
Improving Diagnostic Precision: Phenotype-Driven Analysis Uncovers a Maternal Mosaicism in an Individual with RYR1-Congenital Myopathy
Abstract
Congenital myopathies (CMs) are rare genetic disorders for which the diagnostic yield does not typically exceed 60% . We performed deep phenotyping, histopathological studies, clinical exome and trio genome sequencing and a phenotype-driven analysis of the genomic data, that led to the molecular diagnosis in a child with CM. We identified a heterozygous variant in RYR1 in the affected child, inherited from her asymptomatic mother. Given the alignment of the clinical and histopathological phenotype with RYR1-CM, we considered the potential existence of a missing second variant in trans in the proband, but also hypothesized that the variant might be mosaic in the mother, as subsequently demonstrated. Our study is an example of how heterozygous variants inherited from asymptomatic parents are frequently dismissed. When the genotype-phenotype correlation is strong, it is recommended to consider a parental mosaicism.
INTRODUCTION
Congenital myopathies (CM) are rare genetic disorders that affect skeletal muscle function [1]. The associated variable phenotypes are characterized by genetic heterogeneity [1]. Historically, they have been subclassified based on muscle histopathologic characteristics and specific clinical features [2]. RYR1-related disorders (MIM*180901) account for a significant proportion of CMs, representing 20–30% of cases [3] and are the most common non-dystrophic neuromuscular diseases, with an estimated prevalence of 1:90,000 individuals [4–6]. They are associated to pathogenic variants in RYR1 gene, which encodes the type 1 ryanodine receptor (RyR1) of the skeletal muscle. This is a sarcoplasmic reticulum calcium release channel that also acts as a bridging structure between the sarcoplasmic reticulum and the transverse tubule [4, 7]. Autosomal dominant (AD) inheritance is associated with central core disease (CCD), while autosomal recessive forms are associated with more severe clinical phenotypes: multi-minicore disease (MmD), centronuclear myopathy (CNM), and congenital fiber type disproportion (CFTD) [4, 8].
Individuals with typical clinical and histopathological features of RYR1-related CM often lack a conclusive genetic diagnosis. Despite the use of Next Generation Sequencing (NGS), overall genetic diagnostic rates for CM still rarely exceed 60% [9]. The likely reasons behind this lack of genetic diagnosis are the difficulties in identifying certain types of variants and the challenges of interpreting others. Specifically, the interpretation of variants in RYR1 is challenging due to factors such as a high number of reported VUS, variability within and between families [10], and the known existence of mosaic cases [4].
Mosaicisms have become increasingly recognized as a cause of genetic disorders [11] and they refer to the coexistence of at least two different genetic cell populations within an organism [12]. Detecting mosaicisms poses challenges as they involve a small proportion of cells and exhibit heterogeneous distribution in the different tissues of the organism.
Here, we report the case of an 8-year-old boy with CM and a heterozygous RYR1(NM_000540.3):c.14566G > A(p.Ala4856Thr) variant. The phenotype and the histopathological findings were highly compatible with AD RYR1-related CM. Molecular analyses showed that this causative variant was inherited from his asymptomatic mother, who had somatic mosaicism, explaining her lack of symptoms.
MATERIALS AND METHODS
Clinical examination
The child and his mother underwent clinical examinations at the Neuromuscular Unit of the Hospital Sant Joan de Déu. Blood and muscle samples were collected from the affected child and his parents for diagnostic purposes. Data were collected and analyzed following the ethics guidelines of Hospital Sant Joan de Déu (PIC 98-20). The family provided written informed consent for the study.
Histological analysis
A muscle biopsy specimen was obtained from the index case at 5 years of age. Due to the suspicion of maternal mosaicism, a biopsy was also performed on her at the age of 44. Sections of snap-frozen tissue were processed using hematoxylin & eosin (H&E), modified Gomori trichrome, nicotinamide adenine dinucleotide (NADH), succinate dehydrogenase (SDH), and staining for slow and fast myosin. Ultrathin sections were examined with transmission electron microscopy (JEOL model 1100). Electron micrographs were obtained using the Gatan Orius CCD camera (Olympus Soft Imaging Solutions, Münster, Germany).
