
Meeting Report: 2023 Muscular Dystrophy Association Summit on ‘Safety and Challenges in Gene Therapy of Neuromuscular Diseases’
Abstract
This meeting report summarizes the presentations and discussions held at the summit on Challenges in Gene Therapy hosted by the Muscular Dystrophy Association (MDA) in 2023. Topics covered include safety issues, mitigation strategies and practical considerations pertaining to the clinical translation of gene therapies for neuromuscular disease. The listing of actionable recommendations will assist in overall efforts in the field to achieve safe and efficacious translation of gene therapies for neuromuscular disease patients.
INTRODUCTION
Muscular Dystrophy Association (MDA) held its second annual summit on “Challenges in Gene Therapy using Adeno-Associated Viruses (AAV)”, chaired by Carsten Bönnemann (NINDS, NIH) and Barry Byrne (University of Florida), on February 21–22, 2023. Attendees included 47 researchers/clinicians and 41 industry and 16 patient foundation representatives who convened to discuss clinical and non-clinical topics relevant to the translation of AAV gene therapies in the context of neuromuscular disease. Specific themes covered during this year’s summit included a landscape update of ongoing AAV gene therapy trials, anti-transgene triggered adverse events in Duchenne muscular dystrophy (DMD) gene therapies, durability and redosing, organ toxicities, immune response mitigation strategies, ultra-rare gene therapy programs, and practical considerations in gene therapies. This meeting report broadly covers the presentations as well as important issues raised for each of the above session topics. The complete list of speakers and their presentation titles are outlined in Table 1.
Table 1
Listing of meeting presenters and presentation titles
| Presenter | Affiliation | Presentation title |
| Richard Finkel, MD | St. Jude’s Children’s Research Hospital | Safety update of gene therapy trials in spinal muscular atrophy |
| Tahseen Mozaffar, MD | University of California Irvine | Gene therapies in late-onset Pompe disease |
| Francesco Muntoni, MD, FRCPCH | University College London | Landscape safety update of ongoing gene therapy trials for DMD |
| Serge Braun, PharmD, PhD | AFM Telethon | Overcoming the challenges of transgene immunogenicity |
| Hansell Stedman, MD | University of Pennsylvania | Harnessing central tolerance for AAV vector safety in DMD |
| Jeffrey Chamberlain, PhD | University of Washington | Minimizing transgene immune responses: Expression of enhanced dystrophins via AAV |
| Carrie Miceli, PhD | University of California, LA | Single cell transcriptomics to assess transgene related responses |
| Katherine High, MD | Rockefeller University | Durability of transgene product expression in AAV gene therapy |
| Michael Lawlor, MD, PhD | Diverge Translational Science Laboratory | Gene therapy duration in canines and humans |
| David Mack, PhD | University of Washington | Ten-year durability of AAV8-MTM1 gene transfer in canine model of X-linked myotubular myopathy |
| Dongsheng Duan, PhD | University of Missouri | Long-term microdystrophin gene therapy in murine and canine DMD models |
| Karin Lucas, PhD | Sarepta Therapeutics | Microdystrophin gene therapy delivery and therapeutic plasma exchange in non-human primates |
| Manuela Corti, PhD, PT | University of Florida | Preclinical and clinical data for AAV redosing, durability and pre-existing antibodies |
| Julie Crudele, PhD | University of Washington | Combination therapy reduces existing Anti-AAV antibody by logs and allows for safe and efficacious redosing |
| Christopher Riling, PhD | Spark Therapeutics | Potential combination strategies to remove the barrier of humoral immunity in AAV re-dosing |
| Perry Shieh, MD, PhD | University of California, LA | Hepatotoxicity in MTM1 cases |
| James Dowling, MD, PhD | Hospital for Sick Children | Liver disease and preclinical models of X-linked myotubular myopathy |
| Julie Parsons, MD | Children’s Hospital of Colorado | Liver injury in SMA: Case presentations |
| Kelley Penraat, MS, DVM, MVSc, DACVP | Novartis | Hepatotoxicity, data mining from primate studies |
| Robert Brown Jr., MD, PhD | University of Massachusetts | Neuropathic pain and sensory neuropathy following AAVrh10-antiSOD1-miR therapy |
| Diana Bharucha-Goebel, MD | National Institutes of Health, Children’s National hospital | First-in-human intrathecal gene transfer study for giant axonal neuropathy: Trial updates (focus on safety/dorsal root ganglia) |
| Eloise Hudry, PhD | Novartis | Neurofilament light chain as a potential biomarker for AAV-driven DRG toxicity |
| Juliette Hordeaux, PhD, DVM, DECVP | University of Pennsylvania | Nonhuman primate models of high dose systemic AAV toxicities |
| Fraser Wright, PhD | Stanford University | Potential role of complement responses in viral gene therapy |
| Carl Morris, PhD | Solid Biosciences | Immunogenicity in high dose AAV gene therapy studies |
| Barry Byrne, MD, PhD | University of Florida | Kinetics of innate and adaptive responses to systemic AAV9 gene therapy |
| Jonathan Schwartz, MD | Rocket Pharmaceuticals | Optimizing AAV clinical safety (and efficacy) in Danon disease |
| Brenda Wong, MD | University of Massachusetts | Treatment of a single patient with CRD-TMH-001 |
| Terry Flotte, MD | University of Massachusetts | Adverse event and post-mortem review: CRD-TMH-001 |
| Russell Butterfield, MD, PhD | University of Utah | Comparative analyses between lethal DMD cases |
| Jordan Abbott, MD | University of Colorado | Immunosuppression strategies for gene therapies |
| Philip J. Brooks, PhD | National Institutes of Health | Bespoke gene therapy consortium |
| Sharon Hesterlee, PhD | Muscular Dystrophy Association | MDA’s Kickstart program |
| Lauren Black, PhD | Charles River Laboratories | Regulatory expectations for gene therapy ultra-rare disease |
| Richard Horgan, MBA | Cure Rare Disease | Trial journey and experiences learned from N of 1 case |
| Carsten Bönnemann, MD, habil. | National Institutes of Health | Shared databases and SOPs: Why and how? |
| Brian Tseng, MD, PhD | POLG Foundation | Consent and autopsy considerations |
| Eric Camino, PhD | Parent Project Muscular Dystrophy | Risk/benefit study on gene therapy |
| Paul Melmeyer, MPP | Muscular Dystrophy Association | Reimbursement, economic, advocacy, public policy barriers in gene therapy |
CURRENT STATUS OF CLINICAL AAV MEDIATED GENE THERAPY FOR SPINAL MUSCULAR ATROPHY (SMA), POMPE DISEASE (PD), DMD AND DANON DISEASE
I) Safety update of onasemnogene abeparvovec (OA, Zolgensma®, Novartis) for the treatment of SMA: Richard Finkel (St. Jude Children’s Research Hospital)
Onasemnogene abeparvovec (OA) is now marketed in 47 countries as Zolgensma®. With over 3000 individuals with SMA having been treated, OA has proven to be effective in improving survival and function in children with SMA. Its safety profile, however, is still incompletely established. US [1] and EU labels for OA differ in age, weight, and coverage of SMA types, thereby generating differing risk profiles. While almost all treated patients experience a treatment emergent adverse event (TEAE) such as vomiting, pyrexia, fever, thrombocytopenia or elevated aminotransferases or troponin I elevation [2, 3], about 1% of patients treated with OA have experienced life-threatening serious adverse events (SAEs) such as immune-mediated thrombotic microangiopathy (TMA) and liver failure. Cases of hydrocephalus have also been reported, with one case considered potentially drug related [4]. Deaths have been reported in children after OA administration – three deaths reported in clinical trials and six additional deaths identified post drug approval. While some of these fatalities were attributable to the underlying cause of SMA rather than treatment complications, death due to AAV triggered TMA has been documented as well as secondary hemophagocytic lymphohistiocytosis. Regardless, there is limited guidance for drug makers on how to address post-marketing AAV-related TEAEs. Recently reported TEAEs observed in OA-treated patients that are not currently listed on the label include necrotizing enterocolitis [5] and spinal cord tumor [6]. There are additional concerns for potential long-term side-effects, some of which were raised from preclinical work and not yet documented in dosed patients, such as the potential for late-onset toxic gain-of-function due to SMN overexpression. In mouse studies [7] and following intrathecal delivery in non-human primates [8], dorsal root ganglia and neuronal inflammation and degeneration were seen following intracerebroventicular administration of AAV9-SMN (a vector similar to OA). Even if not yet reported in human patients, careful monitoring over the long term is clearly advisable. Several SMA registries are in existence that can contribute to the collection of real-world data as part of post-marketing surveillance efforts for OA. These include industry-sponsored (RESTORE), academic multi-center (iSMAC, SMArtCARE) and patient advocacy (MDA, CureSMA) registries. Currently there is a lack of alignment between these registries. Data sharing and comparative data analysis would be facilitated by having more uniform assessment and intervention strategies, as well as by standardizing the way TEAEs are captured and reported. Such comparative data sharing would enable the development of improved post gene therapy standards of care and treatment guidelines, including better risk assessment considerations taking into account age, weight, dose and percent of full capsids, mode of administration, and concomitant medications, as well as establishing the basis for gauging durability of drug efficacy. Efforts must include acquiring, collating, and sharing uniform prospective safety and efficacy data, particularly on emerging populations such as neonates identified via newborn screening, older patients who are receiving OA via intrathecal (IT) delivery, one and four-SMN2-copy patients, and those on combination treatments.
II) Ongoing gene therapy programs for late-onset Pompe Disease (LOPD): Tahseen Mozaffar (UC Irvine)
Enzyme replacement therapy (ERT) using recombinant alpha-glucosidase (rhGAA) is currently the only approved therapy for LOPD, but limitations of this approach include insufficient tissue distribution to affected organs, especially in the CNS (only 1% gets to skeletal muscle [9, 10]) and a reduced effectiveness after 36 months [11]. AAV gene replacement therapy for GAA is being investigated as an alternative therapeutic strategy for LOPD [12, 13]. In mouse models, preclinical doses in the range of 2×1011vg/kg resulted in more consistent and durable levels of therapeutic enzyme expressed than in ERT to mitigate the effects of lysosomal glycan accumulation. Clinical programs for LOPD gene therapy are ongoing at five companies with the primary goal of determining impact on levels of functional enzymes in target tissues as well as their safety. The technologies in development differ in target tissue, capsid, promoter and transgene used. Three companies (AskBio, Spark Therapeutics, Astellas) have gene therapy candidates in clinical trials, while two additional companies have candidates in preclinical development (Amicus, Regeneron). AskBio and Spark are testing liver-targeted candidates. Astellas is testing muscle-targeted candidates and Amicus is aiming for candidates that are broadly expressed. AskBio’s liver-targeting gene therapy data so far supports safety and bioactivity of AAV8-LSPhGAA (1.6×1012 vg/kg), with stable levels of serum GAA and an increase in muscle GAA levels at 52 weeks post-treatment, and no reports of SAEs or ALT elevations associated with anti-AAV T-cell response [14]. Astellas’s muscle-targeting gene therapy trial has dosed four patients, three of whom were able to discontinue ERT. A brief clinical hold was placed on the trial following a reported SAE involving an axonal sensory polyneuropathy [15]. Recruitment has now resumed with slight modifications in study protocols. Spark is currently holding recruitment and will restart with enrollment in 2024 with a different, enhanced vector and redesign of the trial. Considerations raised for LOPD gene therapy include the potential for anti-GAA immunity that may develop in patients (as observed in ERT), as well as baseline specific capsid immunity that precludes patients from gene therapy. Given the higher vector dose in adults and especially in Astellas where it is muscle targeted rather than liver targeted, the concern for potential hepatotoxicity and myocardial toxicity is high.
