
A Proposed Roadmap for Parkinson’s Disease Proof of Concept Clinical Trials Investigating Compounds Targeting Alpha-Synuclein
Abstract
The convergence of human molecular genetics and Lewy pathology of Parkinson’s disease (PD) have led to a robust, clinical-stage pipeline of alpha-synuclein (α-syn)-targeted therapies that have the potential to slow or stop the progression of PD and other synucleinopathies. To facilitate the development of these and earlier stage investigational molecules, the Michael J. Fox Foundation for Parkinson’s Research convened a group of leaders in the field of PD research from academia and industry, the Alpha-Synuclein Clinical Path Working Group. This group set out to develop recommendations on preclinical and clinical research that can de-risk the development of α-syn targeting therapies. This consensus white paper provides a translational framework, from the selection of animal models and associated end-points to decision-driving biomarkers as well as considerations for the design of clinical proof-of-concept studies. It also identifies current gaps in our biomarker toolkit and the status of the discovery and validation of α-syn-associated biomarkers that could help fill these gaps. Further, it highlights the importance of the emerging digital technology to supplement the capture and monitoring of clinical outcomes. Although the development of disease-modifying therapies targeting α-syn face profound challenges, we remain optimistic that meaningful strides will be made soon toward the identification and approval of disease-modifying therapeutics targeting α-syn.
INTRODUCTION AND OVERVIEW
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, affecting approximately 1.2% of the world population over the age of 70. In the United States, an estimated 630,000 people had PD in 2010. With anticipated demographic changes due to an aging population, and if no disease-modifying treatment is found, the prevalence is expected to reach 930,000 by 2020 and 1.24 million by 2030 [1–3]. Recent years have seen the approval of several new therapies that address motor or non-motor symptoms of PD, however no therapeutic is available yet to slow disease progression.
In 2017, The Michael J. Fox Foundation (MJFF) convened a group of leaders in the field of PD research from academia and industry, the Alpha-Synuclein Clinical Path Working Group. This working group was tasked to develop a strategic consensus and put forth recommendations on preclinical and clinical research directed at the development of alpha-synuclein (α-syn) targeted, potentially disease-modifying therapies for PD. α-syn has become one of the most attractive targets for new therapeutic agents to slow or arrest the progression of PD and other synucleinopathies, including Dementia with Lewy Bodies (DLB), Multiple System Atrophy (MSA), and Pure Autonomic Failure. A detailed discussion on α-syn as a target for PD is beyond the scope of this consensus paper. However, the interest in targeting α-syn stems from the following three converging lines of evidence that implicate a pathogenic role of α-syn in PD: 1) mutations in the gene encoding α-syn, SNCA, cause autosomal dominant PD, suggesting that a gain of toxic function may underlie the pathogenesis in familial PD cases (reviewed in [4]); 2) several polymorphic variants at the SNCA locus affect the expression of α-syn and are risk factors of sporadic PD, which constitutes over 90% of PD incidence [5–7]; 3) α-syn is the major protein component of Lewy bodies and Lewy neurites, the intracellular inclusions that are the pathological hallmarks of PD [8, 9]. Based on these observations, proposed therapeutic strategies primarily aim to reduce the pathogenic burden of α-syn by decreasing its production, reducing its aggregation, augmenting its clearance, or neutralizing its toxic effects via sequestration (Table 1).
Table 1
Proposed therapeutic strategies targeting alpha-synuclein
| Therapeutic Objective and Mode of Action | Therapeutic Examples | Most Advanced Phase of Drug Development Known |
| Reduce α-syn levels via inhibition of transcription or translation of the SNCA gene | ASOs, siRNA | Preclinical |
| Reduce/prevent formation of α-syn aggregates* | NPT200-11a | Phase I completed |
| Enhance lysosomal or proteosomal enzyme activity to promote clearance of intracellular α-syn | GZ/SAR402671b | Phase II underway |
| Neutralize/clear extracellular α-syn* and/or inhibit cell-to-cell transmission of α-syn | RO7046015 (PRX002)c, BIIB054d | Phase II underway |
ASOs, antisense oligonucleotides; siRNA, small interfering ribonucleic acid. *Presumed toxic species of α-syn. aClinicalTrials.gov Identifier: NCT026066. bClinicalTrials.gov Identifier: NCT02906020. cClinicalTrials.gov Identifier: NCT03100149. dClinicalTrials.gov Identifier: NCT03318523.
In this regard, α-syn-targeting strategies are heavily informed by the amyloid hypothesis of Alzheimer’s disease (AD) that posits, in an analogous manner to the α-syn hypothesis of PD, a toxic gain of function of beta-amyloid (Aβ), an extracellular proteinaceous deposit. In recent years, results of multiple clinical studies testing the amyloid hypothesis have emerged, which provide critical insights into designing clinical trials of α-syn targeted therapies. Although some recent early-phase data on Aβ-targeted therapies for AD are encouraging [10], most have not met their primary efficacy end-points or demonstrated clinically meaningful effects in registration trials [11]. Analyses of the Aβ-targeted AD trials that have failed in the past suggest a number of factors contributing to their failures: 1) insufficient evidence of target engagement [12, 13]; 2) inclusion of patients who did not have Aβ pathology; 3) enrollment of subjects at a stage of the disease that may have been too advanced to permit attaining a meaningful clinical benefit; 4) lack of sensitivity of clinical endpoints used; and 5) selective targeting of a single pathogenic mechanism. Finally, an additional option is that Aβ simply is not a valid therapeutic target in sporadic AD [14], raising the mirror-image possibility that α-syn may not be a suitable therapeutic target for non-SNCA PD (Espay et al., 2018, submitted).
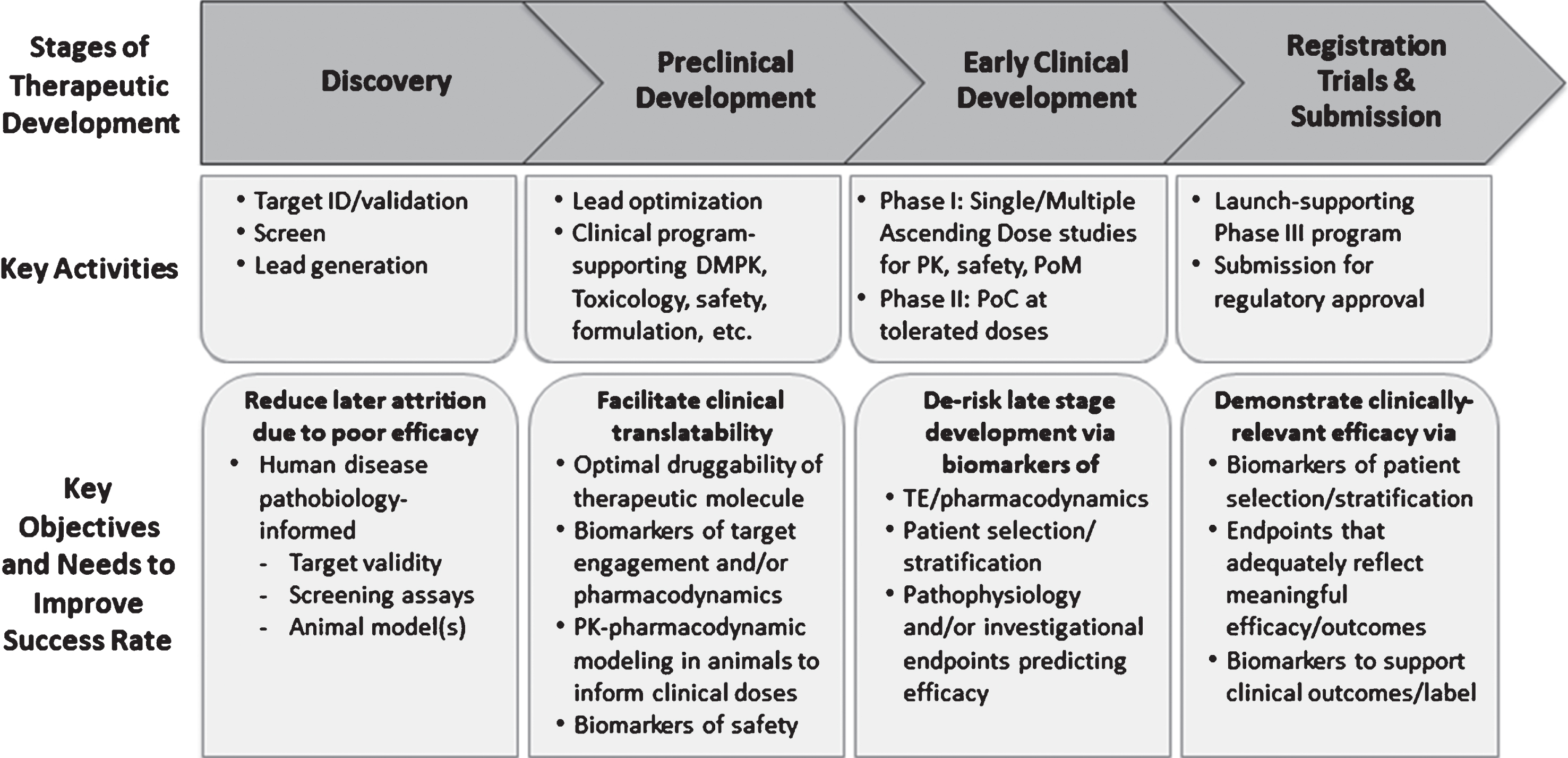
Could past failures in AD have been prevented or the efforts terminated at an earlier stage to re-deploy resources toward alternative targets through improvement in translational research, biomarker use and clinical trial design? Indeed, emerging data from recent early phase clinical trials of AD indicate that a combination of patient enrichment strategy with biomarkers of Aβ pathology could provide a strong data package upon which a decision to initiate registration trials could be founded with greater confidence [10, 15, 16]. In this paper, we will apply lessons from the AD field as well as prior therapeutic trials in PD and other central nervous system (CNS) disorders, to propose a framework for translational research and early stage clinical development of α-syn-targeted therapies. We posit that in addition to ensuring the validity of the selected therapeutic targets to disease biology and drug-like attributes of an investigational therapy, the following five types of biomarker-based evidence (adapted from http://www.ncbi.nlm.nih.gov/books/NBK326791/) generated in both translational animal models and early stage clinical studies could improve success rates (Fig. 1):
Fig. 1.
Key Stages of Therapeutic Discovery and Development. Key stages of therapeutic discovery and development and associated objectives aimed at improving the probability of technical success. Note the specific utility of biomarkers to inform preclinical and clinical decisions during each stage. DMPK, drug metabolism and pharmacokinetics; ID, identification; PK, pharmacokinetics; PoC, proof of concept; PoM, proof of mechanism; TE, target engagement.
1. Target Engagement (TE): a biomarker that indicates that the intervention gets to the site of action and engages its intended target,
2. Pharmacodynamic/Response: a biomarker used to show that a biological response has occurred in an individual who has received an intervention or exposure,
3. Disease Monitoring: a biomarker measured serially and used to detect a change in the degree or extent of disease. For the purpose of this paper, we propose that disease-monitoring biomarkers may be used to generate the so-called Proof of Principle (PoP) or Proof of Concept (PoC) milestones. PoP is achieved when a biomarker is used to indicate that an intervention modifies the known pathology/physiology of PD (e.g., α-synuclein aggregates) whereas PoC is achieved when an intervention produces a clinical response that may be predictive of efficacy in patients. A go/no-go decision on the basis of a PoP endpoint assumes that the PoP biomarker is predictive of clinical efficacy; a Disease Monitoring biomarker may or may not be the same as one that demonstrates the pharmacodynamic effects of a given drug candidate,
4. Safety: a biomarker used to indicate the presence or extent of toxicity related to an intervention or exposure (the toxicity may be related to either on- or off-target pharmacology of the intervention),
5. Patient Selection/Stratification: one or more biomarkers that can be used in early or late stage clinical trials to select patients who both possess the desired target and are at a high likelihood to respond to the drug candidate within the timeframe of the planned study [17]. These biomarkers could be accepted as Predictive Biomarkers with evidence that the biomarker identifies individuals who are more likely than similar patients without the biomarker to experience a favorable or unfavorable effect from a specific intervention or exposure.
Before trust can be placed in biomarkers for decision-making, however, several factors must be taken into account. These include 1) analytical validation of the biomarker assays, 2) understanding predictive value with respect to the intended utility of the biomarker, and 3) rate of change in the biomarker as associated with pathophysiology or natural history of the disease. Importantly, for pharmacodynamic and disease modification biomarkers, it is imperative to assess and establish that a drug candidate can modify a specific biomarker in careful, dose-ranging animal studies. In the following sections of this white paper, we provide a framework for selecting an animal model to generate clinically translatable data, specific biomarkers for α-syn-targeted therapies and clinical PoC factors, and planning that integrates not only pathophysiologic endpoints but also the emerging technology of wearable devices to monitor clinical outcomes.
TRANSLATIONAL RESEARCH
The current pipeline of α-syn-targeted therapies (Table 1) is comprised of both small molecules and biologics such as nucleic acid analogs, proteins, and gene therapy vectors. Independent of the therapeutic modality, a key translational research element in the early discovery and lead optimization stages (Fig. 1) is the use of animal models that recapitulate relevant aspects of disease pathophysiology. Translational research also aims to deliver clinically translatable biomarkers of TE, pharmacodynamics, and PoP which also inform initial clinical study design, including dose and dosing regimens. Hence, in the animal models and subsequent human studies, it is critical to utilize similar, if not identical, biomarkers and/or endpoint methodologies. The following sections provide an overview of animal models and their relevance to the development of α-syn-targeted therapies.
Animal model studies
Several rodent and non-human primate models of α-syn aggregation exist. It is beyond the scope of this review to describe all existing animal models of α-syn pathology, and readers are referred to recent reviews on the topic [18–24]. In this section, we focus on conceptual challenges as well as the utility of current animal models in α-syn-targeted drug discovery.
Conceptual challenges with current models
A major challenge in the field is the lack of consensus on the specific molecular species of α-syn (e.g., oligomer or fibril and specific strains associated with either, or post-translationally modified α-syn species, etc.) responsible for PD pathogenesis, which engenders uncertainty around the construct validity of the models. In addition, definitive evidence of a particular α-syn species causing a specific synucleinopathy (e.g., PD versus MSA) in humans is lacking, even though animal models are beginning to shed light on strain-specific synucleinopathy [25–28]. In section 3 we detail ongoing studies to develop and test α-syn-targeted biomarkers on human biospecimens to garner insights into α-syn species pathogenicity. These insights will undoubtedly refine the generation and characterization of future animal models.
Currently, no animal model faithfully reproduces all the key clinical features of PD. The predictive validity of an animal model can only be established once an α-syn-targeted therapy is found to be effective in humans. Some have argued that because of these factors, testing in animal models is of limited utility in PD drug development. However, as discussed below, animal models of synucleinopathy are critical to establish in vivo proof of mechanism and discovery of translational biomarkers of TE, pharmacodynamics, and PoP for novel α-syn therapies under development.
Commonly used rodent models
Three major types of mammalian models of α-syn aggregation have been utilized for discovery and development of α-syn-targeted therapies: 1) transgenic rodents expressing wild type or mutant α-syn; 2) viral vector-mediated expression of wild type or mutant α-syn; and 3) injection of preformed fibrils (PFFs) of α-syn, Table 2 and the description below briefly highlight the key characteristics of each type of model and their translational utility. Animal models may recapitulate specific features of PD pathology or downstream pathophysiology which can be utilized to assess investigational therapies. In general, investigational therapeutic agents should be tested in multiple animal models to assess efficacy. On the other hand, it is desirable to utilize a single model to establish TE, pharmacodynamic, and PoP endpoints, which can be used to model clinical dosing and define safety margins.
Table 2
Alpha-synucleinopathy-directed therapeutic approaches evaluated in animal models
| Type of Model | Key Characteristics | Exemplary Endpoints to Inform Clinical Translation |
| Transgenic rodents expressing wild-type or mutant human α-syn under the control of different promoters | •Each transgenic strain displays a specific regional pattern and level of α-syn overexpression •None reproduce the development the full repertoire of neuropathology •α-syn expression in these models is frequently much greater than that seen in the human disease | •TE: Therapeutic antibody bound to its targeted α-syn species in the brain and/or accessible biofluids such as CSF and blood compartments •PoP: Reduction in the load of α-syn aggregates |
| Viral vector-mediated over-expression of WT or mutant α-syn in select brain regions | •Labor intensive with relatively high inter-animal variability •Possible to achieve marked α-syn overexpression in nigral dopaminergic neurons •Can lead to impaired striatal DA release, axonal α-syn pathology in the striatum, death of nigral dopaminergic neurons with neuroinflammation and varying degrees of motor deficits | •TE: Therapeutic antibody bound to its targeted α-syn aggregates or species •PoP: Reduction in the load of α-syn aggregates, dopaminergic axonal degeneration measured by DAT density, neostriatal dopamine release, nigral dopaminergic neuroprotection, neuroinflammation •Functional outcomes: Basal ganglia-mediated behavioral deficits (e.g., hypokinesia) |
| Injection of α-syn PFFs injected into rodent brain or administered systemically (IV, IM) | •“Prion-like” propagation of the pathology which follows neural pathways after intracerebral injections | •TE: Therapeutic antibody bound to its targeted α-syn aggregates or species |
| •Following intrastriatal injection: progressive loss of nigral dopaminergic neurons and development of motor deficits | •Pharmacodynamics: α-syn mRNA or protein levels induced by transcription or translation inhibitors | |
| •PFFs injected into the olfactory bulb triggers formation of α-syn aggregates which cause a gradual spread of pathology in multiple brain regions (no reported dopaminergic neuron loss) | •PoP: Reduction in the propagation of α-syn pathology, dopaminergic neuroprotection •Functional efficacy: Behavioral deficits (e.g., hypokinesia, hyposmia) | |
| •Systemic injections of PFFs are also reported to trigger synucleinopathy in the nervous system |
AAV, adeno-associated virus; CSF, cerebrospinal fluid; DA, dopamine; DAT, dopamine transporter; IM, intramuscular; IV, intravenous; mRNA, messenger ribonucleic acid; PFF, preformed fibrils; PoP, proof of principle.
