
An International Retrospective Early Natural History Study of LAMA2-Related Dystrophies
Abstract
Background:
LAMA2-related dystrophies (LAMA2-RDs) represent one of the most common forms of congenital muscular dystrophy and have historically been classified into two subtypes: complete or partial deficiency of laminin-211 (merosin). Patients with LAMA2-RD with the typical congenital phenotype manifest severe muscle weakness, delayed motor milestones, joint contractures, failure to thrive, and progressive respiratory insufficiency.
Objective:
While a comprehensive prospective natural history study has been performed in LAMA2-RD patients over 5 years of age, the early natural history of patients with LAMA2-RD 5 years and younger has not been comprehensively characterized.
Methods:
We extracted retrospective data for patients with LAMA2-RD ages birth through 5 years via the Congenital Muscle Disease International Registry (CMDIR). We analyzed the data using a phenotypic classification based on maximal motor milestones to divide patients into two phenotypic groups: “Sit” for those patients who attained that ability to remain seated and “Walk” for those patients who attained the ability to walk independently by 3.5 years of age.
Results:
Sixty patients with LAMA2-RD from 10 countries fulfilled the inclusion criteria. Twenty-four patients had initiated non-invasive ventilation by age 5 years. Hospitalizations during the first years of life were often related to respiratory insufficiency. Feeding/nutritional difficulties and orthopedic issues were commonly reported. Significant elevations of creatine kinase (CK) observed during the neonatal period declined rapidly within the first few months of life.
Conclusions:
This is the largest international retrospective early natural history study of LAMA2-RD to date, contributing essential data for understanding early clinical findings in LAMA2-RD which, along with the data being collected in international, prospective early natural history studies, will help to establish clinical trial readiness. Our proposed nomenclature of LAMA2-RD1 for patients who attain the ability to sit (remain seated) and LAMA2-RD2 for patients who attain the ability to walk independently is aimed at further improving LAMA2-RD classification.
INTRODUCTION
LAMA2-related dystrophies (LAMA2-RDs), caused by biallelic pathogenic variants in the LAMA2 gene, represent one of the most common forms of congenital muscular dystrophy internationally [1–3]. LAMA2 encodes the α2 subunit of the heterotrimeric laminin-211 protein (α2β1γ1), also known as merosin [4], as well as the α2 subunit of the laminin-221 (α2β2γ1) (formerly s-merosin) [5]. Laminin-211 and laminin-221 are large extracellular basement membrane glycoproteins and the predominant laminin isoforms in skeletal muscle. The laminin α2 subunit is expressed selectively in skeletal muscles, Schwann cells, placental trophoblast, dermis-epidermis junction, and myocardium [6–8] and binds to α-dystroglycan and integrin α7β1D [8, 9], thus playing a key role in connecting the extracellular matrix to the cell surface as part of the protein scaffold [10, 11]. Pathogenic variants in the LAMA2 gene cause either a deficiency of the laminin-211 protein or decreased expression of laminin-211, resulting in LAMA2-RDs [12].
The clinical manifestations of LAMA2-RDs include a wide range of presentations, from severe symptoms presenting congenitally to milder, later-onset symptoms presenting during childhood. The currently used nomenclature classifies these phenotypes according to their degree of laminin-211 (merosin) deficiency as assessed by muscle biopsy immunohistochemical studies [13]. Individuals with partial laminin-211 deficiency may present with milder symptoms during childhood, compared to patients with the more common complete deficiency of laminin-211 who typically present with more severe symptoms, with two thirds with symptoms recognized at birth and one third with symptoms recognized by age 6 months [13–20]. Symptom severity and onset have been found to be correlated with the amount of residual laminin-211 expression and the location and type of pathogenic variants in LAMA2 in addition to other potential disease modifying factors [19, 21, 22]. While residual laminin-211 expression may be correlated with milder symptom severity and a later onset, this is not always the case. Given that the LAMA2-RD diagnosis is currently typically made via next generation sequencing, and thus muscle biopsy is performed less frequently, it is important to acknowledge that the classifications of “complete merosin deficiency” and “partial merosin deficiency” are not as easily identifiable; therefore, use of this traditional nomenclature may complicate efforts to stratify patients both for natural history studies and for future clinical trials.
Independent ambulation is usually not achieved in cases of complete deficiency of laminin-211, although individuals with residual expression of laminin-211 may achieve independent ambulation for variable lengths of time. Contributions to motor function in the LAMA2-RDs include the orthopedic complications of severe, progressive proximal and distal joint contractures, scoliosis, lordosis, and neck rigidity, which are common and occur to varying degrees in both patients who do not achieve independent ambulation and those who do [19, 24].
Respiratory difficulties are common in individuals with LAMA2-RDs and are typically appreciated early in life [13]. Specifically, complications related to progressive, restrictive pulmonary insufficiency due to weakness of respiratory muscles is one of the most frequently noted causes of hospitalization for young children with LAMA2-RD and one of the most common causes of morbidity and mortality in this patient population [13, 22].
Other clinical characteristics seen in patients with LAMA2-RD include abnormal white matter appearance on brain MRI due to increased water signal (increased signal on T2-weighted images and decreased signal on T1-weighted images), which are typically seen in all patients with LAMA2-RD by approximately 6 months of age, sparing compacted white matter tracts [25–27]. Occipital and temporal polymicrogyria can also be seen [7, 19, 26, 28], and has been correlated with an increased risk of epilepsy in patients with LAMA2-RD [26, 28]. Additionally, elevated creatine kinase (CK) (>1,000 U/L) is seen in all patients, which, along with the abnormal white matter appearance on brain MRI can help aid the clinical recognition and diagnosis of LAMA2-RD, although white matter changes may not be appreciated in all patients during the first few months of life [13].
