
Brain-First versus Gut-First Parkinson’s Disease: A Hypothesis
Abstract
Parkinson’s disease (PD) is a highly heterogeneous disorder, which probably consists of multiple subtypes. Aggregation of misfolded alpha-synuclein and propagation of these proteinacious aggregates through interconnected neural networks is believed to be a crucial pathogenetic factor. It has been hypothesized that the initial pathological alpha-synuclein aggregates originate in the enteric or peripheral nervous system (PNS) and invade the central nervous system (CNS) via retrograde vagal transport. However, evidence from neuropathological studies suggests that not all PD patients can be reconciled with this hypothesis. Importantly, a small fraction of patients do not show pathology in the dorsal motor nucleus of the vagus. Here, it is hypothesized that PD can be divided into a PNS-first and a CNS-first subtype. The former is tightly associated with REM sleep behavior disorder (RBD) during the prodromal phase and is characterized by marked autonomic damage before involvement of the dopaminergic system. In contrast, the CNS-first phenotype is most often RBD-negative during the prodromal phase and characterized by nigrostriatal dopaminergic dysfunction prior to involvement of the autonomic PNS. The existence of these subtypes is supported by in vivo imaging studies of RBD-positive and RBD-negative patient groups and by histological evidence— reviewed herein. The present proposal provides a fresh hypothesis-generating framework for future studies into the etiopathogenesis of PD and seems capable of explaining a number of discrepant findings in the neuropathological literature.
INTRODUCTION
During the 20th century, Parkinson’s disease (PD) was thought to be a primary brain disorder characterized mainly by loss of pigmented dopaminergic neurons residing in the substantia nigra. More recently, it has become clear that PD is highly heterogeneous and probably consists of several subtypes [1, 2].
In 2003, it was hypothesized that initial pathological alpha-synuclein (asyn) aggregates appear in the olfactory bulb and dorsal motor nucleus of the vagus (DMV) [3]. This concept immediately gave rise to the idea that PD pathology may in fact originate in synapses of the peripheral nervous system (PNS) and invade the brain from the olfactory epithelium and via retrograde axonal transport through the vagus, termed the dual-hit hypothesis [4]. This concept is supported by epidemiological evidence [5, 6] and the observation of pathological asyn aggregates in the PNS of PD patients up to 20 years prior to diagnosis [7–9]. In addition, the apparent prion-like behavior of asyn aggregation and propagation provides a plausible mechanistic framework [10, 11].
Nevertheless, the “periphery-first” hypothesis is still considered controversial and has been the subject of a large number of recent reviews [12–18]. This particular scientific discussion often takes a dichotomous format, i.e., evidence is examined to support that PD always originates in the PNS, or alternatively, that the pathology in all cases is initiated in the central nervous system (CNS). Rarely has it been considered that both of these scenarios could be true [2, 18, 19].
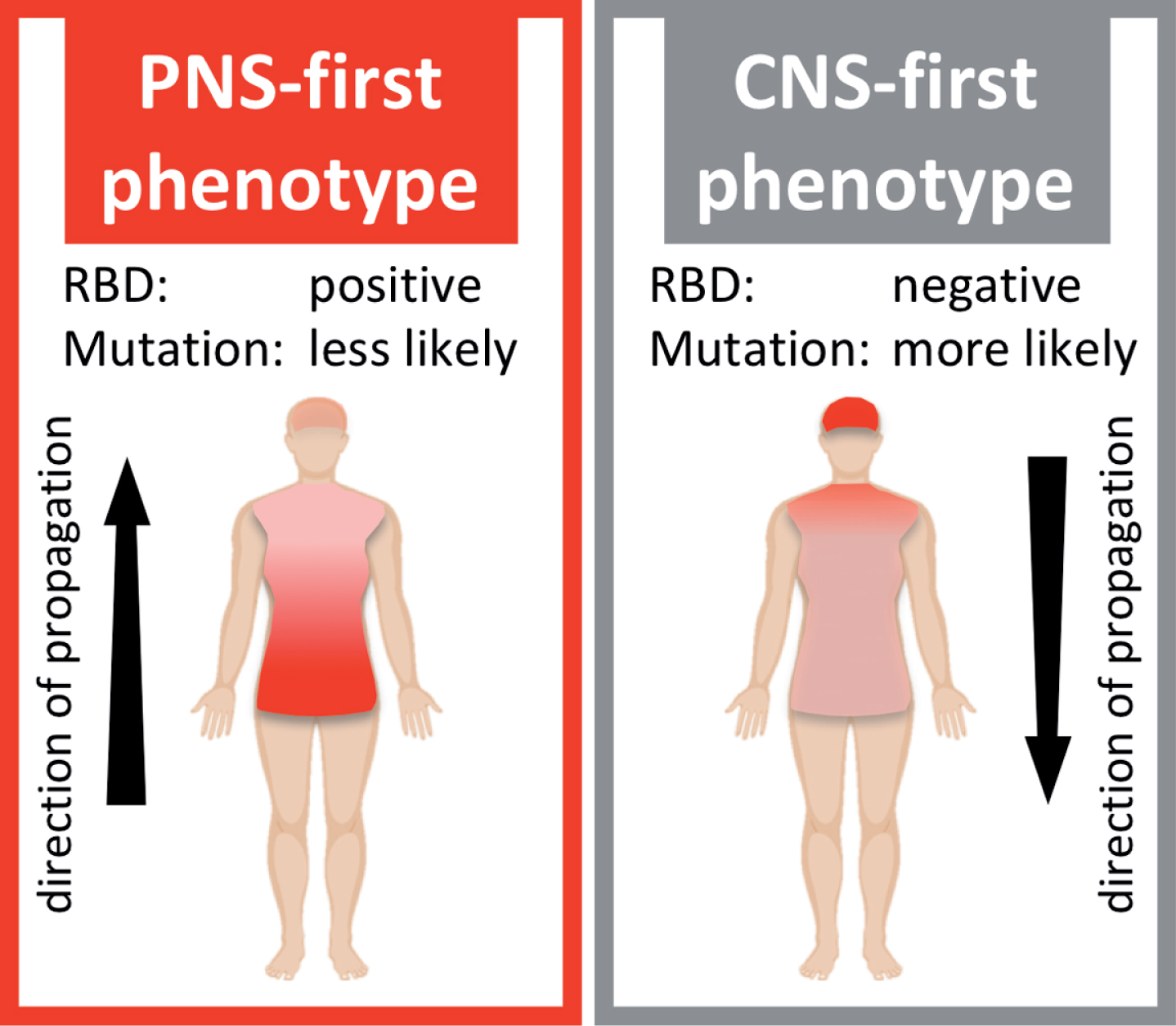
In the present review, we will examine the proposition that Lewy Body disorders (LBD), including PD and dementia with Lewy bodies (DLB), comprise two distinct subtypes: (1) a PNS-first phenotype in which marked damage to the autonomic PNS precedes measurable damage to higher Braak stage structures, including the substantia nigra; (2) a CNS-first phenotype, in which marked damage to the substantia nigra precedes measurable damage to the autonomic PNS.
It is a central component of this hypothesis that the PNS-first phenotype appears to be strongly associated with the presence of REM sleep behavior disorder (RBD) during the prodrome of PD, whereas the CNS-first phenotype is more often RBD-negative during the prodromal phase (Fig. 1).
Fig. 1
Schematic illustration of two hypothetical Lewy body disorder (LBD) phenotypes. The PNS-first phenotype is characterized by early, severe damage to the autonomic PNS. The asyn pathology mainly propagates retrogradely via autonomic connections to the medulla and brainstem. This phenotype is most often RBD-positive during the prodromal phase. The CNS-first phenotype is characterized by early, marked damage to CNS structures, including the substantia nigra, while the autonomic PNS is initially spared. The asyn pathology mainly propagates anterogradely from the CNS to the PNS. Patients are most often initially RBD-negative during the early motor phase.
DEFINITIONS
Idiopathic RBD is a parasomnia defined by loss of REM sleep atonia and dream-enacting behaviors. After 15 years of follow-up, nearly all RBD cases will have converted to a manifest synucleinopathy, including PD, DLB, or rarely multiple system atrophy (MSA) [20–22].
Importantly, idiopathic RBD cases convert to approximately equal numbers of PD and DLB, which are considered part of the same disease spectrum covered by the umbrella term LBD. Most PD patients eventually develop dementia [23], and there is not a single clinical or pathological trait, or molecular characteristic, which uniquely separates PD from DLB at the end stages of disease [24, 25]. Patients with PD or DLB diagnoses comprise a continuous spectrum with a considerable degree of overlap.
Thus, in the present review the concept of PNS-first vs. CNS-first LBD relates mainly to the RBD-status during the prodromal disease phase. In other words, it is hypothesized that patients with PNS-first LBD may eventually get either a PD or a DLB diagnosis, and similarly for the CNS-first subtype.
Also, the PNS- and CNS-first nomenclature presented here is based entirely on a description of the temporal appearance of measurable neuronal dysfunction in the PNS vs. CNS. It does not strictly imply that the initial LBD-related pathology necessarily originates in the PNS vs. CNS, although that seems probable. This is discussed in detail below.