III) Safety updates in ongoing DMD microdystrophin trials: Francesco Muntoni (University College London)
At the time of the meeting there were four DMD gene replacement therapy efforts (Pfizer, Sarepta, Genethon, Solid Biosciences) with a fifth effort that was about to begin (Regenxbio). A combined total of ∼200 DMD patients have received AAV gene therapy across the four programs, and learnings from these lead to a better understanding of emerging common AEs, less common SAEs and the development of mitigating strategies. Typical reactions observed in the first week post-treatment include nausea, vomiting, loss of appetite, fever, malaise, and troponin increase. In the first two weeks, the innate immune response and/or early humoral response against viral capsid can lead to complement activation and cause thrombotic microangiopathy (TMA), with concomitant kidney, liver, and other organ injury. Within the first 3 months, an adaptive immune response against the viral capsid (T-cell mediated) can cause liver inflammation and toxicity, while an adaptive immune response against the transgene can manifest as skeletal and cardiac muscle inflammation (see below). Higher AAV-dose treatment cohorts are linked to more frequent and serious AEs, while the specific capsid used factors in as well. All treated patients experience some form of AE, but only ∼8–10% experience SAEs. Evolving insights into contributing factors towards AEs, such as total AAV capsid load and specific transgene epitopes has led to multiple protocol modifications, including exclusion of at-risk genotypes and the use of immune suppression regimens as a reactive response to AEs or, in some protocols, proactively. Current mitigating strategies for liver injury include use of a universal steroid prophylaxis with plans for increasing oral doses or IV methylprednisolone in cases of severe hepatitis. To address the risk of TMA-triggered atypical hemolytic uremic syndrome (aHUS), Solid and Regenxbio proactively dose with C5b9 inhibitor eculizimab, while other sponsors employ a more reactive protocol. For the occurrence of hepatopathy, myositis and myocarditis, steroids are administered with tacrolimus as an option. Sponsors have moved to employ anti-transgene mitigating strategies and have set the following exon exclusion criteria for further trials: Pfizer [9–13, 29–30, 56–71], Sarepta [9–13, 45], AFM [1–17], Solid [8–11], Regenx [1–17]. Areas for ongoing investigation include understanding susceptibility to SAEs, in the context of host immunological genetic background and other comorbidities.
IV) Optimizing AAV clinical safety and efficacy in Danon disease: Jonathan Schwartz (Rocket Pharmaceuticals)
Rocket pharma conducted a phase 1 clinical trial with RP-A501 (AAV9-LAMP2B) in pediatric and adult patients (6.7×1013 vg/kg - low dose, 1.1×1014 vg/kg - high dose). Results of the trial was consistent with preclinical data generated in the Danon mouse model, which showed efficacy in earlier-stage disease (prevention of end-stage cardiomyopathy/congestive heart failure) as well as in later stages. Although minimal toxicity was observed following administration of the 2–3×1014 vg/kg dose in absence of immunosuppression in mouse and non-human primate (NHP) models, this did not reflect the full spectrum of adverse events observed in patients dosed. The most concerning AE was seen in one patient in the high dose adult cohort in the form of complement-mediated TMA and associated thrombocytopenia and acute kidney injury/renal failure. There were also instances of corticosteroid-related exacerbation of the underlying skeletal myopathy in the adult cohort patients. An immunomodulation regimen used to potentially preempt these events included two doses of rituximab prior to gene therapy and sirolimus and prednisone both pre- and post-therapy, with reduced prednisone doses and earlier taper initiation (starting approximately day 10). This regimen was utilized for the two pediatric cohort patients receiving low dose RP-A501; in these patients there was evidence of limited complement activation, no TMA and no RP-A501 or corticosteroid related serious adverse events. The approach has been accepted as part of the RP-A501 pivotal phase IIB clinical study. These observations contribute to the growing body of evidence that there are disease-, patient- and AAV-specific features that impact safety across all gene therapies.
ANTI-TRANSGENE CONSIDERATIONS IN DMD GENE THERAPY
I) Challenges in transgene-triggered immunogenicity in Genethon’s microdystrophin trial: Serge Braun (AFM-Telethon, Genethon)
Preclinical studies of Genethon’s microdystrophin demonstrate a minimum effective dose range of 1×1013 to 3×1013 vg/kg. The first patient dosed with this construct, despite receiving a relatively low dose (1×1013 vg/kg) compared to other DMD clinical trials, experienced a SAE manifesting as generalized myositis and myocarditis with increased levels of creatine kinase (over baseline) and troponin I at seven weeks post-infusion. Tacrolimus was administered for several months until symptom resolution. Muscle biopsy of the patient after manifestation of symptoms revealed infiltration of inflammatory cells (macrophage, B and T cells). Immunological epitope mapping studies of antibodies identified three IgG epitopes that emerged at week 7, mapping to peptides encoded by exons 10, 12 and 17. Moving forward, a safety mitigation plan was designed to exclude patients with pathogenic mutations in DMD exons 1-17, as well as 28–29. The trial was placed on clinical hold for one year but is now resumed with an added prophylactic immunosuppression using sirolimus. A unique collaborative effort together with the other DMD gene therapy sponsors, as well as academic advisors, point to the location and size of missing protein sequence combined with its epitope content as the most probable cause of immunogenic responses observed in a subset of patients with N-terminal mutations across three of the clinical trials [16]. Factors that may contribute to anti-transgene immune response as they are currently understood involve the patients genotype and resulting cross reactive immunological material (CRIM)-status, the specific transgene epitopes expressed, tolerization through prior exposure to parts of native protein, disease specificity as well as vector type. Ongoing analysis by this collaborative group is focused on identifying at-risk patients based on HLA genotypes as individual susceptibility factors and, so far, points to a probable detrimental T-cell response targeting peptides encoded by exons 8-9. Plans are underway to further investigate the cellular infiltration in muscle using spatial transcriptomics and multiplex proteogenomic approaches to identify involved pathways, while mass spectrometry based immune profiling is applied to identify immune biomarkers. Anti-transgene responses are to be given more consideration for gene therapies given to CRIM-negative patients in whom epitopes of the protein product have never been encountered. The full breadth of anti-transgene responses associated with gene therapies may currently still be under-represented given that FDA-approved gene therapies currently in the clinic such as Luxturna, Glybera and Zolgensma apply to patients with CRIM-positive background and hence expected immune tolerance.
II) Minimizing Transgene Immune Responses in DMD: Jeffrey Chamberlain (University of Washington)
One strategy to overcome anti-transgene responses is to lessen the immunogenic impact of specific regions of microdystrophin constructs. Re-designs of the transgene to generate novel constructs that are less prone to trigger an adaptive immune response is possible without compromising functionality. Software-guided approaches were used to redesign three human dystrophin epitopes previously identified as particularly immunogenic [16] – and confirmed by recent events in the DMD clinical trials. Microdystrophin constructs harboring the redesigned epitopes were tested using a combination of in vitro and in vivo DMD models and were shown to be stable, functional and less immunogenic. Thus, successful redesigns can serve as the basis for construction of next-generation ‘de-immunized’ microdystrophin constructs.
III) Single Cell Transcriptomics to Assess Transgene Related Responses: Carrie Miceli (University of California Los Angeles)
Single nuclear RNA sequencing (snRNA-seq) on muscle biopsies of patients post gene therapy can be used to shed light on transcriptional and cellular responses to AAV mediated micro dystrophin gene transfer. A reference single nuclear transcriptomic dataset of all muscle resident populations has been established from 27 DMD and 5 healthy individuals. Preliminary data on a two-year post gene therapy sample showed an increase in percentage of T cells relative to the reference set. This study will further seek to assess transcriptional responses of cytokines, granzymes, inflammasome components, and Toll-like receptors in treated patient samples to provide insight into the immune responses occurring on a molecular level and thus reveal potential targets for immune modulation.
IV) Harnessing Central Tolerance for AAV Vector Safety in DMD: Hansell Stedman (University of Pennsylvania)
Hansell Stedman (University of Pennsylvania) presented on micro-utrophin gene replacement as an alternative treatment approach to using micro-dystrophins for DMD in the context of mitigating anti-transgenic immunological responses. Studies in a dystrophin deletional-null canine model (GSHPMD) [17] demonstrated that intramuscular (IM) injection of micro-dystrophin, but not micro-utrophin, elicited a robust T cell response to epitopes scattered throughout the entire transgene product, with focal destruction of micro-dystrophin-expressing myocytes. In mdx mice and GRMD dogs, micro-utrophin conferred robust therapeutic benefit, uniquely including protection of MYH16 (+) myocytes which in the untreated GRMD dog are the most severely affected because of the exceptionally high contractile force provided by this “superfast” myosin [18]. These findings point to utrophin-derived therapies as potentially having a more favorable immunologic profile than dystrophin, in particular in the context of a cross reactive immunological material (CRIM)-negative background.
DURABILITY OF AAV GENE THERAPIES
I) Factors affecting durability in hemophilia gene therapy - Katherine High (Rockefeller University)
Although the durability of neuromuscular disease gene therapies remains largely unknown, we can learn from factors that contribute to durability in hemophilia gene therapy. Independent of manufacturing platform, AAV-based therapy for hemophilia B, which delivers factor IX, has shown durability as long as ten years with 1–8% circulating factor IX expression. In contrast, gene therapy for hemophilia A (Roctavian), which delivers factor VIII, has resulted in variable durability [19]. In the Roctavian trials, levels of circulating FVIII appear to decline over time, through a mechanism that is not known [19]. A combination of factors has been identified that may contribute to this clinical outcome. These include both the manufacturing platform, and the size of the expression cassette. In a study in mice, it was shown that for vector manufactured in insect cells using baculovirus, transgene expression gradually declined over the course of a year in mice that received an AAV vector with an oversized insert, but that the decline was much less marked for inserts similar in size to the wild-type genome [20]. Studies of DNA harvested from mouse liver at the point of peak transgene expression and at the end of one year were assessed for areas of open chromatin using ATAC-Seq (assay for transposase-accessible chromatin with high-throughput sequencing). The data show that there is a marked decline in areas of open chromatin for baculoviral-Sf9 vector with oversized inserts between peak expression and one year, and that the level of transposase-accessible chromatin correlates with the ratio of RNA/DNA in the cells. This shutdown of open chromatin does not occur with vector made in HEK293 cells; whether these same changes occur in human liver will require analysis of serial liver biopsies. Age of recipient is another factor that may contribute to loss of expression, as the (predominantly) episomal transgene is lost to one daughter cell with each round of cell division during growth. Thus, young children who are rapidly growing would be expected to experience gradually diminishing levels of expression for vector administered to any tissue where cells are still dividing. This is not expected to be an issue for cells that are no longer dividing at the time of vector administration, for example cells in the retina (Luxturna) or neurons in the spinal cord (Zolgensma).
