
HNRNPA1 de novo Variant Associated with Early Childhood Onset, Rapidly Progressive Generalized Myopathy
Abstract
HNRNPA1 variants are known to cause degenerative motoneuron and muscle diseases which manifests in middle age or later. We report on a girl with early childhood onset, rapidly progressive generalized myopathy including ultrastructural findings in line with a proteinopathy. Proteomics of patient-derived muscle and combined screening of genomic data for copy number variations identified a HNRNPA1 de novo intragenic deletion as causative for the phenotype. Our report expands the spectrum of HNRNPA1-related diseases towards early-childhood onset and adds HNRNPA1 to the growing list of ALS and myopathy genes for which certain mutations may cause severe pediatric phenotypes.
INTRODUCTION
Heterogeneous nuclear ribonucleoprotein A1 (HNRNPA1) is an RNA- and DNA-binding protein, which is involved in RNA transcription, pre-mRNA splicing, mRNA transport, protein translation, microRNA processing, telomere maintenance, regulation of transcription factor activity and organization of nuclear pore complexes [5, 12]. RNA- and DNA-binding proteins are involved in the aetiology and pathogenesis of a variety of neurological disorders [9, 12] and HNRNPA1 interacts with several proteins that are associated with amyotrophic lateral sclerosis (ALS) and degenerative myopathies (TDP-43, FUS, PABPN1 and Matrin-3) [6]. While these findings already supported an important role of HNRNPA1 in neurological diseases, reports of dominant HNRNPA1 variants causing different (overlapping) phenotypes such as ALS, hereditary inclusion body myopathy (hIBM) associated with rimmed vacuoles with or without Paget’s disease of bone, and distal myopathy with rimmed vacuoles [4, 7] provided further solid evidence for a crucial role of HNRNPA1 across the neuromuscular axis. Conditions associated with HNRNPA1 so far are degenerative in nature and typically manifest in middle age or later. Here we identified a previously unreported HNRNPA1 de novo variant in a patient with early childhood onset, rapidly progressive and fatal generalized myopathy.
MATERIALS AND METHODS
The study was performed according to the Declaration of Helsinki. Analytical procedures were undertaken after obtaining parental consent. A detailed methodology description is presented in supplementary document 1.
CASE REPORT
The patient was the only child of healthy non-consanguineous Caucasian parents. Pregnancy, delivery (38 gestational weeks), newborn measurements, and the neonatal period were uneventful. Motor milestones were normal (free sitting at age 8 months, walking independently at 15 months). The patient exhibited tiptoe walking from the start, together with gait instability characterized by a broad-based gait and frequent falls. Absence of deep tendon reflexes was noted at the age of 3 years. Between 3 and 5 years, she developed progressive generalized muscle weakness and atrophy affecting her extremities, limb girdle and trunk muscles (Fig. 1A) accompanied by worsening of gait instability, myopathic facies and difficulties chewing and swallowing. At 5.5 years, she became completely wheelchair bound. At 8 years, she almost entirely lost the ability to lift her arms and exhibited moderate contractures in the elbows and knees, along with external ophthalmoplegia and ptosis of the right eye. At 9 years, bulbar dysfunction progressed to the point where she required a percutaneous gastroenterostomy. At 11 years, she died from a severe bacterial lung infection. Sensory exam was repeatedly normal, and no signs of upper motor neuron involvement were noted. There was a delay in head growth, initially noticed at 4 months. She showed normal cognitive and language development and attended a school for children with physical disabilities.
Fig. 1
A patient with generalized early-onset myopathy. (A) Axial chest CT scans (performed at age 10 years for bacterial pneumonia) showing severe atrophy of shoulder girdle and back muscles. (B–D) Light microscopy of a muscle biopsy specimen revealing muscle fibre atrophy, endomysial fibrosis and presence of rimmed vacuoles. B: H&E; scale bar = 10μm. C: Gömöri trichrome; scale bar = 8μm. D: Semithin section, paraphenylendiamine; scale bar = 30μm. (E & F) Electron microscopy of a muscle biopsy specimen showing subsarcolemmal accumulation of pleomorphic granular and membranous material indicative of abnormal autophagy (in E) and thin (arrows) and thick (arrowheads) tubulofilaments in degenerating muscle fibre nuclei (in F). E: Scale bar = 3.0μm. F: Scale bar = 1.5μm.
