
A Multicenter Cross-Sectional Study of the Swiss Cohort of LAMA2-Related Muscular Dystrophy
Abstract
Background:
LAMA2-related muscular dystrophy (LAMA2-RD) is an autosomal-recessive disorder and one of the most common congenital muscular dystrophies. Due to promising therapies in preclinical development, there is an increasing effort to better define the epidemiology and natural history of this disease.
Objective:
The present study aimed to describe a well-characterized baseline cohort of patients with LAMA2-RD in Switzerland.
Methods:
The study used data collected by the Swiss Registry for Neuromuscular Disorders (Swiss-Reg-NMD). Diagnostic findings were derived from genetics, muscle biopsy, creatine kinase-level and electrophysiological testing, as well as from brain MRIs. Further clinical information included motor assessments (CHOP INTEND, MFM20/32), joint contractures, scoliosis, ophthalmoplegia, weight gain, feeding difficulties, respiratory function, cardiac investigations, EEG findings, IQ and schooling.
Results:
Eighteen patients with LAMA-RD were included in the Swiss-Reg-NMD as of May 2023 (age at inclusion into the registry: median age 8.7 years, range 1 month – 31 years F = 8, M = 10). Fourteen patients presented with the severe form of LAMA2-RD (were never able to walk; CMD), whereas four patients presented with the milder form (present or lost walking capability; LGMD). All patients classified as CMD had symptoms before 12 months of age and 11/14 before the age of six months. 15 carried homozygous or compound heterozygous pathogenic or likely pathogenic variants in LAMA2 and two were homozygous for a variant of unknown significance (one patient unknown). Brain MRI was available for 14 patients, 13 had white matter changes and 11 had additional structural abnormalities, including cobblestone malformations, pontine hypoplasia and an enlarged tegmento-vermial angle not reported before.
Conclusion:
This study describes the Swiss cohort of patients with LAMA2-RD and gives insights into measuring disease severity and disease progression, which is important for future clinical trials, as well as for a better clinical understanding and management of patients with LAMA2-RD.
INTRODUCTION
LAMA2-related muscular dystrophy (LAMA2-RD) is an autosomal-recessive disorder and one of the most common congenital muscular dystrophies. Reported prevalence varies widely between 0.14 : 100 000 (Italy) and 2.5 : 100 000 (Sweden) [1–5]. LAMA2 is located at 6q22-23 and consists of 65 exons. In 60–80% of cases, single nucleotide variants, often premature termination variants [6], are identified as causing the disease; 20–40% of patients harbor single or multiple exon deletions. LAMA2 encodes for the extracellular matrix protein laminin α2, also known as merosin, which is a subunit of laminin-211. Laminin-211 plays an important role as a protein of the basement membrane of muscle cells. The bond between the sarcolemma and the basement membrane is crucial for muscle fiber stability. Laminin-211 is expressed not only in muscle cells and Schwann cells, but also in other tissues including trophoblasts of the placenta, the neuromuscular synapses and the brain [7, 8]. Pathophysiologically, early fibrogenesis results in severe and early contractures of joints and loss of muscle tissue by myofibroblast transdifferentiation [9].
There is a continuum of severities but two types of LAMA2-RD are usually classified: the severe congenital form and a milder phenotype with predominantly limb-girdle-type presentation. Patients with the congenital muscle dystrophy (CMD) phenotype present soon after birth with hypotonia and severe muscle weakness. These children rarely achieve independent ambulation. Typical features are early onset of contractures and the development of rigidity and hyperlordosis of the spine. Respiratory insufficiency and external ophthalmoplegia frequently occur. Feeding difficulties and failure to thrive have a serious impact on daily life [10]. Sensory and motor demyelinating neuropathy is also a known feature [11, 12]. Muscle biopsy usually reveals a complete absence of merosin.
Clinical presentation of patients with the limb-girdle type is milder and has a later onset. Patients may develop proximal muscle weakness, scoliosis, rigid spine, motor difficulties, cardiac and occasionally pulmonary problems [13, 14]. There are case reports of unusual presentations, including cognitive impairment and seizures without muscle weakness, or Charcot-Marie-Tooth-like presentations [15, 16]. Often an unusual clinical pattern is associated with partial merosin staining in muscle biopsies. However, the amount of merosin in muscle biopsy does not enable a clear prediction of clinical phenotype [17].
Brain MRI shows typical periventricular and subcortical white matter changes, which are thought to be due to increased water content [18]. Occasionally, patients may have normal white matter or only patchy changes [18]. Additional structural brain abnormalities (mainly cortical changes) have been reported [19–21]. Usually, cognitive abilities of patients are in the normal range [22]. Children with learning difficulties have been reported but details of cognitive profiles were not provided. Epileptic seizures are relatively frequent [21, 23, 24].
Due to developments in therapeutic options (anti-apoptotic Omigapil [25, 26]) and gene therapy approaches, for example, to introduce a linker protein [7, 27–29], there is an increasing effort to better define the epidemiology and natural history of this rare disease. Previous retrospective studies included cohorts from the United Kingdom, Netherlands, Qatar, Brazil, and China [13, 17, 21, 30, 31]. LAMA2-Europe expert meetings were organized, which highlighted the importance of natural history studies of this patient group [32, 33]. Efforts are also being made to define reliable and valid outcome measures for upcoming clinical studies [34–37]. In Switzerland, we started to include patients with muscular dystrophy due to LAMA2 variants in the national neuromuscular registry (Swiss Registry for Neuromuscular Disorders (Swiss-Reg-NMD)) in 2018. The present study, thus, describes a well-characterized baseline cohort of patients with LAMA2-RD in Switzerland.
METHODS
Study design
This study used data collected by the Swiss-Reg-NMD. The registry was created in 2008 and has been hosted since 2017 at the Institute of Social and Preventive Medicine in Bern. It prospectively collects data on patients of all ages diagnosed with spinal muscular atrophy, dystrophinopathies, LAMA2-RD or collagen 6-related muscle diseases in Switzerland. Patients are identified at regional neuromuscular centers. After consent has been obtained, the treating physicians report the patient’s baseline data to the Swiss-Reg-NMD and regularly provide follow-up data on the clinical status and treatment. Initially this was done via semi-structured reports, since 2021 a predefined case report form has been used for patients with LAMA2-RD. Data for the registry is collected during routine patient visits. Information is entered into a secured database using REDCap electronic data capture tools [38, 39]. The specific aims of registering patients with LAMA2-RD are: (1) to be able to reach patients with LAMA2-RD in Switzerland easily for possible trial recruitment and surveys; (2) to describe the Swiss cohort in detail and produce epidemiological data and (3) to establish a natural history cohort. The Registry is approved by the Cantonal Ethics Committee of Bern (20.06.2018, KEK Bern, 2018-00289).