Transgenic rodent models. Several transgenic mouse lines have been created, expressing wild-type or mutant α-syn under the control of a variety of promoters. It is beyond the scope of this article to describe these models in detail, and their different features have been covered extensively by earlier reviews [23, 29–33]. In short, none of the transgenic α-syn mouse models can be viewed as the “perfect” model for development of therapies targeting α-syn. Different transgenic α-syn mouse models mimic various features of synucleinopathies and have specific advantages depending on the scientific questions that are asked. Here we only briefly mention some of the overriding principles that influence pros and cons with existing transgenic α-syn models. The various transgenic mouse lines display different regional patterns and levels of α-syn expression in the brain or periphery. Most of these models display intraneuronal or intraglial (MSA-like) α-syn accumulation which offer the opportunity to establish TE or PoP for the specific forms of α-syn aggregates being targeted. A disadvantage of most transgenic mouse models is that they rarely exhibit significant neurodegeneration in the substantia nigra. As a consequence, PD-like motor dysfunction is typically not a prominent feature. This precludes the ability to correlate TE or PoP to motor dysfunction that is directly relevant to PD. However, non-motor symptoms of PD such as cognitive or olfactory deficits or gastrointestinal symptoms may be taken advantage of, when present in specific models. Furthermore, the level of α-syn expression in transgenic mouse models is frequently much greater than that seen in the human disease. This caveat needs to be considered in the generation of physiological pharmacokinetic-pharmacodynamic modeling or predictive dose-response modeling.
Viral vector-mediated α-syn overexpression Adeno-associated viruses (AAV) are currently the vector of choice to transduce α-syn in the rodent or non-human primate nigrostriatal system. Although AAV models are relatively more labor-intensive and variable compared to transgenic rodent models, the marked α-syn overexpression in nigral dopaminergic neurons following localized AAV delivery can lead to impaired striatal dopamine (DA) release, axonal α-syn pathology in the nigrostriatal pathway, death of nigral dopaminergic neurons with neuroinflammation, and varying degrees of motor deficits [34–38]. Thus, tests of motor function can be used to assess effects of therapies in these models. MSA-like pathology has also been produced in viral transduction models by using specific promoters to drive expression in oligodendrocytes [39, 40]. As with transgenic α-syn models, concerns remain around the relevance of studying α-syn aggregation or downstream sequelae such as neurodegeneration, motor deficits, or neuroinflammation driven by exceptionally high α-syn levels.
Injections of α-syn preformed fibrils or pathological extracts. Over the past six years, several rodent and non-human primate models based on injection of recombinant α-syn PFFs or extracts of pathogenic human brain-derived α-syn into the brain of rodents have been developed [21, 22]. Typically, PFF injections trigger progressive development of intraneuronal α-syn aggregates with associated neurological deficits, the distribution and behavioral correlates of which depend on the brain regions and the type of α-syn injected [21, 22, 41–43]. The mechanism underlying the PFF models is thought to involve “prion-like”, trans-cellular propagation of the pathology, dependent on endogenous α-syn. Thus, the development of pathology is more rapid when injections are made into transgenic α-syn mice, or animals with viral vector-mediated overexpression of α-syn, than wild-type mice and no pathology develops in α-syn null mice [44–46]. The pathology appears to propagate along connected neuronal pathways in a manner that has been suggested to occur with PD progression [47]. Models where the PFFs are injected into the nigrostriatal system can also show progressive loss of nigral dopaminergic neurons, reduction in striatal DA, and development of motor deficits, allowing for motor testing of functional effects of therapies [44, 45, 48]. More recently, PFFs injected into the olfactory bulb have been shown to produce α-syn aggregation and neurodegeneration in olfactory structures and a gradual development of specific olfactory deficits akin to hyposmia in prodromal PD [49, 50]. Thus, PFF mouse models appear to be relevant when testing therapies intended to mitigate α-syn pathology in early disease by reducing α-syn expression or increasing clearance of extracellular α-syn. Other variations on the PFF models include injections through peripheral routes, (i.e., intravenous or intramuscular) [26, 51] or directly into the gastrointestinal tract [52–54]. Analogous to the PFF injections into the olfactory bulb, targeting the gastrointestinal tract with PFFs or other forms of α-syn has been suggested to be relevant as a model of prodromal PD. Recent reports, however, suggest that brainstem α-syn pathology triggered by PFF injections into the gut of wild type mice is transient and that there is little or no further propagation to other brain regions [53, 54].
What is the value of experiments in non-human primates?
Due to the relatively low cost and the ease at which genetic modifications can be made, rodent models of synucleinopathy are more abundant and frequently used than non-human primate models. Recently, non-human primate models of synucleinopathy that use AAV vectors to overexpress the protein and injections of PFFs have been generated [55–57]. None of these models have been characterized in great detail yet, and therefore, their place in a drug development program targeting α-syn remains to be determined. In the future, non-human primate models might provide the ability to discover and monitor biomarkers in the cerebrospinal fluid (CSF), which remains a major limitation of rodent models. Additionally, their larger blood volume compartment could allow for longitudinal sampling and assessment of biomarkers and pharmacokinetic parameters.
What are relevant outcome measures and biomarkers for clinical translation?
From a translational perspective, the major utility of the animal models of α-syn aggregation described above is to establish TE, pharmacodynamic responses, PoP, and functional efficacy measures (Table 2). By definition, a TE biomarker has to be specific to the therapeutic agent under investigation, making it hard to offer generalizable recommendations. For example, for therapeutic monoclonal antibodies, measurement of antibody-bound α-syn versus free α-syn levels in the brain, as well as in the CSF and peripheral matrices, can provide useful indices of TE. Furthermore, effects on other markers of α-syn pathology (e.g., pS129-modified α-syn or oligomeric α-syn) could be utilized as pharmacodynamic or PoP outcomes. Similarly, therapies that aim to reduce α-syn transcription via antisense mechanisms can rely on direct assessment of α-syn transcripts or protein levels for TE.
Pharmacodynamic biomarkers reflect a change induced by the binding or modulation (activation/inhibition) of the desired target by the therapeutic agent, and as such they provide an additional dimension of drug effect beyond TE. An example of a pharmacodynamic endpoint is the level of the lipid glucosylceramide, the substrate of glucocerebrosidase, for agents that allosterically activate or stabilize this lysosomal enzyme implicated in α-syn clearance.
Neither TE nor pharmacodynamic biomarkers on their own provide insights into the level of TE or pharmacodynamic responses required to produce clinically relevant efficacy. The integration of a PoP biomarker that measures a key pathogenic event in PD brains helps answer this question. For α-syn targeted therapies, immunohistochemistry for α-syn deposits is an important preclinical PoP endpoint, since Lewy pathology is a defining feature of PD. In animal models, deposits of aggregated α-syn are often assessed by immunohistochemistry using antibodies against pS129 α-syn, since pS129 α-syn also tags Lewy pathology in PD brains [58]. Thus, a reduction in pS129 α-syn staining could be viewed as positive preclinical PoP. Other markers for Lewy pathology (e.g., ubiquitination, p62, measurement or staining of proteinase K-resistant α-syn) and biochemical measures of soluble versus insoluble α-syn aggregates have also been employed to verify or extend PoP findings. The status of development of biomarkers to assess Lewy pathology in humans is detailed in sections 3. Animal models that exhibit α-syn aggregation-associated nigral degeneration offer the opportunity to monitor the effect of the therapeutic agent on the second hallmark pathology of PD via stereological cell counts of tyrosine hydroxylase (TH)-immunoreactive neurons.
The dopamine (DA) transporter (DAT), a translational endpoint reflecting integrity of dopaminergic nerve terminals in the striatum, can be measured in the clinic using single-photon emission computed tomography (SPECT) imaging, making assessment of DAT in an animal model, by immunohistochemistry or biochemistry, a particularly relevant preclinical measure. Measuring DA levels or release in the neostriatum also can provide an important functional neurochemical endpoint. Following AAV-mediated expression of α-syn in the substantia nigra, reduced DA release in the striatum (measured using in vivo voltammetry) can be seen in some, but not all animal models [59]. Because changes in striatal dopaminergic terminal function are recognized as an indicator of early stage PD, monitoring perturbations in nigrostriatal neuronal function in AAV α-syn-injected rodents is a pertinent readout. Finally, some models show an increase in neuroinflammation, measured by changes in microglia and increases in the mRNA or protein levels of inflammatory cytokines such as IL-1β, TNFα, and IL-6 [60]. These downstream pathophysiologic markers may be considered for novel therapeutics targeting α-syn.
For behavioral endpoints reflective of basal ganglia dysfunction or olfactory pathology, we refer the readers to several recent reviews (see [18–24]).
Preclinical to human translation
Integrated analysis of animal model data with safety data for translational success
From the discussion above, one can see that it is possible to generate therapeutic mechanism-specific data on TE, pharmacodynamics, PoP, or efficacy in animal models of synucleinopathy. The integration of these outcomes with pharmacokinetic data allow translational pharmacologists to generate physiologically-based pharmacokinetic/pharmacodynamic models to inform dose and dosing regimens for first in human trials. These models can also take into account any dose-limiting safety and toxicity findings observed through incorporation of toxicokinetic data from rigorous preclinical development activities in order to identify therapeutic widows for conducting early stage clinical trials. The pharmacokinetic/pharmacodynamic models should be continuously refined as new clinical pharmacokinetic, safety, tolerability, and biomarker data emerge and inform subsequent clinical decisions.
Considerations for determining target engagement and proof of principle
A recent retrospective review of delayed or denied approval of new medical entities by the Food and Drug Administration (FDA) between 2000 and 2012 indicates that uncertainties related to dose selection is a major factor underlying the failure of drug development programs [61]. As described above, establishing TE in early-phase clinical trials and utilizing pharmacokinetic-TE modeling can minimize the uncertainties in clinical dose selection. In this regard, it is important to take into account the distribution of physiologic and pathologic α-syn in central versus peripheral biofluids and cells. Thus, while Aβ and tau species targeted by investigational therapies of AD are enriched in the brain and CSF as compared to the blood compartment [62–64], α-syn levels are significantly higher in the plasma and red blood cells than the CSF [65, 66]. This raises the concern that peripheral α-syn could act as a sink for a therapeutic antibody to limit the unbound (free) fraction available for entry into the brain; an issue that can be further accentuated by the generally low brain penetration of monoclonal antibodies (CSF/plasma concentration ratio 0.01–0.04; [e.g. 63]. However, this critical concern may be mitigated by data from recent Phase 1 clinical trials of two distinct anti-α-syn therapeutic antibodies. These data demonstrate the presence of unbound α-syn antibody in the CSF of trial subjects [67, 68]. Moreover, therapeutic α-syn antibodies may achieve higher brain distribution, and thus TE, by preferential or even selective binding to brain specific conformational epitopes of α-syn assumed to play a more essential role in α-syn pathophysiology.
Another important TE question when designing α-syn therapies is whether the targeted “pathogenic” α-syn species is in the extracellular or intracellular compartment. Consistent with the Braak hypothesis [47], results of PFF-based animal model studies indicate that α-syn aggregates can indeed be transmitted trans-synaptically [18, 19, 21, 22]. Thus, it may in fact be advantageous for antibody therapies to target an extracellular α-syn species, a concept that is supported by the demonstration that anti-α-syn antibodies reduce the density of α-syn aggregates even in animal models that do not rely upon PFF injection.
As stated above, PoP biomarkers, when combined with TE or pharmacodynamic markers are critical data confirming the validity of clinical dose selection. Thus, prior to initiating large, resource-intensive disease-modifying clinical trials, it would be ideal to establish PoP and, if achieved, its dose dependency in a small PD patient study, as exemplified by the evaluations of amyloid plaque removal with Aβ PET imaging in the Phase 1 studies of the Aβ monoclonal antibody therapeutics Aducanubab [10] and Gantenerumab [15]. Unfortunately, at the time of writing of this review, imaging methods to monitor Lewy pathology or presumed pathogenic α-syn species in the brain or CSF still represent major gaps in our therapeutic development toolbox (see below: “Proof of Concept Clinical Studies”).
In summary, it is most advisable to select an animal model pertinent to the therapeutic mechanism of choice and integrate TE, pharmacodynamic and PoP biomarkers in order to generate PK-TE/pharmacodynamic/PoP modeling to inform clinical dosing. Data from early stage clinical trials could further refine the modeling and provide a rationale-based approach for PoC and registration trials.
CLINICAL BIOMARKERS OF ALPHA-SYNNUCLEIN PATHOLOGY
With at least five α-syn-targeted therapeutics currently under clinical investigation [69], there is an urgent need for robust and validated biomarkers. Here we review the current biomarker landscape, with a particular focus on methodologies to measure α-syn in biofluids and peripheral tissues, as well as imaging modalities and mobile technologies. It is important to note that currently available biomarkers have been validated in clinically-defined cohorts of sporadic PD rather than on non-hypothesis-driven (i.e., agnostic to clinical criteria) cohorts of normal and abnormal aging populations. Thus, data supporting the use of these biomarkers represent statistical separations between patients with PD and age-matched controls, but are fraught with substantial overlap [70].
Biochemical assays in biofluids and tissue
Alpha-synuclein immunoassays
Given that accumulation of aggregated α-syn is a hallmark pathology of PD, perhaps the most obvious choice for a TE, PoP, or disease progression biomarker for α-syn-targeted therapies is various conformers and species of α-syn itself (e.g., native monomer, post-translationally-modified monomers, or assemblies such as oligomers or fibrils). In the absence of an α-syn imaging tracer (discussed in greater detail below), measuring α-syn in CSF assumes it to be a measure of brain pathology (and therefore, a PoP biomarker), but this assumption remains to be tested. A number of studies have measured the so-called “total” α-syn in biofluids such as CSF, plasma, and saliva [71–74]. Although these α-syn assays may offer the ability to establish TE for specific drug candidates, several challenges hamper their utility to establish PoP or monitor disease progression in clinical studies. First, since the precise species of α-syn being measured by current commercially available immunoassays of total α-syn remain unknown, it is not possible to demonstrate that the assay reflects a species of α-syn categorized as pathogenic in animal studies. Furthermore, these assays have shown that the levels of α-syn species measured remain unchanged in 12 month follow-up of the Parkinson’s Progression Markers Initiative (PPMI) cohort (Mollenhauer et al., 2018, submitted). Whether the ongoing, longer follow-up of the PPMI cohort or other similar studies might help remediate this issue and enable the use of total α-syn assays to monitor disease progression remains to be seen. Finally, pre-analytical conditions such as CSF collection, processing, and sample handling appear to influence total α-syn measurements [75], indicating careful studies need to be conducted to establish pre-analytical conditions before the existing assays could be used effectively in clinical trials (see below).
The diagnostic utility of total α-syn has been investigated thoroughly. Numerous studies have demonstrated total α-syn concentrations in the CSF are reduced by an average of 15% at the group level in PD patients compared to healthy controls (reviewed in [76]). However, at the individual level, considerable overlap exists between PD and healthy control subjects which renders the diagnostic use of CSF α-syn problematic [71]. Notably, the reduction in CSF α-syn in PD patients has been demonstrated using a variety of analytically validated immunoassay platforms, including standard chemiluminescent sandwich enzyme-linked immunosorbent assays (ELISA) [71, 72], electrochemiluminescent sandwich immunoassays [73], and bead-based cytometric assays [74]. In an effort to further qualify the total α-syn immunoassays, MJFF sponsored a round robin assessment of four different assay platforms on CSF, whole blood, and saliva samples of 50 PD and 50 healthy control subjects. The four assays selected in this initiative were the Elecsys® Total α-Synuclein Prototype Assay (Roche Diagnostics, Penzberg, Germany), the MSD U-PLEX® Human α-Synuclein Kit (Meso Scale Discovery, Rockville, Maryland), the BioLegend α-Synuclein Immunoassay (BioLegend, San Diego, CA), and the ADx α-Synuclein Immunoassay (ADx Neurosciences, Gent, Belgium). Blinded analyses of the biospecimens were conducted by both the originating laboratory that developed the assay and satellite laboratories equipped to run the assay. The mean intra-platform variance component ranged from 8.12% to 9.81%, an acceptable range. Importantly, the four assays correlated well to each other, even though the absolute concentrations of α-syn measured in the same samples were different (Mollenhauer et al., 2018, in revision). The results of this study highlight the need for common reference materials and methods to control for inter-assay variability. An investigation of peri- and pre-analytical variables affecting assay performance was also undertaken as part of this work, and recommendations from MJFF’s Investigating Synuclein Consortium investigators for ways to reduce α-syn assay variability through controlled and consistent sample handling methods have been published [75].
Immunoassays to detect specific, pathologically-relevant α-syn species have also been generated using isoform-specific antibodies. For example, an increase in pS129 α-syn levels has been reported in the CSF of PD subjects compared to healthy controls, although it is unclear whether this species changes over the course of the disease and correlates with disease severity [72, 77]. Likewise, higher levels of oligomeric α-syn have been reported in PD CSF compared to healthy controls, and they also appear to increase over time [72]. These “modified” α-syn assays have mainly been developed in academic research laboratories, have not been cross-validated by multiple groups in blinded samples, and are not generally available for widespread use by the research community. For this reason, MJFF has undertaken another initiative to analytically validate the pS129 and oligomeric α-syn assays through a broader round robin consortium known as LEAPS (Linked Efforts to Accelerate Parkinson’s Solutions). Research is also in progress to identify novel potentially pathological species of α-syn for which assays could be developed in the future (e.g., truncated, oxidized, phosphorylated at sites other than S129). In addition, biochemical properties of α-syn, such as its ability to bind to lipids, have been investigated as potential differentiating biomarkers, and independent cross-validation and replication efforts are currently underway [78].
Seeding and turnover assays
The immunoassays described above are dependent on the quality and availability of antibodies for specific detection of the relevant form(s) of α-syn. New antibody-independent assay approaches for detection of α-syn aggregates via repeated cycles of seeding, amplification, and break down, similar to DNA amplification by polymerase chain reaction, appear promising. Two variations of this seeding assay technology, protein misfolding cyclic amplification assay (PMCA) and real-time quaking-induced conversion (RT-QuIC), have demonstrated high sensitivity and selectivity in differentiating PD from healthy control subject CSF in initial studies [79, 80]. Efforts are also under way to measure α-syn turnover through stable isotope labeling kinetic (SILK) assay development. This technique measures incorporation of a stable isotope amino acid tracer into newly synthesized α-syn, measured by mass spectrometry in CSF collected from living subjects [81].
Immunohistochemical analysis in the periphery
Given the hypothesis that peripheral α-syn pathology may develop prior to that in the CNS, and in the absence of an α-syn imaging tracer, considerable effort has been devoted to measuring α-syn deposition in peripheral tissues. Several studies (reviewed in [82]) report inconsistent results in colon and skin biopsies, possibly a result of the many permutations in collection and processing protocols utilized or variable distribution of α-syn in the tissues examined. The submandibular salivary gland has emerged as a potentially sensitive and specific peripheral biomarker differentiating PD and healthy control subjects [83], although transfer of this technique and replication by multiple groups, as well as assessment of the feasibility of collection in a multi-site trial, is pending. The ongoing Systemic Synuclein Sampling Study (S4) is designed to evaluate α-syn in three peripheral tissues, skin, colon and submandibular salivary glands, and correlate it with biofluids levels (CSF, saliva, and blood) collected from the same subject [84].
