
Characterization of Clinical Phenotypes in Congenital Myasthenic Syndrome Associated with the c.1327delG Frameshift Mutation in CHRNE Encoding the Acetylcholine Receptor Epsilon Subunit
Abstract
Background:
Congenital myasthenic syndromes (CMS) are a group of rare but often treatable inherited disorders of neuromuscular transmission characterized by fatigable skeletal muscle weakness. In this paper we present the largest phenotypic analysis to date of a cohort of patients carrying the pathogenic variant c.1327delG in the CHRNE gene, leading to CHRNE-CMS.
Objective:
This study aims to identify the phenotypic variability in CMS associated with c.1327delG mutation in the CHRNE gene.
Methods:
Disease specific symptoms were assessed using specific standardized tests for autoimmune myasthenia (Quantitative Myasthenia Gravis score) as well as patient-reported scales for symptom severity. Evaluated clinical manifestations included ocular symptoms (ophthalmoparesis and ptosis), bulbar weakness, axial muscle weakness, proximal and distal muscle weakness, and respiratory function. Patients were allocated into three groups according to clinical impression of disease severity: mild, moderate, and severe.
Results:
We studied 91 Bulgarian Roma patients, carrying the same causative homozygous CHRNE c.1327delG mutation. Bulbar weakness was present in patients throughout all levels of severity of CHRNE-CMS in this study. However, difficulties in eating and swallowing are more prominent characteristics in the moderate and severe clinical phenotypes. Diplopia and ptosis resulting from fatigue of the extraocular muscles were permanent features regardless of disease severity or age. Levels of axial, proximal and distal muscle weakness were variable between disease groups. The statistical analysis showed significant differences between the patients in the three groups, emphasizing a possible variation in symptom manifestation in the evaluated patient population despite the disease originating from the same genetic mutation. Impairment of respiratory function was more prominent in severely affected patients, which might result from loss of compensatory muscle function in those individuals.
Conclusion:
Results from our study indicate significant phenotypic heterogeneity leading to mild, moderate, or severe clinical manifestation in CHRNE-CMS, despite the genotypic homogeneity.
ABBREVIATIONS
CHRNE | Cholinergic receptor nicotinic epsilon subunit |
CMS | Congenital myasthenic syndromes |
NMJ | Neuromuscular junction |
QMG | The Quantitative myasthenia gravis greaterthan grading scale |
FVC | Forced vital capacity |
INTRODUCTION
Congenital myasthenic syndromes (CMS) are a group of hereditary, genetically heterogeneous, often treatable disorders of the neuromuscular junction (NMJ), characterized by fatigable skeletal weakness (e.g. ocular, bulbar, limb muscles). Disease onset is either at or shortly after birth, or in early childhood [1] and patients may show high variability in severity and course of the disease, ranging from minor symptoms to progressive disabling weakness. CMS is considered a rare disease. In different studies prevalence has been reported to be between 9.2 and 22.2 cases per million children [2, 3]. In the advent of modern molecular genetic diagnostics and next generation sequencing, the number of genetic mutations associated with a CMS phenotype has increased dramatically [4], with over 30 genes known to be monogenic causes of different forms of CMS, featuring different pathophysiological mechanisms [5]. The resulting considerable variations in disease severity, course of disease, phenotypic specific manifestations and treatment responses prompt the need for comprehensive classifications, e.g. a recent classification attempt has been provided by Thompson et al. (2018) [6, 7], employing a FAIR data approach [8].
We studied a genetically homogeneous group of CHRNE-CMS patients of Bulgarian Roma origin with the same causative homozygous CHRNE c.1327delG mutation [9]. For clarification, we will refer in this manuscript to the mutation according to the HGVS nomenclature in contrast to the legacy nomenclature, where the mutation is referred to as ɛ1267delG. Amino acid numbers in the legacy nomenclature correspond to those in the crystal structure, whereas HGVS nomenclature includes the nucleotides of the signal peptide [10]. To convert the legacy nomenclature to the HGVS nomenclature 20 codons are added for the AChR ɛ subunit [11]. The biallelic c.1327delG variant in the CHRNE gene is expected to result in the absence of a functional AChR epsilon subunit protein and overall AChR deficiency at the neuromuscular junction. However, there is partial compensation through upregulation of the fetal AChR gamma subunit (CHRNG) expression [12, 13].
The first Bulgarian family with CHRNE-CMS was discovered in Rakitovo in Bulgaria by Tournev et al. [14, 15]. Subsequently, Middleton et al. examined several families of gypsy ethnic origin with CMS from Greece and Turkey and identified the common homozygous frameshift mutation c.1327delG in exon 12 of the CHRNE gene encoding the AChR ɛ subunit in these patients [9, 16]. The same mutation was found in 11 families in Hungary, Serbia, Kosovo, Macedonia, and Turkey [17]. Haplotype analysis shows sharing of mutations, and high carrier rates supported a strong founder effect [18]. Recent demographic analysis found CHRNE mutations to be the most prevalent genetic cause for CMS in the Austrian population [19], with the majority of these patients (9/13) carrying the homozygous or compound heterozygous CHRNE c.1327delG variant. Moreover, CHRNE was the most common type of CMS in a Spanish study including 64 patients with a confirmed genetic diagnosis of CMS [20], with four different CHRNE mutations (c.1327delG, c.1353insG, c.130insG, c.865 C > T) accounting for 26.6% of included patients. Remarkably, the authors of this study are the first to employ the Quantitative Myasthenia Gravis score (QMG score), a standardized disease severity measurement in autoimmune Myasthenia gravis, to assess the fatiguability of different muscle groups in CHRNE patients. Clinical features observed in these patients are fluctuating ptosis, poor cry, weak suck and feeding difficulties at birth or in early childhood. Later on patients present with myasthenia, ptosis and ophthalmoparesis, with occasional episodic exacerbations possibly resulting in respiratory distress and apnea [9]. Nevertheless, vast differences in disease severity despite the genetic homogeneity of this population have been reported [9]. Understanding disease severity and prognosis is crucial in prediction of disease outcomes as well as administration of personalized treatment. The aim of this study is to provide a phenotypic characterization and evaluation of the variable clinical course in this large cohort or genetically homogeneous patient population with the c.1327delG mutation in the CHRNE gene. A sound phenotypic analysis can be used as a baseline for further research, aiming to understand factors resulting in phenotypic diversity, such as possible additional genetic disease modifiers.
