
Genetic Landscape of Amyotrophic Lateral Sclerosis in Czech Patients
Abstract
Background:
Genetic factors are involved in the pathogenesis of familial and sporadic amyotrophic lateral sclerosis (ALS) and constitute a link to its association with frontotemporal dementia (FTD). Gene-targeted therapies for some forms of ALS (C9orf72, SOD1) have recently gained momentum. Genetic architecture in Czech ALS patients has not been comprehensively assessed so far.
Objective:
We aimed to deliver pilot data on the genetic landscape of ALS in our country.
Methods:
A cohort of patients with ALS (n = 88), recruited from two Czech Neuromuscular Centers, was assessed for hexanucleotide repeat expansion (HRE) in C9orf72 and also for genetic variations in other 36 ALS-linked genes via next-generation sequencing (NGS). Nine patients (10.1%) had a familial ALS. Further, we analyzed two subgroups of sporadic patients – with concomitant FTD (n = 7) and with young-onset of the disease (n = 22).
Results:
We detected the pathogenic HRE in C9orf72 in 12 patients (13.5%) and three other pathogenic variants in FUS, TARDBP and TBK1, each in one patient. Additional 7 novel and 9 rare known variants with uncertain causal significance have been detected in 15 patients. Three sporadic patients with FTD (42.9%) were harbouring a pathogenic variant (all HRE in C9orf72). Surprisingly, none of the young-onset sporadic patients harboured a pathogenic variant and we detected no pathogenic SOD1 variant in our cohort.
Conclusion:
Our findings resemble those from other European populations, with the highest prevalence of HRE in the C9orf72 gene. Further, our findings suggest a possibility of a missing genetic variability among young-onset patients.
INTRODUCTION
Amyotrophic lateral sclerosis (ALS) is a heterogeneous group of neurodegenerative conditions, comprising several clinical variants, all leading to various degrees of progressive upper and lower motor neuron loss. Moreover, other related neuronal populations can be affected simultaneously (frontotemporal circuits leading to cognitive disorder or frontotemporal dementia, extrapyramidal system etc.) so that ALS can be regarded as a multisystem degeneration [1]. The pathogenesis of ALS is not fully understood, but there is a genetic component. In 5–10% of patients, there is a family history of ALS (or frontotemporal dementia), usually with autosomal dominant (AD) inheritance [2]. Currently, more than 30 genes have been linked to the pathogenesis of ALS [3]. Mendelian gene variations have been identified in about 50% of those with a family history (fALS) [4, 5] and about 11% of those without, classified as sporadic ALS (sALS) [6]. Every familial ALS gene variant has also been seen in patients with no obvious family history. Furthermore, fALS is often clinically indistinguishable from sALS and even shows the same neuropathological pattern [2]. The most common genetic cause of ALS is an expansion of a GGGGCC (G4C2) – hexanucleotide repeat (HRE) in the first intron of the C9orf72 gene, with a frequency of 33.7% in fALS and 5.6% in sALS among the European population [4], but with some variation among ethnic groups of different ancestry [7–10]. The next most common causal mutations have been found in SOD1, TARDBP, and FUS genes, both in patients with fALS and those with sALS [4]. Furthermore, more than one pathogenic or likely pathogenic variant carried simultaneously by a single individual, suggesting possible oligogenic interaction, has been documented in up to 3% of patients in a recent large-scale study [6].
While robust genetic data regarding mutation frequencies in fALS [4, 5] as well as sALS [6, 7] patients in Europe exist, ethnics with genetic ancestry different from that in the Czech population have been assessed (e.g. German, Dutch, Italian, Portuguese etc.). So far, only one study from a population with predominantly Slavic ethnicity has been published [10], where only four major ALS-associated genes have been assessed. It has been shown that individuals from different populations carry different profiles of rare and common variants and that low-frequency variants show substantial geographic differentiation [11]. Notably, the frequency of HRE in C9orf72 in sALS varies from 0% to 21% [12] and could be specific in the Czech population. Our work could fill a gap in the genetic diversity of the European ALS population.
Thus, considering the abovementioned situation, we designed a study analyzing the genetic architecture in Czech familial, as well as sporadic ALS patients, assessing the frequency of HRE in C9orf72 and also of rare variants in 36 ALS-linked genes via next-generation sequencing in our cohort. With the advance of gene-specific therapies (antisense oligonucleotides; ASOs) specifically for SOD1 and C9orf72-linked ALS, these data will have an increasingly important clinical impact.
MATERIALS AND METHODS
Patients
This was a two-centre, two-cohort study. We recruited 88 Czech patients diagnosed with ALS (9 familial and 79 sporadic cases) according to the revision of the El Escorial criteria – 2015 [13], 71 of them at the European Reference Network (ERN) Neuromuscular Center (NMC), University Hospital Motol and 17 of them at the ERN NMC, University Hospital Brno. The diagnosis of ALS was ascertained by an experienced neuromuscular neurologist (DB, RM, EV) after alternative diagnoses had been excluded. Diagnosis of comorbid frontotemporal dementia was ascertained according to Strong criteria [14]. Familial ALS cases were defined as patients with at least one first or second-degree relative with ALS or FTD. We recorded relevant clinical and demographic features for all patients (Table 1). The study design comprised two distinct cohorts. First one was a prospective longitudinal cohort where we recruited patients and
Table 1
Patient demographics and clinical characteristics (n = 88)
| Age (years) | 51.2 (19; 82) |
| Young-onset patients | 24 (27.3%) |
| Sex | |
| Male | 50 (56.8%) |
| fALS | 9 (10.1%) |
| Site of onset | |
| Spinal | 68 (77.3%) |
| Subtypes | |
| Classic ALS | 57 (83.8%) |
| LMN-predominant | 9 (13.2%) |
| With FTD | 2 (3.0%) |
| Bulbar | 20 (22.7%) |
| Subtypes | |
| Bulbar only | 15 (75.0%) |
| with FTD | 5 (25.0%) |
ALS – amyotrophic lateral sclerosis, Classic ALS – upper and lower motor neuron involvement, fALS – at least one first- or second-degree relative with ALS and/or FTD, FTD – frontotemporal dementia, LMN – lower motor neuron; young-onset:<45 years; Continuous data are presented as median (min; max), categorical data as absolute numbers and relative frequencies.