Molecular genetic analysis
Clinical exome sequencing (CES) was performed in the affected child using the TruSight One Sequencing Panel (Illumina, San Diego, CA, USA) and sequencing data was processed through an in-house pipeline. Segregation studies were performed on blood-derived DNA by Sanger sequencing.
Trio Genome Sequencing (GS) was performed in the framework of the Solve-RD European Project using the RD-Connect Genome Phenome Analysis Platform (https://platform.rd-connect.eu/#/) [13, 14]. The strategy to prioritize candidate variants considered allele frequency in population databases, functional impact predictions, and trio segregation analysis. Selected variants underwent further pathogenicity assessment through software tools: CADD (https://cadd.gs.washington.edu/), Mutation Taster (https://www.mutationtaster.org/, FATHMM-MKL (http://fathmm.biocompute.org.uk/), PROVEAN (https://www.jcvi.org/research/provean) and MetaDome web server (https://stuart.radboudumc.nl/metadome/). We visualized alternative allele frequencies using the Integrative Genomics Viewer (IGV) tool, and Sanger sequencing and droplet digital PCR (ddPCR) confirmed the mosaicism in blood and muscle-derived DNA from the unaffected mother’s samples.
RESULTS
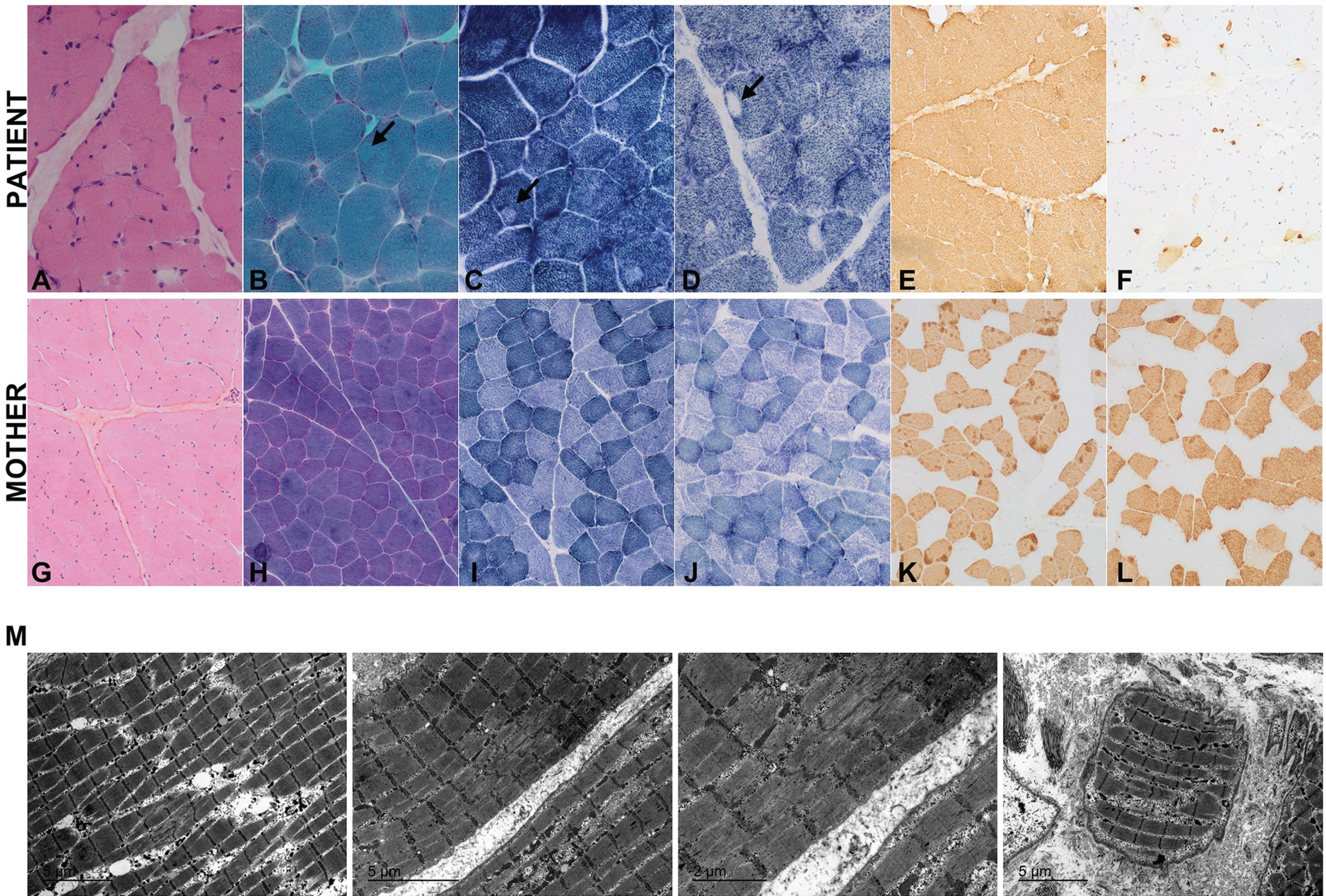
The proband was the first child of non-consanguineous Caucasian parents without a family history of neuromuscular disease. The pregnancy was uneventful, and the child was born at 39 weeks gestation via planned caesarean section. At birth, he weighed 2600g (p4; –1.77 SD) and measured 47 cm in height (p4; –1.78 SD), with a head circumference of 35 cm (p62; + 0.32 SD). The Apgar score was 9 at one minute and 10 at five minutes. Gross motor development was delayed, with head control achieved at 4.5 months of age, independent sitting at 10 months, and walking at 29 months. Examination at 3 years of age showed proximal weakness (4-/5 according to the Medical Research Council (MRC) scale in deltoids, gluteus