MATERIALS AND METHODS
Ninety-one (48 males and 43 females) patients homozygous for the CHRNE c.1327delG mutation were included in the study.
Clinical assessment scales
For determination of disease severity, the following features were assessed: eating and swallowing difficulties (bulbar weakness), respiratory function, ophthalmoparesis as well as weakness of the axial and limb muscles. For assessment of bulbar weakness, a patient-reported scale evaluating tolerated food textures, ability to consume a complete meal as well as swallowing difficulties was used. The scale applied in our study was originally presented as a modified QMG score for assessment of adults and children by Chaouch et al. 2012 and later used by Della Marina et al. 2020 [21, 22] and the present study represents its most comprehensive use in CHRNE-CMS. The maximum reachable scale value is 9 points, indicating that a patient has minimal oral intake and is unable to consume a whole meal as well as shows difficulties with swallowing of saliva. For assessment of children, the same scales for eating and swallowing were used but answers were given by the parents. Single values of the scale can be found in the supplementary data. Additionally, standardized scales for the use in autoimmune Myasthenia gravis were used for symptom quantification. The QMG score was used to assess the clinical severity of fatigable muscle weakness. The QMG score includes several tests focusing on exercise dependent muscle weakness in different body parts: bulbar muscles, extraocular muscles, axial muscles, proximal and distal muscles of upper limbs, including grip strength testing using myometry (measurement obtained in kilograms), proximal muscle groups of lower limbs and respiratory muscles (testing of forced vital capacity (FVC)). For the purposes of the study, we used the well-known QMG score initially developed by Besinger and later modified by Barohn [23]. Muscle weakness was further evaluated by performance of additional exercises. For assessment of lower limb weakness, the maximal number of squat repetitions in 1 minute and the number of times the patient is able to stand up from a seated position (sit-to-stand test) in 1 minute was counted. Additionally, the time the patient needed to rise from the floor prior to and after the described muscle strength tests was collected. The sit-to-stand test was also presented in the modified QMG score by Chaouch et al. 2012 and later used by Della Marina et al. 2020 [21, 22].
Classification of the patients in three disease severity groups was performed by considering the severity of clinical symptoms assessed by the described examinations and scales. Patients with a mild clinical course might suffer from eyelid ptosis and ophthalmoparesis, mild bulbar weakness and/or mild weakness in the proximal muscles of the limbs. A moderate clinical course is characterized by the symptoms of the mild group and additional weakness in the proximal muscles of the limbs whereas patients with a severe clinical course experience eyelid ptosis, ophthalmoparesis, bulbar weakness as well as advanced weakness in the proximal muscles of the limbs, leading to gait disturbances or loss of ambulation.
Ethical approval was obtained prior to genetic testing and examination of the patients.
Statistical analysis
IBM SPSS statistics 21.0 was used to perform the statistical analysis. Mann-Whitney U test, one way ANOVA and Kruskal-Wallis non-parametric test were used for analysis. A p-value of ≤0.05 is considered statistically significant. Patients unable to perform a given task were excluded from the statistical analysis.
RESULTS
Baseline criteria
Ninety-one patients were included in the study, 48 males (age at assessment between 1 year and 11 months and 64 years, mean age was 26.4, SD±15.5), and 43 females (age at assessment between 2 years and 62 years, mean age was 25.8, SD±15.4). Overall, eighteen patients were unable to perform all tests, either due to young age or disability. Patients were divided into three groups by the clinical impression of disease severity: mild, moderate, and severe. Baseline characteristics of the patient population sorted into disease severity groups can be found in Table 1. Bulbar weakness, eyelid ptosis, diplopia and mild proximal muscle weakness were present in all three groups regardless of patients’ age or stage of severity. Proximal and axial muscle weakness appear to be variable symptoms between the groups, as in the first group they are only present in half of the patients whereas in the second group the number of affected patients increased and in the third group half of the patients were unable to perform all of the tasks. In 100% of the cases the diagnosis was genetically confirmed, and all patients carried the c.1327delG mutation in exon 12 in the CHRNE gene encoding the ɛAChR subunit.
Table 1
Baseline characteristics for the different disease severity groups
| Disease Severity | Number of patients (N) | Male (N, [% of total number of males for each disease severity group]) | Female (N, [% of total number of females for each disease severity group]) | Age (mean±SD), median |
| Mild | 44 | 24 [50.0] | 20 [46.5] | 23.7 (±12.3), 26 |
| Moderate | 26 | 16 [33.3] | 10 [23.3] | 20.2 (±14.2), 25.5 |
| Severe | 21 | 8 [16.7] | 13 [30.2] | 38.8 (±16.7), 27 |
| Total | 91 | 48 [100] | 43 [100] | 26.2 (±15.5) |
Twenty-one patients in the first group, 17 in the second group and 13 in the third group received anticholinesterase treatment (pyridostigmine bromide) orally in daily doses between 30 mg and 720 mg. Other therapeutic options were limited as 3,4-diaminopyridine is not available in Bulgaria and off-label prescription of salbutamol is not possible in our country. Two patients in the severely affected group had a history of poliomyelitis and one patient was affected by asthma.
