , Chatamra K , Eaton S , Facheris MF , Hall C , Robieson WZ , Benesh J , Espay AJ ((2018) ) Long-term safety and efficacy of levodopa-carbidopa intestinal gel in advanced Parkinson’s disease. Mov Disord 33: , 928–936. |
[12] | Giladi N , Caraco Y , Gurevich T , Djaldetti R , Adar L , Rachmilewitz Minei T , Oren S ((2017) ) ND0612 (levodopa/carbidopa for subcutaneous infusion) achieves stable levodopa plasma levels when administered in low and high doses in patients with PD [abstract]. Mov Disord 32 (suppl 2). http://www.mdsabstracts.org/abstract/nd0612-levodopacarbidopa-for-subcutaneous-infusion-achieves-stable-levodopa-plasma-levels-when-administered-in-low-and-high-doses-in-patients-with-pd/. Accessed February 15, 2018. |
[13] | Shaltiel-Karyo R , Caraco Y , Zawaznik E , Weinstock I , Nemas M , Oren S , Yacoby-Zeevi O , LeWitt PA ((2019) ) Subcutaneous administration of carbidopa enhances oral levodopa pharmacokinetics: A series of studies conducted in pigs, mice, and healthy volunteers. Clin Neuropharmacol 42: , 111–116. |
[14] | Hughes AJ , Daniel SE , Ben-Shlomo Y , Lees AJ ((2002) ) The accuracy of diagnosis of parkinsonian syndromes in a specialist movement disorder service. Brain 125: , 861–870. |
[15] | Fahn S , Elton RL ((1987) ) Unified Parkinson’s Disease Rating Scale. In Recent Developments in Parkinson’s Disease. MacMillan Healthcare Information, pp. 153-164. |
[16] | Peto V , Jenkinson C , Fitzpatrick R , Greenhall R ((1995) ) The development and validation of a short measure of functioning and well being for individuals with Parkinson’s disease. Qual Life Res 4: , 241–248. |
[17] | Chaudhuri KR , Pal S , DiMarco A , Whately-Smith C , Bridgman K , Mathew R , Pezzela FR , Forbes A , Hogl B , Trenkwalder C ((2002) ) The Parkinson’s disease sleep scale: A new instrument for assessing sleep and nocturnal disability in Parkinson’s disease. J Neurol Neurosurg Psychiatry 73: , 629–635. |
[18] | Johns MW ((1991) ) A new method for measuring daytime sleepiness: The Epworth sleepiness scale.. Sleep 14: , 540–545. |
[19] | Weintraub D , Mamikonyan E , Papay K , Shea JA , Xie SX , Siderowf A ((2012) ) Questionnaire for Impulsive-Compulsive Disorders in Parkinson’s Disease-Rating Scale. Mov Disord 27: , 242–247. |
[20] | Posner K , Brown GK , Stanley B , Brent DA , Yershova KV , Oquendo MA , Currier GW , Melvin GA , Greenhill L , Shen S , Mann JJ ((2011) ) The Columbia-Suicide Severity Rating Scale: Initial validity and internal consistency findings from three multisite studies with adolescents and adults. Am J Psychiatry 168: , 1266–1277. |
[21] | Guilhem I , Leguerrier AM , Lecordier F , Poirier JY , Maugendre D ((2006) ) Technical risks with subcutaneous insulin infusion. Diabetes Metab 32: , 279–284. |
[22] | Deeb A , Abdelrahman L , Tomy M , Suliman S , Akle M , Smith M , Strauss K ((2019) ) Impact of insulin injection and infusion routines on lipohypertrophy and glycemic control in children and adults with diabetes.. Diabetes Ther 10: , 259–267. |
[23] | Lebrun C , Bertagna M , Cohen M ((2011) ) Cutaneous Side-effects of Immunomodulators in MS. Int MS J 17: , 88–94. |
[24] | Bhidayasiri R , Garcia Ruiz PJ , Henriksen T ((2016) ) Practical management of adverse events related to apomorphine therapy. Parkinsonism Relat Disord 33: (Suppl 1), S42–S48. |
[25] | Papapetropoulos SS ((2012) ) Patient diaries as a clinical endpoint in Parkinson’s disease clinical trials. CNS Neurosci Ther 18: , 380–387. |