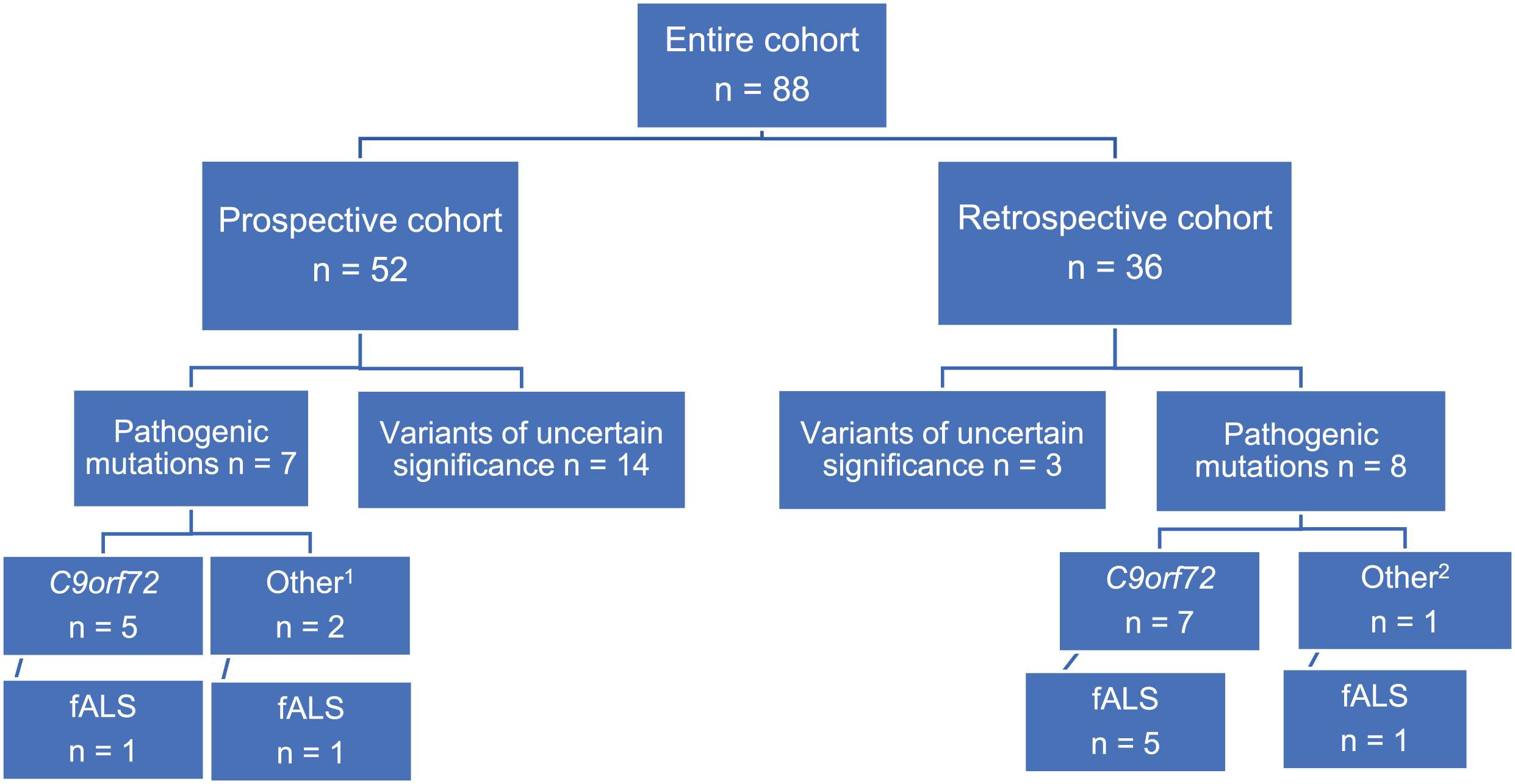
analyzed their samples from 2015 to 2020 in a research setting (n = 52, all at the Neuromuscular Center, Department of Neurology and Department of Biology and Medical Genetics, University Hospital Motol). The second cohort was a retrospective one, enriching the dataset with samples from patients referred for routine genetic testing, mainly due to family history, young age of onset (<45 years) [15] or associated dementia (ERN NMC University Hospital Motol n = 19, ERN NMC University Hospital Brno n = 17; all 36 samples analyzed at the Centre of Molecular Biology and Genetics, University Hospital Brno) (Fig. 1).
Fig. 1
Schematic of patient distribution and variant detection between and within two cohorts. Prospective cohort – longitudinal recruitment and sample analysis between 2015–2020 at the ERN-NMC and Department of Biology and Medical Genetics, both University Hospital Motol; Retrospective cohort – samples from patients, analyzed in a routine setting, all at the Centre of Molecular Biology and Genetics, University Hospital Brno; 1 – pathogenic mutations in other ALS-linked genes: FUS, TBK1; 2 –TARDBP; fALS – at least one first- or second degree relative with ALS and/-or FTD.
Whole blood for DNA extraction was obtained following informed written consent. We obtained approval for this study from the Institutional Ethics Committee of University Hospital Motol.
Molecular methods and analysis
Patient samples from each cohort were processed and analyzed in their respective laboratories, which applied different procedures regarding DNA isolation, C9orf72 analysis, library preparation, data processing and annotation. The final evaluation of the pathogenicity of the variants followed the same rules.
The analysis focused on identifying rare variants in genes, involved in ALS pathogenesis (Table 2), which were selected from Genomics England PanelApp v.1.69 (http://panelapp.genomicsengland.co.uk/panels/263/) and GeneReviews (https://www.ncbi.nlm.nih.gov/books/NBK1450/). The mode of inheritance of each variant was adopted from the abovementioned resources as well.
Table 2
Selected ALS-linked genes, investigated in our study
| ALS2, ANG, ANXA11, AR, C9orf72, CHCHD10*, CHMP2B, DAO**, DCTN1, ERBB4, FIG4, FUS, HNRNPA1, KIF5A, MAPT, MATR3, NEFH, NEK1, OPTN, PFN1, SCFD1**, SETX, SIGMAR1, SLC52A2, SLC52A3, SOD1, SPAST, SPG11, SQSTM1, TARDBP, TBK1, TUBA4A* UBQLN2, UNC13A*, VAPB, VCP |
Those genes, for which analysis could not be performed depending on the use of each library preparation kit (the kits did not contain probes for these genes) are marked with * – TrusightOne and Focus Exome ** – TrusightOne, Focus Exome and the capture method KAPA HyperChoice.
In the prospective cohort, genomic DNA was obtained from peripheral blood using the Gentra Puregene Blood Kit (Qiagen, Hilden, Germany) or MagCore extractor system H16 with a MagCore Genomic DNA Large Volume Whole Blood Kit (RBC Bioscience Corp., Taiwan). All patients were first screened for the presence of an expansion in the C9orf72 gene. The length of the repeat region in the C9orf72 was analysed using previously published repeat-primed polymerase chain reaction (RP-PCR) methods [16].
Every patient in the prospective arm underwent examination through either clinical exome sequencing or whole exome sequencing. DNA library preparation was carried out using three distinct kits, determined by the kit’s availability within the laboratory: 1. TruSight One kit (TSO) from Illumina Inc, San Diego, CA, was employed for 18 samples; 2. SureSelect Focused Exome kit (SSFE) from Agilent, Santa Clara, CA, USA, was used for 22 samples; 3. SeqCap EZ MedExome kit (SCEZME) from Roche, Madison, WI, was utilized for the preparation of 12 samples.DNA libraries were sequenced using the HiSeq 2500 platform / NovaSeq 6000 Sequencer (both from Illumina, USA), with 2x100 base paired-end HiSeq Rapid Cluster kit v2 and Rapid SDS Kit v.2 chemistry / 2x100 base paired-end chemistry NovaSeq 6000 S1 Reagent Kit v1.5 (200 cycles). Following quality control measures, the reads in fastq format were mapped to the human genome reference sequence hg19/GRCh37. This alignment was accomplished through the use of either Novoalign (v4.02.03) or BWA (v4.02.03). Aligned sequence data was processed according to GATK (Genome Analysis Toolkit) best practices workflow. Conversion of the SAM format to BAM and elimination of duplicates were executed using Picard Tools. GATK 3.8 was used for local realignment around indels, base recalibration, variant recalibration and genotyping. Copy number variaton (CNV) analysis was not performed within this study. Variants were annotated using SnfEff4.3T and Ensembl v.75 gene annotation and uploaded to GEMINI database (0.30.2), followed by filtering as describedbellow.