Bulbar weakness
As described above, a patient-reported scale with a maximum sum score of 9 points was used for assessment of eating and swallowing difficulties (bulbar weakness). In the group of patients with a mild clinical presentation, the mean score on this scale was 1.3 points, with individual values between 0 and 5 points. In patients with a moderate clinical course of the disease the maximum scored value individually was 6 points, with a mean score of 2.0 points in this group. In severely affected patients, the group’s mean score was 2.3 points, with individual values reaching from 0 to 7 points.
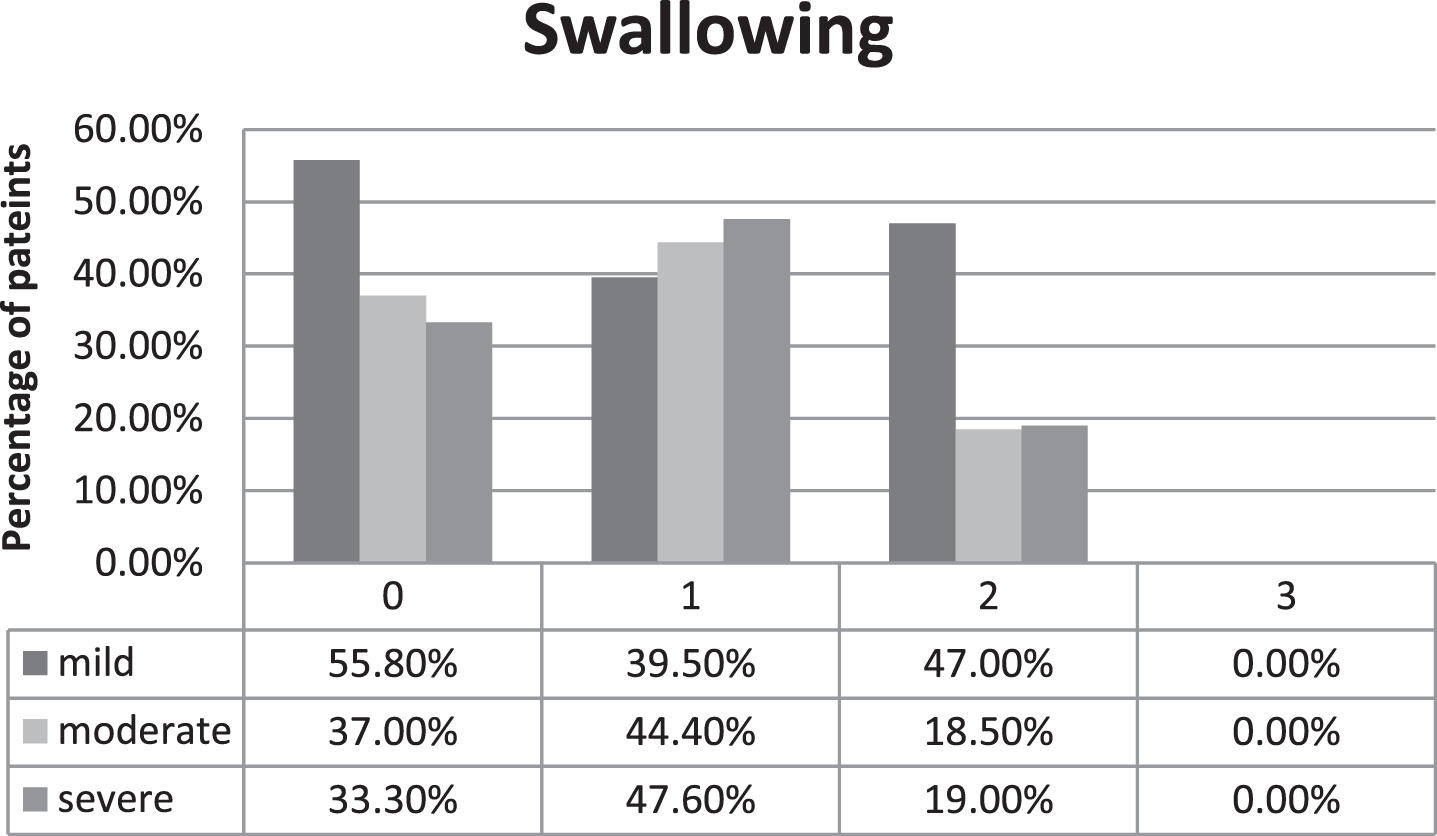
As one item of the described scale, frequency of swallowing difficulties is assessed (scale values 0 to 3 points). Results of this item are presented in Fig. 1, showing the severity of experienced swallowing difficulties for the patients in each group. Most patients with a mild clinical course report no difficulties in swallowing or only occasional choking on certain types of food (<1 per month), equaling 0 points. Only 4.7% of the patients with a mild phenotype report regular troubles swallowing food or drinks (>1 per month, equals 1 point). 18.5% and 19% of patients with a moderate or severe clinical course respectively reported severe swallowing difficulties with regular choking on food/drinks (2 points). None of the patients reported severe difficulties in swallowing like choking on their own saliva or secretions. Kruskal-Wallis non-parametric test did not detect statistically significant difference regarding severity of bulbar weakness between the different groups (p = 0.53). Mann-Whitney U test did not show significant difference regarding the treatment (p = 0.852).
