
Therapies to Slow, Stop, or Reverse Parkinson’s Disease
Abstract
Our understanding of PD pathophysiology is vastly improved compared to the situation 20 years ago. We have identified the major genetic risks for PD, we now have far more representative animal models of the disease, and we can be inspired by the early successes of others using Antisense Oligonucleotide and vaccination approaches in other neurodegenerative diseases. We also have a broad range of repurposed drugs showing the first signals of potential efficacy in the translational pipeline which are being driven forward through the various clinical trial stages. We believe we can be optimistic that the next 20 years will be a time for major breakthroughs towards the discovery of therapies that may slow, stop, or reverse PD.
INTRODUCTION
No one has a crystal ball, but it is increasingly likely that the next 20 years will see great progress in the search for approaches to slow down, stop or reverse the pathological processes of Parkinson’s disease (PD). Our understanding of PD pathophysiology, the development of better laboratory models of PD, combined with data emerging from both the lab and from early phase clinical trials, (in the authors’ view) strongly justifies sending this message of hope to our patients.
The evolution of treatments from the initial laboratory discovery to routine clinical prescription is however frustratingly slow, and a realistic prediction of where we will be in 20 years has to consider this. Investigator initiated trials depend on repetitive cycles of funding application and committee review, and this can add to the time needed to pass through the necessary clinical trial stages. Even evaluation of commercially backed products can seemingly take an excessively long time to run the gauntlet from bench to bedside.
In the search for a symptomatic therapy, it is easy to define a threshold of response and quickly test single and multiple ascending doses over a relatively short duration to identify safety, tolerability and efficacy/futility. In contrast, in the search for effective therapies that may have disease modifying or even neurorestorative properties, it may take months or years for a signal to become detectable in view of the generally slow rate of progression of PD, and therefore can require large sample sizes to identify small but potentially important effect sizes over the longer term. Distinguishing between a transient, i.e., symptomatic effect from a long term, cumulative effect can be extremely challenging. The different trial designs that might be most suitable in the search for disease modifying therapies have been recently highlighted [1], but there is still no agreement about which is the best and most pragmatic way of demonstrating this signal of effect. In short, there is an inevitable compromise between cost, trial duration, expediency and power especially in the search for agents with potentially small effect sizes.
For some approaches, we may however already be a fair distance down the 20 year road towards disease modifying drug discovery and development, which will be the focus of this review. We will highlight the drugs and candidate molecules that appear to be surviving and/or emerging as the most likely disease modifying approaches for PD, despite the challenges described above. Finally we also speculate on how this field will evolve in the next 20 years.
PRECISION MEDICINE MAY HAVE BROAD APPLICATION
LRRK2 (leucine rich repeat kinase 2)
The discovery of genetic risk factors for PD has been effective in elucidating precise pathways to new therapeutic targets. Autosomal dominant mutations in the LRRK2 gene tend to cause PD through a toxic gain of function [2], which in turn has lead to an ongoing focus on agents that inhibit LRRK2 thereby aiming directly at the cause for this population of patients.
However, attempts to exploit LRRK2 inhibitors thus far have been thwarted by the observation that primates exposed to LRRK2 inhibitors develop lung toxicity [3]. Better delivery mechanisms may be one route to overcome this hurdle, or alternatively approaches to LRRK2 may exploit antisense oligonucleotide technology, as has already been shown to be effective in spinal muscular atrophy [4]. This approach (already being pursued commercially) could specifically target brain LRRK2 activity via intrathecal administration (e.g., using minipumps as used in intrathecal Baclofen delivery) or other brain selective delivery mechanisms (e.g., convection enhanced delivery [5]).
But what if brain LRRK2 activity is not actually the relevant target? There are LRRK2 variants associated with an elevated risk of PD in the sporadic PD population, suggesting this enzyme may be a target for a larger population of PD patients [6]. However some of these variants are robustly associated with PD but without appearing to have any impact on brain LRRK2 gene expression [7]. Other factors may influence LRRK2 activity other than gene expression, however reflecting on this and also considering parallel work revealing the relevance of LRRK2 to Crohn’s disease as well as mycobacterial infections [8–10], we might speculate that PD pathogenesis may (in some individuals) follow an interaction between LRRK2 activity in the gut, the gut microbiome, and/or gut alpha synuclein. Further data will be needed to explore this, but if so, our LRRK2 inhibitors may turn out to be effective orally, and maybe not only in patients with Mendelian LRRK2 mutations, but even among those with LRRK2 polymorphisms which may increase the risk for developing sporadic PD.