In the retrospective cohort, genomic DNA was extracted from peripheral blood leukocytes by the standard salting-out method. For the assessment of the length of the repeat region in the C9orf72 gene, we used the AmplideX® PCR/CE C9orf72 Kit (Asuragen, USA). For the identification of sequence variants in the ALS-linked genes (Table 2), we used the capture method KAPA HyperChoice (Roche, USA) followed by next generation sequencing on the NextSeq 500 (Illumina, USA) with NSQ 500/550 Mid Output KT v2.5 (300 cycles). The individual DNA sequence reads were aligned to the published human genome reference hg19/GRCh37 and variants were called using the software CLC Genomics Workbench (QIAGEN). The analysis was designed to detect both small-scale gene variants and large gene deletions/duplications by analysing copy number variations.
In both cohorts, the variants in selected ALS genes with an allele frequency exceeding 0.1% within The Genome Aggregation Database (gnomAD v2.1) were excluded from further analysis, as well as those found in the local Czech genomic variant database with frequency exceeding 0.5%. Intronic and synonymous variants, and variants categorized as benign or likely benign in ClinVar (23/05/2023) were also eliminated from consideration. The remaining variants were checked in databases ClinVar, Human Gene Mutation Database (HGMD Professional 2023.4), gnomAD and other population databases (dbSNP 156 and 1000 Genomes) to supplement variant information.The variants were classified in accordance with the ACMG guideline [17] using Alamut Visual Plus software v.7.1, the VarSome variant classifier (https://varsome.com) and Franklin platform for variant classification (https://franklin.genoox.com). The scores of the bioinformatic predictors of missense variants (PolyPhen-2, SIFT and CADD) and splicing variants (SpliceAl) extracted from Alamut, The Varsome and Franklin were also considered. Crucial for the final decision were the ACMG classification, the mode of inheritance as well as the impact of the particular gene in pathogenesis of ALS, which was crosschecked in the ALS spectrum disorders expert panel (www.clinicalgenome.org) and from the available literature.
All pathogenic/likely pathogenic variants were confirmed by Sanger sequencing using the BigDye Terminator Cycle Sequencing Kit (Applied Biosystems, USA) on the ABI 3130xl Genetic Analyzer (Applied Biosystems, USA).
The statistical analysis (comparison of clinical features) was performed via a two-tailed Fisher exact test, with a level of significance set at p < 0.05.
RESULTS
For the sake of data presentation clarity, we combined the results from the two cohorts together since the applied criteria for variant classification were identical.
Variants with causal significance for ALS
In 15 patients (17%), we detected 15 pathogenic or likely pathogenic variants (Table 3), the most frequent one being the HRE in C9orf72, found in 12 patients (13.5 % of the entire study population, six males, six females). While six patients carrying the C9orf72 HRE had a family history of ALS or frontotemporal dementia (66.7% of fALS patients), the other six had no history of affected relatives (7.55% of sALS patients). Other three patients were carrying one pathogenic or likely pathogenic variant in FUS: p.(Arg521Cys), TBK1 (splice donor c.540 + 1G > T) and TARDBP: p.(Ala382Thr) genes, respectively (every variant in a heterozygous state).The patient with the Arg521Cys mutation in FUS had an affected, already deceased brother from whom we could obtain a stored DNA sample, and the analysis detected the same FUS mutation. The patient with Ala382Thr mutation in TARDBP had multiple affected relatives, but we could not verify the co-segregation in the family, as all affected members had been long dead and no DNA samples were available. Both variants in FUS and TARDBP have already been described [18, 19] and are reported as pathogenic or likely pathogenic in ClinVar without conflicts. Altogether, we detected a pathogenic variant in eight out of nine fALS (88.9%) patients in our study. The splice donor variant detected in TBK1 in an sALS patient from our cohort has already been recorded [20]. Within the young-onset group (24/88, 27.3% of the entire cohort), two fALS patients were harbouring a pathogenic variant, both HRE in C9orf72 (8.33% of the subgroup). Among patients with a standard age of onset (n = 64), there were 13 patients with detected pathogenic variants (20.3%). The difference between these two subgroups was not statistically significant (p = 0.222). Furthermore, among sALS patients, we have observed a significantly higher association of bulbar onset with carrier status of a pathogenic (or likely pathogenic) variant compared to spinal onset (bulbar: 5/17, 29.4%; spinal: 2/62, 2.55%; p = 0.004). The presence of the pathogenic HRE in C9orf72 was significantly associated with bulbar onset in the whole study population (bulbar onset in C9orf72 HRE carriers: 7/12, 58.3%, bulbar onset in rest of the study population: 13/76, 17.1%; p = 0.005). Seven patients (all presented with sALS, 7.95% of the entire cohort) fulfilled the clinical criteria of FTD. In three of these seven patients (42.9%), we detected a pathogenic mutation (all had the HRE in C9orf72). Again, the presence of FTD among the sALS cases (n = 79) was significantly associated with a carrier status of a pathogenic mutation (HRE in C9orf72), compared to other sALS patients (n = 72, pathogenic mutation in 4/72, 5.6%; p = 0.013).
Table 3
Variants with causal significance for ALS: 15 variants in 15 patients
| Patient characteristics | Gene | Inheri-tance | Reference sequence | DNA variant | Protein change | Allele frequency gnomAD v2.1.1 | dbSNP | ACMG criteria | ACMG classification | ClinVar | PolyPhen-2 | SIFT | CADD | Splice Al | Ref. | |||
| Sex | Age of onset | Disease subtype | fALS | |||||||||||||||
| F | 64 | Bulbar + FTD | No | C9orf72 | AD | NG_031977.1 | (G4C2) > 50 | N/A | 0 | - | - | Pathogenic | Pathogenic | N/A | N/A | N/A | N/A | [16] |
| F | 41 | Bulbar | yes | |||||||||||||||
| M | 61 | Bulbar + FTD | no | |||||||||||||||
| M | 65 | Bulbar | no | |||||||||||||||
| F | 69 | Bulbar + FTD | no | |||||||||||||||
| M | 42 | Spinal ALS | yes | |||||||||||||||
| M | 57 | Bulbar | yes | |||||||||||||||
| F | 64 | Spinal ALS | no | |||||||||||||||
| M | 57 | Bulbar | no | |||||||||||||||
| F | 55 | Spinal ALS | yes | |||||||||||||||
| F | 47 | Spinal ALS | yes | |||||||||||||||
| M | 46 | Spinal ALS | yes | |||||||||||||||
| M | 57 | Spinal ALS | yes | FUS | AD | NM_004960.4 | c.1561C > T | p.(Arg521Cys) | 1.4 e–5 | rs121909668 | PS4str, PM2mod, PM5mod, PM1mod, PP5 | Pathogenic | Pathogenic | U (0.94) | D (0) | 24,1 | U(0.12) | [18] |
| F | 68 | Spinal ALS | yes | TARDBP | AD | NM_007375.4 | c.1144G > A | p.(Ala382Thr) | 3 e–5 | rs367543041 | PS4str, PS3sup, PM1mod, PP2sup, PM2mod, PP5 | Pathogenic | Pathogenic | B (0.27) | B(0.23) | 20,7 | B (0) | [19] |
| M | 66 | Spinal ALS | no | TBK1 | AD | NM_013254.4 | c.540 + 1G > T | N/A | 0 | – | PVS1vstr, PM2mod | Likely pathogenic | – | N/A | N/A | N/A | D (1) | [20] |
| NG_046906.1 | g.20024G > A | |||||||||||||||||
All listed variants are heterozygous; AD – autosomal dominant, ALS – amyotrophic lateral sclerosis, fALS – familial ALS, FTD – frontotemporal dementia, F – female; M – Male; Allele frequency gnomAD v2.1.1 shows the aggregated frequency in The Genome Aggregation Database; dbSNP, Single Nucleotide Polymorphism Database reference SNP ID number for the variant; ACMG criteria; ACMG, The American College of Medical Genetics and Genomics; ClinVar – public archive of reports of the relationships among human genetic variations and phenotypes, with supporting evidence, PolyPhen-2 – an automatic tool for prediction of the possible impact of an amino acid substitution on the structure and function of a human protein; SIFT – The Sorting Intolerant from Tolerant (SIFT) algorithm predicts the effect of coding variants on protein function; CADD – the Combined Annotation Dependent Depletion for scoring the deleteriousness of single nucleotide variants, multi-nucleotide substitutions as well as insertion/deletion variants in the human genome, the lower the score, the more benign the substitution; SpliceAl, a deep learning splicing predictor algorithm; D, deleterious; U, uncertain; B, benign; M, moderate; S, strong; N/A, not applicable; ACMG criteria: pathogenic vstr – very strong, str – strong, sup – supporting, mod – moderate.