Given that currently there are multiple, promising therapeutic approaches in preclinical development, including a gene therapy approach using the linker proteins alphaLNNd and mini-agrin [29], a laminin-111 protein therapy [30], and a LAMA1 upregulation therapy [31] which all share the goal of aiming to treat affected children early in the course of disease, the comprehensive study of the early natural history of the LAMA2-RDs is essential for clinical trial readiness. Previous retrospective natural history studies of the LAMA2-RDs include a study of 46 pediatric patients with LAMA2-RD in the United Kingdom [22] and a study of 130 pediatric patients with LAMA2-RD in China [23]. A retrospective study conducted in the UK examined genotype-phenotype correlations in 51 patients with LAMA2-RD [19]. Cross-sectional, single-center, observational studies were conducted in 27 pediatric and adult patients with LAMA2-RD in the Netherlands [32] and 21 pediatric and adult patients in Qatar [33]. A prospective natural history and outcome measures study in patients with LAMA2-RD (ages 4–22 years) was performed in the United States (by our team) over 4 years’ duration and measured longitudinal changes in motor and respiratory clinical outcome measures in 24 children with LAMA2-RD [34]. Despite these efforts to collect both retrospective and prospective natural history data in patients with LAMA2-RD, detailed natural history of the first months and years of life, namely from birth through five years of age, has not been comprehensively studied.
METHODS
We performed an international retrospective cohort study to document the natural history of individuals with LAMA2-RD from birth (0 months) through five years of age (through age 71 months) who were born between the years 2000 and 2017 inclusive (ClinicalTrials.gov Identifier: NCT04299321; Aspire IRB Protocol: PRO-LAMA2-001). A LAMA2-RD diagnosis was necessary for inclusion, defined as the presence of either two LAMA2 pathogenic variants or one LAMA2 pathogenic variant and a supporting clinical phenotype based on two or more of the following: physical examination, brain imaging, muscle imaging, muscle biopsy, and serum CK levels.
Data were entered into the Congenital Muscle Disease International Registry (CMDIR): https://cmdir.org/. The CMDIR is a global registry of individuals affected by congenital muscle disease which is compliant with HIPAA and GDPR laws, operated by individuals from patient organizations (Cure CMD, A Foundation Building Strength, and Team Titin) and overseen by an institutional review board and a scientific advisory board (comprised of physicians, scientists, a genetic counselor and a patient advocacy representative from industry). Parents provided consent to participate in the study on behalf of their child affected with LAMA2-RD. Medical records were either directly provided by parents or parents provided authorization for Cure CMD to obtain medical records directly from healthcare providers. Data from medical records were extracted and entered into an anonymized excel spreadsheet for analysis and review. Of the collected longitudinal variables, the following were considered: CK levels; respiratory complications including the use of noninvasive ventilation (NIV); feeding/nutritional difficulties including swallowing difficulties, G-tube insertion, and failure to thrive; orthopedic complications including joint contractures, lordosis, kyphosis, hip dysplasia, scoliosis, torticollis, pectus excavatum, and fractures; and hospitalization events. The pathogenicity of the reported LAMA2 variants was included in this dataset and had been classified by the laboratory where the genetic testing of the associated patient was conducted. Each patient had genetic testing performed and reported by a Clinical Laboratory Improvement Amendments (CLIA) certified laboratory.
We classified the patients phenotypically based on maximal motor milestones into two subgroups: “Sit” for those patients who attained that ability to remain seated (with or without support) including those who could stand with assistance and “Walk” for those patients who attained the ability to walk, as defined by taking unassisted steps/walking independently by the age of 3.5 years. These maximal motor milestones were defined as the highest level of motor skill ever attained, even if the patient later lost that motor function, as may occur in patients with LAMA2-RD [19]. The oldest patient in this cohort who attained a maximal motor milestone of walking was 3.3 years old, and thus we established 3.5 years as the cut off age for being classified into the “Walk” subgroup in order to capture all the patients in this cohort who achieved the ability to walk independently. Patients for whom we lacked sufficient maximal motor milestone data to assign to either subgroup were categorized as having missing information on ambulation forthis study.
RESULTS
Cohort description
The study cohort included 60 patients from 10 different countries: Argentina, Australia, Bahrain, France, India, New Zealand, Saudi Arabia, Spain, the United Kingdom, and the United States (Supplementary Table 1). Of the patients in this cohort, 31 (52%) were male and 29 (48%) were female with birth dates ranging from September 2001 to November 2017. As classified clinically based on available motor function milestone data, 42 individuals (70%) were classified as part of the “Sit” subgroup, 9 (15%) were classified as part of the “Walk” subgroup, and 9 (15%) had missing information on ambulation. There was one reported death within the cohort which occurred in a patient within the “Sit” subgroup at 46 months of age within the setting of a respiratory tract infection. Clinical images capturing the pattern of weakness observed in a patient within the “Sit” subgroup are included in Fig. 1.
Fig. 1
Classical clinical phenotype of LAMA2-RD in a patient (age 43 months) who obtained a maximal motor milestone of sitting (“Sit” subgroup). Clinical images demonstrating: (A) hyperlordotic sitting posture, (B) facial weakness with mouth maintained in an open position, and (C) axial weakness with neck extension weakness. [Permission to publish images provided by the patient’s family].
![Classical clinical phenotype of LAMA2-RD in a patient (age 43 months) who obtained a maximal motor milestone of sitting (“Sit” subgroup). Clinical images demonstrating: (A) hyperlordotic sitting posture, (B) facial weakness with mouth maintained in an open position, and (C) axial weakness with neck extension weakness. [Permission to publish images provided by the patient’s family].](https://content.iospress.com:443/media/jnd-prepress/jnd--1--1-jnd240048/jnd--1-jnd240048-g001.jpg)
Genetics
Within this cohort of 60 patients, there were 107 total variants reported and 72 distinct variants reported. For each of the 72 distinct variants, there was a range of frequencies at which any variant appeared in the cohort, ranging from being reported once to being reported 14 times. The most common variants reported in this cohort were c.2049_2050delAG which was reported 14 times in this cohort, and c.939_940delAT, c.3630delT, and c.4614del, each of which were reported 4 times in this cohort.