GBA (Glucosidase beta acid)
Along similar lines, the identification of a treatment to combat PD in patients with mutations in the GBA gene, may also have relevance to patients with sporadic forms of PD. The enzyme glucocerebrosidase (GCase), encoded by this gene, plays a role in the normal trafficking and/or processing of alpha synuclein which is reduced in GBA mutation carriers. Exactly how GCase interacts with alpha synuclein to cause PD in some people with GBA mutations is not yet precisely clear but several plausible theories exist [11]. A bidirectional relationship occurs with misfolded alpha synuclein itself impacting on GCase function [12] thus this enzyme may too be a relevant target in patients with sporadic forms of PD, in whom low GCase activity can also be seen [13]. Chaperones that boost this enzyme include Ambroxol [14, 15], a licensed drug for lung surfactant deficiency, and in Europe also available as a cough linctus. Importantly, studies are nearing completion to identify whether PD patients can tolerate the dose required to penetrate the central nervous system and act as a GCase chaperone. Beyond this, the potential efficacy of Ambroxol will undoubtedly be formally explored, but the fact that a GCase chaperone (e.g., Ambroxol) may in parallel also act as an inhibitor of GCase activity could prove to be a stumbling block in developing it as a new therapy for PD [16].
Irrespective of the success or failure of Ambroxol, given the emerging importance of GCase [17] it is important to note that other novel GCase chaperones have also been identified [18]. Some of these may have more favorable profiles of actions and brain penetrance than Ambroxol, and in view of the potential effect on alpha synuclein misfolding, we can expect that they will likely require clinical trial evaluation not only in GBA patients but also in the sporadic PD population.
SLOWING THE SPREAD OF ALPHA SYNUCLEIN PATHOLOGY
There is a mantra that disease modifying treatments must be initiated early if they are to be of any use in preventing disease progression. This is because post mortem specimens suggest that >50% of SNc dopaminergic neurons are already lost by the time that patients present with the classical motor features of the disease [19]. Given that there are many effective symptomatic treatments to help the motor, dopaminergic deficits in PD, it can be argued that the important unmet needs in PD relate to cognitive, speech, gait and balance difficulties and autonomic failure. In this case any treatment that prevented onset/worsening of these symptoms, would be highly relevant even in patients with established motor PD, and would be addressing the major challenges that patients face in its long-term management. From another perspective, since many of these “non-motor” features of PD may precede the onset of motor symptoms, we may have an even earlier window to start therapy, and have an opportunity to slow or stop the development of even the first motor symptoms of PD.
Preventing propagation of pathology from one neuron to the next (and thus in theory preventing the onset of these disabling non-motor symptoms) may be accomplished by the use of antibodies with specificity for pathogenic species of alpha synuclein. This approach could be boosted if publication of the BAN2401 antibody data in Alzheimer’s disease [20] aligns with the results announced at the 2018 Alzheimer’s association International Conference. Biogen, Roche/Prothena and Affiris are all investing in major trials of alpha synuclein antibody or vaccination programs to try and reproduce this type of success in PD. The success of these projects will depend at least in part on 1) ensuring that sufficient antibody accesses the brain parenchyma, 2) that the toxic species of alpha synuclein is visible to the antibody at the trans-synaptic site, and 3) that there is sufficient specificity of the antibody to avoid interference with the synaptic functions of normal alpha synuclein (which have yet to be fully elucidated). This far, it appears that CNS penetration is indeed adequate with the antibodies currently in trials, and that serum alpha synuclein levels fall following sustained antibody administration [21, 22]. As yet the studies currently recruiting are inevitably prioritising safety and tolerability while trying to identify the optimal dose, and are unlikely to be completed until 2021/2022.
Beyond this, the companies will have to look carefully at the emerging data to decide whether a strategy of enrichment (i.e., selecting patients with specific genotypes or according to their predicted rate of progression [23]) might be advantageous, and/or whether to risk extending trial eligibility to patients with more established disease than those currently being recruited.
ENHANCING NEURONAL SURVIVAL PROCESSES
An alternative to stopping the spread of pathology, is to try and help neurons continue to function despite the presence of pathological alpha synuclein, i.e., to provide some form of trophic support. There are several classes of drugs being repurposed (i.e., already confirmed to be safe for human use), which may achieve this.
There has been a lot of publicity surrounding the potential of Glucagon-like peptide 1 (GLP-1) receptor agonists in PD [24, 25]. These drugs are licensed for the treatment of Type 2 diabetes (T2DM) and have neuroprotective properties across the whole range of animal models of PD, including 2 alpha synuclein models. There is some evidence that this action may relate to an improvement in brain insulin signaling (reviewed in [26]) which enhances Akt activity (a critical protein involved in cell survival processes) while additional data indicate these drugs may also act in parallel through a positive effect on neuroinflammation [27]. Indeed, the increased risk of developing PD among T2DM patients may be ameliorated according to the choice of anti-diabetic agent used [28].