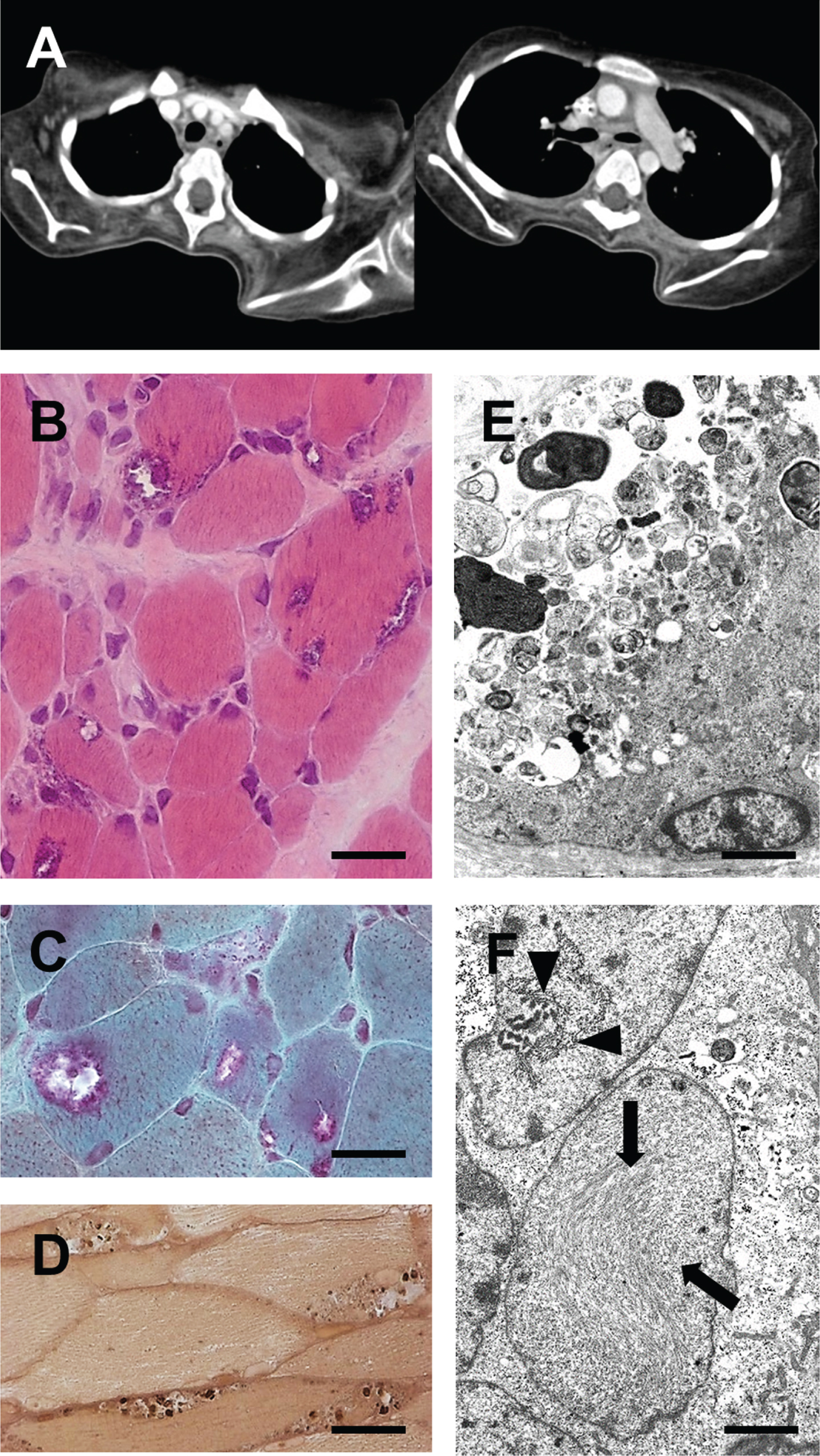
Brain imaging was normal except for small head size. Electroneurography revealed slowed motor nerve conduction at 3 years (28.5 m/sec, peroneal nerve). At 8 years, no CMAPs could be elicited (peroneal and ulnar nerves), whereas sensory nerve conduction (median nerve) was normal. Creatine kinase levels were moderately elevated at 4 years (400 U/l) but had returned to normal when re-assessed at 8 and 9 years, likely due to severe loss of muscle mass.
