
Break Down of the Complexity and Inconsistency Between Levels of Matriglycan and Disease Phenotype in FKRP-Related Dystroglycanopathies: A Review and Model of Interpretation
Abstract
Dystroglycanopathies are a group of muscle degenerative diseases characterized with significant reduction in matriglycan expression critical in disease pathogenesis. Missense point mutations in the Fukutin-related protein (FKRP) gene cause variable reduction in the synthesis of matriglycan on alpha-dystroglycan (α-DG) and a wide range of disease severity. Data analyses of muscle biopsies from patients fail to show consistent correlation between the levels of matriglycan and clinical phenotypes. By reviewing clinical reports in conjunction with analysis of clinically relevant mouse models, we identify likely causes for the confusion. Nearly all missense FKRP mutations retain variable, but sufficient function for the synthesis of matriglycan during the later stage of muscle development and periods of muscle regeneration. These factors lead to a highly heterogenous pattern of matriglycan expression in diseased muscles, depending on age and stages of muscle regeneration. The limited size in clinical biopsy samples from different parts of even a single muscle tissue at different time points of disease progression may well mis-represent the residual function (base-levels) of the mutated FKRPs and phenotypes. We propose to use a simple Multi Point tool from ImageJ to more accurately measure the signal intensity of matriglycan expression on fiber membrane for assessing mutant FKRP function and therapeutic efficacy. A robust and sensitive immunohistochemical protocol would further improve reliability and comparability for the detection of matriglycan.
INTRODUCTION
The dystrophin glycoprotein complex (DGC) is a critical structure of muscle fibers connecting the actin cytoskeleton through the cell membrane to the extracellular matrix (ECM). This linkage is essential to structure stability and prevention from contraction-related fiber damage [1, 2]. The binding of the DGC to ECM components depends crucially on specific O-mannose glycan on alpha-dystroglycan (α-DG) with a repeating disaccharide (-3Xylα1,3-GlcAβ1-)n termed matriglycan. The structure of this matriglycan and the main pathway for its synthesis have now been elucidated [3, 4] (Fig. 1). Matriglycan is a receptor for laminin-G domain-containing proteins such as laminin, agrin, perlecan and neurexin and old-world arenaviruses for infection [5]. Significant reduction of matriglycan (hypoglycosylation) is the common feature of one group of muscular dystrophies, collectively known as dystroglycanopathy, caused by mutations in any genes involved in matriglycan synthesis as well as the dystroglycan gene itself [6–15]. Mutations in the Fukutin-Related Protein (FKRP) gene are among the most common causes of dystroglycanopathy [16, 17]. FKRP functions as a ribitol-5 phosphate (ribitol-5P) transferase, adding ribitol-5P to the glycan core on α-DG and permitting the synthesis of the laminin-binding matriglycan [18–20]. The overwhelming majority of FKRP mutations are missense point mutations within the coding sequence and produce mutant proteins with greatly diminished and variable function depending on the nature of the mutation. Therefore the nature of the mutations in both alleles determines the overall remaining functionality of the gene in individual patients [16, 17, 21]. Different from structural proteins such as dystrophin and dystroglycans, the nature of FKRP as a glycosyltransferase with low levels of expression makes detection of the endogenous FKRP currently impossible by both immunohistochemistry (IHC) and western blot [22]. Fortunately, highly specific antibodies to the matriglycan have long been raised and well established by both detection methods [23]. Since the function of FKRP as a ribitol-5P transferase for matriglycan synthesis has been firmly established [18], expression of matriglycan can be considered the most reliable marker for functionality of FKRP and the levels of matriglycan would be the indicator of remaining function. Furthermore, pathophysiology and significance of matriglycan expression to pathogenesis of all dystroglycanopathies have also been well established. Thus, the levels of matriglycan expression are expected to be correlated to disease severity and could be considered a reliable biomarker for prognostic and therapeutic purposes for FKRP-mutation related diseases [24, 25].
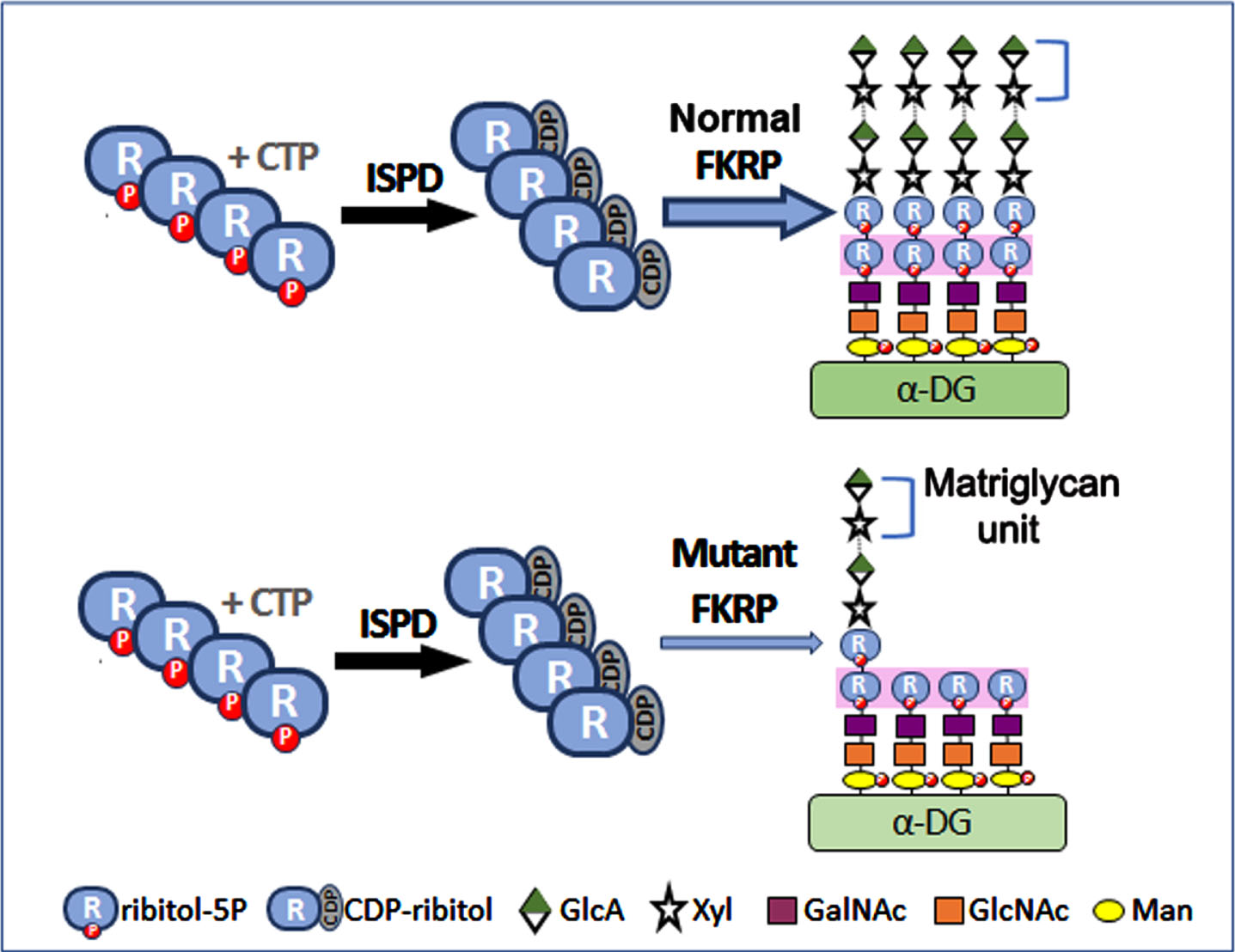
Fig. 1
Effect of Normal and mutant FKRP function on glycosylation of α-DG. The structure of the laminin-binding O-mannosylated glycan of α-DG is delineated with the following chain: (3GlcA-β1-3Xyl-α1) n-3GlcA- β1-4Xyl-Rbo5P-1Rbo5P-3GalNAc- β1-3GlcNAc- β1-4(P-6) Man-1-Thr/ser. POMT1 and POMT2 catalyze the initial O-mannosylation. This is followed by the addition of GlcNAc, GalNAc and the first ribitol-5-phosphate (ribitol-5P) carried out by POMGnT2 (GTDC2), B3GALNT2 and FKTN respectively. FKRP adds a second ribitol-5P. The first Xyl and GlcA are added by TMEM5 and B4GAT1 successively. Finally, LARGE acts as a bifunctional glycosyltransferase having both xylosyltransferase and glucuronyltransferase activities, producing a repeated matriglycan units of 3Xylα1,3-GlcAβ1. ISPD is essential for synthesizing CDP-ribitol, the donor substrate for both FKRP and FKTN. FKRP with missense mutations retains partial function. Abbreviations: CTP, cytidine triphosphate; CDP, cytidine diphosphate; GalNAc, N-acetylgalactosamine; GlcA, glucuronic acid; GlcNAc, N-acetylglucosamine; Man, mannose; P, phosphate; Xyl, xylose; ISPD, isoprenoid synthase domain containing; FKTN, Fukutin; FKRP, Fukutin related protein.
INCONSISTENCY BETWEEN LEVELS OF MATRIGLYCAN EXPRESSION IN PATIENT MUSCLE BIOPSY AND DISEASE SEVERITY