Variants of uncertain significance
We detected 16 rare variants of uncertain significance in 15 patients (Table 4). We performed a literature search for each variant to confirm newly or previously identified ALS-risk variants.
Table 4
Variants with unclear causal significance for ALS
| Patient characteristics | Gene | Inheri-tance | Reference sequence | DNA variant | Protein change | Allele frequency gnomAD v2.1.1 | dbSNP | ACMG criteria | ACMG classification | ClinVar | PolyPhen-2 | SIFT | CADD | Splice Al | Ref. | |||
| Sex | Age of onset | Disease subtype | fALS | |||||||||||||||
| F | 59 | Spinal ALS | no | FIG4L | AD | NM_014845.6 | c.122T > C | p.(Ile41Thr) | 1.0 e–3 | rs121908287 | PM3vstr, PS3sup, PM2mod | Pathogenic | Pathogenic | D (1) | D (0) | 25,8 | B (0) | [21] |
| M | 29 | Spinal ALS | no | FIG4L | AD | NM_014845.6 | c.2095C > T | p.(Arg699Cys) | 3.54 e–5 | rs764799053 | PM2mod, PP3sup | Uncertain | Uncertain | U (0.96) | D (0) | 33 | B (0.01) | [5] |
| F | 59 | Spinal+ FTD | no | KIF5A | AD | NM_004984.4 | c.291 + 5G > A | N/A | 1.2 e–5 | rs775732465 | PM2mod, PP3sup | Uncertain | Uncertain | N/A | N/A | N/A | M (0.33) | [22] |
| NG_088155.1 | g.18635G > A | |||||||||||||||||
| M | 50 | Bulbar | no | NEFHS | AD | NM_021076.4 | c.1084-3C > G | N/A | 1.2 e–5 | rs1347201784 | PM2mod, PP3sup | Uncertain | - | N/A | N/A | N/A | S (0.55) | – |
| NG_008404.1 | g.10529C > G | |||||||||||||||||
| M | 59 | Bulbar | no | NEK1 S | AD | NM_001199397.3 | c.760G > T | p.(Glu254*) | 0 | – | PVS1mod, PM2mod | Uncertain | - | N/A | N/A | N/A | S (0.55) | – |
| F* | 64 | Bulbar+ FTD | no | OPTN | AR | NM_001008212.2 | c.785C > A | p.(Ser262*) | 1.42 e–5 | rs750571210 | PM3str, PVS1vstr, PM2mod, PP5 | Pathogenic | Pathogenic | N/A | N/A | N/A | B (0) | [23] |
| F* | 41 | Bulbar | yes | OPTN | AR | NM_001008212.2 | c.785C > A | p.(Ser262*) | 1.42 e–5 | rs750571210 | PM3str, PVS1vstr, PM2mod, PP5 | Pathogenic | Pathogenic | N/A | N/A | N/A | B (0) | [23] |
| F | 38 | Spinal ALS | no | SETXL | AD | NM_015046.7 | c.4796T > C | p.(Phe1599Ser) | 0 | – | PM2mod | Uncertain | – | D (0.99) | D (0) | 26,8 | B (0) | – |
| M | 64 | Spinal ALS | no | SETXL | AD | NM_015046.7 | c.6865T > C | p.(Ser2289Pro) | 0 | – | PM2mod | Uncertain | – | D (0.99) | U (0.025) | 24,5 | B (0) | – |
| F** | 37 | LMN- predomin. | no | SOD1 | AD/AR | NM_000454.5 | c.378C > G | p.(Asp126Glu) | 0 | – | PM1mod, PP2sup, PM2mod, PM5sup, PP3sup, | Likely pathogenic | - | D (0.99) | D (0) | 25,5 | B (0.03) | – |
| TARDBP | AD | NM_007375.4 | c.169G > A | p.(Val57Ile) | 0 | – | PM2mod, PP2sup | Uncertain | – | B (0.03) | B (0.7) | 21 | B (0) | – | ||||
| F | 57 | Spinal | no | SPAST | AD | NM_014946.4 | c.8C > T | p.(Ser3Phe) | 0 | – | PM2mod, PP2sup | Uncertain | – | D (0.98) | D (0) | 23,4 | B (0.03) | – |
| M | 64 | Bulbar | no | SPG11 | AR | NM_025137.4 | c.2757_2775 del | p.(Leu920Thrfs*10) | 0 | rs1595888828 | PVS1vstr, PM3str, PM2mod, PP5 | Pathogenic | Likely Pathogenic | N/A | N/A | N/A | B (0) | – |
| M | 60 | LMN- predomin. | no | SPG11 | AR | NM_025137.4 | c.3075dup | p.(Glu1026Argfs*4) | 2.79 e–5 | rs312262752 | PVS1vstr, PM3vstr, PM2mod, PP1sup PP5 | Pathogenic | Pathogenic | N/A | N/A | N/A | B (0.06) | [24], [25] |
| M | 29 | Spinal ALS | no | TBK1 | AD | NM_013254.4 | c.2069T > C | p.(Met690Thr) | 0 | – | PM2mod | Uncertain | – | D (0.9) | D (0.1) | 24,9 | B (0.02) | – |
| F | 70 | Bulbar | no | UBQLN2 | XLD | NM_013444.4 | c.1481C > T | p.(Pro494Leu) | 0 | – | PM2mod | Uncertain | – | B (0.02) | B (0) | 22,2 | B (0) | [26] |
All listed variants are heterozygous; LMN – lower motor neuron; * marks 2 related individuals, mother and daughter – the mother was classified as sporadic at the time of inclusion (ad hoc), the daughter developed disease 2 years later; ** marks a single patient, carrying simultaneously 2 variants (in SOD1 and TARDBP); GeneL – limited association with ALS; GeneS – confers susceptibility to ALS; AR – autosomal recessive; XLD – X-linked dominant; Allele frequency gnomAD v2.1.1 shows the aggregated frequency in The Genome Aggregation Database; dbSNP, Single Nucleotide Polymorphism Database reference SNP ID number for the variant; ACMG criteria; ACMG, The American College of Medical Genetics and Genomics; ClinVar – public archive of reports of the relationships among human genetic variations and phenotypes, with supporting evidence, PolyPhen-2 – an automatic tool for prediction of the possible impact of an amino acid substitution on the structure and function of a human protein; SIFT – The Sorting Intolerant from Tolerant (SIFT) algorithm predicts the effect of coding variants on protein function; CADD – the Combined Annotation Dependent Depletion for scoring the deleteriousness of single nucleotide variants, multi-nucleotide substitutions as well as insertion/deletion variants in the human genome, the lower the score, the more benign the substitution; SpliceAl, a deep learning splicing predictor algorithm; D, deleterious; U, uncertain; B, benign; M, moderate; S, strong; N/A, not applicable; ACMG criteria: pathogenic vstr – very strong, str – strong, sup – supporting, mod – moderate.