Population
The present study used data collected up to May 31, 2023. Confirmation of the diagnosis of LAMA2-RD was required for inclusion in the study: (1) homozygous or compound heterozygous pathogenic variants, or by (2) one pathogenic variant and a variant of unknown significance (VUS) with a consistent muscle biopsy result (including dystrophic pattern and absent or reduced staining of merosin) or with typical brain MRI findings, or with typical phenotype (neuromuscular clinician’s rating), or by (3) none, one or two VUS but a typical phenotype, typical biopsy and a typical brain MRI. Genetic findings were reevaluated, and, if from an older report, variants were classified according to current American College of Medical Genetics and Genomics (ACMG) guidelines [40]. The calculation of prevalence was based on all patients with LAMA2 in the registry divided by all people with permanent residence living in Switzerland (reported by the Swiss Federal Statistical Office). One patient was older than 23 years and was excluded from the prevalence estimations.
Data collected in the registry
Data from routine care visits were collected using case report forms. Diagnostic findings were derived from genetic testing, muscle biopsy, creatine kinase (CK)-level, and brain MRI. For few patients electrophysiological assessments were available. Information on the time of onset of the symptoms and the initial symptoms at the time of diagnosis were collected retrospectively for some patients. Recent symptoms were prospectively collected and were derived from information about anthropometric measurements, muscle weakness, joint contractures, scoliosis, spondylodesis, feeding difficulties, epilepsy, cognition, cardiac and respiratory findings. Qualitative motor abilities were recorded in accordance with the TREAT-NMD SMA Patient Registry Dataset, Version 2.1 (https://datasets.treat-nmd.org/sma/groups/motor-function. If motor function was further assessed, the results were collected. For patients younger than 2 years of age and for young, severely affected patients, the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders was used (CHOP INTEND, total score from 0 to 64) [41–43]. For patients older than 2 years the MFM-20/32 or Hammersmith (HMFS, HMFSE) was used to assess motor function, and RULM or PUL for motor function of the upper extremities.
RESULTS
Epidemiology
The Swiss Federal Statistical Office reports a population aged 0–23 years with permanent residence in Switzerland of 2 123 128. As of 15 May 2023, 17 patients aged 0–23 years were included in the registry. Therefore, the prevalence of LAMA2-RD in Switzerland for the age group 0-23 years is at least 17/2 123 128 = 0.8 : 100 000.
Study population
Eighteen patients with LAMA2-RD from 16 families were included in the Swiss-Reg-NMD. Their median age was 8.7 years, ranging between 1 month and 31 years (female = 8, male = 10) at the first entry into the registry. For the various assessments age at the time of assessment is reported. Two patients were preterm (32 weeks gestation) and age was corrected for prematurity. As there is a spectrum of severities of the disease, we defined the phenotype in patients who achieved walking as limb-girdle-like-muscular dystrophy (LGMD), and the phenotype in patients who were never able to walk as CMD.
Diagnostic characteristics
An overview of all diagnostic characteristics is shown in Table 1. Seventeen patients had available genetic testing results, of whom 15 carried homozygous or compound heterozygous pathogenic or likely pathogenic variants in LAMA2. All but two were predicted to be truncating. Two missense VUS were observed in combination with a pathogenic variant. Two individuals were homozygous for a VUS. For more details concerning genetics see Table S1. Muscle biopsy results were available for 11 patients, of whom eight presented with complete merosin deficiency, two with partial merosin deficiency (one LGMD, one CMD), and one with a dystrophic pattern and normal merosin staining. CK level was available for 13 patients and was elevated in all of them (range 676–21160 units per litre); values declined with age at testing. Electrophysiological nerve conduction studies were done in three patients (two with motor and sensory measurements, one patient with only sensory measurement) and were reported as being normal. Electromyography was done in two patients who showed myopathic changes at the age of 4.4 years and 2.6 years respectively.
Table 1
Clinical characteristics of the patient population, including, age, sex, body mass index, and diagnostic findings
| Pat. Nr | Sex | Genetics variants | Type | Pheno-type | Age at biopsy (years; months) | Merosin staining | Age at CK (years; months) | CK level | BMI at entry into the registry | Gastro-stomy | Age at MRI (years; months) | cMRI white matter | cMRI structural |
| 1 | F | Two heterozyg. path. | 1,3 | CMD | 7;4 | c | – | – | <P3 | yes | 7;3 | Diffuse | PH+MCM |
| c.3215delG | |||||||||||||
| c.5235-12G > A | |||||||||||||
| 2 | F | Two heterozyg. path. (in trans) | 1,3 | CMD | No date | c | – | – | <P3 | 12;1 | Diffuse | PH+MCM | |
| c.6993-2A>C | |||||||||||||
| c.2049_2050delAG | |||||||||||||
| 3 | F | Two heterozyg. path. (in trans) | 3,4 | CMD | 2;5 | c | 2;5 | >1000 | <P3 | 2;8 | Diffuse | PH+CM+MCM | |
| c.3085C > T | |||||||||||||
| c.4960-17C > A | |||||||||||||
| 4 | F | Heterozyg. path | 3,4 | LGMD | – | Unknown | 11;0 | 3000 | Normal | 18;1 | Diffuse | – | |
| c.2749 + 2dupT | |||||||||||||
| c.3283C > T | |||||||||||||
| 5 | M | Homozyg. path | 3 | CMD | – | Not done | – | Normal | yes | 6;6 | Patchy | CM+BPC | |
| c.4960-17C > A | |||||||||||||
| 6 | F | Homozyg. path. | 3 | CMD | No date | c | – | – | Normal | 8;7 | Diffuse | PH+CM+MCM | |
| c.4960-17C > A | |||||||||||||
| 7 | M | Two heterozyg. VUS+path. | 1,2 | LGMD | No date | p | 3 | >1000 | Normal | – | Not done | – | |
| c.437C > T | |||||||||||||
| c.7865_7869delGAGAA | |||||||||||||
| 8 | M | Homozyg. path.c.7147C > T | 4 | CMD | Not done | 0;6 | 4607 | <P3 | yes | 1;0 | Delayed myelination | PH+BPC | |
| 9 | F | Homozyg. path.c.5235-12G>A | 2, 3 | LGMD* | 1;5 | n | 0;1 | 3245 | Normal | yes | 23;5 | Delayed myelination | PH+CM+BPC |
| 10 | M | Two heterozyg. path. | 3,4 | CMD | 1;5 | c | 0;3 | 4144 | Normal | 0;6 | Delayed myelination | PH+CM+BPC+HC | |
| c.3976C > T | |||||||||||||
| c.5235-12G > A | |||||||||||||
| 11 | F | Two heterozyg. path. | 1,3 | CMD | 0;3 | p | 0;3 | 2593 | Normal | Not done | – | ||
| c.4692_4695dup | |||||||||||||
| c.8244 + 1G > A | |||||||||||||
| 12 | M | Path. | 4 | CMD | – | Done but results un known | – | – | <P3 | yes | 4;4 | White matter changes | – |
| “E967X” | |||||||||||||
| 13 | M | Homozyg. VUS | dup | CMD | 2;3 | c | 0;11 | 676 | <P3 | 0;1 | Normal | PH | |
| Dupl exons 10–12 | |||||||||||||
| 14 | M | Unknown | CMD | 0;4 | c | 0;3 | 5617 | <P3 | yes | 0;3 | Diffuse | – | |
| 15 | M | Two heterozyg. VUS+path. | 1,2 | CMD | – | Not done | 0;2 | 2915 | Normal | 0;1 | Patchy | PH+CM+MCM | |
| c.3651del | |||||||||||||
| c.8405T>G | |||||||||||||
| 16 | M | Unknown | CMD | No date | c | 0;3 | 2981 | <P3 | yes | Not done | na | ||
| 17 | F | Homozyg. VUS | 2/3 | LGMD | – | Not done | 1;7 | 1468 | Not done | 9;3 | Diffuse | PH | |
| c.2537G > T | |||||||||||||
| 18 | M | Homozyg. path. | 4 | CMD | – | Not done | 0;1 | 21160 | Normal | Not done | na | ||
| c.7147C > T |
Notes: P3 = 3rd percentile, c = complete absence, p = partial absence, n = normal. Type: 1 = frameshift, 2 = missense, 3 = splice, 4 = nonsense, dup = duplication; cMRI: PH = pontine hypoplasia, CM = cobblestone malformation, MCM = megacisterna magna, BPC = Blake’s pouch cyst, HC = hydrocephalus; *lost ability to walk.