Fig. 1
Severity of subjectively experienced swallowing difficulties in the different disease severity groups, 0 points equaling no swallowing difficulties, 3 points equaling difficulties with swallowing saliva, percentages are calculated in relation to absolute number of patients in each disease severity group.
Additionally, bulbar weakness was evaluated with a slurp test, included in the QMG assessment, measuring the time it takes the patient to drink 120 ml water. The mean time for completing this test in the mildly affected group was 11.4 sec (SD±7.5 sec), in the moderate disease group 13.1 sec (SD±5.5 sec) and 15.1 sec (SD±9.1 sec) in patients with a severe disease course. Statistical analysis using Kruskal-Wallis test showed a significant difference between the three groups (p = 0.004). Paired comparison of the groups also showed significant differences between the first and the second group (p = 0.019) and the first and the third group (0.019), but not between the second and the third group (p = 1.0).
Ocular symptoms
Diplopia and ptosis were examined through measurement of the time until appearance of diplopia and ptosis after prolonged lateral and upward gaze, respectively. Points were awarded depending on the length of time (3 points equaling spontaneous symptoms, 0 points equaling no symptoms), summed for each group and divided by the number of patients in the test group, calculating a severity score of ocular symptoms.
The mean severity level of diplopia in the group with mild clinical course was calculated at 0.06 (SD±0.38), at 0.07 (SD±0.26) in the group with moderate clinical course and at 0.19 (SD±0.39) in the group with severe clinical course. The mean severity level of ptosis was found to be 0.58 (SD±0.53) for the first group, 0.69 (SD±0.46) for the second group and 0.85 (±0.46) for the third group. Statistical analysis did not show a significant difference between the groups for diplopia (p = 0.308) and for ptosis (p = 0.532).
Axial weakness
For assessment of axial weakness, the maximum time the patients were able to lift their head at 45° whilst lying on their back was measured (up to 2 minutes). The mean time for keeping this position in the first group was 95 sec (SD±33.4), in the second group 93.2 sec (SD±38.3) and in the third group 56.8 sec (SD±38.0). Fifty-two percent of the patients in the first as well as the second group and 19% of the severely affected group could keep the position for at least 2 min., therefore for these patients a maximum time of 120 sec was used for calculation. According to the values of the QMG score associated with the measured times (0 to 3 points), the mean severity score in the mild and moderate disease groups was 0.5 respectively and increased to 1.1 in the severe disease phenotype group. Kruskal-Wallis non-parametric test showed this difference between the groups to be significant (p = 0.025).
Limb weakness
To assess proximal limb weakness, included in the QMG assessment, patients were asked to outstretch each arm at a 90° degree angle while sitting and keep this position for as long as possible, maximum time was set at 4 minutes. In all the observed patients, the maximum arm holding time decreased with increasing phenotypic severity. The mean time patients with a mild phenotype were able to keep the described position was 180.4 sec (SD±71.9), with a moderate phenotype 169.3 sec (SD±88) and 48.7 sec (SD±54.8) in severely affected patients. Fifty percent of the patients with mild clinical course showed normal results reaching the maximum time measured. In the group with moderate clinical course 48% of patients were able to keep the position for at least 4 min. In the third group only 4.7% of patients could keep their arm outstretched for at least 4 min. For the statistical analysis, the maximum arm holding time for all patients reaching the maximum measured time, independent from disease severity groups, was calculated at 240 seconds. Kruskal-Wallis non-parametric test shows a significant difference between the mean times in each group (p < 0.0001).
For evaluation of the lower extremities, the maximum leg holding time at a 45° degree angle in a supine position was tested. Corresponding to the findings in the arm holding test, maximum time for leg holding also decreased with severity of clinical presentation. The mean time in the first group was 74.9 sec (SD±30.9), in the second group 76.7 sec (SD±31.5), and in the third group 37.6 sec (SD±31.3); Kruskal-Wallis non-parametric test revealed these changes to be significant (p < 0.0001).
Another assessment that was taken into consideration was the maximum number of squats the patients were able to perform in 1 min. While the patients in the first group were able to squat 22 times/min (SD±9.3) on average, moderately affected patients managed 18 squats/min (SD±7.9) and the patients with a clinically severe condition squatted on average 9 times/min (SD±5.2). Nevertheless, 50% of the severely affected patients were unable to perform the task at all. They were excluded from the statistical analysis. The differences between groups were statistically significant (p = 0.002). Moreover, the mean number of times patients were able to stand up from a sitting position in 1 minute was counted (“sit to stand”). The mean number of “sit to stand” actions was 28/minute (SD±9.7), 25/minute (SD±12.8) and 14/minute (SD±7.9) in groups one, two and three respectively. For 50% of the patients in the third group this task was impossible to perform. Statistical analysis showed a significant difference between the groups (p = 0.034). One way ANOVA test was used for these two analyses.
Proximal weakness in the lower limbs was further assessed by the time taken by the patient to rise from the floor twice, as described in the methods section. In the mildly affected group, the mean time needed to rise from the floor was 5.12 sec (SD±3.3) for the first attempt and 5.1 sec (SD±3.1) for the second attempt. The mean time for the first attempt in the second group was 5.4 sec (SD±2.7) and 6.1 sec (SD±3.1) for the second attempt. In the severely affected group the mean time for completing this test was 9.2 sec (SD±7.1) on the first attempt and 10.6 sec (SD±7.1) on the second attempt. Two patients (7.6%) with moderate clinical course of the disease and 5 patients (23.8%) with severe clinical course were unable to perform this test, due to the muscle weakness. These patients were excluded from the statistical analysis. Overall, time needed to complete the exercise increased with severity of disease. Kruskal-Wallis non-parametric test showed a significant difference in the time for the first (p = 0.031) and for the second attempt (p < 0.0001) between the groups.