Within a few years, we will know whether this class of drug fulfills its promise as plans for a phase 3 trial of exenatide are afoot, and there is growing interest in exploring other drugs in this class for their potential disease modifying properties (NCT02953665, NCT03439943, NCT03659682). Enhancing the potency of existing anti-diabetic medications is therefore an appealing proposition for a number of commercial entities. Neuraly have modified the original GLP-1 receptor agonist, exenatide to enhance its brain penetration and reduce its immunogenicity, while Peptron have developed a separate novel slow release exenatide formulation.
Other drugs which may enhance neuronal survival are also becoming major avenues of interest. Ursodeoxycholic acid (UDCA) is a licensed treatment for primary biliary cirrhosis and emerged as the most promising agent from a drug screen using fibroblasts from parkin and LRRK2 patients [29, 30]. Similar to exenatide, UDCA appears to have an action on the protein kinase Akt, with a consequent mitochondrial rescue effect. Whether simply recommending a specific programme of high intensity training might be an alternative way of engaging the same pathways is also of some interest [31]. Another agent, Nilotinib, a c-abl inhibitor used in chronic myelocytic leukemia also has actions on alpha synuclein toxicity and apoptotic pathways of relevance to PD [32, 33], and is the subject of formal safety/tolerability evaluation while trials of Inosine and Isradipine will soon report whether any signals of efficacy associated with their use may encourage their further development.
Attention has also been drawn to agonists of the beta adrenoceptor (Salbutamol, Clenbuterol) following the observation that 1) patients on these drugs appear to have a lower risk of developing PD, 2) patients on beta receptor antagonists appear to have a higher risk of PD, 3) alpha synuclein transcription can be modulated by beta agonists [34]. Whether these data fully account for the known influence of smoking on PD risk (smoking may lead to increased use of beta agonists) remains a subject of debate, indeed the epidemiological evidence has failed to be replicated in a recent study [35], however given the long term known safety data, if these drugs indicate any evidence of neuroprotection in early clinical trials, rapid progression to phase 3 could be easily envisaged.
CONFRONTING The PROBLEM “AT SOURCE”
Immunomodulation
Neuroinflammation has been on the radar as a factor involved in PD on and off for the last 30 years, and it is yet again gaining attention. Interestingly, there is evidence that alpha synuclein may turn out to be a central player in this immune response. Indeed T cells from patients with PD recognize alpha synuclein peptides [36], suggesting that an individual’s immune response to pathogenic forms of alpha synuclein could play a key role in disease progression. On a related note, alpha synuclein itself may even play a role in the innate immune response in the gut [37]. Either way, there is increasing interest in immunomodulatory therapies to prevent PD, and there are a host of existing immunosuppressive agents including, e.g., Azathioprine and Sargramostim [38] that are being considered as potential candidates for slowing disease progression.
The gut microbiome
At the same time, there is growing evidence of an infectious trigger for this immune-dysregulation. For example, alpha synuclein transgenic mice can be spared onset of pathology if bred in germ free environments or if exposed to broad spectrum antibiotics. Furthermore colonization of the gut from fecal bacteria from PD patients greatly exacerbates alpha synuclein pathology in the brains of these animals [39]. A link between metabolites produced by gut bacteria and brain inflammation has also been identified [40], so shouldn’t we already be actively trying to interfere with this gut/brain axis to prevent neurodegeneration? Giving encapsulated fecal transplants from healthy individuals to people with PD are currently being considered (this is already an accepted treatment for gastrointestinal illnesses like C-difficile). Extending the parallel interests between diabetes processes and neurodegeneration, it is interesting to observe that encapsulated fecal transplants have also be shown to improve peripheral insulin sensitivity [41] possibly offering another clue regarding the genesis of PD.
How this might ultimately translate to PD therapeutics needs to be considered. Useful responses must depend somewhat on whether the alpha synuclein pathology is already established to the extent that “the horse has already bolted” by this stage? However an alternative possibility is that ongoing gut pathology may continue to seed abnormal alpha synuclein to the brain via vagal transmission or short chain fatty acid signalling and contribute to ongoing neurodegeneration over the longer term. Given that the use of fecal transplants to treat diseases is already being explored in patients with motor neuron disease, it is therefore highly likely that there will be a range of attempts at manipulating the gut microbiome over the coming years in PD patients, to try and close the stable door, lest more alpha synuclein oligomeric horses will be racing to other areas of the central and peripheral autonomic nervous systems.