IN VIVO IMAGING STUDIES
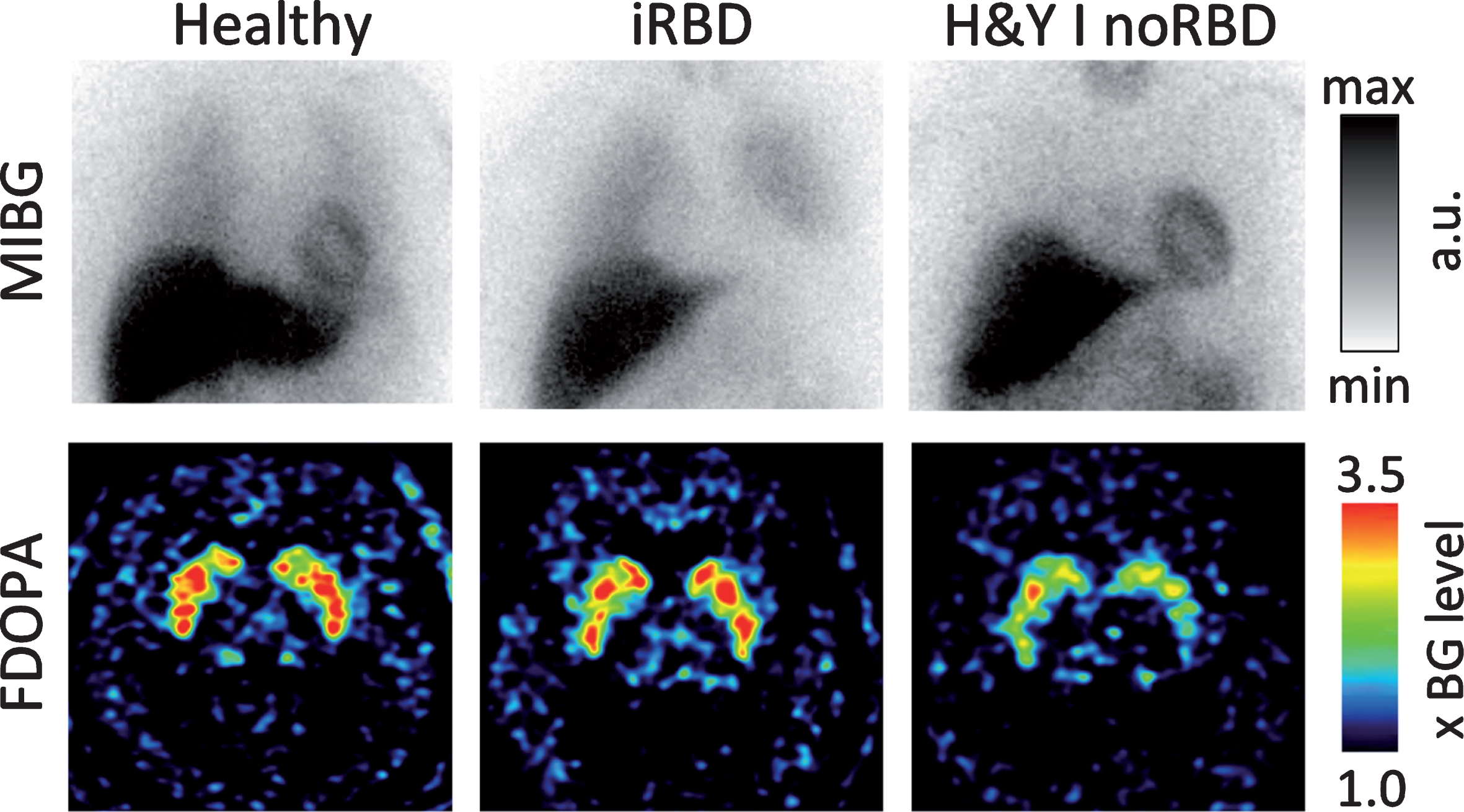
The importance of idiopathic RBD as a marker of the PNS-first phenotype is supported by a number of observations from in vivo human imaging studies. Figure 2 depicts representative cases from our imaging center as an illustration of this literature.
Fig. 2
The cardiac sympathetic innervation imaged with 123I-MIBG scintigraphy (top row) and the nigrostriatal dopamine storage capacity image by 18F-DOPA PET (bottom row) in a healthy control (left column), a patient with idiopathic RBD (iRBD; middle), and a de novo H&Y stage I PD patient without RBD (right). Note that the iRBD patient shows severely reduced sympathetic cardiac innervation but almost normal striatal dopaminergic storage capacity. In contrast, the de novo PD patient without RBD shows normal cardiac innervation but markedly reduced striatal dopamine storage capacity. These cases represent the hypothesized PNS-first and CNS-first phenotypes, respectively. [MIBG heart/mediastinum ratios are scaled arbitrarily. The FDOPA images are scaled from 1 to 3.5 times the cortical background intensity.
RBD-positive prodromal LBD
Five studies used 123I-MIBG scintigraphies to investigate the cardiac sympathetic system in a total of 70 idiopathic RBD cases [26–30]. Of these cases, 94.3% displayed markedly reduced presynaptic functional tone of the cardiac sympathetic nerves compared to study-specific healthy control subjects.
Several studies have investigated the nigrostriatal dopamine system of idiopathic RBD using 123I-FP-CIT SPECT (reviewed in [31]) and 18F-FDOPA PET scans [26]. In summary, this literature finds that a large fraction of idiopathic RBD cases, often more than 50% in each study, still have nigrostriatal dopamine innervation within normal limits.
Thus, idiopathic RBD cases, which almost always represent prodromal PD or DLB, are characterized by nearly obligatory sympathetic denervation, while the nigrostriatal dopamine system is fully or nearly intact in a large proportion of cases. Idiopathic RBD can therefore be considered a PNS-first phenotype (Fig. 1 left).
RBD-negative prodromal LBD
Several large MIBG studies stratified PD patients according to Hoehn & Yahr (H&Y) stages, and reported concurrent findings [28, 32–34]. At H&Y stage I, a large fraction of PD patients (approximately 40–50%) still had cardiac sympathetic innervation within normal limits. In contrast, by H&Y stage III-IV nearly all scintigraphies disclosed severely reduced cardiac innervation.
The RBD status of PD patients in these studies was unknown. However, as mentioned above, more than 94% of idiopathic RBD cases display marked pathology on MIBG examinations, meaning that they will also have pathological MIBG scintigraphies, once they convert to PD or DLB. It can therefore be inferred that those de novo H&Y stage I PD patients with normal MIBG scans must be primarily RBD-negative. On the other hand, H&Y stage I PD patients nearly always show marked nigrostriatal dopaminergic denervation [35].
Thus, a large fraction of de novo H&Y I PD patients without RBD are characterized by obligatory marked damage to the nigrostriatal dopamine system, while their cardiac sympathetic innervation is relatively normal. Such cases may therefore represent a CNS-first phenotype (Fig. 1 right).
IMAGING A GRADIENT OF PATHOLOGY
In a recently published study [26], we aimed to image neuronal dysfunction in Braak stage I, II, and III structures of patients with idiopathic RBD [3]. We assessed Braak stage I using 11C-donepezil PET, a measure of cholinergic (parasympathetic) gut innervation [36, 37], and 123I-MIBG scintigraphy to measure cardiac sympathetic innervation. Neuromelanin-sensitive MRI quantifies the integrity of pigmented locus coeruleus neurons, and 11C-methylreboxetine (MeNER) PET visualizes noradrenergic nerve terminals originating from the coeruleus, both measures of Braak stage II. Finally, stage III was assessed with 18F-DOPA PET.
We hypothesized that if the initial asyn pathology originates in the enteric nervous system (ENS) or autonomic PNS of RBD-positive LBD, we would see a bottom-up gradient of damage using this imaging battery. The duration of the prodromal phase in PD is conservatively set at 20 years [38], so any PNS-first pathology has plenty of time to propagate throughout the autonomic connectome and inflict measurable damage on these neuronal systems years before the appearance of nigrostriatal damage. Indeed, we did find that patients with idiopathic RBD had fully developed pathology in the autonomic PNS and the locus coeruleus, equal to that seen in moderate stage PD. In contrast, most RBD patients had normal putaminal dopamine storage capacity confirming the presence of the hypothesized gradient.
Figure 3 extends this concept by summarizing findings from several imaging studies - grouped such that they reflect damage to specific Braak stage structures. Each study included a group of PD patients and/or a group of idiopathic RBD patients in addition to a representative healthy control group. Within each individual study, the imaging parameter of the healthy control group was set to 100% and the percentage reduction in the patient groups was calculated.
Fig. 3
Summary of imaging modalities used to measure relevant neuronal systems. The healthy control mean (blue) was set to 100% in each study, and the percentage reduction in the patient groups calculated. In cases with more than one study, the standard deviation is depicted by whiskers. Cardiac sympathetic innervation was measured with 123I-MIBG scintigraphy; intestinal cholinergic (parasympathetic) innervation with 11C-donepezil PET; integrity of pigmented locus coeruleus neurons with neuromelanin-sensitive MRI; nigrostriatal dopaminergic synaptic function with 123I-FP-CIT SPECT or 18F-DOPA PET; cortical glucose metabolism with 18F-FDG and principal component analysis to quantify the PD related network (PDRP) z-score. The corresponding Braak stages are shown in the bottom row. Patients with idiopathic RBD (red) show marked loss of autonomic and locus coeruleus imaging parameters, but only minor dopaminergic terminal loss and only slight perturbation of cortical metabolism. The opposite pattern is seen for H&Y stage I-II PD patients. Concerning annotations to PD data: “HY” shows data from different H&Y stage data sets; “PD meta” shows the mean % reduction in PD patients’ DaTscan putamen binding from a metaanalysis [31], and “PD PPMI” the % putaminal reduction seen in early PD patients from the PPMI data set [35]. [Note that except for PDRP z-scores, the imaging parameters approximate loss of specific binding, i.e. heart/mediastinum ratio – 1 for MIBG; SUV – 1 for donepezil; locus coeruleus/pons – 1 for neuromelanin MRI; putamen/occipital cortex – 1 for FP-CIT & FDOPA. For didactic purposes the PDRP z-scores are listed as negative in this figure in contrast to common practice. See main text for study references.]
![Summary of imaging modalities used to measure relevant neuronal systems. The healthy control mean (blue) was set to 100% in each study, and the percentage reduction in the patient groups calculated. In cases with more than one study, the standard deviation is depicted by whiskers. Cardiac sympathetic innervation was measured with 123I-MIBG scintigraphy; intestinal cholinergic (parasympathetic) innervation with 11C-donepezil PET; integrity of pigmented locus coeruleus neurons with neuromelanin-sensitive MRI; nigrostriatal dopaminergic synaptic function with 123I-FP-CIT SPECT or 18F-DOPA PET; cortical glucose metabolism with 18F-FDG and principal component analysis to quantify the PD related network (PDRP) z-score. The corresponding Braak stages are shown in the bottom row. Patients with idiopathic RBD (red) show marked loss of autonomic and locus coeruleus imaging parameters, but only minor dopaminergic terminal loss and only slight perturbation of cortical metabolism. The opposite pattern is seen for H&Y stage I-II PD patients. Concerning annotations to PD data: “HY” shows data from different H&Y stage data sets; “PD meta” shows the mean % reduction in PD patients’ DaTscan putamen binding from a metaanalysis [31], and “PD PPMI” the % putaminal reduction seen in early PD patients from the PPMI data set [35]. [Note that except for PDRP z-scores, the imaging parameters approximate loss of specific binding, i.e. heart/mediastinum ratio – 1 for MIBG; SUV – 1 for donepezil; locus coeruleus/pons – 1 for neuromelanin MRI; putamen/occipital cortex – 1 for FP-CIT & FDOPA. For didactic purposes the PDRP z-scores are listed as negative in this figure in contrast to common practice. See main text for study references.]](https://content.iospress.com:443/media/jpd/2019/9-s2/jpd-9-s2-jad191721/jpd-9-jpd191721-g003.jpg)
In brief, the figure demonstrates that idiopathic RBD patients show more pronounced reduction on measures of sympathetic (MIBG scintigraphy) and parasympathetic (donepezil PET) integrity compared to H&Y stage I-II PD patients with unknown RBD status [26–28, 32]. Idiopathic RBD cases also show reduced neuromelanin content (MRI) in the locus coeruleus, which is on average similar to that seen in PD patients with RBD [26, 39, 40], and more pronounced than that seen in PD patients without RBD [40, 41].