Physiological biomarkers
Physiological biomarkers provide additional opportunities for segregating PD and healthy control subjects and may be relevant as phenoconversion markers of prodromal PD (reviewed in [85]). Functional, non-invasive techniques such as heart rate variability, electroencephalography, electromyography, optical coherence tomography, and skin conductance have shown potential utility as relevant functional biomarkers of α-syn pathology. Future replication through natural history studies of large cohorts of PD as well as non-PD neurodegenerative Parkinsonisms may elucidate whether a combinatory physiological “PD signature” will emerge, sufficiently distinct from PD-like disorders. There is also ongoing development of new physiological biomarker platforms, including pilomotor reflex [86], breath volatile organic compounds [87], eye tracking [88] and corneal confocal microscopy [89], facial expressivity [90], and acoustic speech [91]. Correlation of all of these physiological measures to α-syn pathology will be an important determinant of potential integration of these measures into clinical trials. Wearable and interactive devices (see 3.3) may also be considered a type of physiological biomarker.
Imaging
While other biomarkers are evolving, the most robust markers for assessing PD progression (and presumably, the effects of disease-modifying interventions) are based on neuroimaging and, for now, particularly those that assess dopaminergic neuron function. Dopaminergic imaging, discussed in further detail below, may not be optimal for a variety of reasons. Here we consider other possible imaging approaches to studying PD progression (Table 3).
Table 3
Potential imaging biomarkers for Parkinson’s disease
| Modality | Indication | Caveats | Other comments | |
| Structural MRI | High-resolution T1, T2*/GRE, FLAIR | Exclusion of atypical Parkinson syndromes | Differences may be subtle, limiting the utility | – |
| Subcortical volumetry | Exclude atypical Parkinson syndromes | – | – | |
| Cortical thickness, hippocampal volume | Thinning/atrophy associated with cognitive decline | Patterns likely to be heterogeneous depending on clinical phenotype | Pattern of atrophy linked to nigral connectivity and may progress according to a defined network of propagation | |
| Neuromelanin | Assessment of dopaminergic and noradrenergic cell bodies | Small volume of aminergic nuclei makes reliable quantitation difficult | Need data from longitudinal studies to understand potential as progression marker | |
| Diffusion weighted | Free water | Initial studies not independently reproduced | Nigral free water increases with disease progression but unclear how robust after early time points | |
| Susceptibility weighted or T2* | Iron sensitive | Increase may not be robust enough to be useful | Increased iron with disease progression | |
| Functional Imaging | Resting state fMRI | Connectivity of pre- and post-central gyri, occipital cortex and cuneus | Difficult to control quality of images in multicenter studies | Altered connectivity correlates with cognitive decline and with CSF α-synuclein |
| FDG-PET | Parkinson Disease Related Pattern | PDRP is suppressed by symptomatic therapies; requires sophisticated statistical analysis; some controversy regarding global normalization | Diagnostic value and changes with disease progression | |
| Parkinson Disease Cognitive Pattern | (As above) | PDCP is linked to cognitive decline, a greater source of disability in advanced disease | ||
| Task-related activity | Motor activation | Difficult to standardize across centers. Primary utility is for research studies rather than clinical trials | Provides evidence of functional integration | |
| Cognitive tasks | Difficult to standardize across centers. Primary utility is for research studies rather than clinical trials | Alterations in prodromal disease reflect compensation; unclear role in studying disease progression | ||
| Molecular Imaging | DA systems: DAT SPECT or PET | Assessment of dopamine nerve terminal integrity, primarily in striatum | DAT may be subject to pharmacological and compensatory regulation | Widely available. Different tracers have varying selectivity and kinetic properties |
| DA systems: VMAT2 binding | More closely approximates monoamine nerve terminal density | Less widely available than DaT, not specific for DA | May track disease progression but additional data are needed. | |
| DA systems: F-dopa uptake | Assessment of presynaptic dopamine nerve terminals and dopamine synthesis rates | Not widely available; decarboxylase is subject to regulation and not specific to dopaminergic neurons | Historical ‘gold standard’. Prolonged scans can be used to assess DA turnover | |
| Non-DA systems | Cholinergic (cholinesterase, VAChT); serotonergic (SERT) | Cholinesterase activity cannot be quantitated in striatum, but VAChT can | Cholinergic dysfunction relates to several aspects of disability, especially cognition | |
| PoP Biomarkers | α-syn | Quantification of alpha-synuclein pathology in brain | No specific tracers to date; intra-cellular localization and limited brain density suggests signal will be limited; possibility of peripheral consumption | ‘Holy grail’ of synucleinopathy imaging biomarker |
| TSPO | Microglial activation | Classical ligand has low signal; binding of newer agents varies according to polymorphism; quantitation difficult | Unclear how microglial activation progresses with disease and whether it is harmful or protective |
CSF, cerebrospinal fluid; DAT, dopamine transporter; fMRI, functional magnetic resonance imaging; F-dopa, fluorodopa; FDG, fludeoxyglucose; MRI, magnetic resonance imaging; NM, neuromelanin; PD, Parkinson’s disease; PDCP, Parkinson’s disease cognitive pattern; PDRP, Parkinson’s disease related pattern; PET, positron emission tomography; PoP, proof of principle; SERT, serotonin transporter; SPECT, single-photon emission computed tomography; TSPO, translocator protein; VAChT, vesicular acetylcholine transporter; VMAT2, vesicular monoamine transporter 2.
Structural imaging
Standard diagnostic MRI is of limited use for confirming a PD clinical diagnosis but may be helpful to exclude atypical forms of Parkinsonism such as MSA or progressive supranuclear palsy (PSP) [92]. This is an important consideration, as inclusion of patients with atypical disease could dilute the results of any interventional study. More sophisticated approaches to structural MRI may prove informative. Striatal volume declines over time, particularly in the early stages of PD [93], while cortical thinning and hippocampal atrophy are associated with cognitive decline [94]. The pattern of atrophy in PD may progress according to a defined network of connectivity to the substantia nigra, compatible with propagation along anatomical pathways [95]. Susceptibility weighted or T2* imaging provide insight into brain levels of iron. Nigral iron signal increases in association with clinical measures of disease severity in PD [96, 97], and this may accordingly represent a relatively inexpensive and widely available approach to quantify disease state. T2* weighted imaging at high field strengths detects a dorsal nigral hyperintensity (DNH) deemed analogous to the calbindin-negative nigrosome 1 [98]. This DNH is lost both in PD and in a majority of subjects with REM sleep behavior disorder (RBD) [99], is preserved in non-degenerative forms of Parkinsonism, and predicts loss of DAT binding [100].
More experimental MRI techniques include neuromelanin imaging and tract-tracing techniques such as diffusion tensor imaging (DTI). Reduced neuromelanin signal has been reported in PD [101], may decline as disease progresses [102, 103], and may indeed correlate in some hands (but not others, [104]) with striatal DAT binding [105], but this is currently not an approach in wide use. DTI examines the movement of protons and is widely used to assess fiber tracts. This technique has revealed reduced fractional anisotropy (FA) in the posterior substantia nigra in PD [106], and changes in FA in the nigrostriatal tract appear to correlate with clinical measures of disease severity [107]. Furthermore, reduced nigral FA correlates with increased free water in PD, and, when assessed using a bitensor model, increases with disease progression [108].
Functional imaging
Resting state. Resting state functional MRI (rs-fMRI) has revealed reductions in connectivity of the pre- and post-central gyri, occipital cortex, and cuneus in PD that progresses over the course of illness and correlates with cognitive decline [109]. The potential validity of using a change in patterns of rs-fMRI connectivity to assess disease progression is highlighted by a study demonstrating a correlation between reduced sensorimotor connectivity and reduced CSF levels of α-syn [110]. However, it should be noted that all rs-fMRI studies to date have been small and single-site. Feasibility in multicenter trials is still a concern.
Task-related activity. PD is typically associated with reduced activation of the supplementary motor cortex (SMA) during performance of a simple motor sequence, and activation can be restored by symptomatic medication such as apomorphine [111]. While this may seem a relatively non-specific outcome measure, it may be important to demonstrate the functional relevance of improvement in other markers. Increased dopaminergic activity was seen within six months after fetal mesencephalic transplantation for PD, but maximal clinical improvement did not occur until later, associated with increased activation of the SMA, presumably reflecting a gradual and protracted integration of the graft into the cortico-striatal-pallidothalamo-cortical loop [112]. Cognitive activation studies may also provide insight into disease state, but interpretation is challenging. Performance of an executive task should be associated with suppression of activity in the Default Mode Network (DMN). In patients with PD, there is failure of suppression in the DMN [113], but the relationship to disease progression and to symptomatic therapy is unknown. Changes in activation patterns can be seen even in presymptomatic carriers of LRRK2 mutations during performance of a motor imagery task or while performing the Stroop interference task [114, 115]. It is unclear how these alterations change with disease progression, but they are thought to represent compensatory shifts and highlight the challenges in interpreting such data.
The pattern of cerebral glucose metabolism, as measured by fluorodeoxyglucose positron emission tomography (FDG-PET) imaging, is altered in PD in a characteristic fashion, i.e., relatively increased metabolism in the basal ganglia, thalamus, pons and cerebellum, with concomitant relative reductions of metabolism in the premotor and parietal cortices (the PD-related pattern, PDRP). PDRP expression increases with disease progression and is inversely correlated to DAT binding in the putamen [116]. However, the utility of the PDRP as an imaging biomarker of disease progression is unclear because the pattern is suppressed by symptomatic therapies including levodopa and deep brain stimulation [117]. While the PDRP correlates with motor dysfunction in PD, trials of α-syn-based therapies should also take into consideration the cognitive decline that represents a significant source of disability for a substantial number of patients. The PDCP (PD cognitive pattern) is a distinct network of altered metabolism that has been found to correlate with cognitive change and with DAT binding in the caudate nucleus rather than the putamen [118]. PDRP and PDCP may be measured in the same set of images. FDG-PET may serve to differentiate PD from other akinetic rigid syndromes such as MSA or PSP [119]. This potential diagnostic utility may be highly relevant to the conduct of trials of disease-modifying therapies, as these conditions can be difficult to diagnose in early stages, are not readily differentiated by dopaminergic tracer studies, and inclusion of such patients may reduce the chances of detecting therapeutic benefit.
Molecular imaging
Molecular imaging of the nigrostriatal dopaminergic system is still likely the most widely accepted biomarker to assess PD progression and, by extension, the impact of potential disease-modifying therapies. The most extensively used approach is some form of imaging presynaptic dopaminergic neuron integrity. While this clearly addresses the most obvious deficit in PD, it is probably not the only outcome of interest, as progression of PD and major sources of disability are associated with changes in several non-dopaminergic neurotransmitter systems, and neurochemical studies provide only one measure of the underlying pathology.
Dopaminergic measures. The most widely used approaches utilize PET or SPECT to assess expression of the membrane DAT, the vesicular monoamine transporter type 2 (VMAT2), or activity of L-aromatic amino acid decarboxylase (L-AAAD). All three approaches provide similar, although not identical, information on the integrity of dopaminergic nerve terminals in PD. All can demonstrate the characteristic asymmetric reduction of tracer uptake in PD, with a rostro-caudal gradient in which the posterior putamen is maximally affected. All approaches have, to varying degrees, demonstrated a correlation between tracer uptake and nigral dopaminergic cell counts [120–124]. Furthermore, the progression of abnormalities using any of the three approaches [125] is remarkably similar to the pattern of striatal TH and nigral dopaminergic neuron loss demonstrated in post-mortem PD studies [126], with an exponential pattern of decline that reaches an asymptote at four-to-five years into the disease. Nonetheless, considerable caution is required in the application of these approaches to study disease modification. Although the practical significance is uncertain, both L-AAAD activity and DAT are subject to pharmacological and compensatory regulation, particularly evident in early PD stages [127, 128]. While there is a general correlation between clinical measures of PD severity and imaging markers, it has been increasingly difficult to demonstrate a correlation between changes over time in the two parameters, regardless of which tracer is used (see [129]). Furthermore, there are several examples of discordance between imaging and clinical measures of improvement, particularly related to trials of putative disease modification [130–133]. Thus, while dopaminergic neuron imaging undoubtedly serves as a useful adjunctive biomarker to assess the effects of potential disease-modifying therapies, findings must be interpreted in the light of other available data, and it is unlikely to ever become a surrogate endpoint.
An interesting molecular imaging technique involves uptake of 6-18F-fluoro-L-dopa (F-dopa) which reflects not only the activity of L-AAAD (a marker for the health of dopaminergic nerve terminals), but also trapping of 6-18F-dopamine (F-DA) in synaptic vesicles, prolonged scans with F-dopa can therefore be used to assess DA turnover [134]. However, F-dopa itself is only available at a few centers worldwide, and turnover studies are technically challenging, requiring scan times of up to four hours, arterial sampling, and high-performance liquid chromatography for metabolite analysis.
Non-dopaminergic systems. Significant disability in PD is attributable to non-motor complications, many of which presumably reflect changes in non-DA systems. Serotonergic neurons of the raphe are affected in PD, but binding of the serotonin transporter (SERT) ligand 11C-3-Amino-4-(2-dimethylaminomethyl-phenylsulfaryl)-benzonitrile (DASB) is only minimally reduced, with no clear correlation to disease severity. In fact, increased 11C-DASB binding is seen in PD patients with depression [135, 136], and relative preservation of 11C-DASB binding in the putamen is associated with levodopa-induced dyskinesias [137, 138]. DASB binding is also increased in multiple brain regions prior to clinical evidence of disease onset in subjects with LRRK2 mutations [139]. It is therefore unlikely SERT binding will be useful as a marker of treatment efficacy, although a dramatic increase in SERT binding might be associated with treatment-related complications.
Cholinergic dysfunction is common in PD and is associated with cognitive impairment, postural instability, olfactory impairment, and RBD [140–143]. Cholinergic dysfunction with these disabling non-motor features suggests that it may be a meaningful measure of disease activity, particularly with respect to therapies targeting α-syn. To date, most studies of cholinergic function in PD have been conducted using PET with tracers for acetylcholinesterase [144]. This provides a less direct measure of cholinergic neuronal integrity, and recent development of tracers for the vesicular acetylcholine transporter [145] may offer a more reliable measure and permit easier quantitation, particularly in the striatum.
Both sympathetic and parasympathetic nervous systems are affected in PD, albeit to somewhat varying degrees (see [146] for review). The unequivocal presence of cardiac sympathetic denervation as detected by 123I-metalodobenzylguanidine (MIBG) or 11C-hydroxyephedrine (HED) SPECT, or 6-18F-fluorodopamine PET, might help differentiate between PD and other atypical parkinsonian conditions such as MSA or PSP, in which cardiac sympathetic denervation is not thought to be present, but even this is controversial. There are similarly recent reports of gastrointestinal cholinergic denervation in early PD, which would not be expected in the atypical Parkinsonisms. However, in both cases, the degree of denervation is highly variable, and there is insufficient evidence of a clear relationship to disease progression that would justify the use of such approaches to monitor either disease progression or the effects of a disease modifying therapy. Furthermore, these peripheral abnormalities are present long before motor manifestations are expressed, as is evident from studies conducted in idiopathic RBD [147], thus suggesting the likelihood of a floor effect in many patients.
Pathology or pathophysiology markers for proof of principle and patient enrichment
The most useful imaging biomarker to establish or confirm the impact of α-syn-based therapies would be a probe for α-syn itself. Thus, a PET ligand to measure α-syn deposition in the brain is highly coveted as a PoP biomarker, analogous to the utility of Aβ PET tracers in clinical trials of amyloid-targeted therapies [15, 148, 149]. If an α-syn PET tracer would become available for clinical use, there are some important considerations for using it as a PoP biomarker for drug development. Most importantly, one has to take the drug’s mechanism of action into account; i.e., would the PET signal need to detect effects of the drug on progressive aggregation and propagation of misfolded α-syn, or is a decrease of already established α-syn pathology expected (clearance effect)? In either case, quantitative methods will have to be developed, with the identification of relevant regions of interest and reference region. Once the directionality of the effect and quantitative methods have been established, a pre-set target for the desired effect size has to be defined. Each factor discussed above could have a major influence on the sample size and treatment duration, and consequently the ability to obtain a robust PoP based on a PET readout. Additionally, a meaningful go-decision based on α-syn PET imaging, would also have to assume a relationship between the treatment-induced directionality and extent of the effect on the PET signal with a clinical benefits. This critical aspect is well illustrated by the use of amyloid PET imaging in AD programs. Thus, treatment with BACE inhibitor, that reduced CSF Aβ peptide levels [150] and prevented further increase in amyloid deposition, as demonstrated by PET imaging, did not provide any clinical benefit [16]. On the other hand, there are findings suggesting that treatment with an anti-Aβ monoclonal antibody leads to amyloid clearance (reduction from baseline) accompanied with a potential delay in clinical disease progression [10]. What further complicates the possibility translate PoP findings to potential clinical benefit is that the stage of disease at treatment may impact the outcome, with stronger effects presumably attainable at earlier disease stages [151]. Beyond the PoP utility of an α-syn PET ligand, it could provide a critical tool for patient enrichment via assessment of α-syn pathology load as well as to monitor the progression of PD pathology in natural history and therapeutic intervention studies.
The discovery and validation of an α-syn-targeted PET ligand has proved elusive for several reasons [152]: 1) while a number of PET ligands have been shown to bind α-syn, these compounds bind to proteins that take on a β-pleated conformation, thus selectivity is generally poor (e.g., BF227 [153] and SIL23 [154] bind to Aβ and tau, although BF227 did not bind in Aβ-negative DLB); 2) unlike Aβ, α-syn is predominantly intracellular and expressed in small quantities, thus adequate cellular penetration and signal-to-noise ratio will be difficult to accomplish; 3) greater selectivity might be afforded by radiolabeled antibodies, but passage across the blood-brain barrier will be quite limited; and 4) kinetics may be challenging, as suggested by the time required (several days) for a molecule to achieve equilibrium [155]. As α-syn is highly expressed in tissues other than the brain, the question arises as to whether a PET tracer would achieve a significant signal in the brain or whether the signal would be consumed peripherally. The fact that one can detect presumed off-label binding of the tau ligand PBB3 to α-syn in brain (at least in high concentrations) [156] suggests this may not be an issue. Other 11C and 18F tracers that have demonstrated brain penetrance with rapid washout kinetics are in development [157], but selectivity for α-syn and utility in transgenic animal models or humans with synucleinopathies have yet to be demonstrated in vivo.
As noted above, the emergence of cognitive impairment is a major clinical aspect of PD and could potentially be an important measure of a clinically meaningful impact of disease-modifying therapies targeting α-syn. Several studies have used PET to determine the presence of abnormal Aβ deposition, but the prevalence is overall too low [158] to make Aβ PET a useful biomarker for this purpose.