NANOTECHNOLOGY: SCIENCE FICTION OR FACT?
The grand vision would be to develop a therapy that would accurately target alpha synuclein pathology, dissolve the toxic aggregates and push the equilibrium back towards normal monomeric alpha synuclein. Would this sci-fi dream ultimately be the definitive cure for PD? With the aptly named, graphene quantum dots (GQDs), perhaps this dream is right in from of us. These nanoparticles have been shown to penetrate the blood brain barrier in rodents and protect dopaminergic neurons exposed to alpha synuclein preformed fibrils. In vitro data suggests these GQDs can in fact interact with alpha synuclein fibrils, blocking their formation and even promoting their disaggregation [42]. If safe and sufficiently selective, might these GQDs be the magic bullet for PD?
DISCUSSION
Research in PD has moved away from the traditional approach of testing potential neuroprotective agents in laboratory mouse models of non-progressive dopamine toxicity. With efficacy being demonstrated in models of progressive alpha synuclein associated neurodegeneration combined with positive data from early phase clinical trials, we can have greater optimism that laboratory results will translate to the clinic. It is of the highest importance therefore that we not only robustly confirm consistent signals of efficacy in phase 3 designs, but also that we make appropriate preparations for success.
Drugs being repurposed for their potential neuroprotective properties frequently lack the commercial support required to apply for licensing. The burden of responsibility to bring positive clinical trial data to the attention of the regulatory authorities must be shared among all parties with a vested interest, i.e., academics, charities, patient groups, and industry. A recent initiative called “Linked Clinical Trials”, set up by the Cure Parkinson’s Trust has involved representatives from each sector to evaluate and prioritize which agents are the most important to take from the laboratory to the clinical trial setting. Bearing in mind the eventual need to deliver successful agents to the clinical setting, there needs to be appropriate discussion of the magnitude of any beneficial effect size, whether this is has a static or cumulative nature, the long term safety and tolerability of the intervention, and its overall health economic impact.
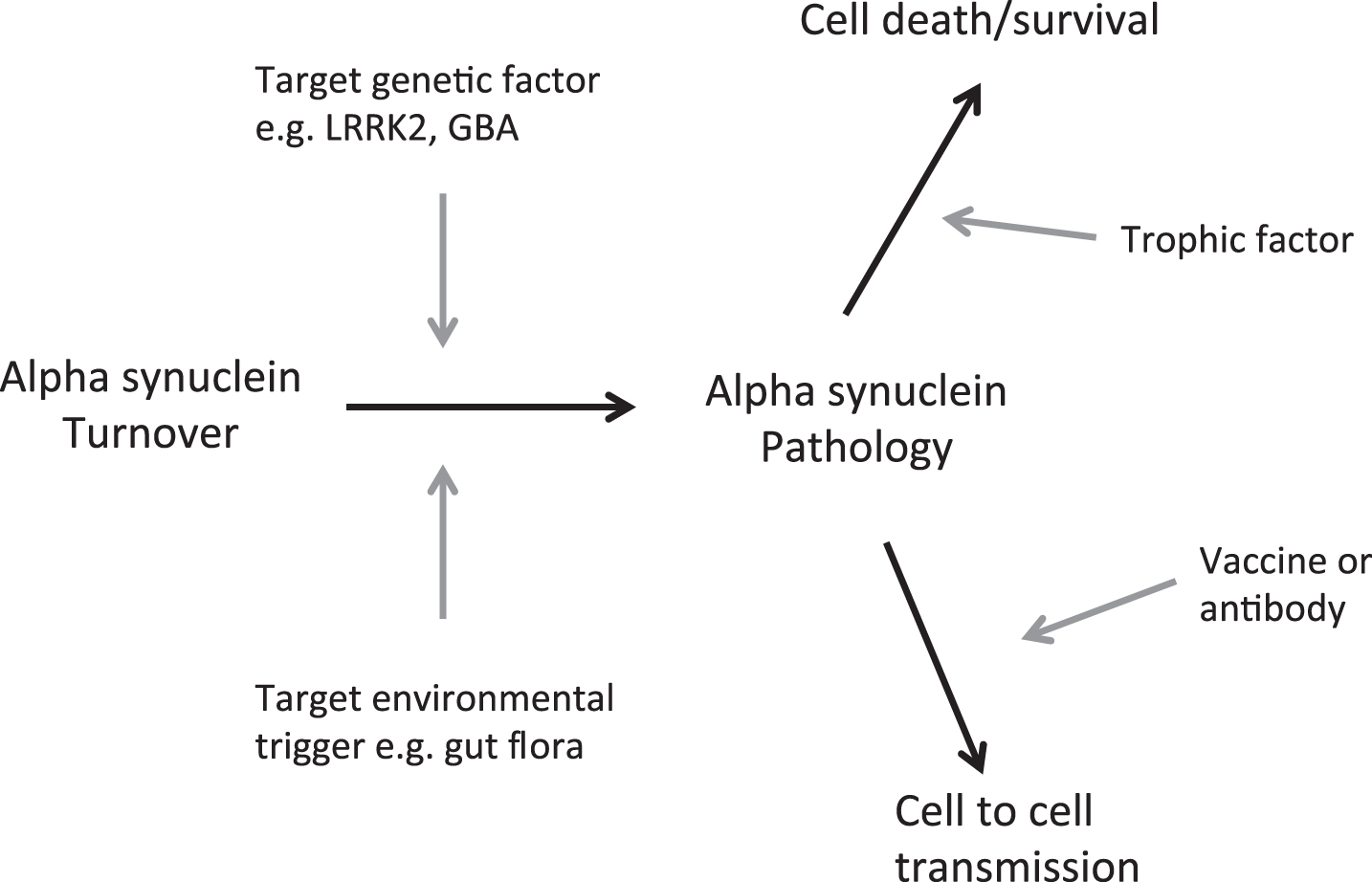
The divergent approaches being brought into clinical trials, if any signal of effect can be robustly confirmed, will likely also ultimately require testing in combination to explore additive/synergistic effects (Fig. 1). It is tempting to speculate that the future patient may be recruited into research reminiscent of the current state of play in HIV/cancer fields, e.g., where following genotyping/ microbiome testing, they are either given the curative enzyme corrective therapy or randomised to receive combination therapies rather than any/each of these alone.
Fig. 1.
Opportunities to impact on PD progression.
CONFLICTS OF INTEREST
The authors have no conflict of interest to report.
REFERENCES