In contrast, idiopathic RBD patients show on average only a 20–25% reduction in the level of putaminal dopaminergic presynaptic imaging markers compared to the more than 50% reduction seen in H&Y I-II PD patients [26, 31, 42]. Of note, more than 400 newly diagnosed PD patients and 193 matched controls have been included into the Parkinson’s Progression Marker Initiative (PPMI) cohort. Forty-four percent of patients in the PPMI cohort were H&Y I and 56 percent were H&Y II. Their baseline dopamine transporter (DaT) SPECT scan showed an average 67% reduction of putaminal specific binding ratios compared to the matched controls [35].
Put together, these imaging measures of Braak stage I-III structures clearly demonstrate that RBD-positive prodromal LBD is characterized by preferential damage to the autonomic PNS and the coeruleus, while initially leaving the nigra relatively intact.
The opposite gradient is seen in H&Y I-II PD patients with unknown RBD status. These patients are characterized by preferential damage to the dopamine system while the Braak stage I structures are relatively spared. Note that approximately 30% of H&Y I-II patients are expected to be RBD-positive [43]. Thus, it is expected that a group of pure RBD-negative H&Y I patients would show even less PNS pathology and therefore an even more striking, inverted gradient of pathology. However, no suitable studies have been conducted in such enriched patient groups. Nevertheless, the contrasting gradients shown in Fig. 3 are already fully compatible with the hypothesis that RBD-positive LBD represents a PNS-first phenotype, whereas RBD-negative LBD is a CNS-first phenotype.
Whereas damage to Braak stage I-III structures can be objectively measured with some degree of confidence, it is more difficult to unambiguously image the higher, cortical Braak stages. Figure 3 includes data from 18F-fluoro-deoxyglucose (FDG) PET scans of cerebral metabolism. The PD related pattern (PDRP) is a well-validated measure to assess the severity of cortical metabolic perturbations in PD based on FDG PET scans [44, 45]. Although, there has been some controversies as to the physiological interpretation of the PDRP [46–48], there is no doubt that cortical hypometabolism progressively worsens with increasing disease severity [49, 50], and that the PDRP to some degree reflects this progression. Principal component analysis is used to determine the degree to which a single patient’s FDG scan resembles the prototypical PDRP and a z-score is calculated. For that reason, the healthy control group always has a mean z-score of zero, and high z-scores signify more extreme perturbations of cerebral metabolism.
Two FDG PET studies investigated groups of idiopathic RBD patients and diagnosed PD and DLB, in addition to healthy controls [51, 52]. From these studies it is evident that the cortical metabolism is less perturbed in idiopathic RBD compared to H&Y I PD patients, and far less than higher stage PD patients and DLB patients. It is unknown to what extent the PDRP specifically reflects pathology to Braak stage IV–VI structures. It is certainly possible that the expression of the PDRP could in part be caused by damage to modulatory brainstem nuclei, including the locus coeruleus, raphe nuclei, substantia nigra, and nucleus basalis - all Braak stage II-III nuclei [3]. Nevertheless, the important point here is that idiopathic RBD showed relatively more normal cortical metabolism compared to H&Y I PD patients, once again reinforcing the claim that idiopathic RBD represents a PNS-first phenotype.
FUTURE MULTI-MODALITY IMAGING STUDIES
As mentioned above, no studies have simultaneously investigated the loss of dopaminergic innervation and the loss of autonomic innervation in prodromal LBD without RBD, or in de novo H&Y I PD patients without RBD. Figure 4A depicts the projected distribution of such data and illustrates how the hypothesis could be investigated by means of multi-modality imaging studies.
Fig. 4
A) Schematic illustration of the two hypothetical routes. The PNS-first (RBD-positive) patients initially show cardiac sympathetic denervation followed by secondary nigrostriatal dopaminergic denervation. The opposite temporal pattern is seen in the CNS-first (RBD-negative) phenotype. B) Red squares depict 18F-DOPA PET and 123I-MIBG cardiac scintigraphy data from idiopathic RBD cases. Healthy control reference data are shown with grey squares (top right) and H&Y II–IV PD data with grey circles (bottom left). Vertical and horizontal lines denote cut-off thresholds based on the healthy reference data. [data from Knudsen et al. [26].
![A) Schematic illustration of the two hypothetical routes. The PNS-first (RBD-positive) patients initially show cardiac sympathetic denervation followed by secondary nigrostriatal dopaminergic denervation. The opposite temporal pattern is seen in the CNS-first (RBD-negative) phenotype. B) Red squares depict 18F-DOPA PET and 123I-MIBG cardiac scintigraphy data from idiopathic RBD cases. Healthy control reference data are shown with grey squares (top right) and H&Y II–IV PD data with grey circles (bottom left). Vertical and horizontal lines denote cut-off thresholds based on the healthy reference data. [data from Knudsen et al. [26].](https://content.iospress.com:443/media/jpd/2019/9-s2/jpd-9-s2-jad191721/jpd-9-jpd191721-g004.jpg)
Figure 4B shows our previously published data from idiopathic RBD, healthy controls, and PD patients [26]. It is evident that these RBD patients fall convincingly along the projected PNS-first route, and substantiates the concept of idiopathic RBD being a PNS-first phenotype. We are currently studying de novo PD patients without RBD with an identical imaging battery to test whether those patients follow the CNS-first route depicted with a grey arrow in Fig. 4A.
EVIDENCE FROM HUMAN HISTOLOGY
It seems clear that RBD is, in general, a marker of a more malignant PD phenotype with faster progression of motor and non-motor symptoms [53]. At autopsy, RBD-positive PD patients had significantly more asyn pathology in 9 of 10 investigated brain regions when compared to RBD-negative PD [54]. However, limited research has been conducted with respect to contrasting the amount of PNS asyn pathology in RBD-negative and -positive PD.
A recent study reported that 18 of 28 (64%) RBD-positive PD cases had pathological asyn aggregates in colonic biopsies compared to only 2 of 15 (13%) RBD-negative PD patients [55]. Another study reported asyn pathology in colonic submucosal biopsies from 4 of 17 (24%) idiopathic RBD patients but only in 1 of 19 (5%) PD patients with unknown RBD status [56].
In a seminal paper on the use of submandibular gland biopsies [57], 8 of 9 (89%) idiopathic RBD cases showed asyn pathology, when glandular tissue was present in the biopsy, and 52% of iRBD cases were asyn positive when including all biopsies. In diagnosed PD cases, 8 of 12 (67%) were positive in the glandular tissue, and 54% were positive when including biopsies without glandular tissue. Importantly, 92% of those PD patients with a clinical history of RBD were asyn positive, compared to 55% in PD patients without a clinical history of RBD (p = 0.06).
Put together, it appears that asyn pathology in the PNS is more frequent in RBD-positive PD compared to RBD-negative cases. But as mentioned above, asyn pathology is also more severe in the brain of RBD-positive PD patients, so it is possible that RBD-positive patients are simply characterized by more widespread asyn pathology in general.
Future studies will be needed to quantify the frequency and severity of asyn pathology in both the PNS and the CNS of idiopathic RBD compared to de novo PD patients without RBD. The development of an asyn-specific PET ligand would greatly facilitate such research. Based on the hypothesized PNS-first and CNS-first phenotypes in the present review, it is predicted that idiopathic RBD cases will show marked asyn pathology in the autonomic PNS but relatively minor pathology in the mesencephalon and limbic regions, whereas de novo RBD-negative PD patients have relatively little PNS pathology, but more pathology in higher Braak stage structures.
A total of 602 whole body autopsies have been performed as part of the Arizona PD consortium. In a recent review paper, the investigators mentioned that not a single case of “gut-only” asyn pathology has been found in this cohort [17], but the full data set and methodology has not yet been published. Of note, other studies have reported a high prevalence of aggregated asyn in the colon and appendix of normal controls [8, 58] and in the small intestine of children infected by norovirus [59], so differences in detection sensitivity needs clarification [60].
Still, a lack of gut-only cases would seem to be a strong argument against the dual-hit hypothesis, but several factors need to be addressed. First, the Arizona study aims to include cases at risk of neurodegenerative disorders, and 46% of cases had some degree of asyn pathology in the CNS [61]. These CNS-positive cases are therefore not eligible in a study of “gut-only” pathology cutting the overall sample size nearly in half. Second, it is conceivable that initial ENS or PNS asyn pathology could be highly localized – perhaps covering only a few cm2. The human gastrointestinal canal measures approximately 8–10 meters at post mortem and has a geometric surface area of at least 7000 cm2. Thus, many hundreds of microscopy slides are required to rule out such localized gut pathology with any degree of confidence. Of note, the terminal end field of a parasympathetic motor neuron probably covers only a few cm2 [62]. It is certainly possible that initially highly localized gut pathology could immediately propagate to the DMV, giving rise to the first CNS pathology, and thereby disqualifying this patient from being categorized as a gut-only case. Also, asyn aggregates are probably transported through the vagus relatively fast [63], so the time window for identifying gut-only pathology may be quite narrow (a few weeks).
Taken together, the reported lack of gut only cases therefore does not rule out that LBD could start in the ENS or PNS, but rather that such initial asyn pathology would probably be very localized or the time window for its detection very narrow. Finally, if the current hypothesis about PNS-first vs. CNS-first LBD is correct, only a fraction of LBD patients will have an initial “gut only” stage of asyn pathology. Finding these cases would therefore be even more difficult.
EVIDENCE FROM ANIMAL STUDIES
The concept of PNS-to-CNS propagation of asyn pathology is supported by several animal studies. It has been shown that asyn assemblies are efficiently transported both retrogradely and anterogradely through the vagus [63–65]. Oral challenge as well as intra-peritoneal, intra-muscular, and intra-venous injections with asyn fibrils lead to widespread asyn pathology in the CNS of transgenic mice with A53T mutations [66–68]. Also, oral gavage with the pesticide rotenone in wild-type mice lead to the formation of pathological asyn assemblies in the DMV and medullary preganglionic neurons of the intermediolateral column, and possibly also in the substantia nigra [69]. This progressive pathology was prevented by hemi-vagotomy and partial sympatectomy [70].