Each of the reported variants was classified by a CLIA certified laboratory that conducted the genetic testing as pathogenic, likely pathogenic, variant of unknown significance (VUS), likely benign, or benign. Of the 60 patients, 51/60 (85%) had two reported pathogenic/likely pathogenic LAMA2 variants and 9/60 (15%) had one reported pathogenic/likely pathogenic LAMA2 variant. Of the 9 patients with only one reported pathogenic/likely pathogenic variant, 2 patients reported an additional VUS which was interpreted as PM2 (recommended for absence/rarity criterion), based on extremely low frequency in gnomAD population databases. Both of the patients with a VUS in compound heterozygosity with a pathogenic/likely pathogenic variant were clinically classified as being in the “Sit” subgroup. In the other 7 patients with only one reported LAMA2 variant, the single variant was classified as pathogenic/likely pathogenic variant in each (Supplemental Table 2), and the carefully reviewed medical records described a clinical phenotype consistent with LAMA2-related dystrophy.
Onset of symptoms and age at genetic diagnosis
The age at which the first symptom was recognized was recorded for 31 of the 60 patients in this cohort (52%). Eleven of these 31 (35%) had their first symptom recognized at birth, 13/31 (42%) had their first symptom recognized between 1–6 months of life, 3/31 (10%) had their first symptom recognized between 7–12 months of life, and 4/31 (13%) had their first symptom recognized after the first 12 months of life. Of the 11 patients for whom the first symptom was recognized at birth, 4 (36%) were diagnosed at birth, 2 (18%) were diagnosed between 1–6 months of age, 1 (9%) was diagnosed between 7–12 months of age, 3 (27%) were diagnosed after 12 months of age, and the age at diagnosis was not provided for 1 individual. Of the 13 individuals for whom the first symptom was recognized at 1–6 months of age, genetic diagnosis was made in 3 (23%) between 1–6 months of age, in 6 (46%) between 7–12 months of age, and in 3 (23%) after 12 months of age, with the age at diagnosis not provided for one patient. In 3/3 individuals for whom the first symptom was recognized at 7–12 months of age and 4/4 individuals for whom the first symptom was recognized after 12 months of age were all diagnosed after 12 months of age (Table 1, Supplementary Table 3).
Table 1
Clinical characteristics by clinical classification
| Total | “Sit” | “Walk” | Missing | |
| Information on | ||||
| Ambulation | ||||
| Onset of Symptoms | ||||
| Symptom recognition before 1 year | n = 27/31 | n = 19/19 | n = 3/7 | n = 5/5 |