[1] | Thibault L , Rascol O , Corvol JC , Ferreira JJ , Defebvre L , Deplanque D , Bordet R , Moreau C , Devos D ((2017) ) New perspectives on study designs for evaluating neuroprotection in Parkinson’s disease. Mov Disord 32: , 1365–1370. |
[2] | Dachsel JC , Behrouz B , Yue M , Beevers JE , Melrose HL , Farrer MJ ((2010) ) A comparative study of Lrrk2 function in primary neuronal cultures. Parkinsonism Relat Disord 16: , 650–655. |
[3] | Fuji RN , Flagella M , Baca M , S Baptista MA , Brodbeck J , Chan BK , Fiske BK , Honigberg L , Jubb AM , Katavolos P , Lee DW , Lewin-Koh S-C , Lin T , Liu X , Liu S , Lyssikatos JP , O'Mahony J , Reichelt M , Roose-Girma M , Sheng Z , Sherer T , Smith A , Solon M , Sweeney ZK , Tarrant J , Urkowitz A , Warming S , Yaylaoglu M , Zhang S , Zhu H , Estrada AA , Watts RJ ((2015) ) Effect of selective LRRK2 kinase inhibition on nonhuman primate lung. Sci Transl Med 7: , 273ra15. |
[4] | Mercuri E , Darras BT , Chiriboga CA , Day JW , Campbell C , Connolly AM , Iannaccone ST , Kirschner J , Kuntz NL , Saito K , Shieh PB , Tulinius M , Mazzone ES , Montes J , Bishop KM , Yang Q , Foster R , Gheuens S , Bennett CF , Farwell W , Schneider E , De Vivo DC , Finkel RS ((2018) ) Nusinersen versus sham control in later-onset spinal muscular atrophy. N Engl J Med 378: , 625–635. |
[5] | Lewis O , Woolley M , Johnson D , Rosser A , Barua NU , Bienemann AS , Gill SS , Evans S ((2016) ) Chronic, intermittent convection-enhanced delivery devices. J Neurosci Methods 259: , 47–56. |
[6] | Simon-Sanchez J , Schulte C , Bras JM , Sharma M , Gibbs JR , Berg D , Paisan-Ruiz C , Lichtner P , Scholz SW , Hernandez DG , Kruger R , Federoff M , Klein C , Goate A , Perlmutter J , Bonin M , Nalls MA , Illig T , Gieger C , Houlden H , Steffens M , Okun MS , Racette BA , Cookson MR , Foote KD , Fernandez HH , Traynor BJ , Schreiber S , Arepalli S , Zonozi R , Gwinn K , Van der Brug M , Lopez G , Chanock SJ , Schatzkin A , Park Y , Hollenbeck A , Gao J , Huang X , Wood NW , Lorenz D , Deuschl G , Chen H , Riess O , Hardy JA , Singleton AB , Gasser T ((2009) ) Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat Genet 41: , 1308–1312. |
[7] | Trabzuni D , Ryten M , Emmett W , Ramasamy A , Lackner KJ , Zeller T , Walker R , Smith C , Lewis PA , Mamais A , De Silva R , Vandrovcova J , Hernandez D , Nalls MA , Sharma M , Garnier S , Lesage S , Simon-Sanchez J , Gasser T , Heutink P , Brice A , Singleton A , Cai H , Schadt E , Wood NW , Bandopadhyay R , Weale ME , Hardy J , Plagnol V ((2013) ) Fine-mapping, gene expression and splicing analysis of the disease associated LRRK2 locus. PLoS One 8: , e70724. |
[8] | Hartlova A , Herbst S , Peltier J , Rodgers A , Bilkei-Gorzo O , Fearns A , Dill BD , Lee H , Flynn R , Cowley SA , Davies P , Lewis PA , Ganley IG , Martinez J , Alessi DR , Reith AD , Trost M , Gutierrez MG ((2018) ) LRRK2 is a negative regulator of Mycobacterium tuberculosis phagosome maturation in macrophages. EMBO J 37: , e98694. |
[9] | Fava VM , Manry J , Cobat A , Orlova M , van Thuc N , Ba NN , Thai VH , Abel L , Alcaïs A , Schurr E , Brown E , Gibbings D , Hayley S , Park D , Philpott DC , Rioux JD , Schlossmacher M ((2016) ) A missense LRRK2 variant is a risk factor for excessive inflammatory responses in leprosy. PLoS Negl Trop Dis 10: , e000441. |