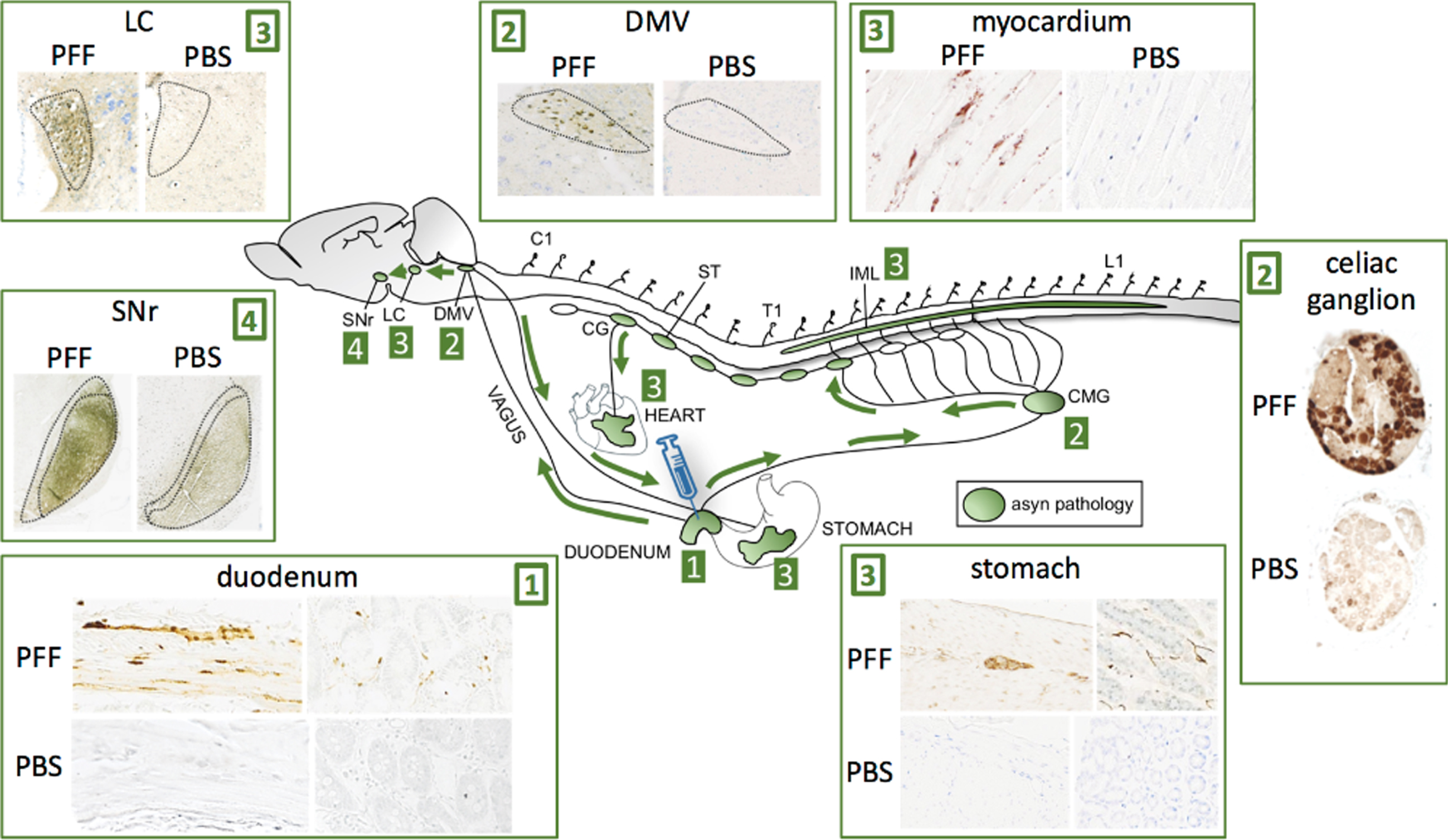
In a recent study, we performed injections with preformed asyn fibrils into the duodenum of transgenic rats, which overexpress human full-length wild-type asyn (Fig. 5) [71]. At 2- and 4 months post-injection, we detected very robust propagation of asyn pathology via the vagus nerve to the DMV, and via the sympathetic connectome to the coeliac ganglion and the intermediolateral cell column (IML). The pathology propagated rostrally in the brainstem with involvement of the locus coeruleus and subtantia nigra pars reticulata. Furthermore, we saw clear pathology in the cardiac sympathetic nerves probably derived from the sympathetic trunk, and also in the stomach, which could signify bi-directional vagal propagation (duodenum-to-DMV-to-stomach). Overall, this propagation pattern demonstrates the feasibility of the gut-first hypothesis, and substantiates that the cardiac sympathetic system can show early prodromal involvement even though the initial pathology originated in the ENS. A similar pattern of asyn propagation after duodenal injection was reported in 3-month-old wild-type mice after duodenal injection [72]. These authors also reported progressive dopaminergic damage associated with the appearance of motor symptoms.
Fig. 5
Injection of asyn preformed fibrils (PFF) into the duodenum of transgenic rats leads to robust propagation of phosphorylated, aggregated asyn through the sympathetic nervous system via the celiac ganglion (CMG) to the IML, and via the vagus nerve to the DMV. Caudo-rostral propagation was seen in the brainstem with involvement of the LC and substantia nigra pars reticulata (SNr). Striking asyn pathology was seen in the sympathetic nerves of the myocardium indicative of anterograde propagation from the cervical ganglia (CG) of the sympathetic trunk (ST). Pathology was also seen in the ENS of the stomach several centimeters from the injection site indicative of anterograde propagation from the DMV or celiac ganglion. No pathological asyn was seen in control transgenic animals injected with phosphate-buffered saline (PBS) in the duodenum.
Of note, almost identical patterns of propagation through the autonomic connectome have been reported in animal models of true prion disorders, including bovine spongiform encephalopathy and scrapie [12, 73–75].
However, other studies have failed to show convincing gut-to-brain propagation after peripheral seeding with asyn fibrils. Injection into the colon wall of young wild-type rats and non-human primates lead to persistent asyn pathology in the ENS, but only transient pathology in the DMV and locus coeruleus, which disappeared after 1 month [76]. Fibril injections into the stomach wall of young wild-type mice similarly lead to persistent ENS pathology, and marked asyn pathology in the DMV at 1.5 months post-injection, but the number of asyn aggregates in the DMV decreased significantly at the 12 month time point and no further caudo-rostral propagation was observed [77].
A potential caveat of these studies was the use of young wild-type animals. Age is the greatest risk factor for PD [78], and it is conceivable that young wild-type mammals, including humans, are capable of mounting a sufficient defense against the toxic insult applied in these studies, thereby minimizing or altogether preventing neuroinvasion of the peripherally administered seeds. Of note, the effects of age on the efficiency of asyn aggregation and propagation have received almost no attention so far, and nearly all studies employ young wild-type or young transgenic animals.
Interestingly, the Fischer-344 wild-type rat strain develops spontaneous asyn pathology in the ENS at old age [79]. When these aged rats were exposed to E. coli bacteria producing the functional bacterial amyloid curli, pathological asyn deposits were seen in both the ENS and brain, suggesting that a high age per se could be an underappreciated permissive factor for gut-to-brain propagation of asyn pathology [80]. The rotenone mouse model cited above also employed 1-year old wild-type animals, which may have facilitated the formation of asyn pathology in that model.
In summary, efficient gut-to-brain propagation of pathological asyn assemblies has mainly been seen in transgenic animals characterized by increased propensity to formation of asyn aggregates, whereas young wild-type animals appear to be more resistant. Important future research goals include studying whether different asyn strains might show differential capacity for neuroinvasion upon peripheral seeding [81], as well as determining the importance of aging as an aggravating factor in this type of study.
IS PAF A VERY EARLY PNS-FIRST SUBTYPE?
Pure autonomic failure (PAF) is considered part of the LBD spectrum. Originally, PAF was defined by severe autonomic dysfunction in the absence of any CNS symptoms [82]. This definition may be in need of revision, since it has now been shown that a large fraction of PAF patients have RBD and olfactory dysfunction [83, 84]. Alternatively, patients with severe dysautonomia but also signs of CNS involvement should not be labeled as PAF.
Nearly all patients with PAF show severe loss of cardiac sympathetic innervation [83, 85, 86], in addition to widespread asyn pathology in autonomic nerve fibres of the skin [85]. However, the vast majority of cases have normal presynaptic dopamine imaging status [85, 86].
Only few patients with PAF have been examined at post mortem. The majority of these cases had Lewy pathology in relevant brainstem structures, including the DMV, locus coeruleus, substantia nigra, and nucleus basalis [87–91]. These data underscore a close relationship between PAF and the common LBDs, and indeed suggest that PAF may be a subtype of LBD characterized by especially severe, early autonomic dysfunction. In support, recent longitudinal studies reported that PAF patients show a high conversion rate to manifest PD or DLB [83]. Importantly, the vast majority (96%) of these converters were RBD-positive. Thus, it seems probable that PAF and idiopathic RBD are closely linked manifestations of prodromal, PNS-first LBD.
Of the 42 patients who had not yet converted to PD, DLB, or MSA, 30 showed clear signs of CNS involvement including probable RBD, impaired olfaction, or subtle motor signs. Only 12 cases remained free of any signs of CNS involvement including RBD [83]. These observations raise the intriguing possibility that PAF may be the most pristine form of PNS-first LBD. In short, PAF may in some or most cases be characterized by damage initially restricted to the autonomic PNS (Braak stage I). Once the pathology has spread and inflicted sufficient damage to the pons (Braak stage II), RBD emerges. Eventually, higher Braak stages are involved giving rise to either parkinsonism or cognitive decline and consequently a diagnosis of PD or DLB. This hypothesis could be tested by following longitudinally a group of RBD-negative PAF patients to study whether such patients first develop RBD, which is then followed by conversion to PD or DLB. This would lend additional support to the concept of a PNS-first phenotype.