MRI findings
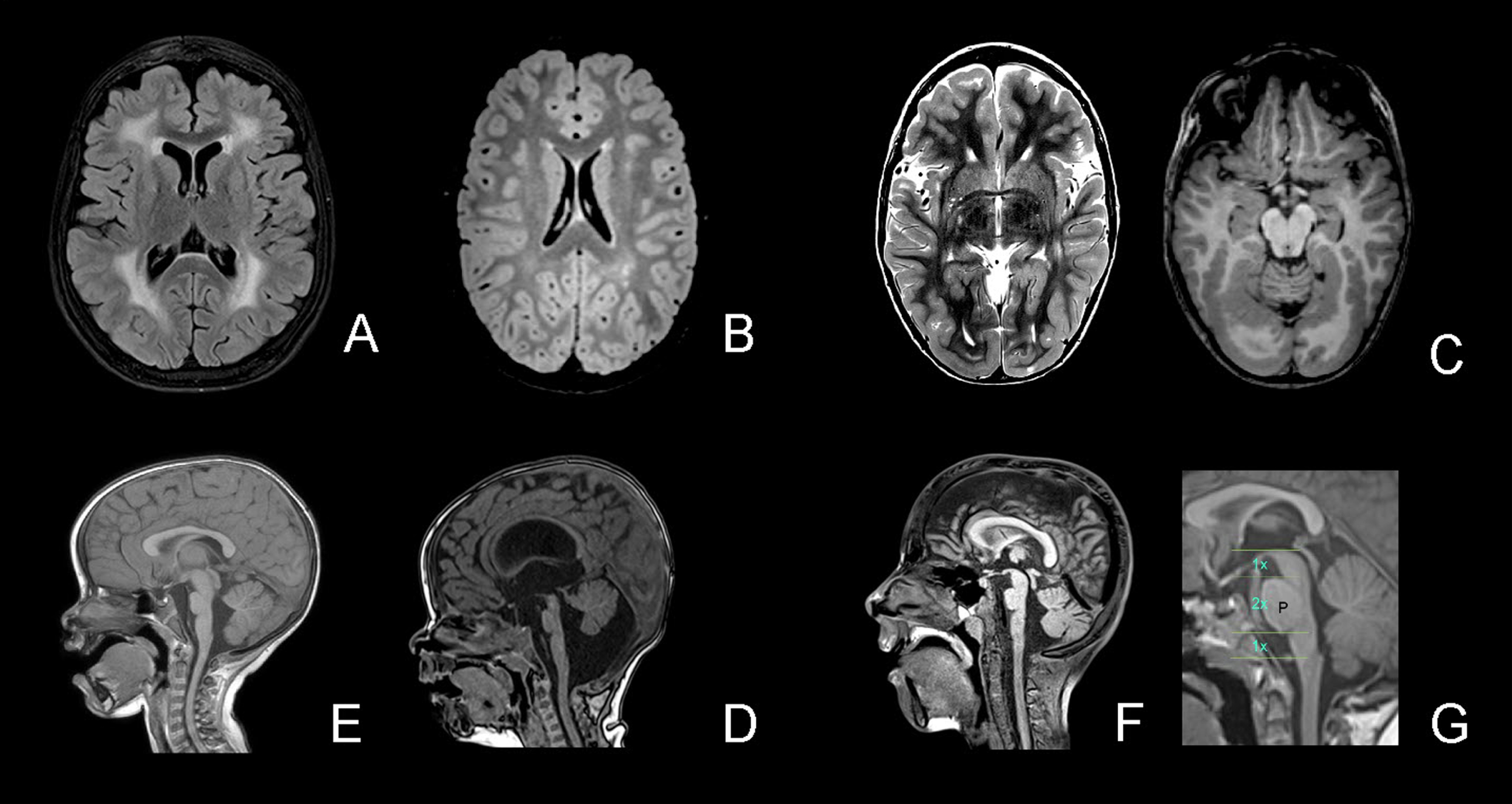
Brain MRI results were available for 14 patients and MRI images were reevaluated in 12. In one patient, the brain MRI was normal at age 1 month. In thirteen patients white matter changes were found (diffuse = 7; patchy = 2; delayed myelination = 3; WM changes without further information = 1). The youngest age at which MRI showed clear white matter changes was 3 months. In three MRIs from children aged below 6 months, the typical white matter changes were not yet visible but delayed myelination was observed. In 7 patients, additional cortical abnormalities were found: parieto-occipital cobblestone malformation (six patients) and/or polymicrogyria (three patients). Furthermore, nine of the 12 reevaluated patients had structural abnormalities of the brainstem-vermis angle: four patients had a Blake’s pouch cyst, one of whom needed a ventriculo-peritoneal shunt; and five patients had a megacisterna magna (brainstem-vermis-angle 10–18°)[44]. In ten of the 12 reevaluated MRIs we found pontine hypoplasia, as well as four with a large interthalamic adhesion. MRIs were available for 3/4 patients in the LGMD group, all of whom had white matter changes and one patient (who had lost ambulation) had cobblestone malformation.
Clinical symptoms
History of clinical symptoms
For patients with the LGMD phenotype (n = 4), delayed motor milestones, failure to thrive, muscular hypotonia, and feeding difficulties were reported as initial symptoms. Onset of symptoms was for one patient at 12 months and for three patients the time of onset of the symptoms was not reported in the registry.