| (87%) | (100%) | (43%) | (100%) | |
| Diagnosis established before 1 year | n = 16/29 | n = 13/18 | n = 1/6 | n = 2/5 |
| (55%) | (72%) | (17%) | (40%) | |
| Orthopedic Complications | ||||
| Joint Contractures | 16 | 16 | 0 | 0 |
| Lordosis or Kyphosis | 19 | 13 | 6 | 0 |
| Hip Dysplasia or Subluxation | 16 | 14 | 1 | 1 |
| Scoliosis | 15 | 12 | 2 | 1 |
| Torticollis | 11 | 8 | 3 | 0 |
| Pectus Excavatum | 11 | 9 | 2 | 0 |
| Fractures | 6 | 6 | 0 | 0 |
| Total Orthopedic Complication | 94 | 78 | 14 | 2 |
| Feeding/Nutritional Complications | ||||
| Feeding Difficulties | 21 | 16 | 2 | 3 |
| Swallowing Difficulties | 18 | 14 | 1 | 3 |
| G-tube | 12 | 10 | 0 | 2 |
| Failure to Thrive | 9 | 8 | 0 | 1 |
| Total Feeding/Nutritional Complications | 60 | 48 | 3 | 9 |
Creatine kinase
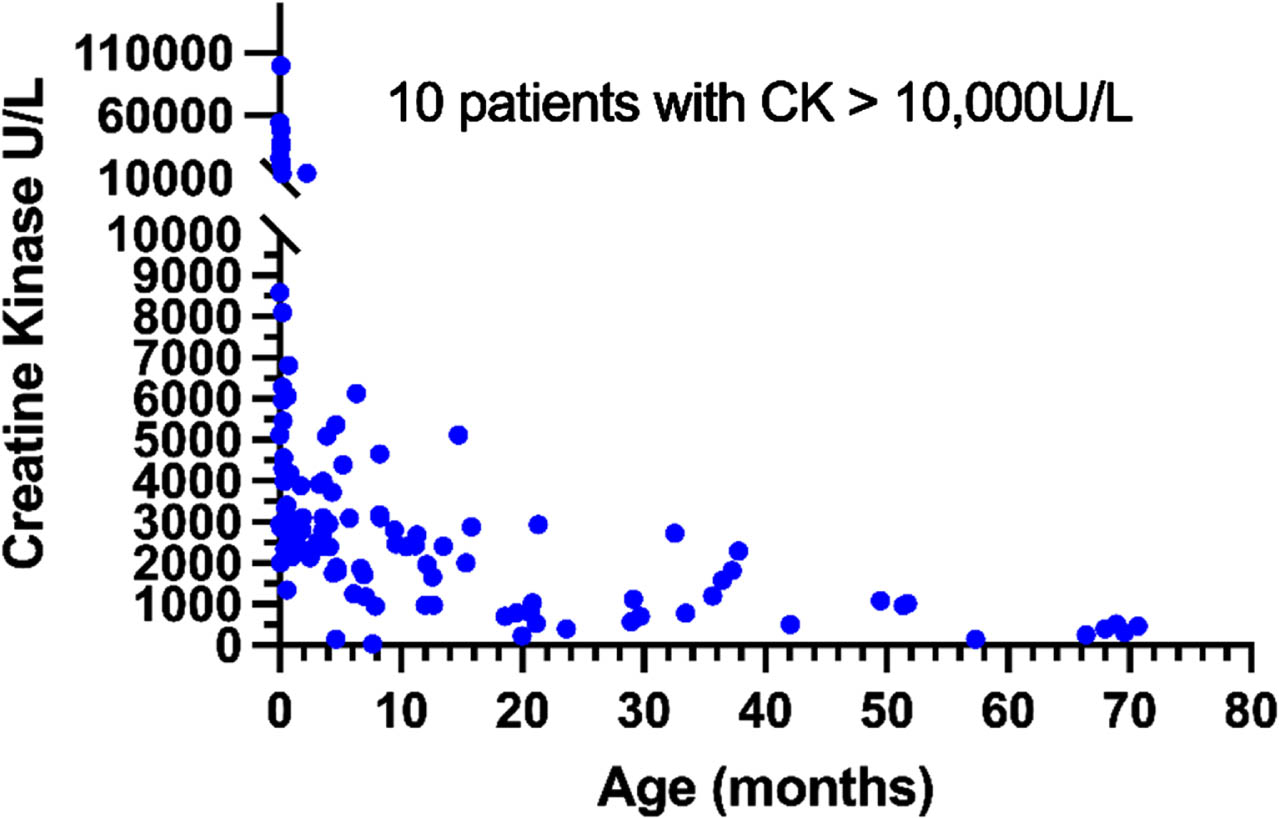
Serum CK values demonstrated a consistent pattern notable for significant elevation around the time of birth/during the first month of life: 9 out of 14 samples from patients under the age of 7 days had elevated CK levels of more than 10,000 U/L, with a median CK value for 0 to 1 month of age of 5,456 U/L (n = 30, range 1,343–100,000 U/L). During the first year of life, CK levels significantly declined, so that the median CK level among patients older than 12 months was 969 U/L (n = 36, range 152–5,121 U/L) (Fig. 2). Accordingly, the Spearman rank correlation test showed a strong and statistically significantnegative correlation between age and CK levels (r = –0.76; two-tailed p < 0.0001; 95% confidence interval: –0.83 to –0.67), and the Kruskal-Wallis test showed significant differences between the four age groups: birth –1 month, 1–6 months, 6–12 months and >12 months (p < 0.0001). CK levels were not significantly different between age-matched (±1 month) patients in the “Sit” and “Walk” subgroups (two-tailed Wilcoxon matched-pairs signed ranktest, n = 7).
Fig. 2
CK vs Age.
Respiratory involvement
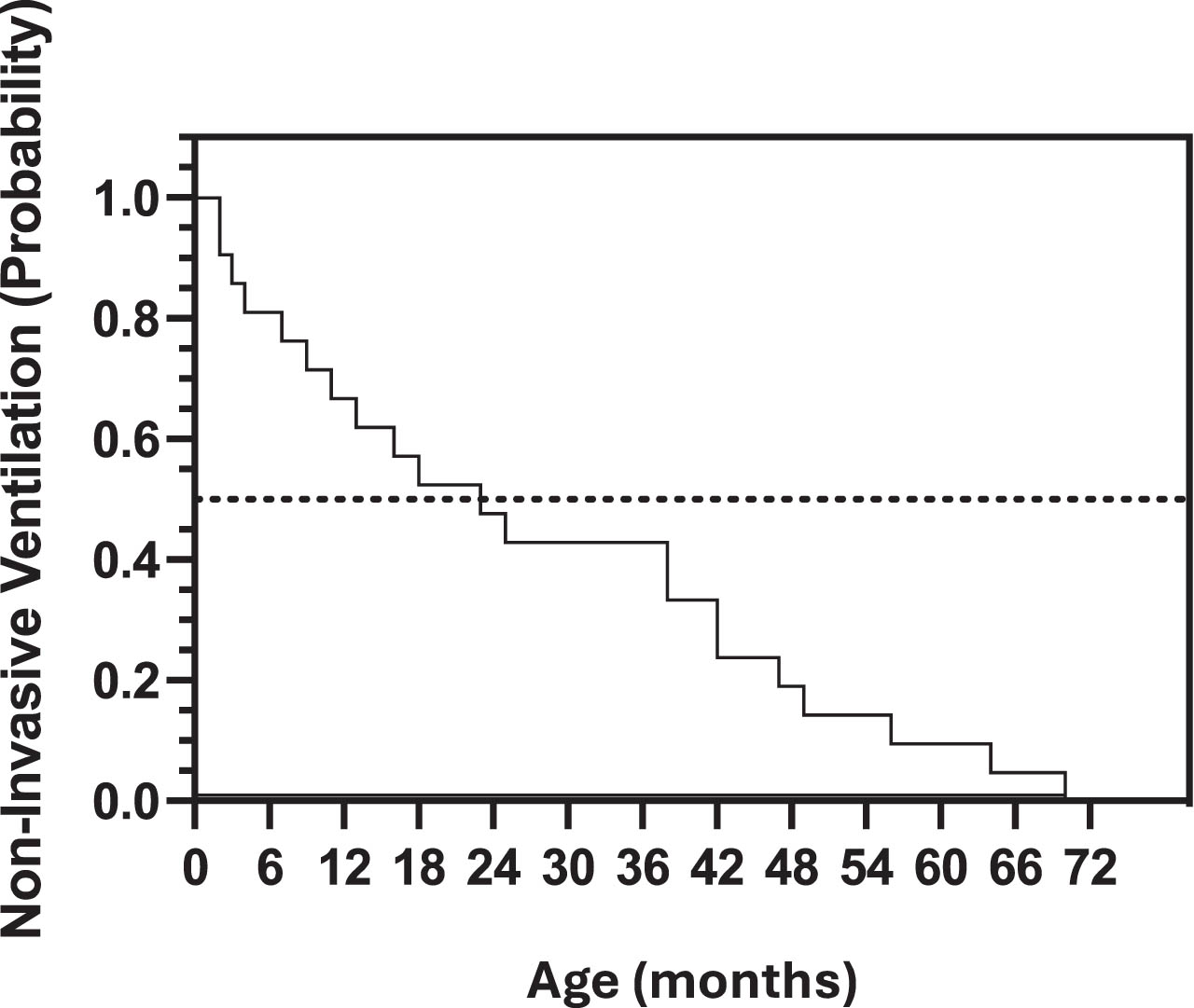
Of the 24 individuals in this cohort who reported the use of NIV in the form of bilevel positive airway pressure (BiPAP) or continuous positive airway pressure (CPAP), 12/24 (50%) had initiated BiPAP or CPAP by the age of 24 months, 22/24 (92%) had initiated the use of BiPAP or CPAP by 50 months of age, and 24/24 (100%) had initiated the use of BiPAP or CPAP before 72 months of age (Fig. 3). Of these 24 individuals, 18 (75%) were classified as part of the “Sit” subgroup, 2 (8%) were classified as part of the “Walk” subgroup, and 4 (17%) had missing information on ambulation.