THE CNS-FIRST TYPE AND NEUROPATHOLOGICAL STAGING
Skepticism concerning the dual-hit hypothesis and the Braak staging system derives in part from a number of discrepant findings in the neuropathology literature. Several studies have reported that ∼20–50% of PD patients do not conform to the Braak staging scheme [19, 92–94]. Importantly, 7–17% of cases do not exhibit asyn pathology in the lower brainstem including the DMV [92–95]. Also, many of the regions showing Lewy pathology have never been rigorously assessed for cellular dysfunction and degeneration. It was recently suggested that Braak staging seems valid for young onset PD patients with an extended motor-phase, but less so for late onset, rapid course PD [96].
These findings argue against a uniform etiology and site of onset in all LBD patients. However, the diverging neuropathological findings fit perfectly with the concept of separate CNS-first and PNS-first phenotypes. In this scenario, such variation would be expected, especially given the cross sectional nature of post mortem studies, which most often include heterogeneous patient groups at various clinical and neuropathological stages.
Finally, it is important to note that approximately 10–20% of DLB patients initially have normal DaT SPECT scans. With time the majority of these scans become abnormal [97, 98]. Such DLB patients could represent CNS-first cases, in whom the pathology possibly started in the limbic system and caused cognitive decline prior to marked dopaminergic damage. However, they could also represent PNS-first individuals, who have a particularly resistant nigrostriatal dopamine system thereby delaying the onset of parkinsonism.
FAMILIAL PD
More than 20 familial PD mutations have been characterized [99, 100]. The majority of these mutations are very rare and few imaging studies have been performed. Most patients with the Parkin mutation have normal MIBG scans [30, 101–104]. Quattrone et al. reported that one of two patients with the DJ-1 mutation had normal MIBG, and one of two patients with the PINK-1 mutation had normal MIBG [104]. Thus, a large percentage of these autosomal recessive mutation carriers had relatively normal cardiac sympathetic innervation.
Approximately half of 28 European patients with LRRK2 mutations had normal MIBG [30, 104], and most Japanese patients with the I2020T mutation also had normal MIBG [105].
In contrast, all examined symptomatic carriers (n = 4) and 2 of 3 asymptomatic carriers of the E46K PARK-1 mutation had markedly abnormal MIBG [106, 107]. In patients with PARK-4 (multiplication of the asyn gene SNCA), two symptomatic patients with SNCA triplications had reduced cardiac sympathetic innervation, whereas two asymptomatic carriers had normal innervation [108]. Two symptomatic carriers of SNCA duplications also had reduced cardiac innervation [109]. Finally, ten of eleven patients with mutations in the beta-glucocerebrocidase (GBA) gene had pathological MIBG [110–112].
In summary, it seems that relative conservation of cardiac innervation is more common in some types of familial PD than in idiopathic PD. Exceptions include PD patients with GBA mutations and SNCA multiplications, in whom reduced innervation is seen in nearly all symptomatic cases and in some asymptomatic SNCA mutation carriers. These findings suggest that the pathogenic mechanism introduced by some mutations may facilitate a CNS-first subtype, whereas the GBA and SNCA mutation carriers seem to be characterized by early, marked sympathetic denervation resembling more the PNS-first phenotype.
However, given the limited data no firm conclusions can presently be drawn. Longitudinal multi-modality imaging studies initiated during the asymptomatic phase is needed to elucidate whether autonomic denervation or dopaminergic denervation is first to appear in different types of familial PD.
PNS-START OR CELLULAR VULNERABILITY
Based on the evidence presented in this review, the existence of the PNS-first and CNS-first phenotypes seems difficult to dispute. However, for the moment these phenotypes refer only to the observed temporal appearance of measurable neuronal dysfunction. The available evidence does not prove that the initial asyn pathology, or any other pathology relevant for development of LBDs, arises initially in the PNS vs. the CNS, although this seems probable. Nevertheless, alternative explanations are possible.
The observed patterns of PNS-first vs. CNS-first damage, striking as they may be, could have arisen from inter-individual variations in cellular vulnerability factors. It is theoretically possible that the observed inverted gradients depicted in Fig. 3 are caused by differential vulnerability profiles in the autonomic PNS and relevant CNS structures including the nigral dopamine system. At the moment there is no evidence to support this alternative hypothesis and future research is therefore needed.
Fortunately, it will be possible to test a number of relevant hypotheses in human tissue samples. In short, different vulnerability profiles should be evident in easily identifiable RBD-positive vs. RBD-negative de novo LBD patients. If such studies fail to produce evidence of neuronal vulnerability patterns that mirror the observed gradients of neuronal dysfunction, it must be considered increasingly likely that these gradients simply reflect the site of origin of LBD pathology.
THE OLFACTORY BULB
Although the olfactory bulb is an integral part of the dual-hit hypothesis, it will be only briefly discussed in this review. Hyposmia is a frequent, but not universal finding in newly diagnosed PD patients. On average, 70–80% of early cases show decreased olfaction test scores [113–115], and autopsy studies have reported that asyn pathology restricted to olfactory bulb is not uncommon [17, 116]. Interestingly, a recent study reported that only 48% of de novo PD patients with normal MIBG were hyposmic, compared to 71% of those patients with abnormal MIBG scintigraphy [113]. This finding is compatible with the interpretation that the olfactory system is less affected in CNS-first PD patients with normal MIBG, which again might suggest that the pathology in some of these cases did not originate in the olfactory bulb.
Recent rodent studies have demonstrated that projection neurons located in the SN and the LC directly innervate the olfactory bulb [117, 118]. If such projections exist in humans, asyn pathology could rapidly propagate from the olfactory bulb to these key brainstem structures. However, anterograde propagation from the brainstem to the olfactory bulb would also be possible. A recent study showed that injection of asyn fibrils into the olfactory bulb of wild-type mice leads to recruitment of endogenous α-syn into pathological aggregates that spread trans-neuronally to remote brain regions [119]. However, to our knowledge anterograde asyn propagation from brainstem nuclei to the bulb has not been studied.
In summary, the olfactory bulb shows clear asyn pathology in the vast majority of PD cases, and it was hypothesized that the bulb might be one possible entry point of asyn pathology originating in the periphery. Inhaled pathogens or toxins might initiate the first asyn pathology in the olfactory epithelium of susceptible individuals. Swallowed nasal secretions would then expose the gastrointestinal lining to the same pathogens, thus setting the stage for the dual-hit hypothesis.
However, in line with the ideas presented in this review, this pathogenic process may occur in only a subset of PD cases – the PNS-first subtype. Conceivably, the olfactory bulb could sometimes be affected without involvement of the ENS, and vice versa. And in other patients, the initial pathology could arise within the CNS and reach the olfactory bulb via anterograde spreading. In support, some incidental Lewy body cases showed isolated asyn pathology in the amygdala, LC, or dorsal medulla without involvement of the olfactory bulb [17, 116]. Such cases may represent prodromal CNS-first patients, in whom olfactory bulb pathology would arise later.
CONCLUSIONS
Patients with PD display highly heterogeneous symptoms and variable involvement of different neuronal systems during the early disease phases. Here we have proposed that this heterogeneity can be explained in part by dividing PD into a PNS-first and a CNS-first subtype. The former is tightly associated with RBD during the prodromal phase and is characterized by marked autonomic damage before involvement of the dopaminergic system. In contrast, the CNS-first subtype is most often RBD-negative during the prodromal phase and characterized by nigrostriatal dopaminergic dysfunction prior to involvement of the autonomic PNS.
It is possible that these subtypes are determined by the location of the initial asyn pathology, which may originate in the ENS or autonomic PNS in the PNS-first subtype, and in the brainstem or limbic system in the CNS-first subtype. Alternatively, the two subtypes may be caused by different patterns of cellular vulnerability.