For the CDM group (N = 14), the most commonly reported initial symptoms included muscular hypotonia (n = 13), muscle weakness (n = 9), and delayed motor milestones (n = 6), followed by joint contractures (n = 4), rigidity of the spine (n = 2), feeding difficulties (n = 1) and respiratory difficulties (n = 1). Onset of symptoms was reported to have been at birth for six patients and for eight patients between 1 and 12 months (1, 1, 1, 3, 4, 10, 12, 12 months).
Joint contractures
Descriptions of joint contractures based on the neurological examination (yes/no without further grading) were recorded in the CRF and were available for 18 patients and most frequently observed in the elbow (N = 10), hip (N = 9), long finger flexors (N = 12), knee (N = 13) and ankle (N = 13) (Fig. 1). However, contractures at other joints might have been overlooked by the treating physicians. Rigid spine was documented in 8/18 patients; however, flexibility of the spine could not be assessed in five of them as they had undergone spondylodesis. There were five patients without rigid spine (three with CMD and two with LGMD). All five patients with neck contractures had a rigid spine or had undergone spondylodesis. The youngest patient with neck contractures was 5y10 m old. None of the patients able to walk had neck contractures and one had a rigid spine.
Fig. 1
White matter changes and structural abnormalities. Legend. A, White matter changes – diffuse (FLAIR); B, White matter changes – patchy (T2); C, Cobblestone malformation occipital (T2 left and T1 right); D, Brainstem – pons hypoplasia and vermis rotation (T1); E, Pons hypoplasia and large interthalamic adhesion; F, Pons hypoplasia, Midbrain-Pons-Medulla Proportions (ac. Barkovitch); G: normal midbrain-pons-medulla proportions.
Scoliosis and scoliosis surgery
15 patients were non-ambulatory and 10 of these had scoliosis; the youngest patient with scoliosis was 2y10 m old. Five underwent surgery at the age of 7 to 12 years. None of the three ambulant patients had scoliosis.
Motor assessments
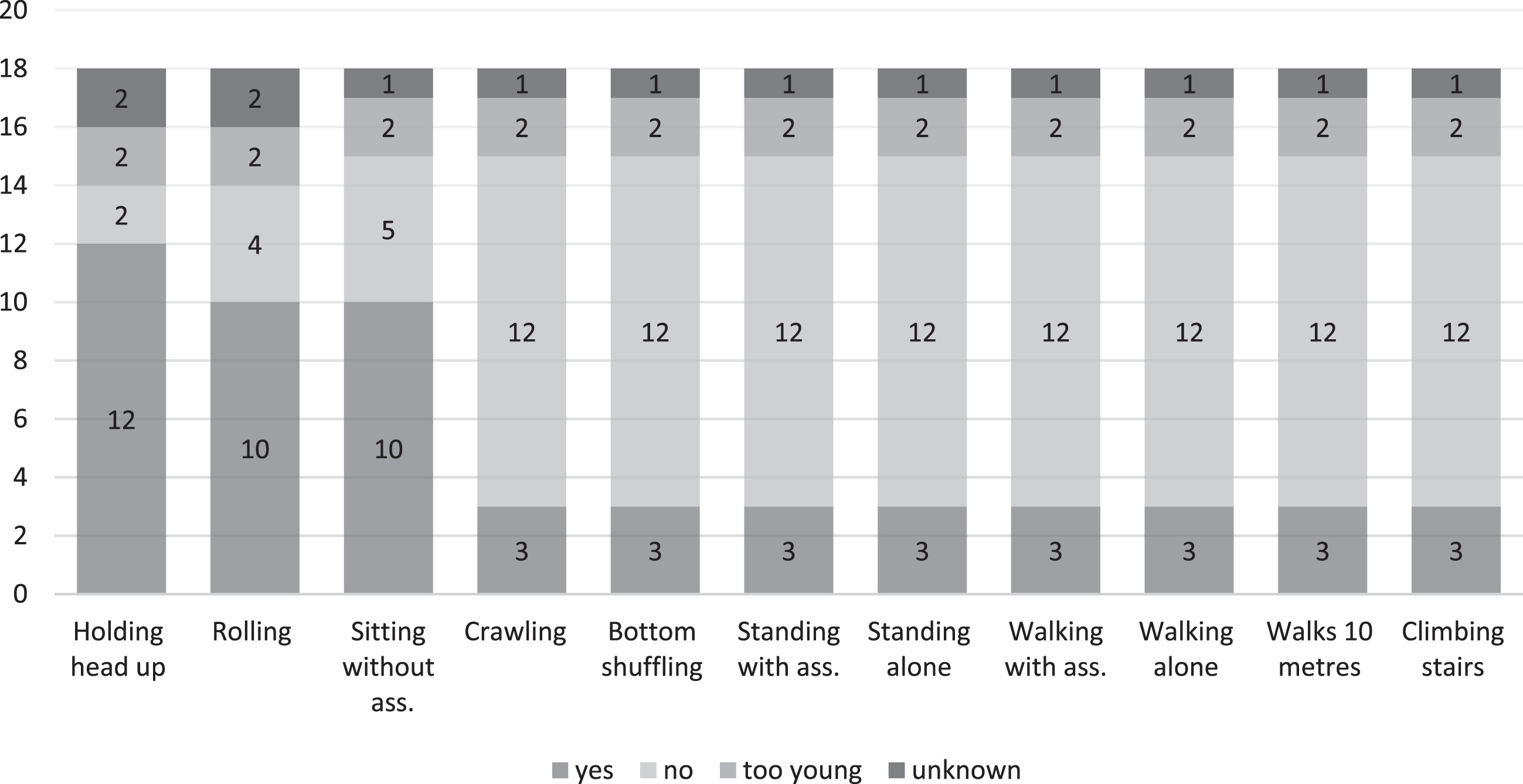
Motor functions
A detailed qualitative overview of motor functions at the time of the study was available for 18 patients and illustrated in Fig. 2. Head control was the motor function most frequently achieved and maintained (12/14); 10 patients were able to roll to the side and sit. All other items were only reported to be possible for three patients with the milder form LGMD. Retrospective data about gaining motor milestones were available for some patients. In the group of the non-ambulant patients (CMD), all patients gained the ability to sit without support after the age of 12 months (range was between 1- 7 years) (see table S2). In the group of patients with LGMD (N = 4), they gained walking between 1.8 to 3.5 years (for details see table S2). One patient lost the ability to sit without support at the age of 5 years, and one lost the ability to walk at age 8 years (patient classified as LGMD).
Fig. 2
Qualitative measurements of motor functions for eighteen patients. Legend. Qualitative motor abilities were recorded in accordance with the TREAT-NMD SMA Patient Registry Dataset, Version 2.1 (https://datasets.treat-nmd.org/sma/groups/motor-function).