α-syn interacts with phosphorylated tau [159], and both proteins appear to be co-deposited (along with Aβ) in PD with cognitive impairment [160], raising the possibility that tau imaging might provide indirect insights into α-syn deposition. Several PET ligands for misfolded tau have been recently been developed. However, the most widely used PET tau tracer, 18F-AV 1451, only binds to a limited extent in discrete cerebral cortex regions in patients with cognitive dysfunction associated with PD or DLB [161]. Emerging evidence suggests that another preferential tau ligand, 11C-PBB3, binds to α-syn, but likely only where there is a high concentration of aggregates, such as in MSA or individuals with SNCA multiplication [156, 162].
PD and other neurodegenerative disorders are associated with microglial activation [163, 164]. The ‘peripheral benzodiazepine receptor’ (now referred to as Translocator Protein, TSPO) ligand 11C-PK 11195 has been used to study PD, with variable results. One study showed localized binding in the substantia nigra that correlated with disease severity [165], whereas another found more widespread inflammation that did not correlate with clinical severity [166]. 11C-PK 11195 is a difficult ligand to work with, as the signal-to-noise ratio is low, and the lack of a clear reference region makes modeling challenging. Several newer TSPO ligands have been developed, but while the signal is better compared to 11C-PK 11195, the binding of these agents varies, subject to genetic polymorphism [167]. Thus, interpretation requires polymorphism testing, and low affinity binders, representing ≤10% of the population, cannot be studied. Only limited data are available on TSPO binding in PD using these later generation ligands, and it is unclear how useful they will be [168]. Furthermore, the relationship between microglial activation and disease progression is unclear.
Application of molecular imaging approaches
There is no single imaging measure that can reliably provide a measure of treatment effectiveness. Nonetheless, molecular imaging may prove useful in PoC studies. The ability to demonstrate blood-brain barrier passage and TE would be an important first step in deciding whether or not to proceed with a potential therapy and in the interpretation of failed therapeutic efficacy.
As noted above, measures of nigrostriatal dopaminergic system integrity will inevitably be regarded as necessary to exclude non-DA-dependent Parkinsonism (SWEDD, scans without evidence of dopaminergic deficit), and they would likely constitute a secondary outcome measure in PD trials. Indeed, DAT SPECT has recently been approved by the European Medicines Agency as an enrichment marker for inclusion in clinical trials (draft qualification opinion - [169]), and the FDA has also issued a letter of support for this use [170]. However, although in one study, density of DAT in striatum did correlate inversely with density of α-syn staining, 1) the relationship between change in a dopaminergic marker and change in clinical function is inconsistent [129]; 2) meaningful disability associated with PD progression arises from non-dopaminergic mechanisms; 3) the treatment may have a pharmacological effect on the dopaminergic marker, independent of changes in disease severity; and 4) it may take considerable time and a large patient cohort to demonstrate a convincing effect on the DA-related marker. If robust imaging measures of disease pathology were available, there is still no guarantee that an effect of the treatment on the imaging marker would correspond to an impact on disease progression.
Digital and wearable technologies
Mobile technology-derived measures in human anti-alpha-synuclein trials
Assessment of human behavior using technology placed on the surface or inside the body of a person, or in the immediate environment, has been increasingly recognized as a powerful source of data that may enhance diagnosis and management in medicine (Fig. 2). “Technology” here means detection systems for biological, chemical, and other processes. Mobile technology is worn on the body (so-called “wearables”), and its development has been accelerated, fueled by collaborations with the health and fitness sector industry. This technology allows for the detection of subtle changes in human activity and behavior, changes that may escape detection by traditional measurements, such as diaries, questionnaires, or even in-clinic assessments by trained clinicians. Mobile technology-derived measures are thus attractive options to include as outcome measures in trials of symptomatic and disease-modulating compounds. Due to the nature of PD (associated with predominantly motor symptoms including mobility limitations, falls, and sleep problems; exhibiting a chronic and progressive course), this disease is an interesting “target disease” for the assessment of mobile technology-derived measures. This interest is reflected by many observational studies recently published (for a review, see [171]).
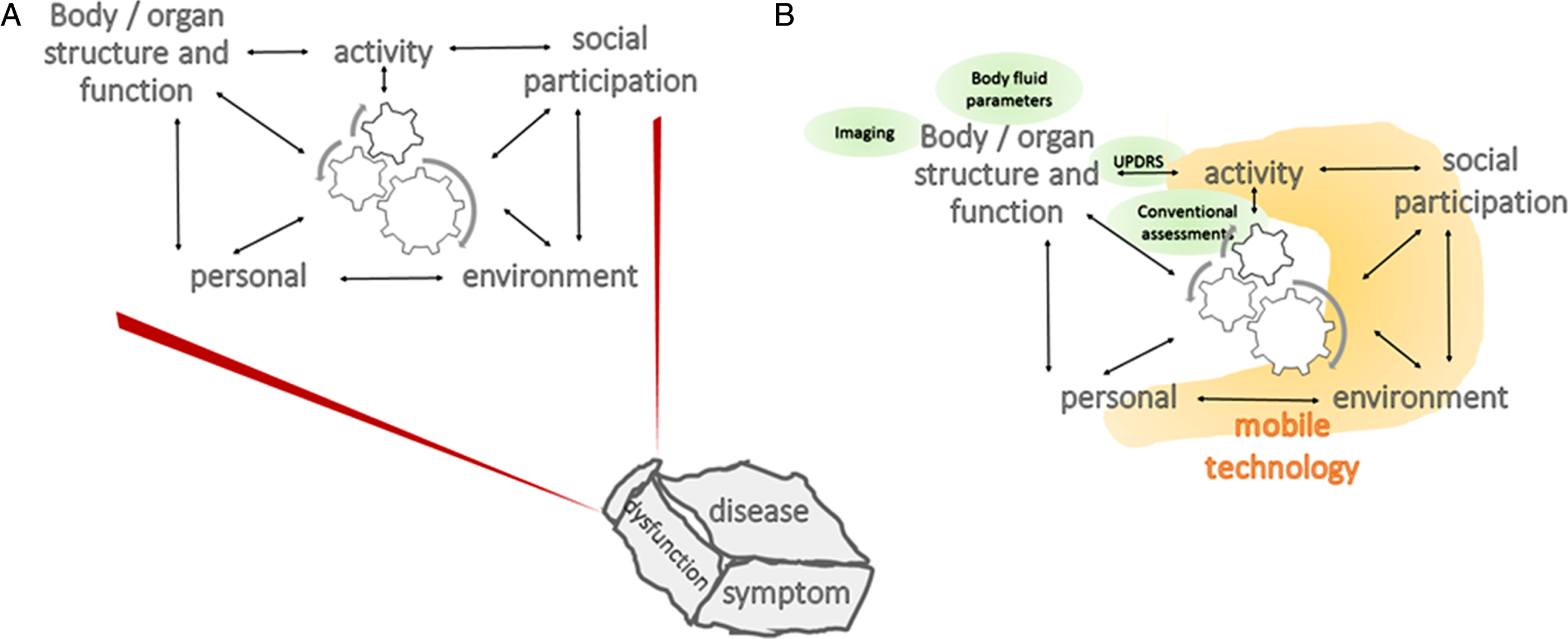
Fig. 2.
Adapted Illustration of the International Classification of Functioning, Disability and Health Model of the World Health Organization. A) The description (and treatment) of a disease, dysfunction or symptom require(s) consideration of multiple domains of an individual (more specifically, body/organ structure and function, activity, social participation, as well as personal and environmental factors), and the corresponding interactions across domains. B) Traditional outcome measures, such as clinical scales, body fluid parameters and imaging (green circles) document variables about the body/organ (here: brain) structure and function domain and to some extent about the activity domain. Mobile technology (orange) has the potential to collect information over most of the ICF domains. ICF, International Classification of Functioning, Disability, and Health; UPDRS, Unified Parkinson’s Disease Rating Scale.
Potential of mobile technology-derived measures
Objective data from mobile technologies can quantify single and recurrent events of interest, extended continuously over a timeframe that captures their frequency in the background of expected fluctuations, or intermittently, offering the opportunity to reliably include a virtually infinite number of highly accurate “visits” and data points. Moreover, measures obtained from mobile technologies can support supervised assessments in the clinic and laboratory, where outcome measures mainly reflect functional capacity (“How well can you perform?”) and easily can be used under unsupervised conditions in ecologically natural environments, where the outcome measures reflect functional activity (“How do you regularly perform?”) [172, 173].
The high accuracy and inter-event reliability in the measurement of endpoints of interest by mobile technologies promises to increase the signal-to-noise ratio, thereby reducing the sample size required by clinical trials [174]). It could also lower trial costs by shortening the duration and lowering the burden on patients whose efforts no longer require answering diaries or questionnaires [175, 176]. Furthermore, use of mobile and interactive technologies may reduce the need for in-clinic assessments in clinical trials, further reducing burden, complexity and expense. A condensed overview of short-, medium- and long-term efficacy, efficiency, and patient centricity benefits expected from mobile technology-based measures, integrated in a battery of assessments built into clinical trials are available [177].
Why is it so difficult to develop mobile technology-derived measures?
The integration of mobile technology in clinical trials has been difficult for a number of reasons. First, there is a lack of studies investigating mobile technology-derived measures in PD that convincingly describe endpoints of interest. Importantly, as a field we lack consensus defining the optimal concepts of interest and contexts of use for technologies in PD clinical trials. This is due in large part to the fact that the maturity of mobile technology in terms of medical use is low. A recent review found that only 5 percent of about 850 original studies applying technology-derived measures in PD used systems that were close to or already positioned on the market [171]. Furthermore, only a small proportion of studies provide qualitatively acceptable data in terms of cohort and algorithm description, sensitivity to change, specificity, accuracy, and predictive value of investigated measures [171]. Finally, technological development does not follow clinical decision-making, and often the opposite is true. Researchers often focus on seemingly easy-to-detect symptoms or signs with questionable clinical meaningfulness or allow the technology at hand to dictate the questions being asked of subjects.
The multidimensional and protean nature of PD’s clinical manifestations further complicates the selection of concepts of interest and contexts of use for the application of technologies; distinct features of PD occur at different time points and deteriorate with different slopes of progression during distinct PD phases. As an example, fine motor movement of the upper extremities may be a useful progression parameter in prodromal and early, but not late PD phases [178–180]. Moreover, our “pre-technology era” clinical view may hinder us from discovering the clinical relevance and usefulness of PD-related features particularly sensitive to technology-based assessment strategies: candidate measures could be loss or preservation of variability (e.g., of movement “episodes” and activity patterns during the day and during the night); loss or preservation of similarity of comparable movements; phenomena associated with multitasking-related deficits, and a range of non-motor behaviors (e.g., heart rate variability, sleep architecture, etc.). Fourth, we have not yet solved retention issues regarding the use of mobile technology over long periods, especially when they require active participation of the study subjects (e.g., entering data). For example, only two percent of those who had downloaded the mPower app during the first six months of availability used it more than five times [181]. Other studies have reported similarly poor long-term adherence to technology requiring active participation. Thus, such assessments not only suffer from low retention but also from a potential selection bias, as the study participants adopting and using mobile technologies may not be representative of a population of interest. Such lack of adherence could create issues for data completeness and integrity in clinical studies. Fifth, access (see below) and regulatory issues make it difficult for investigators to integrate these new tools. Raw data is often not freely accessible, and is only interpretable by using company-owned algorithms, and there are often separate proprietary platforms required for devices from different companies, preventing the time-locked integration of their data. Importantly, the field currently lacks data and metadata standards that would allow sharing and common interpretability of device data. Finally, legal (privacy) and regulatory barriers exist and often differ across countries.
In spite of these challenges, the potential of mobile technology-based measures as exploratory endpoints in anti-α-syn trials is enormous, specifically if we consider the following recommendations.
Recommendations for the integration of mobile technology-derived measures in human anti-alpha-synuclein trials
The International Parkinson and Movement Disorders Society Task Force on Technology [176] and the Clinical Trials Transformation Initiative recently published general recommendations for the development of mobile technology-derived measures and for the implementation of mobile technology in PD clinical trials [177]. There is broad consensus that every ongoing clinical PD trial should integrate mobile technology-derived measures, initially on an exploratory level and targeting endpoints previously validated in clearly defined cohorts.
– There is no convincing evidence currently available that TE of pathological α-syn can lead to improvements in specific PD symptoms; as such, mobile technology should gather data about a wide range of symptoms, using different modalities (e.g., combinations of accelerometers, gyroscopes, magnetometers, GPS, heart rate) and body surfaces instead of assessing single symptoms and applying single sensors for data collection. Shorter assessment periods (e.g., days rather than weeks of assessment) may compensate for the higher effort on the part of the patients.
– Data obtained from the habitual environment likely have a higher potential to add novel information to a battery of measurements than mobile technology-derived data collected in the clinic and laboratory. Note that traditional assessments mostly examine functional capacity, which do not necessarily reflect behaviors in habitual environments, under unobserved conditions. We recommend performing combined assessments covering functional capacity and functional activity.
– Data collection approaches that do not require (much) active engagement of the study participants during the active data collection phase should mitigate the current rate of attrition. Translation of these data into understandable information to the technology wearers during or after the active data collection phase may enhance adherence [182].
– If the aim of an α-syn targeted trial is to reduce the progression of progressively worsening symptoms rather than improving potentially non-progressive symptoms, there should be enough evidence that the target symptoms exist in an as large as possible proportion of study participants, have a high chance to be in a “progressive phase” of the disease, and can be measured by technology without the risk of ceiling or floor effects. Interesting candidate features in prodromal and early PD are fine motor movements of the hands and arms and axial symptoms, such as gait and postural instability, at later stages.
– Mobile technology collecting and providing free access to raw data (i.e., the sensor signal) has the advantage that the PIs can contribute to independent data analysis approaches as soon as new algorithms and statistical strategies are available. Moreover, PIs may benefit from open-access platforms for mobile technology that may become available in the near future.
PROOF OF CONCEPT CLINICAL STUDIES
As a key mediator of PD pathogenesis, therapeutic modulation of α-syn-associated pathogenic mechanisms are anticipated to produce clinical efficacy reflective of slowing of disease progression (i.e., disease-modification). The scope of the effort required to unequivocally demonstrate slowing of disease progression, with an appropriate safety and tolerability profile, will require the design, conduct, interpretation, and regulatory review of data generated from multiple clinical trials. The breadth of such a development program, which will be highly dependent upon the size of the beneficial clinical effect observed, is likely to require hundreds of biomarker-enriched (and potentially thousands) of patients studied over relatively long time periods (months to years). Such efforts, as exemplified in recent AD therapeutic development efforts, will be resource intensive, the bulk of which expended at the latter stages of clinical evaluation (i.e., pivotal or registration studies). Therefore, the development of an α-syn-based therapeutic should progress in a step-wise manner in which advancement decisions are guided by the strength of the aggregate preclinical, non-clinical and clinical data.
Previous sections in this white paper have covered translational approaches, tools and measures currently available to assess the therapeutic potential of a given α-syn-directed drug candidate. It is imperative to continue to characterize and refine these tools and outcomes in a translational manner to optimally design initial clinical studies. The focus of this section is to discuss key early clinical development objectives and clinical trial elements for α-syn-based therapeutics, with specific emphasis on advancement to PoP and PoC. The challenges and issues faced in latter stages of clinical development (pivotal/registration trials) of an α-syn-based therapeutic (or any other potentially disease-modifying therapy for a neurodegenerative disease) (recently reviewed in [69]) will not be addressed herein.
What defines proof of principle and proof of concept for alpha-synuclein-based therapeutics?
PoP and PoC relate to the level of confidence one assigns to an available dataset at key decision points during development. In the absence of a uniformly accepted definition, the present discussion will emphasize that PoP is defined by data generated in humans demonstrating that a potential therapeutic can be administered safely at dosages (or, more accurately from a translational perspective, exposures) that lead to a measurable change in a pathological or pathophysiological endpoint assessed by physiological, biochemical, or imaging biomarker. Similarly, PoC is defined by data generated in humans that a potential therapeutic favorably impacts a clinical outcome, or a validated biomarker that may reliably predict clinical efficacy when studied in a larger, perhaps more diverse, patient population. For instance, lowering α-syn levels in a central or peripheral tissue compartment after short-term exposure (days to weeks) without seeing a beneficial clinical outcome may constitute PoP. On the other hand, demonstrating slowing of motoric decline after longer-term (likely months/years) exposure, preferably with a reduction in brain α-syn load via a PET tracer, may constitute PoC. When considered in these broad terms, the goals of any early clinical evaluation of an α-syn-based therapeutic should be to initially establish robust PoP as a foundation to support the latter demonstration of clinical PoC. As we have argued earlier, it is difficult to envision achievement of clinical PoC for an α-syn-based intervention without first demonstrating PoP.
Symptomatic effects versus disease modification
Clinical assessments for symptomatic outcomes should be included in all initial, short-duration treatment studies as it is a confounding factor that influences the design and interpretation of later, larger pivotal studies seeking to demonstrate disease modification. Given the difficulty in modeling symptomatic outcomes in α-syn-based animal models discussed above, uncertainty exists as to what clinical signs and symptoms of a given synucleinopathy may improve following exposure to an α-syn-modulating intervention (e.g., motor, cognitive, autonomic). Modalities like wearable sensors that capture broad and unbiased scopes of function appear particularly well-suited for use in these clinical studies. Demonstration that modulating α-syn produces improvements in a patient’s symptomatology or function, not resulting from a drug-drug interaction with co-incidentally administered symptomatic therapies, would constitute PoC.
Choosing the optimal patient population
The known clinical heterogeneity in PD and the variable motor and non-motor manifestations represent a challenge to optimal patient selection. Importantly, the ability to establish initial PoP (or clinical PoC) may depend heavily on the ability to select an experimental population characterized by a nearly uniform underlying biology/pathophysiology or other biological trait – even if such molecularly homogenous population remains clinically heterogeneous.
The ideal α-syn patient population carries point mutations, duplication, or triplication of the α-syn gene, SNCA [183, 184]. Unlike in sporadic PD, where the pathogenicity of α-syn is assumed, in these populations, the pathogenicity of α-syn is confirmed. Unfortunately, such patients are very rare and are difficult to effectively recruit into clinical trials. Lacking available SNCA-PD cohorts, one could argue that a “patient enrichment” strategy using biological and/or imaging endpoints to identify patients with high expression of pathological α-syn being targeted by a specific therapy, may improve the probability of success in establishing PoP.