Fig. 3
Kaplan Meier curve depicting the probability of non-invasive ventilation (in the form of BiPAP or CPAP) for the patients with LAMA2-RD in this cohort.
Nutritional challenges
Reported feeding/nutritional difficulties in this cohort include feeding difficulties, swallowing difficulties, gastrostomy-tube insertion, and failure to thrive, all of which were reported more frequently in those who were classified as part of the “Sit” subgroup as compared to those classified as part of the “Walk” subgroup or those who had missing information on ambulation (Table 1).
Orthopedic complications
Orthopedic complications are a well-known feature of LAMA2-RD. It is notable that such complications are part of the early natural history of LAMA2-RD, including joint contractures, lordosis/kyphosis, hip dysplasia, scoliosis, torticollis, pectus excavatum, and fractures. All of these complications were reported more frequently in those patients who were classified as part of the “Sit” subgroup as compared to those patients classified as part of the “Walk” subgroup or those who had missing information on ambulation (Table 1). The youngest age at which scoliosis was reported in this cohort was 18 months, and the mean age at which scoliosis was first reported in this cohort was 51.9 months (n = 8).
Hospitalization events
Retrospective data on hospitalization events revealed that the average age at the time of the first reported hospitalizations in this cohort, which were due to poor oral intake, oxygen desaturation, respiratory infection, or hypoglycemia, were all less than 24 months of age (Supplementary Table 4). For the other reported hospitalizations, which were for respiratory distress, respiratory infection, severe cough, lung collapse or dehydration, the average age at the time of first hospitalization was less than 36 months of age. Comparing the number of patients represented and the number of total events reported, we also see that there are many hospitalization events, including for pneumonia, respiratory distress, and hypoglycemia among others, for which individual patients had experienced multiple hospitalization events. Of the 21 distinct patients who reported any hospitalization event related to reparatory involvement, 8 (38%) did not report any use of NIV. Of the 25 distinct patients who reported any hospitalization events, 21/25 (84%) were part of the “Sit” subgroup, 4/25 (16%) were missing information on ambulation, and 0 were part of the “Walk” subgroup.
DISCUSSION
Here, we present a retrospective early natural history study of a large international pediatric cohort of 60 patients with LAMA2-RD with data collected from medical records. We confirm and quantify early symptom manifestations in the LAMA2-RDs, including elevated CK, progressive respiratory insufficiency requiring noninvasive ventilatory support, feeding/nutritional difficulties, and orthopedic complications. The early symptoms and complications observed in children with LAMA2-RD reported here were often underlying causes of hospitalization events during the first five years of life.
All 60 individuals in this cohort have a clinical diagnosis of LAMA2-RD with at least one pathogenic/likely pathogenic variant and a supporting clinical phenotype. In particular, 51/60 patients have two pathogenic/likely pathogenic variants, and 9/60 patients have one identified pathogenic/likely pathogenic variant and a clinical phenotype consistent with LAMA2-RD. It is important to note that in the LAMA2-RDs the identification of a second pathogenic variant is often challenging since copy number variants (CNVs) and intronic variants may not be captured unless analyses for CNVs and evaluations of intronic regions are performed [35]. Using classifications provided by the CLIA certified laboratories where LAMA2 genetic sequencing was performed, we identified 105/107 total reported LAMA2 variants as pathogenic/likely pathogenic and 2 as VUS. Our cohort’s data demonstrate a lag between the age at symptom recognition (87% of this cohort with symptoms recognized before the end of the first year) and age at diagnosis (55% of this cohort diagnosed before the end of the first year). Given the increasing availability of next generation sequencing (NGS) technology which enables the rapid sequencing of many congenital muscle disease genes simultaneously, including the availability in some centers of NGS for patients in the neonatal intensive care unit (NICU), we expect that the time to diagnosis, particularly in patients presenting with congenital symptoms, will decrease. We also expect that the number of patients with LAMA2-RD with genetic confirmation will increase overall.
Early recognition of clinical symptoms could also contribute to an earlier diagnosis of LAMA2-RD. Of the 31 individuals in this cohort who had a reported age at first symptom recognition, in 24 (77%) the first symptoms were recognized within the first six months of life. When stratified into the maximal motor milestone phenotype subgroups, patients in this cohort who were classified as part of the “Sit” subgroup more frequently had a reported age at first suspected LAMA2-RD symptoms of 12 months or younger, while the patients with first suspected symptoms occurring after 12 months of age were classified as part of the “Walk” subgroup.
Hospitalization data collected in this study suggest that respiratory insufficiency, orthopedic complications, and feeding/nutritional difficulties are leading causes of morbidity in children with LAMA2-RD during the first years of life, which is important to note as there are specific interventions for many of these complications which can minimize symptoms and improve quality of life. Respiratory insufficiency remains a leading cause of morbidity for patients with LAMA-2RD, and when it manifests, requires the use of NIV. While 8/21 (38%) of patients in this cohort with reported respiratory related hospital events did not report the use of NIV, this does not necessarily mean that NIV was not clinically indicated for those patients or that they would not have benefited from the use of NIV. NIV was initiated at the discretion of the local medical team, and thus the threshold for NIV initiation was not standardized across medical centers in this multinational, multicenter, retrospective study. Of note, while BiPAP is the recommended form of NIV in patients with LAMA2-RD, some patients in this cohort were started on CPAP by local medical providers. In this cohort, 24 patients had initiated NIV by five years of age, with over half of these patients initiating non-invasive ventilation for at least part of the day and/or night by two years of age. This data suggests that noninvasive ventilatory support may be required earlier than previously thought in patients with LAMA2-RD, especially for those in the “Sit” subgroup, who made up 75% of the patients in this cohort requiring NIV. This is essential information for clinicians caring for patients with LAMA2-RD, given that timely initiation of NIV reduces morbidity and improves quality of life.