[10] | Hui KY , Fernandez-Hernandez H , Hu J , Schaffner A , Pankratz N , Hsu NY , Chuang LS , Carmi S , Villaverde N , Li X , Rivas M , Levine AP , Bao X , Labrias PR , Haritu-nians T , Ruane D , Gettler K , Chen E , Li D , Schiff ER , Pontikos N , Barzilai N , Brant SR , Bressman S , Cheifetz AS , Clark LN , Daly MJ , Desnick RJ , Duerr RH , Katz S , Lencz T , Myers RH , Ostrer H , Ozelius L , Payami H , Peter Y , Rioux JD , Segal AW , Scott WK , Silverberg MS , Vance JM , Ubarretxena-Belandia I , Foroud T , Atz-mon G , Pe'er I , Ioannou Y , McGovern DPB , Yue Z , Schadt EE , Cho JH , Peter I ((2018) ) Functional variants in the LRRK2 gene confer shared effects on risk for Crohn's disease and Parkinson’s disease. Sci Transl Med 10: , 423. |
[11] | Gegg ME , Schapira AHV ((2018) ) The role of glucocere-brosidase in Parkinson disease pathogenesis. FEBS J 285: , 3591–360. |
[12] | Mazzulli JR , Xu YH , Sun Y , Knight AL , McLean PJ , Caldwell GA , Sidransky E , Grabowski GA , Krainc D ((2011) ) Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 146: , 37–52. |
[13] | Gegg ME , Burke D , Heales SJR , Cooper JM , Hardy J , Wood NW , Schapira AHV ((2012) ) Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann Neurol 72: , 455–463. |
[14] | Magalhaes J , Gegg ME , Migdalska-Richards A , Schapira AH ((2018) ) Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci Rep 8: , 1385. |
[15] | McNeill A , Magalhaes J , Shen C , Chau KY , Hughes D , Mehta A , Foltynie T , Cooper JM , Abramov AY , Gegg M , Schapira AHV ((2014) ) Ambroxol improves lysosomal biochemistry in glucocerebrosidase mutation-linked Parkinson disease cells. Brain 137: , 1481–1495. |
[16] | Maegawa GHB , Tropak MB , Buttner JD , Rigat BA , Fuller M , Pandit D , Tang L , Kornhaber GJ , Hamuro Y , Clarke JTR , Mahuran DJ ((2009) ) Identification and characterization of ambroxol as an enzyme enhancement agent for Gaucher disease. J Biol Chem 284: , 23502–23516. |
[17] | Papadopoulos VE , Nikolopoulou G , Antoniadou I , Karachaliou A , Arianoglou G , Emmanouilidou E , Sardi SP , Stefanis L , Vekrellis K ((2018) ) Modulation of ß-glucocerebrosidase increases α-synuclein secretion and exosome release in mouse models of Parkinson’s disease. HumMol Genet 27: , 1696–1710. |
[18] | Richter F , Fleming SM , Watson M , Lemesre V , Pellegrino L , Ranes B , Zhu C , Mortazavi F , Mulligan CK , Sioshansi PC , Hean S , Rosa KD La , Khanna R , Flanagan J , Lockhart DJ , Wustman BA , Clark SW , Chesselet M ((2014) ) A GCase chaperone improves motor function in a mouse model of synucleinopathy. Neurotherapeutics 11: , 840–56. |
[19] | Kordower JH , Olanow CW , Dodiya HB , Chu Y , Beach TG , Adler CH , Halliday GM , Bartus RT ((2013) ) Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136: , 2419–2431. |
[20] | Logovinsky V , Satlin A , Lai R , Swanson C , Kaplow J , Oss-wald G , Basun H , Lannfelt L ((2016) ) Safety and tolerability of BAN2401 - A clinical study in Alzheimer’s disease with a protofibril selective Aß antibody. Alzheimers Res Ther 8: ,14. |