Motor function measurement with the CHOP INTEND and MFM-20/32
CHOP INTEND scores and MFM-30/scores are illustrated in Figure S2. The two patients with the milder form, LGMD, displayed the highest scores. For the patients with CMD (N = 3), no age-related pattern of the MFM-20/32 scores was observed (Figure S2).
Lung function
Lung function data were only available for four patients in the baseline CRF (follow-up CRF not reported). Note that 12 patients were younger than 6 years old (lung function test is usually not performed in children under 6). One patient with the LGMD phenotype showed normal values (forced vital capacity (FVC) 93% predicted, no absolute value available); three patients with CMD had each one abnormal lung function measurement (FVC 0,5 l (19% pred) at the age of 12y1 m; 1,46 l (38% pred) at the age of 14y 3 m; 0,36 l (21% pred) at the age of 6y2 m). Non-invasive ventilation was reported for five patients, all of whom presented with the CMD phenotype and were older than 5 years. Start of non-invasive ventilation was at 5y1 m, 7y0 m, 7y0 m, 8y0 m, 14y8 m (four patients were ventilated overnight and one patient needed ventilation >16 h/day after the age of 13y). Tracheostomy and invasive ventilation were reported for two patients, tracheotomy was performed at the age of 9y (followed by full time ventilation at the age of 14y), resp 20y (ventilation during the night).
Cardiological examinations
Fourteen out of the 18 patients had a cardiological examination (electrocardiogram and echocardiography); the results were normal in 13 and showed abnormalities in one (conduction block; V1, partial right bundle branch block with RSr’ picture). Four of 18 patients had a Holter ECG with normal findings.
Nutrition, dysphagia and BMI
Patients with LGMD had a normal BMI. Of the group with the CMD phenotype, seven needed feeding via a gastrostomy tube starting at the age of six months to 20 years. Four of these patients still had a BMI < 3rd percentile (see Table 1).
Epilepsy
Seizures were reported in four patients with age of onset ranging between 7 and 31 years. Two of these patients had normal EEG and were seizure-free with medication (Table 2). One had continuous spike-waves during sleep (CSWS) and one had drug-resistant frequent focal seizures with impaired awareness (cortical malformation in one patient). Two were treated with levetiracetam, one with a combination of levetiracetam and oxcarbazepine and one with levetiracetam, valproate and ethosuximide. One other patient was reported to have had febrile seizures in infancy.
Table 2
Epilepsy, brain MRI findings, cognitive assessments, and schooling
| Sex | Cognitive test | Score | Schooling | Epilepsy | Febrile seizures | Pheno-type | MRI |
| M | WPPSI III | 74 | Mainstream with support | CMD | WM changes (patchy)+2 + 4 | ||
| F | na | na | Mainstream with support | Yes | CMD | WM changes (diffuse) 1 + 2+3 | |
| M | WISC V | 96 | Mainstream | Yes | LGMD | No MRI available | |
| F | WISC V | 91 | Special school | Yes | LGMD | WM changes (diffuse)+1 + 2+4 | |
| M | na | na | Mainstream | Yes | CMD | WM changes | |
| M | Bayley | Cog:100 | Not yet in education | CMD | MRI normal | ||
| F | SON-R | <50 | Special school | Yes | LGMD | WM changes (diffuse) + 1 |
Notes. 1 = pontine hypoplasia, 2 = cobblestone malformation, 3 = mega cisterna magna, 4 = Blake’s pouch cyst. F = female, M = male, WPPSI = Wechsler Preschool and Primary Scale of Intelligence, WISC = Wechsler Intelligence Scale for Children, SON-R=Snijders-Oomen non-verbal intelligence test, CMD = congenital muscular dystrophy, LGMD = limb-girdle-like-muscular dystrophy, MRI = magnetic resonance imaging, WM = white matter.
Cognition
Eleven patients were in education at the time at entry into the registry, of whom one was in mainstream school without dedicated support, five were in mainstream school with dedicated support, and five were in special schools or in an apprenticeship in a special institution. The other seven patients were either too young or too old for school at the time of entry into the registry. Cognitive assessments were done in five patients. IQ and cognitive development were assessed with different scales and ranged from <50 to 100 (Table 2).
DISCUSSION
Based on the present study, the prevalence of LAMA2-RD in Switzerland is at least 0.8/100 000, which is in line with previous studies (5, 30, 42), but higher than the estimated prevalence in Italy of 0.14/100 000 [4]. It might be even higher considering that milder phenotypes may not have been diagnosed. Regarding genetic testing, 15 out of 18 patients were compound heterozygous or homozygous for pathogenic or likely pathogenic variants (according to current ACMG classification guidelines) in LAMA2, two were homozygous for a VUS but had typical clinical and imaging or biopsy findings. Two intronic, potential splice variants were recurrently found in unrelated patients thus supporting their pathogenicity (c.5235-12G > A, not listed in LOVD/ClinVar; c.4960-17C > A, ClinVar: VUS, but reported by [21]). We confirmed aberrant splicing for the c.4960-17C > A variant (see Figure S1 and Table S1).
In our cohort there was no clear correlation between the muscle biopsy findings and the severity of the clinical phenotype. Of the four initially ambulant patients, two patients had a muscle biopsy: one (still ambulatory) showed partial absence of merosin. The patient, who had lost ambulation, had a dystrophic pattern but normal merosin staining (unfortunately reevaluation was not possible). In contrast, two patients with the CMD phenotype had muscle biopsies with partial merosin staining. Therefore, we decided to classify our cohort according to the clinical phenotype. Patients, who achieved walking were classified as “LGMD phenotype”. There is currently not enough knowledge about the natural history of early presenting children who later develop a milder phenotype. For future clinical trials, it would be very important to predict the phenotype as early as possible. All patients classified as CMD had symptoms before 12 months of age and 11/14 before the age of six months. Also sitting was not achieved before the age of 12 months for the CMD phenotype. This might help to distinguish between the typical CMD phenotype and a milder phenotype. Unfortunately we lack early milestone information of ambulant older patients. In the prospective part of our study, we currently assess infants every 4 to 6 months with the CHOP intend, these results might help to predict motor outcome in the future. Concerning CHOP intend results (Figure S2 in the supplement), some of the infants gained points, probably reflecting development rather than increasing strength. After the age of 1.5 years the scores remained stable. For the MFM-20/32, the scores stayed relatively stable within patients (Figure S2), but there is not enough data yet to draw conclusions from these values.
The most frequently reported initial symptoms for the CMD group included muscular hypotonia, muscle weakness, and delayed motor milestones, which appeared before the age of 1 year. Joint contractures are one of the main clinical symptoms of this disease, starting at a young age. In four patients with CMD, contractures were reported as the initially presenting symptom. The longer the disease duration, the more frequent and more severe the contractures. In our cohort five out of ten patients with CMD needed a surgical intervention for their severe scoliosis. These findings are in line with studies in other cohorts [17, 30].