As evidenced by this data, feeding/nutritional difficulties are characteristic of the early natural history of the LAMA2-RDs and include feeding and swallowing difficulties leading to failure to thrive and the need for gastrostomy-tube insertion. It is important that feeding/nutritional difficulties are recognized and monitored in individuals with LAMA2-RD to maximize nutritional status, which is important for overall general health and growth especially in the first few years of life. Furthermore, hypoglycemic episodes, which can occur in the LAMA2-RDs related to patients’ reduced muscle mass and most typically occur during the first few years of life, can be triggered by prolonged fasting or illness, especially in the setting of failure to thrive, and often require hospitalization [36]. Recognizing the risk of hypoglycemic episodes in the LAMA2-RDs is essential for identifying and promptly treating this medical emergency as well as for providing appropriate patient education and resources, including access to a home glucometer.
The recognition of early orthopedic complications in patients with LAMA2-RD can enable the optimization of clinical monitoring, such as surveillance of the spine, the tailoring of interventions, such as the provision of supportive seating, and careful consideration for potential bracing and the assessment of when surgical scoliosis repair may be indicated.
The reported serum CK values in this cohort validate previous reports of elevated CK in LAMA2-RD patients. Both the strong negative correlation between CK values and age and the significant decline of CK values after the first year of life suggest that CK may have the potential to serve as a surrogate serum biomarker of disease that can be used to both aid clinical recognition of the LAMA2-RDs as well as follow disease progression. It is important to note, however, that given the natural history of decline in CK during the first year of life, CK would not be suitable as a biomarker for a potential therapeutic response. Our data further support the diagnostic importance of highly elevated CK (in the thousands) in the neonatal period in the LAMA2-RDs and thus may contribute to the early recognition of LAMA2-RDs internationally. For example, early recognition of high CK is highly suggestive of LAMA2-RDs, as well as alpha-dystroglycanopathies, LMNA-CMD, and Duchenne muscular dystrophy (DMD). With promising therapies for the LAMA2-RDs in development, neonatal CK elevation may serve as a robust screening tool. In fact, newborn screening programs for DMD which are using newborn CK levels as a means to screen for DMD would likely identify patients with LAMA2-RD [37].
Historically, the nomenclature used for the congenital muscular dystrophy subtype due to laminin-211 (merosin) deficiency included congenital muscular dystrophy type 1A (MDC1A) and merosin deficient CMD, as well as subclassifications of merosin deficiency as “complete” or “partial” merosin deficiency. We suggest that the field move towards a nomenclature which is based on the distinct motor phenotypes seen in patients with LAMA2-RD and propose that moving forward, we consider using the gene-related nomenclature terms LAMA2-RD1 to correspond to the maximal motor milestone of sitting, as defined as those patients who attained the ability to remain seated (with or without support), including those who could stand with assistance, and LAMA2-RD2 to correspond to the maximal motor milestones of walking for those patients who attained the ability to walk, as defined by taking unassisted steps/walking independently by age 3.5 years. (There have been rare reports of a few patients who attained the ability to walk independently after 3.5 years of age [22, 23]; however, we chose 3.5 years based on the data of this large international cohort.) In this way, patients with LAMA2-RD are more clearly classified in a manner which more accurately characterizes overall motor function, keeping in mind that even within such subgroup classifications there may be variations in motor function abilities. For the purpose of both prospective natural history studies in progress and future clinical trials, classifying patients into subgroups with comparatively similar motor function can help the design of clinical trials and the potential of such trials to capture meaningful changes in function by applying appropriate outcome measures for patients’ motor function abilities.
Due to the retrospective nature of this cohort study, there are inherent limitations related to the collection, analysis, and interpretation of the data. One of these limitations is that these retrospective data had been collected over a timeframe of 18 years, and thus the current age range of the patients included in this cohort is wide. While the year 2000 was selected as the beginning timepoint of the study due to relative advances in the recognition and clinical standard of care for individuals with LAMA2-RD around that time, the data from the earlier years of this study may be limited when compared to data collected during more recent timepoints.
Additionally, as data from this study were collected via review of medical records, there are limitations to the data related to the unstructured documentation provided by local medical teams, which often included inconsistent or incomplete information. Consequently, we were not able to conduct meaningful analysis on aspects of the LAMA2-RDs including brain MRI findings, cardiac involvement, and more detailed scoliosis assessments. Also, each finding and event represented in this data was defined and documented by local medical teams, limiting our ability to describe specific definitions or criteria for these findings and events. Clinician and parent symptom recognition may vary greatly and may have contributed to the timeframe between patient age at first symptom recognition and patient age at diagnosis, which this study identified as the date of the positive genetic testing report. Furthermore, given that genetic testing relies on recognition of symptoms as well as the complexities of obtaining genetic testing via local medical teams (which varies internationally), the date of genetic diagnosis may be many months after the initial clinical suspicion of LAMA2-RD.