II) Durability of XLMTM gene therapy in canines – David Mack (University of Washington)
Astellas’s AAV8-MTM1 gene therapy has been extensively studied in mouse [21] and canine models [22, 23], demonstrating reversal of disease pathology at the functional and molecular levels [24–26]. Results from these Investigational New Drug (IND) studies demonstrated prolonged survival, restored muscle strength, reversal of muscle pathology and no immune response to the transgene. Importantly, no disease-related liver dysfunction comparable to that encountered in patients was observed in these models. Long-term durability data were collected on a cohort of treated dogs approaching their ten-year mark post-treatment. All functional metrics, including gait, diaphragmatic strength, hindlimb force output and yearly neurological physical exams remain indistinguishable from normal controls. Histology on biopsies from three muscles showed hallmarks of aging but no structural abnormalities suggesting the reemergence of MTM pathology. Vector copy number in the biceps brachii, vastus lateralis and cranial tibialis in years 4-8 post-treatment showed left-right and forelimb versus hindlimb variability between dogs. mRNA levels appeared more consistent, with 10-20 fold higher levels of transgenic MTM1 RNA than endogenous levels in normal controls. Transgenic myotubularin protein expression was consistent from one year to the next at levels approximately 25% of normal but somewhat variable between the three muscles biopsied and between dogs. These data confirm that less-than-normal levels of myotubularin protein are sufficient to completely restore function and demonstrate that AAV-mediated gene transfer for a congenital myopathy can last for at least a decade in this model.
III) Durability considerations in animals and humans - Michael Lawlor (Diverge Translational Science Laboratory and Medical College of Wisconsin)
Analysis of tissues from AT132-treated XLMTM patients illuminated differences in the timing and extent of pathological recovery between experimental animals and humans. That is, for mice pathology reversal is noted within 8 weeks of treatment; whereas in patients, improvement in histology is typically noted between 6–12 months post-treatment. Assessment of efficacy and duration in gene therapy is likely not fully comparable between animals and humans, or between humans with different disease indications. AAV delivered episomes remain extrachromosomal for the majority and therefore are not replicated with cell division, resulting in their eventual dilution and loss. Diseases that cause damage in a mitotically active tissue, as is the case for DMD, may thus have different therapeutic duration than those where degeneration/regeneration is not a major feature (XLMTM), or where the target cell is postmitotic (motor neurons in SMA). However, in cases where successful gene therapy prevents further myofiber degeneration, therapeutic duration could ideally be similar between degenerative- and non-degenerative disorders. In addition, duration of gene therapy may depend on the age of treatment if target tissues have not reached maximal volume, i.e. is still growing by nuclear division or addition. Collection of several hundred biopsies to model growth curves for predicting fiber size is under way and will be important for assessing the impact of myofiber size change on therapeutic duration.
Assessment of muscle tissues from Solid Bioscience’s IGNITE study on DMD patients highlights the variability of transgene encoded micro-dystrophin protein expression. Western blot protein quantitation at 3 months post-treatment of two patients resulted in micro-dystrophin restoration to 17.5% and 8%, with one below detection limit. In these same patients past one-year post-treatment, two patients showed an increase of micro-dystrophin to 69.8% and 20.3%, respectively. Another patient in this cohort had microdystrophin expression below the level of quantification (5%) at both 3 and 12 months. Future follow-up protocols should consider that three-months post treatment does not yet reflect a plateau of efficacy for DMD gene therapy as assessed by transgene encoded protein expression.
The overall durability of gene therapy efficacy in humans thus remains unclear. Given that animal models do not accurately predict timeline for maximum efficacy in humans, their ability to predict duration remains questionable, with many studies not hitting their ideal efficacy target with the patients that have been dosed. Variables relating to efficacy and duration noted so far include disease pathophysiology and age at dosing. Current clinical trial studies are not well suited to answer issues of therapeutic duration.
IV) Long-term micro-dystrophin expression in animal models - Dongsheng Duan (University of Missouri)
In mdx mice, systemic AAV-delivered micro-dystrophin gene therapy resulted in life-long protection. That is, a single dose at 3 months of age resulted in reduction of fibrosis in both skeletal muscle and heart, reduction in creatine kinase, improved grip force and treadmill running, and improved ECG and cardiac hemodynamics which were maintained until at least 18 months post-treatment [27]. In affected GRMD dogs, long-term systemic micro-dystrophin gene therapy did not cause acute or delayed toxicity, and improved muscle force, muscle blood perfusion, whole body activity, and heart function. Regardless of initial dose level, micro-dystrophin RNA and protein levels persisted through 42 months, despite notable loss in vector genomes over time. The mechanisms underlying the loss of the vector genome remain to be clarified but may relate to fiber degeneration and turnover inherent to the dystrophic phenotype of the model.
RE-DOSING STRATEGIES FOR AAV GENE THERAPIES
I) Therapeutic Plasma Exchange in Non-Human Primates - Karin Lucas (Sarepta)
Two consecutive studies were performed dosing non-human primates (NHPs) with 2×1014 vg/kg delandistrogene moxeparvovec. The first study sought to investigate the impact of various immunosuppression strategies on the safety and efficacy of a single dose of gene therapy, with the hypothesis that the duration and regimen of steroids could lead to higher vector genome copy numbers. In this study, different strategies involving combinations of prednisone, rituximab, and sirolimus were investigated. Transient elevations in liver enzyme ALT and AST were observed in NHPs that received only prednisone. The triple immune-suppression cohort experienced vomiting and transient elevation in heart rate after the final rituximab dose as well as hives 2–3 weeks post-treatment. Ultimately, the results of this study showed no difference in vector genome copies among NHP with different immune strategies 12 weeks after a single dose of gene therapy.
NHPs from the study above were further evaluated in a second study which sought to investigate the impact of performing therapeutic plasma exchange (TPE) prior to re-dosing, with the hypothesis that TPE will reduce anti-AAVrh74 antibody titer. TPE involves the physical removal of antibodies using plasmapheresis. The triple immune suppression regimen did not effectively prevent anti-capsid antibody production. NHPs from this cohort were re-dosed without TPE as a control. NHPs from the prednisone-only cohorts underwent TPE which was well tolerated with no abnormal clinical or immunological observations. NHPs that received 2–3 rounds of TPE demonstrated significant albeit variable reduction of circulating antibody titers and were safely re-dosed. It was noted that in some NHPs, TPE did not reduce titers below the eligibility threshold of 1 : 400 used for dosing SRP-9001 in clinical trials. Redosing resulted in transient elevations in liver enzymes (ALT and AST), as was observed following the first exposure. NHPs re-dosed in the presence of high antibody titers experienced the following AEs: increased heart rate and ventilation rate, vomiting, rash near delivery site, paleness of the skin, and shallow breathing. These resolved after administration of diphenhydramine and dexamethasone. When antibody titers were reduced with TPE, only minimal safety issues were observed. These results highlight the importance of reducing anti-AAV antibodies before dosing. The data presented suggests that TPE may be a safe and efficacious strategy to consider lowering anti-AAV antibodies, however a practical consideration is that the short duration of antibody lowering following TPE presents challenges to using this approach for commercial therapy. Further studies are needed to evaluate the safety and efficacy of GT dosing with pre-existing immunity to AAV.
II) Preclinical and Clinical Data for AAV Redosing, Durability and Pre-existing Antibodies - Manuela Corti (University of Florida)
Long term clinical data from AAV trials has shown a decline of transgene expression over time, which may be due to immune responses and vector dilution in muscle [28]. Reduced transgene expression in newborn NHPs dosed with systemic AAV-GAA was observed over the first year by muscle biopsy. Factors that may reduce durability include non-immunological as well as immunological factors. Non-immunological factors include epigenetic processes (transgene silencing), cell turnover in nontarget and target cells, cellular stress, patient lifestyle factors [28]. Immunological factors include innate immune response to CpG motifs and dsRNA, adaptive immune response to vector/transgene protein (cellular and humoral), and bystander activation. A study using a canine model for early onset Pompe (GAA-null) demonstrated that liver-targeted vector delivery with AAV-LSPcoGAA along with systemic delivery AAV-DES-GAA was successful in preventing the anti-hGAA antibody response that is typically noted at day 90 post-infusion. This demonstrated that that liver-targeted vector delivery prior the therapeutic muscle directed vector was successful in inducing transgene tolerance in the setting of a CRIM-negative model. Re-dosing was successfully investigated using immune suppression protocols based on B-cell depletion (rituximab) and mTOR inhibitor (sirolimus) agents in primates and in two Pompe patients (NCT02240407). The immune suppression effectively mitigated the rise of antibodies against AAV, thereby enabling transgene expression following the second dose of AAV in the setting of naïve status. Strategies for removal of anti-capsid antibodies were also tested. The window of IgM and IgG reduction following one round of plasmapheresis was 24–48 hours in a subject who received a systemic dose of AAV. Various pharmaceutical strategies for reduction of anti-capsid antibody were explored in preimmuned mice, employing combinations of immune suppression drugs, including rituximab, sirolimus, darzalex, velcade, and vyvgart. All combinations of immune modulation drugs tested in the study resulted in a reduction of anti-capsid antibodies ranging from 33% to 88% compared to baseline. Further studies are needed to determine a meaningful threshold cut-off for antibody titers (neutralizing (nAb) or total binding (tAb)) for dosing in clinical trials. Additionally, further research is required to identify the most effective immune modulation regimen for decreasing preexisting AAV immunity in humans.
III) Strategy to reduce anti-AAV antibodies for safe and efficacious re-dosing in XLMTM canine model – Julie Crudele (University of Washington)
XLMTM is caused by mutations in the myotubularin 1 gene (MTM1). The gene therapy used for this study was created at Genethon and consisted of AAV8 delivering the canine MTM1 under the human desmin promoter. A first dose of 8×1013 vg/kg of AAV was administered to two 10.9 week-old XLMTM dogs. No immune suppression other than prednisolone was given to prevent anti-AAV antibody formation. Dogs were given a combination of immune suppression drugs (prednisone, sirolimus and rituximab), along with 3 rounds of therapeutic plasma exchange (TPE), given 4, 2, and 0 days prior to second dose of gene therapy. The second dose was 1.4×1014 vg/kg of the same vector used for dose one and was given 112 or 119 days after the first dose, depending on the dog. Dogs were necropsied 243 days after the first dose. One dog had a slight drop in anti-AAV total antibodies and both dogs had a drop in anti-AAV neutralizing antibodies with pharmacologic intervention alone. Both dogs total and neutralizing antibodies dropped by more than one log following TPE. Neutralizing antibodies were < 1 : 5 at baseline and peaked at 1 : 5120 before intervention in both dogs. After intervention, the neutralizing antibodies fell to 1 : 320 and 1 : 160, depending on the dog. The investigators predict that additional rounds of TPE would lower antibodies even further. Each dog showed signs of re-dosing efficacy, with improved muscle forces and neurologic assessments. One dog developed a urinary tract infection during the week of TPE that was successfully treated with antibiotics. There were no indications of anaphylaxis, nephritis or other hypersensitivity reactions, which might be expected when administering AAV in the context of high anti-AAV antibodies without interventions. These studies represent a proof of concept that AAV can be readministered safely and efficaciously using a combined approach to reduce anti-AAV antibodies in a large animal model.
IV) Protein-based strategies to remove barriers to humoral immunity - Chris Riling (Spark Tx)
IdeS is an endopeptidase that degrades human IgGs (removes effector domain from antigen-binding), resulting in rapid clearance of IgGs [29]. It is used in the European Union for adult kidney transplant patients. Neonatal Fc receptor (FcRn) facilitates IgG recycling and extends IgG half-life in circulation. Monoclonal antibodies that bind FcRN can interfere with IgG recycling and can reduce IgG levels in circulation. In preclinical models, anti-FcRN and IdeS dosed consecutively as a combination therapy enables AAV transduction to the liver in the presence of neutralizing antibody titers. Thus, this combination may be used as a complimentary approach to anti-AAV IgG reduction to allow vector re-administration.