FKRP mutations are associated with huge variation in disease phenotype from mild Limb Girdle Muscular Dystrophy (LGMD) 2I (R9) to more severe Duchenne-like muscular dystrophy and most severe congenital muscular dystrophy (CMD) with brain and eye involvement. There are clear phenotype and genotype correlations [24]. In general, the common mutation c.826C > A (L276I) homozygote is associated with mild LGMD2I, whereas mutations such as c.1343C > T (P448L) and other relatively rare mutations, including nonsense mutations, are associated with severe CMD types [17, 26, 27]. Compound heterozygotes of L276I mutation and CMD-related mutations often present as a Duchenne-like phenotype. Studies of FKRP mutant animal models with the mutations found in humans show a similar correlation between genotype and disease severity in humans [28, 29]. L276I homozygotes present with mild LGMD2I phenotype, whereas P448L homozygotes present a more severe muscular dystrophy. The c.928G > T (E310X) homozygotes are embryonic lethal and all compound heterozygotes showed intermediate phenotypes between the homozygotes of each mutation. Consistently, muscles of L276I homozygotes express widespread, but reduced levels of matriglycan in the majority of muscle fibers [29]. By contrast, muscles from P448L homozygotes often lack clearly detectable matriglycan in almost all fibers except some revertant fibers (RF) expressing matriglycan with signal intensity up to that of normal muscle fibers [28, 29].
However, clear discrepancies exist between the levels of matriglycan expression and disease severity in FKRP-related muscular dystrophy from clinical studies. Brown et al. examined the expression patterns of matriglycan in dystrophic muscles from FKRP mutation-related patients by both immunostaining and western blots [31]. The authors report a clear correlation between the residual expression of matriglycan and the phenotypes. Patients at the severe end of the clinical spectrum (MDC1C) caused by compound heterozygotes with nonsense and/or missense mutations displayed a profound depletion of matriglycan. Patients with a Duchenne-like severity caused by compound heterozygotes with a common L276I and either a missense or a nonsense mutation typically had a moderate reduction in matriglycan. Individuals with the milder form of LGMD2I mostly caused by homozygous L276I mutations showed variable but detectable matriglycan by immunostaining. They conclude that the correlation between a reduction in matriglycan and the clinical phenotype supports the notion that dystroglycan plays a central role in the pathogenesis of the FKRP-related dystroglycanopathy. The only outlier is the observation that relatively abundant remaining matriglycan is detected in muscle biopsy from a MDC1C patient. Jimenez-Mallebrera C et al. conducted a comparative study of α-DG glycosylation in dystroglycanopathies including 10 cases of FKRP-related diseases and concluded that the hypoglycosylation of α-DG, specifically in FKRP-related diseases, does not consistently correlate with clinical severity [32]. This was supported by the observation that one patient (P15 in the original report) diagnosed as LGMD with homozygous p.Pro89Leu mutation without brain involvement has profound depletion of matriglycan. This patient presented as severe Duchenne-like course evidenced by the fact that the biopsy was performed at the age of 2 years. A few longitudinal muscle fibers showed barely detectable membrane staining with IIH6 antibody although high background cytoplasmic staining was also noticed. These muscle fibers show no clear degeneration and are not centrally nucleated, as judged from the image from the original report. Another more recent report by Maisoon Alhamidi et al. studied 25 LGMD2I patients homozygous for the common L276I mutation. They reported that, while matriglycan is reduced in all the patients, no close correlation was detected between clinical severity and matriglycan levels in the group of patients [33]. Specifically, when the patients were grouped into most severe DMD-like (loss of ambulation at the age of 11), typical Becker-like and mild-end LGMD2I, expression of matriglycan follows a trend with disease severity with the DMD-like muscles having severe depletion of matriglycan while the mild-end cases show well preserved matriglycan. However, matriglycan detection varied from 20% to 100% in the same subgroups. For an example, 2 brothers (Case #2 and #19) have disease onset at the age of 12 and 27, but with 100% of the fibers matriglycan positive in case #2, whereas only 30% of the fibers were matriglycan positive in case #19. The study also shows great variation in matriglycan expression between individual patients and within a single biopsy presenting as a mosaic pattern with matriglycan positive and negative fibers. Interestingly, areas of biopsy from cases with moderate to mild disease phenotypes, such as the one (case #17) reported by Maisoon Alhamidi, can show moderate to high signal for matriglycan in fibers of almost normal size without clear fibrosis and degeneration. However, most of these fibers do contain centrally localized nuclei. Inconsistency was also reported by Boito et al. in patients of L276I homozygotes showing variable reduction in matriglycan expression by immunostaining and only trace amount of matriglycan by western blot, even in one asymptomatic patient [34]. All these reports raise fundamental questions about the role matriglycan plays in the pathogenesis of the disease and more importantly, whether the levels of matriglycan can be considered a useful biomarker for prognosis and particularly for evaluation of therapeutic efficacy with the aim to restore the expression of this glycan receptor.