In our cohort, we detected seven novel variants (all in a heterozygous state) in six sporadic patients. The NEK1 variant p.(Glu254*) was a novel loss-of-function (LoF) variant (nonsense type mutation – premature termination codon), but since the role of LoF variants in NEK1 for pathogenesis of ALS remains debated, we classified the variant as of unclear significance. There were two novel variants in SETX: p.(Phe1599Ser) and p. (Ser2289Pro) in two patients (one with young onset). Both variants were missense, and based on in silico analysis, both were assessed as of uncertain significance. Other novel missense variants were identified in SPAST p.(Ser3Phe) and TBK1 p.(Met690Thr). As parental DNA was unavailable in these patients for segregation analysis, we could not further elucidate the pathogenicity of these variants.∥In one young-onset patient with LMN-predo-minant phenotype, we identified simultaneously two novel variants of uncertain significance: p. (Asp126Glu) in SOD1 and p. (Val57Ile) in TARDBP. After performing a segregation analysis, we learned that the SOD1 variant was inherited from her father, while the TARDBP variant was inherited from her mother; both parents were unaffected.∥Besides that, we detected nine previously described variants in other ALS-linked genes (Table 4). Notably, there were two related patients – a mother with bulbar onset at 64 years with associated FTD and a daughter with bulbar onset at 41 years (only two years after the diagnosis in the mother), both carrying simultaneously the C9orf72 HRE and a previously recorded heterozygous, autosomal recessive pathogenic mutation in OPTN p.(Ser262*) [23].
DISCUSSION
To the best of our knowledge, this was the first comprehensive assessment of the genetic landscape of ALS in Czech patients. We aimed to bring pilot data on the genetic findings among familial and sporadic ALS patients from our country. Familial ALS cases were slightly overrepresented in our study (10.1%) due to the intentional enrichment of these individuals in the retrospective cohort. As this was not a population-based study, this percentage probably does not reflect the actual proportion of fALS cases in the Czech population (based on our estimates, the actual proportion will be around 5% of the ALS population). The unusually high proportion of pathogenic mutations detected in our fALS cases (88%) stems probably from the small sample size and would rather be lower in larger cohorts of patients, as other studies documented [4, 5]. On the other hand, the proportion of cases with pathogenic mutations among sALS (10.1%) was very similar to a recent, large-scale study (6013 sALS cases) [6] and could, therefore, be a realistic representation of the situation in the Czech population.
The most frequent mutation was the C9orf72 repeat expansion, detected in 12 patients (13.5%) from the entire study population. This data is in line with many previous cohorts. Six out of the 12 patients with the C9orf72 HRE had no family history of ALS or FTD, making this mutation the most common finding among sporadic cases (7.6%). Again, the proportion was relatively similar to other cohorts from European populations [6, 7]. With regards to the clinical phenotype of the C9orf72 HRE carriers, bulbar onset significantly predominated (seven out of 12 patients, 58.3%) – which is in line with findings from substantially larger cohorts from Europe [27, 28] – and frontotemporal dementia cases were overrepresented (3 out of 12 patients, 25%), compared to the rest of the study population, although the latter difference was not statistically significant. Notably, among sALS patients, the presence of ALS-FTD phenotype was associated with a significantly higher likelihood of detecting a pathogenic variant. However, the generalizability of this finding is limited due to the small size of our study population.
Young-onset ALS patients were overrepresented in our study (27.3% of the entire cohort, compared to about 10% referred in the literature [15]). Contrary to expectations, based on predictions from a validated model of pathogenesis [29], as well as findings from larger cohorts [30], the detection rate of pathogenic (or likely pathogenic) variants in this subgroup was not higher than in the rest of the study population. Notably, there were four young-onset patients in our study, carrying five variants of uncertain significance in FIG4 p. (Arg699Cys), SETX p. (Phe1599Ser), SOD1 p.(Asp126Glu), TARDBP p.(Val57Ile) and TBK1 p.(Met690Thr), where pathogenicity seems possible, but could not be confirmed. One plausible explanation for the low detection rate within the young-onset group could have been the overall small population of our study. Nevertheless, even with the overrepresentation of young-onset patients, most of the pathogenic variants were detected in the group with standard age of onset, suggesting a possibility of a missing genetic variability in the former subgroup. Interestingly, another study focused on young-onset sporadic patients reported a low detection rate of pathogenic mutations in established ALS-linked genes in this subgroup as well [31]. The authors focused on the burden of rare variants in genes coding RNA-binding proteins that are not yet established as causal in ALS pathogenesis but are involved in common functional pathways. This study gave some evidence for oligogenic interaction and synergistic effect among variants in this functional cluster of genes in the group of young-onset patients. As such a broad gene analysis was beyond the scope of our study, which was limited mainly by the sample size, we can only speculate about the applicability of this explanation to our cohort. Nevertheless, this seems to be a promising direction to elucidate the so far unexplained genetic component among young-onset patients in our cohort.
Mutations in the FUS and TARDBP genes are among the more commonly detected in ALS patients [4, 6]. Besides the two documented pathogenic variants, detected in two fALS patients from our cohort – p.(Arg521Cys) in the FUS gene and p.(Ala382Thr) in TARDBP - we found a novel variant of uncertain significance in TARDBP, p.(Val57Ile). This variant was detected in a sporadic young patient with a LMN-predominant phenotype and was inherited from her unaffected mother. Interestingly, this affected individual harboured a second heterozygous variant, a likely pathogenic, autosomal recessive missense mutation p.(Asp126Glu) in SOD1, inherited from her unaffected father. Although the segregation analysis ruled out a causal effect of neither of these variants, we speculated a possible synergistic effect, when present simultaneously in one individual, based on documented interaction between SOD1 and TDP-43 proteins in an experimental study [32]. Notably, an oligogenic interaction with other co-segregating rare variants has been documented for another variant in SOD1, p.(Asp91Ala) [33], which is also remarkable for its reduced penetrance and variable inheritance (recessive or dominant) [34].
We also noticed the absence of causal mutations in SOD1 among fALS, as well as sALS patients in our cohort, which very probably stems from its small sample size., Data from a large-scale study documented SOD1 mutation detection in only 0.63 % of sporadic ALS patients [6]. The size of our fALS cohort was small and unequally represented geographic regions of our country. Thus, a regional family cluster of SOD1-positive cases could have easily been missed.
In the last decade, several studies brought evidence for a suggested oligogenic model of inheritance in ALS [35, 36]. On the other hand, some studies documented the C9orf72 HRE alone to be sufficient to cause ALS, irrespective of the co-occurrence of other variants in ALS-linked genes [37, 38]. In our study, besides the abovementioned young-onset patient harbouring variants both in SOD1 and TARDBP, we further detected a variant p.(Ser262*) in OPTN in two related patients (mother and daughter), both harbouring C9orf72 HRE. This variant in OPTN has been detected in an autosomal recessive form of ALS only [23]. As a matter of fact, the phenotype differed to some degree between the two patients (much younger age of onset in the daughter, the presence of FTD in the mother, but not in the daughter). However, since they were both harbouring the same OPTN variant, this seems not to be a plausible genetic modifier and we therefore deemed its causal significance in our patients as rather unlikely.
We identified a novel variant in the NEK1 gene p.(Glu254*) in a sporadic, bulbar onset patient. For this variant p.(Glu254*), we assume the loss of function (LoF) effect. The LoF variants in the NEK1 gene, coding NIMA-associated kinase, have been suggested to play a direct pathogenic role by some authors [39], while others consider them to only confer significant susceptibility to disease development and hypothesize a necessary oligogenic interaction with other co-segregating rare variants in ALS-linked genes [40, 41]. In our patient, we haven't found any such additional variant in the extent of examined genes. Taken together, we categorized this variant as of uncertain significance.