ORGAN TOXICITIES OBSERVED IN AAV GENE THERAPIES
I) Liver toxicity in XLMTM cases – Perry Shieh (University of California Los Angeles)
The four patient deaths who received AT132 in Astella’s ASPIRO trial were attributed to severe cholestatic liver failure [30]. All participants met the trial eligibility criteria of ALT/AST< =5x upper limit of normal at the time of dosing and there was no evidence of hepatic peliosis on liver ultrasound. Timelines of clinical events in all four patients were variable. In an effort to reverse the progressive liver dysfunction all received immunomodulating therapies, without apparent benefit: After initial prophylactic prednisolone regimen per protocol, escalating regimens varied by subject and included increased prednisolone, methylprednisolone pulses, and in some patients anakinra, tocilizumab and ruxolitinib. Five additional participants experienced hepatobiliary SAEs with non-fatal outcomes. Three of the four participants with a fatal SAE, and who had received the high dose of 3.5×1014 vector copy number per kilogram (vg/kg) had substantially higher vector load in the liver compared to skeletal muscle and heart. In the one patient who had experienced a fatal SAE after receiving the low-dose of 1.3×1014 vg/kg, the liver vector copy number was less than the higher-dose participants, though substantially higher than in other tissues. In all four of these patients there was no transgene protein expression in the liver as expected for vector delivery under the control of the muscle-specific desmin promoter. Immune response assessments performed include humoral adaptive immune response (anti-AAV8 neutralizing antibody and anti-MTM2 antibody), T-cell immune reactivity (IFN-g ELISPOT) to AAV8 and myotubular peptides, complement factors and cytokines. These measures do not clearly indicate the immune-mediated response as directly contributing to the hepatobiliary SAEs in ASPIRO trial. Careful reanalysis of XLMTM natural history reveals that underlying hepatobiliary disease is an under-recognized feature of the disease itself. This had not been observed in mouse, canine or NHP models in the absence or presence of treatment but has recently been recapitulated in the Mtm1 zebrafish model [31]. The ASPIRO trial remains on hold while investigations continue into the deaths resulting from the consequences of acute exacerbation of this hepatobiliary disease after AAV dosing, to further understand the mechanism of AAV hepatotoxicity and susceptibility due to underlying liver disease in XLMTM patients. Plans for patient identification and risk mitigation for hepatotoxicity related SAEs include further experiments to understand mechanism of cholestasis in XLMTM and exacerbation by AAV; reduction of empty capsids to enable lower total capsid dose; and modification of participant eligibility criteria to reduce risk of hepatic toxicities.
II) Preclinical Models of X-linked Myotubular Myopathy - Jim Dowling (Hospital for Sick Children)
Zebrafish model of XLMTM was characterized to better understand the liver disease associated with XLMTM and its relationship to gene therapy [31]. Mtm1 deficient zebrafish have both fatty liver and changes consistent with liver cholestasis. Feeding fish with a fluorescent lipid dye results in its abnormal localization due to defects in bile transport which is mechanistically attributed to loss of the bile salt transporters Bsep and Mdr1. Interestingly Mtm1 deficient zebrafish livers show a characteristic transcriptional profile with changes in immune-related genes, different from other liver disease models. Re-expression of MTM1 in hepatocytes rescue the cholestatic defects in mtm zebrafish. Drug screening using this model revealed that dynamin2 inhibitors have the potential to rescue the cholestatic phenotype of mtm zebrafish. Other XLMTM models currently explored in the Dowling lab include ‘sensitized’ XLMTM mice and iPSC-derived hepatocytes with MTM1 mutations. Nutritional sensitization of the XLMTM mouse results in a reproducible liver phenotype – testing of AAV exposure on this background is now under way. XLMTM iPSC-derived hepatocytes have abnormalities in the bile canalicular structure that can be rescued using dynamin2 inhibition. Strategies to prevent the AAV dependent liver toxicity in MTM1 disease therefore include small molecule treatments to downregulate dynamin 2, re-introducing MTM1 prior to AAV therapy using non-AAV strategies, and preventing AAV from entering liver.
III) Liver Injury in SMA: Julie Parsons (Children’s Hospital of Colorado)
There have been clinical cases of unexpected liver injury in children treated with Onasemnogene abeparvovec (Zolgensma®) for SMA as reflected by 3 cases observed at Children’s Hospital of Colorado). Case 1 presented with mild hepatitis with blood eosinophils; Case 2 presented with chronic hepatitis with inflammatory cells (CD3 and T cells), moderate fibrosis and focal hepatocyte necrosis; Case 3 presented with hepatic degeneration with balloon cells, extensive inflammation (CD8 T cells), marked bile ductular reaction, and moderate fibrosis. Laboratory findings in all cases include increases in transaminases and inflammatory markers. Treatment regimens include high dose methylprednisolone, prednisolone, anakinra, and tacrolimus. It was pointed out that neurologists do not have a ‘game plan’ for dealing with unexpected liver failure in response to Onasemnogene abeparvovec. Timing of consultation with hepatologist, and important lab parameters need to be established, as well as standard treatment protocols for how much prednisolone/methylprednisolone to administer. Administration of gene therapies requires engagement of a multi-disciplinary clinical team that may consist of immunologists, hepatologists, cardiologists and nephrologists. In general, neurologists are the specialists prescribing Onasemnogene abeparvovec. When unexpected liver failure occurs after dosing, currently there is no standard protocol for when to consult a hepatologist or how to treat effectively using steroids (methylprednisolone/prednisone) or other immune modulating therapies. Safe and effective administration of gene transfer therapies requires access to a knowledgeable multidisciplinary team of subspecialists including immunology, hematology, cardiology, nephrology and hepatology.
IV) Hepatotoxicity, Data Mining from Primate Studies: Kelley Penraat (Novartis)
Hepatotoxicity is frequently reported in the preclinical and clinical setting in the context of AAV gene therapies–most AAV serotypes traffic to the liver regardless of route of administration resulting in high capsid load to the liver and depending on the transgene promoter creating heterogeneous expression patterns (varying from low to high expression) in individual hepatocytes. Risk of hepatotoxicity is particularly elevated after IV delivery of large amounts of vector but can also arise when AAV is delivered within the IT space as the bulk of capsids rapidly egress from the cerebrospinal fluid (CSF) to the periphery. Liver effects observed have ranged from asymptomatic transient elevations in aminotransferases to acute liver failure. Novartis has generated a large data set (>100,000 data points) in NHPs using scAAV9 therapeutic and reporter constructs under the CBA ubiquitous promoter, which was used to mine data on hepatoxicity. The time course of events with intravenous dosing shows an increase in ALT and decrease in platelets within 3 days after dosing, followed by a gradual decline in ALT and subtle increase in platelets, which occurs as vector plasma load decreases. These events are followed by an increase in monocytes. After two weeks, livers have no toxicity signs in necropsy. The robust anti-AAV9 antibody response has no apparent correlation to increased transaminase activity and monocytes or decreased platelets with intravenous dosing. Liver toxicity occurs prior to a measurable adaptive immune response and prednisolone at 1 mg/kg does not appear to mitigate the increase in transaminases. Overall, NHP studies suggest that increased vector load and increased heterogeneous overexpression of the transgene in liver may be inciting events for acute liver injury with scAAV9. NHP studies offer insight into the mechanism of acute (<1 month post dose) liver injury with scAAV9 gene therapy, suggesting that high vector load and early innate immune responses (prior to the adaptive responses) may be important initial alterations setting up a proinflammatory microenvironment that can predispose for downstream liver injury.
V) Neuropathic Pain and Sensory Neuropathy Following AAVrh10-antiSOD1-miR Therapy: Robert Brown (University of Massachusetts)
Neuropathic pain and sensory neuropathy was reported following intrathecal AAVrh10-anti-SOD1 miRNA therapy in an ALS patient [32]. In a single case, IT administration of AAVrh10-antiSOD1-miR provoked a focal, transient pain syndrome concurrently with transient elevations of liver enzymes followed by evidence for a generalized sensory neuropathy. In this case, serum T cell reactivity to AAV capsid was elevated, CSF revealed pleocytosis and elevations of neurofilament levels. Autopsy confirmed the presence of a T-cell infiltrate in some dorsal root ganglia and peripheral nerves. In a second case, in which the gene therapy recipient was immunosuppressed for 6 months, neither hepatotoxicity, sensory symptoms nor neuropathy were noted.
VI) Intrathecal Gene Transfer Study for Giant Axonal Neuropathy: Diana Bharucha-Goebel (NIH, Children’s National Hospital)
In the first-in-human trial for Giant Axonal Neuropathy (GAN), scAAV9-JeT-GAN (single site, phase I dose escalation) was administered with immune modulation (IV methylprednisolone, prednisone) as well as rapamycin (at the 3.5×1014 vg per patient level) and tacrolimus (for patients predicted to be CRIM negative) were administered to minimize anti-transgenic product responses in CRIM negative patients. Patients were dosed at ages 6 years and older when they were already symptomatic. There was no reported clinical evidence of Dorsal Root Ganglion (DRG) toxicity following IT gene therapy with scAAV9-JeT-GAN and no abnormal sensory findings. Further, there was no evidence of inflammation in the spinal cord or DRG in a single autopsy case from a trial participant. The frequency of SAEs noted in this trial appeared to be linked to baseline disease severity. The most frequent related adverse event is CSF pleocytosis/WBC elevation (asymptomatic). There was persistence of and/or reappearance of upper extremity sensory SNAP (Sensory Nerve Action Potential) responses occurring in 6 participants, indicating preservation or restoration of DRG function and adding further support against DRG toxicity due to the gene.
VII) Neurofilament Light Chain as a Potential Biomarker for AAV-driven DRG Toxicity: Eloise Hudry (Novartis)
Neurofilament light chain can be used as a potential biomarker for AAV-driven dorsal root ganglia (DRG) toxicity [33]. Neurosensory toxicity with AAV gene therapy has been noted in preclinical models (non-human primates, pigs, and rats) and in a clinical trial for SOD1 related ALS. Most AAV serotypes traffic to the DRG in non-human primates (NHPs) after intravenous (IV) or intrathecal (IT) dosing, creating heterogeneous expression patterns in individual sensory neurons. Therefore, AAV-driven DRG toxicity is a platform safety concern for AAV as a therapeutic modality. The AAV-induced toxicity takes the form of a lesion that is generally asymptomatic, multifocal and sporadic, making its identification and characterization challenging. Key histopathological features include neuronal degeneration and mononuclear cell inflammation which decreases in incidence and severity after 12 weeks post-dosing and appears to be non-progressive at 52 weeks. The study reported included 260 NHPs (after either IV or IT delivery of AAV9) and demonstrated the role of transgene expression for the neuronal injury in contrast to a paucity of changes following the administration of empty capsids and promoter-less constructs. Neurofilament light chain (NfL) emerged as a promising biomarker of DRG injury in NHP dosed with AAV9. The time course of changes in blood and CSF NfL correlated with severity and time course of the histopathological changes observed in tissue. Blood (plasma and serum) NfL correlated well with CSF values and therefore the sampling of blood may suffice to detect this otherwise clinically silent injury in the primate. However, NfL is constitutively released at lesser concentrations during normal physiologic processes and at greater concentrations with neuronal injury that are not disease specific and could be related to the dosing procedure itself. Longitudinal time course sampling is recommended when interpreting asymptomatic transient elevations of NfL in blood and CSF as a potential biomarker of AAV-induced asymptomatic DRG injury. The potential usefulness of NfL measurement to monitor AAV-driven DRG toxicity in the clinical setting, especially in patient populations affected by neurodegenerative disorders, warrants further investigation.
VIII) Systemic AAV Toxicities: Juliette Hordeaux (University of Pennsylvania)
Studies of high-dose systemic AAV toxicities in non-human primate models suggest three stages of toxicity:
Acute toxicity (0–14 days) is represented by an innate immune response and early over-expression. Day 3–7 is characterized by transaminitis, thrombocytopenia, while day 6–12 is characterized by TMA, DIC, cytokine release syndrome, generalized edema, pericardial effusion, and cardiac event. This stage can be summarized as endothelial injury syndrome.