Distal muscle weakness in upper limbs was examined through measurement of hand gripping strength of the dominant hand. Results showed mean muscle strength in the mild group was 17.7 kg (SD±22.1), 8.9 kg (SD±6.8) in the moderate group and 5.1 kg (SD±22.1) in the severe group. Median values were 9.4 kg, 6.7 kg, 5.7 kg, respectively. Statistical analysis of myometry results proved a significant difference between the groups (p = 0.021).
Respiratory function
For assessment of respiratory function, spirometry was performed in a sitting as well as a supine position. In the sitting position, results for forced vital capacity (FVC) were measured between 36% and 151%, the mean FVC was calculated at 77.92% (SD±21.15). When spirometry was performed in a supine position, FVC values between 15% and 130% were achieved, with a FVC mean of 71.95% (SD±19.7). Table 2 presents the FVC values in the different disease severity groups. Statistical analysis shows a significant difference in the results for FVC between the groups for both sitting (p1) and supine (p2) positions (p1 < 0.001, p2 < 0.001). Additional pairwise comparison between the groups shows a significant difference between the first and third group (p1 < 0.001, p2 < 0.001) and the second and third group (p1 = 0.005, p2 = 0.003). If FVC values between examinations in a sitting and a supine position are compared, 14 patients showed more than a 10% difference in FVC, with reduced values in the supine position. Nine (20%) of these 14 patients had a mild, 3 (11%) a moderate and 2 (9%) a severe clinical course of the disease. Only one of the 91 patients had a temporary tracheostomy (duration for 1 month) associated with a respiratory crisis during pneumonia. This patient presented with a severe clinical phenotype. None of the other patients was dependent on non-invasive or invasive ventilation or has had a respiratory crisis prior to conduction of this study.
Table 2
FVC mean values in the different clinical groups
| Clinical course | FVC mean (sitting position) | FVC mean (supine) |
| Mild | 85.40% | 77.50% |
| Moderate | 78% | 75.40% |
| Severe | 62.80% | 59.50% |
DISCUSSION
In this study, we examined 91 patients carrying the c.1327delG variant in the CHRNE gene in order to evaluate the levels of diversity in their clinical course. Disease specific symptoms were assessed using specific clinical tests as well as patient reported scales for symptoms severity. Clinical manifestations evaluated included ocular symptoms (ophthalmoparesis and ptosis), bulbar weakness, axial muscle weakness, proximal and distal muscles weakness, and respiratory function. Our results show a significant clinical variability regarding severity of symptoms in this genetically homogeneous population, classifying affected individuals into three different phenotypic groups according to disease severity. This finding is in line with previous comparable studies, for example in a population with 84 CHRNE patients [24], variability in the severity of the disease manifestation has been shown.
In our study, the 91 patients were allocated into three groups depending on the clinical impression of severity of their symptoms. Bulbar weakness was present in patients throughout all levels of severity of CHRNE-CMS in this study. Although no statistically significant differences in the results for “food texture, eating a meal and swallowing” scales were detected, the score in this scale for bulbar weakness increased from mild to severe clinical manifestation. It must be noted that the results from these scales are subjective as they rely on the answers of the patients. Therefore, comparable severity of symptoms can be judged differently depending on the individual. Nevertheless, a significant increase in symptom severity was observable in the additionally performed slurp test, which provides an objective measure for analysis of differences in bulbar weakness between the groups. Overall, bulbar weakness may be detected in patients with all levels of disease severity, but the manifestation of symptoms and the daily life impact can vary. On average, increased difficulties in eating and swallowing are characteristic for a moderate or severe clinical phenotype of CMS.
Regarding the severity of diplopia and ptosis, no significant difference between the groups was detectable. A probable reason for this finding could be a lack of variability in these symptoms of the disease, as has been described in previous genotype-phenotype correlations [9]. Lack of diplopia can be explained due to the presence of external ophthalmoplegia since infancy, enabling compensation for the altered visual axis [5]. Our results suggest that the ocular symptoms resulting from fatigue of the extraocular muscles are permanent features of CHRNE-CMS, that can appear independently from disease severity at all ages. Ptosis and ophthalmoparesis do not improve after puberty [12], underlining their role as a prominent clinical disease marker.
Levels of axial, proximal and distal muscle weakness were variable between disease groups. Overall, the ability to perform the required tasks varied between patients, from being able to complete the test within normal time limits to full inability to perform the task at all. Especially in patients with a severe clinical phenotype performance of some muscle strength tests was impossible. The statistical analysis showed significant differences between the patients in the three groups, emphasizing a possible variation in symptom manifestation in the evaluated patient population despite the disease originating from the same genetic mutation.