In the cranial MRIs, white matter anomalies were commonly observed. The youngest age in our cohort at which white matter changes could be detected was 3 months, and delayed myelination was seen in three additional patients with an MRI performed before the age of 6 months. In one patient, the brain MRI was normal at age 1 month. This is consistent with previous reports of unremarkable white matter appearance in young infants up to 6 months of age [10, 46]. Cortical malformations are known to be common in patients with LAMA2-RD [21, 30] and were present in half of the MRIs of our cohort. In 12 reevaluated MRIs we found abnormalities of the brainstem/pons and the posterior fossa. Pontine hypoplasia was also mentioned by Jayakody et al. [47], Salvati et al. [23] and Philpot et al. [19]. Although, to our knowledge, not previously reported, we observed the frequent occurrence of a large tegmento-vermian angle (9/12) classified as megacisterna magna or Blake’s pouch cyst (3/12), causing a hydrocephalus and requiring a ventriculoperitoneal shunt in one patient. Other authors reported cerebellar hypoplasia or cerebellar cleft [23] or mild brainstem and cerebellar atrophy [24]. Geranmayeh et al. [48] reported two patients with hydrocephalus (not specified) and one patient with “subtle abnormalities pons medulla”.
Neuropathic changes were not documented in the electrophysiological investigation, but this was only available for three patients. The electromyography available for two patients showed myopathic changes. Severe hind-limb paralysis due to demyelinating and secondary axonal neuropathy has been seen in different murine models for LAMA2 [27, 49]. Several papers provide evidence of neuropathic findings in humans [11, 12] and neurophysiological investigations should be performed in this patient group to elucidate the extent of neuropathy in LAMA2-RD.
Regarding lung function, we only had four values to report. Due to facial hypotonia, difficulties closing the mouth, young age or severe phenotype, lung function assessment is often not feasible. Also, in clinical practice, pneumologists stop doing lung function tests in severely affected patients and rely on clinical data and polysomnography. In our cohort, especially in the older patients, the start of ventilation was sometimes delayed. A possible explanation could be the earlier reluctance in Switzerland to offer ventilation to severely affected patients with neuromuscular disorders with respiratory insufficiency and a very limited life expectancy in an era without disease-modifying treatments. In recent years the age at start of ventilation of the younger children has been decreasing, also as the experience with ventilation of young children with neuromuscular disorders has increased. Our data concerning ventilation are comparable with the cohort studied by Abdel Aleem et al. [30] (29% BiPAP Aleem, 27% Swiss/14% invasive Aleem, Swiss 11%) and Bouman et al. [13] (ventilation in total in 37%, Swiss 39%).
The cardiological examinations in our cohort did not reveal any meaningful pathologies. Ventricular arrhythmias have been described in other cohorts with LAMA2-RD [13, 17]. Bouman et al. [13] found that 19 out of 21 patients had ECG abnormalities. In contrast, in the Swiss cohort only 1/14 patients showed ECG abnormalities, which might reflect the different distribution of age and severity of the two cohorts. On the other hand, Tan et al. [31] reported few cardiac problems, which is in line with our findings. Cardiac abnormalities are rare in the paediatric group with LAMA2-RD but have recently been described [30, 50, 51] and Holter ECGs should also be performed regularly in children. Failure to thrive is a known feature of LAMA2-RD. In our cohort 9/18 or 64% of the CMD patients had a BMI < P3, even if they had a feeding tube. This is in line with other cohort studies and an important topic to address in clinical care.
Epilepsy is also reported in the literature on LAMA2-RD. In our cohort, four out of 18 (22%) patients had been diagnosed with epilepsy. This is similar to previous reports (36% [24]; 17.4% [48]; 9.5% in CMD, 35.7% in LGMD [31]). Three out of four patients with epilepsy showed structural abnormalities on brain MRI (pontine hypoplasia = 3, Blake’s pouch cyst = 1, and mega cisterna magna = 1) but these abnormalities are not associated with epilepsy; only two of the patients had cortical malformations. Standardized cognitive testing was done in five patients, who showed a large variance in cognitive performance (IQ < 50 to 100). The child with an IQ score below 50, is a walker, and had diffuse white matter changes, pontine hypoplasia and a treatment resistant epilepsy (Epileptic Encephalopathy with Continuous Spike-and-Wave during Sleep CSWS). Cognitive impairment in patients with LAMA2 CMD has been reported previously in isolated cases [22, 52, 53]. In our cohort, there were insufficient data to confirm the previous evidence that cognitive impairment is more frequently associated with structural brain changes.
A limitation of the study is that the registry acquired data from routine clinical visits, and before 2018 medical reports were used as source data in five patients. The findings reflect local standards, which adds information but also leads to missing data points. The completeness of the whole national cohort of paediatric patients, the wide range in age and disease severity and the broad range of clinical and functional assessments are the major strengths of this study.
CONCLUSION
Our data summarize a well-characterized baseline cohort of patients with LAMA2-RD, which is important for informing future clinical trials. In our cohort, reevaluation of MRIs frequently revealed further brain abnormalities, especially of the pons and posterior fossa not previously reported. The clinical meaning of these findings needs to be further investigated. Cardiac investigations are still not routinely performed and should be stressed in standards of care recommendations for this disease. Furthermore, our study provides insights into measuring disease severity and the difficulties to measure disease progression in severely affected individuals for longitudinal studies. To gather uniformly collected information on larger cohorts international cooperation is very important [32, 33].
ACKNOWLEDGMENTS
We thank all the participants and their families for making this study possible. We also want to thank the Swiss-Reg-NMD Group members for their clinical and scientific support.
FUNDING
This study was supported by a grant from fsrmm (foundation suisse de recherche sur les maladies musculaires; www.fsrmm.ch). The Swiss-Reg-NMD is funded by the national patient organisations ‘Schweizerische Muskelgesellschaft’, ‘Association Suisse Romande Intervenant contre les Maladies neuro-Musculaires’, ‘Associazione Malattie Genetiche Rare Svizzera italiana’, ‘SMA Schweiz’, ‘Duchenne Schweiz’ and by the ‘Schweizerische Stiftung für die Erforschung der Muskelkrankheiten’. And received unconditional and research grants from AveXis EU Limited, Sarepta International, PTC Therapeutics Switzerland GmbH, Pfizer AG, Biogen Switzerland AG, Novartis Gene Therapies, Roche Pharma (Schweiz) AG. None of the funders were involved in the study design, data collection, analysis, interpretation of data or writing the article.
CONFLICT OF INTEREST
No conflict of interest.
DATASETS/DATA
All data included in this study are recorded in the Swiss-Reg-NMD registry. Data can be obtained anonymized and aggregated upon reasonable request to the corresponding author (L.S. or A.K.) and after approval of registry’s steering committee.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article: https://dx.doi.org/10.3233/JND-240023.