Subacute toxicity (2–4 wks) is represented by an adaptive immune response and overexpression. This stage is characterized by delayed transaminitis, liver failure, cardiac inflammation, and skeletal muscle inflammation.
Chronic toxicity (>4 wks) is represented by an adaptive immune response and is capsid or transgene related. Toxicities occurring from 2 week onwards can be summarized as T-cell mediated cytotoxicity due to capsid/transgene.
Acute toxicities observed in humans from the Onasemnogene abeparvovec experience and in the current DMD trials show both similarities to and differences from the NHP data. Similarities include transaminitis, thrombocytopenia, generalized edema, DIC/coagulopathy, troponin I elevation, and alternative pathway complement activation. Aspects unique to humans are vomiting, hypertension, kidney failure, cytokine release syndrome, cardiac events, and classical immune-complex mediated complement activation. There appears to be a dose threshold effect for liver toxicity and complement activation after systemic AAV, which does seem to translate from NHP to humans (this threshold is reached at doses ≥5×1013 vg/kg). Conclusions from the NHP studies that are relevant to human applications therefore include predicting potential toxicities and target organs of toxicities (liver, endothelial cells, DRG sensory neurons, heart); predicting dose-limiting toxicities (risk zone above 5×1013vg/kg); and insight into the actual mechanism of toxicities. Gaps that remain include extrapolating risk related to patient disease when testing toxicity in healthy adolescent NHPs in terms of baseline liver and cardiac function as well as their reserve capacity.
COMPLEMENT-RELATED ADVERSE EVENTS
I) Potential Role of Complement Responses in Viral Gene Therapy: Fraser Wright (Stanford University)
The classical immune response pathway requires antibodies (IgM, IgG) binding to the target antigen to activate the complement cascade. Antibody binding to AAV capsids subsequently blocks transduction and potentiates inflammatory responses. Complement activation by immune complexes is correlated to total rAAV capsid dose (the sum of “fulls” – the actual product, plus “partial and “empty” capsid impurities). Results from several clinical trials that use systemic administration of high doses of rAAV support a correlation between capsid dose and complement activation leading to SAEs [34, 35]. In addition to AAV dose dependent complement activation, other attributes of AAV products have been shown to contribute to potentiation of unwanted immune response. In particular, activation of the pathogen associated molecular pattern sensor Toll-like Receptor (TLR)-9 in recipient immune cells by AAV expression cassettes containing a high density of unmethylated CpG dinucleotides, as is the case in microbial DNA for example, also correlates with adverse innate and adaptive immune response to AAV. The combination of high doses of an AAV vector and a high CpG containing genome in that vector is predicted to lead to inflammatory responses representing an additive or synergistic response to these two innate immune agonists. Efforts are underway to develop a clinically relevant assay to quantify TLR9 activation using human PBMCs to differentiate AAV vectors as a function of dose and unmethylated CpG content by INFa1 secretion.
II) Complement activation in IGNITE-DMD trial: Carl Morris (Solid Biosciences)
Activation of the terminal pathway (sC5b9) of the classical complement system was observed in all subjects dosed, resulting in 3 clinically serious adverse events (two cases of systemic inflammatory response syndrome and one case of thrombocytopenia). Most common lab abnormalities reported include: thrombocytopenia (decreased platelets), anemia, proteinuria, increases in fibrin D-dimer, soluble C5b9 and LDH. There is now a predictable and measurable sequence of events post-dosing: thrombocytopenia, humoral response, complement activation, cytokine elevation, kidney injury and anemia. Following high dose gene therapy with AAV9, all components needed to induce the classical complement pathway are present. Mitigation strategies to reduce complement activation include blocking immunocomplex triggers by lowering anti-AAV antibodies, AAV capsid levels or capsid binding to antibodies. Prophylactic inhibition of terminal complement or C1 esterase was evaluated with limited changes observed in either C3 and C4 depletion or formation of terminal complement (sC5b9). An in vitro whole blood model can be used as a translational model to study innate immune response to AAV vectors. Solid is now prioritizing SGT-003, a muscle-tropic next-gen microdystrophin vector (IND clearance by FDA in 2023) that may reduce the dose required for muscle targeting and thus the systemic viral load.
III) Kinetics of Innate and Adaptive Responses to Systemic AAV9 Gene Therapy: Barry Byrne (University of Florida)
Immune responses to AAV capsid and transgene are known universal or drug-class effects. Immune modulation using agents such as rituximab (B-cell depletion) and sirolimus (inhibits B-cell proliferation, downregulates cytotoxic T-cells) prevents the humoral response to AAV capsid and enhances safety and efficacy, allowing for repeat dosing and preventing immune responses to the transgene in null mutations (CRIM-negative) [36]. Fifteen study participants in several studies of AAV9 systemic gene therapy received pre-treatment with rituximab and sirolimus and a reduced steroid regimen of one week post dosing. In the rituximab group, IgM and IgG response was attenuated and there was no activation of complement other than low levels due to the alternative pathway. All individuals (n = 23) who received only glucocorticoid treatment had evidence of classical complement pathway activation and production of C3b and C5b which are important mediators of inflammation and tissue edema. At higher total viral load, the downstream activation of complement was more severe and associated with cardiovascular adverse events in the setting of DMD with underlying cardiomyopathy.
LETHAL SAE IN DMD GENE THERAPY TRIALS
Case 1: Russell Butterfield (University of Utah)
The first case is a 16-year-old non-ambulant trial participant in the Pfizer Phase 1B microdystrophin (AAV9) gene therapy trial (NCT03362502) who weighed 43 kg and was dosed at 2×1014 vg/kg. Screening cardiac MRI showed LVEF of 56% and areas of myocardial fibrosis and septal and LV wall hypokinesis. The participant met the study inclusion criteria of absent baseline neutralizing antibodies to AAV9. Immunomodulatory drugs included steroids (IV methylprednisolone and oral prednisone) and sirolimus. Cardiac troponin I was elevated on day 2 (0.49 ng/ml, ULN = 0.03 ng/ml), however this result was not available to the study team until day 5. Nausea and vomiting began on day 3. Cardiac troponin was noted to be elevated to 2.13 ng/ml on day 6. Echocardiogram on day 6 showed marked loss of LV function and increase in LV mass. Treatment for cardiogenic shock was administered. CPR measures and mechanical support were declined, and the patient died on day 7. An autopsy was declined. Labs revealed unremarkable liver function (ALT/AST, GGT, GLDH) but significant increases in troponin I, pro-BNP and CK. C3 and C4 values along with platelet levels were also unremarkable – suggesting no evidence of TMA. Hemoglobin levels increased steadily from day 5 to day 7, suggesting possible loss of endothelial integrity. This case demonstrates that LVEF alone may be an insufficient screening criterium for safety of AAV gene therapies in DMD and closer attention to cardiac fibrosis, LV wall motion, and other parameters should be considered. In addition, in a clinical trial setting, critical safety labs in the first week post-infusion should be collected locally and available for rapid review (point-of-service) rather than sent out to central labs, which often have significant delays to report critical values.
Case 2: Terence Flotte (University of Massachusetts) and Brenda Wong (University of Massachusetts)
The second case presented was from an n-of-1 gene therapy using a CRISPR-transactivator approach [37]. The 27-year-old non-ambulant patient was dosed at 1×1014 vg/kg of AAV9 with a dCas9-VP64 construct targeted at upregulating the full-length cortical dystrophin isoform (Dp427 C). The patient’s baseline FVC was 36% with LVEF at 55%, and a lean muscle mass of 45%. The patient received prophylactic and reactive immune suppression (sirolimus, prednisolone) and was negative for neutralizing antibodies to AAV9. Increasing BNP and decreasing platelets were noted on day 2 post infusion. Five days after gene therapy, the patient developed worsening cardiac function (LVEF 45-50%; troponin I level of 0.59 ng/ml, normal range: 0.01-0.04) and pericardial effusion with tamponade physiology on echocardiography. The patient developed sudden acute respiratory distress at day 6 with chest X-ray findings supportive of acute respiratory distress syndrome (ARDS). The patient had a sudden decompensation and cardiopulmonary arrest and was placed on extracorporeal membrane oxygenation (ECMO), yet died on day 8 from multi-organ failure and neurological injury. The patient’s labs showed mildly elevated complement components, and only a slight elevation of serum IL-6. Pericardial fluid analysis showed evidence of cellular inflammation and elevated cytokines (IL-6, MCP-1), with IL-6 levels 100X higher than in serum. Post-mortem tissue analysis did not show signs of mononuclear cell infiltrate/myocarditis or complement deposition, yet the heart weight was significantly increased. The lungs were noted to be heavy and edematous with diffuse alveolar damage, consistent with ARDS and or heart failure and contained a relatively large amount of vector DNA (119 vg/diploid genome). SaCas9 transcript and protein expression were not detected in skeletal or cardiac muscle and only trace amounts were detected in the liver. No AAV9 capsid or Cas9 transgene specific T-cell responses were detected in patient’s PBMC. The CRISPR transgene product is not thought to have contributed to the patient’s death due to the timing of clinical events. Instead, the post-mortem findings suggest diffuse capillary leakage as the primary event, either due to cytokine release, heart failure or to some other mechanism of direct endothelial injury that has yet to be elucidated.
IMMUNOSUPPRESSION STRATEGIES
Summary of immune response and strategies: Jordan Abbott (University of Colorado)
The prototypical sequence of immune responses to AAV gene therapy consists of three waves (Fig. 1), all of which have only been partially characterized.
Fig. 1
Timeline of immune response and affected tissues following AAV gene therapy in neuromuscular disease patients. The three waves of immune responses to AAV gene therapy as triggered by capsid and transgene products. Timing of adverse events in specific tissues in response to neuromuscular disease gene therapies are shown.