ANIMAL MODEL STUDY PROVIDES EXPLANATION
The inconsistency between matriglycan levels and disease severity is indicated by the association of clearly detectable matriglycan with severe CMD phenotype and barely detectable matriglycan in mild LGMD2I as well as the mosaic pattern in matriglycan expression in muscles with a wide-range of disease severity. To explain these observed variabilities, we clearly need to have a comprehensive understanding of how matriglycan expression is modulated during the disease progression. However, highly random sampling of muscle tissue biopsy with limited size at a single time point from a patient pool with individual mutations affecting the gene’s function differentially provide us with only a fragmented picture of the matriglycan expression in diseased muscles. This leads to the current confusion and often conflicted conclusion, affecting patient care and therapy development. Fortunately, several mouse models have been reported and analysis of whole muscle samples from animal models with clinically relevant FKRP mutations at different time points provide opportunity to establish a comprehensive pattern of matriglycan expression in muscles in relation to disease progression and pathology [35, 36].
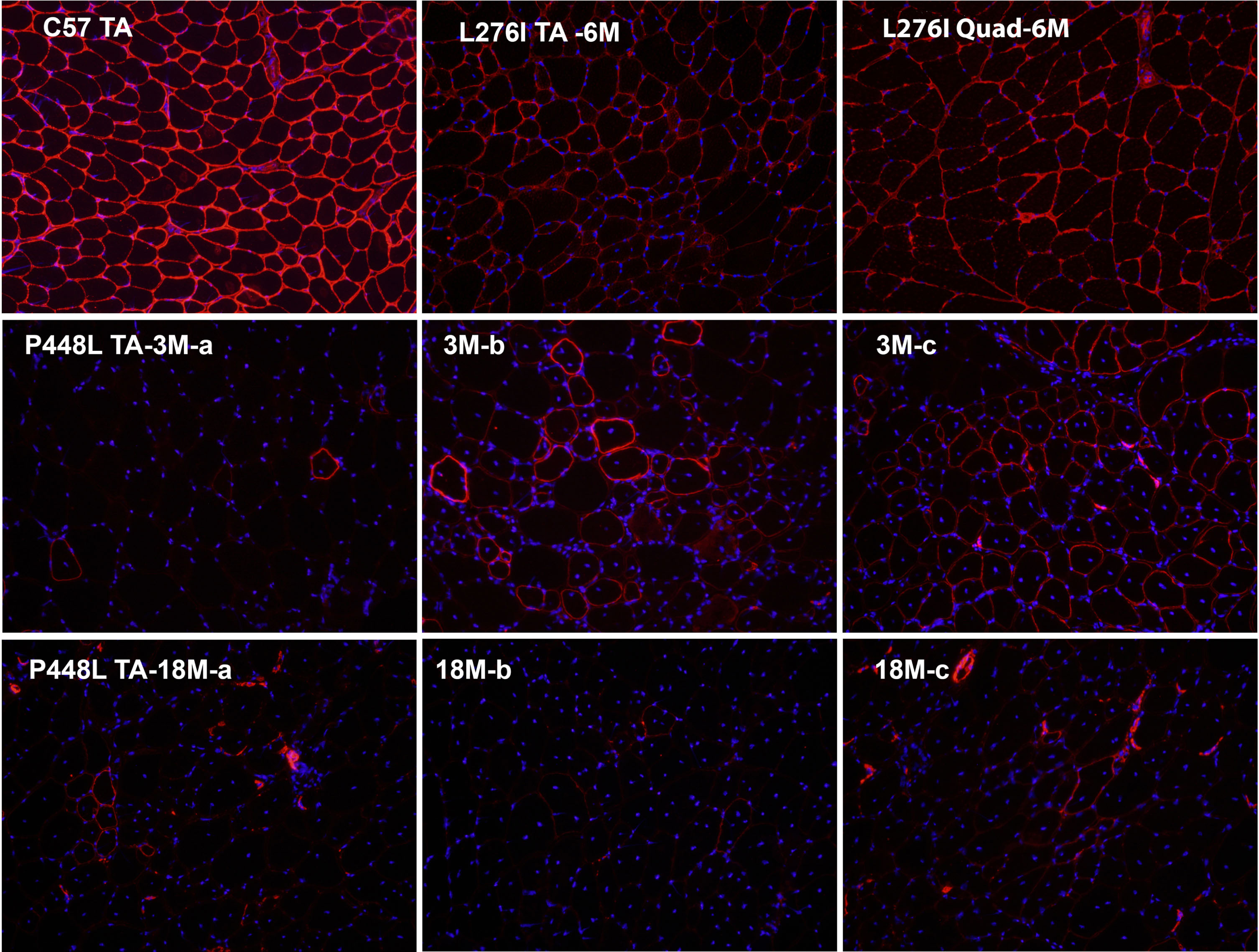
Our group developed several mouse models bearing FKRP mutations as identified in patients with CMD and LGMD2I phenotypes. As expected, the homozygotes of the common L276I mutations with very mild phenotype and only a few central nucleated fibers were observed sporadically within diseased muscles after 3 months of age [30]. Consistently, almost all muscle fibers from the mice express low level, but detectable matriglycan. Few fibers can be identified with higher signal intensity above the generally weak background staining with the IIH6 antibody against matriglycan (Fig. 2). This is consistent with limited degeneration and regeneration in the muscle [29]. However, the homozygote of P448L mutation, clinically associated with a CMD phenotype, shows barely detectable matriglycan by western blot and in nearly all muscle fibers by IHC with the IIH6 antibody from weaning to old age. Interestingly, RFs expressing strong matriglycan start to emerge in the diseased muscles at weaning age and become especially prominent during the first 6 months of life. Most of the RFs present as a small cluster with variable sizes and central nucleation (Fig. 2). Large clusters of muscle fibers of relatively normal size can also be detected with relatively strong, but still weaker than normal matriglycan expression. These large clusters are mostly located in the area near the epimysium and these areas often lack clear degeneration and fibrosis (Fig. 2). However, most of the fibers within the clusters are centrally nucleated, indicating that they are newly regenerated. Restoration of matriglycan expression in diseased muscle is associated with muscle regeneration which is supported by our examination of muscle regeneration after notexin treatment. Beginning from the emergence of small regenerating myofibers within 3-4 days after notexin treatment, strong matriglycan expression is detected with the same signal intensity as that of regenerating normal muscle fibers. Signal intensity remains strong by 2 weeks post treatment and then starts to decline to barely detectable levels by about 4 weeks post treatment (Fig. 3). By this time, most newly regenerated fibers show normal size in diameter, but a proportion of them have lost the central nucleation [36]. We also observed that the number of RF and the size of clusters decrease as the mouse ages (Fig. 2). This is consistent with decreased muscle degeneration and regeneration seen in many muscular dystrophy models [38]. Overall, the presence of RF at different stages of maturation (variation in muscle fiber diameter) and in clusters with different sizes mixed with a large proportion of fibers lacking clearly detectable matriglycan create the mosaic pattern similar to what have been described in clinical biopsy samples from muscles with FKRP mutations. The highly variable distribution of fibers with matriglycan expression in muscles of mouse models provides a solid base for result interpretation of clinical observation. Importantly, human muscles are significantly larger than the mouse equivalent, and the limited size of biopsy can only sample a tiny area of a muscle. This sampled area therefore would most likely contain the part of muscle either with a large cluster of RF, or fibers only with barely detectable matriglycan, or a mixed population of matriglycan positive and negative fibers. From this perspective, the inconsistency between detected matriglycan from individual biopsy and genotype or disease severity is a result of poor representation of a small biopsy to the status of overall matriglycan expression in the musculature and function of the mutant FKRP.