REFERENCES
[1] | Sframeli M , Sarkozy A , Bertoli M , Astrea G , Hudson J , Scoto M , et al. Congenital muscular dystrophies in the UK population: Clinical and molecular spectrum of a large cohort diagnosed over a 12-year period. Neuromuscul Disord. (2017) ;27: (9):793–803. |
[2] | Mostacciuolo ML , Miorin M , Martinello F , Angelini C , Perini P , Trevisan CP . Genetic epidemiology of congenital muscular dystrophy in a sample from north-east Italy. Hum Genet. (1996) ;97: (3):277–9. |
[3] | Darin N , Tulinius M . Neuromuscular disorders in childhood: A descriptive epidemiological study from western Sweden. Neuromuscul Disord. (2000) ;10: (1):1–9. |
[4] | Graziano A , Bianco F , D’Amico A , Moroni I , Messina S , Bruno C , et al. Prevalence of congenital muscular dystrophy in Italy: A population study. Neurology. (2015) ;84: (9):904–11. |
[5] | Lake NJ , Phua J , Liu W , Moors T , Axon S , Lek M . Estimating the Prevalence of LAMA2 Congenital Muscular Dystrophy using Population Genetic Databases. J Neuromuscul Dis. (2023) ;10: (3):381–7. |
[6] | Oliveira J , Gruber A , Cardoso M , Taipa R , Fineza I , Gonçalves A , et al. LAMA2 gene mutation update: Toward a more comprehensive picture of the laminin-α2 variome and its related phenotypes. Hum Mutat. (2018) ;39: (10):1314–37. |
[7] | Yurchenco PD , McKee KK , Reinhard JR , Rüegg MA . Laminin-deficient muscular dystrophy: Molecular pathogenesis and structural repair strategies. Matrix Biol. (2018) ;71-72: :174–87. |
[8] | Mohassel P , Foley AR , Bönnemann CG . Extracellular matrix-driven congenital muscular dystrophies. Matrix Biol. (2018) ;71-72: :188–204. |
[9] | Accorsi A , Cramer ML , Girgenrath M . Fibrogenesis in LAMA2-Related Muscular Dystrophy Is a Central Tenet of Disease Etiology. Front Mol Neurosci. (2020) ;13: :3. |
[10] | Sarkozy A , Foley AR , Zambon AA , Bönnemann CG , Muntoni F . LAMA2-Related Dystrophies: Clinical Phenotypes, Disease Biomarkers, and Clinical Trial Readiness. Front Mol Neurosci. (2020) ;13: :123. |
[11] | Verma S , Goyal P , Guglani L , Peinhardt C , Pelzek D , Barkhaus PE . COL6A and LAMA2 Mutation Congenital Muscular Dystrophy: A Clinical and Electrophysiological Study. J Clin Neuromuscul Dis. (2018) ;19: (3):108–16. |
[12] | Previtali SC , Zambon AA . LAMA2 Neuropathies: Human Findings and Pathomechanisms From Mouse Models. Front Mol Neurosci. (2020) ;13: :60. |
[13] | Bouman K , Groothuis JT , Doorduin J , van Alfen N , Udink Ten Cate FEA , van den Heuvel FMA , et al. LAMA2-Related Muscular Dystrophy Across the Life Span: A Cross-sectional Study. Neurol Genet. (2023) ;9: (5):e200089. |
[14] | Magri F , Brusa R , Bello L , Peverelli L , Del Bo R , Govoni A , et al. Limb girdle muscular dystrophy due to LAMA2 gene mutations: New mutations expand the clinical spectrum of a still challenging diagnosis. Acta Myol. (2020) ;39: (2):67–82. |
[15] | Marques J , Duarte ST , Costa S , Jacinto S , Oliveira J , Oliveira ME , et al. Atypical phenotype in two patients with LAMA2 mutations. Neuromuscul Disord. (2014) ;24: (5):419–24. |
[16] | Chan SH , Foley AR , Phadke R , Mathew AA , Pitt M , Sewry C , et al. Limb girdle muscular dystrophy due to LAMA2 mutations: Diagnostic difficulties due to associated peripheral neuropathy. Neuromuscul Disord. (2014) ;24: (8):677–83. |
[17] | Zambon AA , Ridout D , Main M , Mein R , Phadke R , Muntoni F , et al. LAMA2-related muscular dystrophy: Natural history of a large pediatric cohort. Ann Clin Transl Neurol. (2020) ;7: (10):1870–82. |
[18] | Alkan A , Sigirci A , Kutlu R , Aslan M , Doganay S , Yakinci C . Merosin-negative congenital muscular dystrophy: diffusion-weighted imaging findings of brain. J Child Neurol. (2007) ;22: (5):655–9. |
[19] | Philpot J , Cowan F , Pennock J , Sewry C , Dubowitz V , Bydder G , et al. Merosin-deficient congenital muscular dystrophy: The spectrum of brain involvement on magnetic resonance imaging. Neuromuscul Disord. (1999) ;9: (2):81–5. |
[20] | Pini A , Merlini L , Tomé FM , Chevallay M , Gobbi G . Merosin-negative congenital muscular dystrophy, occipital epilepsy with periodic spasms and focal cortical dysplasia. Report of three Italian cases in two families. Brain Dev. (1996) ;18: (4):316–22. |
[21] | Camelo CG , Artilheiro MC , Martins Moreno CA , Ferraciolli SF , Serafim Silva AM , Fernandes TR , et al. Brain MRI Abnormalities, Epilepsy and Intellectual Disability in LAMA2 Related Dystrophy - a Genotype/Phenotype Correlation. J Neuromuscul Dis. (2023) ;10: (4):483–92. |
[22] | Messina S , Bruno C , Moroni I , Pegoraro E , D’Amico A , Biancheri R , et al. Congenital muscular dystrophies with cognitive impairment. A population study. Neurology. (2010) ;75: (10):898–903. |
[23] | Salvati A , Bonaventura E , Sesso G , Pasquariello R , Sicca F . Epilepsy in LAMA2-related muscular dystrophy: A systematic review of the literature. Seizure. (2021) ;91: :425–36. |
[24] | Natera-de Benito D , Muchart J , Itzep D , Ortez C , González-Quereda L , Gallano P , et al. Epilepsy in LAMA2-related muscular dystrophy: An electro-clinico-radiological characterization. Epilepsia. (2020) ;61: (5):971–83. |
[25] | Erb M , Meinen S , Barzaghi P , Sumanovski LT , Courdier-Früh I , Rüegg MA , et al. Omigapil ameliorates the pathology of muscle dystrophy caused by laminin-alpha2 deficiency. J Pharmacol Exp Ther. (2009) ;331: (3):787–95. |
[26] | Yu Q , Sali A , Van der Meulen J , Creeden BK , Gordish-Dressman H , Rutkowski A , et al. Omigapil treatment decreases fibrosis and improves respiratory rate in dy(2J) mouse model of congenital muscular dystrophy. PLoS One. (2013) ;8: (6):e65468. |
[27] | Reinhard JR , Lin S , McKee KK , Meinen S , Crosson SC , Sury M , et al. Linker proteins restore basement membrane and correct LAMA2-related muscular dystrophy in mice. Sci Transl Med. (2017) ;9: (396). |