Muscle biopsies obtained from the gastrocnemius and quadriceps muscles showed marked muscle fibre atrophy, characterized by numerous rounded atrophic fibres and few regenerating fibres, an increased number of centralized myonuclei and substantial endomysial fibrosis. Rimmed vacuoles were observed in numerous fibres (Fig. 1B–D). Ultrastructural examination revealed autophagic vacuoles containing pleomorphic, often electron-dense membranous and granular material (Fig. 1E). Moreover, tubulofilamentous inclusions were identified, often near myonuclei (Fig. 1F). Diagnostic multiplex Western blotting for muscular dystrophy proteins did not yield significant findings. A biopsy taken from the pure sensory sural nerve at 3 years revealed only minimal reduction in myelinated nerve fibre density and no signs of ongoing nerve fiber degeneration and regeneration (data not shown).
RESULTS AND DISCUSSION
Several attempts to identify the genetic cause of the disease were unsuccessful. Conventional karyotyping, comparative genomic hybridisation, subtelomere analysis and phenotype-oriented analysis of disease genes (MYH2, MYH7, COL6A2, FHL1, FKRP, TRIM32, GAA, DES, LAMA2, SEPN1, GNE, DNAJB6) did not clarify the aetiology. Trio exome sequencing of child and parental DNA also failed to identify (likely) pathogenic single nucleotide substitutions (SNV) or small insertions or deletions (indel, 1–50 nucleotides) in genes known to cause a neuromuscular or neurodegenerative disorder. Proteomic profiling was carried out as the next omics-based step in the diagnostic work-up of this patient in the context of NMD-GPS (nmd-gps.net), a translational research project aiming to investigate the power of mass spectrometry-based protein quantification to improve diagnostic concepts in rare diseases such as neuromuscular disorders. Thus, a small portion of the protein extract obtained from the patient’s muscle biopsy, initially utilized for diagnostic western blotting, was used for relative protein quantification through mass spectrometry (Suppl. Document 1) revealing significant protein dysregulations (Suppl. Document 1 & 2, Table 1). We then reanalysed exome sequencing data for the corresponding genes, expanding the analysis beyond SNVs and small indels using Pindel software, developed to identify deletions, insertions, inversions, tandem duplications and other structural variants in short-read next-generation sequencing data [13]. Pindel revealed a suspicious finding in the gene encoding HNRNPA1, one of the proteins highlighted by the proteomic approach (Fig. 2A). This anomaly was a heterozygous 85-bp de novo deletion, validated through polymerase chain reaction: primers flanking the putative deletion yielded a product of the expected size in the parents and the same amplicon as well as an additional shorter amplicon in the proband (Fig. 2B). Sanger sequencing of these amplicons confirmed a deletion of 85 base-pairs, starting at the 8th nucleotide of exon 7 and extending 17 nucleotides into intron 7 (chr12 : 54,282,807-54,282,891 (GRCh38/hg38), NM_002136.4: c.684_751 + 17del; Fig. 1H). This variant was absent from more than 125,000 control chromosomes in gnomAD 4.0 (CNVs and SVs) and more than 70,000 alleles from an in-housedatabase.