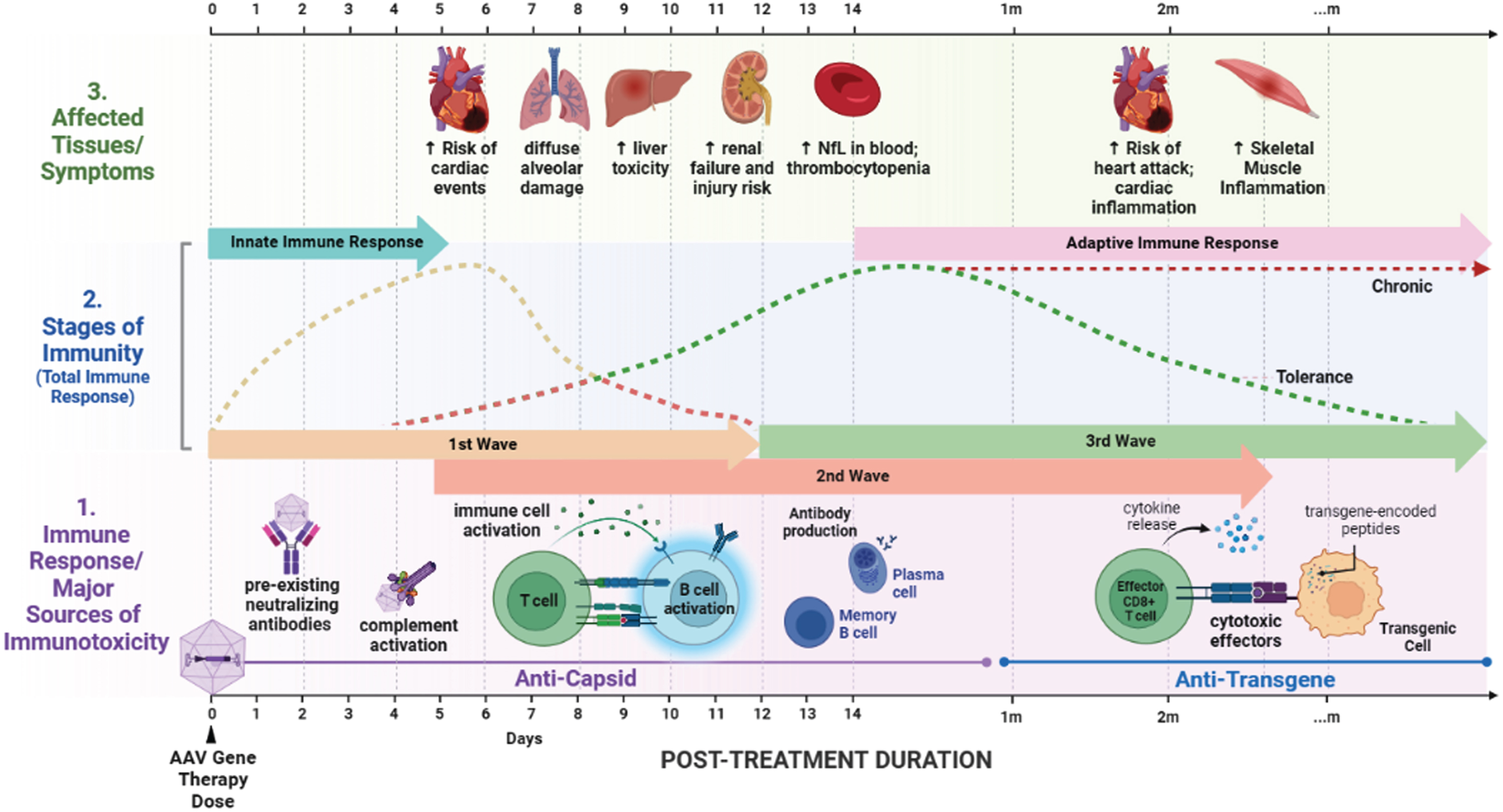
First wave immunity: Immediately following systemic introduction of AAV vectors, viral capsid encounters fluid phase immune molecules including immunoglobulin and complement molecules. Simultaneously, capsids adhere to cell surface glycans fostering interaction with toll-like receptors (TLRs) in cells expressing those receptors. Viral particles that are internalized through endosomes continue to interact with TLRs, and released DNA can interact with DNA-specific TLRs. Particles that escape endosomes can be processed by proteasomal machinery and presented on the surface by Major Histocompatibility Complex (MHC) class I molecules. Broken capsids are presumed to release free DNA into the cytosol allowing for interaction with innate DNA sensing molecules such as cGAS (cyclic GMP-AMP Synthase), resulting in STING (Stimulator of Interferon Genes) activation. Similar innate immune interactions are proposed to occur within the nucleus.
Second wave immunity: Following innate immune reactions, antigen presenting cells that are sufficiently activated foster the initial adaptive immune response in secondary lymphoid structures such as lymph nodes. APC-activated T cells interact with capsid-presenting B cells, spawning germinal centers specific to generating high-affinity antibodies to capsid epitopes. Initial output from these interactions includes short-lived antibody-secreting plasmablasts that primarily produce IgM. Simultaneously, the effector functions of predisposed memory T cells are activated through interaction with MHC I presented peptides.
Germinal center reactions result in the production of memory B cells, memory T cells, and long-lived plasma cells that home to various protected niches within the body. These responses are specific to both the capsid and the transgene product. The cells produced as a result of this coordinated interaction persist for long times, allowing for rapid clearance of subsequent exposures to the same or similar capsid particles.
Third wave immunity: In the long term, innate responses to the capsid have resolved and the capsid itself no longer exists within the body. The only remaining components of the gene therapy product are transgene episomes and the transgene protein product. If transgene-specific memory T cells have been formed, there is a risk of ongoing T-cell response to cells expressing the transgene, particularly in CRIM negative patients that can present transgene peptides through MHC class I.
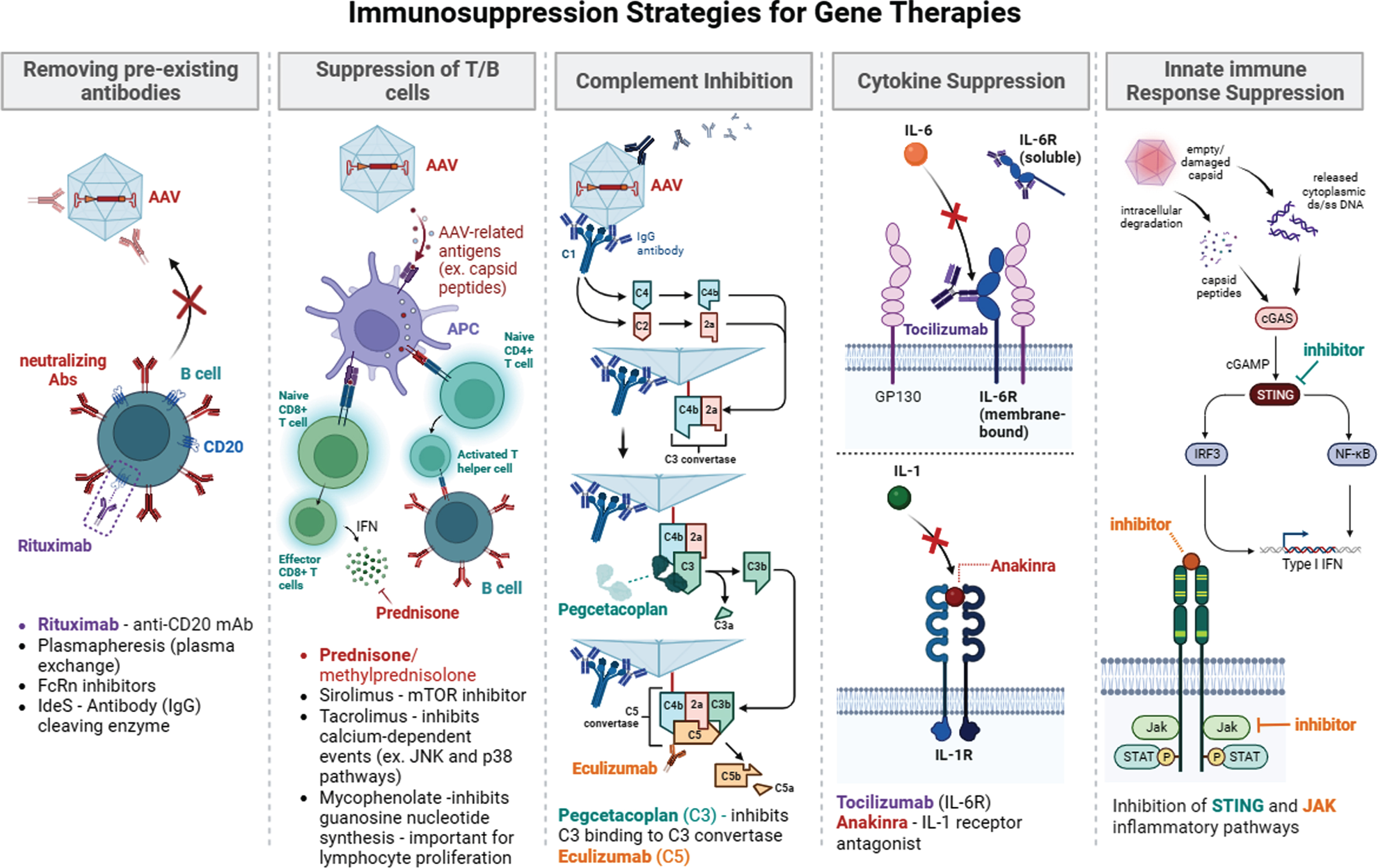
Given this model of the immune response, several immune suppression strategies have been explored to overcome immunoreactivity in response to gene therapy (Fig. 2). Other strategies to minimize immunotoxicity can also include patient selection and vector modification and supporting Treg development and expansion to confer adaptive tolerance.
Fig. 2
Summary of immunosuppression strategies for gene therapy. Immunosuppression approaches in current use or being explored include removing pre-existing antibody (plasmapheresis, rituximab, FcRn, IdeS), suppression of T/B cells (pred/methylpred, sirolimus, tacrolimus, mycophenolate, jak inhibitor), complement inhibition (eculizumab (C5), pegcetacoplan (C3)), cytokine suppression (tocilizumab (Il-6 R), and Anakinra (Il-1)), and innate response suppression (STING inhibition and JAK inhibition).
ULTRA-RARE GENE THERAPY PROGRAMS
I) MDA Kickstart Program: Sharon Hesterlee (Muscular Dystrophy Association)
The Kickstart program is MDA’s in-house drug development program that aims to de-risk an ultra-rare indication by investing resources and personnel towards the very early stages of drug development. The program will assemble a project team and outsource necessary studies to obtain sufficient proof-of-concept data for a given gene therapy indication. MDA selected a congenital myasthenic syndrome due to ChAT (choline acetyltransferase) deficiency as the pilot indication to advance for Kickstart [38, 39]. The condition affects infants with fatal episodes of apnea (9 in 1 million individuals) and is caused by pathogenic mutations in the ChAT enzyme that affect the synthesis of acetylcholine at the neuromuscular junction [40]. The plan is to also document roadblocks and lessons learned to benefit advocacy efforts around ultra-rare disease, as well as build an infrastructure of CROs, regulatory consultants, manufacturers and project managers. End goals of the project are to in-license relevant IP from collaborating academic institutes and conduct a successful pre-IND meeting for the project with the FDA. MDA has assembled an overall program committee as well as specific project advisors.
II) Bespoke Gene Therapy Consortium: Philip J. Brooks (National Institutes of Health, National Center for Advancing Translational Sciences)
The goal of this program is to make AAV technology more accessible to a broader range of diseases by optimizing AAV vector production, streamlining preclinical and product testing and facilitating regulatory advances. BGTC is composed of two arms: a basic science arm that looks to enhance vector generation and therapeutic gene expression; and a clinical application arm that streamlines the process of clinical translation. The collaborative effort currently consists of 13 industry partners, 12 government agencies/NIH institutes and cross-institute programs and 10 non-profits. Standardized protocols or ‘playbooks’ will be developed for manufacturing critical quality attributes, preclinical testing requirements for GLP/tox study and clinical trials. Eight rare diseases have been selected for the BGTC consortium clinical trial portfolio, including 3 using ocular delivery, 3 using CNS delivery, and two using systemic delivery. All of the diseases are so rare that they are currently of no commercial interest. However, the streamlining efforts and increased efficiencies of the regulatory process resulting from the BGTC could have substantial positive impacts on the larger gene therapy field, including changing the consideration of what constitutes a disease of commercial interest in the future. See AMP® Bespoke Gene Therapy Consortium (BGTC) | FNIH for more information.
CONSIDERATIONS FOR TRIALS WITH SMALL N’S
I) Regulatory expectations for ultra-rare gene therapies: Lauren Black (Charles River Laboratories)
Investigators interested in translation of genetic therapies should seek advice from the CBER regulatory webpage, which contains vital information pertinent to AAV gene therapy, clinical trial phase, patient population and route of administration. Additional suggested research topics include surrogate endpoints, expanded access guidelines, and the pediatric rule. Investigators may need to seek help with their institute’s resources or external regulatory affairs agency to prepare IND documentation, and to handle communications and logging of documents to FDA. Steps towards a successful IND include familiarization with regulatory guidelines and identification of phase 1 and 2 needs; planning of studies including POC (Proof of Concept), non-clinical and clinical toxicology; proposing plans in a preIND meeting and seeking FDA buy-in on proposed animal model and manufacturing plans. Other tips include being ready to defend the selected animal species, relevant pathways and read-outs in selected models (what does and doesn’t match human disease), and also building a toxicology plan using the selected animal model and planning biodistribution studies.