and psoas), but not facial weakness or ophthalmoparesis. Electromyography of tibialis anterior performed at 3 years of age showed myopathic features, and peripheral nerve conduction studies found no abnormalities. Creatine phosphokinase levels were in the normal range. Whole-body magnetic resonance imaging (WBMRI) performed at 3 years of age showed fatty infiltration in gluteus maximus, anterior and posterior compartments of thighs, and the posterior compartments of both calves. Rectus femoris muscle is relatively spared as related to the vastus lateralis (Figure 1). Muscle biopsy at 5 years of age (right quadriceps) showed variability in fiber size, with some internalized nuclei and occasional small, round fibers. Oxidative stains showed eccentric cores, more often seen in type I fibers. Myosin heavy chain immunohistochemistry confirmed slow fiber predominance (Fig. 1, A-F). Ultrastructural examination showed the presence of Z-line streaming affecting a focal area of the sarcomere (Fig. 1, M). The clinical picture improved over the years, with proximal weakness persisting at the last examination at 8 years of age (4-/5 according to the MRC scale in deltoids, gluteus and psoas). However, the child was able to climb steps without holding on and to rise from the floor without support.
Fig. 1
Muscle biopsy from the right quadriceps of a child with congenital myopathy at 5 years of age (upper panel) and his unaffected mother at 44 years of age (lower panel). A, G: hematoxylin-eosin; B, H: Gomori modified trichrome; C, I: NADH; D, J: SDH; E, K: slow myosin and F, L: fast myosin staining. In staining B, C and D eccentric cores can be observed and are indicated with an arrow. M: ultrastructural examination of the child’s muscle. Although the mother’s biopsy showed a highly preserved muscle, with no variability in fiber size or increase of connective tissue, sarcoplasma with moth-eaten of the intermyofibrillar pattern and eccentric cores were observed in some fibers.