CHRNE-CMS affects the respiratory muscles including the intercostal muscles and the diaphragm. Performance of spirometry in a sitting and a supine position allowed indirect assessment of the strength of these muscles. The statistical analysis revealed a significant difference for FVC between the groups in both sitting and supine positions. The results between FVC measured in a seated position compared to FVC in a supine position varied more than 10% of FVC in 14 individuals, probably due to weakness of the diaphragm [25]. This observation was made in patients throughout all three clinical groups, indicating possible diaphragmatic weakness at all described clinical phenotypes. Pronounced weakness of diaphragm has been reported in other CMS variants as well [26]. Nevertheless, when comparing FVC across the groups, impairment of respiratory function was more prominent in severely affected patients, which might result from loss of compensatory muscle function in these individuals. Despite the wide variability observed in the clinical course of CHRNE-CMS and the detectable impairment of respiratory function, respiratory insufficiency is rare, unlike in the CMS caused by a mutation in DOK7 or CHAT genes [27]. Although none of the patients in our study used permanent mechanical ventilation and only one patient had developed respiratory failure as a complication during pneumonia in the past, monitoring of respiratory function should not be underestimated. Awareness is especially required, since previous studies suggested that respiratory failure predominantly appears as a sudden apneic crisis rather than with a gradual development [28], emphasizing that patients with severe weakness in particular are at risk of FVC reduction and possible complications in case of diaphragmatic weakness.
Interestingly, in our study, patients with a more severe disease phenotype on average were older than patients with a mild or moderate clinical course. However, this finding does not necessarily suggest an increase of symptom burden with higher age, due to the median ages in the three groups being similar. In CHRNE associated CMS long-term follow-up studies even observed improvement of neonatal symptoms such as bulbar weakness and intermittent worsening over the disease course, if patients were adequately treated [22, 29]. Nevertheless, despite the regularly observed improvement under therapy, cases with insufficient treatment responses have been reported [30–32]. Our observations indicate that the severely affected patients in our cohort respond poorly to treatment. Thus, we believe that severity is rather a phenotypic characteristic than a result of treatment or age. It must be noted that other factors can influence the clinical phenotype appearance with increasing age, such as comorbidities leading to reduced mobility, difficulties with swallowing or respiratory weakness. Moreover, delayed diagnosis associated with delayed implementation of sufficient treatment could result in increased severity of phenotypes in patients of older age.
Although we were able to gain an in-depth understanding of different phenotypes in this cohort of CMS patients with CHRNE c.1327delG, our study has several limitations related to the study design with a one-time-point evaluation as well as due to the inherently small cohort sign, limited by the prevalence of this disease. Confounding factors, related to comorbidities, differences in treatment plans or age, influencing the clinical presentation and test performance cannot be eliminated. Assessment of individual disease progression with intraindividual comparison at different time-points would be of interest, to evaluate further how the clinical appearance progresses over time to provide deeper insights into prediction of clinical course. Inclusion of patients with more balanced baseline characteristics is not possible, due to the small number of patients affected by this specific mutation, since especially the phenotypic evaluation in a genetically homogeneous cohort is the key focus point of this study. Nevertheless, all patients included in this study were evaluated at the same clinical center by the same physicians, providing strong comparability between individuals, due to standardized procedure and stable levels of clinical experience. Therefore, our study results provide, to our knowledge, the largest phenotypic cohort analysis to date for CHRNE c.1327delG associated CMS. Therefore, this data can serve as a baseline for further studies, evaluating factors influencing clinic presentation additional to the causing gene mutation in this homogeneous cohort.
CONCLUSION
CMS caused by a homozygous frameshift mutation c.1327delG in CHRNE is a rare condition, predominantly described among the European, Indian, and Pakistani populations. The clinical course of the disease is generally benign in comparison with some other CMS subtypes, but variability in the clinical course and phenotype is possible. Results from our study indicate significant phenotypic heterogeneity leading to mild, moderate, or severe clinical manifestation, despite the genotypic homogeneity. While ocular symptoms appear consistently throughout all severity levels of the described phenotypes, axial, proximal and distal muscle weakness are more pronounced in patients with moderate and severe disease course. Respiratory function is generally preserved but weakness of the diaphragm can be present at any level of clinical severity, underlining the relevance of monitoring of respiratory function. Diverging clinical phenotypes despite genetic homogeneity suggest additional, yet unknown factors influencing disease severity and clinical course in this patient population. Therefore, further exploration of these assumed factors, especially regarding the existence of modifying genes is needed to further facilitate diagnosis, genetic counselling, and treatment. The careful phenotypic workup in this large patient cohort can present a baseline for future studies evaluating these factors, and such work is already underway on this cohort through advanced bioinformatic strategies developing multilayer networks to elucidate relationships between severity and potential modifiers found in omics data [33].
ACKNOWLEDGMENTS
We are very grateful to the patients who participated with interest in this study. SM is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) within the Clinician Scientist Program “Cell Dynamics in Disease and Therapy” at the University Medical Center Goettingen (project number 413501650). VG was a research fellow of the Alexander von Humboldt Foundation. VM was awarded a BAYHOST Scholarship. HL receives support from the Canadian Institutes of Health Research (CIHR) for Foundation Grant FDN-167281 (Precision Health for Neuromuscular Diseases), Transnational Team Grant ERT-174211 (ProDGNE) and Network Grant OR2-189333 (NMD4 C), from the Canada Foundation for Innovation (CFI-JELF 38412), the Canada Research Chairs program (Canada Research Chair in Neuromuscular Genomics and Health, 950-232279), the European Commission (Grant # 101080249) and the Canada Research Coordinating Committee New Frontiers in Research Fund (NFRFG-2022-00033) for SIMPATHIC, and from the Government of Canada Canada First Research Excellence Fund (CFREF) for the Brain-Heart Interconnectome. RT received a CIHR postdoctoral fellowship award under grant no. MFE-171275.
FUNDING
The authors received no financial support for the research, authorship, and/or publication of this article. This study is financed by the European Union-NextGenerationEU, through the National Recovery and Resilience Plan of the Republic of Bulgaria, project No. BG-RRP-2.004-0004-C01.