II) Trial journey and experiences learned from N of 1 case: Richard Horgan (Cure Rare Disease)
Cure Rare Disease assembled a collaborative team consisting of partners at Yale University, University of Massachusetts and Charles River Labs to develop a CRISPR-based gene therapy for an N-of-1 DMD trial. Proof-of-concept studies were completed in spring 2019, exploratory dosing began in spring 2020 and a preIND meeting was held in fall 2020. GMP manufacturing was committed in 2021 with GLP toxicology performed in early 2022. Full IND was approved in summer of 2022 with the patient dosed in fall 2022 [37]. GLP toxicology was performed on wildtype hDMD D2/mdx mice dosed with 1×1014 vg/kg (final dose in patient) and 2×1014 vg/kg – with no deaths or test-article related clinical observations on body weight, hematology, cytokine analysis, splenocyte ELISPOT analysis or immunochemistry. The GMP-grade vector was verified for potency to upregulate cortical dystrophin (Dp427c) in skeletal and cardiac muscle. Changes were made to the proposed immune suppression protocol as suggested by the FDA. That is, the multiple rituximab dosing planned was reduced to a single dose, and planned corticosteroid dosing up to 60 days post-treatment was reduced to 30 days. A long term safety monitoring plan of 15 years was also mandated. Early FDA involvement was important to achieve rapid advancement of this first-in-human technology.
PRACTICAL CONSIDERATIONS IN GENE THERAPY
I) Informed consent and autopsy considerations: Brian Tseng (The POLG Foundation) and Mike Lawlor (Diverge Translational Science Laboratory)
It is important for the field to recognize the risk of death and need for autopsies/post-mortem studies to plan early for all contingencies including unexpected and rare tragedies. Trial participant deaths can occur due to non-trial related tragic events, and post-mortem analysis can show positive trial intervention effects. Post-mortem tissues can provide key insights on therapeutic mechanism of action, durability, biodistribution and pharmacodynamic effect [41]. Collection of post-mortem tissue and analysis involves communication between many stakeholders – study PIs, research labs, shipping, funeral home, pathologist and patient’s family. Study sponsors need to plan for this contingency and complexity before the trial starts and ideally assign a pre-designated point person to coordinate.
Although mentioning pre-consent is not sufficient permission to perform an autopsy, the risk of death and tissue collection should be raised by the study PI during the initial informed consent process and mandated by the Institutional Review Board (IRB). The initial informed pre-consent should mention ‘risk of death’ as this has occurred with AAV trials and ‘consent for autopsy’ is a critical need to ensure that further deaths can be avoided if cause of death is understood. Clinicians may give a presumptive cause of death, but literature shows it is the pathologists who can uncover the actual and full cause of death [42]. The legal guardians or next of kin reserve the right to decline consent for autopsy. Seeking such consent in time of imminent death with highest emotional turmoil is far from a good practice to respect the family and participant. Documenting a study participant’s interest in contributing to autopsy studies can assist the family in understanding the participant’s wishes, which can significantly unburden the family when making decisions around autopsy study participation.
It can be difficult to anticipate the right person or hospital for the autopsy and there may be hesitation or unexpected hospital and/or state bureaucracy or family cultural or religious convictions to navigate. Note that autopsies performed by a hospital versus medical examiner have different implications - specimens collected by medical examiners cannot be distributed for legal reasons, and medical examiner/coroner offices often do not have the storage equipment necessary to appropriately store some types of samples. It must be recognized that a trial participant may not necessarily be at the study site medical center at time of death. Also note that while hospital consent forms are sufficient for tissue collection, distribution and release for a study sponsor or other laboratories may require additional funds and consent. Typically, tissue collection during autopsies will include sampling of most major organs and organs of interest related to the patient history and disease state. Muscle tissue may not be collected in all cases, and the standard procedure is formalin fixation which may undermine the performance of important studies including assessments of vector biodistribution in muscle. Subsequently, pathologist reporting will focus on findings that are present but will not likely address issues that the FDA/EMA will consider important and further studies will need to be undertaken on collected tissues. Typical FDA/EMA requests from gene therapy related deaths include: biodistribution of vector, transgene expression, comparison to other deaths in the field, exploratory studies to further study the biological impact of the treatment, and interpretation of findings by other specialists. It is not common practice to perform the broad collection of frozen tissue necessary to address some of these endpoints, so it is necessary to coordinate tissue collection with the autopsy team before the procedure has occurred. Finally, it should be noted that the FDA/EMA may require a comprehensive assessment of autopsy findings in the context of safety concerns or other deaths in other patients in the trial. Such a comparison will require detailed knowledge of multiple patients and likely across multiple sites, and this again is outside the scope of what the traditional hospital autopsy is meant to accomplish. While the official hospital autopsy report will be critical in determining the pathological findings in a specific patient and understanding that patient’s clinical course and terminal events, appropriately addressing a patient death in a trial will require extensive partnership between the autopsy team, PI, sponsor, patient family, and potentially other pathology experts to learn from participant deaths and maximize the safety of other trial participants.
II) Risk tolerance for gene therapy: Eric Camino (Parent Project Muscular Dystrophy)
The BRAVE program aims to quantify how DMD patients and caregivers think and feel about emerging gene therapies and their preferences for treatments. This study was conducted in 2017-2018 through a community-engagement approach, and involved data collected from 320 DMD adult patients and parents/guardians of DMD children [43, 44]. Participants were asked to complete a quantitative survey on gene therapy which included clinical trial decision making, participation interest and maximal associated risk of death. Survey results conveyed that caregivers and adults with DMD have similar tolerance for risk and uncertainty. Risk tolerance was highest in the last year of being able to bring arms to mouth, followed by last year of walking well, followed by present time. Caregivers of newborns with DMD were not as risk tolerant. Thus, timing of disease state/progression is an important consideration for risk tolerance, with non-ambulatory patients and their caregivers willing to accept more risk for therapy. PPMD has since completed a follow-up survey given that there have been new developments and outcomes in the gene therapy field over the past few years, and manuscripts on updated risk tolerance and decision making are being drafted. The survey will identify whether risk tolerance levels have changed given a broader knowledge by the community of adverse events and reported deaths associated with DMD gene therapies, as well as the knowledge that persistence of the potential benefits of gene therapy are not known.
III) Reimbursement, economic, advocacy, public policy barriers in gene therapies: Paul Melmeyer (Muscular Dystrophy Association)
Public policy barriers with the potential to influence the clinical translation and access to gene therapies were discussed. Several incentives currently exist for clinical development of gene therapies in rare disease which include exclusivity periods, tax credits, user fee waivers, 340B incentives. These incentives have yet to prove their adequacy to successfully encourage the commercial development and subsequent marketing of gene therapies for rare neuromuscular diseases, and alternate incentives may be required to support ultra-rare therapy development. Stakeholders may need to advocate for FDA to take a more flexible approach to their regulatory consideration of gene therapies. Potential topics for advocacy may include review and guidance around the type and amount of preclinical data needed for an IND approval, what trial structures and expedited approval pathways are appropriate, as well as ensuring that FDA has sufficient expertise and capacity to review applications. The topic of coverage and reimbursement for gene therapies was also raised with respect to private and public health coverage, including the need for state Medicaid programs covering these gene therapies affordably and expeditiously. Also, reimbursement may not necessarily cover diagnostics and ancillary care associated with gene therapies. Concerns were also raised about workforce shortages that can affect access to gene therapies. Clinic sites looking to administer gene therapies need to be appropriately staffed by supportive professionals, including an expert gene therapy team, a pharmacy team, adverse event specialists, and genetic counselors. The clinical translation of gene therapies is a multi-faceted process requiring alignment of the many stakeholders who are involved, and public policy collaborations may prove fruitful to tackle these complex challenges.
CONCLUDING POINTS
The breadth of topics discussed during this meeting reflects the many stakeholders involved in the clinical translation of gene therapies in neuromuscular disease. Alignment of goals and incentives for all these stakeholders are necessary to achieve the overall goal of safe and efficacious translation of gene therapies in patients. It has become clear that safety and efficacy of gene therapies are influenced by factors that are often interlinked. These include disease-specific features, disease state, AAV properties, AAV dose, patient age/size, target organ, and individual patient susceptibilities. Together, these factors can ideally result in an individualized risk profile for each patient seeking to undergo gene therapy and should thus be factored into the risk/benefit calculation by clinicians. Case studies and statistics presented in this meeting demonstrate that adverse events are to be expected and planned for each patient undergoing AAV gene therapy. As a field, we have converged on a set of predictable and measurable sequence of events post-dosing in terms of thrombocytopenia, elevation of transaminases, complement activation, cytokine elevation, antibody response and capsid biodistribution. Although the kinetics of innate and adaptive responses have been relatively well mapped, more insight is required into anti-transgene responses in the context of gene therapies for CRIM negative patients, with implications for long term immunosuppression strategies. Currently, the durability of AAV gene therapy in humans remains unclear, with disease state and age at dosing acting as contributing factors. Concerns were raised around animal models lacking the predictive power to determine efficacy window and durability of therapy in humans. Reduction of anti-AAV antibodies for safe redosing is shown to be possible in large animal models and in a limited group of human subjects, thus may be used as a strategy to compensate for lack of durability.
Although some insight has been gained into the two lethal DMD gene therapy cases, the underlying cause of deaths are still not entirely understood. Risk tolerance for uncertainty associated with gene therapy is an individual choice, however it is clear that probabilities of serious adverse events increase with age and disease progression in the context of DMD. Autopsy and tissue collection should be raised during the informed consent process by the investigator. Plans for tissue collection and preservation should be at hand in the event of unexpected deaths following gene therapy to enable a thorough investigation.
Additionally, there is a role for NIH and patient-advocacy organizations to actively participate and de-risk development of gene therapy programs for ultra-rare conditions that otherwise have little commercial interest. Steps involved and lessons learned from such efforts should be documented and shared with the community to enable other organizations to pursue similar efforts for their respective diseases. There are also advocacy-related opportunities pertaining to the translation of gene therapies. These include addressing barriers as they relate to coverage, access and reimbursement of diagnostics and gene therapy and follow-up care. Table 2 summarizes a list of actionable meeting recommendations for implementation by relevant stakeholders in the field of gene therapy for neuromuscular disorders.
Table 2
Actionable meeting recommendations
| •Development of systematic protocols to evaluate and report innate as well as anti-capsid and anti-transgene adaptive immune responses at different timepoints post dosing |
| •Comparative evaluation of proactive and reactive immune modulation protocols |
| •Establishment of working groups and/or virtual platform to share data on evolving best clinical practices and adverse event reporting |
| •Increased involvement of specialists with experience in immunology, cardiology and hepatology to help address adverse events with gene therapies |
| •Establishment and evaluation of clinically predictive in vitro immune assays and in-silico immunogenicity prediction tools |
| •Identification of a list of appropriate tissues/organs and best handling and preservation methods for autopsy, and protocols for informative post-mortem tests |
| •Collection and databasing of comprehensive genotyping of all patients dosed with AAV mediated gene therapies |
ACKNOWLEDGMENTS
The Muscular Dystrophy Association would like to acknowledge Sarepta Therapeutics, Vertex, AskBio, Pfizer, Astellas and Regenxbio for contributing to the meeting sponsorship and all presenters as well as Sujatha Gurunathan for helping with manuscript edits.