A clinical exome sequencing was performed from the blood-derived DNA of the affected child. The variant RYR1(NM_000540.3):c.14566G > A(p.Ala4856Thr) was identified in heterozygosity. It had strong in-silico predictions of pathogenicity (Table 1) and was classified as “likely pathogenic” following the American College of Medical Genetics and Genomics guidelines (PM1, PP2, PM2, PP3) [15]. The variant affected a highly conserved residue that was predicted to be intolerant to variations (Figure 2) and was absent from population databases [16]. Due to its correlation with the individual’s phenotype, it was considered a relevant candidate variant and segregation analysis was performed. Sanger sequencing analysis of blood-derived DNA showed that it was inherited from his unaffected mother (Fig. 2, A), ruling it out as causative at this point. The mother underwent a comprehensive examination at the age of 44 years conducted by specialists in neuromuscular disorders, revealing no signs of any neuromuscular disease. Muscle MRI or muscle ultrasound were not performed.
Fig. 2
Molecular analysis of the RYR1 genetic variant. (A) The family pedigree and Sanger sequencing analysis. The electropherogram shows that the affected child is heterozygous for the RYR1(NM_000540.3):c.14566G > A variant (arrow). The asymptomatic mother was found to be also heterozygous when blood-derived DNA was analyzed. Nevertheless, the mosaicism is reflected in the small green peak not present in the father. The analysis of a second tissue (muscle-derived DNA) in the mother confirmed the mosaicism (again, a small green peak). (B) IGV screenshot of genome sequencing of the results from blood-derived DNA of the affected child and his mother, including the variant position Chr19:39,071,064 (hg19). Mother mosaicism is observed in the lower AAF (0.23) when compared to the child with congenital myopathy (0.60). (C) Droplet digital PCR results confirmed the mosaic condition of the mother. Abbreviations: AAF, alternative allele frequency; DP, depth.
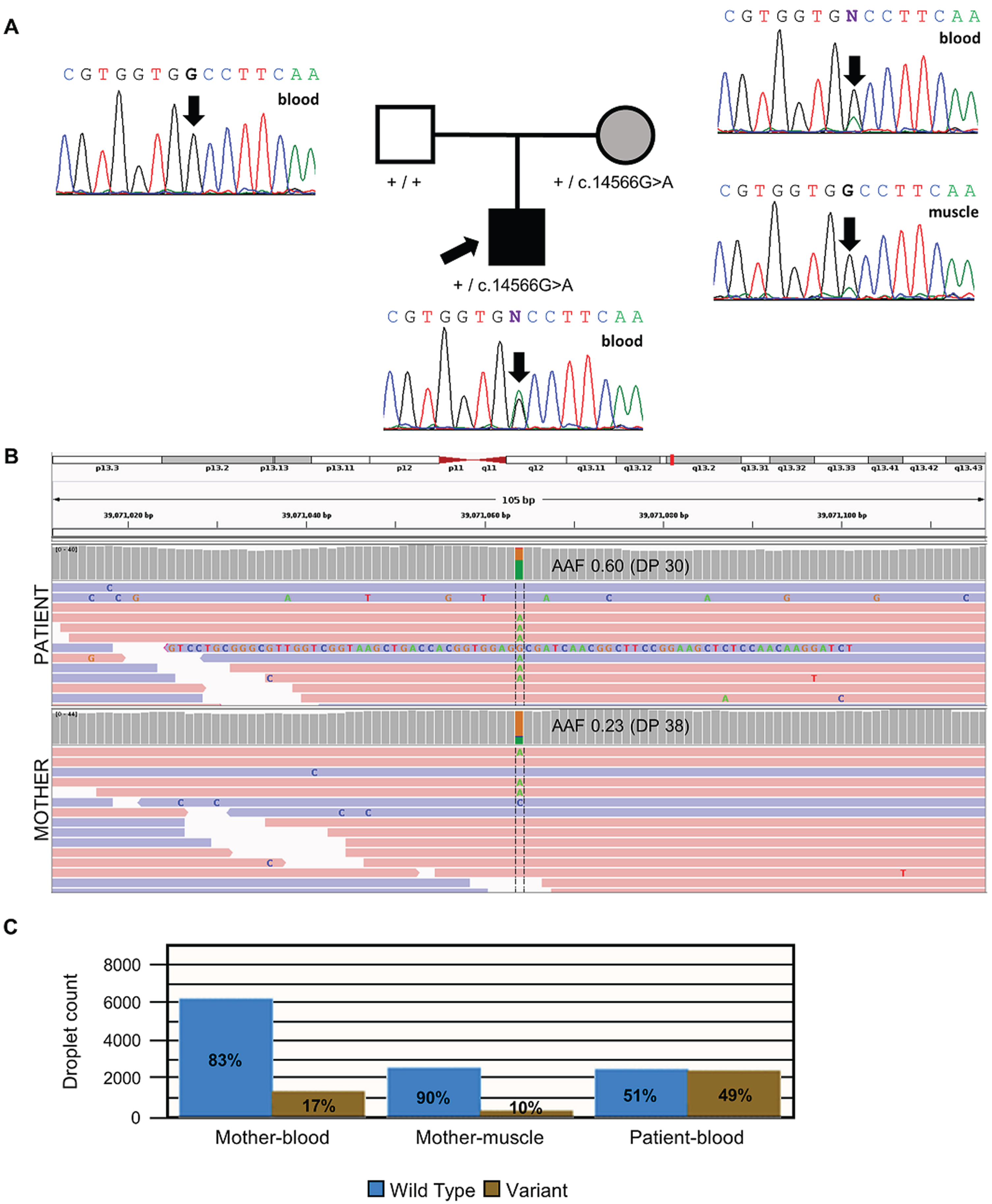
Later, trio genome sequencing was performed, detecting the same candidate variant in RYR1 and no other candidate variants in RYR1 or other known neuromuscular genes. Intronic regions of RYR1 were explored, without detecting any further candidate variant. Trio GS confirmed the heterozygous state of the unaffected mother and the affected child. Given the strong correlation of the clinical and histopathological phenotype of the affected child with an RYR1-CM, we hypothesized the variant might be mosaic in the mother. Therefore, a muscle biopsy was performed on her, revealing highly preserved muscle tissue (Fig. 1, G-L).