CONFLICT OF INTEREST
HL is an Editorial Board Member of this journal, but was not involved in the peer-review process nor had access to any information regarding its peer-review.
AVAILABILITY OF DATA AND MATERIALS
The data that support the findings of this study are available from the corresponding author, [KK], upon reasonable request.
REFERENCES
[1] | Abicht A , Müller JS , Lochmüller H . Congenital Myasthenic Syndromes Overview, 2023 May 9 [updated 2021 Dec 23]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2023. Available from: https://pubmed.ncbi.nlm.nih.gov/20301347/. |
[2] | Parr JR , Andrew MJ , Finnis M , Beeson D , Vincent A , Jayawant S . How common is childhood myasthenia? The UK incidence and prevalence of autoimmune and congenital myasthenia. Arch Dis Child. (2014) ;99: (6):539–42. doi:10.1136/archdischild-2013-304788. |
[3] | Gergeli AT , Neubauer D , Golli T , Butenko T , Loboda T , Maver A , et al. Prevalence and genetic subtypes of congenital myasthenic syndromes in the pediatric population of Slovenia. Euro J Paediatr Neurol. (2020) ;1: (26):34–8. doi:10.1016/j.ejpn.2020.02.002. |
[4] | Rodríguez Cruz PM , Palace J , Beeson D . The Neuromuscular Junction and Wide Heterogeneity of Congenital Myasthenic Syndromes. Int J Mol Sci. (2018) ;19: (6):1677. doi:10.3390/ijms19061677. |
[5] | Ohno K , Ohkawara B , Shen XM , Selcen D , Engel AG . Clinical and Pathologic Features of Congenital Myasthenic Syndromes Caused by 35 Genes-A Comprehensive Review. Int J Mol Sci. (2023) ;24: (4):3730. doi:10.3390/ijms24043730. |
[6] | Thompson R , Abicht A , Beeson D , Engel AG , Eymard B , Maxime E , et al. A nomenclature and classification for the congenital myasthenic syndromes: preparing for FAIR data in the genomic era. Orphanet J Rare Dis. (2018) ;13: (1):211. doi:10.1186/s13023-018-0955-7. |
[7] | Spendiff S , Dong Y , Maggi L , Rodríguez Cruz PM , Beeson D , et al. ENMC 260th workshop study grou260th ENMC International Workshop: Congenital myasthenic syndromes 11–13 March Hoofddorp, The Netherlands. Neuromuscul Disord. (2023) ;33: (9):111–118. doi:10.1016/j.nmd.2022.12.006. |
[8] | Wilkinson MD , Dumontier M , Aalbersberg IJ , Appleton G , Axton M , Baak A , et al. The FAIR Guiding Principles for scientific data management and stewardshiSci Data. 18. (2016) ;3: :160018. doi:10.1038/sdata.2016.18. |
[9] | Abicht A , Stucka R , Karcagi V , Herczegfalvi A , Horváth R , Mortier W , et al. A common mutation (epsilondelG) in congenital myasthenic patients of Gypsy ethnic origin. Neurology. (1999) ;53: (7):1564–9. doi:10.1212/wnl.53.7.1564. |
[10] | Shen XM , Milone M , Wang HL , Banwell B , Selcen D , Sine SM , et al. Slow-channel myasthenia due to novel mutation in M2 domain of AChR delta subunit. Ann Clin Transl Neurol. (2019) ;6: (10):2066–78. doi:10.1002/acn3.50902. |
[11] | Shen XM , Okuno T , Milone M , Otsuka K , Takahashi K , Komaki H , et al. Mutations Causing Slow-Channel Myasthenia Reveal That a Valine Ring in the Channel Pore of Muscle AChR is Optimized for Stabilizing Channel Gating. Hum Mutat. (2016) ;37: (10):1051–9. doi:10.1002/humu.23043. |
[12] | Witzemann V , Schwarz H , Koenen M , Berberich C , Villarroel A , Wernig A , et al. Acetylcholine receptor ɛ-subunit deletion causes muscle weakness and atrophy in juvenile and adult mice. PNAS USA. (1996) ;93: (23):13286–91. doi:10.1073/pnas.93.23.13286. |
[13] | Cetin H , Beeson D , Vincent A , Webster R . The Structure, Function, and Physiology of the Fetal and Adult Acetylcholine Receptor in Muscle. Front Mol Neurosci. (2020) ;13: :581097. doi:10.3389/fnmol.2020.581097. |
[14] | Guergueltcheva V , Litvinenko I , Cherninkova S , Bojinova V , Mihailova V , Ishpekova B , et al. Congenital Myasthenic Syndrome Type Ia (Familial Infantile Myasthenia) in Bulgarian Roma. Bulgarska nevrologia. (2007) ;7: (5):242–245. |
[15] | Karcagi V , Tournev I , Schmidt C , Herczegfalvi A , Guergueltcheva V , Litvinenko I , et al. Congenital Myasthenic Syndrome in southeastern European Roma (Gypsies). Acta Myol. (2001) ;20: (3):231–237. |
[16] | Middleton LT , Christodoulou K , Deymeer F , Serdaroglu P , Ozdemir C , al-Qudah AK , et al. Congenital myasthenic syndrome. (CMS) type Ia. Clinical and genetic diversity. Ann N Y Acad Sci. (1998) ;841: :157–66. doi:10.1111/j.1749-6632.1998.tb10922.x. |