Fig. 2
Matriglycan detection by immunohistochemical staining with IIH6 antibody in Tibialis Anterior (TA) and quadriceps (Quad) muscles from C57 control, FKRP L276I homozygote and P448L homozygote. 6M, 6 months of age. Variation in signal intensity can be viewed between muscles and within the same muscle from a L276I mouse. However, most of the fibers are clearly stained. a, b and c are from the same TA muscle of a P448L mouse. Three different distributions in matriglycan expression are clearly demonstrated in the muscle of 3-month-old P448L mouse: a, only one isolated revertant fiber is viewed in the entire field; b, a cluster of revertant fibers with significant variation in staining intensity in matriglycan and in fiber sizes; c, a large cluster of fibers with most of them clearly stained with the antibody IIH6 and most of the fibers normal sized, but with central nucleation. The differential distribution of matriglycan expression is also perpetuated in muscle of 18-month-old P448L mouse, though with a reduced number of positive fibers. IIH6 antibody is visualized by AlexaFluor 594 goat anti-mouse IgM. Original magnification×20. Nuclei are stained with DAPI.
The appearance of RFs expressing detectable to near normal levels of matriglycan in most muscle biopsy from clinical samples and in our P448L mutant mouse with CMD phenotype suggests that almost all mutant FKRPs retain sufficient function for matriglycan synthesis in muscle during the early stage of fiber regeneration and maturation [36]. Our studies with the P448L mutant model also demonstrate that matriglycan expression is retained during the late stage of muscle development until soon after birth. At post-natal day 1, matriglycan level in the P448L mutant muscle is similar to the muscle from normal mice of the same age. However, the level gradually decreases and becomes hardly detectable by post-natal day 7 with both IHC and western blot [39]. This observation may partially explain the detection of a significant amount of matriglycan in muscles from infants with FKRP mutations although the age for the loss of matriglycan expression in developing human muscle remains to be determined. Retained function of mutant FKRP was directly demonstrated with the delivery of P448L mutant FKRP as a transgene by AAV gene therapy [40].
PROPOSAL FOR MATRIGLYCAN ANALYSIS IN DISEASED MUSCLES FOR GENOTYPE-PHENOTYPE ANALYSIS AND THERAPEUTIC EFFICACY EVALUATION
The data from both animal model and human study clearly suggest that simple measurement of either the total amount of matriglycan or the percentage of matriglycan positive fibers from a small biopsy does not reliably reflect the base level of matriglycan expression and functionality of mutant FKRP. How then should matriglycan expression in biopsy from muscles of FKRP mutations be analyzed and results interpreted? Here we propose to take the following factors into consideration. First, RFs are the consequence of degeneration and regeneration and have been observed in a majority of muscle biopsy samples from CMD to LGMD2I phenotypes. The number of these fibers does not represent the base level of matriglycan expression and functionality of mutant FKRP, and therefore should be excluded from analysis of genotype-phenotype association and for disease progression prediction. The clear association of RFs with muscle regeneration and later stage of muscle development provides useful morphological and biochemical characteristics for determination. RFs are generally smaller in sizes with central nucleation and/or expressing neonatal or embryonic myosin. These 3 features should be used to define RFs. Expression of neonatal myosin and the presence of central nucleation can be conveniently achieved by co-staining or staining in serial sections with antibody to matriglycan (Fig. 3). Considering RFs may express matriglycan longer than 1 month from the start of regeneration and the clustering nature of regeneration, matriglycan positive fibers without central nucleation and neonatal myosin, but within and surrounding a RF cluster, should also be considered as RF and excluded from analysis. Nevertheless, the number and signal intensity of those RFs may be analyzed separately for disease progression alongside the measurement in base level of matriglycan.