While the affected child had an alternative allele frequency (AAF) of 0.60, the mother had an AAF of only 0.23 (Fig. 2, B). This was consistent with a mosaic status in the mother, which was also confirmed by reviewing the Sanger electropherogram peaks corresponding to the amplification from both blood–and muscle-derived DNA (Fig. 2 A).
To confirm the mosaicism hypothesis, quantification through ddPCR from blood and muscle-derived DNA was performed. The analysis revealed that in the maternal blood-derived DNA, only 17% of droplet counts corresponded to the alternative allele, whereas in the individual’s sample, 49% was observed, confirming maternal mosaicism for this variant. Notably, the maternal muscle-derived DNA exhibited an even lower quantification of the alternative allele (Fig. 2, C).
DISCUSSION
Mosaicism is defined as the existence of two or more genetically distinct populations of cells in a particular organism [17], and in recent years has been suggested as the cause of CM in an increasing number of individuals. In apparently sporadic probands, born to asymptomatic parents, mosaicism can also be a hindrance in reaching a genetic diagnosis and a major concern due to its implications for genetic counseling and overall management of the condition.
Moreover, some bioinformatics analysis pipelines may interpret a mosaic variant as a standard heterozygous call if the alternative allelic frequency (AAF) exceeds the established thresholds. This occurrence was observed in the case of the asymptomatic mother, where the default minimum AAF in our analysis was set at 0.2. This situation can pose a problem for interpretation since it is often a reason to dismiss a variant when it is inherited from an asymptomatic parent. When detected, confirmation of the mosaic variant by a quantitative technique and DNA derived from a different tissue is necessary for conclusive interpretation. Another potential scenario may arise when the AAF of mosaic variants is low and does not exceed the established threshold: it might be interpreted that an asymptomatic parent is homozygous wild-type and that the offspring carries a de novo variant.