This study is the largest international retrospective early natural history study of the LAMA2-RDs to date, which provides data essential for understanding the clinical history during the first weeks, months, and years of life in children with LAMA2-RD. This age group will be of considerable importance in future clinical trials that aim to treat children with LAMA2-RD early in the disease course. This study highlights that symptoms of respiratory insufficiency, feeding/nutritional difficulties, and orthopedic involvement are prevalent during the first few years of life (Table 2), requiring multidisciplinary surveillance and care. With multiple promising therapeutic approaches for LAMA2-RDs currently in preclinical development, it is essential that the early natural history of the LAMA2-RDs be comprehensively understood to ensure clinical trial readiness. We propose a new nomenclature aimed at improving the clarity of clinical classification for the LAMA2-RDs based on maximal motor milestones, namely LAMA2-RD1 corresponding to the maximal motor milestone of sitting and LAMA2-RD2, corresponding to the maximal motor milestones of walking. We believe that the use of this classification will help to ensure that natural history data collection and evaluation as well as the design of future clinical trials clearly and consistently classify patients with distinct motor phenotypes.
Table 2
Demographics and outcomes of this cohort and other published large retrospective natural history cohorts in LAMA2-RD
| This Cohort | Zambon et al., 2020 | Tan et al., 2021 | |
| Total number of patients | n = 60 | n = 46 | n = 130 |
| Country | International | UK | China |
| Age Range | 0–5 years | 0–22.7 years | 0–27.3 years |
| Median age at last follow up | 5 years | 12.1 years | 6.6 years |
| Number of patients using NIV | 24 (40%) | 22 (48%) | 4 (3%) |
| Median age when NIV started | 24 months | 11.7 years | 12.7 years |
| Number of patients with G-tube or NG-tube | 12 (20%) | 20 (43%) | 2 (2%) |
| Number of patients who developed scoliosis | 15 (25%) | 34 (74%) | 45 (40.5%) |
| Median age when scoliosis first observed | 4.5 years | 6.8 years | 6.0 years |
This study of the early natural history of the LAMA2-RDs further highlights the need for, and the feasibility of, prospective early natural history studies of the LAMA2-RDs, a number of which are already in progress internationally, including at our site. The intended goal of these prospective early natural history studies in LAMA2-RD is to be able to elucidate clinical findings during the first months of life which may be predictive of future maximal motor function. For example, the age when a child may attain the ability to remain seated when placed in a seated position may be predictive of future motor function. Good quality motor function data with this level of granularity and standardized definitions of motor achievements can be obtained from the prospective natural history studies in progress, which entail formal motor development assessments performed by specialized pediatric neuromuscular physical therapists. The collective data gathered internationally from these early natural history studies will help to optimize clinical care and validate outcome measures, further improving clinical trial readiness for patients with LAMA2-RD, for whom the therapies currently in development are seeking to address an unmet therapeutic need.
ACKNOWLEDGMENTS
We thank the affected individuals with LAMA2-RD and their families for sharing clinical information via the Congenital Muscle Disease International Registry (CMDIR) for the purpose of this study. We thank Cure CMD for its support of this study and its ongoing management of the CMDIR. We thank Tristen Moors, Karima Alui, Marc Giles and Patrick Rush for help in extracting data. We thank Tonya Marmon for help with data analyses. We thank Nicole Acquaye for her help with initial data extraction.
FUNDING
This work was supported by intramural funds from the National Institute of Neurological Disorders and Stroke, NIH (grant to C.G.B) and Cure CMD. Data extraction and analyses were supported by Prothelia Therapeutics.
CONFLICT OF INTEREST
Carsten G. Bönnemann is an Editorial Board Member of this journal but was not involved in the peer-review process nor had access to any information regarding its peer-review.
DATA AVAILABILITY STATEMENT
Anonymized data can be shared with qualified investigators upon request.
AUTHOR CONTRIBUTIONS
RA established the exact data fields that would be extracted from medical records. CGB and ARF reviewed the initial data entry forms.
GD and RA designed the study protocol and obtained IRB approval.
JP, RA, and GD conducted participant screening, consenting, medical records collection and collation, and contributed to data curation.
CGB and ARF contributed to the design of the study and review of the data analyses.
LH, RO, CGB and ARF contributed to the drafting and revision of the manuscript.
LH and RO conducted data analysis.
All authors reviewed the manuscript.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-240048.
REFERENCES
[1] | Ge L , Zhang C , Wang Z , et al. Congenital muscular dystrophies in China. Clin Genet. (2019) ;96: :207–15. |
[2] | Sframeli M , Sarkozy A , Bertoli M , et al. Congenital muscular dystrophies in the UK population: Clinical and molecular spectrum of a large cohort diagnosed over a 12-year period. Neuromuscular Disorders. (2017) ;27: :793–803. |
[3] | Muntoni F , Voit T . The congenital muscular dystrophies in A century of exciting progress. Neuromuscular Disorders. (2004) ;14: :635–49. |
[4] | Ehrig K , Leivo I , Argraves WS , et al. Merosin, a tissue-specific basement membrane protein, is a laminin-like protein. Proc Natl Acad Sci U S A. (1990) ;87: (9), 3264–8. |
[5] | Vuolteenaho R , Nissinen M , Sainio K , et al. Human laminin M chain (merosin): complete primary structure, chromosomal assignment, and expression of the M and A chain in human fetal tissues. J Cell Biol. (1994) ;124: (3), 381–94. |