[17] | Croxen R , Beeson D , Newland C , Betty M , Vincent A , Newsom-Davis J . A single nucleotide deletion in the epsilon subunit of the acetylcholine receptor (AChR) in five congenital myasthenic syndrome patients with AChR deficiency. Ann N Y Acad Sci. (1998) ;841: :195–8. doi:10.1111/j.1749-6632.1998.tb10927.x. |
[18] | Morar B , Gresham D , Angelicheva D , Tournev I , Gooding R , Guergueltcheva V , et al. Mutation history of the roma/gypsies. Am J Hum Genet. (2004) ;75: (4):596–609. doi:10.1086/424759. |
[19] | Krenn M , Sener M , Rath J , Zulehner G , Keritam O , Wagner M , et al. The clinical and molecular landscape of congenital myasthenic syndromes in Austria: a nationwide study. J Neurol. (2023) ;270: (2):909–16. doi:10.1007/s00415-022-11440-0. |
[20] | Natera-de Benito D , Töpf A , Vilchez JJ , González-Quereda L , Domínguez-Carral J , Díaz-Manera J . et al. Molecular characterization of congenital myasthenic syndromes in Spain. Neuromuscul Dis. (2017) ;27: (12):1087–98. doi:10.1016/j.nmd.2017.08.003. |
[21] | Chaouch A , Beeson D , Hantaï D , Lochmüller H . 186th ENMC international workshop: congenital myasthenic syndromes 24–26 June Naarden, The Netherlands. Neuromuscul Disord. (2012) ;22: :566–76. doi:10.1016/j.nmd.2011.12.004. |
[22] | Della Marina A , Wibbeler E , Abicht A , Kölbel H , Lochmüller H , Roos A , et al. Long Term Follow-Up on Pediatric Cases With Congenital Myasthenic Syndromes-A Retrospective Single Centre Cohort Study. Front Hum Neurosci. (2020) ;14: :560860. doi:10.3389/fnhum.2020.560860. |
[23] | Barohn RJ , McIntire DD , Herbelin L , Wolfe GI , Nations SP , Bryan WW . Reliability Testing of the Quantitative Myasthenia Gravis Scorea. Ann N Y Acad Sci. (1998) ;841: (1 MYASTHENIA GR):1769–772. doi:10.1111/j.1749-6632.1998.tb11015.x. |
[24] | Burke G , Cossins J , Maxwell S , Robb S , Nicolle M , Vincent A , et al. Distinct phenotypes of congenital acetylcholine receptor deficiency. Neuromuscul Disord. (2004) ;14: (6):356–64. doi:10.1016/j.nmd.2004.03.005. |
[25] | Lechtzin N , Wiener CM , Shade DM , Clawson L , Diette GB . Spirometry in the supine position improves the detection of diaphragmatic weakness in patients with amyotrophic lateral sclerosis. Chest. (2002) ;121: (2):436–42. doi:10.1378/chest.121.2.436. |
[26] | Kramer JJ , Boon HTM , Leijten QH , Ter Laak H , Eshuis L , Kusters B , et al. Dystrophic Myopathy of the Diaphragm with Recurrent Severe Respiratory Failure is Congenital Myasthenic Syndrome 11. J Neuromuscul Dis. (2023) ;10: (2):271–7. doi:10.3233/JND-221542. |
[27] | Ohno K , Tsujino A , Brengman JM , Harper CM , Bajzer Z , Udd B , et al. Choline acetyltransferase mutations cause myasthenic syndrome associated with episodic apnea in humans. Proc Natl Acad Sci U S A. (2001) ;98: (4):2017–22. doi:10.1073/pnas.98.4.2017. |
[28] | Robb SA , Muntoni F , Simonds AK . Respiratory management of congenital myasthenic syndromes in childhood: Workshop 8th December UCL Institute of Neurology, London, UK. Neuromuscul Disord. (2010) ;20: (12):833–8. doi:10.1016/j.nmd.2010.08.002. |
[29] | McMacken G , Whittaker RG , Evangelista T , Abicht A , Dusl M , Lochmüller H . Congenital myasthenic syndrome with episodic apnoea: clinical, neurophysiological and genetic features in the long-term follow-up of 19 patients. J Neurol. (2018) ;265: (1):194–203. doi:10.1007/s00415-017-8689-3. |
[30] | Durmus H , Shen XM , Serdaroglu-Oflazer P , Kara B , Parman-Gulsen Y , Ozdemir C , et al. Congenital myasthenic syndromes in Turkey: Clinical clues and prognosis with long term follow-up. Neuromuscul Disord. (2018) ;28: (4):315–22. doi:10.1016/j.nmd.2017.11.013. Epub 2017 Nov 28. Erratum in: Neuromuscul Disord. 2018;28(10):896. |
[31] | Kao JC , Milone M , Selcen D , Shen XM , Engel AG , Liewluck T . Congenital myasthenic syndromes in adult neurology clinic: A long road to diagnosis and therapy. Neurology. (2018) ;91: (19):e1770–7. doi:10.1212/WNL.0000000000006478. |
[32] | Natera-de Benito D , Domínguez-Carral J , Muelas N , Nascimento A , Ortez C , Jaijo T , et al. Phenotypic heterogeneity in two large Roma families with a congenital myasthenic syndrome due to CHRNE delG mutation. A long-term follow-up. Neuromuscul Disord. (2016) ;26: (11):789–95. doi:10.1016/j.nmd.2016.08.005. |
[33] | Nuñez Carpintero I , O’Connor E , Rigau M , Bosio M , Azuma Y , Topf A , et al. Rare disease research workflow using multilayer networks elucidates the molecular determinants of severity in Congenital Myasthenic Syndromes. Nature Communications [In Press] . https://doi.org/10.1038/s41467-024-45099-0 |