One could envision designing a clinical PoP study in PD patients with evidence of synucleinopathy, where the primary goal is to establish that the drug-targeted pathologic α-syn levels are reduced after treatment. Potential trial subjects could be screened for high α-syn levels using analysis of serum, peripheral tissues, and CSF, and potentially via α-syn molecular imaging, once developed. In the absence of a means of identifying the presence of α-syn pathology in living patients, DAT imaging, discussed above, may be used to differentiate Parkinsonian patients with a higher risk of symptom progression [185]. The ability to extend the demonstration of PoP to clinical PoC in a given sub-population, and clinical PoC to the broadest population, will depend on measuring change in clinical status as a result of lowering a relevant α-syn species. As a subset of “responders” may be statistically drowned in an overall negative study, it will be important to anticipate the possibility that a clinical PoC can be confirmed only in a subset of the cohort. Understanding the biological differences between the responders and the rest of the cohort may help fine-tune patient selection for a subsequent phase of development. Implementation of such an approach will require identification of clinical, biomarker and/or genetic indicators associated with responder population for recruitment of patients in the next phase. The identification of such markers to enable personalization of disease-modifying therapy will require that all studies, both interventional and observational, collect harmonized clinical and biomarker measures, and that the field redoubles its efforts to encourage all subjects to provide biospecimens and consent to concurrent and future analyses.
Outcome measures – safety
Non-clinical safety studies are necessary to identify potential liabilities to be monitored in humans and to identify safe starting doses for first-in-human studies. Safety (bio)markers should be sought in non-clinical studies and are particularly useful if they can be used to reduce risk in a clinical program. At present, it is unknown if lowering levels of physiological (“normal”) α-syn species has any inherent liabilities in humans. Optimally, α-syn-based therapeutics should selectively target pathological forms and spare normal species. Several of the current α-syn-based approaches spare “normal” α-syn species and are specific for α-syn (versus β- and γ-synuclein). Because of our poor understanding of the normal function of the synucleins, no guidance can yet be made regarding “target-mediated” side effects of anti-α-syn therapies.
Outcome measures – proof of principle
The decision to initiate human studies in healthy subjects versus synucleinopathy patients will be specific to each therapeutic modality (small molecule, antibody, gene therapy, antisense oligonucleotide), based on a risk/benefit estimate as determined by non-clinical safety experiments. Therefore, we do not provide a specific “class” recommendation for α-syn-based therapies. It is anticipated that either starting first in human studies in a synucleinopathy patient population or advancing to such a population early in development will be advantageous. The advantages include early assessments of safety issues inherent to the target patient population and enabling assessments of physiologically-based pharmacokinetic/pharmacodynamic relationships that may rely on the presence of underlying α-syn pathophysiology.
Demonstrating a relationship between dose and TE in the brain is important where the TE measure could be specific to a drug candidate. A clear dose-response effect on TE, pharmacodynamic, and safety markers is key to interpretation of results of early stage clinical trials and to the initiation of pivotal trials. Taking advantage of expression of the target α-syn species in the CSF and peripheral biospecimens is an attractive approach to establishing measures for TE. Although extrapolation of exposures and TE from the peripheral to the central compartment has its challenges, evidence for peripheral TE may be highly informative, especially if a relationship is established in animal models and a valid physiologically-based pharmacokinetic/pharmacodynamic model can be built. Additionally, many existing CNS imaging and physiological assessment tools (structural MRI, DTI, fMRI, 2-deoxyglucose imaging, and electroencephalograms) have limited utility as direct TE markers but may be suitable as pharmacodynamic or PoP outcome measures.
Reduction of α-syn protein levels, comparing baseline levels to multiple post-exposure assessments, in biological fluids (serum and CSF), and potentially in accessible cells like peripheral blood mononuclear cells [186, 187] would be an informative outcome. In order for these modalities to be useful, they must have robust assay metrics (e.g., low test/retest variability). Establishing PoP by demonstrating reductions in α-syn expression in small, relatively short-duration clinical studies will be a critical step, and will help translate PoP into clinical efficacy through the establishment of clinical PoC.
Current challenges in translation proof of principle to proof of concept
In order to support investment in large-scale, long, and expensive pivotal clinical trials, the objective of a PoC trial should be to increase confidence that the previously demonstrated TE/PoP outcomes translate into a clinically meaningful outcome. For PD, such outcomes include assessment of motor function utilizing the Movement Disorders Society Unified Parkinson’s Disease Rating Scale (MDS-UPDRS). As such development programs advance, continued assessments of safety and tolerability remain critical.
Ideally, the population upon which PoP was demonstrated can also serve as the population to establish PoC and eventual approval. As discussed above, this may not always be the case, therefore the synucleinopathy patient population is one of the key variables to address in a PoC study. Whereas biological factors may weigh heavily in the choice of patient population to study for PoP, clinical characteristics are critically important in determining an optimal PoC study population. Clinical signs and symptoms differ between synucleionpathies, and inclusion of patients who manifest disease progression with specific clinical characteristics (e.g., predominantly motor versus cognitive versus peripheral/autonomic signs and symptoms) will dictate what clinical outcome measures should be included for use in these studies. For PoC studies, the primary endpoint does not necessarily have to be an approved endpoint, or even the planned pivotal study endpoint (such as MDS-UPDRS, see Table 4). In fact, ideal endpoints for PoC should be more sensitive, more precise, and correlate with or predict outcomes on the primary endpoint planned for pivotal trials (if the primary endpoints are not the same). Planned pivotal study outcome measures should be included in the PoC study, even if the study is underpowered to detect a statistically significant difference between groups. Modeling and simulation techniques should be used to estimate probability of success in future pivotal trials.
Table 4
Alternate outcomes to be considered for alpha-synuclein proof of concept studies
| Modality | Examples |
| Neuroimaging | DAT, VMAT2 or other tracers capable of visualizing structural or functional abnormalities in PD patients |
| Wearable sensors | More granularity and reliable assessments than clinical rating scales (do not need to correlate with or predict change in existent clinical rating scales)* |
| Non-motor endpoints (time to milestones) | Gait, cognition, dysautonomia* |
*If these measures correlate with or predict change on validated clinical rating scales they may be more valuable for PoC studies. DAT, dopamine transporter; PD, Parkinson’s disease; PoC, proof of concept; VMAT2, vesicular monoamine transporter 2.
There are, however, several challenges inherent in this approach. If subjects are taking medications for control of disease symptoms, one should carefully consider their effect on chosen outcome measures. One must also consider the current state of validation or available natural history data on novel outcomes (e.g., wearables, new imaging methods) when incorporating them into studies, and must prepare a background dossier to support any go-forward decisions based thereon. There are also several analysis issues. Currently, there is a lack of clarity on the necessary effect size of change in MDS-UPDRS and other outcome scales, as they have not yet been used in a sufficient number of completed trials. We do not yet understand whether existing scales are suitable to be a global measure. Novel composite scales may need to be developed to combine non-motor and cognitive aspects into a single global measure of disease progression in PD. Another outstanding question is whether our statistical methods for declaring PoC are antiquated. Do we really need results of a “delayed start” study to declare PoC, or can modern methods such as modeling and simulation approaches or adaptive and Bayesian study designs maximize our ability to interpret results of studies in relatively small populations, since PoC trials are often underpowered for traditional endpoints? An example of the utility of an adaptive dose randomization strategy with Bayesian statistical approach was reported recently for an AD trial [149]. When coupled with more sensitive clinical outcome measures, if available, it may be possible to reduce the sample size while evaluating PoC at a range of doses.
Many factors influence the choice of clinical outcomes including ease of measurement, metrics of the clinical scales used to assess a particular sign or symptom, and sensitivity to detect change. Progression rate-specific clinical signs and symptoms differ, not only between synucleinopathies but also within given disease state at specific stages of disease (progression can be non-linear). Existing data from longitudinal clinical cohorts for PD such as DATATOP [188], PreCEPT/PostCEPT [189], PPMI [190] and other natural history studies, as well as studies in other synucleinopathies such as MSA and DLB, can be used to model disease progression to optimize trial design.
Although unlikely to be of current utility in PoC study design, the use of prodromal patient populations is likely to become more relevant over time. There are unique challenges in stratification of a de novo or prodromal populations. Strategies for how to handle medication use, baseline disease severity, and even accounting for regional practice differences must be devised to minimize interpatient variability. When designing studies to be carried out in prodromal or pre-motor populations, characteristics of the at-risk population will need to be carefully defined. For example, should the initial population be defined by individual symptoms (e.g., RBD) versus symptom complexes (multi-modal approach); obviously genetics and other risk factors will also need to be taken into account and could further help enrich the at-risk population. For the field to be ready to undertake trials in prodromal patients, we will need to improve our understanding of prodromal symptoms and rate of progression to meet existing disease diagnostic criteria in order to reduce the number of patients to be screened for these trials. Conducting a longitudinal, non-interventional clinical study in an enriched prodromal population would generate critical data to support the use of such populations in drug development efforts.
Finally, application of available clinical, imaging, and physiological tools to enrich a given synucleinopathy population will be critical to achieving success in PoC. Stratifying the target synucleinopathy population during randomization or analysis for key clinical variables such as concomitant medication usage, baseline disease severity, genetic factors, and clinical operational variability (e.g., geographic, standard of care) will increase the probability of detecting a clinical effect of the therapeutic intervention under study. In the absence of an α-syn PET ligand, studies are currently relying on DAT SPECT scans to serve this purpose (see above).
Given the breadth of α-syn-based therapeutic modalities under study and the complexity and variability of the clinical synucleinopathy diseases, PoC trial designs will need to be optimized for each specific therapeutic under study. Sponsors are encouraged to share as much non-competitive α-syn therapeutic PoC trial designs and data as possible, perhaps within clinical trial consortia or harmonized databases such as the Alzheimer’s Disease Neuroimaging Initiative (ADNI) [191] and the Critical Path for Parkinson’s Initiative [192]. Such collaborative efforts will be critically important to achieve the shared goal of lowering disease burden and improving the lives of existing and future patients suffering from synuclein-based neurodegenerative diseases.
General perspective
That α-syn is the major drug development target for the treatment of human synucleinopathies, and PD in particular, is supported by the depth and mechanistic breadth of the approaches currently being pursued (Table 1). Multiple therapeutics have recently entered clinical trials, and critical human data that will inform all α-syn-based therapeutic development programs is on the horizon. Although these efforts face many profound challenges, including the lack of key tools such as an α-syn-based imaging agent and the inherent difficulty of demonstrating clinical efficacy in slowly progressive neurodegenerative diseases, we remain optimistic that meaningful strides toward the ultimate identification and approval of α-syn-based disease-modifying therapeutics will be made in the near future.
CONFLICT OF INTEREST
KMM is a Senior Advisor to The Michael J. Fox Foundation for Parkinson’s Research. She serves on the Scientific Advisory Board and/or provides consulting services to AbbVie, Acumen Pharmaceuticals, Epiodyne, Lysosomal Therapeutics, Neuroinitiative, Pfizer, Sinopia Biosciences and private equity investors. She also serves on the NCATS Advisory Council and CAN Review Board, NIA’s Division of Neuroscience Review Committee, and Wellcome Trust Review Board.
JC is an employee and shareholder of Biogen.
PB has received commercial support as a consultant from Renovo Neural, Inc., Cellular Dynamics International, Axial Biotherapeutics, Roche, Teva Inc, Lundbeck A/S, NeuroDerm, AbbVie, ClearView Healthcare, FCB Health, IOS Press Partners and Capital Technologies, Inc. He is conducting sponsored research on behalf of Roche and Lundbeck A/S. He has ownership interests in Acousort AB.
KDD is an employee of The Michael J. Fox Foundation for Parkinson’s Research and has no conflicts of interest to report.
JE is an employee of The Michael J. Fox Foundation for Parkinson’s Research and has no conflicts of interest to report.
AJE has received grant support from the NIH, Great Lakes Neurotechnologies and The Michael J. Fox Foundation for Parkinson’s Research; personal compensation as a consultant/scientific advisory board member for Abbvie, TEVA, Impax, Acadia, Acorda, Cynapsus/Sunovion, Lundbeck, and USWorldMeds; publishing royalties from Lippincott Williams & Wilkins, Cambridge University Press, and Springer; and honoraria from Abbvie, UCB, USWorldMeds, Lundbeck, Acadia, the American Academy of Neurology, and the Movement Disorders Society.
SJH is an employee The Michael J. Fox Foundation for Parkinson’s Research and has no conflicts of interest to report.
MJ has no conflicts of interest to report.
JL is a full time employee of Eisai Inc, USA. He owns shares in AstraZeneca and Merck Co.
WM receives or received funding from the European Union, The Michael J. Fox Foundation for Parkinson’s Research, Robert Bosch Foundation, Neuroalliance, Lundbeck and Janssen, and holds part of a patent for the assessment of dyskinesias (German patent office, 102015220741.2). He received speaker honoraria from GlaxoSmithKline, Abbvie, UCB, Licher MT and Rölke Pharma, and was invited to Advisory Boards of Market Access & Pricing Strategy GmbH and Abbvie.
LM is an employee The Michael J. Fox Foundation for Parkinson’s Research and has no conflicts of interest to report.
ANR is an employee of The Michael J. Fox Foundation for Parkinson’s Research and has no conflicts of interest to report.
AJS is an employee of the University of British Columbia and is supported by a Canada Research Chair and by research grants from the Michael J. Fox Foundation and Weston Brain Institute. He is Deputy Editor of Movement Disorders. He has received consulting and speaking honoraria from AbbVie and is an advisor for Cellular Dynamics International and on a Data Safety Monitoring Board for Voyager Inc.
DMW is an Advisor to The Michael J. Fox Foundation for Parkinson’s Research.