Fig. 2
Identification of an HNRNPA1 deletion variant. (A) Proteomic profiling of patient-derived and control muscle tissue. Extracted ion chromatograms (XICs) of the unique HNRNPA1 peptide 285SSGPYGGGGQYFAKPR300 +++ at 543.5990 m/z indicated increased abundance in the patient sample. (B) Detection of the HNRNPA1 deletion variant in genomic DNA. Primers placed in introns 6 and 7 yielded a fragment of 221 bp in case of the wild-type sequence. An additional shorter 136 bp fragment was obtained when using patient’s DNA as a template. The sequencing chromatogram corresponding to the larger amplicon (top) shows the wild-type sequence for the intron 6 exon 7 junction. The chromatogram corresponding to the smaller amplicon (bottom) shows a partial deletion of exon 7 and intron 7. MW = molecular weight size standard. (C) Schematic overview of HNRNPA1 protein. HNRNPA1 contains two N-terminal RNA-recognition motifs (RRMs) and a C-terminal prion-like domain (PrLD), containing an arginine-glycine-glycine (RGG) motif and a nuclear localization signal (NLS). Known disease-causing variants are shown and color-coded for associated phenotypes. The deletion identified in our patient is highlighted in blue.
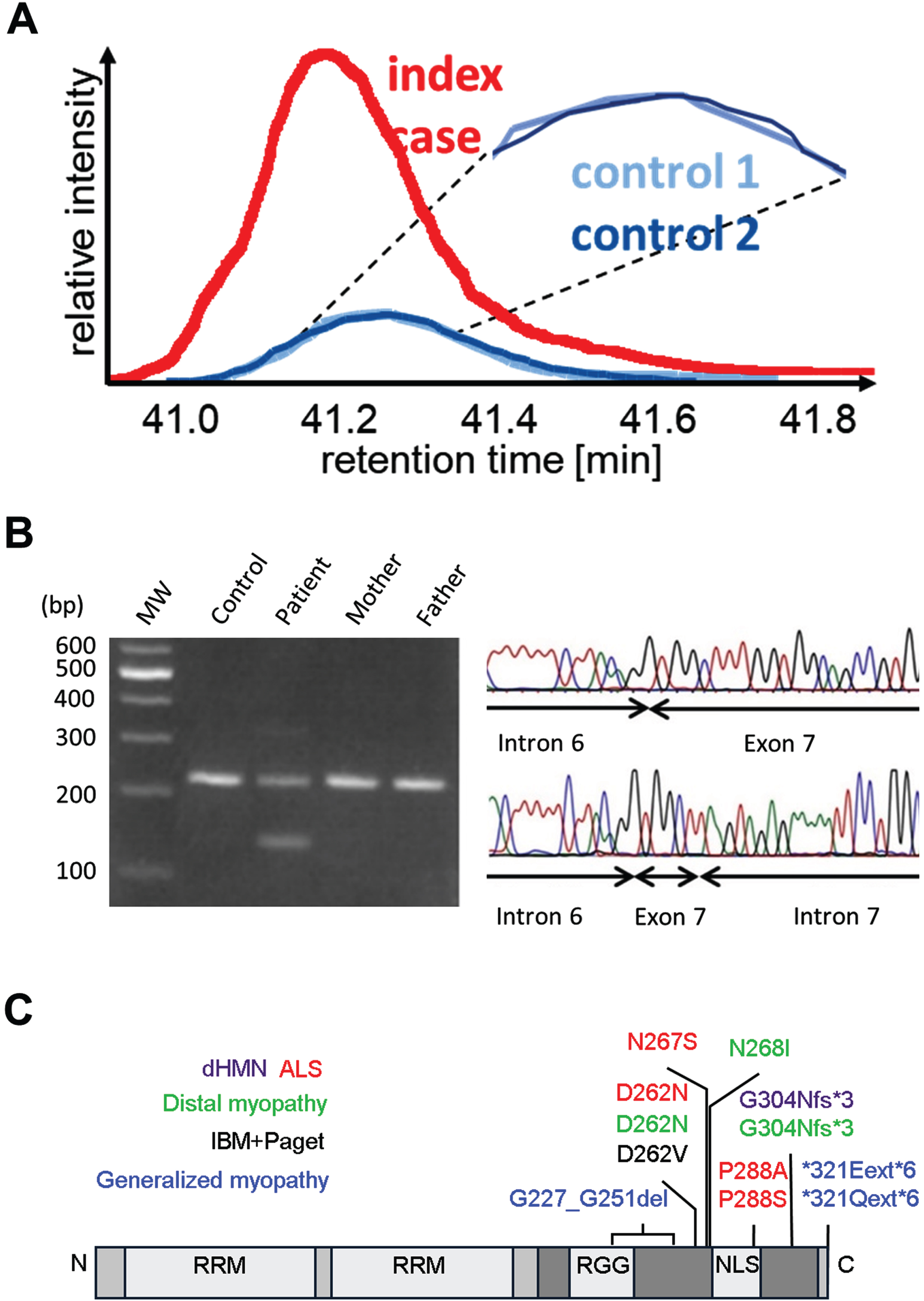
At the transcript level, we predicted that this variant leads to skipping of exon 7, which would result in in-frame splicing of exon 6 to exon 8 and an HNRNPA1 protein lacking 25 amino acids of the PrLD domain (NP_002127.1: p.Gly227_Gly251del). Unfortunately, no suitable tissue samples or cells were obtainable for mRNA isolation to prove this hypothesis and other potential outcomes (e.g., activation of cryptic splice sites) are also possible. It is presently unclear how any of the potential consequences of the HNRNPA1 deletion variant lead to increased levels of HNRNPA1 in patient muscle as observed in the proteome analysis. It is conceivable that the mutant protein exhibits increased stability, as suggested by findings in overexpression