Furthermore, we detected two variants in TBK1 in two sporadic patients with spinal onset ALS without FTD. First, a previously recorded [20], likely pathogenic splice-donor variant c.540 + 1G > T, which probably disrupts the donor splice site and could lead to haploinsufficiency via loss-of-function mechanism – a likely molecular genetic mechanism of toxicity in TBK1 mutations. The other one was a novel, missense variant p.(Met690Thr), assessed by in silico analysis as of uncertain significance. While the mechanism through which nonsense mutations lead to pathogenesis via haploinsufficiency has been elucidated, the mechanisms underlying the disease due to missense mutations are still largely unclear [43]. Our data add to the growing knowledge that mutations in TBK1 are an important contributor to the genetics of sporadic ALS.
Moreover, we identified two variants in FIG4. Although biallelic pathogenic variants in FIG4 have been established as causative for AR-Charcot-Marie-Tooth disease 4J, several studies have brought some (case level) evidence of its involvement in the pathogenesis of autosomal dominant ALS type 11 [21, 35, 44]. The variant p.(Ile41Thr) in the heterozygous state has previously been reported in cohorts from European [21] and Northern American [30] populations and is reported in ClinVar with conflicting results. However, due to its relatively high prevalence in the gnomAD control database (0.002), we do not think that it alone causes ALS in accordance with the conclusion from another recent study [44]. The other detected variant in FIG4- p.(Arg699Cys) – has been reported in a German fALS cohort [5]. Without co-segregation data, it could not be conclusively confirmed as disease-causing.
Further, we found two novel missense variants in SETX, a gene associated with early-onset, autosomal dominant ALS type 4, in two sporadic patients. This rare form of ALS has been described as a very slowly progressive one without the involvement of respiratory or bulbar muscles [45]. Notably, a broader phenotypic variation among ALS patients with novel SETX variants has been reported in more recent studies [46]. The patient with the p.(Phe1599Ser) variant was a young-onset (38 years) sporadic case with a marked upper-motor neuron involvement, developing a prominent bulbar involvement later on in the course of the disease. The other patient, carrying the variant p.(Ser2289Pro) presented with sporadic ALS with onset at 60 years and a rapid course of the disease. Without segregation analysis and further evidence, we cannot conclude regarding the pathogenicity of these variants. Analogically, this was the case of a rare variant p.(Pro494Leu) in UBQLN2, detected in a sALS patient in our cohort and previously described in a French fALS patient [26].
We detected two previously recorded, likely pathogenic frameshift mutations in SPG11 in a heterozygous state: p. (Leu920Thrfs*10) and p.(Glu1026Argfs*4) [24, 25] and a novel missense variant p.(Ser3Phe) in SPAST. Although mutations in both SPG11 and SPAST have been associated with hereditary spastic paraplegia, biallelic variants in SPG11 are also causal in recessive juvenile ALS [47]. Furthermore, heterozygous variants in SPG11 have been reported from cohorts of fALS and sALS patients [48, 49]. Nevertheless, the latter studies haven't established any causal relationship between these novel variants and ALS. Association between SPAST and ALS has been reported only on a case level so far [50, 51]. As for the variants in SPG11, detected in our cohort, it is also possible that we failed to reveal the second variant (e.g. deep-intronic or CNV). However, the phenotype of the patients (onset > 60 years, bulbar / LMN-predominant sALS) does not resemble previously described cases. The clinical significance of these variants is, therefore, uncertain.
We are aware of the limitations of our study due to the different approaches to library preparation and, thus, different coverage of relevant ALS genes between the two laboratories. The small size of the patient cohort may have led to a possible omission of variants in less common ALS-associated genes and limits the generalizability of the phenotypic features, observed in our cohort, due to low statistical power. Moreover, as a result of the hospital-based design of our study, as well as of the intentional enrichment of specific subgroups of patients (young-onset, fALS), the obtained data might be skewed and not entirely representative regarding the frequency and proportion of the most common variants, as compared to a population-based study.
In summary, our data has shown the frequency of C9orf72 HRE among Czech ALS patients to be very similar to other European populations. Furthermore, the data suggest some consistent phenotypic features (bulbar onset or predominance, higher proportion of FTD) even among apparently sporadic patients with the C9orf72 HRE, providing altogether (regarding its relatively high prevalence) some guidance for targeted first-line genetic testing. Pathogenic mutations in TBK1, FUS, and TARDBP, commonly detected among other cohorts, are present in the Czech population as well. On the other hand, we detected no causal variants in SOD1 in our study. We encountered a low yield of pathogenic or likely pathogenic mutations among young-onset patients in our cohort, contrary to our assumptions, upon which we intentionally recruited this subgroup. From another perspective, these findings suggest a high probability of a missing genetic variability at the level of as yet not established (and, therefore, not examined in our study) genes. In our opinion, exploring this subgroup of patients for new groups of genes is highly warranted, holding a promise of a novel option for potential gene-modifying treatment in ALS. Regarding the variants of uncertain significance found in patients of our cohort, no definite conclusion regarding their pathogenicity can be drawn, and further functional, genetic, and clinical studies in large independent cohorts are required to fully understand and establish their contribution to ALS.
ACKNOWLEDGMENTS
The four (DB, RM, EV, LJ) authors of this publication are members of the European Reference Network for Neuromuscular Diseases— Project ID No. 870177. We thank The National Center for Medical Genomics (LM2023067) for providing allelic frequencies in ethnically matched populations. We acknowledge the Core Facility Genomics supported by the NCLG research infrastructure (LM2018132 funded by MEYS CR) for their support with obtaining scientific data presented in this paper.
FUNDING
This study was supported by NF-CZ11-PDP-3-2014-Norway grants and by the Ministry of Health,
Czech Republic— conceptual development of research organization, University Hospital Motol, Prague, Czech Republic grant no. 00064203 and University Hospital Brno, Brno, Czech Republic no. 65269705.
AUTHOR CONTRIBUTIONS
ZM, PH, JZ and LF carried out the experiments and the clinical and molecular investigations; PV and TK carried out the experiments; VS and JG performed the clinical and molecular investigations. DB, RM, EV, LJ recruited the patients, performed the clinical evaluation, and acquired clinical data. DB, ZM and JZ drafted the manuscript. DB, RM and ZM conceptualized and designed the study. All authors had full access to the data in the study, critically revised, and approved the final version of the manuscript.
CONFLICT OF INTEREST
On behalf of all authors, the corresponding author states that there is no conflict of interest.
DATA AVAILABILITY
The dataset used and/or analysed during the current study is available from the corresponding author on reasonable request. The data are not publicly available due to privacy or ethical restrictions. All novel variants have been submitted to the Global Variome shared LOVD v.3 – Leiden Open Variation Database.
ETHICAL APPROVAL AND PATIENT CONSENT
Informed consent was obtained from the patients who participated in the study. The study was approved by the Ethics Committee of the University Hospital Motol, Prague (Reference No.: EK-849/23) and has, therefore, been performed in accordance with the ethical standards laid down in the 1975 Declaration of Helsinki.