REFERENCES
[1] | Kowal SL , Dall TM , Chakrabarti R , Storm MV , Jain A ((2013) ) The current and projected economic burden of Parkinson’s disease in the United States. Mov Disord 28: , 311–318. |
[2] | Dorsey ER , Bloem BR ((2018) ) The Parkinson pandemic-a call to action. JAMA Neurol 75: , 9–10. |
[3] | Marras C , Beck JC , Bower JH , Roberts E , Ritz B , Ross GW , Abbott RD , Savica R , Van Den Eeden SK , Willis AW , Tanner CM , Parkinson’s Foundation P4 Group ((2018) ) Prevalence of Parkinson’s disease across North America. NPJ Parkinsons Dis 4: , 21. |
[4] | Cookson MR , Hardy J , Lewis PA ((2008) ) Genetic neu-ropathology of Parkinson’s disease. Int J Clin Exp Pathol 1: , 217–231. |
[5] | Nalls MA , Pankratz N , Lill CM , Do CB , Hernandez DG , Saad M , DeStefano AL , Kara E , Bras J , Sharma M , Schulte C , Keller MF , Arepalli S , Letson C , Edsall C , Stefansson H , Liu X , Pliner H , Lee JH , Cheng R , International Parkinson’s Disease Genomics Consortium (IPDGC), Parkinson’s Study Group (PSG) Parkinson’s Research: The Organized GENetics Initiative (PROGENI), 23andMe, Gene PD, NeuroGenetics Research Consortium (NGRC), Hussman Institute of Human Genomics (HIHG), The Ashkenazi Jewish Dataset Investigator, Cohorts for Health and Aging Research ©in Genetic Epidemiology (CHARGE), North American Brain Expression Consortium (NABEC), United Kingdom Brain Expression Consortium (UKBEC), Greek Parkinson’s Disease Consortium, Alzheimer Genetic Analysis Group, Ikram MA , Ioannidis JP , Hadjigeorgiou GM , Bis JC , Martinez M , Perlmutter JS , Goate A , Marder K , Fiske B , Sutherland M , Xiromerisiou G , Myers RH , Clark LN , Stefansson K , Hardy JA , Heutink P , Chen H , Wood NW , Houlden H , Payami H , Brice A , Scott WK , Gasser T , Bertram L , Eriksson N , Foroud T , Singleton AB ((2014) ) Large-scale meta-analysis of genome-wide association data identifies six new risk loci for Parkinson’s disease. Nat Genet 46: , 989–993. |
[6] | Devine MJ , Gwinn K , Singleton A , Hardy J ((2011) ) Parkinson’s disease and alpha-synuclein expression. Mov Disord 26: , 2160–2168. |
[7] | Soldner F , Stelzer Y , Shivalila CS , Abraham BJ , Latourelle JC , Barrasa MI , Goldmann J , Myers RH , Young RA , Jaenisch R ((2016) ) Parkinson-associated risk variant in distal enhancer of alpha-synuclein modulates target gene expression. Nature 533: , 95–99. |
[8] | Spillantini MG , Crowther RA , Jakes R , Hasegawa M , Goedert M ((1998) ) alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. Proc Natl Acad Sci USA 95: , 6469–6473. |
[9] | Toulorge D , Schapira AH , Hajj R ((2016) ) Molecular changes in the postmortem parkinsonian brain. J Neurochem 139: (Suppl 1), 27–58. |
[10] | Sevigny J , Chiao P , Bussiere T , Weinreb PH , Williams L , Maier M , Dunstan R , Salloway S , Chen T , Ling Y , O'Gorman J , Qian F , Arastu M , Li M , Chollate S , Bren-nan MS , Quintero-Monzon O , Scannevin RH , Arnold HM , Engber T , Rhodes K , Ferrero J , Hang Y , Mikul-skis A , Grimm J , Hock C , Nitsch RM , Sandrock A ((2016) ) The antibody aducanumab reduces Abeta plaques in Alzheimer’s disease. Nature 537: , 50–56. |
[11] | Amanatkar HR , Papagiannopoulos B , Grossberg GT ((2017) ) Analysis of recent failures of disease modifying therapies in Alzheimer’s disease suggesting a new methodology for future studies. Expert Rev Neurother 17: , 7–16. |
[12] | Karran E , Hardy J ((2014) ) A critique of the drug discovery and phase 3 clinical programs targeting the amyloid hypothesis for Alzheimer disease. Ann Neurol 76: , 185–205. |
[13] | Toyn JH , Ahlijanian MK ((2014) ) Interpreting Alzheimer’s disease clinical trials in light of the effects on amyloid-beta. Alzheimers Res Ther 6: , 14. |
[14] | Herrup K ((2015) ) The case for rejecting the amyloid cascade hypothesis. Nat Neurosci 18: , 794–799. |
[15] | Ostrowitzki S , Lasser RA , Dorflinger E , Scheltens P , Barkhof F , Nikolcheva T , Ashford E , Retout S , Hofmann C , Delmar P , Klein G , Andjelkovic M , Dubois B , Boada M , Blennow K , Santarelli L , Fontoura P , SCarlet RoAD Investigators ((2017) ) A phase III randomized trial of gantenerumab in prodromal Alzheimer’s disease. Alzheimers Res Ther 9: , 95. |
[16] | Egan MF , Kost J , Tariot PN , Aisen PS , Cummings JL , Vellas B , Sur C , Mukai Y , Voss T , Furtek C , Mahoney E , Harper Mozley L , Vandenberghe R , Mo Y , Michelson D ((2018) ) Randomized trial of verubecestat for mild-to-moderate Alzheimer’s disease. N Engl J Med 378: , 16911703. |
[17] | Lang AE , Espay AJ ((2018) ) Disease modification in Parkinson’s disease: Current approaches, challenges, and future considerations. Mov Disord 33: , 660–677. |
[18] | Volpicelli-Daley LA , Kirik D , Stoyka LE , Standaert DG , Harms AS ((2016) ) How can rAAV-alpha-synuclein and the fibril alpha-synuclein models advance our understanding of Parkinson’s disease? J Neurochem 139 Suppl 1: , 131–155. |
[19] | Recasens A , Ulusoy A , Kahle PJ , Di Monte DA , Dehay B ((2018) ) In vivo models of alpha-synuclein transmission and propagation. Cell Tissue Res 373: , 183–193. |
[20] | Dehay B , Fernagut PO ((2016) ) Alpha-synuclein-based models of Parkinson’s disease. Rev Neurol (Paris) 172: , 371–378. |
[21] | Dehay B , Vila M , Bezard E , Brundin P , Kordower JH ((2016) ) Alpha-synuclein propagation: New insights from animal models. Mov Disord 31: , 161–168. |
[22] | Rey NL , George S , Brundin P ((2016) ) Review: Spreading the word: Precise animal models and validated methods are vital when evaluating prion-like behaviour of alpha-synuclein. Neuropathol Appl Neurobiol 42: , 51–76. |
[23] | Koprich JB , Kalia LV , Brotchie JM ((2017) ) Animal models of alpha-synucleinopathy for Parkinson disease drug development. Nat Rev Neurosci 18: , 515–529. |
[24] | Jiang P , Dickson DW ((2018) ) Parkinson’s disease: Experimental models and reality. Acta Neuropathol 135: , 13–32. |
[25] | Melki R ((2015) ) Role of different alpha-synuclein strains in synucleinopathies, similarities with other neurodegenerative diseases. J Parkinsons Dis 5: , 217–227. |
[26] | Peelaerts W , Bousset L , Van der Perren A , Moskalyuk A , Pulizzi R , Giugliano M , Van den Haute C , Melki R , Baekelandt V ((2015) ) alpha-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 522: , 340–344. |
[27] | Peng C , Gathagan RJ , Covell DJ , Medellin C , Stieber A , Robinson JL , Zhang B , Pitkin RM , Olufemi MF , Luk KC , Trojanowski JQ , Lee VM ((2018) ) Cellular milieu imparts distinct pathological alpha-synuclein strains in alpha-synucleinopathies. Nature 557: , 558–563. |
[28] | Peng C , Gathagan RJ , Lee VM ((2018) ) Distinct alpha-Synuclein strains and implications for heterogeneity among alpha-Synucleinopathies. Neurobiol Dis 109: , 209–218. |
[29] | Kahle PJ ((2008) ) alpha-Synucleinopathy models and human neuropathology: Similarities and differences. Acta Neuropathol 115: , 87–95. |
[30] | Hatami A , Chesselet MF ((2015) ) Transgenic rodent models to study alpha-synuclein pathogenesis, with a focus on cognitive deficits. Curr Top Behav Neurosci 22: , 303–330. |
[31] | McDowell K , Chesselet MF ((2012) ) Animal models of the non-motor features of Parkinson’s disease. Neurobiol Dis 46: , 597–606. |
[32] | Chesselet MF ((2008) ) In vivo alpha-synuclein overexpression in rodents: A useful model of Parkinson’s disease? Exp Neurol 209: , 22–27. |
[33] | Fernagut PO , Chesselet MF ((2004) ) Alpha-synuclein and transgenic mouse models. Neurobiol Dis 17: , 123–130. |
[34] | Decressac M , Mattsson B , Lundblad M , Weikop P , Bjorklund A ((2012) ) Progressive neurodegenerative and behavioural changes induced by AAV-mediated overexpression of alpha-synuclein in midbrain dopamine neurons. Neurobiol Dis 45: , 939–953. |
[35] | Oliveras-Salva M , Van der Perren A , Casadei N , Stroobants S , Nuber S , D'Hooge R , Van den Haute C , Baekelandt V ((2013) ) rAAV2/7 vector-mediated overexpression of alpha-synuclein in mouse substantia nigra induces protein aggregation and progressive dose-dependent neurodegeneration. Mol Neurodegener 8: , 44. |
[36] | Chung CY , Koprich JB , Siddiqi H , Isacson O ((2009) ) Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J Neurosci 29: , 3365–3373. |
[37] | Kirik D , Rosenblad C , Burger C , Lundberg C , Johansen TE , Muzyczka N , Mandel RJ , Bjorklund A ((2002) ) Parkinson-like neurodegeneration induced by targeted overexpression of alpha-synuclein in the nigrostriatal system. J Neurosci 22: , 2780–2791. |
[38] | St Martin JL , Klucken J , Outeiro TF , Nguyen P , Keller-McGandy C , Cantuti-Castelvetri I , Grammatopoulos TN , Standaert DG , Hyman BT , McLean PJ ((2007) ) Dopaminergic neuron loss and up-regulation of chaperone protein mRNA induced by targeted over-expression of alpha-synuclein in mouse substantia nigra. J Neurochem 100: , 1449–1457. |
[39] | Mandel RJ , Marmion DJ , Kirik D , Chu Y , Heindel C , McCown T , Gray SJ , Kordower JH ((2017) ) Novel oligodendroglial alpha synuclein viral vector models of multiple system atrophy: Studies in rodents and nonhuman primates. Acta Neuropathol Commun 5: , 47. |
[40] | Bassil F , Guerin PA , Dutheil N , Li Q , Klugmann M , Meissner WG , Bezard E , Fernagut PO ((2017) ) Viral-mediated oligodendroglial alpha-synuclein expression models multiple system atrophy. Mov Disord 32: , 1230–1239. |
[41] | Rey NL , Petit GH , Bousset L , Melki R , Brundin P ((2013) ) Transfer of human alpha-synuclein from the olfactory bulb to interconnected brain regions in mice. Acta Neuropathol 126: , 555–573. |
[42] | Luk KC , Covell DJ , Kehm VM , Zhang B , Song IY , Byrne MD , Pitkin RM , Decker SC , Trojanowski JQ , Lee VM ((2016) ) Molecular and biological compatibility with host alpha-synuclein influences fibril pathogenicity. Cell Rep 16: , 3373–3387. |
[43] | Polinski NK , Volpicelli-Daley LA , Sortwell CE , Luk KC , Cremades N , Gottler LM , Froula J , Duffy MF , Lee VMY , Martinez TN , Dave KD ((2018) ) Best practices for generating and using alpha-synuclein pre-formed fibrils to model Parkinson’s disease in rodents. J Parkinsons Dis 8: , 303–322. |
[44] | Luk KC , Kehm V , Carroll J , Zhang B , O'Brien P , Tro-janowski JQ , Lee VM ((2012) ) Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 338: , 949–953. |
[45] | Luk KC , Kehm VM , Zhang B , O'Brien P , Trojanowski JQ , Lee VM ((2012) ) Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med 209: , 975–986. |
[46] | Thakur P , Breger LS , Lundblad M , Wan OW , Mattsson B , Luk KC , Lee VMY , Trojanowski JQ , Bjorklund A ((2017) ) Modeling Parkinson’s disease pathology by combination of fibril seeds and alpha-synuclein overexpression in the rat brain. Proc Natl Acad Sci USA 114: , E8284–E8293. |
[47] | Braak H , Del Tredici K , Rub U , de Vos RA , Jansen Steur EN , Braak E ((2003) ) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 24: , 197–211. |
[48] | Paumier KL , Luk KC , Manfredsson FP , Kanaan NM , Lip-ton JW , Collier TJ , Steece-Collier K , Kemp CJ , Celano S , Schulz E , Sandoval IM , Fleming S , Dirr E , Polinski NK , Trojanowski JQ , Lee VM , Sortwell CE ((2015) ) Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol Dis 82: , 185–199. |
[49] | Rey NL , George S , Steiner JA , Madaj Z , Luk KC , Trojanowski JQ , Lee VM , Brundin P ((2018) ) Spread of aggregates after olfactory bulb injection of alpha-synuclein fibrils is associated with early neuronal loss and is reduced long term. Acta Neuropathol 135: , 65–83. |
[50] | Rey NL , Steiner JA , Maroof N , Luk KC , Madaj Z , Trojanowski JQ , Lee VM , Brundin P ((2016) ) Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J Exp Med 213: , 1759–1778. |
[51] | Sacino AN , Brooks M , Thomas MA , McKinney AB , Lee S , Regenhardt RW , McGarvey NH , Ayers JI , Notterpek L , Borchelt DR , Golde TE , Giasson BI ((2014) ) Intramuscular injection of alpha-synuclein induces CNS alpha-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc Natl Acad Sci USA 111: , 10732–10737. |
[52] | Holmqvist S , Chutna O , Bousset L , Aldrin-Kirk P , Li W , Bjorklund T , Wang ZY , Roybon L , Melki R , Li JY ((2014) ) Direct evidence of Parkinson pathology spread from the gastrointestinal tract to the brain in rats. Acta Neuropathol 128: , 805–820. |
[53] | Manfredsson FP , Luk KC , Benskey MJ , Gezer A , Garcia J , Kuhn NC , Sandoval IM , Patterson JR , O'Mara A , Yonkers R , Kordower JH ((2018) ) Induction of alpha-synuclein pathology in the enteric nervous system of the rat and non-human primate results in gastrointestinal dysmotility and transient CNS pathology. Neurobiol Dis 112: , 106–118. |
[54] | Uemura N , Yagi H , Uemura MT , Hatanaka Y , Yamakado H , Takahashi R ((2018) ) Inoculation of alpha-synuclein preformed fibrils into the mouse gastrointestinal tract induces Lewy body-like aggregates in the brainstem via the vagus nerve. Mol Neurodegener 13: , 21. |
[55] | Shimozawa A , Ono M , Takahara D , Tarutani A , Imura S , Masuda-Suzukake M , Higuchi M , Yanai K , Hisanaga SI , Hasegawa M ((2017) ) Propagation of pathological alpha-synuclein in marmoset brain. Acta Neuropathol Commun 5: , 12. |
[56] | Kirik D , Annett LE , Burger C , Muzyczka N , Mandel RJ , Bjorklund A ((2003) ) Nigrostriatal alpha-synucleinopathy induced by viral vector-mediated overexpression of human alpha-synuclein: A new primate model of Parkinson’s disease. Proc Natl Acad Sci USA 100: , 2884–2889. |
[57] | Eslamboli A , Romero-Ramos M , Burger C , Bjorklund T , Muzyczka N , Mandel RJ , Baker H , Ridley RM , Kirik D ((2007) ) Long-term consequences of human alpha-synuclein overexpression in the primate ventral midbrain. Brain 130: , 799–815. |
[58] | Oueslati A ((2016) ) Implication of alpha-synuclein phosphorylation at S129 in synucleinopathies: What have we learned in the last decade? J Parkinsons Dis 6: , 39–51. |
[59] | Lundblad M , Decressac M , Mattsson B , Bjorklund A ((2012) ) Impaired neurotransmission caused by overexpression of alpha-synuclein in nigral dopamine neurons. Proc Natl Acad Sci USA 109: , 3213–3219. |
[60] | Theodore S , Cao S , McLean PJ , Standaert DG ((2008) ) Targeted overexpression of human alpha-synuclein triggers microglial activation and an adaptive immune response in a mouse model of Parkinson disease. J Neuropathol Exp Neurol 67: , 1149–1158. |
[61] | Sacks LV , Shamsuddin HH , Yasinskaya YI , Bouri K , Lan-thier ML , Sherman RE ((2014) ) Scientific and regulatory reasons for delay and denial of FDA approval of initial applications for new drugs, 2000-2012. JAMA 311: , 378–384. |
[62] | Paterson RW , Slattery CF , Poole T , Nicholas JM , Magdali-nou NK , Toombs J , Chapman MD , Lunn MP , Heslegrave AJ , Foiani MS , Weston PSJ , Keshavan A , Rohrer JD , Rossor MN , Warren JD , Mummery CJ , Blennow K , Fox NC , Zetterberg H , Schott JM ((2018) ) Cerebrospinal fluid in the differential diagnosis of Alzheimer’s disease: Clinical utility of an extended panel of biomarkers in a specialist cognitive clinic. Alzheimers Res Ther 10: , 32. |
[63] | Keshavan A , Heslegrave A , Zetterberg H , Schott JM ((2017) ) Blood biomarkers for Alzheimer’s disease: Much promise, cautious progress. Mol Diagn Ther 21: , 13–22. |
[64] | Lue LF , Guerra A , Walker DG ((2017) ) Amyloid beta and tau as Alzheimer’s disease blood biomarkers: Promise from new technologies. Neurol Ther 6: , 25–36. |
[65] | Goldman JG , Andrews H , Amara A , Naito A , Alcalay RN , Shaw LM , Taylor P , Xie T , Tuite P , Henchcliffe C , Hogarth P , Frank S , Saint-Hilaire MH , Frasier M , Arnedo V , Reimer AN , Sutherland M , Swanson-Fischer C , Gwinn K , The Fox Investigation of New Biomarker Discovery, Kang UJ ((2018) ) Cerebrospinal fluid, plasma, and saliva in the BioFIND study: Relationships among biomarkers and Parkinson’s disease Features. Mov Disord 33: , 282–288. |
[66] | Barbour R , Kling K , Anderson JP , Banducci K , Cole T , Diep L , Fox M , Goldstein JM , Soriano F , Seubert P , Chilcote TJ ((2008) ) Red blood cells are the major source of alpha-synuclein in blood. Neurodegener Dis 5: , 55–59. |
[67] | Brys M , Ellenbogen A , Fanning L , Penner N , Yang M , Welch M , Koenig E , David E , Fox T , Makh S , Aldred J , Goodman I , Graham D , Weihofen A , Cedarbaum J ((2018) ) Randomized, double-blind, placebo-controlled, single ascending dose study of anti-alpha-synuclein antibody BIIB054 in patients with Parkinson’s disease. The American Academy of Neurology (AAN) 70th Annual Meeting, Los Angeles, California. |
[68] | Jankovic J , Goodman I , Safirstein B , Marmon TK , Schenk DB , Koller M , Zago W , Ness DK , Griffith SG , Grundman M , Soto J , Ostrowitzki S , Boess FG , Martin-Facklam M , Quinn JF , Isaacson SH , Omidvar O , Ellenbogen A , Kinney GG ((2018) ) Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-alpha-synuclein monoclonal antibody, in patients with Parkinson disease: A randomized clinical trial. JAMA Neurol. doi: 10.1001/jamaneurol.2018.1487 |
[69] | Brundin P , Dave KD , Kordower JH ((2017) ) Therapeutic approaches to target alpha-synuclein pathology. Exp Neurol 298: , 225–235. |