studies for certain HNRNPA1 variants [1] and consistent with computational predictions [10]. Alternatively, or in addition, the mutant protein might enhance the stability of the wildtype protein produced by the normal allele. It is worth noting that HNRNPA1 has the capability to form homodimers in the presence of DNA [2].
Diseases previously linked to HNRNPA1 typically manifest as adult-onset, slowly progressive conditions, leading to weakness and wasting of leg and arm muscles and bulbar features in some ALS cases (Fig. 2C). However, cases with earlier onset have been reported in a recent case series [1]. The clinical presentation of our patient bears some resemblance to two patients of this series (families C and F), who displayed distal-onset myopathy at the age of 8–12 years, followed by the development of proximal, facial, and bulbar weakness as the condition progressed [1]. Nevertheless, our case is unique in terms of the even earlier onset of the disease, earlier generalization, faster progression, and early fatality. No additional manifestations of MSP such as ALS, Paget’s disease of bone or frontotemporal dementia were noted; however, given the progressive course of the disease, it seems possible that these features might have emerged later in life if the patient had not died prematurely. Microcephaly was an additional finding, though not associated with intellectual disability, developmental delay or other symptoms indicating central nervous system involvement. As no non-genetic cause of microcephaly was obvious in the patient’s medical history and no variants in known microcephaly genes were found by exome sequencing, we cannot exclude the possibility that HNRNPA1 is also involved in the control of brain and head size. As besides the myopathy the phenotype of our patient also involves motor neuropathy, the concept of a multisystem proteinopathy (MSP) is not totally misplaced even if brain, upper motor neuron or bone disease were not shown.