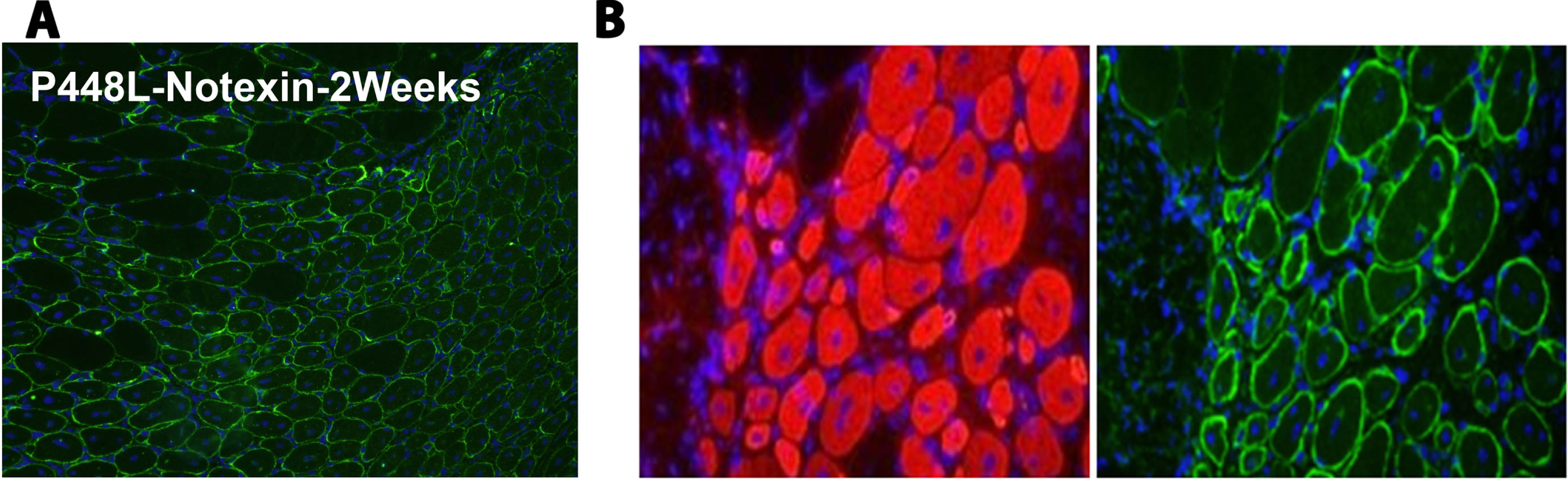
Fig. 3
Matriglycan detection by immunohistochemical staining with IIH6 antibody in notexin-treated Tibialis Anterior (TA) muscles from P448L homozygote mouse. (A) IIH6 staining of TA from P448L homozygote 2 weeks post notexin treatment. The majority of the muscle fibers express strong matriglycan and remain centrally nucleated. (B) Staining of embryonic myosin heavy chain (F1-652, Developmental Studies Hybridoma Bank, Iowa City, IA) (left) and matriglycan (right) in serial sections from TA muscle 2 weeks after notexin treatment. IIH6 (Millipore, Temecula, CA) and F1-652 are visualized by AlexaFluor 488 goat anti-mouse IgM and AlexaFluor 594 goat anti-mouse IgG respectively. Original magnification×20. Nuclei are stained with DAPI.
The second important factor impeding detection of weak matriglycan in diseased muscles with FKRP mutation is the background staining from muscle degeneration, fibrosis, fat deposition and inflammatory response in the biopsy area. Increasing background staining is always correlated with a decrease in sensitivity of specific signal detection. With the currently available antibodies to matriglycan, IIH6 and VIA4, these elements within a biopsy sample produce weak but visible overall background staining such as presented in case #16 and #17 reported by Jimenez-Mallebrera C et al. [32]. Such staining does not cause difficulty for strong signal identification but is sufficient to mask any weak specific staining on the cell membrane with the available antibodies to matriglycan. Difficulty in staining assessment and comparison is also closely related to the available antibodies, IIH6 and VIA4, which are known to be quite variable in affinity from batch to batch and from different sources. Furthermore, different procedures employed by different groups also affect sensitivity and background levels, leading to incomparable results between different studies. We also found that tissue preservation and storage can reduce the sensitivity for the detection of matriglycan, especially when its levels are low as in the FKRP mutant muscles. Therefore, a simple and robust procedure needs to be established and widely adopted for meaningful analysis and comparison.
An appropriate means for measuring the base levels of matriglycan expression in diseased muscles should represent the functionality of mutant FKRP and can be used to reliably predict disease progression as well. Based on the analysis above, we propose the following protocol as a principle for base levels of matriglycan determination. First, properly stored samples are cut into same thickness with normal control muscle samples and the freshly cut sections (within 2 weeks and stored in -80 freezer) are stained with the available antibody against matriglycan together with an antibody against neonatal or embryonic myosin. After the staining, the sections should be counter-stained with nuclear dye such as DAPI for analysis. After confirming the positive staining in normal control sections and acceptable background staining in diseased muscles with fibrosis, infiltration, or degenerating fibers software such as NIH ImageJ can be used to measure the signal intensity of each muscle fiber excluding those RFs as described earlier. We recommend using the Multi Point tool in NIH ImageJ for measurement of signal intensity. At least 100 muscle fibers should be measured for a segment of each fiber membrane representing the general intensity of the fiber membrane (illustrated in Fig. 4). The size and shape of the Multi Point tool can be adjusted to fit the width of the stained membrane. Measuring signal intensity in a segment has its advantages as it can avoid segments of high background in individual fibers due to damage and other artifacts close to the membrane. This is important for diseased muscles as their higher background often makes the overall staining look stronger in severely damaged muscles than better preserved muscles. Signal intensity on fiber membrane less than that of cytoplasm often indicates a poor quality of the tissue, or degenerating fibers. Such fibers should not be included for analysis. Since each measurement by the Multi Point tool is recorded precisely, it constitutes a verifiable document. The excel data can then be analyzed with appropriate means as illustrated in Fig. 4. Again, RFs should be counted separately for analysis.