[70] | Espay AJ , Schwarzschild MA , Tanner CM , Fernandez HH , Simon DK , Leverenz JB , Merola A , Chen-Plotkin A , Brundin P , Kauffman MA , Erro R , Kieburtz K , Woo D , Macklin EA , Standaert DG , Lang AE ((2017) ) Biomarker-driven phenotyping in Parkinson’s disease: A translational missing link in disease-modifying clinical trials. Mov Disord 32: , 319–324. |
[71] | Kang JH , Mollenhauer B , Coffey CS , Toledo JB , Weintraub D , Galasko DR , Irwin DJ , Van Deerlin V , Chen-Plotkin AS , Caspell-Garcia C , Waligorska T , Taylor P , Shah N , Pan S , Zero P , Frasier M , Marek K , Kieburtz K , Jennings D , Tanner CM , Simuni T , Singleton A , Toga AW , Chowdhury S , Trojanowski JQ , Shaw LM , Parkinson’s Progression Marker Initiative ((2016) ) CSF biomarkers associated with disease heterogeneity in early Parkinson’s disease: The Parkinson’s Progression Markers Initiative study. Acta Neuropathol 131: , 935–949. |
[72] | Majbour NK , Vaikath NN , van Dijk KD , Ardah MT , Varghese S , Vesterager LB , Montezinho LP , Poole S , Safieh-Garabedian B , Tokuda T , Teunissen CE , Berendse HW , van de Berg WD , El-Agnaf OM ((2016) ) Oligomeric and phosphorylated alpha-synuclein as potential CSF biomarkers for Parkinson’s disease. Mol Neurodegener 11: , 7. |
[73] | Kruse N , El-Agnaf OM , Mollenhauer B ((2017) ) Validation of electrochemiluminescence assays for highly sensitive and reproducible quantification of alpha-synuclein in cerebrospinal fluid. Bioanalysis 9: , 621–630. |
[74] | Hong Z , Shi M , Chung KA , Quinn JF , Peskind ER , Galasko D , Jankovic J , Zabetian CP , Leverenz JB , Baird G , Montine TJ , Hancock AM , Hwang H , Pan C , Bradner J , Kang UJ , Jensen PH , Zhang J ((2010) ) DJ-1 and alpha-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain 133: , 713–726. |
[75] | Mollenhauer B , Batrla R , El-Agnaf O , Galasko DR , Lashuel HA , Merchant KM , Shaw LM , Selkoe DJ , Umek R , Vanderstichele H , Zetterberg H , Zhang J , Caspell-Garcia C , Coffey C , Hutten SJ , Frasier M , Taylor P ((2017) ) A user's guide for alpha-synuclein biomarker studies in biological fluids: Perianalytical considerations. Mov Dis-ord 32: , 1117–1130. |
[76] | Simonsen AH , Kuiperij B , El-Agnaf OM , Engelborghs S , Herukka SK , Parnetti L , Rektorova I , Vanmechelen E , Kapaki E , Verbeek M , Mollenhauer B ((2016) ) The utility of alpha-synuclein as biofluid marker in neurodegenerative diseases: A systematic review of the literature. Biomark Med 10: , 19–34. |
[77] | Stewart T , Sossi V , Aasly JO , Wszolek ZK , Uitti RJ , Hasegawa K , Yokoyama T , Zabetian CP , Leverenz JB , Stoessl AJ , Wang Y , Ginghina C , Liu C , Cain KC , Auinger P , Kang UJ , Jensen PH , Shi M , Zhang J ((2015) ) Phosphorylated alpha-synuclein in Parkinson’s disease: Correlation depends on disease severity. Acta Neuropathol Commun 3: ,7. |
[78] | Abd-Elhadi S , Honig A , Simhi-Haham D , Schechter M , Linetsky E , Ben-Hur T , Sharon R ((2015) ) Total and proteinase K-resistant alpha-synuclein levels in erythrocytes, determined by their ability to bind phos-pholipids, associate with Parkinson’s disease. Sci Rep 5: , 11120. |
[79] | Shahnawaz M , Tokuda T , Waragai M , Mendez N , Ishii R , Trenkwalder C , Mollenhauer B , Soto C ((2017) ) Development of a biochemical diagnosis of Parkinson disease by detection of alpha-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol 74: , 163–172. |
[80] | Fairfoul G , McGuire LI , Pal S , Ironside JW , Neumann J , Christie S , Joachim C , Esiri M , Evetts SG , Rolinski M , Baig F , Ruffmann C , Wade-Martins R , Hu MT , Parkkinen L , Green AJ ((2016) ) Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol 3: , 812–818. |
[81] | Bateman RJ , Munsell LY , Morris JC , Swarm R , Yarasheski KE , Holtzman DM ((2006) ) Human amyloid-beta synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med 12: , 856–861. |
[82] | Lee JM , Derkinderen P , Kordower JH , Freeman R , Munoz DG , Kremer T , Zago W , Hutten SJ , Adler CH , Serrano GE , Beach TG ((2017) ) The search for a peripheral biopsy indicator of alpha-synuclein pathology for Parkinson disease. J Neuropathol Exp Neurol 76: , 2–15. |
[83] | Beach TG , Adler CH , Serrano G , Sue LI , Walker DG , Dugger BN , Shill HA , Driver-Dunckley E , Caviness JN , Intorcia A , Filon J , Scott S , Garcia A , Hoffman B , Belden CM , Davis KJ , Sabbagh MN , Arizona Parkinson’s Disease Consortium ((2016) ) Prevalence of submandibular gland synucleinopathy in Parkinson’s disease, dementia with Lewy bodies and other Lewy body disorders. J Parkinsons Dis 6: , 153–163. |
[84] | Visanji NP , Mollenhauer B , Beach TG , Adler CH , Coffey CS , Kopil CM , Dave KD , Foroud T , Chahine L , Jennings D , Systemic Synuclein Sampling Study ((2017) ) The Systemic Synuclein Sampling Study: Toward a biomarker for Parkinson’s disease. Biomark Med 11: , 359–368. |
[85] | Postuma RB , Berg D ((2016) ) Advances in markers of prodromal Parkinson disease. Nat Rev Neurol 12: , 622–634. |
[86] | Siepmann T , Pinter A , Buchmann SJ , Stibal L , Arndt M , Kubasch AS , Kubasch ML , Penzlin AI , Frenz E , Zago W , Horvath T , Szatmari S, Jr. , Bereczki D , Takats A , Ziemssen T , Lipp A , Freeman R , Reichmann H , Barlinn K , Illigens BM ((2017) ) Cutaneous Autonomic Pilomotor Testing to Unveil the Role of Neuropathy Progression in Early Parkinson’s Disease (CAPTURE PD): Protocol for a multicenter study. Front Neurol 8: , 212. |
[87] | Patterson SG , Bayer CW , Hendry RJ , Sellers N , Lee KS , Vidakovic B , Mizaikoff B , Gabram-Mendola SG ((2011) ) Breath analysis by mass spectrometry: A new tool for breast cancer detection? Am Surg 77: , 747–751. |
[88] | Gitchel GT , Wetzel PA , Baron MS ((2012) ) Pervasive ocular tremor in patients with Parkinson disease. Arch Neurol 69: , 1011–1017. |
[89] | Petropoulos IN , Kamran S , Li Y , Khan A , Ponirakis G , Akhtar N , Deleu D , Shuaib A , Malik RA ((2017) ) Corneal confocal microscopy: An imaging endpoint for axonal degeneration in multiple sclerosis. Invest Ophthalmol Vis Sci 58: ,3677–3681. |
[90] | Bandini A , Orlandi S , Escalante HJ , Giovannelli F , Cin-cotta M , Reyes-Garcia CA , Vanni P , Zaccara G , Manfredi C ((2017) ) Analysis of facial expressions in Parkinson’s disease through video-based automatic methods. J Neurosci Methods 281: , 7–20. |
[91] | Hlavnicka J , Cmejla R , Tykalova T , Sonka K , Ruzicka E , Rusz J ((2017) ) Automated analysis of connected speech reveals early biomarkers of Parkinson’s disease in patients with rapid eye movement sleep behaviour disorder. Sci Rep 7: , 12. |
[92] | Whitwell JL , Hoglinger GU , Antonini A , Bordelon Y , Boxer AL , Colosimo C , van Eimeren T , Golbe LI , Kas-subek J , Kurz C , Litvan I , Pantelyat A , Rabinovici G , Respondek G , Rominger A , Rowe JB , Stamelou M , Josephs KA , Movement Disorder Society-endorsed PSP Study Group ((2017) ) Radiological biomarkers for diagnosis in PSP: Where are we and where do we need to be? Mov Disord 32: , 955–971. |
[93] | Lewis MM , Du G , Lee EY , Nasralah Z , Sterlin NW , Zhang L , Wagner D , Kong L , Tröster AI , Styner M , Eslinger PJ , Mailman RB , Huang X ((2016) ) The pattern of gray matter atrophy in Parkinson’s disease differs in cortical and subcortical regions. J Neurol 263: , 68–75. |
[94] | Mak E , Su L , Williams GB , Firbank MJ , Lawson RA , Yarnall AJ , Duncan GW , Owen AM , Khoo TK , Brooks DJ , Rowe JB , Barker RA , Burn DJ , O'Brien JT ((2015) ) Baseline and longitudinal grey matter changes in newly diagnosed Parkinson’s disease: ICICLE-PD study. Brain 138: , 2974–2986. |
[95] | Zeighami Y , Ulla M , Iturria-Medina Y , Dadar M , Zhang Y , Larcher KM , Fonov V , Evans AC , Collins DL , Dagher A ((2015) ) Network structure of brain atrophy in de novo Parkinson’s disease. Elife 4: , e08440. |
[96] | Martin WR , Wieler M , Gee M ((2008) ) Midbrain iron content in early Parkinson disease: A potential biomarker of disease status. Neurology 70: , 1411–1417. |
[97] | Du G , Liu T , Lewis MM , Kong L , Wang Y , Connor J , Mailman RB , Huang X ((2016) ) Quantitative susceptibility mapping of the midbrain in Parkinson’s disease. Mov Disord 31: , 317–324. |
[98] | Blazejewska AI , Schwarz ST , Pitiot A , Stephenson MC , Lowe J , Bajaj N , Bowtell RW , Auer DP , Gowland PA ((2013) ) Visualization of nigrosome 1 and its loss in PD: Pathoanatomical correlation and in vivo 7 T MRI. Neurology 81: , 534–540. |
[99] | De Marzi R , Seppi K , Hogl B , Muller C , Scher-fler C , Stefani A , Iranzo A , Tolosa E , Santamaria J , Gizewski E , Schocke M , Skalla E , Kremser C , Poewe W ((2016) ) Loss of dorsolateral nigral hyperintensity on 3.0 tesla susceptibility-weighted imaging in idiopathic rapid eye movement sleep behavior disorder. Ann Neurol 79: , 1026–1030. |
[100] | Mahlknecht P , Krismer F , Poewe W , Seppi K ((2017) ) Meta-analysis of dorsolateral nigral hyperintensity on magnetic resonance imaging as a marker for Parkinson’s disease. Mov Disord 32: , 619–623. |
[101] | Sasaki M , Shibata E , Tohyama K , Takahashi J , Otsuka K , Tsuchiya K , Takahashi S , Ehara S , Terayama Y , Sakai A ((2006) ) Neuromelanin magnetic resonance imaging of locus ceruleus and substantia nigra in Parkinson’s disease. NeuroReport 17: , 1215–1218. |
[102] | Matsuura K , Maeda M , Tabei KI , Umino M , Kajikawa H , Satoh M , Kida H , Tomimoto H ((2016) ) A longitudinal study of neuromelanin-sensitive magnetic resonance imaging in Parkinson’s disease. Neurosci Lett 633: , 112–117. |
[103] | Schwarz ST , Xing Y , Tomar P , Bajaj N , Auer DP ((2016) ) In vivo assessment of brainstem depigmentation in Parkinson disease: Potential as a severity marker for multicenter studies. Radiology 283: , 789–798. |
[104] | Kawaguchi H , Shimada H , Kodaka F , Suzuki M , Shino-toh H , Hirano S , Kershaw J , Inoue Y , Nakamura M , Sasai T , Kobayashi M , Suhara T , Ito H ((2016) ) Principal component analysis of multimodal neuromelanin MRI and dopamine transporter PET data provides a specific metric for the nigral dopaminergic neuronal density. PLoS One 11: , e0151191. |
[105] | Isaias IU , Trujillo P , Summers P , Marotta G , Mainardi L , Pezzoli G , Zecca L , Costa A ((2016) ) Neuromelanin imaging and dopaminergic loss in Parkinson’s disease. Front Aging Neurosci 8: , 196. |
[106] | Vaillancourt DE , Spraker MB , Prodoehl J , Abraham I , Corcos DM , Zhou XJ , Comella CL , Little DM ((2009) ) High-resolution diffusion tensor imaging in the substantia nigra of de novo Parkinson disease. Neurology 72: , 1378–1384. |
[107] | Zhang Y , Wu IW , Buckley S , Coffey CS , Foster E , Mendick S , Seibyl J , Schuff N ((2015) ) Diffusion tensor imaging of the nigrostriatal fibers in Parkinson’s disease. Mov Disord 30: , 1229–1236. |
[108] | Ofori E , Pasternak O , Planetta PJ , Li H , Burciu RG , Snyder AF , Lai S , Okun MS , Vaillancourt DE ((2015) ) Longitudinal changes in free-water within the substantia nigra of Parkinson’s disease. Brain 138: , 2322–2331. |
[109] | Olde Dubbelink KT , Schoonheim MM , Deijen JB , Twisk JW , Barkhof F , Berendse HW ((2014) ) Functional connectivity and cognitive decline over 3 years in Parkinson disease. Neurology 83: , 2046–2053. |
[110] | Campbell MC , Koller JM , Snyder AZ , Buddhala C , Kotzbauer PT , Perlmutter JS ((2015) ) CSF proteins and resting-state functional connectivity in Parkinson disease. Neurology 84: , 2413–2421. |
[111] | Jenkins IH , Fernandez W , Playford ED , Lees AJ , Frack-owiak RS , Passingham RE , Brooks DJ ((1992) ) Impaired activation of the supplementary motor area in Parkinson’s disease is reversed when akinesia is treated with apomor-phine. Ann Neurol 32: , 749–757. |
[112] | Piccini P , Lindvall O , Bjorklund A , Brundin P , Hagell P , Ceravolo R , Oertel W , Quinn N , Samuel M , Rehn-crona S , Widner H , Brooks DJ ((2000) ) Delayed recovery of movement-related cortical function in Parkinson’s disease after striatal dopaminergic grafts. Ann Neurol 48: ,689–695. |
[113] | van Eimeren T , Monchi O , Ballanger B , Strafella AP ((2009) ) Dysfunction of the default mode network in Parkinson disease: A functional magnetic resonance imaging study. Arch Neurol 66: , 877–883. |
[114] | Thaler A , Mirelman A , Helmich RC , van Nuenen BF , Rosenberg-Katz K , Gurevich T , Orr-Urtreger A , Marder K , Bressman S , Bloem BR , Giladi N , Hendler T , the LRRK2 Ashkenazi Jewish consortium ((2013) ) Neural correlates of executive functions in healthy G2019S LRRK2 mutation carriers. Cortex 49: , 2501–2511. |
[115] | van Nuenen BF , Helmich RC , Ferraye M , Thaler A , Hendler T , Orr-Urtreger A , Mirelman A , Bressman S , Marder KS , Giladi N , van de Warrenburg BP , Bloem BR , Toni I , LRRK2 Ashkenazi Jewish Consortium ((2012) ) Cerebral pathological and compensatory mechanisms in the premotor phase of leucine-rich repeat kinase 2 parkin-sonism. Brain 135: , 3687–3698. |
[116] | Huang C , Tang C , Feigin A , Lesser M , Ma Y , Pourfar M , Dhawan V , Eidelberg D ((2007) ) Changes in network activity with the progression of Parkinson’s disease. Brain 130: , 1834–1846. |
[117] | Hirano S , Eckert T , Flanagan T , Eidelberg D ((2009) ) Metabolic networks for assessment of therapy and diagnosis in Parkinson’s disease. Mov Disord. 24: Suppl 2, S725–S731. |
[118] | Niethammer M , Tang CC , Ma Y , Mattis PJ , Ko JH , Dhawan V , Eidelberg D ((2013) ) Parkinson’s disease cognitive network correlates with caudate dopamine. Neuroimage 78: , 204–209. |
[119] | Tang CC , Poston KL , Eckert T , Feigin A , Frucht S , Gudes-blatt M , Dhawan V , Lesser M , Vonsattel JP , Fahn S , Eidelberg D ((2010) ) Differential diagnosis of parkinson-ism: A metabolic imaging study using pattern analysis. Lancet Neurol. 9: , 149–158. |
[120] | Snow BJ , Tooyama I , McGeer EG , Yamada T , Calne DB , Takahashi H , Kimura H ((1993) ) Human positron emission tomographic [18F] fluorodopa studies correlate with dopamine cell counts and levels. Ann Neurol 34: , 324–330. |
[121] | Pate BD , Kawamata T , Yamada T , McGeer EG , Hewitt KA , Snow BJ , Ruth TJ , Calne DB ((1993) ) Correlation of striatal fluorodopa uptake in the MPTP monkey with dopaminergic indices. Ann Neurol 34: , 331–338. |
[122] | Kraemmer J , Kovacs GG , Perju-Dumbrava L , Pirker S , Traub-Weidinger T , Pirker W ((2014) ) Correlation of striatal dopamine transporter imaging with post mortem substantia nigra cell counts. Mov Disord 29: , 1767–1773. |
[123] | Karimi M , Tian L , Brown CA , Flores HP , Loftin SK , Videen TO , Moerlein SM , Perlmutter JS ((2013) ) Validation of nigrostriatal positron emission tomography measures: Critical limits. Ann Neurol 73: , 390–396. |
[124] | Blesa J , Pifl C , Sánchez-González MA , Juri C , García-Cabezas MA , Adanez R , Iglesias E , Collantes M , Peñuelas I , Sánchez-Hernández JJ , Rodríguez-Oroz MC , Avendaflo C , Hornykiewicz O , Cavada C , Obeso JA ((2012) ) The nigrostriatal system in the presymptomatic and symptomatic stages in the MPTP monkey model: A PET, histological and biochemical study. Neurobiol Dis 48: , 79–91. |
[125] | Nandhagopal R , Kuramoto L , Schulzer M , Mak E , Cragg J , Lee CS , McKenzie J , McCormick S , Samii A , Troiano A , Ruth TJ , Sossi V , de la Fuente-Fernandez R , Calne DB , Stoessl AJ ((2009) ) Longitudinal progression of sporadic Parkinson’s disease: A multi-tracer positron emission tomography study. Brain 132: , 2970–2979. |
[126] | Kordower JH , Olanow CW , Dodiya HB , Chu Y , Beach TG , Adler CH , Halliday GM , Bartus RT ((2013) ) Disease duration and the integrity of the nigrostriatal system in Parkinson’s disease. Brain 136: , 2419–2431. |
[127] | Lee CS , Samii A , Sossi V , Ruth TJ , Schulzer M , Holden JE , Wudel J , Pal PK , de la Fuente-Fernandez R , Calne DB , Stoessl AJ ((2000) ) In vivo positron emission tomo-graphic evidence for compensatory changes in presynaptic dopaminergic nerve terminals in Parkinson’s disease. Ann Neurol 47: , 493–503. |
[128] | Nandhagopal R , Kuramoto L , Schulzer M , Mak E , Cragg J , McKenzie J , McCormick S , Ruth TJ , Sossi V , de la Fuente-Fernandez R , Stoessl AJ ((2011) ) Longitudinal evolution of compensatory changes in striatal dopamine processing in Parkinson’s disease. Brain 134: , 3290–3298. |
[129] | Simuni T , Siderowf A , Lasch S , Coffey CS , Caspell-Garcia C , Jennings D , Tanner CM , Trojanowski JQ , Shaw LM , Seibyl J , Schuff N , Singleton A , Kieburtz K , Toga AW , Mollenhauer B , Galasko D , Chahine LM , Weintraub D , Foroud T , Tosun D , Poston K , Arnedo V , Frasier M , Sherer T , Chowdhury S , Marek K , Parkinson’s Progression Marker Initiative ((2018) ) Longitudinal change of clinical and biological measures in early Parkinson’s disease: Parkinson’s Progression Markers Initiative Cohort. Mov Disord 33: , 771–782. |
[130] | Marek K , Seibyl J , Shoulson I , Holloway R , Kieburtz K , Mc Dermott M , Kamp C , Shinaman A , Fahn S , Lang A , Weiner W , Welsh M ((2002) ) Dopamine transporter brain imaging to assess the effects of pramipexole vs levodopa on Parkinson disease progression. JAMA 287: , 1653–1661. |