The relationship between HNRNPA1 variants and clinical outcomes remains puzzling (Fig. 1I). For instance, the p.Asp262Asn variant was initially identified in ALS patients; however, it was later discovered to also cause inclusion body myopathy. Moreover, certain myopathy-related variants can enhance the propensity for HNRNPA1 fibrillization, have no effect on fibril formation, or even slow down fibrillization [1]. Thus, despite elucidation of numerous cellular and molecular pathways involving HNRNPA1 [12], the precise mechanisms by which variants in HNRNPA1 lead to disease remain incompletely understood. Beyond its role in aiding genetic diagnosis, proteomic profiling of the patient’s muscle biopsy has also provided a resource that can be further utilized to investigate the pathophysiology of HNRNPA1-related disorders. The proteomic results support among others the involvement of protein aggregation and cytoskeleton damage (Fig. 3A & B, Supplementary document 2) and show a functional interdependence among dysregulated proteins (Fig. 3C).
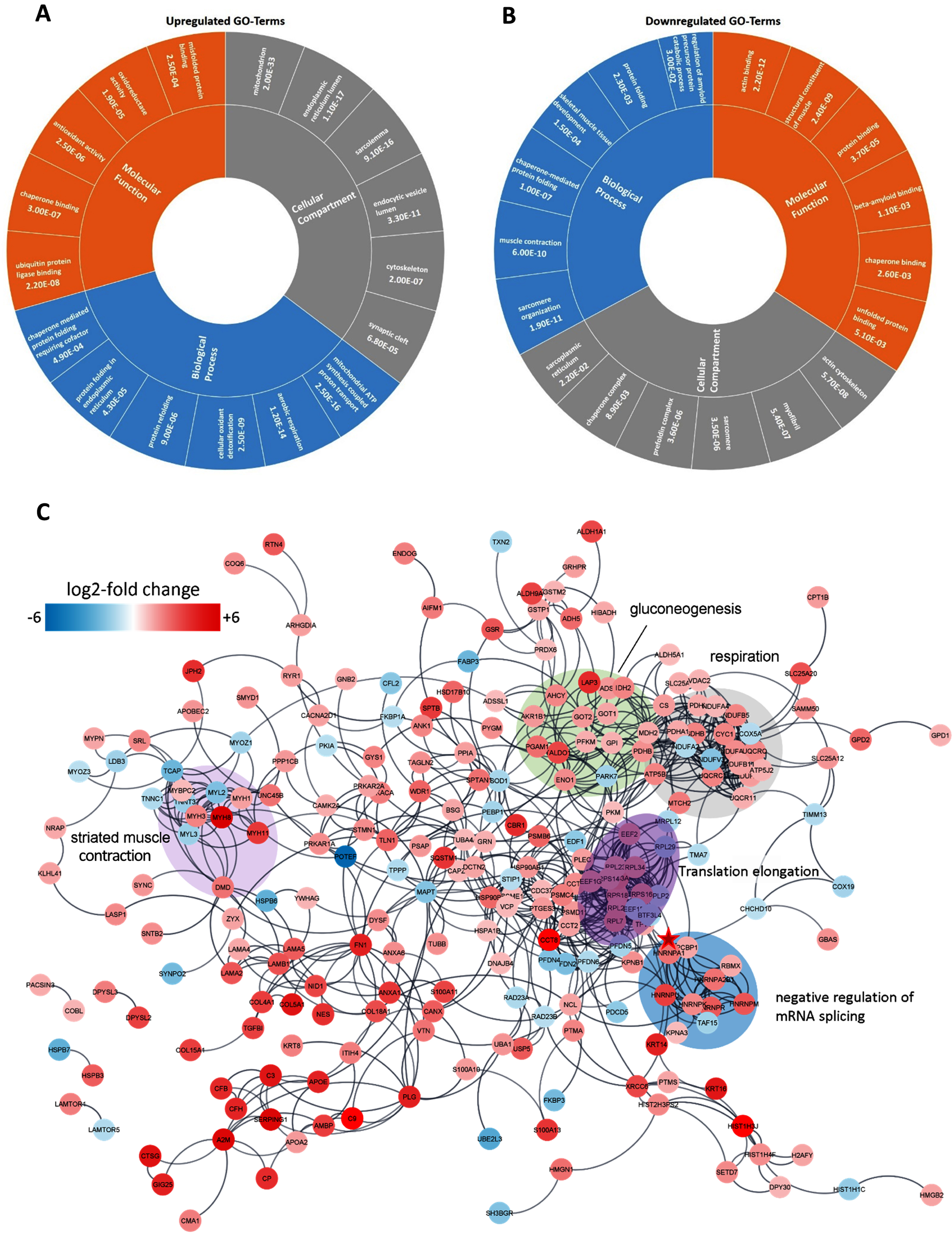
Fig. 3
In silico analyses of proteomic data obtained from HNRNPA1 patient muscle. (A) The six top-enriched molecular functions, biological processes and sub-cellular compartments identified by GO term analysis of proteins with increased abundance in patient-derived muscle tissue. (B) The six top-enriched molecular functions, biological processes and sub-cellular compartments identified by GO term analysis of proteins with decreased abundance in patient-derived muscle tissue. Numbers in A & B represent p-values from the respective GO terms based on EASE score (a modified Fisher exact p-value test). (C) High confidence STRING protein/protein interaction network of proteins that had a relative standard deviation <30% in the 2 control samples and were at least 2-fold up/down-regulated in the patient sample compared to the controls. The position of HNRNPA1 in the network is highlighted by a red star.