[21] | Jankovic J , Goodman I , Safirstein B , Schenk D , Kinney G , Koller M , Ness D , Griffith S , Grundman M , Soto J , Ostrow-itzki S , Boess F , Martin-Facklam M , Quinn J , Isaacson S , Jennings D , Omidvar O , Ellenbogen A ((2017) ) Results from a phase 1b multiple ascending-dose study of prx002, an anti-alpha-synuclein monoclonal antibody, in patients with Parkinson’s disease. Mov Disord 32: , 964. |
[22] | Kutzelnigg A , Volc D , Hendrix S , Thun-Hohenstein C , Zimmermann J , Winter D , Schmidhuber S , Galabova G , Staffler G , Schneeberger A , Bürger V ((2017) ) Safe boosting of an existing affitope® PD01A vaccine induced immune response in patients with early stage parkinson's disease. Neurodegener Dis 17: , 1487. |
[23] | Williams-Gray CH , Barker RA ((2017) ) Parkinson disease: Defining PD subtypes - A step toward personalized management? Nat Rev Neurol 13: , 454–455. |
[24] | Athauda D , Maclagan K , Skene SS , Bajwa-Joseph M , Letchford D , Chowdhury K , Hibbert S , Budnik N , Zampedri L , Dickson J , Li Y , Aviles-Olmos I , Warner TT , Limousin P , Lees AJ , Greig NH , Tebbs S , Foltynie T ((2017) ) Exe-natide once weekly versus placebo in Parkinson’s disease: A randomised, double-blind, placebo-controlled trial. Lancet 390: , 1664–1675. |
[25] | Aviles-Olmos I , Dickson J , Kefalopoulou Z , Djamshidian A , Ell P , Soderlund P , Whitton P , Wyse R , Isaacs T , Lees A , Limousin P , Foltynie T ((2013) ) Exenatide and the treatment of patients with Parkinson’s disease. J Clin Invest 123: ,2370–2736. |
[26] | Athauda D , Foltynie T ((2016) ) Insulin resistance and Parkinson’s disease: A new target for disease modification? Prog Neurobiol 145-146: , 98–120. |
[27] | Yun SP , Tam T-I , Panicker N , KIm S , Oh Y , Jong-Sung P , Kwon S-H , Park YJ , Karuppagounder SS , Park H , Kim S , Oh N , Kim N , Lee S , Brahmachari S , Mao X , Lee JH , Kumar M , An D , Kang S-U , Lee Y , Lee KC , Na DH , Kim D , Lee SH , Roschke VV , Liddelow S , Mari Z , Barres B , Dawson VL , Lee S , Dawson TM , Ko HS ((2018) ) Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med 24: , 931–938. |
[28] | Svenningsson P , Wirdefeldt K , Yin L , Fang F , Markaki I , Efendic S , Ludvigsson JF ((2016) ) Reduced incidence of Parkinson’s disease after dipeptidyl peptidase-4 inhibitors—A nationwide case-control study. Mov Disord 31: , 1422–1423. |
[29] | Mortiboys H , Aasly J , Bandmann O ((2013) ) Ursocholanic acid rescues mitochondrial function in common forms of familial Parkinson’s disease. Brain 136: , 3038–3050. |
[30] | Mortiboys H , Furmston R , Bronstad G , Aasly J , Elliott C , Bandmann O ((2015) ) UDCA exerts beneficial effect on mitochondrial dysfunction in LRRK2 G2019S carriers and in vivo. Neurology 85: , 846–852. |
[31] | Schenkman M , Moore CG , Kohrt WM , Hall DA , Delitto A , Comella CL , Josbeno DA , Christiansen CL , Berman BD , Kluger BM , Melanson EL , Jain S , Robichaud JA , Poon C , Corcos DM ((2018) ) Effect of high-intensity treadmill exercise on motor symptoms in patients with de novo Parkinson disease a phase 2 randomized clinical trial. JAMA Neurol 75: , 219–226. |
[32] | Karuppagounder SS , Brahmachari S , Lee Y , Dawson VL , Dawson TM , Ko HS ((2014) ) The c-Abl inhibitor, nilotinib, protects dopaminergic neurons in a preclinical animal model of Parkinson’s disease. Sci Rep 4: , 4874. |