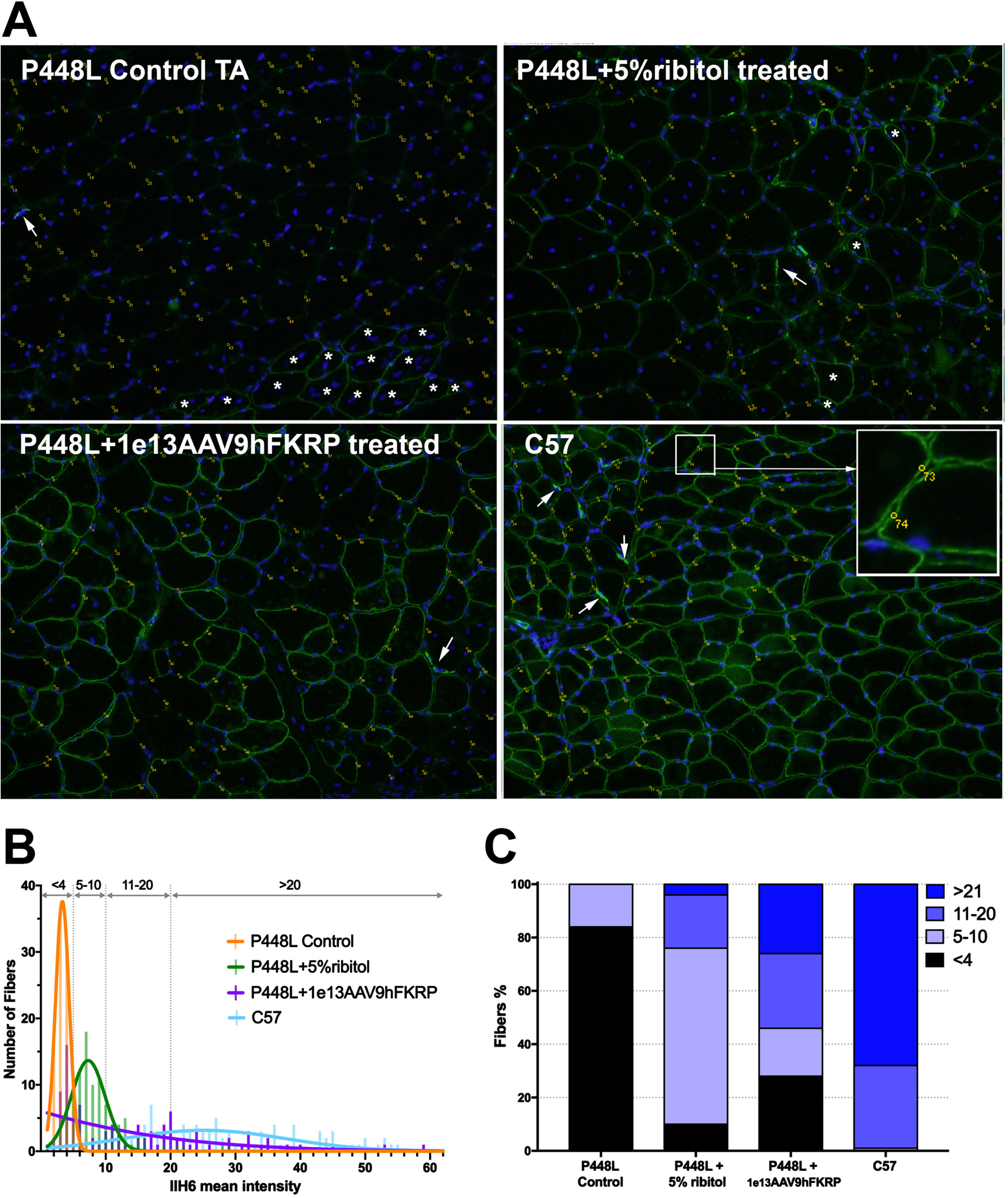
Fig. 4
Illustration of matriglycan measurement by ImageJ Multi Point tool for comparison. (A) Matriglycan is detected with IIH6 antibody and visualized with Goat anti-mouse IgM Alexa 488 (green) and nuclei are counterstained with DAPI (blue). Tibialis Anterior (TA) muscles from control P448L mutant mouse (top left), P448L mutant mouse treated with 5% ribitol for 6 months (top right), P448L mutant mouse treated with AAV9hFKRP at 1e13vg/KG for 6 months (bottom left), C57 (bottom right). Yellow dots and numbers indicate the measured area of the membrane. Arrows indicate the staining of neuromuscular junction, and * indicates the RFs expressing strong matriglycan signal with central nucleation. Amplified area in the square illustrates the measured sites in circles on fiber membrane. (B) Distribution of matriglycan signal intensity measured from the above images. Bin width = 1. A Gaussian nonlinear regression curve is shown for each treatment and untreated mice. Dotted lines represent thresholds for IIH6 intensity < 4, 5 to 10, 11 to 20, and > 21 and graphed as a percentage of fibers in “C”. (C) Staked-bar graph representing the percentage of fibers with IIH6 fluorescence intensity showed in “B”. The detail of the measurement is listed in the Excel file in the Supplementary data. For illustration only with no statistical analysis performed.
The importance of correctly evaluating the base levels of matriglycan expression in diseased muscles also extends to assessing efficacy of experimental therapies such as gene therapy. Currently, two clinical trials for FKRP mutation related LGMD2I have dosed patients with AAV9 virus expressing normal human FKRP protein (ClinicalTrials.gov. GNT0006 Gene Therapy Trial in Patients With LGMDR9; A Study to Evaluate the Safety of LION-101 in Subjects With Genetic Confirmation of LGMD2I/R9). The aim of the trials is to provide sufficient FKRP to restore the expression of matriglycan, therefore preventing disease progression. While the treatment is expected to show a significant restoration in matriglycan expression, results from animal model studies with the same virus serotype and expression vector also suggest a high likelihood that the expression of matriglycan will be heterogenous, especially when dose is limited by potential toxicity [41]. The patients enrolled in these trials are ambulant LGMD2I with relatively slow progression. The muscle biopsies from such patients are expected to also have a mosaic pattern of matriglycan expression with fibers of very weak matriglycan and clusters of RFs with strong matriglycan expression. When matriglycan expression is equal to or higher than normal levels by western blot after AAV gene therapy such data can be considered strong evidence of therapeutic effect. However, it will be difficult to draw conclusions when the levels of matriglycan after treatment are lower than normal muscle. In this instance, immunostaining with specific antibody in combination with muscle histology could be of significant help. The majority of fibers with strong positive signal, but lacking regeneration markers such as central nucleation and neonatal myosin would indicate an enhanced basal level of matriglycan and constitute strong evidence supporting the efficacy of gene therapy. Furthermore, matriglycan levels may be lower by western blot in muscle biopsy after treatment than that in pretreatment biopsy due to sampling variation. Therefore comparison in basal levels of matriglycan before and after treatment by immunostaining could still be valuable for assessing therapeutic effect [42].