Since parental mosaicism has been previously reported in individuals with RYR1-CM [4], our study reinforces the idea that this phenomenon may be a challenge for diagnosing RYR1–related CM, since causative variants inherited from asymptomatic parents with mosaicisms are likely to be overlooked.
Our study highlights the importance of phenotype-driven analysis and careful interrogation of the likely relevant variants, even when the initial segregation data can be somehow contradictory.
ACKNOWLEDGMENTS
We are indebted to the “Biobanc de l’Hospital Infantil Sant Joan de Déu per a la Investigació” for the sample and data procurement. AN, BEA, CJ, CO, DNdB, JEE, LCG and HL are members of the European Reference Network for Neuromuscular Diseases –Project ID N° 870177. FP is member of the European Reference Network for Rare Malformation Syndromes, Intellectual and Other Neurodevelopmental Disorders.
ETHICS DECLARATION
Data were collected and analyzed following the ethics guidelines of Hospital Sant Joan de Déu (PIC 98-20). The family provided written informed consent for the study.
FUNDING STATEMENT
This study has been funded by Instituto de Salud Carlos III (ISCIII) through the project “CP22/00141” and co-funded by the European Union. FP is supported by the Spanish State Research Agency (AEI, grant PID2020-114655RB-100), CIBERER (grant ACCI2021-20), the EU Horizon Europe Health Programme, Fundación Ramón Areces and AGAUR (grant 2021-SGR-01610)). JH is supported by the Instituto de Salud Carlos III (grant PI22-00680), IRSJD Carmen de Torres 2023 grant and Torró Solidari-RAC1 i Torrons Vicens. HL receives support as principal investigator from the Canadian Institutes of Health Research (CIHR) for his Foundation Grant (FDN-167281) on Precision Health for Neuromuscular Diseases and Network Grant for NMD4C, the neuromuscular network for Canada, from the Canada Foundation for Innovation for CFI-JELF 38412, and the Canada Research Chairs program for his Canada Research Chair in Neuromuscular Genomics and Health (950-232279). He receives support as co-investigator from CIHR for ProDGNE (Transnational Team Grant), from the European Commission (Grant # 101080249) and the Canada Research Coordinating Committee New Frontiers in Research Fund (NFRFG-2022-00033) for SIMPATHIC, and from the Government of Canada Canada First Research Excellence Fund (CFREF) for the Brain-Heart Interconnectome. This project has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement No. 779257 (Solve-RD). This study makes use of data and tools shared/provided through the RD-Connect GPAP, which received funding originally from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement No. 305444. The remaining authors report no disclosures.
CONFLICT OF INTEREST
The authors declare no conflict09/03/2024s of interest.
DATA AVAILABILITY
Any data not published within the article will be shared by the corresponding author upon reasonable request.
SUPPLEMENTARY MATERIAL
[1] The supplementary material is available in the electronic version of this article:https://dx.doi.org/10.3233/JND-230216.