[6] | Vainzof M , Richard P , Herrmann R , et al. Prenatal diagnosis in laminin α2 chain (merosin)-deficient congenital muscular dystrophy: A collective experience of five international centers. Neuromuscular Disorders. (2005) ;15: :588–94. |
[7] | Taratuto AL , Lubieniecki F , Díaz D , et al. Merosin-deficient congenital muscular dystrophy associated with abnormal cerebral cortical gyration: an autopsy study. Neuromuscul Disord. (1999) ;9: (2), 86–94. |
[8] | Colognato H , Yurchenco PD . Form and function: The laminin family of heterotrimers. Developmental Dynamics. (2000) ;218: :213–34. |
[9] | Ervasti JM , Campbell KP . A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol. (1993) ;122: (4), 809–23. |
[10] | McKee KK , Harrison D , Capizzi S , et al. Role of laminin terminal globular domains in basement membrane assembly. Journal of Biological Chemistry. (2007) ;282: :21437–47. |
[11] | Yurchenco PD , McKee KK , Reinhard JR , et al. Laminin-deficient muscular dystrophy: Molecular pathogenesis and structural repair strategies. Matrix Biology. (2018) ;71–72: :174–87. |
[12] | Helbling-Leclerc A , Zhang X , Topaloglu H , et al. Mutations in the laminin alpha 2-chain gene (LAMA2) cause merosin-deficient congenital muscular dystrophy. Nat Genet. (1995) ;11: (2):216–8. |
[13] | Sarkozy A , Foley AR , Zambon AA , et al. LAMA2-Related Dystrophies: Clinical Phenotypes, Disease Biomarkers, and Clinical Trial Readiness. Front Mol Neurosci. (2020) ;13: :123. |
[14] | Naom I , D’Alessandro M , Sewry CA , et al. Laminin alpha 2-chain gene mutations in two siblings presenting with limb-girdle muscular dystrophy. Neuromuscul Disord. (1998) ;8: (7), 495–501. |
[15] | Pegoraro E , Marks H , Garcia CA , et al. Laminin α2 muscular dystrophy: Genotype/phenotype studies of 22 patients. Neurology. (1998) ;51: :101–10. |
[16] | Topaloğlu H , Talim B , Vignier N , et al. Merosin-deficient congenital muscular dystrophy with severe mental retardation and normal cranial MRI: a report of two siblings. Neuromuscul Disord. (1998) ;8: (3-4):169–74. |
[17] | Tezak Z , Prandini P , Boscaro M , et al. Clinical and molecular study in congenital muscular dystrophy with partial laminin α2 (LAMA2) deficiency. Hum Mutat. (2003) ;21: :103–11. |
[18] | Oliveira J , Santos R , Soares-Silva I , et al. LAMA2 gene analysis in a cohort of 26 congenital muscular dystrophy patients. Clin Genet. (2008) ;74: :502–12. |
[19] | Geranmayeh F , Clement E , Feng LH , et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscular Disorders. (2010) ;20: :241–50. |
[20] | Xiong H , Tan D , Wang S , et al. Genotype/phenotype analysis in Chinese laminin-α2 deficient congenital muscular dystrophy patients. Clin Genet. (2015) ;87: :233–43. |
[21] | Prandini P , Berardinelli A , Fanin M , et al. LAMA2 loss-of-function mutation in a girl with a mild congenital muscular dystrophy. Neurology. (2004) ;63: :1118–21. |
[22] | Zambon AA , Ridout D , Main M , et al. LAMA2-related muscular dystrophy: Natural history of a large pediatric cohort. Ann Clin Transl Neurol. (2020) ;7: :1870–82. |
[23] | Tan D , Ge L , Fan Y , et al. Natural history and genetic study of LAMA2-related muscular dystrophy in a large Chinese cohort. Orphanet J Rare Dis. (2021) ;16: (1):319. |
[24] | Prandini P , Berardinelli A , Fanin M , et al. LAMA2 loss-of-function mutation in a girl with a mild congenital muscular dystrophy. Neurology. (2004) ;63: :1118–21. |
[25] | Philpot J , Cowan F , Pennock J , et al. Merosin-deficient congenital muscular dystrophy: the spectrum of brain involvement on magnetic resonance imaging. Neuromuscul Disord. (1999) ;9: (2), 81–85. |
[26] | Camelo CG , Artilheiro MC , Martins Moreno CA , et al. Brain MRI abnormalities, epilepsy and intellectual disability in LAMA2 related dystrophy - a genotype/phenotype correlation. J Neuromuscul Dis. (2023) ;10: :483–92. |
[27] | Leite CC , Lucato LT , Martin MGM , et al. Merosin-deficient congenital muscular dystrophy (CMD): A study of 25 Brazilian patients using MRI. Pediatr Radiol. (2005) ;35: :572–9. |
[28] | Natera-de Benito D , Muchart J , Itzep D , et al. Epilepsy in LAMA2-related muscular dystrophy: An electro-clinico-radiological characterization. Epilepsia. (2020) ;61: :971–83. |
[29] | Reinhard JR , Lin S , McKee KK , et al. Linker proteins restore basement membrane and correct LAMA2-related muscular dystrophy in mice. Sci Transl Med. (2017) ;9: (396):eaal4649. |
[30] | Rooney JE , Knapp JR , Hodges BL , et al. Laminin-111 protein therapy reduces muscle pathology and improves viability of a mouse model of merosin-deficient congenital muscular dystrophy. American Journal of Pathology. (2012) ;180: :1593–602. |
[31] | Kemaladewi DU , Bassi PS , Erwood S , et al. A mutation-independent approach for muscular dystrophy via upregulation of a modifier gene. Nature. (2019) ;572: :125–30. |
[32] | Bouman K , Groothuis JT , Doorduin J , et al. LAMA2-Related Muscular Dystrophy Across the Life Span: A Cross-sectional Study. Neurol Genet. (2023) ;9: (5):e200089. |
[33] | Abdel Aleem A , Elsaid MF , Chalhoub N , et al. Clinical and genomic characteristics of LAMA2 related congenital muscular dystrophy in a patients’ cohort from Qatar. A population specific founder variant. Neuromuscular Disorders. (2020) ;30: :457–71. |
[34] | Jain MS , Meilleur K , Kim E , et al. Longitudinal changes in clinical outcome measures in COL6-related dystrophies and LAMA2-related dystrophies. Neurology. (2019) ;93: :E1932–43. |
[35] | Cauley ES , Pittman A , Mummidivarpu S , et al. Novel mutation identification and copy number variant detection via exome sequencing in congenital muscular dystrophy. Mol Genet Genomic Med. (2020) ;8: (11):e1387. |
[36] | Hayes LH , Yun P , Mohassel P , et al. Hypoglycemia in patients with congenital muscle disease. BMC Pediatr. (2020) ;20: (1):57. |
[37] | Tavakoli NP , Gruber D , Armstrong N , et al. Newborn screening for Duchenne muscular dystrophy: A two-year pilot study. Ann Clin Transl Neurol. (2023) ;10: :1383–96. |