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Sauerbier A , Jenner P , Todorova A , Chaudhuri KR ((2016) ) Non motor subtypes and Parkinson’s disease. Parkinsonism Relat Disord 22: (Suppl 1), S41–46. |
[2] | Marras C , Chaudhuri KR ((2016) ) Nonmotor features of Parkinson’s disease subtypes. Mov Disord 31: , 1095–1102. |
[3] | Braak H , Del Tredici K , Rub U , de Vos RA , Jansen Steur EN , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[4] | Braak H , Rub U , Gai WP , Del Tredici K ((2003) ) Idiopathic Parkinson’s disease: Possible routes by which vulnerable neuronal types may be subject to neuroinvasion by an unknown pathogen. J Neural Transm (Vienna) 110: , 517–536. |
[5] | Svensson E , Horvath-Puho E , Thomsen RW , Djurhuus JC , Pedersen L , Borghammer P , Sorensen HT ((2015) ) Vagotomy and subsequent risk of Parkinson’s disease. Ann Neurol 78: , 522–529. |
[6] | Liu B , Fang F , Pedersen NL , Tillander A , Ludvigsson JF , Ekbom A , Svenningson P , Chen H , Wirdefeldt K ((2017) ) Vagotomy and Parkinson disease: A Swedish register-based matched-cohort study. Neurology 88: , 1996–2002. |
[7] | Shannon KM , Keshavarzian A , Dodiya HB , Jakate S , Kordower JH ((2012) ) Is alpha-synuclein in the colon a biomarker for premotor Parkinson’s disease? Evidence from 3 cases. Mov Disord 27: , 716–719. |
[8] | Stokholm MG , Danielsen EH , Hamilton-Dutoit SJ , Borghammer P ((2016) ) Pathological alpha-synuclein in gastrointestinal tissues from prodromal Parkinson disease patients. Ann Neurol 79: , 940–949. |
[9] | Hilton D , Stephens M , Kirk L , Edwards P , Potter R , Zajicek J , Broughton E , Hagan H , Carroll C ((2014) ) Accumulation of alpha-synuclein in the bowel of patients in the pre-clinical phase of Parkinson’s disease. Acta Neuropathol 127: , 235–241. |
[10] | Uchihara T , Giasson BI ((2016) ) Propagation of alpha-synuclein pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol 131: , 49–73. |
[11] | Goedert M , Masuda-Suzukake M , Falcon B ((2017) ) Like prions: The propagation of aggregated tau and alpha-synuclein in neurodegeneration. Brain 140: , 266–278. |
[12] | Borghammer P ((2018) ) How does parkinson’s disease begin? Perspectives on neuroanatomical pathways, prions, and histology. Mov Disord 33: , 48–57. |
[13] | Brundin P , Melki R ((2017) ) Prying into the prion hypothesis for Parkinson’s disease. J Neurosci 37: , 9808–9818. |
[14] | Surmeier DJ , Obeso JA , Halliday GM ((2017) ) Parkinson’s disease is not simply a prion disorder. J Neurosci 37: , 9799–9807. |
[15] | Engelender S , Isacson O ((2017) ) The threshold theory for Parkinson’s disease. Trends Neurosci 40: , 4–14. |
[16] | Breen DP , Halliday GM , Lang AE ((2019) ) Gut-brain axis and the spread of alpha-synuclein pathology: Vagal highway or dead end? Mov Disord 34: , 307–316. |
[17] | Adler CH , Beach TG ((2016) ) Neuropathological basis of nonmotor manifestations of Parkinson’s disease. Mov Disord 31: , 1114–1119. |
[18] | Shannon K , Vanden Berghe P ((2018) ) The enteric nervous system in PD: Gateway, bystander victim, or source of solutions. Cell Tissue Res 373: , 313–326. |
[19] | Beach TG , Adler CH , Lue L , Sue LI , Bachalakuri J , Henry-Watson J , Sasse J , Boyer S , Shirohi S , Brooks R , Eschbacher J , White CL , 3rd , Akiyama H , Caviness J , Shill HA , Connor DJ , Sabbagh MN , Walker DG , Arizona Parkinson’s Disease Consortium ((2009) ) Unified staging system for Lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117: , 613–634. |
[20] | Iranzo A , Tolosa E , Gelpi E , Molinuevo JL , Valldeoriola F , Serradell M , Sanchez-Valle R , Vilaseca I , Lomena F , Vilas D , Llado A , Gaig C , Santamaria J ((2013) ) Neurodegenerative disease status and post-mortem pathology in idiopathic rapid-eye-movement sleep behaviour disorder: An observational cohort study. Lancet Neurol 12: , 443–453. |
[21] | Postuma RB , Gagnon JF , Bertrand JA , Genier Marchand D , Montplaisir JY ((2015) ) Parkinson risk in idiopathic REM sleep behavior disorder: Preparing for neuroprotective trials. Neurology 84: , 1104–1113. |
[22] | Hogl B , Stefani A , Videnovic A ((2018) ) Idiopathic REM sleep behaviour disorder and neurodegeneration - an update. Nat Rev Neurol 14: , 40–55. |
[23] | Aarsland D , Andersen K , Larsen JP , Lolk A , Kragh-Sorensen P ((2003) ) Prevalence and characteristics of dementia in Parkinson disease: An 8-year prospective study. Arch Neurol 60: , 387–392. |
[24] | De Pablo-Fernandez E , Lees AJ , Holton JL , Warner TT ((2019) ) Prognosis and neuropathologic correlation of clinical subtypes of Parkinson disease. JAMA Neurol 76: , 470–479. |
[25] | Walker L , Stefanis L , Attems J ((2019) ) Clinical and neuropathological differences between Parkinson’s disease, Parkinson’s disease dementia and dementia with Lewy bodies - current issues and future directions. J Neurochem, doi: 10.1111/jnc.14698. |
[26] | Knudsen K , Fedorova TD , Hansen AK , Sommerauer M , Otto M , Svendsen KB , Nahimi A , Stokholm MG , Pavese N , Beier CP , Brooks DJ , Borghammer P ((2018) ) In-vivo staging of pathology in REM sleep behaviour disorder: A multimodality imaging case-control study. Lancet Neurol 17: , 618–628. |
[27] | Miyamoto T , Miyamoto M , Inoue Y , Usui Y , Suzuki K , Hirata K ((2006) ) Reduced cardiac 123I-MIBG scintigraphy in idiopathic REM sleep behavior disorder. Neurology 67: , 2236–2238. |
[28] | Kashihara K , Imamura T , Shinya T ((2010) ) Cardiac 123I-MIBG uptake is reduced more markedly in patients with REM sleep behavior disorder than in those with early stage Parkinson’s disease. Parkinsonism Relat Disord 16: , 252–255. |
[29] | Sakakibara R , Tateno F , Aiba Y , Ogata T , Kishi M , Terada H , Inaoka T , Nakatsuka T , Matsuoka K ((2019) ) MIBG myocardial scintigraphy identifies premotor PD/DLB during a negative DAT scan period: Second report. Mov Disord Clin Pract 6: , 46–50. |
[30] | Gabilondo I , Llorens V , Rodriguez T , Fernandez M , Concha TP , Acera M , Tijero B , Murueta-Goyena A , Del Pino R , Cortes J , Gomez-Esteban JC ((2019) ) Myocardial MIBG scintigraphy in genetic Parkinson’s disease as a model for Lewy body disorders. Eur J Nucl Med Mol Imaging 46: , 376–384. |
[31] | Bauckneht M , Chincarini A , De Carli F , Terzaghi M , Morbelli S , Nobili F , Arnaldi D ((2018) ) Presynaptic dopaminergic neuroimaging in REM sleep behavior disorder: A systematic review and meta-analysis. Sleep Med Rev 41: , 266–274. |
[32] | Nagayama H , Hamamoto M , Ueda M , Nagashima J , Katayama Y ((2005) ) Reliability of MIBG myocardial scintigraphy in the diagnosis of Parkinson’s disease. J Neurol Neurosurg Psychiatry 76: , 249–251. |
[33] | Satoh A , Serita T , Seto M , Tomita I , Satoh H , Iwanaga K , Takashima H , Tsujihata M ((1999) ) Loss of 123I-MIBG uptake by the heart in Parkinson’s disease: Assessment of cardiac sympathetic denervation and diagnostic value. J Nucl Med 40: , 371–375. |
[34] | Hamada K , Hirayama M , Watanabe H , Kobayashi R , Ito H , Ieda T , Koike Y , Sobue G ((2003) ) Onset age and severity of motor impairment are associated with reduction of myocardial 123I-MIBG uptake in Parkinson’s disease. J Neurol Neurosurg Psychiatry 74: , 423–426. |
[35] | Marek K , Chowdhury S , Siderowf A , Lasch S , Coffey CS , Caspell-Garcia C , Simuni T , Jennings D , Tanner CM , Trojanowski JQ , Shaw LM , Seibyl J , Schuff N , Singleton A , Kieburtz K , Toga AW , Mollenhauer B , Galasko D , Chahine LM , Weintraub D , Foroud T , Tosun-Turgut D , Poston K , Arnedo V , Frasier M , Sherer T , Parkinson’s Progression Markers Initiative ((2018) ) The Parkinson’s progression markers initiative (PPMI) - establishing a PD biomarker cohort. Ann Clin Transl Neurol 5: , 1460–1477. |
[36] | Gjerloff T , Fedorova T , Knudsen K , Munk OL , Nahimi A , Jacobsen S , Danielsen EH , Terkelsen AJ , Hansen J , Pavese N , Brooks DJ , Borghammer P ((2015) ) Imaging acetylcholinesterase density in peripheral organs in Parkinson’s disease with 11C-donepezil PET. Brain 138: , 653–663. |
[37] | Fedorova TD , Seidelin LB , Knudsen K , Schacht AC , Geday J , Pavese N , Brooks DJ , Borghammer P ((2017) ) Decreased intestinal acetylcholinesterase in early Parkinson disease: An 11C-donepezil PET study. Neurology 88: , 775–781. |
[38] | Savica R , Bradley BF , Mielke MM ((2018) ) When do alpha-synucleinopathies start? An epidemiological timeline: A review. JAMA Neurol 75: , 503–509. |
[39] | Ehrminger M , Latimier A , Pyatigorskaya N , Garcia-Lorenzo D , Leu-Semenescu S , Vidailhet M , Lehericy S , Arnulf I ((2016) ) The coeruleus/subcoeruleus complex in idiopathic rapid eye movement sleep behaviour disorder. Brain 139: , 1180–1188. |
[40] | Sommerauer M , Fedorova TD , Hansen AK , Knudsen K , Otto M , Jeppesen J , Frederiksen Y , Blicher JU , Geday J , Nahimi A , Damholdt MF , Brooks DJ , Borghammer P ((2018) ) Evaluation of the noradrenergic system in Parkinson’s disease: An 11C-MeNER PET and neuromelanin MRI study. Brain 141: , 496–504. |
[41] | Garcia-Lorenzo D , Longo-Dos Santos C , Ewenczyk C , Leu-Semenescu S , Gallea C , Quattrocchi G , Pita Lobo P , Poupon C , Benali H , Arnulf I , Vidailhet M , Lehericy S ((2013) ) The coeruleus/subcoeruleus complex in rapid eye movement sleep behaviour disorders in Parkinson’s disease. Brain 136: , 2120–2129. |