CONFLICT OF INTEREST
The authors have no financial conflicts of interest to report.
REFERENCES
[1] | https://www.novartis.com/us-en/sites/novartis_us/files/zolgensma.pdf. |
[2] | Day JW , Mendell JR , Mercuri E , Finkel RS , Strauss KA , Kleyn A , et al. Clinical Trial and Postmarketing Safety of Onasemnogene Abeparvovec Therapy. Drug Saf. (2021) ;44: (10):1109–19. |
[3] | Yang D , Ruan Y , Chen Y . Safety and efficacy of gene therapy with onasemnogene abeparvovec in the treatment of spinal muscular atrophy: A systematic review and meta-analysis. J Paediatr Child Health. (2023) ;59: (3):431–8. |
[4] | Day JW , Finkel RS , Chiriboga CA , Connolly AM , Crawford TO , Darras BT , et al. Onasemnogene abeparvovec gene therapy for symptomatic infantile-onset spinal muscular atrophy in patients with two copies of SMN2 (STR1VE): an open-label, single-arm, multicentre, phase 3 trial. Lancet Neurol. (2021) ;20: (4):284–93. |
[5] | Gaillard J , Gu AR , Neil Knierbein EE . Necrotizing Enterocolitis following Onasemnogene Abeparvovec for Spinal Muscular Atrophy: A Case Series. J Pediatr. (2023) ;260: :113493. |
[6] | Retson L , Tiwari N , Vaughn J , Bernes S , Adelson PD , Mansfield K , et al. Epithelioid neoplasm of the spinal cord in a child with spinal muscular atrophy treated with onasemnogene abeparvovec. Molecular Therapy. (2023) ;31: (10):2991–8. |
[7] | Van Alstyne M , Tattoli I , Delestrée N , Recinos Y , Workman E , Shihabuddin LS , et al. Gain of toxic function by long-term AAV9-mediated SMN overexpression in the sensorimotor circuit. Nat Neurosci. (2021) ;24: (7):930–40. |
[8] | Tukov FF , Mansfield K , Milton M , Meseck E , Penraat K , Chand D , et al. Single-Dose Intrathecal Dorsal Root Ganglia Toxicity of Onasemnogene Abeparvovec in Cynomolgus Monkeys. Hum Gene Ther. (2022) ;33: (13-14):740–56. |
[9] | Stevens D , Milani-Nejad S , Mozaffar T . Pompe Disease: a Clinical, Diagnostic, and Therapeutic Overview. Vol. 24, Current Treatment Options in Neurology. (2022) . |
[10] | DoH V, KhannaR, GotschallR. Challenges in treating Pompe disease: an industry perspective. Ann Transl Med. (2019) ;7: (13):291–291. |
[11] | Harlaar L , Hogrel JY , Perniconi B , Kruijshaar ME , Rizopoulos D , Taouagh N , et al. Large variation in effects during 10 years of enzyme therapy in adults with Pompe disease. Neurology. (2019) ;93: (19). |
[12] | Ronzitti G , Collaud F , Laforet P , Mingozzi F . Progress and challenges of gene therapy for Pompe disease. Ann Transl Med. (2019) ;7: (13):287–287. |
[13] | Salabarria SM , Nair J , Clement N , Smith BK , Raben N , Fuller DD , et al. Advancements in AAV-mediated Gene Therapy for Pompe Disease. J Neuromuscul Dis. (2020) ;7: (1):15–31. |
[14] | Smith EC , Hopkins S , Case LE , Xu M , Walters C , Dearmey S , et al. Phase I study of liver depot gene therapy in late-onset Pompe disease. Molecular Therapy. (2023) ;31: (7):1994–2004. |
[15] | |
[16] | Mendell JR , Campbell K , Rodino-Klapac L , Sahenk Z , Shilling C , Lewis S , et al. Dystrophin Immunity in Duchenne’s Muscular Dystrophy. New England Journal of Medicine. (2010) ;363: (15):1429–37. |
[17] | Song Y , Morales L , Malik AS , Mead AF , Greer CD , Mitchell MA , et al. Non-immunogenic utrophin gene therapy for the treatment of muscular dystrophy animal models. Nat Med. (2019) ;25: (10):1505–11. |
[18] | Stedman HH , Kozyak BW , Nelson A , Thesier DM , Su LT , Low DW , et al. Myosin gene mutation correlates with anatomical changes in the human lineage. Nature. (2004) ;428: (6981):415–8. |
[19] | Samelson-Jones BJ , George LA . Adeno-Associated Virus Gene Therapy for Hemophilia. Annu Rev Med. (2023) ;74: (1):231–47. |
[20] | Handyside B , Ismail AM , Zhang L , Yates B , Xie L , Sihn CR , et al. Vector genome loss and epigenetic modifications mediate decline in transgene expression of AAV5 vectors produced in mammalian and insect cells. Molecular Therapy. (2022) ;30: (12):3570–86. |
[21] | Buj-Bello A , Fougerousse F , Schwab Y , Messaddeq N , Spehner D , Pierson CR , et al. AAV-mediated intramuscular delivery of myotubularin corrects the myotubular myopathy phenotype in targeted murine muscle and suggests a function in plasma membrane homeostasis. Hum Mol Genet. (2008) ;17: (14):2132–43. |
[22] | Childers MK , Joubert R , Poulard K , Moal C , Grange RW , Doering JA , et al. Gene Therapy Prolongs Survival and Restores Function in Murine and Canine Models of Myotubular Myopathy. Sci Transl Med. (2014) ;6: (220). |
[23] | Elverman M , Goddard MA , Mack D , Snyder JM , Lawlor MW , Meng H , et al. Long-term effects of systemic gene therapy in a canine model of myotubular myopathy. Muscle Nerve. (2017) ;56: (5):943–53. |
[24] | Mack DL , Poulard K , Goddard MA , Latournerie V , Snyder JM , Grange RW , et al. Systemic AAV8-Mediated Gene Therapy Drives Whole-Body Correction of Myotubular Myopathy in Dogs. Molecular Therapy. (2017) ;25: (4):839–54. |
[25] | Dupont JB , Guo J , Renaud-Gabardos E , Poulard K , Latournerie V , Lawlor MW , et al. AAV-Mediated Gene Transfer Restores a Normal Muscle Transcriptome in a Canine Model of X-Linked Myotubular Myopathy. Molecular Therapy. (2020) ;28: (2):382–93. |
[26] | Ross JA , Tasfaout H , Levy Y , Morgan J , Cowling BS , Laporte J , et al. rAAV-related therapy fully rescues myonuclear and myofilament function in X-linked myotubular myopathy. Acta Neuropathol Commun. (2020) ;8: (1):167. |
[27] | Wasala NB , Yue Y , Hu B , Shin JH , Srivastava A , Yao G , et al. Lifelong Outcomes of Systemic Adeno-Associated Virus Micro-Dystrophin Gene Therapy in a Murine Duchenne Muscular Dystrophy Model. Hum Gene Ther. (2023) ;34: (9-10):449–58. |
[28] | Muhuri M , Levy DI , Schulz M , McCarty D , Gao G . Durability of transgene expression after rAAV gene therapy. Molecular Therapy. (2022) ;30: (4):1364–80. |
[29] | Jordan SC , Lorant T , Choi J , Kjellman C , Winstedt L , Bengtsson M , et al. IgG Endopeptidase in Highly Sensitized Patients Undergoing Transplantation. New England Journal of Medicine. (2017) ;377: (5):442–53. |
[30] | Shieh PB , Kuntz NL , Dowling JJ , Müller-Felber W , BönnemannCG , Seferian AM , et al. Safety and efficacy of gene replacementtherapy for X-linked myotubular myopathy (ASPIRO): a multinational,open-label, dose-escalation trial. Lancet Neurol. (2023) ;22: (12):1125–39. |
[31] | Karolczak S , Deshwar AR , Aristegui E , Kamath BM , Lawlor MW , Andreoletti G , et al. Loss of Mtm1 causes cholestatic liver disease in a model of X-linked myotubular myopathy. Journal of Clinical Investigation. (2023) ;133: (18). |
[32] | Mueller C , Berry JD , McKenna-Yasek DM , Gernoux G , Owegi MA , Pothier LM , et al. SOD1 Suppression with Adeno-Associated Virus and MicroRNA in Familial ALS. New England Journal of Medicine. (2020) ;383: (2):151–8. |
[33] | Johnson EW , Sutherland JJ , Meseck E , McElroy C , Chand DH , Tukov FF , et al. Neurofilament light chain and dorsal root ganglia injury after adeno-associated virus 9 gene therapy in nonhuman primates. Mol Ther Methods Clin Dev. (2023) ;28: :208–19. |
[34] | Wright JF . Codon Modification and PAMPs in Clinical AAV Vectors: The Tortoise or the Hare? Molecular Therapy. (2020) ;28: (3):701–3. |
[35] | Nicole Paulk. Gene Therapy: It’s Time to Talk about High-Dose AAV. Genetic Engineering and Biotechnology News. 2020. |
[36] | Salabarria SM , Corti M , Coleman KE , Wichman MB , Berthy JA , D’Souza P , et al. Thrombotic microangiopathy following systemic AAV administration is dependent on anti-capsid antibodies. Journal of Clinical Investigation. (2023) . |
[37] | Lek A , Wong B , Keeler A , Blackwood M , Ma K , Huang S , et al. Death after High-Dose rAAV9 Gene Therapy in a Patient with Duchenne’s Muscular Dystrophy. New England Journal of Medicine. (2023) ;389: (13):1203–10. |
[38] | Maselli RA , Chen D , Mo D , Bowe C , Fenton G , Wollmann RL . Choline acetyltransferase mutations in myasthenic syndrome due to deficient acetylcholine resynthesis. Muscle Nerve. (2003) ;27: (2):180–7. |
[39] | Arredondo J , Lara M , Gospe SM , Mazia CG , Vaccarezza M , Garcia-Erro M , et al. Choline Acetyltransferase Mutations Causing Congenital Myasthenic Syndrome: Molecular Findings and Genotype-Phenotype Correlations. Hum Mutat. (2015) ;36: (9):881–93. |
[40] | McMacken G , Whittaker RG , Evangelista T , Abicht A , Dusl M , Lochmüller H . Congenital myasthenic syndrome with episodic apnoea: clinical, neurophysiological and genetic features in the long-term follow-up of 19 patients. J Neurol. (2018) ;265: (1):194–203. |
[41] | Ramos DM , d’Ydewalle C , Gabbeta V , Dakka A , Klein SK , Norris DA , et al. Age-dependent SMN expression in disease-relevant tissue and implications for SMA treatment. Journal of Clinical Investigation. (2019) ;129: (11):4817–31. |
[42] | Shojania KG , Burton EC , McDonald KM , Goldman L . Changes in Rates of Autopsy-Detected Diagnostic Errors Over Time. JAMA. (2003) ;289: (21):2849. |
[43] | Paquin RS , Fischer R , Mansfield C , Mange B , Beaverson K , Ganot A , et al. Priorities when deciding on participation in early-phase gene therapy trials for Duchenne muscular dystrophy: a best–worst scaling experiment in caregivers and adult patients. Orphanet J Rare Dis. (2019) ;14: (1):102. |
[44] | Peay HL , Fischer R , Mange B , Paquin RS , Smith EC , Sadosky A , et al. Patients’ and caregivers’ maximum acceptable risk of death for non-curative gene therapy to treat Duchenne muscular dystrophy. Mol Genet Genomic Med. (2021) ;9: (5). |