Our report expands the spectrum of HNRNPA1-related diseases towards early-childhood-onset, generalized and rapidly progressive myopathy caused by a de novo intragenic deletion. Hence, our findings add HNRNPA1 to the growing list of ALS and myopathy genes for which certain mutations may cause a severe pediatric phenotype as recently demonstrated for HNRNPA2B1 [8], MATR3 [14], SOD1 [3] and VCP [11].
ACKNOWLEDGMENTS
The authors thank Stephan Buchkremer and Hannelore Mader for expert technical support.
FUNDING
AR acknowledges the support of the Germany Society of Muscular Diseases (DGM), the French Muscular Dystrophy Association (AFM-Téléthon grant # 21644) and the European Regional Development Fund (NMD-GPS; https://nmd-gps.net). LK and RZ are supported by the Ministerium für Innovation, Wissenschaft und Forschung des Landes Nordrhein-Westfalen and the Federal Ministry of Education and Research, Germany (BMBF). HL receives support from the Canadian Institutes of Health Research (CIHR) for Foundation Grant FDN-167281 (Precision Health for Neuromuscular Diseases), Transnational Team Grant ERT-174211 (ProDGNE) and Network Grant OR2-189333 (NMD4C), from the Canada Foundation for Innovation (CFI-JELF 38412), the Canada Research Chairs program (Canada Research Chair in Neuromuscular Genomics and Health, 950-232279), the European Commission (Grant # 101080249), the Canada Research Coordinating Committee New Frontiers in Research Fund (NFRFG-2022-00033) for SIMPATHIC, and from the Government of Canada Canada First Research Excellence Fund (CFREF) for the Brain-Heart Interconnectome. JS receives support from the Fritz-Thyssen-Stiftung (10.15.1.021MN) and the BMBF through the CMT-NET network (01GM1511B).
CONFLICT OF INTEREST
The authors have no conflict of interest to report.
DATA AVAILABILITY
Proteomic data are available on request.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-240050.