[131] | Whone AL , Watts RL , Stoessl AJ , Davis M , Reske S , Nahmias C , Lang AE , Rascol O , Ribeiro MJ , Remy P , Poewe WH , Hauser RA , Brooks DJ , REAL-PET Study Group ((2003) ) Slower progression of Parkinson’s disease with ropinirole versus levodopa: The REAL-PET study. Ann Neurol 54: , 93–101. |
[132] | Freed CR , Greene PE , Breeze RE , Tsai WY , DuMouchel W , Kao R , Dillon S , Winfield H , Culver S , Trojanowski JQ , Eidelberg D , Fahn S ((2001) ) Transplantation of embryonic dopamine neurons for severe Parkinson’s disease. N Engl J Med 344: , 710–719. |
[133] | Olanow CW , Goetz CG , Kordower JH , Stoessl AJ , Sossi V , Brin MF , Shannon KM , Nauert GM , Perl DP , Godbold J , Freeman TB ((2003) ) A double-blind controlled trial of bilateral fetal nigral transplantation in Parkinson’s disease. Ann Neurol 54: , 403–414. |
[134] | Sossi V , Holden JE , de la Fuente-Fernandez R , Ruth TJ , Stoessl AJ ((2003) ) Effect of dopamine loss and the metabolite 3-O-methyl-[18F]fluoro-dopa on the relation between the 18F-fluorodopa tissue input uptake rate constant Kocc and the [18F]fluorodopa plasma input uptake rate constant Ki. J Cereb Blood Flow Metab 23: , 301–309. |
[135] | Boileau I , Warsh JJ , Guttman M , Saint-Cyr JA , Mc Cluskey T , Rusjan P , Houle S , Wilson AA , Meyer JH , Kish SJ ((2008) ) Elevated serotonin transporter binding in depressed patients with Parkinson’s disease: A preliminary PET study with [11C]DASB. Mov Disord 23: , 1776–1780. |
[136] | Politis M , Wu K , Loane C , Turkheimer FE , Molloy S , Brooks DJ , Piccini P ((2010) ) Depressive symptoms in PD correlate with higher 5-HTT binding in raphe and limbic structures. Neurology 75: , 1920–1927. |
[137] | Politis M , Wu K , Loane C , Brooks DJ , Kiferle L , Turkheimer FE , Bain P , Molloy S , Piccini P ((2014) ) Serotonergic mechanisms responsible for levodopa-induced dyskinesias in Parkinson’s disease patients. J Clin Invest 124: , 1340–1349. |
[138] | Lee JY , Seo S , Lee JS , Kim HJ , Kim YK , Jeon BS ((2015) ) Putaminal serotonergic innervation: Monitoring dyskine-sia risk in Parkinson disease. Neurology 85: , 853–860. |
[139] | Wile DJ , Agarwal PA , Schulzer M , Mak E , Dinelle K , Shahinfard E , Vafai N , Hasegawa K , Zhang J , McKenzie J , Neilson N , Strongosky A , Uitti RJ , Guttman M , Zabetian CP , Ding YS , Adam M , Aasly J , Wszolek ZK , Farrer M , Sossi V , Stoessl AJ ((2017) ) Serotonin and dopamine transporter PET changes in the premotor phase of LRRK2 parkinsonism: Cross-sectional studies. Lancet Neurol 16: , 351–359. |
[140] | Bohnen NI , Kaufer DI , Ivanco LS , Lopresti B , Koeppe RA , Davis JG , Mathis CA , Moore RY , DeKosky ST ((2003) ) Cortical cholinergic function is more severely affected in parkinsonian dementia than in Alzheimer disease: An in vivo positron emission tomographic study. Arch Neurol 60: , 1745–1748. |
[141] | Bohnen NI , Muller ML , Koeppe RA , Studenski SA , Kil-bourn MA , Frey KA , Albin RL ((2009) ) History of falls in Parkinson disease is associated with reduced cholinergic activity. Neurology 73: , 1670–1676. |
[142] | Bohnen NI , Muller ML , Kotagal V , Koeppe RA , Kilbourn MA , Albin RL , Frey KA ((2010) ) Olfactory dysfunction, central cholinergic integrity and cognitive impairment in Parkinson’s disease. Brain 133: , 1747–1754. |
[143] | Kotagal V , Albin RL , Muller ML , Koeppe RA , Chervin RD , Frey KA , Bohnen NI ((2012) ) Symptoms of rapid eye movement sleep behavior disorder are associated with cholinergic denervation in Parkinson disease. Ann Neurol 71: , 560–568. |
[144] | Muller ML , Bohnen NI ((2013) ) Cholinergic dysfunction in Parkinson’s disease. Curr Neurol Neurosci Rep 13: , 377. |
[145] | Petrou M , Frey KA , Kilbourn MR , Scott PJ , Raffel DM , Bohnen NI , Muller ML , Albin RL , Koeppe RA ((2014) ) In vivo imaging of human cholinergic nerve terminals with (-)-5-(18)F-fluoroethoxybenzovesamicol: Biodistribution, dosimetry, and tracer kinetic analyses. JNucl Med 55: , 396–404. |
[146] | Borghammer P , Knudsen K , Fedorova TD , Brooks DJ ((2017) ) Imaging Parkinson’s disease below the neck. NPJ Parkinsons Dis 3: , 15. |
[147] | Knudsen K , Fedorova TD , Hansen AK , Sommerauer M , Otto M , Svendsen KB , Nahimi A , Stokholm MG , Pavese N , Beier CP , Brooks DJ , Borghammer P ((2018) ) In-vivo staging of pathology in REM sleep behaviour disorder: A multimodality imaging case-control study. Lancet Neurol 17: ,618–628. |
[148] | Sevigny J , Suhy J , Chiao P , Chen T , Klein G , Purcell D , Oh J , Verma A , Sampat M , Barakos J ((2016) ) Amyloid PET screening for enrichment of early-stage Alzheimer disease clinical trials: Experience in a Phase 1b clinical trial. Alzheimer Dis Assoc Disord 30: , 1–7. |
[149] | Satlin A , Wang J , Logovinsky V , Berry S , Swanson C , Dhadda S , Berry DA ((2016) ) Design of a Bayesian adaptive phase 2 proof-of-concept trial for BAN2401, a putative disease-modifying monoclonal antibody for the treatment of Alzheimer’s disease. Alzheimers Dement (N Y) 2: , 1–12. |
[150] | Kennedy ME , Stamford AW , Chen X , Cox K , Cumming JN , Dockendorf MF , Egan M , Ereshefsky L , Hodgson RA , Hyde LA , Jhee S , Kleijn HJ , Kuvelkar R , Li W , Mattson BA , Mei H , Palcza J , Scott JD , Tanen M , Troyer MD , Tseng JL , Stone JA , Parker EM , Forman MS ((2016) ) The BACE1 inhibitor verubecestat (MK-8931) reduces CNS beta-amyloid in animal models and in Alzheimer’s disease patients. Sci Transl Med 8: , 363ra150. |
[151] | Sperling R , Mormino E , Johnson K ((2014) ) The evolution of preclinical Alzheimer’s disease: Implications for prevention trials. Neuron 84: , 608–622. |
[152] | Eberling JL , Dave KD , Frasier MA ((2013) ) a-synuclein imaging: A critical need for Parkinson’s disease research. J Parkinsons Dis 3: , 565–567. |
[153] | Fodero-Tavoletti MT , Mulligan RS , Okamura N , Furu-moto S , Rowe CC , Kudo Y , Masters CL , Cappai R , Yanai K , Villemagne VL ((2009) ) In vitro characterisation of BF227 binding to alpha-synuclein/Lewy bodies. Eur J Pharmacol 617: , 54–58. |
[154] | Bagchi DP , Yu L , Perlmutter JS , Xu J , Mach RH , Tu Z , Kotzbauer PT ((2013) ) Binding of the radioligand SIL23 to alpha-synuclein fibrils in Parkinson disease brain tissue establishes feasibility and screening approaches for developing a Parkinson disease imaging agent. PLoS One 8: , e55031. |
[155] | Shah M , Seibyl J , Cartier A , Bhatt R , Catafau AM ((2014) ) Molecular imaging insights into neurodegeneration: Focus on a-synuclein radiotracers. J Nucl Med, 1397–1400. |
[156] | Perez-Soriano A , Arena JE , Dinelle K , Miao Q , McKenzie J , Neilson N , Puschmann A , Schaffer P , Shinotoh H , Smith-Forrester J , Shahinfard E , Vafai N , Wile D , Wszolek Z , Higuchi M , Sossi V , Stoessl AJ ((2017) ) PBB3 imaging in Parkinsonian disorders: Evidence for binding to tau and other proteins. Mov Disord 32: , 1016–1024. |
[157] | Zhang X , Jin H , Padakanti PK , Li J , Yang H , Fan J , Mach RH , Kotzbauer P , Tu Z ((2014) ) Radiosynthesis and in vivo evaluation of two PET radioligands for imaging a-synuclein. Appl Sci (Basel) 4: , 66–78. |
[158] | Petrou M , Dwamena BA , Foerster BR , MacEachern MP , Bohnen NI , Muller ML , Albin RL , Frey KA ((2015) ) Amyloid deposition in Parkinson’s disease and cognitive impairment: A systematic review. Mov Disord 30: , 928–935. |
[159] | Giasson BI , Forman MS , Higuchi M , Golbe LI , Graves CL , Kotzbauer PT , Trojanowski JQ , Lee VM ((2003) ) Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 300: , 636–640. |
[160] | Compta Y , Parkkinen L , Kempster P , Selikhova M , Lashley T , Holton JL , Lees AJ , Revesz T ((2014) ) The significance of a-synuclein, amyloid-ß and tau pathologies in Parkinson’s disease progression and related dementia. Neurodegener Dis 13: , 154–156. |
[161] | Gomperts SN , Locascio JJ , Makaretz SJ , Schultz A , Caso C , Vasdev N , Sperling R , Growdon JH , Dickerson BC , Johnson K ((2016) ) Tau positron emission tomographic imaging in the Lewy body diseases. JAMA Neurol 73: , 1334–1341. |
[162] | Koga S , Ono M , Sahara N , Higuchi M , Dickson DW ((2017) ) Fluorescence and autoradiographic evaluation of tau PET ligand PBB3 to a-synuclein pathology. Mov Disord 32: , 884–892. |
[163] | McGeer PL , Itagaki S , Boyes BE , McGeer EG ((1988) ) Reactive microglia are positive for HLA-DR in the sub-stantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 38: , 1285–1291. |
[164] | McGeer PL , McGeer EG ((2004) ) Inflammation and neurodegeneration in Parkinson’s disease. Parkinsonism Relat Disord 10: Suppl 1, S3–S7. |
[165] | Ouchi Y , Yoshikawa E , Sekine Y , Futatsubashi M , Kanno T , Ogusu T , Torizuka T ((2005) ) Microglial activation and dopamine terminal loss in early Parkinson’s disease. Ann Neurol 57: , 168–175. |
[166] | Gerhard A , Pavese N , Hotton G , Turkheimer F , Es M , Hammers A , Eggert K , Oertel W , Banati RB , Brooks DJ ((2006) ) In vivo imaging of microglial activation with [11C](R)-PK11195 PET in idiopathic Parkinson’s disease. NeurobiolDis 21: , 404–412. |
[167] | Owen DR , Yeo AJ , Gunn RN , Song K , Wadsworth G , Lewis A , Rhodes C , Pulford DJ , Bennacef I , Parker CA , StJean PL , Cardon LR , Mooser VE , Matthews PM , Rabiner EA , Rubio JP ((2012) ) An 18-kDa translocator protein (TSPO) polymorphism explains differences in binding affinity of the PET radioligand PBR28. J Cereb Blood Flow Metab 32: , 1–5. |
[168] | Koshimori Y , Ko JH , Mizrahi R , Rusjan P , Mabrouk R , Jacobs MF , Christopher L , Hamani C , Lang AE , Wilson AA , Houle S , Strafella AP ((2015) ) Imaging striatal microglial activation in patients with Parkinson’s disease. PLoS One 10: , e0138721. |
[169] | Minakaki G , Menges S , Kittel A , Emmanouilidou E , Scha-effner I , Barkovits K , Bergmann A , Rockenstein E , Adame A , Marxreiter F , Mollenhauer B , Galasko D , Buzas EI , Schlotzer-Schrehardt U , Marcus K , Xiang W , Lie DC , Vekrellis K , Masliah E , Winkler J , Klucken J ((2018) ) Autophagy inhibition promotes SNCA/alpha-synuclein release and transfer via extracellular vesicles with a hybrid autophagosome-exosome-like phenotype. Autophagy 14: , 98–119. |
[170] | Woodcock J. FDA Letter of Support: Exploratory Prognostic Biomarkers for Enrichment in Early Stage Parkinson’s Disease Clinical Trials. Food and Drug Administration, https://www.fda.gov/downloads/Drugs/DevelopmentApprovalProcess/DrugDevelopmentToolsQualificationProgram/BiomarkerQualificationProgram/UCM605357.pdf, Accessed March 28. |
[171] | Sanchez-Ferro A , Elshehabi M , Godinho C , Salkovic D , Hobert MA , Domingos J , van Uem JM , Ferreira JJ , Maetzler W ((2016) ) New methods for the assessment of Parkinson’s disease (2005 to 2015): A systematic review. Mov Disord 31: , 1283–1292. |
[172] | Artusi CA , Mishra M , Latimer P , Vizcarra JA , Lopiano L , Maetzler W , Merola A , Espay AJ ((2018) ) Integration of technology-based outcome measures in clinical trials of Parkinson and other neurodegenerative diseases. Parkin-sonism Relat Disord 46: Suppl 1, S53–S56. |
[173] | van Lummel RC , Walgaard S , Pijnappels M , Elders PJ , Garcia-Aymerich J , van Dieen JH , Beek PJ ((2015) ) Physical performance and physical activity in older adults: Associated but separate domains of physical function in old age. PLoS One 10: , e0144048. |
[174] | Wolz R , Schwarz AJ , Gray KR , Yu P , Hill DL , Alzheimer’s Disease Neuroimaging Initiative ((2016) ) Enrichment of clinical trials in MCI due to AD using markers of amyloid and neurodegeneration. Neurology 87: , 1235–1241. |
[175] | Del Din S , Godfrey A , Mazza C , Lord S , Rochester L ((2016) ) Free-living monitoring of Parkinson’s disease: Lessons from the field. Mov Disord 31: , 1293–1313. |
[176] | Espay AJ , Bonato P , Nahab FB , Maetzler W , Dean JM , Klucken J , Eskofier BM , Merola A , Horak F , Lang AE , Reilmann R , Giuffrida J , Nieuwboer A , Horne M , Little MA , Litvan I , Simuni T , Dorsey ER , Burack MA , Kubota K , Kamondi A , Godinho C , Daneault JF , Mitsi G , Krinke L , Hausdorff JM , Bloem BR , Papa-petropoulos S , Movement Disorders Society Task Force on Technology ((2016) ) Technology in Parkinson’s disease: Challenges and opportunities. Mov Disord 31: , 1272–1282. |
[177] | CTTI Recommendations: Developing Novel Endpoints Generated by Mobile Technology for Use in Clinical Trials, Clinical Trials Transformation Initiative, https://www.ctti-clinicaltrials.org/files/novelendpoints-recs.pdf, Accessed October 30. |
[178] | Haaxma CA , Bloem BR , Borm GF , Horstink MW ((2008) ) Comparison of a timed motor test battery to the Unified Parkinson’s Disease Rating Scale-III in Parkinson’s disease. Mov Disord 23: , 1707–1717. |
[179] | Heinzel S , Bernhard FP , Roeben B , Nussbaum S , Heger T , Martus P , Hobert MA , Maetzler W , Berg D ((2017) ) Progression markers of motor deficits in Parkinson’s disease: A biannual 4-year prospective study. Mov Disord 32: , 1254–1256. |
[180] | Postuma RB , Lang AE , Gagnon JF , Pelletier A , Mont-plaisir JY ((2012) ) How does parkinsonism start? Prodromal parkinsonism motor changes in idiopathic REM sleep behaviour disorder. Brain 135: , 1860–1870. |
[181] | Bot BM , Suver C , Neto EC , Kellen M , Klein A , Bare C , Doerr M , Pratap A , Wilbanks J , Dorsey ER , Friend SH , Trister AD ((2016) ) The mPower study, Parkinson disease mobile data collected using ResearchKit. Sci Data 3: , 160011. |
[182] | van Uem JM , Maier KS , Hucker S , Scheck O , Hobert MA , Santos AT , Fagerbakke O , Larsen F , Ferreira JJ , Maetzler W ((2016) ) Twelve-week sensor assessment in Parkinson’s disease: Impact on quality of life. Mov Disord 31: , 1337–1338. |
[183] | Chartier-Harlin MC , Kachergus J , Roumier C , Mouroux V , Douay X , Lincoln S , Levecque C , Larvor L , Andrieux J , Hulihan M , Waucquier N , Defebvre L , Amouyel P , Farrer M , Destee A ((2004) ) Alpha-synuclein locus duplication as a cause of familial Parkinson’s disease. Lancet 364: , 1167–1169. |
[184] | Singleton AB , Farrer M , Johnson J , Singleton A , Hague S , Kachergus J , Hulihan M , Peuralinna T , Dutra A , Nussbaum R , Lincoln S , Crawley A , Hanson M , Maraganore D , Adler C , Cookson MR , Muenter M , Baptista M , Miller D , Blancato J , Hardy J , Gwinn-Hardy K ((2003) ) alpha-Synuclein locus triplication causes Parkinson’s disease. Science 302: , 841. |
[185] | Conrado DJ , Nicholas T , Tsai K , Macha S , Sinha V , Stone J , Corrigan B , Bani M , Muglia P , Watson IA , Kern VD , Sheveleva E , Marek K , Stephenson DT , Romero K , Critical Path for Parkinson’s (CPP) Parkinson’s Disease Modeling and Simulation Working Group ((2018) ) Dopamine transporter neuroimaging as an enrichment biomarker in early Parkinson’s disease clinical trials: A disease progression modeling analysis. Clin Transl Sci 11: , 63–70. |
[186] | Shin EC , Cho SE , Lee DK , Hur MW , Paik SR , Park JH , Kim J ((2000) ) Expression patterns of alpha-synuclein in human hematopoietic cells and in Drosophila at different developmental stages. Mol Cells 10: , 65–70. |
[187] | Gardai SJ , Mao W , Schule B , Babcock M , Schoebel S , Lorenzana C , Alexander J , Kim S , Glick H , Hilton K , Fitzgerald JK , Buttini M , Chiou SS , McConlogue L , Anderson JP , Schenk DB , Bard F , Langston JW , Yednock T , Johnston JA ((2013) ) Elevated alpha-synuclein impairs innate immune cell function and provides a potential peripheral biomarker for Parkinson’s disease. PLoS One 8: , e71634. |
[188] | Parkinson Study Group ((1989) ) DATATOP: A multicenter controlled clinical trial in early Parkinson’s disease. Arch Neurol 46: , 1052–1060. |
[189] | Ravina B , Tanner C , Dieuliis D , Eberly S , Flagg E , Galpern WR , Fahn S , Goetz CG , Grate S , Kurlan R , Lang AE , Marek K , Kieburtz K , Oakes D , Elliott R , Shoulson I , Parkinson Study Group LABS-PD Investigators ((2009) ) A longitudinal program for biomarker development in Parkinson’s disease: A feasibility study. Mov Disord 24: , 2081–2090. |
[190] | Parkinson Progression Marker Initiative ((2011) ) The Parkinson Progression Marker Initiative (PPMI). Prog Neurobiol 95: , 629–635. |
[191] | Weiner MW , Veitch DP , Aisen PS , Beckett LA , Cairns NJ , Cedarbaum J , Donohue MC , Green RC , Harvey D , Jack CR, Jr., Jagust W , Morris JC , Petersen RC , Saykin AJ , Shaw L , Thompson PM , Toga AW , Trojanowski JQ , Alzheimer’s Disease Neuroimaging Initiative ((2015) ) Impact of the Alzheimer’s Disease Neuroimaging Initiative, 2004 to 2014. Alzheimers Dement 11: , 865–884. |
[192] | Stephenson D , Hu MT , Romero K , Breen K , Burn D , Ben-Shlomo Y , Bhattaram A , Isaac M , Venuto C , Kubota K , Little MA , Friend S , Lovestone S , Morris HR , Grosset D , Sutherland M , Gallacher J , Williams-Gray C , Bain LJ , Aviles E , Marek K , Toga AW , Stark Y , Forrest Gordon M , Ford S ((2015) ) Precompetitive data sharing as a catalyst to address unmet needs in Parkinson’s disease. J Parkinsons Dis 5: , 581–594. |