[42] | Rolinski M , Griffanti L , Piccini P , Roussakis AA , Szewczyk-Krolikowski K , Menke RA , Quinnell T , Zaiwalla Z , Klein JC , Mackay CE , Hu MT ((2016) ) Basal ganglia dysfunction in idiopathic REM sleep behaviour disorder parallels that in early Parkinson’s disease. Brain 139: , 2224–2234. |
[43] | Zhang J , Xu CY , Liu J ((2017) ) Meta-analysis on the prevalence of REM sleep behavior disorder symptoms in Parkinson’s disease. BMC Neurol 17: , 23. |
[44] | Eidelberg D , Moeller JR , Dhawan V , Spetsieris P , Takikawa S , Ishikawa T , Chaly T , Robeson W , Margouleff D , Przedborski S , Fahn S ((1994) ) The metabolic topography of parkinsonism. J Cereb Blood Flow Metab 14: , 783–801. |
[45] | Huang C , Tang C , Feigin A , Lesser M , Ma Y , Pourfar M , Dhawan V , Eidelberg D ((2007) ) Changes in network activity with the progression of Parkinson’s disease. Brain 130: , 1834–1846. |
[46] | Borghammer P , Cumming P , Aanerud J , Forster S , Gjedde A ((2009) ) Subcortical elevation of metabolism in Parkinson’s disease–a critical reappraisal in the context of global mean normalization. Neuroimage 47: , 1514–1521. |
[47] | Borghammer P , Cumming P , Aanerud J , Gjedde A ((2009) ) Artefactual subcortical hyperperfusion in PET studies normalized to global mean: Lessons from Parkinson’s disease. Neuroimage 45: , 249–257. |
[48] | Ma Y , Tang C , Moeller JR , Eidelberg D ((2009) ) Abnormal regional brain function in Parkinson’s disease: Truth or fiction? Neuroimage 45: , 260–266. |
[49] | Borghammer P ((2012) ) Perfusion and metabolism imaging studies in Parkinson’s disease. Dan Med J 59: , B4466. |
[50] | Bohnen NI , Koeppe RA , Minoshima S , Giordani B , Albin RL , Frey KA , Kuhl DE ((2011) ) Cerebral glucose metabolic features of Parkinson disease and incident dementia: Longitudinal study. J Nucl Med 52: , 848–855. |
[51] | Meles SK , Vadasz D , Renken RJ , Sittig-Wiegand E , Mayer G , Depboylu C , Reetz K , Overeem S , Pijpers A , Reesink FE , van Laar T , Heinen L , Teune LK , Hoffken H , Luster M , Kesper K , Adriaanse SM , Booij J , Leenders KL , Oertel WH ((2017) ) FDG PET, dopamine transporter SPECT, and olfaction: Combining biomarkers in REM sleep behavior disorder. Mov Disord 32: , 1482–1486. |
[52] | Huang Z , Jiang C , Li L , Xu Q , Ge J , Li M , Guan Y , Wu J , Wang J , Zuo C , Yu H , Wu P ((2019) ) Correlations between dopaminergic dysfunction and abnormal metabolic network activity in REM sleep behavior disorder. J Cereb Blood Flow Metab, doi: 10.1177/0271678X19828916. |
[53] | Fereshtehnejad SM , Romenets SR , Anang JB , Latreille V , Gagnon JF , Postuma RB ((2015) ) New clinical subtypes of parkinson disease and their longitudinal progression: A prospective cohort comparison with other phenotypes. JAMA Neurol 72: , 863–873. |
[54] | Postuma RB , Adler CH , Dugger BN , Hentz JG , Shill HA , Driver-Dunckley E , Sabbagh MN , Jacobson SA , Belden CM , Sue LI , Serrano G , Beach TG ((2015) ) REM sleep behavior disorder and neuropathology in Parkinson’s disease. Mov Disord 30: , 1413–1417. |
[55] | Leclair-Visonneau L , Clairembault T , Coron E , Le Dily S , Vavasseur F , Dalichampt M , Pereon Y , Neunlist M , Derkinderen P ((2017) ) REM sleep behavior disorder is related to enteric neuropathology in Parkinson disease. Neurology 89: , 1612–1618. |
[56] | Sprenger FS , Stefanova N , Gelpi E , Seppi K , Navarro-Otano J , Offner F , Vilas D , Valldeoriola F , Pont-Sunyer C , Aldecoa I , Gaig C , Gines A , Cuatrecasas M , Hogl B , Frauscher B , Iranzo A , Wenning GK , Vogel W , Tolosa E , Poewe W ((2015) ) Enteric nervous system alpha-synuclein immunoreactivity in idiopathic REM sleep behavior disorder. Neurology 85: , 1761–1768. |
[57] | Vilas D , Iranzo A , Tolosa E , Aldecoa I , Berenguer J , Vilaseca I , Marti C , Serradell M , Lomena F , Alos L , Gaig C , Santamaria J , Gelpi E ((2016) ) Assessment of alpha-synuclein in submandibular glands of patients with idiopathic rapid-eye-movement sleep behaviour disorder: A case-control study. Lancet Neurol 15: , 708–718. |
[58] | Killinger BA , Madaj Z , Sikora JW , Rey N , Haas AJ , Vepa Y , Lindqvist D , Chen H , Thomas PM , Brundin P , Brundin L , Labrie V ((2018) ) The vermiform appendix impacts the risk of developing Parkinson’s disease. Sci Transl Med 10: , eaar5280. |
[59] | Stolzenberg E , Berry D , Yang , Lee EY , Kroemer A , Kaufman S , Wong GCL , Oppenheim JJ , Sen S , Fishbein T , Bax A , Harris B , Barbut D , Zasloff MA ((2017) ) A role for neuronal alpha-synuclein in gastrointestinal immunity. J Innate Immun 9: , 456–463. |
[60] | Corbille AG , Letournel F , Kordower JH , Lee J , Shanes E , Neunlist M , Munoz DG , Derkinderen P , Beach TG ((2016) ) Evaluation of alpha-synuclein immunohistochemical methods for the detection of Lewy-type synucleinopathy in gastrointestinal biopsies. Acta Neuropathol Commun 4: , 35. |
[61] | Shprecher DR , Adler CH , Zhang N , Hentz JG , Serrano GE , Dugger BN , Shill HA , Savica R , Caviness JN , Sabbagh MN , Belden CM , Driver-Dunckley E , Mehta SH , Sue LI , Davis KJ , Zamrini E , Beach TG ((2018) ) Predicting alpha-synuclein pathology by REM sleep behavior disorder diagnosis. Parkinsonism Relat Disord 55: , 92–96. |
[62] | Holst MC , Kelly JB , Powley TL ((1997) ) Vagal preganglionic projections to the enteric nervous system characterized with Phaseolus vulgaris-leucoagglutinin. J Comp Neurol 381: , 81–100. |
[63] | Holmqvist S , Chutna O , Bousset L , Aldrin-Kirk P , Li W , Bjorklund T , Wang ZY , Roybon L , Melki R , Li JY ((2014) ) Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol 128: , 805–820. |
[64] | Ulusoy A , Phillips RJ , Helwig M , Klinkenberg M , Powley TL , Di Monte DA ((2017) ) Brain-to-stomach transfer of alpha-synuclein via vagal preganglionic projections. Acta Neuropathol 133: , 381–393. |
[65] | Ulusoy A , Rusconi R , Perez-Revuelta BI , Musgrove RE , Helwig M , Winzen-Reichert B , Di Monte DA ((2013) ) Caudo-rostral brain spreading of alpha-synuclein through vagal connections. EMBO Mol Med 5: , 1051–1059. |
[66] | Breid S , Bernis ME , Babila JT , Garza MC , Wille H , Tamguney G ((2016) ) Neuroinvasion of alpha-synuclein prionoids after intraperitoneal and intraglossal inoculation. J Virol 90: , 9182–9193. |
[67] | Ayers JI , Brooks MM , Rutherford NJ , Howard JK , Sorrentino ZA , Riffe CJ , Giasson BI ((2017) ) Robust central nervous system pathology in transgenic mice following peripheral injection of alpha-synuclein fibrils. J Virol 91: , e02095–16. |
[68] | Lohmann S , Bernis ME , Tachu BJ , Ziemski A , Grigoletto J , Tamguney G ((2019) ) Oral and intravenous transmission of alpha-synuclein fibrils to mice. Acta Neuropathol, doi: 10.1007/s00401-019-02037-5 |
[69] | Pan-Montojo F , Anichtchik O , Dening Y , Knels L , Pursche S , Jung R , Jackson S , Gille G , Spillantini MG , Reichmann H , Funk RH ((2010) ) Progression of Parkinson’s disease pathology is reproduced by intragastric administration of rotenone in mice. PLoS One 5: , e8762. |
[70] | Pan-Montojo F , Schwarz M , Winkler C , Arnhold M , O’Sullivan GA , Pal A , Said J , Marsico G , Verbavatz JM , Rodrigo-Angulo M , Gille G , Funk RH , Reichmann H ((2012) ) Environmental toxins trigger PD-like progression via increased alpha-synuclein release from enteric neurons in mice. Sci Rep 2: , 898. |
[71] | Van Den Berge N , Ferreira N , Gram H , Mikkelsen T , Alstrup AK , Casadei N , Tsung-Pin P , Riess O , Nyengaard JR , Tamgüney G , Jensen PH , Borghammer P ((2019) ) Evidence for bidirectional and trans-synaptic parasympathetic and sympathetic propagation of alpha-synuclein in rats. Acta Neuropathol, doi: 10.1007/s00401-019-02040-w |
[72] | Kim S , Kwon SH , Kam TI , Panicker N , Karuppagounder SS , Lee S , Lee JH , Kim WR , Kook M , Foss CA , Shen C , Lee H , Kulkarni S , Pasricha PJ , Lee G , Pomper MG , Dawson VL , Dawson TM , Ko HS ((2019) ) Transneuronal propagation of pathologic alpha-synuclein from the gut to the brain models Parkinson’s disease. Neuron, doi: 10.1016/j.neuron.2019.05.035 |
[73] | van Keulen LJ , Vromans ME , Dolstra CH , Bossers A , van Zijderveld FG ((2008) ) Pathogenesis of bovine spongiform encephalopathy in sheep. Arch Virol 153: , 445–453. |
[74] | McBride PA , Schulz-Schaeffer WJ , Donaldson M , Bruce M , Diringer H , Kretzschmar HA , Beekes M ((2001) ) Early spread of scrapie from the gastrointestinal tract to the central nervous system involves autonomic fibers of the splanchnic and vagus nerves. J Virol 75: , 9320–9327. |
[75] | Hoffmann C , Ziegler U , Buschmann A , Weber A , Kupfer L , Oelschlegel A , Hammerschmidt B , Groschup MH ((2007) ) Prions spread via the autonomic nervous system from the gut to the central nervous system in cattle incubating bovine spongiform encephalopathy. J Gen Virol 88: , 1048–1055. |
[76] | Manfredsson FP , Luk KC , Benskey MJ , Gezer A , Garcia J , Kuhn NC , Sandoval IM , Patterson JR , O’Mara A , Yonkers R , Kordower JH ((2018) ) Induction of alpha-synuclein pathology in the enteric nervous system of the rat and non-human primate results in gastrointestinal dysmotility and transient CNS pathology. Neurobiol Dis 112: , 106–118. |
[77] | Uemura N , Yagi H , Uemura MT , Hatanaka Y , Yamakado H , Takahashi R ((2018) ) Inoculation of alpha-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol Neurodegener 13: , 21. |
[78] | GBD 2016 Parkinson’s Disease Collaborators ((2018) ) Global, regional, and national burden of Parkinson’s disease, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol 17: , 939–953. |