[33] | Hebron ML , Lonskaya I , Moussa CEH ((2013) ) Nilotinib reverses loss of dopamine neurons and improvesmotor-behavior via autophagic degradation of a-synuclein in parkinson's disease models. HumMol Genet 22: ,3315–3328. |
[34] | Mittal S , Bjørnevik K , Im DS , Flierl A , Dong X , Locascio JJ , Abo KM , Long E , Jin M , Xu B , Xiang YK , Rochet J-C , Engeland A , Rizzu P , Heutink P , Bartels T , Selkoe DJ , Caldarone BJ , Glicksman MA , Khurana V , Schiile B , Park DS , Riise T , Scherzer CR ((2017) ) Beta2-Adrenoreceptor is a regulator of the alpha-synuclein gene driving risk of Parkinson’s disease. Science 357: , 891–898. |
[35] | Searles Nielsen S , Gross A , Camacho-Soto A , Willis A , Racette B ((2018) ) ß2-adrenoreceptor medications and risk of Parkinson disease. Ann Neurol, doi:10.1002/ana.25341.0 |
[36] | Sulzer D , Alcalay RN , Garretti F , Cote L , Kanter E , Agin-Liebes J , Liong C , McMurtrey C , Hildebrand WH , Mao X , Dawson VL , Dawson TM , Oseroff C , Pham J , Sidney J , Dillon MB , Carpenter C , Weiskopf D , Phillips E , Mallal S , Peters B , Frazier A , Lindestam Arlehamn CS , Sette A ((2017) ) T cells from patients with Parkinson’s disease recognize α-synuclein peptides. Nature 546: , 656–661. |
[37] | Labrie V , Brundin P ((2017) ) Alpha-synuclein to the rescue: Immune cell recruitment by alpha-synuclein during gastrointestinal infection. J Innate Immun 9: , 437–440. |
[38] | Gendelman HE , Zhang Y , Santamaria P , Olson KE , Schutt CR , Bhatti D , Shetty BLD , Lu Y , Estes KA , Standaert DG , Heinrichs-Graham E , Larson L , Meza JL , Follett M , Forsberg E , Siuzdak G , Wilson TW , Peterson C , Mosley RL ((2017) ) Evaluation of the safety and immunomodulatory effects of sargramostiminarandomized, double-blind phase 1 clinical Parkinson’s disease trial. NPJ Parkinsons Dis 3: , 10. |
[39] | Sampson TR , Debelius JW , Thron T , Janssen S , Shastri GG , Ilhan ZE , Challis C , Schretter CE , Rocha S , Gradinaru V , Chesselet MF , Keshavarzian A , Shannon KM , Krajmalnik-Brown R , Wittung-Stafshede P , Knight R , Mazmanian SK ((2016) ) Gut microbiota regulate motor deficits and neuroinflammation in a model of Parkinson’s disease. Cell 167: , 1469–1480.e12. |
[40] | Wekerle H ((2018) ) Brain inflammatory cascade controlled by gut-derived molecules. Nature 557: , 642–643. |
[41] | Kootte RS , Levin E , Salojarvi J , Smits LP , Hartstra A V. , Udayappan SD , Hermes G , Bouter KE , Koopen AM , Holst JJ , Knop FK , Blaak EE , Zhao J , Smidt H , Harms AC , Hanke-meijer T , Bergman JJGHM , Romijn HA , Schaap FG , Olde Damink SWM , Ackermans MT , Dallinga-Thie GM , Zoe-tendal E , De Vos WM , Serlie MJ , Stroes ESG , Groen AK , Nieuwdorp M ((2017) ) Improvement of insulin sensitivity after lean donor feces in metabolic syndrome is driven by baseline intestinal microbiota composition. Cell Metab 26: , 611–619.e6. |
[42] | Kim D , Yoo JM , Hwang H , Lee J , Lee SH , Yun SP , Park MJ , Lee MJ , Choi S , Kwon SH , Lee S , Kwon SH , Kim S , Park YJ , Kinoshita M , Lee YH , Shin S , Paik SR , Lee SJ , Lee S , Hong BH , Ko HS ((2018) ) Graphene quantum dots prevent α-synucleinopathy in Parkinson’s disease. Nat Nanotech-nol, doi:10.1038/s41565-018-0179-y |