CONCLUSION AND FUTURE DIRECTIONS
Mutations in the FKRP gene cause variable and significant reduction in function for matriglycan synthesis, ranging from hardly to clearly detectable matriglycan in muscles. The highly heterogenous matriglycan expression in diseased muscles reflects the variable degrees of function retained by individual mutation and is more prominently imposed by the appearance of RFs in association with regeneration, later stages of muscle development and the limited sensitivity of detection methods. The small size in clinic muscle biopsy poorly represents the overall status of musculature and is the source of inconsistency between the biomarker and disease severity, although other disease modifying factors and possible FKRP functions other than ribitol-5P-transferase could also affect disease progression [43]. While western blot remains essential for quantification of matriglycan, a robust and sensitive IHC protocol is required for meaningful measurement of base level matriglycan for comparison and for efficacy evaluation of experimental therapies. We recommend use of the Multi Point tool from ImageJ to measure membrane signal intensity for base level matriglycan expression and for comparison before and after experimental treatments, although validation and further refinement may be required for clinical sample evaluation. The same protocol can be adapted for measurement of other membrane proteins detected by immunostaining.
AUTHOR CONTRIBUTIONS
Conceptualization, Q.L.L.; Methodology, M.P.C.; A.B; M.C.H. and Q.L.L.; Writing - Original Draft, Q.L.L; Writing -Review & Editing, M.P.C., A.B., M.C.H. and Q.L.L.; Funding Acquisition, Q.L.L.
ACKNOWLEDGMENTS
The authors have no acknowledgments to report.
FUNDING
This work was supported by the Carolinas Muscular Dystrophy Research Endowment at the Atrium Health Foundation.
CONFLICT OF INTEREST
The authors declare no conflict of interest
DATA AVAILABILITY
The data supporting the findings of this study are available within the article and/or its supplementary material.
REFERENCES
[1] | Endo T . Mammalian O-mannosyl glycans: Biochemistry and glycopathology. Proc Jpn Acad Ser B Phys Bio Sci (2019) ;95: :39–51. |
[2] | Yoshida-Moriguchi T , Campbell KP . Matriglycan: A novel polysaccharide that links dystroglycan to the basement membrane. Glycobiology (2015) ;25: :702–13. |
[3] | Kanagawa M . Dystroglycanopathy: From Elucidation of Molecular and Pathological Mechanisms to Development of Treatment Methods. Int J Mol Sci (2021) ;22: :13162. |
[4] | Walimbe AS , Okuma H , Joseph S , Yang T , Yonekawa T , Hord JM et al. POMK regulates dystroglycan function via LARGE1-mediated elongation of matriglycan. ELife (2020) ;9: :e61388. |
[5] | Kunz S , Rojek JM , Perez M , Spiropoulou CF , Oldstone MBA . Characterization of the interaction of lassa fever virus with its cellular receptor α-dystroglycan. J Virol (2005) ;79: :5979–87. |
[6] | Manya H , Chiba A , Yoshida A , Wang XH , Chiba Y , Jigami Y et al. Demonstration of mammalian protein O-mannosyltransferase activity: Coexpression of POMT1 and POMT2 required for enzymatic activity. Proc Natl Acad Sci USA (2004) ;101: :500–5. |
[7] | Yoshida-Moriguchi T , Willer T , Anderson ME , Venzke D , Whyte T , Muntoni F et al. SGK196 is a glycosylation-specific O-mannose kinase required for dystroglycan function. Science (2013) ;341: :896–9. |
[8] | Manzini MC , Tambunan DE , Hill RS , Yu TW , Maynard TM , Heinzen EL et al. Exome sequencing and functional validation in zebrafish identify GTDC2 mutations as a cause of Walker-Warburg syndrome. Am J Hum Genet (2012) ;91: :541–7. |
[9] | Hiruma T , Togayachi A , Okamura K , Sato T , Kikuchi N , Kwon Y et al. A novel human beta1,3-N-acetylgalactosaminyltransferase that synthesizes a unique carbohydrate structure, GalNAcbeta1-3GlcNAc. J Biol Chem (2004) ;279: :14087–95. |
[10] | Manya H , Yamaguchi Y , Kanagawa M , Kobayashi K , Tajiri M , Akasaka-Manya K et al. The Muscular Dystrophy Gene TMEM5 Encodes a Ribitol beta1,4-Xylosyltransferase Required for the Functional Glycosylation of Dystroglycan. J Biol Chem (2016) ;291: :24618–27. |
[11] | Willer T , Inamori K , Venzke D , Harvey C , Morgensen G , Hara Y et al. The glucuronyltransferase B4GAT1 is required for initiation of LARGE-mediated alpha-dystroglycan functional glycosylation. ELife (2014) ;3: :e03941. |
[12] | Inamori K , Yoshida-Moriguchi T , Hara Y , Anderson ME , Yu L , Campbell KP . Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science (2012) ;335: :93–6. |
[13] | Hara Y , Balci-Hayta B , Yoshida-Moriguchi T , Kanagawa M , Beltrán-Valero de Bernabé D , Gündesli H , et al. A Dystroglycan Mutation Associated with Limb-Girdle Muscular Dystrophy. N Engl J Med (2011) ;364: :939–46. |
[14] | Riemersma M , Froese DS , vanTol W , Engelke UF , Kopec J , van Scherpenzeel M et al. Human ISPD Is a cytidyltransferase required for dystroglycan O-mannosylation. Chemistry & Biology (2015) ;22: :1643–52. |
[15] | Longman C , Brockington M , Torelli S , Jimenez Mallebrera C , Kennedy C , Khalil N et al. Mutations in the human LARGE gene cause MDC1D, a novel form of congenital muscular dystrophy with severe mental retardation and abnormal glycosylation of alpha-dystroglycan. Hum Mol Genet (2003) ;12: :2853–61. |
[16] | Brockington M , Blake DJ , Prandini P , Brown SC , Torelli S , Benson MA et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet (2001) ;69: :1198–209. |
[17] | Beltran-Valero de Bernabé D , Voit T , Longman C , Steinbrecher A , Straub V , Yuva Y et al. Mutations in the FKRP gene can cause muscle-eye-brain disease and Walker-Warburg syndrome. J Med Genet (2004) ;41: :e61. |
[18] | Kanagawa M , Kobayashi K , Tajiri M , Manya H , Kuga A , Yamaguchi Y et al. Identification of a Post-translational Modification with Ribitol-Phosphate and Its Defect in Muscular Dystrophy. Cell Rep (2016) ;14: :2209–23. |