REFERENCES
[1] | Verde F , Del Tredici K , Braak H , Ludolph A . The multisystem degeneration amyotrophic lateral sclerosis – neuropathological staging and clinical translation. Arch Ital Biol. (2017) ;155: (4):118–130. doi: 10.12871/00039829201746. |
[2] | Shatunov A , Al-Chalabi A . The genetic architecture of ALS. Neurobiol Dis. (2021) ;147: :105156. doi: 10.1016/j.nbd.2020.105156. |
[3] | Mathis S , Goizet C , Soulages A , Vallat JM , Le Masson G . Genetics of amyotrophic lateral sclerosis: A review. J Neurol Sci. (2019) ;399: :217–226. doi: 10.1016/j.jns.2019.02.030. |
[4] | Zou ZY , Zhou ZR , Che CH , Liu CY , He RL , Huang HP . Genetic epidemiology of amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. (2017) ;88: (7):540–549. doi: 10.1136/jnnp-2016-315018. |
[5] | Müller K , Brenner D , Weydt P , Meyer T , Grehl T , Petri S , et al.; German ALS network MND-NET. Comprehensive analysis of the mutation spectrum in 301 German ALS families. J Neurol Neurosurg Psychiatry. (2018) ;89: (8):817–827. doi: 10.1136/jnnp-2017-317611. |
[6] | Van Daele SH , Moisse M , van Vugt JJFA , Zwamborn RAJ , van der Spek R , van Rheenen W , et al. Genetic variability in sporadic amyotrophic lateral sclerosis. Brain. (2023) ;146: (9):3769. doi: 10.1093/brain/awad120. |
[7] | Yilmaz R , Grehl T , Eckrich L , Marschalkowski I , Weishaupt K , Valkadinov I , et al. Frequency of C9orf72 and SOD1 mutations in 302 sporadic ALS patients from three German ALS centers. Amyotroph Lateral Scler Frontotemporal Degener. (2023) ;24: (5-6):414–419. doi: 10.1080/21678421.2023.2165946. |
[8] | Gromicho M , Pinto S , Gisca E , Pronto-Laborinho AC , Andersen PM , de Carvalho M . Frequency of C9orf72 hexanucleotide repeat expansion and SOD1 mutations in Portuguese patients with amyotrophic lateral sclerosis. Neurobiol Aging. (2018) ;70: :325.e7–325.e15. doi: 10.1016/j.neurobiolaging.2018.05.009. |
[9] | Tripolszki K , Gampawar P , Schmidt H , Nagy ZF , Nagy D , Klyvényi P , et al. Comprehensive Genetic Analysis of a Hungarian Amyotrophic Lateral Sclerosis Cohort. Front Genet. (2019) ;10: :732. doi: 10.3389/fgene.2019.00732. |
[10] | Vrabec K , Koritnik B , Leonardis L , Dolenc-Grošelj L , Zidar J , Smith B , et al. Genetic analysis of amyotrophic lateral sclerosis in the Slovenian population. Neurobiol Aging. (2015) ;36: (3):1601e17–20. doi: 10.1016/j.neurobiolaging.2014.11.011. |
[11] | Abecasis GR , Auton A , Brooks LD , DePristo MA , Durbin RM , Handsaker RE , Kang HM , Marth GT , McVean GA . An integrated map of genetic variation from 1,092 human genomes. Nature. (2012) ;491: (7422):56–65. doi: 10.1038/nature11632. |
[12] | Majounie E , Renton AE , Mok K , Dopper EG , Waite A et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. (2012) ;11: (4):323–30. doi: 10.1016/S1474-4422(12)70043-1. |
[13] | Ludolph A , Drory V , Hardiman O , Nakano I , Ravits J , Robberecht W , Shefner J; WFN Research Group On ALS/MND A revision of the El Escorial criteria – 2015. Amyotroph Lateral Scler Frontotemporal Degener. (2015) ;16: (5-6):291–2. doi: 10.3109/21678421.2015.1049183. |
[14] | Strong MJ , Grace GM , Freedman M , Lomen-Hoerth C , Wooley S , Goldstein LH , et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioural syndromes in amyotrophic lateral sclerosis, Amyotrophic Lateral Sclerosis. (2009) ;10: (3):131–146. doi: 10.1080/17482960802654364. |
[15] | Turner MR , Barnwell J , Al-Chalabi A , Eisen A . Young-onset amyotrophic lateral sclerosis: historical and other observations. Brain. (2012) ;135: (Pt 9):2883–91. doi: 10.1093/brain/aws144. |
[16] | DeJesus-Hernandez M , Mackenzie IR , Boeve BF , Boxer AL , Baker M , Rutherford NJ , et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. (2011) ;72: (2):245–56. doi: 10.1016/j.neuron.2011.09.011. |
[17] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) ;17: (5):405–24. doi: 10.1038/gim.2015.30. |
[18] | Kwiatkowski TJ Jr , Bosco DA , Leclerc AL , Tamrazian E , Vanderburg CR , Russ C , et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science. (2009) ;323: (5918):1205–8. doi: 10.1126/science.1166066. |
[19] | Chiò A , Borghero G , Pugliatti M , Ticca A , Calvo A , Moglia C , et al. Large proportion of amyotrophic lateral sclerosis cases in Sardinia due to a single founder mutation of the TARDBP gene. Arch Neurol. (2011) ;68: (5):594–8. doi: 10.1001/archneurol.2010.352. |
[20] | Cirulli ET , Lasseigne BN , Petrovski S , Sapp PC , Dion PA , Leblond CS ,et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. (2015) ;347: (6229):1436–41. doi: 10.1126/science.aaa3650. |
[21] | Osmanovic A , Rangnau I , Kosfeld A , Abdulla S , Janssen C , Auber B , et al. FIG4 variants in central European patients with amyotrophic lateral sclerosis: a whole-exome and targeted sequencing study. Eur J Hum Genet. (2017) ;25: (3):324–331. doi: 10.1038/ejhg.2016.186. |
[22] | Nicolas A , Kenna KP , Renton AE , Ticozzi N , Faghri F , Chia R , et al. Genome-wide Analyses Identify KIF5A as a Novel ALS Gene. Neuron. (2018) ;97: (6):1283–e6. doi: 10.1016/j.neuron.2018.02.027. |
[23] | Pottier C , Rampersaud E , Baker M , Wu G , Wuu J , McCauley JL , et al. Identification of compound heterozygous variants in OPTN in an ALS-FTD patient from the CReATe consortium: a case report. Amyotroph Lateral Scler Frontotemporal Degener. (2018) ;19: (5-6):469–471. doi: 10.1080/21678421.2018.1452947. |
[24] | Elert-Dobkowska E , Stepniak I , Krysa W , Ziora-Jakutowicz K , Rakowicz M , Sobanska A et al. Next-generation sequencing study reveals the broader variant spectrum of hereditary spastic paraplegia and related phenotypes. Neurogenetics. (2019) ;20: (1):27–38. doi: 10.1007/s10048-019-00565-6. |
[25] | Günther S , Elert-Dobkowska E , Soehn AS , Hinreiner S , Yoon G , Heller R , Hellenbroich Y , Hübner CA , Ray PN , Hehr U , Bauer P , Sulek A , Beetz C . High Frequency of Pathogenic Rearrangements in SPG11 and Extensive Contribution of Mutational Hotspots and Founder Alleles. Hum Mutat. (2016) ;37: (7):703–9. doi: 10.1002/humu.23000. |
[26] | Teyssou E , Chartier L , Amador MD , Lam R , Lautrette G , Nicol M , et al. Novel UBQLN2 mutations linked to amyotrophic lateral sclerosis and atypical hereditary spastic paraplegia phenotype through defective HSP70-mediated proteolysis. Neurobiol Aging. (2017) ;58: :239.e11–239.e20. doi: 10.1016/j.neurobiolaging.2017.06.018. |