[79] | Phillips RJ , Walter GC , Ringer BE , Higgs KM , Powley TL ((2009) ) Alpha-synuclein immunopositive aggregates in the myenteric plexus of the aging Fischer 344 rat. Exp Neurol 220: , 109–119. |
[80] | Chen SG , Stribinskis V , Rane MJ , Demuth DR , Gozal E , Roberts AM , Jagadapillai R , Liu R , Choe K , Shivakumar B , Son F , Jin S , Kerber R , Adame A , Masliah E , Friedland RP ((2016) ) Exposure to the functional bacterial amyloid protein curli enhances alpha-synuclein aggregation in aged Fischer 344 rats and Caenorhabditis elegans. Sci Rep 6: , 34477. |
[81] | Peelaerts W , Bousset L , Baekelandt V , Melki R ((2018) ) a-Synuclein strains and seeding in Parkinson’s disease, incidental Lewy body disease, dementia with Lewy bodies and multiple system atrophy: Similarities and differences. Cell Tissue Res 373: , 195–212. |
[82] | ((1996) ) Consensus statement on the definition of orthostatic hypotension, pure autonomic failure, and multiple system atrophy. The Consensus Committee of the American Autonomic Society and the American Academy of Neurology. Neurology 46: , 1470. |
[83] | Kaufmann H , Norcliffe-Kaufmann L , Palma JA , Biaggioni I , Low PA , Singer W , Goldstein DS , Peltier AC , Shibao CA , Gibbons CH , Freeman R , Robertson D , Autonomic Disorders Consortium ((2017) ) Natural history of pure autonomic failure: A United States prospective cohort. Ann Neurol 81: , 287–297. |
[84] | Miglis MG , Muppidi S , During E , Jaradeh S ((2017) ) A case series of REM sleep behavior disorder in pure autonomic failure. Clin Auton Res 27: , 41–44. |
[85] | Donadio V , Incensi A , Piccinini C , Cortelli P , Giannoccaro MP , Baruzzi A , Liguori R ((2016) ) Skin nerve misfolded alpha-synuclein in pure autonomic failure and Parkinson disease. Ann Neurol 79: , 306–316. |
[86] | Goldstein DS , Sewell L , Sharabi Y ((2011) ) Autonomic dysfunction in PD: A window to early detection? J Neurol Sci 310: , 118–122. |
[87] | Arai K , Kato N , Kashiwado K , Hattori T ((2000) ) Pure autonomic failure in association with human alpha-synucleinopathy. Neurosci Lett 296: , 171–173. |
[88] | Hague K , Lento P , Morgello S , Caro S , Kaufmann H ((1997) ) The distribution of Lewy bodies in pure autonomic failure: Autopsy findings and review of the literature. Acta Neuropathol 94: , 192–196. |
[89] | Miura H , Tsuchiya K , Kubodera T , Shimamura H , Matsuoka T ((2001) ) [An autopsy case of pure autonomic failure with pathological features of Parkinson’s disease]. Rinsho Shinkeigaku 41: , 40–44. |
[90] | Terao Y , Takeda K , Sakuta M , Nemoto T , Takemura T , Kawai M ((1993) ) Pure progressive autonomic failure: A clinicopathological study. Eur Neurol 33: , 409–415. |
[91] | van Ingelghem E , van Zandijcke M , Lammens M ((1994) ) Pure autonomic failure: A new case with clinical, biochemical, and necropsy data. J Neurol Neurosurg Psychiatry 57: , 745–747. |
[92] | Kalaitzakis ME , Graeber MB , Gentleman SM , Pearce RK ((2008) ) The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease: A critical analysis of alpha-synuclein staging. Neuropathol Appl Neurobiol 34: , 284–295. |
[93] | Zaccai J , Brayne C , McKeith I , Matthews F , Ince PG , Mrc Cognitive Function ANS ((2008) ) Patterns and stages of alpha-synucleinopathy: Relevance in a population-based cohort. Neurology 70: , 1042–1048. |
[94] | Parkkinen L , Pirttila T , Alafuzoff I ((2008) ) Applicability of current staging/categorization of alpha-synuclein pathology and their clinical relevance. Acta Neuropathol 115: , 399–407. |
[95] | Attems J , Jellinger KA ((2008) ) The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson’s disease. Neuropathol Appl Neurobiol 34: , 466–467. |
[96] | Jellinger KA ((2019) ) Is Braak staging valid for all types of Parkinson’s disease? J Neural Transm (Vienna) 126: , 423–431. |
[97] | McKeith I , O’Brien J , Walker Z , Tatsch K , Booij J , Darcourt J , Padovani A , Giubbini R , Bonuccelli U , Volterrani D , Holmes C , Kemp P , Tabet N , Meyer I , Reininger C ((2007) ) Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: A phase III, multicentre study. Lancet Neurol 6: , 305–313. |
[98] | van der Zande JJ , Booij J , Scheltens P , Raijmakers PG , Lemstra AW ((2016) ) [(123)]FP-CIT SPECT scans initially rated as normal became abnormal over time in patients with probable dementia with Lewy bodies. Eur J Nucl Med Mol Imaging 43: , 1060–1066. |
[99] | Langston JW , Schule B , Rees L , Nichols RJ , Barlow C ((2015) ) Multisystem Lewy body disease and the other parkinsonian disorders. Nat Genet 47: , 1378–1384. |
[100] | Tomiyama H , Lesage S , Tan EK , Jeon BS ((2015) ) Familial Parkinson’s disease/parkinsonism. Biomed Res Int 2015: , 736915. |
[101] | Suzuki M , Hattori N , Orimo S , Fukumitsu N , Abo M , Kono Y , Sengoku R , Kurita A , Honda H , Inoue K ((2005) ) Preserved myocardial [123I]metaiodobenzylguanidine uptake in autosomal recessive juvenile parkinsonism: First case report. Mov Disord 20: , 634–636. |
[102] | Orimo S , Amino T , Yokochi M , Kojo T , Uchihara T , Takahashi A , Wakabayashi K , Takahashi H , Hattori N , Mizuno Y ((2005) ) Preserved cardiac sympathetic nerve accounts for normal cardiac uptake of MIBG in PARK2. Mov Disord 20: , 1350–1353. |
[103] | Yoritaka A , Shimo Y , Shimo Y , Inoue Y , Yoshino H , Hattori N ((2011) ) Nonmotor Symptoms in Patients with PARK2 Mutations. Parkinsons Dis 2011: , 473640. |
[104] | Quattrone A , Bagnato A , Annesi G , Novellino F , Morgante L , Savettieri G , Zappia M , Tarantino P , Candiano IC , Annesi F , Civitelli D , Rocca FE , D’Amelio M , Nicoletti G , Morelli M , Petrone A , Loizzo P , Condino F ((2008) ) Myocardial 123metaiodobenzylguanidine uptake in genetic Parkinson’s disease. Mov Disord 23: , 21–27. |
[105] | Hasegawa K , Stoessl AJ , Yokoyama T , Kowa H , Wszolek ZK , Yagishita S ((2009) ) Familial parkinsonism: Study of original Sagamihara PARK8 (I2020T) kindred with variable clinicopathologic outcomes. Parkinsonism Relat Disord 15: , 300–306. |
[106] | Tijero B , Gomez-Esteban JC , Llorens V , Lezcano E , Gonzalez-Fernandez MC , de Pancorbo MM , Ruiz-Martinez J , Cembellin JC , Zarranz JJ ((2010) ) Cardiac sympathetic denervation precedes nigrostriatal loss in the E46K mutation of the alpha-synuclein gene (SNCA). Clin Auton Res 20: , 267–269. |
[107] | Orimo S , Ghebremedhin E , Gelpi E ((2018) ) Peripheral and central autonomic nervous system: Does the sympathetic or parasympathetic nervous system bear the brunt of the pathology during the course of sporadic PD? Cell Tissue Res 373: , 267–286. |
[108] | Singleton A , Gwinn-Hardy K , Sharabi Y , Li ST , Holmes C , Dendi R , Hardy J , Singleton A , Crawley A , Goldstein DS ((2004) ) Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain 127: , 768–772. |
[109] | Nishioka K , Hayashi S , Farrer MJ , Singleton AB , Yoshino H , Imai H , Kitami T , Sato K , Kuroda R , Tomiyama H , Mizoguchi K , Murata M , Toda T , Imoto I , Inazawa J , Mizuno Y , Hattori N ((2006) ) Clinical heterogeneity of alpha-synuclein gene duplication in Parkinson’s disease. Ann Neurol 59: , 298–309. |
[110] | Itokawa K , Tamura N , Kawai N , Shimazu K , Ishii K ((2006) ) Parkinsonism in type I Gaucher’s disease. Intern Med 45: , 1165–1167. |
[111] | Lebouvier T , Clairembault T , Devos D , Pallardy A , Coron E , Neunlist M , Derkinderen P ((2014) ) Peripheral autonomic nervous system involvement in Gaucher-related parkinsonism. J Parkinsons Dis 4: , 29–32. |
[112] | Li Y , Sekine T , Funayama M , Li L , Yoshino H , Nishioka K , Tomiyama H , Hattori N ((2014) ) Clinicogenetic study of GBA mutations in patients with familial Parkinson’s disease. Neurobiol Aging 35: , 935 e933–938. |
[113] | Kim JS , Park HE , Park IS , Oh YS , Ryu DW , Song IU , Jung YA , Yoo IR , Choi HS , Lee PH , Lee KS ((2017) ) Normal ‘heart’ in Parkinson’s disease: Is this a distinct clinical phenotype? Eur J Neurol 24: , 349–356. |
[114] | Lee HR , Park JH , Han SW , Baik JS ((2018) ) Cognition, Olfaction and Uric Acid in Early de novo Parkinson’s Disease. J Mov Disord 11: , 139–144. |
[115] | Swallow DM , Lawton MA , Grosset KA , Malek N , Smith CR , Bajaj NP , Barker RA , Ben-Shlomo Y , Burn DJ , Foltynie T , Hardy J , Morris HR , Williams N , Wood NW , Grosset DG , PRoBaND Clinical Consortium ((2016) ) Variation in recent onset Parkinson’s disease: Implications for prodromal detection. J Parkinsons Dis 6: , 289–300. |
[116] | Sumikura H , Takao M , Hatsuta H , Ito S , Nakano Y , Uchino A , Nogami A , Saito Y , Mochizuki H , Murayama S ((2015) ) Distribution of alpha-synuclein in the spinal cord and dorsal root ganglia in an autopsy cohort of elderly persons. Acta Neuropathol Commun 3: , 57. |
[117] | Hoglinger GU , Alvarez-Fischer D , Arias-Carrion O , Djufri M , Windolph A , Keber U , Borta A , Ries V , Schwarting RK , Scheller D , Oertel WH ((2015) ) A new dopaminergic nigro-olfactory projection. Acta Neuropathol 130: , 333–348. |
[118] | Kebschull JM , Garcia da Silva P , Reid AP , Peikon ID , Albeanu DF , Zador AM ((2016) ) High-throughput mapping of single-neuron projections by sequencing of barcoded RNA. Neuron 91: , 975–987. |
[119] | Rey NL , Wesson DW , Brundin P ((2016) ) The olfactory bulb as the entry site for prion-like propagation in neurodegenerative diseases. Neurobiol Dis 109(Pt B): , 226–248. |