REFERENCES
[1] | Gineste C , Laporte J . Therapeutic approaches in different congenital myopathies. Curr Opin Pharmacol. (2023) ;68: :102328. doi: 10.1016/J.COPH.2022.102328 |
[2] | Gonorazky HD , Bönnemann CG , Dowling JJ . The genetics of congenital myopathies. Handb Clin Neurol. (2018) ;148: :549–64. doi: 10.1016/B978-0-444-64076-5.00036-3 |
[3] | Natera-de Benito D , Ortez C , Jou C , Jimenez-Mallebrera C , Codina A , Carrera-García L , et al. The Phenotype and Genotype of Congenital Myopathies Based on a Large Pediatric Cohort. Pediatr Neurol. (2021) ;115: :50–65. doi: 10.1016/J.PEDIATRNEUROL.2020.11.002 |
[4] | Marks S , van Ruitenbeek E , Fallon P , Johns P , Phadke R , Mein R , et al. Parental mosaicism in RYR1-related Central Core Disease. Neuromuscul Disord. (2018) ;28: :422–6. doi: 10.1016/J.NMD.2018.02.011 |
[5] | Lawal TA , Todd JJ , Witherspoon JW , Bönnemann CG , Dowling JJ , Hamilton SL , et al. Ryanodine receptor 1-related disorders: An historical perspective and proposal for a unified nomenclature. Skelet Muscle. (2020) ;10: . doi: 10.1186/S13395-020-00243-4 |
[6] | Amburgey K , McNamara N , Bennett LR , McCormick ME , Acsadi G , Dowling JJ . Prevalence of congenital myopathies in a representative pediatric united states population. Ann Neurol. (2011) ;70: :662–5. doi: 10.1002/ANA.22510 |
[7] | MacKenzie AE , Korneluk RG , Zorzato F , Fujii J , Phillips M , Iles D , et al. The human ryanodine receptor gene: Its mapping to 19q13. 1, placement in a chromosome 19 linkage group, and exclusion as the gene causing myotonic dystrophy. Am J Hum Genet.(1990) ;46: ;1082. Available:/pmc/articles/PMC814/?report=abstract. |
[8] | Todd JJ , Sagar V , Lawal TA , Allen C , Razaqyar MS , Shelton MS , et al. Correlation of phenotype with genotype and protein structure in RYR1-related disorders. J Neurol. (2018) ;265: :2506–24. doi: 10.1007/S00415-018-9033-2 |
[9] | Yubero D , Natera-De Benito D , Pijuan J , Armstrong J , Martorell L , Fernàndez G , et al. Molecular Sciences The Increasing Impact of Translational Research in the Molecular Diagnostics of Neuromuscular Diseases. 2021 [cited 19 Jan 2023]. doi: 10.3390/ijms22084274 |
[10] | Klein A , Lillis S , Munteanu I , Scoto M , Zhou H , Quinlivan R , et al. Clinical and genetic findings in a large cohort of patients with ryanodine receptor 1 gene-associated myopathies. Hum Mutat. (2012) ;33: :981–8. doi: 10.1002/HUMU.22056 |
[11] | Campbell IM , Yuan B , Robberecht C , Pfundt R , Szafranski P , McEntagart ME , et al. Parental somatic mosaicism is underrecognized and influences recurrence risk of genomic disorders. Am J Hum Genet. (2014) ;95: :173–82. doi: 10.1016/J.AJHG.2014.07.003 |
[12] | Mohiuddin M , Kooy RF , Pearson CE . De novo mutations, genetic mosaicism and human disease. Front Genet. (2022) ;13: :2732. doi: 10.3389/FGENE.2022.983668/BIBTEX |
[13] | Laurie S , Piscia D , Matalonga L , Corvó A , Fernández-Callejo M , Garcia-Linares C , et al. The RD-Connect Genome-Phenome Analysis Platform: Accelerating diagnosis, research, and gene discovery for rare diseases. Hum Mutat. (2022) ;43: :717–33. doi: 10.1002/HUMU.24353 |
[14] | Zurek B , Ellwanger K , Vissers LELM , Schüle R , Synofzik M , Töpf A , et al. Solve-RD: Systematic pan-European data sharing and collaborative analysis to solve rare diseases. European Journal of Human Genetics. (2021) ;29: :9. 2021;29:1325–31. doi: 10.1038/s41431-021-00859-0 |
[15] | Richards S , Aziz N , Bale S , Bick D , Das S , Gastier-Foster J , et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. (2015) ;17: :405–24. doi: 10.1038/GIM.2015.30 |
[16] | Karczewski KJ , Francioli LC , Tiao G , Cummings BB , Alföldi J , Wang Q , et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) ;581: :434–43. doi: 10.1038/S41586-020-2308-7 |
[17] | Biesecker LG , Spinner NB . A genomic view of mosaicism and human disease. Nat Rev Genet. (2013) ;14: :307–20. doi: 10.1038/NRG3424 |