[28] | McKee KK , Yurchenco PD . Amelioration of muscle and nerve pathology of Lama2-related dystrophy by AAV9-laminin-αLN linker protein. JCI Insight. (2022) ;7: (13). |
[29] | McKee KK , Yurchenco PD . Dual transgene amelioration of Lama2-null muscular dystrophy. Matrix Biol. (2023) ;118: :1–15. |
[30] | Abdel Aleem A , Elsaid MF , Chalhoub N , Chakroun A , Mohamed KAS , AlShami R , et al. Clinical and genomic characteristics of LAMA2 related congenital muscular dystrophy in a patients’ cohort from Qatar. A population specific founder variant. Neuromuscul Disord. (2020) ;30: (6):457–71. |
[31] | Tan D , Ge L , Fan Y , Chang X , Wang S , Wei C , et al. Natural history and genetic study of LAMA2-related muscular dystrophy in a large Chinese cohort. Orphanet J Rare Dis. (2021) ;16: (1):319. |
[32] | Smeets H , Verbrugge B , Bulbena X , Hristova L , Vogt J , van Beckhoven I . European Joint Programme on Rare Diseases workshop: LAMA2-muscular dystrophy: Paving the road to therapy March 17–19, 2023, Barcelona, Spain. Neuromuscul Disord. (2024) ;36: :16–22. |
[33] | Smeets HJM , Verbrugge B , Springuel P , Voermans NC . Merosin deficient congenital muscular dystrophy type 1A: An international workshop on the road to therapy 15–17 November 2019, Maastricht, the Netherlands. Neuromuscul Disord. (2021) ;31: (7):673–80. |
[34] | Meilleur KG , Jain MS , Hynan LS , Shieh CY , Kim E , Waite M , et al. Results of a two-year pilot study of clinical outcome measures in collagen VI- and laminin alpha2-related congenital muscular dystrophies. Neuromuscul Disord. (2015) ;25: (1):43–54. |
[35] | Bendixen RM , Butrum J , Jain MS , Parks R , Hodsdon B , Nichols C , et al. Upper extremity outcome measures for collagen VI-related myopathy and LAMA2-related muscular dystrophy. Neuromuscul Disord. (2017) ;27: (3):278–85. |
[36] | Jain MS , Meilleur K , Kim E , Norato G , Waite M , Nelson L , et al. Longitudinal changes in clinical outcome measures in COL6-related dystrophies and LAMA2-related dystrophies. Neurology. (2019) ;93: (21):e1932–e43. |
[37] | Le Goff L , Meilleur KG , Norato G , Rippert P , Jain M , Fink M , et al. Responsiveness and Minimal Clinically Important Difference of the Motor Function Measure in Collagen VI-Related Dystrophies and Laminin Alpha2-Related Muscular Dystrophy. Arch Phys Med Rehabil. (2021) ;102: (4):604–10. |
[38] | Harris PA , Taylor R , Minor BL , Elliott V , Fernandez M , O’Neal L , et al. The REDCap consortium: Building an international community of software platform partners. Journal of Biomedical Informatics. (2019) ;95: :103208. |
[39] | Harris PA , Taylor R , Thielke R , Payne J , Gonzalez N , Conde JG . Research electronic data capture (REDCap)—A metadata-driven methodology and workflow process for providing translational research informatics support. Journal of Biomedical Informatics. (2009) ;42: (2):377–81. |
[40] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) ;17: (5):405–24. |
[41] | Glanzman AM , Mazzone E , Main M , Pelliccioni M , Wood J , Swoboda KJ , et al. The Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND): Test development and reliability. Neuromuscul Disord. (2010) ;20: (3):155–61. |
[42] | Glanzman AM , McDermott MP , Montes J , Martens WB , Flickinger J , Riley S , et al. Validation of the Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP INTEND). Pediatr Phys Ther. (2011) ;23: (4):322–6. |
[43] | De Sanctis R , Pane M , Coratti G , Palermo C , Leone D , Pera MC , et al. Clinical phenotypes and trajectories of disease progression in type 1 spinal muscular atrophy. Neuromuscul Disord. (2018) ;28: (1):24–8. |
[44] | Osborn AG , Hedlund GL , Salzman KL . Osborn’s Brain: Imaging, Pathology, and Anatomy: Elsevier; (2017) . |
[45] | Norwood FL , Harling C , Chinnery PF , Eagle M , Bushby K , Straub V . Prevalence of genetic muscle disease in Northern England: In-depth analysis of a muscle clinic population. Brain. (2009) ;132: (Pt 11):3175–86. |
[46] | Tan E , Topaloglu H , Sewry C , Zorlu Y , Naom I , Erdem S , et al. Late onset muscular dystrophy with cerebral white matter changes due to partial merosin deficiency. Neuromuscul Disord. (1997) ;7: (2):85–9. |
[47] | Jayakody H , Zarei S , Nguyen H , Dalton J , Chen K , Hudgins L , et al. Cobblestone Malformation in LAMA2 Congenital Muscular Dystrophy (MDC1A). J Neuropathol Exp Neurol. (2020) ;79: (9):998–1010. |
[48] | Geranmayeh F , Clement E , Feng LH , Sewry C , Pagan J , Mein R , et al. Genotype-phenotype correlation in a large population of muscular dystrophy patients with LAMA2 mutations. Neuromuscul Disord. (2010) ;20: (4):241–50. |
[49] | Barraza-Flores P , Hermann HJ , Bates CR , Allen TG , Grunert TT , Burkin DJ . Human laminin-111 and laminin-211 protein therapy prevents muscle disease progression in an immunodeficient mouse model of LAMA2-CMD. Skelet Muscle. (2020) ;10: (1):18. |
[50] | Nguyen Q , Lim KRQ , Yokota T . Current understanding and treatment of cardiac and skeletal muscle pathology in laminin-α2 chain-deficient congenital muscular dystrophy. Appl Clin Genet. (2019) ;12: :113–30. |
[51] | Wang Y , Fang Y , Zhang D , Li Y , Luo S . A rare case of arrhythmogenic right ventricular cardiomyopathy associated with LAMA2 mutation: A case report and literature review. Front Med (Lausanne). (2022) ;9: :922347. |
[52] | Brett FM , Costigan D , Farrell MA , Heaphy P , Thornton J , King MD . Merosin-deficient congenital muscular dystrophy and cortical dysplasia. Eur J Paediatr Neurol. (1998) ;2: (2):77–82. |
[53] | Mercuri E , Gruter-Andrew J , Philpot J , Sewry C , Counsell S , Henderson S , et al. Cognitive abilities in children with congenital muscular dystrophy: Correlation with brain MRI and merosin status. Neuromuscul Disord. (1999) ;9: (6-7):383–7. |