[27] | Wiesenfarth M , Günther K , Müller K , Witzel S , Weiland U , Mayer K et al. Clinical and genetic features of amyotrophic lateral sclerosis patients with C9orf72 mutations. Brain Commun. (2023) ;5: (2):fcad087. doi: 10.1093/braincomms/fcad087. |
[28] | Chiò A , Borghero G , Restagno G , Mora G , Drepper C , Traynor BJ et al. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain. (2012) ;135: (Pt 3):784–93. doi: 10.1093/brain/awr366. |
[29] | Chiò A , Mazzini L , D’Alfonso S , Corrado L , Canosa A , Moglia C , et al. The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology. (2018) ;91: (7):e635–e642. doi: 10.1212/WNL.0000000000005996. |
[30] | Cady J , Allred P , Bali T , Pestronk A , Goate A , Miller TM , et al. Amyotrophic lateral sclerosis onset is influenced by the burden of rare variants in known amyotrophic lateral sclerosis genes. Ann Neurol. (2015) ;77: (1):100–13. doi: 10.1002/ana.24306. |
[31] | Cooper-Knock J , Robins H , Niedermoser I , Wyles M , Heath PR , Higginbotom A , et al. Targeted Genetic Screen in Amyotrophic Lateral Sclerosis Reveals Novel Genetic Variants with Synergistic Effect on Clinical Phenotype. Front Mol Neurosci. (2017) ;10: :370. doi: 10.3389/fnmol.2017.00370. |
[32] | Jeon GS , Shim YM , Lee DY , Kim JS , Kang MJ , Ahn SH , et al. Pathological Modification of TDP-43 in Amyotrophic Lateral Sclerosis with SOD1 Mutations. Mol Neurobiol. (2019) ;56: (3):2007–2021. doi: 10.1007/s12035-018-1218-2. |
[33] | Gentile G , Perrone B , Morello G , Simone IL , Andò S , Cavallaro S , Conforti FL . Individual Oligogenic Background in p.D91A-SOD1 Amyotrophic Lateral Sclerosis Patients. Genes (Basel). (2021) ;12: (12):1843. doi: 10.3390/genes12121843. |
[34] | Robberecht W , Aguirre T , Van den Bosch L , Tilkin P , Cassiman JJ , Matthijs G . D90A heterozygosity in the SOD1 gene is associated with familial and apparently sporadic amyotrophic lateral sclerosis. Neurology. (1996) ;47: (5):1336–9. doi: 10.1212/wnl.47.5.1336. |
[35] | van Blitterswijk M , van Es MA , Hennekam EA , Doojies D , van Rheenen W , Medic J , et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet. (2012) ;21: (17):3776–84. doi: 10.1093/hmg/dds199. |
[36] | McCann EP , Henden L , Fifita JA , Zhang KY , Grima N , Bauer DC , et al. Evidence for polygenic and oligogenic basis of Australian sporadic amyotrophic lateral sclerosis. J Med Genet. (2020) :jmedgenet-2020-106866. doi: 10.1136/jmedgenet-2020-106866. |
[37] | Keogh MJ , Wei W , Aryaman J , Wilson I , Talbot K , Turner MR , et al. Oligogenic genetic variation of neurodegenerative disease genes in 980 postmortem human brains. J Neurol Neurosurg Psychiatry. (2018) ;89: (8):813–816. doi: 10.1136/jnnp-2017-317234. |
[38] | Ross JP , Leblond CS , Laurent SB , Spiegelman D , Dione-Laporte A , Camu W , et al. Oligogenicity, C9orf72 expansion, and variant severity in ALS. Neurogenetics. (2020) ;21: (3):227–242. doi: 10.1007/s10048-020-00612-7. |
[39] | David Brenner , Kathrin Müller , Thomas Wieland , Patrick Weydt , Sarah Böhm , Dorothée Lulé et al. NEK1 mutations in familial amyotrophic lateral sclerosis, Brain. (2016) ;139: (5):e28, https://doi.org/10.1093/brain/aww033. |
[40] | Riva N , Pozzi L , Russo T , Pipitone GB , Schito P , Domi T et al. NEK1 Variants in a Cohort of Italian Patients With Amyotrophic Lateral Sclerosis. Front Neurosci. (2022) ;16: :833051. doi: 10.3389/fnins.2022.833051. |
[41] | Lattante S , Doronzio PN , Conte A , Marangi G , Martello F , Bisogni G et al. Novel variants and cellular studies on patients’ primary fibroblasts support a role for NEK1 missense variants in ALS pathogenesis. Hum Mol Genet. (2021) ;30: (1):65–71. doi: 10.1093/hmg/ddab015. |
[42] | Lu YQ , Chen JM , Lin H , Feng SY , Che CH , Liu CY , et al. Novel Intronic Mutations of TBK1 Promote Aberrant Splicing Modes in Amyotrophic Lateral Sclerosis. Front Mol Neurosci. (2022) ;15: :691534. doi: 10.3389/fnmol.2022.691534. |
[43] | Gurfinkel Y , Polain N , Sonar K , Nice P , Mancera RL , Rea SL . Functional and structural consequences of TBK1 missense variants in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Neurobiol Dis. (2022) ;174: :105859. doi: 10.1016/j.nbd.2022.105859. |
[44] | Scarlino S , Domi T , Pozzi L , Romano A , Pipitone GB , Falzone YM et al. Burden of Rare Variants in ALS and Axonal Hereditary Neuropathy Genes Influence Survival in ALS: Insights from a Next Generation Sequencing Study of an Italian ALS Cohort. Int J Mol Sci. (2020) ;21: (9):3346. doi: 10.3390/ijms21093346. |
[45] | Bennett CL , Dastidar SG , Ling SC , Malik B , Ashe T , Wadhwa M , et al. Senataxin mutations elicit motor neuron degeneration phenotypes and yield TDP-43 mislocalization in ALS4 mice and human patients. Acta Neuropathol. (2018) ;136: (3):425–443. doi: 10.1007/s00401-018-1852-9. |
[46] | Tripolszki K , Török D , Goudenège D , Farkas K , Sulák A , Török N , et al. High-throughput sequencing revealed a novel SETX mutation in a Hungarian patient with amyotrophic lateral sclerosis. Brain Behav. (2017) ;7: (4):e00669. doi: 10.1002/brb3.669. |
[47] | Orlacchio A , Babalini C , Borreca A , Patrono C , Massa R , Basaran S , et al. SPATACSIN mutations cause autosomal recessive juvenile amyotrophic lateral sclerosis. Brain. (2010) ;133: (Pt 2):591–8. doi: 10.1093/brain/awp325. |
[48] | Couthouis J , Raphael AR , Daneshjou R , Gitler AD . Targeted exon capture and sequencing in sporadic amyotrophic lateral sclerosis. PLoS Genet. (1004) ;10: (10):e1004704. doi: 10.1371/journal.pgen.1004704. |
[49] | Kenna KP , McLaughlin RL , Byrne S , Elamin M , Heverin M , Kenny EM , et al. Delineating the genetic heterogeneity of ALS using targeted high-throughput sequencing. J Med Genet. (2013) ;50: (11):776–83. doi: 10.1136/jmedgenet-2013-101795. |
[50] | Meyer T , Schwan A , Dullinger JS , Brocke J , Hoffmann KT , Nolte CH , et al. Early-onset ALS with long-term survival associated with spastin gene mutation. Neurology. (2005) ;65: (1):141–3. doi: 10.1212/01.wnl.0000167130.31618.0a. |
[51] | Münch C , Rolfs A , Meyer T . Heterozygous S44L missense change of the spastin gene in amyotrophic lateral sclerosis. Amyotroph Lateral Scler. (2008) ;9: (4):251–3. doi: 10.1080/17482960801900172. |