[19] | Praissman JL , Willer T , Sheikh MO , Toi A , Chitayat D , Lin YY et al. The functional O-mannose glycan on alpha-dystroglycan contains a phospho-ribitol primed for matriglycan addition. ELife (2016) ;5: :e14473. |
[20] | Gerin I , Ury B , Breloy I , Bouchet-Seraphin C , Bolsée J , Halbout M et al. ISPD produces CDP-ribitol used by FKTN and FKRP to transfer ribitol phosphate onto alpha-dystroglycan. Nat Commun (2016) ;7: :11534–543. |
[21] | Michele DE , Barresi R , Kanagawa M , Saito F , Cohn RD , Satz JS et al. Post-translational disruption of dystroglycan-ligand interactions in congenital muscular dystrophies. Nature (2002) ;418: :417–22. |
[22] | Vannoy CH , Xiao W , Lu P , Xiao X , Lu QL . Efficacy of Gene Therapy Is Dependent on Disease Progression in Dystrophic Mice with Mutations in the FKRP Gene. Mol Ther Methods & clinical development (2017) ;5: :31–42. |
[23] | Ervasti JM , Campbell KP . A role for the dystrophin-glycoprotein complex as a transmembrane linker between laminin and actin. J Cell Biol (1993) ;122: :809–23. |
[24] | Krag TO , Hauerslev S , Sveen ML , Schwartz M , Vissing J . Level of muscle regeneration in limb-girdle muscular dystrophy type 2I relates to genotype and clinical severity. Skeletal Muscle (2011) ;1: :31–40. |
[25] | Sheikh MO , Capicciotti CJ , Liu L , Praissman J , Ding D , Mead DG et al. Cell surface glycan engineering reveals that matriglycan alone can recapitulate dystroglycan binding and function. Nat Commun (2022) ;13: :3617–29. |
[26] | Esapa CT , Benson MA , Schröder JE , Martin-Rendon E , Brockington M , Brown SC et al. Functional requirements for fukutin-related protein in the Golgi apparatus. Hum Mol Genet (2002) ;11: :3319–31. |
[27] | Frosk P , Greenberg CR , Tennese AAP , Lamont R , Nylen E , Hirst C et al. The most common mutation in FKRP causing limb girdle muscular dystrophy type 2I (LGMD2I) may have occurred only once and is present in Hutterites and other populations. Hum Mutat (2005) ;25: :38–44. |
[28] | Chan YM , Keramaris-Vrantsis E , Lidov HG , Norton JH , Zinchenko N , Gruber HE et al. Fukutin-related protein is essential for mouse muscle, brain and eye development and mutation recapitulates the wide clinical spectrums of dystroglycanopathies. Hum. Mol. Genet (2010) ;19: :3995–4006. |
[29] | Blaeser A , Keramaris E , Chan YM , Sparks S , Cowley D , Xiao X et al. Mouse models of fukutin-related protein mutations show a wide range of disease phenotypes. Hum Genet (2013) ;132: :923–34. |
[30] | Qiao C , Wang CH , Zhao C , Lu P , Awano H , Xiao B et al. Muscle and Heart Function Restoration in a Limb Girdle Muscular Dystrophy 2I (LGMD2I) Mouse Model by Systemic FKRP Gene Delivery. Mol Ther (2014) ;22: :1890–9. |
[31] | Brown SC , Torelli S , Brockington M et al. Abnormalities in alpha-dystroglycan expression in MDC1C and LGMD2I muscular dystrophies. Am J Pathol (2004) ;164: :727–37. |
[32] | Jimenez-Mallebrera C , Torelli S , Feng L , Kim J , Godfrey C , Clement E et al. A comparative study of alpha- dystroglycan glycosylation in dystroglycanopathies suggests that the hypoglycosylation of alpha-dystroglycan does not consistently correlate with clinical severity. Brain Pathol (2009) ;19: :596–611. |
[33] | Alhamidi M , Brox V , Stensland E , Liset M , Lindol S , Nilssen Q . Limb girdle muscular dystrophy type 2I: No correlation between clinical severity, histopathology and glycosylated alpha-dystroglycan levels in patients homozygous for common FKRP mutation. Neuromuscul Disord (2017) ;27: :619–26. |
[34] | Boito CA , Melacini P , Vianello A , Prandini P , Gavassini BF , Bagattin A et al. Clinical and molecular characterization of patients with limb-girdle muscular dystrophy type 2I. Arch Neurol (2005) ;62: :1894–9. |
[35] | Whitmore C , Fernandez-Fuente M , Booler H , Parr C , Kavishwar M , Ashraf A et al. The transgenic expression of LARGE exacerbates the muscle phenotype of dystroglycanopathy mice. Hum Mol Genet (2014) ;23: :1842–55. |
[36] | Gicquel E , Maizonnier N , Foltz SJ , Martin WJ , Bourg N , Svinartchouk F et al. AAV-mediated transfer of FKRP shows therapeutic efficacy in a murine model but requires control of gene expression. Hum Mol Genet (2017) ;26: :1952–65. |
[37] | Awano N , Blaeser A , Keramaris E , Xu L , Tucker J , Wu B et al. Restoration of Functional Glycosylation of alpha-Dystroglycan in FKRP Mutant Mice Is Associated with Muscle Regeneration. The American Journal of Pathology (2015) ;185: :2025–37. |
[38] | Echigoya Y , Lee J , Rodrigues M , Nagata T , Tanihata J , Nozohourmehrabad A et al. Mutation types and aging differently affect revertant fiber expansion in dystrophic mdx and mdx52 mice. PLoS One.e (2013) ;8: :69194. |
[39] | Keramaris E , Lu PJ , Tucker J , Lu QL . Expression of glycosylated alpha-dystroglycan in newborn skeletal and cardiac muscles of fukutin related protein (FKRP) mutant mice. Muscle & Nerve (2017) ;55: :582–90. |
[40] | Tucker JD , Lu PJ , Xiao X , Lu QL . Overexpression of Mutant FKRP Restores Functional Glycosylation and Improves Dystrophic Phenotype in FKRP Mutant Mice. Mol Ther Nucleic Acids (2018) ;11: :216–27. |
[41] | Vannoy CH , Leroy V , Lu QL . Dose-Dependent Effects of FKRP Gene Replacement Therapy on Functional Rescue and Longevity in Dystrophic Mice Mol Ther Methods Clin Dev (2018) ;11: :106–20. |
[42] | Cataldi MP , Vannoy CH , Blaeser A , Tucker JD , Leroy V , Rawls R , Killilee J et al. Improved efficacy of FKRP AAV gene therapy by combination with ribitol treatment for LGMD2I. Mol Ther. 2023;S1525-0016(23)00599-3. |
[43] | Wood AJ , Lin CH , Li M , Nishtala K , Alaei S , Rossello F et al. FKRP-dependent glycosylation of fibronectin regulates muscle pathology in muscular dystrophy. Nat Commun (2021) ;12: :2951–60. |