REFERENCES
[1] | Beijer D , Kim HJ , Guo L , O’Donovan K , Mademan I , Deconinck T , Van Schil K , Fare CM , Drake LE , Ford AF , et al. Characterization of HNRNPA1 mutations defines diversity in pathogenic mechanisms and clinical presentation. JCI Insight. (2021) ;6. doi: 10.1172/jci.insight.148363 |
[2] | Ding J , Hayashi MK , Zhang Y , Manche L , Krainer AR , Xu RM . Crystal structure of the two-RRM domain of hnRNP A1 (UP1) complexed with single-stranded telomeric DNA. Genes Dev. (1999) ;13: :1102–15. doi: 10.1101/gad.13.9.1102 |
[3] | Ezer S , Daana M , Park JH , Yanovsky-Dagan S , Nordström U , Basal A , Edvardson S , Saada A , Otto M , Meiner V , et al. Infantile SOD1 deficiency syndrome caused by a homozygous SOD1 variant with absence of enzyme activity. Brain. (2022) ;145: :872–8. doi: 10.1093/brain/awab416 |
[4] | Hackman P , Rusanen SM , Johari M , Vihola A , Jonson PH , Sarparanta J , Donner K , Lahermo P , Koivunen S , Luque H , et al. Dominant Distal Myopathy 3 (MPD3) Caused by a Deletion in the HNRNPA1 Gene. Neurol Genet. (2021) ;7: :e632. doi: 10.1212/nxg.0000000000000632 |
[5] | Izumi R , Ikeda K , Niihori T , Suzuki N , Shirota M , Funayama R , Nakayama K , Warita H , Tateyama M , Aoki Y , et al. Nuclear pore pathology underlying multisystem proteinopathy type 3-related inclusion body myopathy. Annals of Clinical and Translational Neurology. (2023) . doi: 10.1002/acn3.51977 |
[6] | Kamelgarn M , Chen J , Kuang L , Arenas A , Zhai J , Zhu H , Gal J . Proteomic analysis of FUS interacting proteins provides insights into FUS function and its role in ALS. Biochimica et Biophysica Acta. (2016) ;1862: :2004–14. doi: 10.1016/j.bbadis.2016.07.015 |
[7] | Kim HJ , Kim NC , Wang YD , Scarborough EA , Moore J , Diaz Z , MacLea KS , Freibaum B , Li S , Molliex A , et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. (2013) ;495: :467–73. doi: 10.1038/nature11922 |
[8] | Kim HJ , Mohassel P , Donkervoort S , Guo L , O’Donovan K , Coughlin M , Lornage X , Foulds N , Hammans SR , Foley AR , et al. Heterozygous frameshift variants in HNRNPA2B1 cause early-onset oculopharyngeal muscular dystrophy. Nature Communications. (2022) ;13: :2306. doi: 10.1038/s41467-022-30015-1 |
[9] | Korb MK , Kimonis VE , Mozaffar T . Multisystem proteinopathy: Where myopathy and motor neuron disease converge. Muscle Nerve. (2021) ;63: :442–54. doi: 10.1002/mus.27097 |
[10] | Krebs BB , De Mesquita JF . Amyotrophic Lateral Sclerosis Type 20 – In Silico Analysis and Molecular Dynamics Simulation of hnRNPA1. PloS One. (2016) ;11: :e0158939. doi: 10.1371/journal.pone.0158939 |
[11] | Mah-Som AY , Daw J , Huynh D , Wu M , Creekmore BC , Burns W , Skinner SA , Holla ØL , Smeland MF , Planes M , et al. An autosomal-dominant childhood-onset disorder associated with pathogenic variants in VCP. Am J Hum Genet. (2023) ;110: :1959–75. doi: 10.1016/j.ajhg.2023.10.007 |
[12] | Picchiarelli G , Dupuis L . Role of RNA Binding Proteins with prion-like domains in muscle and neuromuscular diseases. Cell Stress. (2020) ;4: :76–91. doi: 10.15698/cst2020.04.217 |
[13] | Ye K , Schulz MH , Long Q , Apweiler R , Ning Z . Pindel: a pattern growth approach to detect break points of large deletions and medium sized insertions from paired-end short reads. Bioinformatics. (2009) ;25: :2865–71. doi: 10.1093/bioinformatics/btp394 |
[14] | Zech M , Seibt A , Zumbaum B , Klee D , Meitinger T , Winkelmann J , Mayatepek E , Wagner M , Distelmaier F . MATR3 haploinsufficiency and early-onset neurodegeneration. Brain. (2021) ;144: :e72. doi: 10.1093/brain/awab240 |


